

Abstract
Viral myocarditis is characterized by infiltration of mononuclear cells essential for virus elimination. GPR15 has been identified as a homing receptor for regulatory T cells in inflammatory intestine diseases, but its role in inflammatory heart diseases is still elusive. Here we show that GPR15 deficiency impairs coxsackievirus B3 elimination, leading to adverse cardiac remodeling and dysfunction. Delayed recruitment of regulatory T cells in GPR15-deficient mice was accompanied by prolonged persistence of cytotoxic and regulatory T cells. In addition, RNA sequencing revealed prolonged inflammatory response and altered chemotaxis in knockout mice. In line, we identified GPR15 and its ligand GPR15L as an important chemokine receptor–ligand pair for the recruitment of regulatory and cytotoxic T cells. In summary, the insufficient virus elimination might be caused by a delayed recruitment of T cells as well as delayed interferon-γ expression, resulting in a prolonged inflammatory response and an adverse outcome in GPR15-deficient mice.
Subject terms: Cardiovascular diseases, Infectious diseases, Inflammation
Stoffers, Wolf et al. report that, in virus-mediated myocarditis, GPR15 deficiency delays the recruitment of T cells into cardiac tissue, which impairs viral elimination and leads to delayed but also prolonged inflammation and, thus, adverse cardiac outcomes.
Main
Myocarditis is an inflammatory disease of the myocardium characterized by mononuclear cell infiltration1. It is predominantly caused by infectious agents, such as the cardiotropic enterovirus coxsackievirus B3 (CVB3)2. Especially in young adults, myocarditis is a major source of sudden cardiac arrest3–5. However, its clinical course has a broad spectrum of outcomes, ranging from mild symptoms and complete recovery to cardiac dysfunction and dilated cardiomyopathy (DCM)2,6. In more than 65% of patients with DCM of unknown etiology, viral genomes were detected in the myocardium6. DCM after acute viral myocarditis arises due to sustained inflammation predominantly caused by insufficient virus clearance and subsequent virus persistence in the cardiac tissue3,7,8. Thus, virus elimination and dampening cardiac inflammation are eminent steps toward complete recovery for patients suffering from myocarditis. As therapeutic strategies for viral myocarditis are limited, regulators orchestrating virus elimination and cardiac inflammation may display innovative targets for future treatment.
Chemoattractant G-protein-coupled receptors (GPCRs) are critical regulators in recruiting lymphocyte subsets from blood and secondary lymphoid organs to peripheral tissues, such as the heart. Furthermore, they are involved in the migration of these immune cells between and within these organs and tissues in both homeostatic and inflammatory states9,10.
Based on sequence similarities, G-protein-coupled receptor 15 (GPR15) was discovered in 1996 as a chemokine receptor11,12. Functionally, it has been identified as a T cell homing receptor in the context of inflammatory intestine and skin diseases13–17. Because GPR15 controls the specific homing of anti-inflammatory FOXP3+ regulatory T (Treg) cells to the large intestine in mice, GPR15-deficient mice developed more severe inflammation during colitis15. Furthermore, GPR15 is important not only for recruiting Treg cells to the mouse colon but also for effector T cell recruitment in both homeostatic and inflammatory conditions13. Two ligands are known for GPR15: a subunit of the membrane protein thrombomodulin and the chemokine-like ligand GPR15L18–20.
