

Summary
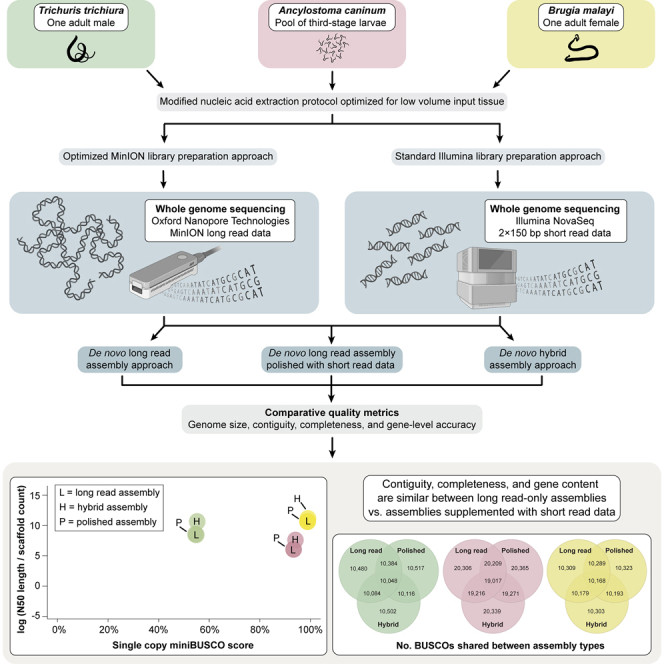
In this study, we assessed the quality of de novo genome assemblies for three species of parasitic nematodes (Brugia malayi, Trichuris trichiura, and Ancylostoma caninum) generated using only Oxford Nanopore Technologies MinION data. Assemblies were compared to current reference genomes and against additional assemblies that were supplemented with short-read Illumina data through polishing or hybrid assembly approaches. For each species, assemblies generated using only MinION data had similar or superior measures of contiguity, completeness, and gene content. In terms of gene composition, depending on the species, between 88.9 and 97.6% of complete coding sequences predicted in MinION data only assemblies were identical to those predicted in assemblies polished with Illumina data. Polishing MinION data only assemblies with Illumina data therefore improved gene-level accuracy to a degree. Furthermore, modified DNA extraction and library preparation protocols produced sufficient genomic DNA from B. malayi and T. trichiura to generate de novo assemblies from individual specimens.
Subject areas: genomics, techniques in genetics, parasitology, genomic analysis, biological science instrumentation
Graphical abstract
Highlights
-
•
Reference-quality helminth genomes are assembled using only MinION data
-
•
Polishing MinION genomes with Illumina data moderately improves gene-level accuracy
-
•
Modified protocols allow for whole genome sequencing and assembly from single helminths
Genomics; Techniques in genetics; Parasitology; Genomic analysis; Biological science instrumentation
Introduction
Parasitic nematodes represent an enormous burden of disease. It is estimated that 1.5 billion people are infected with soil-transmitted helminths, while approximately 120 million and 15 million people suffer from filariasis and onchocerciasis, respectively.1 Helminths are responsible for nearly 12 million disability-adjusted life years lost annually.1 In addition to the morbidity caused by nematodes themselves, infection also exacerbates the disease burden of other existing conditions such as malaria, HIV, and tuberculosis, and can reduce immunological response to important vaccines.2,3,4,5,6 Reference genomes for parasitic nematodes have proven to be an invaluable resource for increasing our understanding of helminth biology, treatment, and control. For example, the landmark comparative genomic study by the International Helminth Genomes Consortium7 analyzed dozens of complete genomes representing all major helminth lineages. This extensive dataset allowed for the identification of multiple gene family expansions in various groups that are targets for novel drug and vaccine development. Additional more focused comparative genomic work has revealed differences between helminth and host metabolic pathways and extracellular vesicle protein content that represent potential druggable targets, evidence for positive selection in gene families that are uniquely expanded in flatworms and implicated in the biology of endoparasitism, conserved immunomodulatory proteins that are potential vaccine targets for soil-transmitted helminths, and patterns of coevolution between parasitic roundworms and their mammalian hosts.8,9,10,11,12,13,14 These advances further highlight helminth reference genomes as indispensable tools for comparative biological and biomedical research.
Reductions in the cost and improved ease of access of next-generation sequencing (NGS) have made the assembly of whole genomes from helminths more feasible.15 As a result, reference genomes have now been generated for the majority of species of medical and veterinary importance.16,17 Most of these species, however, are still represented by only a single reference genome. These solitary references are often generated from laboratory models that have been maintained for decades and therefore cannot represent the biological diversity observed in natural helminth populations.18,19 Generating multiple reference genomes per species from contemporary and geographically disparate populations would begin to capture the genomic diversity of helminths, and allow for characterization of differences in, for example, genome organization, gene copy number and structural variants, and adaptation to local selective pressures. When generating a genome from a natural population, using genomic DNA from one specimen is ideal to avoid complications in the assembly process. Therefore, streamlined laboratory and computational workflows that allow high-quality assemblies to be generated from an individual specimen using a single data source would be invaluable. The Oxford Nanopore Technologies (ONT) MinION is a single molecule sequencing platform capable of generating long reads ideal for de novo genome assembly. Lower read-level accuracy of ONT data have previously required long-read assemblies to be error-corrected with more accurate short-read data. Recent updates to ONT chemistries have decreased the error rate of MinION reads, allowing for the potential to generate helminth reference genomes using only long-read data. In fact, Lee et al. (2023) were able to generate complete genomes from individual free-living nematodes using ONT and transcriptome data.20 However, similar approaches have not been explored in parasitic nematodes.
This study aims to assess the contiguity, completeness, gene content, and gene composition of whole genome de novo assemblies of parasitic nematodes using MinION data only compared to assemblies supplemented with accurate short-read Illumina data through polishing and/or hybrid assembly approaches. Three species that collectively span the breadth of parasitic nematode diversity were utilized for sequencing: the filarial worm Brugia malayi (Spirurina), the whipworm Trichuris trichiura (Trichinellida), and the dog hookworm Ancylostoma caninum (Rhabditina). Assemblies generated using only ONT MinION data were compared to reference genomes for each species, as well as to assemblies supplemented with short-read Illumina data through polishing or hybrid assembly approaches that were generated as a part of this study. This work highlights a straightforward approach for generating high-quality de novo genome assemblies from parasitic nematodes using only data generated from the ONT MinION.
Results
Data generation
Total gDNA was successfully extracted from a single adult worm for each of B. malayi and T. trichiura, and from a pool of L3 larvae for A. caninum. For each species, both MinION and Illumina sequencing libraries were prepared from the same source of gDNA. The bioinformatic pipeline used to generate each assembly type is outlined in Figure 1. Total amounts sequence data generated for each species prior to basecalling and/or quality control were as follows: ∼15.9 Gb (MinION) and ∼7.9 Gb (Illumina) for B. malayi; ∼26.8 Gb (MinION) and ∼4.5 Gb (Illumina) for T. trichiura; and ∼33.1 Gb (MinION) and ∼20.8 Gb (Illumina) for A. caninum (Table S1). For all three species, a median quality score value of >Q16 was achieved for MinION reads, and for MinION reads aligned to final MinION data only assemblies (Figure S1). Values for average depth of coverage of quality-controlled reads mapped to species-specific reference assemblies were as follows: 124.85× (MinION) and 82.95× (Illumina) for B. malayi; 249.91× (MinION) and 48.58× (Illumina) for for T. trichiura; and 49.42× (MinION) 38.64× (Illumina) for A. caninum.
Figure 1.

Bioinformatic pipelines used to generate de novo whole genome assemblies
The MinION data only pipeline is provided at left and the hybrid pipeline is provided at right.
Selection of assembly approaches
A total for four MinION-only assembly approaches and three hybrid assembly approaches were compared to determine which assembly algorithms would produce the highest quality genome assemblies from our datasets. For the MinION data only assembly of the B. malayi genome, Canu outperformed WTDBG2, Shasta, and Flye for contiguity and completeness metrics. For hybrid genome assembly of the B. malayi genome, MaSuRCA outperformed WENGAN and HASLR for contiguity and completeness metrics (Table S2). All additional comparative analyses for B. malayi, T. trichiura, and A. caninum were conducted using the MinION data only assembly generated from Canu and the hybrid assembly from MaSuRCA, respectively.
Assembly size, heterozygosity, and contiguity
All assemblies generated in this study were shorter in terms of total length compared to current reference genomes, with the exception of the MinION data only assembly for B. malayi (Table 1). Genome sizes estimated by GenomeScope were similarly shorter than those of existing references (Table S1; Figure S2). The best-fit GenomeScope model was that of the B. malayi dataset, which estimated low heterozygosity. GenomeScope models for T. trichiura and A. caninum each show two peaks in the frequency spectra, consistent with estimates of elevated levels of heterozygosity. Of the assemblies generated in this study, hybrid assemblies were consistently the most contiguous as represented by higher N50 values (Table 1). The most improvement in terms of contiguity was observed in A. caninum, where each assembly approach resulted in a shorter genome and more contiguous assembly compared to the current reference genome. Within a species, all assemblies had similar levels of GC content.
Table 1.
Comparative quality metrics output by QUAST for the assemblies generated as part of this study and the reference assemblies available for each species
| Length (bp) | No. contigs | N50 (bp) | GC content | N content | |
|---|---|---|---|---|---|
| Brugia malayi | |||||
| Ghedin et al. (2007) reference assemblya | 88,235,797 | 197 | 14,214,749 | 28.5% | 0.30% |
| MinION data only assembly | 88,513,498 | 88 | 4,666,641 | 28.43% | 0% |
| MinION assembly polished with Illumina data | 88,428,546 | 88 | 4,667,161 | 28.45% | 0% |
| Hybrid assembly | 84,528,932 | 49 | 3,943,761 | 28.57% | <0.001% |
| Trichuris trichiura | |||||
| Foth et al. (2014) reference assemblyb | 75,474,068 | 4,086 | 70,602 | 42.3% | 0.30% |
| Doyle et al. (2022) reference assembly | 80,573,711 | 113 | 11,299,416 | 42.3% | 0.31% |
| MinION data only assembly | 73,094,541 | 174 | 806,679 | 42.22% | 0% |
| MinION assembly polished with Illumina data | 72,959,001 | 174 | 805,117 | 42.23% | 0% |
| Hybrid assembly | 72,705,678 | 63 | 2,773,302 | 41.15% | 0% |
| Ancylostoma caninum | |||||
| International Helminth Genomes Consortium (2019) reference assemblyc | 465,750,606 | 25,339 | 256,700 | 42.8% | 13.50% |
| MinION data only assembly | 358,159,610 | 1,578 | 638,901 | 43.36% | 0% |
| MinION assembly polished with Illumina data | 357,986,713 | 1,578 | 637,973 | 43.38% | 0% |
| Hybrid assembly | 348,034,555 | 652 | 1,095,227 | 43.24% | <0.001% |
Abbreviations: bp = base pairs.
Reference assembly included PacBio, optical mapping, and Sanger sequencing data.
Reference assembly generated using Illumina data.
Reference assembly generated using 454 sequencing.
Assembly completeness
For all species and assembly types, complete mitochondrial genomes were concurrently generated. For B. malayi, the complete genome for the Wolbachia endosymbiont was also assembled in a single circular genome for both the MinION data only assembly and the hybrid genome assembly. BUSCO scores were nearly identical among the three assembly types generated for all three species and matched or exceeded completeness scores of current reference assemblies (Tables 2 and S3). For the Nematoda reference ortholog database, specifically, proportions of single copy orthologs identified in each assembly generated ranged from 98.79 to 98.88% (B. malayi), 56.47–56.72% (T. trichiura), and 91.50–92.40% (A. caninum). For A. caninum, BUSCO scores indicate that the assemblies generated here are more complete than the existing International Helminth Genomes Consortium7 reference assembly. All assemblies compared for T. trichiura, including both available references, had high proportions of missing BUSCOs when analyzed with the Nematoda reference ortholog database (i.e., ∼39%; see Table 2). According to the assessment of contiguity versus completeness, the assemblies generated here for B. malayi and A. caninum can be classified as “tier 1” genomes sensu the International Helminth Genomes Consortium7 (i.e., >85% single copy BUSCO score and >1.6 log value for the defined contiguity metric; Figure 2). For T. trichiura, the contiguity of assemblies generated here is sufficient to qualify them each as “tier 1”, but, as mentioned previously, high proportions of BUSCO missingness prevented any trichurid assembly assessed from achieving “tier 1” status (see Figure 2). For B. malayi and A. caninum, four contigs totaling 49,030 bp and four contigs totaling 116,599 bp, respectively, were confidently identified by BlobTools as potential contamination and were removed from final assemblies (see Table S4). Blobplots for all species are presented in Figures S3–S5 (Minion data only assemblies) and Figures S6–S8 (hybrid assemblies).
Table 2.
Scores from compleasm for the assemblies generated as part of this study and the reference assemblies available for each species
| Single copy | Duplicated | Fragmented | Missing | |
|---|---|---|---|---|
| Brugia malayi | ||||
| Ghedin et al. (2007) reference assembly | 3,097 (98.91%) | 16 (0.51%) | 10 (0.32%) | 8 (0.26%) |
| MinION data only assembly | 3,096 (98.88%) | 17 (0.54%) | 10 (0.32%) | 8 (0.26%) |
| MinION assembly polished with Illumina data | 3,096 (98.88%) | 17 (0.54%) | 10 (0.32%) | 8 (0.26%) |
| Hybrid assembly | 3,093 (98.79%) | 17 (0.54%) | 11 (0.35%) | 10 (0.32%) |
| Trichuris trichiura | ||||
| Foth et al. (2014) reference assembly | 1,746 (55.76%) | 55 (1.76%) | 99 (3.16%) | 1,231 (39.32%) |
| Doyle et al. (2022) reference assembly | 1,746 (55.76%) | 55 (1.76%) | 99 (3.16%) | 1,231 (39.32%) |
| MinION data only assembly | 1,768 (56.47%) | 26 (0.83%) | 93 (2.97%) | 1,244 (39.73%) |
| MinION assembly polished with Illumina data | 1,768 (56.47%) | 26 (0.83%) | 94 (3.00%) | 1,243 (39.70%) |
| Hybrid assembly | 1,776 (56.72%) | 27 (0.86%) | 97 (3.10%) | 1,231 (39.32%) |
| Ancylostoma caninum | ||||
| International Helminth Genomes Consortium (2019) reference assembly | 2,657 (84.86%) | 242 (7.73%) | 146 (4.66%) | 86 (2.75%) |
| MinION data only assembly | 2,868 (91.60%) | 103 (3.29%) | 67 (2.14%) | 93 (2.97%) |
| MinION assembly polished with Illumina data | 2,865 (91.50%) | 105 (3.35%) | 69 (2.20%) | 92 (2.94%) |
| Hybrid assembly | 2,893 (92.40%) | 110 (3.51%) | 54 (1.72%) | 74 (2.36%) |
Scores are presented as number of BUSCOs recovered in each assembly followed in parentheses by proportion of the total number of nematode orthologs assessed by miniBUSCO (n = 3,131).
Figure 2.

Plot of single copy BUSCO score versus the log value of N50 length divided by contig (or scaffold) count for the assemblies generated as part of this study, and for all genomes of nematodes that parasitize animals as adults available from WormBase ParaSite
Dotted lines indicate the cutoff for “tier 1” genome status sensu the International Helminth Genomes Consortium (i.e., >85% single copy BUSCO score and >1.6 log value for the contiguity metric). A key to symbolic designations is provided at top left. The Nematoda ortholog database (n = 3,131 BUSCOs) was used for comparison.
Gene content and composition
For organelle genomes, within a species, the mitochondrial and Wolbachia genomes generated were nearly identical (i.e., >99.9% pairwise nucleotide identity) across assembly types (Table S4). Genome-wide nucleotide-level pairwise identity varied across species and assembly type comparisons, ranging from 99.04 to 99.74% for A. caninum to 99.22–99.86% for T. trichiura to 99.57–99.89% for B. malayi (Table S5). For gene datasets produced by GeMoMa, the number of predicted genes was roughly equal across assembly types for each species (Figure 3). Additionally, the majority of these genes were shared between assembly types. Within a species, genes predicted across assembly types were similar or identical in mean gene length, mean length of introns, exons, and coding sequences, and mean number of exons per coding sequence (Table 3). Pairwise nucleotide comparisons of genes shared between MinION only data assemblies and MinION assemblies polished with Illumina data showed 88–98% of these genes to be identical at the nucleotide level (Figure 4A). The majority of differences in gene composition were the result of single SNPs or single indels, with few shared genes demonstrating greater than ten mismatches (Figure 4B).
Figure 3.

Venn diagrams of the number of genes predicted by GeMoMa that were shared among the assembly types generated
(A) Brugia malayi, (B) Trichuris trichiura, and (C) Ancylostoma caninum.
Table 3.
Comparative summary statistics output by AGAT for genes predicted by GeMoMa in assembly types for each species
| Mean gene length | Mean exon length | Mean intron length | Mean CDS length | Mean no. exons per CDS | |
|---|---|---|---|---|---|
| Brugia malay | |||||
| MinION data assembly | 4,299 | 148 | 346 | 1,394 | 9.4 |
| MinION assembly polished with Illumina data | 4,291 | 148 | 346 | 1,394 | 9.4 |
| Hybrid assembly | 4,267 | 148 | 344 | 1,389 | 9.4 |
| Trichuris trichiura | |||||
| MinION data assembly | 2,916 | 212 | 325 | 1,280 | 6.0 |
| MinION assembly polished with Illumina data | 2,909 | 212 | 325 | 1,279 | 6.0 |
| Hybrid assembly | 2,918 | 212 | 324 | 1,283 | 6.0 |
| Ancylostoma caninum | |||||
| MinION data assembly | 2,663 | 125 | 332 | 822 | 6.5 |
| MinION assembly polished with Illumina data | 2,655 | 125 | 330 | 821 | 6.5 |
| Hybrid assembly | 2,681 | 125 | 332 | 825 | 6.6 |
Mean lengths are presented as number of base pairs. Abbreviations: CDS = coding sequence.
Figure 4.

Summary of differences in gene composition identified in predicted genes shared between MinION data assemblies and MinION assemblies polished with Illumina data
(A) Proportions of shared genes identified at the nucleotide level as identical, differing by SNPs only, differing by indels only, or differing by both SNPS and indels for Brugia malayi (left), Trichuris trichiura (center), and Ancylostoma caninum (right).
(B) Number of differences identified between non-identical shared genes for Brugia malayi (left), Trichuris trichiura (center), and Ancylostoma caninum (right). A key to bar plot colors is provided at bottom center.