GPR15-mediated recruitment of Treg cells to the gut is essential for dampening inflammation during colitis in mice15, but the role of GPR15 in inflammatory heart diseases is still elusive. In this study, we used the murine model of viral myocarditis of C57Bl/6J (B6) mice, using cardiotropic CVB3 (ref. 21). The B6 mouse strain has a low susceptibility to CVB3 infection, which predominantly results in virus elimination after acute myocarditis22,23. In B6 mice, CVB3-induced myocarditis typically takes place in a three-phased manner1,24. During the first days (days 2–4) post infection (p.i.), CVB3 itself exerts direct cardiotoxic damage, followed by a highly inflammatory phase of immune cell infiltration (days 4–14). In this latter phase, host immunity mostly eliminates the virus from cardiac tissue23. The subsequent stage is characterized by the recovery and reversion of cardiac remodeling. Employing GPR15-deficient mice, we investigated the subacute phase (16 days p.i.) and found that GPR15 deficiency affected the outcome of CVB3-induced myocarditis. To elucidate the patho-mechanistical function of GPR15 during myocarditis, we thoroughly investigated its acute phase (5 days, 6 days and 7 days p.i.). Thereby, we focused on the following questions. Does GPR15 deficiency (1) lead to differences in immune cell infiltrate composition or cytokine expression in the heart during the acute phase of myocarditis; (2) affect the efficient elimination of CVB3 in the heart, thereby altering the outcome; and (3) have an impact on chemotactic migration, adhesion or functionality of T cells?
Results
Poor recovery from myocarditis in GPR15-deficient mice
We examined the regulation of GPR15 in various heart failure entities (Fig. 1a). During the acute phase of CVB3-mediated myocarditis, Grp15 expression was 7.3-fold increased, which was the strongest increase in the heart failure models investigated. Therefore, we aimed to examine the role of GPR15 during viral myocarditis.
Fig. 1. Impact of GPR15 deficiency during the subacute phase of CVB3-induced myocarditis 16 days p.i.

a, Gpr15 expression in LV tissue of WT mice with angiotensin II-induced hypertension (AngII), myocardial infarction (MI) and myocarditis (CVB3). Ct values were normalized to the mean of 18S and Cdkn1b and to the corresponding sham controls (ΔΔCt). Unpaired two-tailed t-test (AngII and CVB3) or ordinary one-way ANOVA with Holm–Sidak correction (MI). n numbers are represented by data points. b, Study design: CVB3-injected mice, which revealed viral RNA neither in blood (4 days) nor in LV (16 days), were excluded from subsequent analyses. Within the arrow: n numbers per group, with CVB3+ mice and the original n number separated by a backslash. c, Virus load in blood and LV tissue. Ct values were normalized to Cdkn1b (ΔCt). Unpaired two-tailed t-test. n numbers according to b. d, Number of mice with verified viremia (4 days) or virus persistence in LV (16 days). n numbers according to b are plotted as stacked bar charts. Two-tailed Fisher’s exact test was performed on the underlying contingency table. e, In situ hybridization visualized a representative region with viral RNA (purple) in LV tissue from two CVB3+ Gpr15gfp/gfp mice with co-stained nuclei (blue). Two technical replicates. f, Body weight in relation to individual initial weight (mean ± s.e.m.). Two-tailed multiple t-tests with Holm–Sidak correction. g, Ratio of biventricular weight to tibia length (mean ± 95% CI). Unpaired two-tailed t-test with Bonferroni correction. h, Representative histological PSR staining of biventricular tissue. j, Quantification of fibrotic areas (mean ± 95% CI). Unpaired two-tailed t-test with Bonferroni correction. k, Ccl2, Ifnγ, Cd8a and Foxp3 expression in LV. Symbols filled in gray and light green indicate mice that cleared virus from LV tissue. Ct values were normalized to the mean of 18S and Cdkn1b and corresponding sham controls (ΔΔCt). Unpaired two-tailed t-test with Bonferroni correction. a,d,k, Gene expression data were plotted as 2−ΔCt or 2−ΔΔCt (geo-mean ± 95% CI). Significant, compared * to sham of the same genotype, # between similarly treated groups of different genotypes. (*, ***, ****; P < 0.05, 0.001, 0.0001). d, days; sh, sham; wk, weeks.
To explore whether GPR15 has an impact on progression of or recovery from viral myocarditis, we first investigated the subacute phase 16 days p.i. Mice with GPR15 deficiency were employed as depicted in Fig. 1b. To prove infection, we verified viremia in small blood samples at day 4 p.i. As depicted in Fig. 1b,d, mice without verified viremia were excluded from subsequent analyses.
Albeit virus load in blood 4 days p.i. was not different between both genotypes (Fig. 1c), significantly more GPR15-deficient mice had persisting virus in left ventricular (LV) tissue at 16 days p.i. Remarkably, 73% of CVB3-infected WT mice (eight of 11) but only 13% of CVB3-infected GPR15-deficient mice (two of 15) cleared the virus from the heart after 16 days (Fig. 1d). In situ hybridization on LV tissue sections revealed that viral RNA was hardly detectable in the subacute phase. As exemplarily shown in Fig. 1e, viral RNA was mainly localized in non-myocyte cells. Despite the inefficient virus elimination from LV tissue, gene expression of chemokine (Ccl2) or T cell markers (Cd8a, cytotoxic T (TC) cells / Foxp3, Treg cells) was not different compared to either infected wild-type (WT) mice or corresponding sham controls (Fig. 1k). Furthermore, CVB3-infected mice, with or without effective virus clearance, revealed similar gene expression of inflammatory markers compared to the respective sham group. However, the significantly increased gene expression of anti-viral Ifnγ in infected GPR15-deficient mice is in line with the detected virus persistence. Decreasing body weight was observed in both infected groups until 6 days p.i. Although WT mice stabilized their body weight afterwards, the weight loss further proceeded in GPR15-deficient mice, construed as a sign of aggravating disease severity (Fig. 1f). This divergent timecourse of body weight started on day 7. In line with this finding, GPR15-deficient mice revealed significantly lower heart weights compared to WT mice at 16 days p.i. (Fig. 1g). This was accompanied by larger lesions, presumably owing to cardiomyocyte death, resulting in more reparative cardiac fibrosis in GPR15-deficient mice (Fig. 1h,j). Interestingly, two of the three WT mice without virus clearance had clearly larger fibrotic lesions than those that had cleared the virus from LV tissue.
Additionally, pressure–volume (PV) loops revealed cardiac dysfunction in both infected groups, indicating that they did not restore their cardiac function in the subacute phase of myocarditis (Table 1). However, GPR15-deficient mice showed a tendency to more impaired cardiac function. Analyzing parameters that specifically characterize systolic function, solely CVB3-infected GPR15-deficient mice showed significantly impaired cardiac function compared to their sham controls, whereas comparison of WT groups did not reach significance. This is displayed, for example, by impaired preload-independent and preload-dependent cardiac contractility (preload-recruitable stroke work (PRSW) −23%, P = 0.0256 and ΔP/Δtmax −29%, P = 0.0272) in GPR15-deficient mice but almost unaffected contractility in WT mice. Analysis of diastolic function revealed significantly slowed relaxation and increased myocardial stiffness (ΔP/Δtmin +27%, P = 0.0151 and Tau +21%, P = 0.0373) in GPR15-deficient mice, which is in line with increased cardiac fibrosis determined by histology (Fig. 1h,j). Additionally, comparing both infected groups normalized to their sham controls, significantly impaired parameters, such as preload-independent cardiac contractility (PRSW −19%, P = 0.0063), maximal pressure (Pmax −11%, P = 0.0175) and relaxation (ΔP/Δtmin +24%, P = 0.0016), were determined in GPR15-deficient mice (Table 1). Even virus clearance and resolved cardiac inflammation, mainly observed in infected WT mice, did not result in preserved cardiac function but may prevent more severe cardiac dysfunction, as detected in GPR15-deficient mice with predominant virus persistence.
Table 1.