Discussion
Sample processing and sequencing
The DNA extraction and MinION library preparation protocols described here were optimized for low input gDNA extracted from individual parasitic nematodes, allowing us to retain the majority of input gDNA through library preparation. Genomic DNA from a single individual is the ideal input for de novo whole genome assembly for diploid organisms. This allows assembly pipelines to contend with only two potential haplotypes, leading to more accurate assemblies with less haplotypic duplication. An alternative method would be to sequence multiple individuals from a highly inbred homozygous line. Many species cannot be maintained in a laboratory setting; however, making the establishment of inbred lines for these taxa challenging or impossible.21 Additionally, sampling from natural, rather than laboratory-maintained, populations is preferred for many genomic studies, further highlighting the advantage of whole-genome sequencing from single individuals.22 For B. malayi and T. trichiura, which both have relatively large adult stages (∼3–5 cm in length) and tractable genome sizes (<100 Mb), optimized protocols allowed for the generation of both short- and long-read data from a single adult worm. Adults of A. caninum, however, tend not to exceed ∼1.5 cm in length, and have the largest genome size of the three focal species in this study (i.e., ∼400 Mb). Thus, for A. caninum, even if adult worms had been accessible, these protocols would likely still not have allowed for generation of sufficient genomic data from a single adult. Despite using a pooled sample, however, copy number spectrum plots suggest that incorporation of a purging step produces results similar to those for single individuals (see Figure S9). For nematodes where gDNA from single individuals does not meet the requisite quantity for sequencing on the ONT MinION, additional strategies such as whole genome amplification can be pursued.
There was an association between the average Q score of MinION read datasets and the total amount of data generated by a flow cell: more efficient MinION sequencing runs generated data with higher average Q scores (see Table S1; Figure S1). For example, MinION libraries for both B. malayi and T. trichiura generated ∼10.8–11.8 Gb of data per flow cell post-basecalling with comparably high Q scores. Conversely, libraries for A. caninum generated ∼7.7–8.5 Gb of data per flow cell post-basecalling, and these data had the lowest average Q score of the three species sequenced (see Table S1; Figure S1). We observed that flow cells that were used to sequence the A. caninum libraries experienced the most rapid decline in pore availability. This observation highlights the variability in data generation and quality depending on the sample type when using the latest ONT chemistries.
Estimation of genome size and heterozygosity
There were appreciable differences in overall size of assemblies produced in this study compared to reference genomes (see Table 1). These differences were notable for both T. trichiura and A. caninum, but most dramatic for A. caninum, where assemblies produced here were >100 Mb smaller than the existing reference. This is likely due to the fact that a purging step to remove haplotypic duplication was included in the bioinformatic pipeline for all assembly types generated in this study (see Figure 1). This was not the case for the reference genome for A. caninum7, and thus it likely includes haplotypic duplication that artificially inflates genome size. Three lines of evidence support that the genomes assembled here more accurately represent the true haplotypic genome size for both T. trichiura and A. caninum. First, the lengths of these assemblies more closely match k-mer based genome size estimates than do those of the existing reference genomes (see Table 1). Second, BUSCO scores were largely indistinguishable across all assembly types (including existing reference assemblies), indicating that all are comparably complete despite ranging in size. In fact, the Doyle et al. reference assembly for T. trichiura23 and the International Helminth Genomes Consortium reference assembly for A. caninum7 had higher proportions of duplicated BUSCOs as compared to the assemblies generated herein (see Tables 2 and S3). Finally, for hybrid assemblies, copy number spectrum plots suggest they were not “over-purged” and thus missing genomic content (see Figure S9). As previously advocated by other authors, purging haplotypic duplication is an important step in genome curation regardless of assembly approach.24,25 Unsurprisingly, purging becomes increasingly important for highly heterozygous sample types, as observed here for A. caninum.
Results from GenomeScope (see Table S1; Figure S2) and copy number spectrum plots (see Figure S9) indicate low heterozygosity for B. malayi and elevated heterozygosity for T. trichiura and A. caninum. These results are congruent with the fact that the single individual of B. malayi sequenced came from a long-maintained inbred laboratory strain while the individual of T. trichiura sequenced came from a naturally infected human host, and sequence data for A. caninum were generated from a pool of individuals. When sequencing adult females that are potentially gravid, as was the case for B. malayi, analyses like GenomeScope and KAT that utilize k-mer spectra can be altered by the presence of paternal haplotypes in eggs. Given that the female of B. malayi sequenced came from an inbred strain, however, maternal and paternal haplotypes are likely to be identical, or highly similar, ameliorating this concern. It is worth noting that the k-mer based approaches used to estimate genome size and heterozygosity and to assess the effectiveness of purging rely on short-read data as input, and therefore cannot be used to evaluate long-read only assemblies at this time.
Wolbachia genome assembly from Brugia malayi
Complete circular Wolbachia genomes 1.08 Mb in length were assembled alongside the B. malayi genome for both assembly approaches. Wolbachia genomes generated in this study were highly similar to one another (>99.9%; 19 nucleotide mismatches) at the nucleotide level upon comparison via multisequence alignment. This was expected, given they were generated from the same individual sample. This high level of nucleotide conservation extends to the publicly available Wolbachia genomes generated from the FR3 strain of B. malayi (GenBank: CP034333.1, AE017321.1; Table S4).26,27 The majority of nucleotide mismatches identified between the Wolbachia genomes generated from the MinION data only and hybrid assembly occur in homopolymer regions with >5 nucleotide repeats, and are most commonly represented by a single base insertion in these genomes as compared to the references. The hybrid genome contained more nucleotide mismatches compared to the reference genomes (185 and 185 mismatches, respectively) than the MinION data only assembly (23 and 18 mismatches, respectively). These discrepancies are due to two sections of the genome that contain a large number of multiple base pair indels and large (>5bp) runs of SNPs, which is likely the result of a misassembly event. Overall, the high level of nucleotide identity between all Wolbachia genomes assessed in this study is expected given that they are generated from the same laboratory strain of B. malayi that has been in isolation since 1970.28
Assembly contiguity
We assessed a number of different MinION data only and hybrid assembly approaches to assess which program gave us the highest quality genomes for our datasets generated in the study in order to compare genomes generated with MinION data only compared to genomes supplemented with accurate short read data. The reference genome assemblies for both B. malayi and T. trichiura are highly contiguous and nearing chromosome scale. For B. malayi and T. trichiura, the N50 values for the assemblies generated in this study were substantially lower compared to those of the highly contiguous reference genome. This is due in part to the additional data sources used to generate these references, which for B. malayi included optical mapping29 and for T. trichiura included unspecified updates that resulted in improvements to contiguity.23 For A. caninum, however, the contiguity of the assemblies generated here represents marked improvements as compared to the reference genome. Hybrid assemblies were in all cases more contiguous than MinION data assemblies (see Table 1). This result was somewhat unexpected given that including short-read data in whole genome assembly has traditionally been touted to increase accuracy rather than contiguity.30,31,32 Increased contiguity of hybrid assemblies over long read-only assemblies has been observed in bacteria whole genome sequencing33,34 but does not appear to have been reported previously for eukaryotes. It is worth noting that the long reads generated for each species here did not approach the read lengths that MinION devices are capable of sequencing. Across species, read length N50 values ranged from ∼2.7 to 8.6 kb (see Table S1), which are relatively small. This may be due to the optimized MinION library preparation approach used here, which relied on washing final libraries with a titration of short and long fragment buffer to retain some short fragments of gDNA in addition to long fragments (see STAR Methods). Rigorous size selection which could have improved read lengths was therefore sacrificed to enable sequencing from low quantities of input gDNA. Had it been possible, generation of ultra-long reads would have likely lessened the gap in contiguity metrics between MinION data and hybrid assemblies. Additionally, a single assembly algorithm was chosen to generate long-read and hybrid assemblies (i.e., Canu and MaSuRCA, respectively). Given the same read-level data, different assembly algorithms may have produced assemblies with different contiguity metrics.
Assembly completeness
In general, completeness values as measured by BUSCO scores from compleasm are indistinguishable between assembly types for B. malayi and T. trichiura, regardless of which reference ortholog database is used for comparison. Assemblies generated here for A. caninum showed an improvement in the number of single copy genes identified and a reduction in the number of duplicated and fragmented genes identified as compared to the reference genome across comparative databases. Results from compleasm indicate a high proportion of missingness for all trichurid assemblies when compared to the Nematoda reference ortholog database (see Table 2; Figure 2). Underrepresentation in the BUSCO “nematoda_odb10” dataset has been noted to result in biases and underestimation in ortholog detection for some species of nematodes.35 When comparing to higher-level Metazoa and Eukaryota databases, elevated proportions of missing BUSCOs were still found in the trichurid assemblies (∼25% for Metazoa and ∼7–8% for Eukaryota; see Table S3), albeit not high as those found for the Nematoda database. This presents a challenge when using BUSCO scores as a completeness metric to assess trichurid genome assemblies.
Gene content and composition
In terms of gene content, within a species, similar numbers of genes were predicted by GeMoMA for each of the three assembly types generated (see Figure 3). Though not all predicted genes were identified in all three assemblies, large proportions were found to be shared among them (i.e., ∼93–98%, depending on the species). In particular, MinION only assemblies and MinION assemblies polished with Illumina data were highly similar in terms of gene content, with ∼98.7–99.7% of predicted genes shared between them within a species. Furthermore, predicted genes sets had, by species, identical or nearly identical mean genes lengths, mean intron, exon, and coding sequence lengths, and mean numbers of exons per coding sequence (see Table 3), providing further evidence for a high degree of similarity in gene content across assembly types.