Cardiac function in the subacute phase (16 days p.i.) of myocarditis characterized by PV loops
| Sham groups | CVB3 myocarditis groups | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| WT | Gpr15gfp/gfp | WT | vs. Sh | Gpr15gfp/gfp | vs. Sh | Gpr15gfp/gfp vs. WT | ||||
| Change (%) | P valuea | Change (%) | P valuea | Change (%) | P valueb | |||||
| Global function | ||||||||||
| Heart rate (bpm) | 571 ± 16 | 613 ± 13 | 553 ± 19 | −3 | 1.0000 | 552 ± 19 | −10 | 0.0544 | −7 | 0.1514 |
| Cardiac output (ml min−1) | 18.3 ± 1 | 19.2 ± 1.8 | 14.3 ± 1.9 | −22 | 0.3406 | 13.2 ± 1.3 | −31 | 0.0243 | −12 | 0.4487 |
| Stroke volume (µl) | 32 ± 1 | 31 ± 3 | 26 ± 3 | −20 | 0.2492 | 24 ± 2 | −23 | 0.0876 | −4 | 0.7987 |
| Stroke work (µl × mmHg) | 2,699 ± 146 | 2,737 ± 270 | 2,046 ± 270 | −24 | 0.2218 | 1,707 ± 172 | −38 | 0.006 | −18 | 0.2533 |
| Systolic function | ||||||||||
| Ejection fraction (%) | 80 ± 7 | 74 ± 4 | 59 ± 6 | −26 | 0.1042 | 59 ± 5 | −21 | 0.1056 | +6 | 0.6219 |
| PRSW (mmHg) | 75.2 ± 3.2 | 78.6 ± 6.4 | 71.6 ± 3.3 | −5 | 0.9513 | 60.3 ± 3.3 | −23 | 0.0256 | −19 | 0.0063 |
| Pmax (mmHg) | 101 ± 4 | 103 ± 3 | 93 ± 3 | −8 | 0.2366 | 84 ± 3 | −19 | 0.0001 | −11 | 0.0175 |
| ΔP/Δtmax (mmHg s−1) | 10,786 ± 563 | 12,379 ± 1225 | 9,943 ± 1053 | −8 | 1.0000 | 8,759 ± 726 | −29 | 0.0272 | −23 | 0.0642 |
| Diastolic function | ||||||||||
| Ped (mmHg) | 5 ± 1 | 3.9 ± 1 | 3.6 ± 0.7 | −28 | 0.5618 | 3.6 ± 0.9 | −7 | 1.0000 | +29 | 0.4593 |
| ΔP/Δtmin (mmHg s−1) | −9,001 ± 453 | −10,246 ± 906 | −8,684 ± 428 | +4 | 1.0000 | −7,531 ± 436 | +27 | 0.0151 | +24 | 0.0016 |
| Tau (ms) | 4.9 ± 0.1 | 4.5 ± 0.3 | 5.1 ± 0.3 | +4 | 1.0000 | 5.4 ± 0.2 | +21 | 0.0373 | +16 | 0.0648 |
Values are given as mean ± s.e.m. aTwo-tailed t-test with Bonferroni correction. bComparison between infected genotypes was based on values normalized to the corresponding sham group. Unpaired two-tailed t-test. Due to normalization, only one comparison was made, and P value was, therefore, not corrected for multiple testing. P values indicting significance are highlighted in bold. WT sh/CVB3 (n = 6/11); Gpr15gfp/gfp sh/CVB3 (n = 9/13).
Pmax, maximum pressure; ΔP/Δtmax, maximal rate of rise of LV pressure; Ped, end-diastolic pressure; ΔP/Δtmin, maximal rate of decrease of LV pressure; Sh, sham.
Persistently high virus load in Gpr15gfp/gfp mice
Because we hypothesized that the observed differences arise from the acute phase of myocarditis, we investigated CVB3-induced myocarditis in more detail on days 5, 6 and 7 p.i., as depicted in Fig. 2a. Virus load in blood at day 4 p.i. and virus load in LV tissue at 5 days and 6 days p.i. revealed no differences between genotypes (Fig. 2b). At day 7 p.i., however, the virus load in LV tissue of WT mice was significantly lower than in Gpr15gfp/gfp mice, which remained at a high level. Timecourse of body weight showed significant weight loss for both infected groups but no differences between genotypes (Fig. 2c). Compared to sham controls, biventricular heart weight tended to be lower in CVB3-infected mice 6 days and 7 days p.i. in both genotypes (Fig. 2d) but reached significance only in Gpr15gfp/gfp mice on day 6 p.i. With respect to the significantly lower biventricular weight in GPR15-deficient mice at 16 days p.i. (Fig. 1g), we suggest that a disease progression in GPR15-deficient mice becomes apparent at a later timepoint, which would be in line with the decrease in body weight after day 7. Viral RNA visualized by in situ hybridization was localized in cell infiltrates stained with wheat germ agglutinin (WGA) and frequently in cardiomyocytes identified by troponin staining (Fig. 2e), which was in contrast to the subacute phase.
Fig. 2. Impact of GPR15 deficiency during acute phase of CVB3-induced myocarditis 5 days, 6 days and 7 days p.i.

a, Study design to investigate the impact of GPR15 in acute myocarditis. n numbers per group and timepoint are specified within the arrow. CVB3-injected mice, which revealed viral RNA neither in blood (4 days) nor in LV (5 days, 6 days or 7 days), were excluded from subsequent analyses. n numbers of CVB3+ mice included is given, with the original n number after the backslash. b, Virus load was determined in blood and LV tissue. Ct values were normalized to Cdkn1b (ΔCt). c, Body weight plotted as percentage to the individual initial weight (mean ± s.e.m.). Two-tailed multiple t-tests with Holm–Sidak correction. d, Biventricular weight normalized to tibia length (mean ± 95% CI). Unpaired two-tailed t-test with Bonferroni correction. e, In situ hybridization visualized viral RNA (red) in cardiac tissue from a CVB3-infected WT mouse co-stained with WGA (green) and troponin (white). Arrows point to two infected cardiomyocytes (left) or infected infiltrated cells (right). n = 2 mice in two independent experiments. f,g, Gene expression was determined in LV tissue of WT and GPR15-deficient mice (f) from selected chemokines (Ccl2, Ccl5 and Cxcl10) (g), from selected anti-viral interferons (Ifnβ and Ifnγ) and from the anti-inflammatory interleukin Il10. Ct values were normalized to the mean of 18S and Cdkn1b and the corresponding sham controls (ΔΔCt). b,f,g, Gene expression data were plotted as 2−ΔCt or 2−ΔΔCt (geo-mean ± 95% CI). Unless stated otherwise, significance was tested using an unpaired two-tailed t-test with Bonferroni correction. Significant, compared * to sham of the same genotype, # between similarly treated groups of different genotypes. (*, **, ***, ****; P < 0.05, 0.01, 0.001, 0.0001). d, days; sh, sham; wk, week.
Prolonged cardiac inflammation in Gpr15gfp/gfp mice
To investigate the innate and adaptive immune responses in the LV tissue after CVB3 infection, gene expression of chemotactic, anti-viral and anti-inflammatory cytokines was determined (Fig. 2f,g). Overall, these cytokines were significantly increased in the infected LV tissue at 5 days, 6 days and 7 days p.i. compared to respective sham groups, revealing strongly induced cytokine expression during acute myocarditis in both genotypes. Comparing the infected genotypes, no significant differences were observed at 6 days p.i. in contrast to 5 days and 7 days p.i. At 5 days p.i., GPR15-deficient mice showed significantly lower gene expression of Ifnγ. At 7 days p.i., however, infected GPR15-deficient mice exhibited significantly higher gene expression of Ccl2, Ccl5, Cxcl10, Ifnβ, Ifnγ and Il10. This might be caused by declined cytokine levels in infected WT mice but still increased cytokine levels in infected GPR15-deficient mice on day 7.