In terms of gene composition, a major goal of this study was to assess the potential of Illumina data to correct errors that may be present in the final assembly as a result of less accurate MinION data. We took multiple approaches to evaluate this, including calculating pairwise nucleotide-level identity between different assembly approaches at both the genome and gene level. Genome level nucleotide identities ranged from 99.04% to 99.89% (Table S5). Gene datasets predicted by GeMoMa for MinION data only assemblies and for MinION assemblies polished with Illumina data were pairwise aligned and assessed for mismatches. Comparisons between MinION data only assemblies and hybrid assemblies are not tenable because long-read assemblers like Canu and hybrid assemblers like MaSuRCA utilize assembly algorithms that handle read-level data dissimilarly.36,37 Given that gene predictions for each assembly were based on existing reference genomes and their corresponding annotations (see STAR Methods), plus these reference genomes were generated from different biological samples, comparison to reference genomes was also not salient.
The most direct comparison to determine whether short reads improve gene accuracy in final assemblies are the MinION data only assembly versus that same assembly polished with Illumina short reads. If Illumina polishing substantially improves accuracy, homology-based gene prediction would be expected to result in a low proportion of genes shared between these two assembly types, and or/the majority of shared genes to differ at the nucleotide level. This was not the case as all three species showed high proportions of predicted genes shared between assembly types, and the majority of the genes shared between the two assembly types were identical (see Figures 3 and 4A). For those that were not identical, most differed by only a single SNP or indel. These single differences are presumably corrections made during Illumina polishing (Figure 4B). Despite this, a substantial number of gene comparisons still contained 10–100+ mismatches (Figure 4B). It is unlikely these differences result from polishing errors in MinION data only assemblies; rather, they are more likely the result of homology-based gene prediction models identifying incongruent genes between assembly types in a small number of cases. In summary, depending on the species, correcting with short reads did not change nucleotide calls in coding sequences for ∼88–98% of the genes compared.
Conclusions
For this study, de novo whole genome assemblies were generated for three species of parasitic nematodes (Brugia malayi, Trichuris trichiura, and Ancylostoma caninum) using only MinION long-read data, using MinION data polished with Illumina short reads, and using a combined hybrid approach. For B. malayi and T. trichiura, optimized gDNA extraction and library preparation protocols allowed for the generation of complete genomes from individual adult worms. For all species sequenced, MinION data only assemblies had similar, or superior, measures of contiguity and completeness as compared to existing reference genomes. The most substantial improvements in quality metrics were observed for A. caninum, which was the only one of the three focal species for which the existing reference genome is not a near-chromosome scale assembly. Among the three assembly types generated, predicted gene content was nearly identical with a species, and the vast majority of predicted genes shared between MinION data only assemblies and MinION assemblies polished with short reads were identical at the nucleotide level. For some genes, however, polishing did result in the correction of SNPs or indels, indicating that the inclusion of short reads results in more accurate final genome assemblies. Although additional data types beyond MinION long reads are needed to produce reference-quality, chromosome-scale assemblies, the results of this study demonstrate that MinION data alone can be used to generate highly contiguous and complete de novo whole genomes from parasitic helminths. Importantly, these approaches are accessible and can use individual worms as input, allowing for the generation of more genome assemblies that will ultimately increase our understanding of genomic diversity and facilitate population-level genomic analyses for these important parasites.
Limitations of the study
This study has multiple limitations. For two of the three focal species, gDNA was generated from adult worms. For most species, and particularly those that infect humans, adult nematodes are difficult or impossible to obtain, somewhat limiting the utility of this approach in practice. The assemblies generated here remain incomplete in terms of modern reference genome standards. They are not chromosome-scale, they are not annotated, structural variation has not been explored, and additional data types (e.g., RNA-seq) are required to complete them. For B. malayi and T. trichiura, existing reference genomes were already exceptionally high-quality “tier 1”-status assemblies sensu the International Helminth Genomes Consortium,7 and the assemblies generated here using a single data source approached their quality in contiguity and matched their quality in completeness. The comparison in which the added value of long-read MinION data are most evident is that of A. caninum. Using gDNA from the same strain and sample type on which the existing reference assembly was based, the genomic resources available for this species were improved considerably.
Comparisons of MinION data only versus hybrid assemblies generated from the same sample were limited to genomes assembled using a single approach. There are dozens of comparable tools available for each step of the genome assembly pipeline (e.g., de novo assemblers, methods for purging duplication, polishing algorithms, pipelines for identifying contamination, etc.) and comparing all combinations of these many tools was outside of the scope of this study. Benchmarking studies typically rely on prokaryotic genomes with “truth datasets” in which base calls at all positions are known with confidence. In contrast, the nematodes sequenced here are non-model eukaryotes with comparably large and complex genomes for which there are no truth datasets available for comparison. Benchmarking assembly pipelines was therefore not feasible, nor indeed was it the goal of this study. Rather, we sought to evaluate the feasibility and quality of whole genome assemblies generated from an accessible NGS data type. Ultimately, we constructed a straightforward pipeline of well-validated tools (Figure 1) that provided the best results for our data, establishing a roadmap that others seeking to generate high-quality helminth genomes using similar data types can follow moving forward.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Brugia malayi adults | NIH/NIAID Filariasis Research Reagent Resource Center via BEI Resources (Michalski et al.28) | FR3 strain |
| Trichuris trichiura adults | Peter Nejsum, Department of Clinical Medicine, Aarhus University | N/A |
| Ancylostoma caninum third-stage larvae | John M. Hawdon, Department of Microbiology, Immunology, and Tropical Medicine, The George Washington University | Baltimore strain |
| Chemicals, peptides, and recombinant proteins | ||
| DNA/RNA Shield | Zymo Research, Irvine, CA, USA | Cat#R1200-25 |
| Critical commercial assays | ||
| Quick-DNA HMW MagBead Kit | Zymo Research, Irvine, CA, USA | Cat#D6060 |
| Monarch® Pestle Set single-use microtube pestle | New England Biolabs® Inc., Ipswich, MA, USA | Cat#T3000L |
| Qubit™ 4 1X dsDNA High Sensitivity (HS) Assay | ThermoFisher Scientific, Waltham, MA, USA | Cat#Q33231 |
| Agilent 2200 TapeStation System Genomic DNA ScreenTape and Reagents | Agilent, Santa Clara, CA, USA | Cat#5067-5365; 5067-5366 |
| AMPure XP | Beckman Coulter, Brea, CA, USA | Cat#A63880 |
| DNA Clean & Concentrator Magbee Kit | Zymo Research, Irvine, CA, USA | Cat#D4012 |
| Ligation Sequencing Kit | Oxford Nanopore Technologies, Oxford, United Kingdom | Cat#SQK-LSK114 |
| NEBNext® Companion Module for Oxford Nanopore Technologies® Ligation Sequencing | New England Biolabs® Inc., Ipswich, MA, USA | Cat#E7180S |
| xGen™ cfDNA & FFPE DNA Library Prep Kit | Integrated DNA Technologies, Newark, NJ, USA | Cat#10010207 |
| Deposited data | ||
| Quality-controlled MinION and Illumina read data for Brugia malayi | National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) | BioProject accession ID PRJNA1074771; BioSample accession no. SAMN39888962 |
| Quality-controlled MinION and Illumina read data for Trichuris trichiura | National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) | BioProject accession ID PRJNA1074771; BioSample accession no. SAMN39888963 |
| Quality-controlled MinION and Illumina read data for Ancylostoma caninum | National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) | BioProject accession ID PRJNA1074771; BioSample accession no. SAMN39888964 |
| Final assemblies generated for Brugia malayi | National Center for Biotechnology Information (NCBI) Genome database | Accession nos. GCA_037903335.1; GCA_037903345.1; GCA_037903365.1 |
| Final assemblies generated for Trichuris trichiura | National Center for Biotechnology Information (NCBI) Genome database | Accession nos. GCA_037903355.1; GCA_037903375.1;GCA_037903465.1 |
| Final assemblies generated for Ancylostoma caninum | National Center for Biotechnology Information (NCBI) Genome database | Accession nos. GCA_037903475.1; GCA_037903715.1; GCA_037903745.1 |
| Reference genome for Brugia malayi | Tracy et al.29 | BioProject accession ID PRJNA10729 |
| Reference genome for Trichuris trichiura | Foth et al.38 | BioProject accession ID PRJEB535 |
| Reference genome for Trichuris trichiura | Doyle et al.23 | https://github.com/stephenrdoyle/ancient_trichuris/tree/master/02_data |
| Reference genome for Ancylostoma caninum | International Helminth Genomes Consortium7 | BioProject accession ID PRJNA72585 |
| Mitochondrial reference genome for Ancylostoma caninum | Xie et al.39 | GenBank accession ID MN215971 |
| Reference genomes for nematodes that parasitize animals | WormBase ParaSite16,17 | https://parasite.wormbase.org/species.html#Nematoda |
| Software and algorithms | ||
| MinKNOW v. 22.10.7; 22.10.10; 22.12.7 | Oxford Nanopore Technologies | https://community.nanoporetech.com/docs/prepare/library_prep_protocols/experiment-companion-minknow/v/mke_1013_v1_revdc_11apr2016/installing-minknow-on-linu |
| Guppy v. 6.3.4 | Oxford Nanopore Technologies | https://community.nanoporetech.com/downloads?from=support |
| NanoPlot v. 1.40.2 | De Coster40 | https://github.com/wdecoster/NanoPlot |
| Mash v. 2.2.2 | Ondov et al.41 | https://mash.readthedocs.io/en/latest/# |
| Minimap2 v. 2.16 | Li42 | https://github.com/lh3/minimap2 |
| SAMtools v. 1.9 | Danecek et al.43 | https://github.com/samtools/samtools |
| fastp v. 0.23.2 | Chen et al.44 | https://github.com/OpenGene/fastp |
| BWA v. 0.7.17 | Li and Durbin45 | https://github.com/lh3/bwa |
| Jellyfish v. 2.3.0 | Marçais and Kingsford46 | https://github.com/gmarcais/Jellyfish |
| GenomeScope web-based graphical user interface | Ranallo-Benavidez et al.44 | https://github.com/schatzlab/genomescope |
| Canu v. 2.1 | Koren et al.36 | https://github.com/marbl/canu |
| wtdbg2 v. 0.0 | Ruan and Li47 | https://github.com/ruanjue/wtdbg2 |
| Shasta 0.11.1 | Shafin et al.48 | https://github.com/chanzuckerberg/shasta |
| Flye v. 2.9.1-b1780 | Kolmogorov et al.49 | https://github.com/mikolmogorov/Flye |
| SeqKit v 0.10.1 | Shen et al.50 | https://bioinf.shenwei.me/seqkit/ |
| BBMap v.38.84 | Bushnell51 | https://github.com/BioInfoTools/BBMap |
| purge_dups v. 1.2.5 | Guan et al.52 | https://github.com/dfguan/purge_dups |
| Racon v. 1.5.0 | Vaser et al.53 | https://github.com/isovic/racon |
| Medaka v. 1.7.2 | Oxford Nanopore Technologies | https://github.com/nanoporetech/medaka |
| Pilon v. 1.24 | Walker et al.54 | https://github.com/broadinstitute/pilon |
| MaSuRCA v. 4.1.0 | Zimin et al.37 | https://github.com/alekseyzimin/masurca |
| WENGAN v. 0.2 | Di Genova et al.55 | https://github.com/adigenova/wengan |
| HASLR v. 0.8a1 | Haghshenas et al.56 | https://github.com/vpc-ccg/haslr |
| K-mer Analysis Toolkit (KAT) v. 2.4.0 | Mapleson et al.57 | https://github.com/TGAC/KAT |
| BlobTools v. 1.1.1 | Challis et al.58 | https://github.com/DRL/blobtools |
| BLAST v. 2.14.0 | Altschul et al.59 | https://blast.ncbi.nlm.nih.gov/doc/blast-help/downloadblastdata.html#downloadblastdata |
| ggplot2 R package | Wickham60 | https://ggplot2-book.org/ |
| R v. 4.3.0; 4.2.3 | Core R Team61 | https://cran.rstudio.com/ |
| RStudio v. 2023.03.1; 2023.06.1 | N/A | https://posit.co/download/rstudio-desktop/ |
| Geneious Prime v. 2022.0.1 | Kearse et al.62 | https://www.geneious.com/updates |
| QUAST v. 5.0.2 | Gurevich et al.63 | https://github.com/ablab/quast |
| compleasm v. 0.2.6 | Huang and Li64 | https://github.com/huangnengCSU/compleasm |
| GeMoMA v. 1.9 | Keilwagen et al.65,66 | https://www.jstacs.de/index.php/GeMoMa |
| AGAT v. 0.9.1 | Dainat et al.67 | https://github.com/NBISweden/AGAT |
| Biostrings R package v. 2.66.0 | Pagès et al.68 | https://bioconductor.org/packages/release/bioc/html/Biostrings.html |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Joseph R. Fauver (jfauver@unmc.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
Quality-controlled MinION and Illumina data for each species are deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under the BioProject accession ID PRJNA1074771 and under BioSample accession nos. SAMN39888962 (Brugia malayi), SAMN39888963 (Trichuris trichiura) and SAMN39888964 (Ancylostoma caninum), and are publicly available as of the date of publication. Final assemblies for each species are deposited in the NCBI Genome database under the accession numbers GCA_037903335.1, GCA_037903345.1, and GCA_037903365.1 (Brugia malayi), GCA_037903355.1, GCA_037903375.1, and GCA_037903465.1 (Trichuris trichiura), and GCA_037903475.1, GCA_037903715.1, and GCA_037903745.1 (Ancylostoma caninum), and are publicly available as of the date of publication.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Method details
Specimen acquisition
The specimens sequenced were obtained from the same strains previously used to generate existing reference genomes for each species. Adults of B. malayi (FR3 strain) were acquired through the NIH/NIAID Filariasis Research Reagent Resource Center via BEI Resources.28 Worms were received frozen and stored at -80°C. Prior to sequencing, worms were thawed at 4°C and subsequently preserved in DNA/RNA Shield (Zymo Research, Irvine, CA, USA). Adults of T. trichiura were obtained from a Danish patient after anthelmintic treatment for an infection acquired in Uganda. Worms were received preserved in 70% ethanol and were stored at 4°C prior to sequencing. Pooled third-stage (L3) larvae of A. caninum (Baltimore strain) were obtained from experimental infection in laboratory-maintained canines. Pooled L3s were received frozen, stored at -80°C, and thawed at 4°C prior to sequencing.
DNA extraction
Total nucleic acid was extracted separately from each of a single adult female (B. malayi), a single adult male (T. trichiura), and a pool of ∼10,000 L3 larvae (A. caninum) using a Quick-DNA HMW MagBead Kit (Zymo Research, Irvine, CA, USA) and modified Solid Tissue extraction protocol (Table S1). Prior to extraction, adult worms were placed in clean 1.5 mL DNA LoBind® tubes (Eppendorf® North America, Enfield, CT, USA) using sterilized forceps, while pooled L3s were centrifuged at 6,540 g for 3 min to allow for the removal of excess supernatant. All samples were then homogenized mechanically using a Monarch® Pestle Set single-use microtube pestle (New England Biolabs® Inc., Ipswich, MA, USA). Homogenization was performed both prior to, and immediately following, the addition of Proteinase K. Samples were then digested in a digital dry bath at 55°C for 24 hr with occasional flick mixing. The protocol for DNA purification was then followed, with these modifications: After the addition of 33 μL of MagBinding Beads, samples were placed on a benchtop rotating mixer for 40 min to 1.5 hr at RT; and after the addition of 52 μL DNA Elution Buffer, samples were incubated in a digital dry bath at 37°C for 2 hr with occasional flick mixing, then incubated at RT overnight prior to collecting eluted DNA. To avoid DNA fragmentation, samples were mixed by flicking rather than pipette mixing wherever possible. The concentration of each extraction was measured using a Qubit™ 4 1X dsDNA High Sensitivity (HS) Assay (ThermoFisher Scientific, Waltham, MA, USA) and fragment size distribution was determined using an Agilent 2200 TapeStation System and associated protocol for genomic DNA ScreenTape analysis (Agilent, Santa Clara, CA, USA). For T. trichiura, TapeStation analysis was not performed.
For A. caninum, initial attempts to sequence ONT libraries resulted in low sequencing efficiency, poor data quality, and rapid pore loss. To ameliorate concerns of protein contamination, extracted genomic DNA (gDNA) was purified via bead-based cleanup prior to final ONT library preparation. This cleanup was performed as follows: Two aliquots of extracted gDNA were each bound to a 0.5× volume of AMPure XP beads (Beckman Coulter, Brea, CA, USA) on a benchtop rotator mixer for 1 hr at RT. Beads were then washed twice with 80% EtOH, air-dried, and gDNA was eluted in 52 μL Zymo DNA Elution Buffer for 2 hr at 37°C with occasional flick mixing, followed by overnight elution at RT. This cleanup process was repeated a second time for both aliquots. Purified extractions were then quantified using a Qubit™ 4 1X dsDNA HS Assay and the Agilent 2200 TapeStation System.
MinION library preparation
Aliquots of gDNA extracted for each species were used to prepare one or more libraries for whole genome sequencing on an ONT MinION desktop sequencer (see Table S1). Libraries were prepared using a combination of the DNA Clean & Concentrator MagBead Kit (Zymo Research, Irvine, CA, USA), the ONT SQK-LSK114 Ligation Sequencing Kit (Oxford Nanopore Technologies, Oxford, United Kingdom), the NEBNext® Companion Module for Oxford Nanopore Technologies® Ligation Sequencing (New England Biolabs® Inc., Ipswich, MA, USA), and a modified hybrid protocol. First, gDNA was incubated with ONT DNA repair and end preparation reagents for 10 min at RT followed by 10 min at 65°C. End preparation reactions were then bound to 20 μL Zymo MagBinding Beads in 4× volumes of Zymo DNA MagBinding Buffer on a benchtop rotating mixer for 30 min to 2 hr. Beads were then washed twice with Zymo DNA Wash Buffer, air-dried for 10 min, eluted in 51 μL Zymo DNA Elution Buffer via manual agitation for 10 min, and quantified via Qubit™ 1X dsDNA HS Assay. Sequencing adaptors were then ligated via a 15 min incubation at RT, and libraries were bound to a 0.4× volumes of AMPure XP beads on a benchtop rotator mixer at RT for 1 hr. Beads were then washed twice with either ONT Long Fragment Buffer (A. caninum) or a titrated wash mix of 1:3 ONT Short Fragment Buffer:ONT Long Fragment Buffer (B. malayi and T. trichiura). After washing, beads were air-dried, then libraries were eluted in 15–17 μL of ONT Elution Buffer for 2 hr at 37°C with occasional flick mixing followed by overnight elution at RT. Final libraries were quantified using a Qubit™ 4 1X dsDNA HS Assay and the Agilent 2200 TapeStation System. For T. trichiura, TapeStation analysis was not performed. Additionally, for T. trichiura, a portion of ONT library reserved after sequencing on one flow cell was re-washed prior to sequencing on a second flow cell (see Table S1). This aliquot of library was bound to a 0.4× volume of AMPure XP beads on a benchtop rotator mixer for 1 hr, washed twice with ONT Long Fragment Buffer, air-dried, then eluted in 17 μL of ONT Elution Buffer for 2 hr at 37°C with occasional flick mixing followed by overnight elution at RT.
MinION sequencing and basecalling
Portions of ONT libraries were sequenced for ∼62–80 hr each on one (B. malayi), two (T. trichiura), or three (A. caninum) ONT MinION R10.4.1 flow cells. The amount of library loaded onto each flow cell and the total number of pores available at the start of sequencing are provided in Table S1. MinKNOW software (ONT) v. 22.10.7 (B. malayi), v. 22.10.10 (T. trichiura), or v. 22.12.7 (A. caninum) was used to run each flow cell with pore scans every 1.5 hr. After sequencing, signal data (i.e., fast5 files) from each flow cell were basecalled using Guppy v. 6.3.4 (ONT). The bioinformatic pipeline used to generate MinION data assemblies is outlined in Figure 1. Sequencing adaptors were simultaneously removed by specifying the “--trim_adapters” flag. The results of each Guppy run were summarized using NanoPlot v. 1.40.2.40 Fastq files that passed basecalling were input to Mash v. 2.2.241 to confirm there was no significant contamination in the read-level data prior to assembly. Estimated genome sizes provided to Mash were based on the lengths of the existing reference assemblies available for each species, and were thus set as 88 Mb for B. malayi,29 465 Mb for A. caninum7 and 75 Mb for T. trichiura23 (https://github.com/stephenrdoyle/ancient_trichuris/tree/master/02_data). Average depth of coverage of reads across the species-specific reference genome and proportion of reads mapped were estimated using Minimap2 v. 2.1642 and SAMtools v. 1.9.43
Illumina library preparation and sequencing
Aliquots of gDNA extracted for each species were sent to the Yale Center for Genome Analysis (YCGA) for Illumina whole genome library preparation and sequencing (see Table S1). Libraries were prepared using an xGen™ cfDNA & FFPE DNA Library Prep Kit with unique dual indexing (Integrated DNA Technologies, Newark, NJ, USA) and standard protocol. Libraries were subjected to 5–7 cycles of PCR to increase concentration (see Table S1) and multiplexed for 2×150 paired-end sequencing on an Illumina NovaSeq 6000 targeting 50× depth of coverage genome-wide for each species. Reads were demultiplexed by the YCGA and subsequently filtered for remaining adaptor contamination, quality, and length using fastp v. 0.23.2.44 The bioinformatic pipeline used to process Illumina data and generate hybrid assemblies is outlined in Figure 1. Fastq files that passed fastp were input to Mash to confirm there was no significant contamination in the read-level data prior to assembly. Estimated genome sizes provided to Mash were based on the lengths of the existing reference assemblies available for each species (see above). Average depth of coverage across the species-specific reference genome and proportion of reads mapped were estimated using BWA v. 0.7.1745 and SAMtools v. 1.9. A second round of fastp was run on the raw demultiplexed Illumina data to remove sequencing adapters, only, by specifying “--detect_adapter_for_pe”, “--disable_length_filtering”, and “--disable_quality_filtering”. These data were the used for hybrid genome assembly, as MaSuRCA utilizes built-in error correction and cleaning.
Estimation of genome size and heterozygosity
Quality-controlled Ilumina data were used to estimate genome size and heterozygosity for each species. First, Jellyfish v. 2.3.046 was used to generate k-mer spectra, specifying a k-mer size of 21 and an initial hash size of 1,000,000,000. Histograms from Jellyfish were then provided to the GenomeScope web-based graphical user interface69 to visualize k-mer spectra and estimate heterozygosity.
MinION data genome assembly
FASTQ files that passed basecalling by Guppy were concatenated and used as input for whole genome assembly. A total of four long-read only assembly algorithms were tested using the MinION data for B. malayi to determine which approach provided the most contiguous and complete genome assembly, including Canu v. 2.1,36 wtdbg2 v. 0.0,47 Shasta v. 0.11.1,48 and Flye v. 2.9.1-b1780.49 Assembly metrics included genome size, N50, contig count, GC content, and BUSCO scores. Following comparisons, we determined that Canu provided the highest quality genome assemblies from our data (see Table S2) and it was used to generate MinION data assemblies for all three species sequenced. For T. trichiura and A. caninum, a 3 kb minimum read length requirement was specified to Canu, and for B. malayi, a 5 kb minimum read length requirement was specified. Estimated genome sizes provided to Canu were based on the lengths of the existing reference assemblies available for each species (see above). Contigs in the resulting Canu assemblies that were indicated as potential alternative alleles (i.e., with FASTA headers including “suggestBubble=yes”) were removed using a combination of SeqKit v 0.10.150 and the filterbyname.sh script of BBMap v.38.84.51 Resulting “popped” assemblies were used as input to purge_dups v. 1.2.552 to remove false duplications. These “popped and purged” assemblies were then polished with one round of Racon v. 1.5.053 followed by one round of Medaka v. 1.7.2 (https://github.com/nanoporetech/medaka) to generate “popped, purged, and polished” assemblies. Copies of these final assemblies were also separately polished with Illumina data using Pilon v. 1.2454 and BWA. Three iterations of Pilon were iteratively run specifying the “--diploid” flag.
Hybrid genome assembly
A total of three hybrid genome assembly algorithms were tested using sequence data for B. malayi to determine which approach provided the most contiguous and complete assembly, including MaSuRCA v. 4.1.0,37 WENGAN v. 0.2,55 and HASLR v. 0.8a1.56 Assembly metrics included genome size, N50, contig count, GC content, and BUSCO scores. Following comparisons, we determined that MaSuRCA provided the highest quality hybrid genome assemblies from our data (see Table S2) and it was used to generate hybrid assemblies for all three species sequenced. FASTQ files that passed basecalling by Guppy and Illumina data from which only sequencing adaptors were removed by fastp were used as input to MaSuRCA. Default settings were used except for setting “LHE_COVERAGE” to “35” and “cgwErrorRate=0.15”, and setting “JF_SIZE” to a value ten times the genome size of the species-specific reference assembly. Assemblies output by MaSuRCA were input to purge_dups v. 1.2.544 to remove false duplications.
Refinement of final assemblies
To ensure “purged” MaSuRCA hybrid assemblies were not over- or under-purged, copy number spectrum plots were generated using the K-mer Analysis Toolkit (KAT) v. 2.4.057 specifying a k-mer size of 31. “Popped, purged, and polished” Canu MinION data assemblies and “purged” MaSuRCA hybrid assemblies were input to BlobTools v. 1.1.158 to identify potential contaminants. First, the reads used to generate each assembly were mapped to the assembly using Minimap2 (MinION data) and/or BWA (Illumina data), and SAMtools was used to generate sorted BAM files for each mapping. The “blastn” function in BLAST v. 2.14.059 was then used to compare all contigs to reference sequences available in the National Center for Biotechnology Information (NCBI)’s nucleotide databases, specifying “-evalue 1e-25”, “-max_target_seqs 10” and “-max_hsps 1”. BLAST outputs and sorted BAM files were then used as input to BlobTools. Contigs that could be confirmed as host, human, or bacterial contamination were excluded from final assemblies. Quality scores (i.e., Q scores) of basecalled MinION reads, and Q scores of reads when mapped to the final “popped, purged, and polished” MinION data assemblies, were measured using NanoPlot and plotted using the “geom_density” function in the ggplot2 package60 in R v. 4.3.061 in RStudio v. 2023.03.1.
Mitogenome and Wolbachia genome assembly
For B. malayi and A. caninum, contigs containing the mitogenomes—and, for B. malayi, the genome of its Wolbachia endosymbiont—were extracted from “popped” MinION and unpurged MaSuRCA assemblies. For T. trichiura, mitogenomes were extracted from the original “unpopped” Canu assembly and the unpurged MaSuRCA assembly. These organelle genome contigs were then mapped to species-specific reference organelle genomes using Geneious Prime v. 2022.0.162 and manually reconfigured to match the linear orientation of the reference. For B. malayi and T. trichiura, reference organelle genomes were extracted from the reference genome assemblies for each species (see above). For A. caninum, for which a mitochondrial contig is not labeled in the reference assembly, a reference mitogenome was downloaded from GenBank (accession no. MN215971).39 Contigs containing complete organelle genomes were then manually added to the appropriate final version of each assembly.
Assembly contiguity and completeness
Contiguity and completeness were assessed for each assembly using QUAST v. 5.0.263 and compleasm v. 0.2.6,64 respectively. For compleasm, the Nematoda, Metazoa, and Eukaryota ortholog databases (nematoda_odb10, metazoa_odb10, and eukaryota_odb10, respectively) were chosen for comparison. Compleasm was also run on the aforementioned reference genomes for each species, as well as for all genomes of nematodes that parasitize animals as adults available from WormBase ParaSite.16,17 For each assembly, the proportion of single copy BUSCOs present was plotted against the log value of the assembly’s N50 length divided by its scaffold or contig count using R v. 4.2.3 via RStudio v. 2023.06.1. Cutoff values of >85% (for proportion of single copy BUSCOs present) and >1.6 (for the log value of the contiguity metric) were used to assess whether each assembly could be classified as a “tier 1” genome following the standard for helminth genomes established by the International Helminth Genomes Consortium.7
Gene content and composition
Homology-based gene prediction was performed for each assembly generated here using GeMoMA v. 1.9.65,66 Predictions were made using default settings and the species-specific reference genome and annotation as input. Resulting GFF files were summarized using AGAT v. 0.9.1.67 For each species, the predicted genes shared between the MinION data assembly and the MinION assembly polished with Illumina data were pairwise aligned using the “pairwiseAlignment” function in Biostrings package v. 2.66.068 in R v. 4.2.3 via RStudio v. 2023.06.1 specifying the alignment type as “global”. Biostrings was then used to characterize each pairwise alignment in terms of percent identity, alignment length, gene lengths, and number of single nucleotide polymorphisms (i.e., SNPS) and insertions/deletions (i.e., indels). Summary figures for all comparisons were generated in R v. 4.3.0 via RStudio v. 2023.03.1 using the ggplot2 package.
Acknowledgments
The authors thank the BEI Resources NIH/NIAID Filariasis Research Reagent Resource Center for providing specimens of Brugia malayi for sequencing. Thanks are also due to Dr. Michael Wiley, Dr. Shaun Cross, Kristen Bernhard, and Mahmood Al-Haehm (University of Nebraska Medical Center) for providing access and support for use of select laboratory equipment. The authors additionally thank Christopher Castaldi and Irina Tikhonova (Yale Center for Genome Analysis, Yale School of Medicine) for generation of, and correspondence regarding, Illumina sequence data. Bioinformatic analysis was enabled by the University of Nebraska-Lincoln Holland Computing Center computing cluster. The authors are grateful to three anonymous reviewers whose feedback greatly improved this manuscript.
Author contributions
Conceptualization: K.S.H. and J.R.F.; methodology: K.S.H. and J.R.F.; investigation; K.S.H., R.W., J.M.H., P.N., and J.R.F.; writing: K.S.H. and J.R.F.; review and editing: K.S.H., J.M.H., P.N., and J.R.F.; resources: J.M.H., P.N., and J.R.F.
Declaration of interests
The authors declare no competing interests.
Published: July 30, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.110614.
Supplemental information
References
- 1.Vos T., Lim S.S., Abbafati C., Abbas K.M., Abbasi M., Abbasifard M., Abbasi-Kangevari M., Abbastabar H., Abd-Allah F., Abdelalim A., et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. 2020;396:1204–1222. doi: 10.1016/S0140-6736(20)30925-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Biraro I.A., Egesa M., Toulza F., Levin J., Cose S., Joloba M., Smith S., Dockrell H.M., Katamba A., Elliott A.M. Impact of Co-Infections and BCG Immunisation on Immune Responses among Household Contacts of Tuberculosis Patients in a Ugandan Cohort. PLoS One. 2014;9 doi: 10.1371/journal.pone.0111517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kizito D., Tweyongyere R., Namatovu A., Webb E.L., Muhangi L., Lule S.A., Bukenya H., Cose S., Elliott A.M. Factors affecting the infant antibody response to measles immunisation in Entebbe-Uganda. BMC Publ. Health. 2013;13:619. doi: 10.1186/1471-2458-13-619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morawski B.M., Yunus M., Kerukadho E., Turyasingura G., Barbra L., Ojok A.M., DiNardo A.R., Sowinski S., Boulware D.R., Mejia R. Hookworm infection is associated with decreased CD4+ T cell counts in HIV-infected adult Ugandans. PLoS Negl. Trop. Dis. 2017;11 doi: 10.1371/journal.pntd.0005634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nash S., Mentzer A.J., Lule S.A., Kizito D., Smits G., van der Klis F.R.M., Elliott A.M. The impact of prenatal exposure to parasitic infections and to anthelminthic treatment on antibody responses to routine immunisations given in infancy: Secondary analysis of a randomised controlled trial. PLoS Negl. Trop. Dis. 2017;11 doi: 10.1371/journal.pntd.0005213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ndyomugyenyi R., Kabatereine N., Olsen A., Magnussen P. Malaria and hookworm infections in relation to haemoglobin and serum ferritin levels in pregnancy in Masindi district, western Uganda. Trans. R. Soc. Trop. Med. Hyg. 2008;102:130–136. doi: 10.1016/j.trstmh.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 7.International Helminth Genomes Consortium Comparative genomics of the major parasitic worms. Nat. Genet. 2019;51:163–174. doi: 10.1038/s41588-018-0262-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bennett A.P.S., de la Torre-Escudero E., Robinson M.W. Helminth genome analysis reveals conservation of extracellular vesicle biogenesis pathways but divergence of RNA loading machinery between phyla. Int. J. Parasitol. 2020;50:655–661. doi: 10.1016/j.ijpara.2020.04.004. [DOI] [PubMed] [Google Scholar]
- 9.Collington E., Lobb B., Mazen N.A., Doxey A.C., Glerum D.M. Phylogenomic Analysis of 155 Helminth Species Reveals Widespread Absence of Oxygen Metabolic Capacity. Genome Biol. Evol. 2023;15 doi: 10.1093/gbe/evad135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu Y., Yu L., Fan H., Huang G., Wu Q., Nie Y., Liu S., Yan L., Wei F. Genomic Signatures of Coevolution between Nonmodel Mammals and Parasitic Roundworms. Mol. Biol. Evol. 2021;38:531–544. doi: 10.1093/molbev/msaa243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luo G., Gong R., Li P., Li Q., Wei X. Comparative genomic analysis of Echinococcus multilocularis with other tapeworms. Biologia. 2022;77:2743–2750. [Google Scholar]
- 12.Montaño K.J., Cuéllar C., Sotillo J. Rodent Models for the Study of Soil-Transmitted Helminths: A Proteomics Approach. Front. Cell. Infect. Microbiol. 2021;11 doi: 10.3389/fcimb.2021.639573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosa B.A., Choi Y.-J., McNulty S.N., Jung H., Martin J., Agatsuma T., Sugiyama H., Le T.H., Doanh P.N., Maleewong W., et al. Comparative genomics and transcriptomics of 4 Paragonimus species provide insights into lung fluke parasitism and pathogenesis. GigaScience. 2020;9 doi: 10.1093/gigascience/giaa073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J. Genomics of the Parasitic Nematode Ascaris and Its Relatives. Genes. 2021;12 doi: 10.3390/genes12040493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doyle S.R. Improving helminth genome resources in the post-genomic era. Trends Parasitol. 2022;38:831–840. doi: 10.1016/j.pt.2022.06.002. [DOI] [PubMed] [Google Scholar]
- 16.Howe K.L., Bolt B.J., Shafie M., Kersey P., Berriman M. WormBase ParaSite- a comprehensive resource for helminth genomics. Mol. Biochem. Parasitol. 2017;215:2–10. doi: 10.1016/j.molbiopara.2016.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Howe K.L., Bolt B.J., Cain S., Chan J., Chen W.J., Davis P., Done J., Down T., Gao S., Grove C., et al. WormBase 2016: expanding to enable helminth genomic research. Nucleic Acids Res. 2016;44:D774–D780. doi: 10.1093/nar/gkv1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Valiente-Mullor C., Beamud B., Ansari I., Francés-Cuesta C., García-González N., Mejía L., Ruiz-Hueso P., González-Candelas F. One is not enough: On the effects of reference genome for the mapping and subsequent analyses of short-reads. PLoS Comput. Biol. 2021;17 doi: 10.1371/journal.pcbi.1008678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang X., Lee W.-P., Ye K., Lee C. One reference genome is not enough. Genome Biol. 2019;20:104. doi: 10.1186/s13059-019-1717-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee Y.-C., Ke H.-M., Liu Y.-C., Lee H.-H., Wang M.-C., Tseng Y.-C., Kikuchi T., Tsai I.J. Single-worm long-read sequencing reveals genome diversity in free-living nematodes. Nucleic Acids Res. 2023;51:8035–8047. doi: 10.1093/nar/gkad647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Solares E.A., Tao Y., Long A.D., Gaut B.S. HapSolo: an optimization approach for removing secondary haplotigs during diploid genome assembly and scaffolding. BMC Bioinf. 2021;22:9. doi: 10.1186/s12859-020-03939-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adams M., McBroome J., Maurer N., Pepper-Tunick E., Saremi N.F., Green R.E., Vollmers C., Corbett-Detig R.B. One fly-one genome: chromosome-scale genome assembly of a single outbred Drosophila melanogaster. Nucleic Acids Res. 2020;48:e75. doi: 10.1093/nar/gkaa450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doyle S.R., Søe M.J., Nejsum P., Betson M., Cooper P.J., Peng L., Zhu X.-Q., Sanchez A., Matamoros G., Sandoval G.A.F., et al. Population genomics of ancient and modern Trichuris trichiura. Nat. Commun. 2022;13:3888. doi: 10.1038/s41467-022-31487-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Howe K., Chow W., Collins J., Pelan S., Pointon D.-L., Sims Y., Torrance J., Tracey A., Wood J. Significantly improving the quality of genome assemblies through curation. GigaScience. 2021;10 doi: 10.1093/gigascience/giaa153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rhie A., McCarthy S.A., Fedrigo O., Damas J., Formenti G., Koren S., Uliano-Silva M., Chow W., Fungtammasan A., Kim J., et al. Towards complete and error-free genome assemblies of all vertebrate species. Nature. 2021;592:737–746. doi: 10.1038/s41586-021-03451-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lefoulon E., Vaisman N., Frydman H.M., Sun L., Voland L., Foster J.M., Slatko B.E. Large Enriched Fragment Targeted Sequencing (LEFT-SEQ) Applied to Capture of Wolbachia Genomes. Sci. Rep. 2019;9:5939. doi: 10.1038/s41598-019-42454-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Foster J., Ganatra M., Kamal I., Ware J., Makarova K., Ivanova N., Bhattacharyya A., Kapatral V., Kumar S., Posfai J., et al. The Wolbachia genome of Brugia malayi: endosymbiont evolution within a human pathogenic nematode. PLoS Biol. 2005;3 doi: 10.1371/journal.pbio.0030121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Michalski M.L., Griffiths K.G., Williams S.A., Kaplan R.M., Moorhead A.R. The NIH-NIAID Filariasis Research Reagent Resource Center. PLoS Negl. Trop. Dis. 2011;5 doi: 10.1371/journal.pntd.0001261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tracey A., Foster J.M., Paulini M., Grote A., Mattick J., Tsai Y.-C., Chung M., Cotton J.A., Clark T.A., Geber A., et al. Nearly Complete Genome Sequence of Brugia malayi Strain FR3. Microbiol. Resour. Announc. 2020;9 doi: 10.1128/MRA.00154-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bashir A., Klammer A., Robins W.P., Chin C.-S., Webster D., Paxinos E., Hsu D., Ashby M., Wang S., Peluso P., et al. A hybrid approach for the automated finishing of bacterial genomes. Nat. Biotechnol. 2012;30:701–707. doi: 10.1038/nbt.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.English A.C., Richards S., Han Y., Wang M., Vee V., Qu J., Qin X., Muzny D.M., Reid J.G., Worley K.C., Gibbs R.A. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS One. 2012;7 doi: 10.1371/journal.pone.0047768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koren S., Schatz M.C., Walenz B.P., Martin J., Howard J.T., Ganapathy G., Wang Z., Rasko D.A., McCombie W.R., Jarvis E.D., Phillippy A.M. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 2012;30:693–700. doi: 10.1038/nbt.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neal-McKinney J.M., Liu K.C., Lock C.M., Wu W.-H., Hu J. Comparison of MiSeq, MinION, and hybrid genome sequencing for analysis of Campylobacter jejuni. Sci. Rep. 2021;11:5676. doi: 10.1038/s41598-021-84956-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.George S., Pankhurst L., Hubbard A., Votintseva A., Stoesser N., Sheppard A.E., Mathers A., Norris R., Navickaite I., Eaton C., et al. Resolving plasmid structures in Enterobacteriaceae using the MinION nanopore sequencer: assessment of MinION and MinION/Illumina hybrid data assembly approaches. Microb. Genom. 2017;3 doi: 10.1099/mgen.0.000118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rödelsperger C. The community-curated Pristionchus pacificus genome facilitates automated gene annotation improvement in related nematodes. BMC Genom. 2021;22:216. doi: 10.1186/s12864-021-07529-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koren S., Walenz B.P., Berlin K., Miller J.R., Bergman N.H., Phillippy A.M. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zimin A.V., Marçais G., Puiu D., Roberts M., Salzberg S.L., Yorke J.A. The MaSuRCA genome assembler. Bioinformatics. 2013;29:2669–2677. doi: 10.1093/bioinformatics/btt476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Foth B.J., Tsai I.J., Reid A.J., Bancroft A.J., Nichol S., Tracey A., Holroyd N., Cotton J.A., Stanley E.J., Zarowiecki M., et al. Whipworm genome and dual-species transcriptome analyses provide molecular insights into an intimate host-parasite interaction. Nat. Genet. 2014;46:693–700. doi: 10.1038/ng.3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie Y., Xu Z., Zheng Y., Li Y., Liu Y., Wang L., Zhou X., Zuo Z., Gu X., Yang G. The mitochondrial genome of the dog hookworm Ancylostoma caninum (Nematoda, Ancylostomatidae) from Southwest China. Mitochondrial DNA. B Resour. 2019;4:3002–3004. doi: 10.1080/23802359.2019.1666048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Coster, W. NanoPlot: Plotting Scripts for Long Read Sequencing Data (Github).
- 41.Ondov B.D., Treangen T.J., Melsted P., Mallonee A.B., Bergman N.H., Koren S., Phillippy A.M. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016;17:132. doi: 10.1186/s13059-016-0997-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H. New strategies to improve minimap2 alignment accuracy. Bioinformatics. 2021;37:4572–4574. doi: 10.1093/bioinformatics/btab705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwham A., Keane T., McCarthy S.A., Davies R.M., Li H. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10 doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen S., Zhou Y., Chen Y., Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H., Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marçais G., Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–770. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ruan J., Li H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods. 2020;17:155–158. doi: 10.1038/s41592-019-0669-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shafin K., Pesout T., Lorig-Roach R., Haukness M., Olsen H.E., Bosworth C., Armstrong J., Tigyi K., Maurer N., Koren S., et al. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat. Biotechnol. 2020;38:1044–1053. doi: 10.1038/s41587-020-0503-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kolmogorov M., Yuan J., Lin Y., Pevzner P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019;37:540–546. doi: 10.1038/s41587-019-0072-8. [DOI] [PubMed] [Google Scholar]
- 50.Shen W., Le S., Li Y., Hu F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS One. 2016;11 doi: 10.1371/journal.pone.0163962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bushnell B. Lawrence Berkeley National Lab.(LBNL); 2014. BBMap: A Fast, Accurate, Splice-Aware Aligner. [Google Scholar]
- 52.Guan D., McCarthy S.A., Wood J., Howe K., Wang Y., Durbin R. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics. 2020;36:2896–2898. doi: 10.1093/bioinformatics/btaa025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vaser R., Sović I., Nagarajan N., Šikić M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017;27:737–746. doi: 10.1101/gr.214270.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Walker B.J., Abeel T., Shea T., Priest M., Abouelliel A., Sakthikumar S., Cuomo C.A., Zeng Q., Wortman J., Young S.K., Earl A.M. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9 doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Di Genova A., Buena-Atienza E., Ossowski S., Sagot M.-F. Efficient hybrid de novo assembly of human genomes with WENGAN. Nat. Biotechnol. 2021;39:422–430. doi: 10.1038/s41587-020-00747-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haghshenas E., Asghari H., Stoye J., Chauve C., Hach F. HASLR: Fast Hybrid Assembly of Long Reads. iScience. 2020;23 doi: 10.1016/j.isci.2020.101389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mapleson D., Garcia Accinelli G., Kettleborough G., Wright J., Clavijo B.J. KAT: a K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics. 2017;33:574–576. doi: 10.1093/bioinformatics/btw663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Challis R., Richards E., Rajan J., Cochrane G., Blaxter M. BlobToolKit - Interactive Quality Assessment of Genome Assemblies. G3. 2020;10:1361–1374. doi: 10.1534/g3.119.400908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 60.Wickham, H. ggplot2 (Springer New York).
- 61.R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/.
- 62.Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., Buxton S., Cooper A., Markowitz S., Duran C., et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gurevich A., Saveliev V., Vyahhi N., Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang N., Li H. compleasm: a faster and more accurate reimplementation of BUSCO. Bioinformatics. 2023;39 doi: 10.1093/bioinformatics/btad595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Keilwagen J., Hartung F., Paulini M., Twardziok S.O., Grau J. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinf. 2018;19:189. doi: 10.1186/s12859-018-2203-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Keilwagen J., Wenk M., Erickson J.L., Schattat M.H., Grau J., Hartung F. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 2016;44:e89. doi: 10.1093/nar/gkw092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dainat J. AGAT: Another Gff Analysis Toolkit to Handle Annotations in Any GTF/GFF Format Version v0.7.0. [DOI]
- 68.Pagès H., Aboyoun P., Gentleman R., DebRoy S. 2020. Biostrings: Efficient Manipulation of Biological Strings.https://rdrr.io/bioc/Biostrings/ [Google Scholar]
- 69.Ranallo-Benavidez T.R., Jaron K.S., Schatz M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020;11:1432. doi: 10.1038/s41467-020-14998-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Quality-controlled MinION and Illumina data for each species are deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under the BioProject accession ID PRJNA1074771 and under BioSample accession nos. SAMN39888962 (Brugia malayi), SAMN39888963 (Trichuris trichiura) and SAMN39888964 (Ancylostoma caninum), and are publicly available as of the date of publication. Final assemblies for each species are deposited in the NCBI Genome database under the accession numbers GCA_037903335.1, GCA_037903345.1, and GCA_037903365.1 (Brugia malayi), GCA_037903355.1, GCA_037903375.1, and GCA_037903465.1 (Trichuris trichiura), and GCA_037903475.1, GCA_037903715.1, and GCA_037903745.1 (Ancylostoma caninum), and are publicly available as of the date of publication.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.