During the immune response, various immune cells of the adaptive immune system, as well as monocytes and macrophages, are recruited to the infected LV tissue. Therefore, gene expression of specific immune cell markers was examined in LV tissue (Fig. 3a). Although the gene expression of the B cell marker Cd19 was not altered compared to sham controls, the T cell marker Cd3 and the macrophage marker Cd68 were significantly increased in all infected groups. When comparing the infected genotypes, no significant differences were observed for Cd68, but Cd3 was significantly different at 5 days and 7 days p.i. While Cd3 expression at 5 days p.i. was significantly lower in LV tissue of GPR15-deficient mice, it was significantly higher at 7 days p.i. compared to WT mice.
Fig. 3. Analysis of immune cell infiltration in LV tissue during the acute phase 5 days, 6 days and 7 days after CVB3 infection.

a,b, Gene expression was determined in LV tissue of WT and GPR15-deficient mice (n numbers stated in Fig. 2a) from specific immune cell markers for T cells (Cd3), B cells (Cd19) and macrophages (Cd68) (a) and markers for the T cell subpopulations TH (Cd4), TC (Cd8) and Treg cells (Foxp3) (b). Ct values were normalized to the mean of 18S and Cdkn1b and the corresponding sham controls (ΔΔCt). 2−ΔΔCt values were plotted (geo-mean ± 95% CI). c, For all genes that were significantly regulated at any of the timepoints examined, the FC of gene expression compared to sham treatment is shown as a heat map. Significance was tested using a paired two-tailed t-test with Bonferroni correction. d, CD3+ and CD8+ cells were stained on tissue sections from CVB3-infected mice (n numbers stated in Fig. 2a) and then quantified and normalized to tissue area (mean ± 95% CI). Representative images show immune cell infiltrates in GPR15-deficient mice at 7 days p.i. Significance was tested using an unpaired two-tailed t-test with Bonferroni correction unless otherwise stated. Significant, compared * to sham of the same genotype, # between similarly treated groups of different genotypes. (*, **, ***, ****; P < 0.05, 0.01, 0.001, 0.0001). NS, not significant; sh, sham.
Because the expression of the T-cell-specific marker Cd3 revealed significant differences between both genotypes on days 5 and 7 p.i., we subsequently examined markers for the T cell subpopulations T helper (TH, Cd4), TC (Cd8) and Treg cells (Foxp3). In general, CVB3 infection resulted in a slightly increased gene expression of Cd4 but a strong increase of Cd8 and Foxp3 compared to sham controls (Fig. 3b). However, Foxp3 was not increased in GPR15-deficient mice at 5 days p.i. When comparing the infected genotypes, significant differences were detected in the expression of Foxp3 (on days 5 and 7 p.i.) and Cd8 (on day 7 p.i.) but not in the expression of Cd4. Furthermore, none of these markers was differentially expressed on day 6 p.i. This indicates genotype-specific and temporal differences in the infiltration of Treg and TC cells. Although at day 5 p.i. Foxp3 expression remained at basal levels in GPR15-deficient mice but was significantly increased in WT mice, on day 7 p.i. it was significantly higher in GPR15-deficient mice than in WT mice. This suggests delayed recruitment and prolonged persistence of Treg cells. Furthermore, Cd8 was not differentially expressed on day 5 and day 6, but it was significantly lower in WT mice at 7 days p.i. than in GPR15-deficient mice. The latter finding indicates prolonged persistence of TC cells in the infected LV tissue of GPR15-deficent mice. We could strengthen the gene expression data by staining T cell subpopulations on LV tissue sections. Although no significant differences were detected between the two genotypes, significantly more CD3+ and CD8+ cells were detected in the infected GPR15-deficient mice at day 7 and at days 6 and 7, respectively. Representative images of immune cell infiltrates are shown (Fig. 3d and Extended Data Fig. 1a).
Extended Data Fig. 1. Analysis of immune cells in blood, lymph nodes and LV tissue sections.

(a) Representative staining of CD3 (left panel) or CD8 (right panel) in immune cell infiltrates during the acute phase of myocarditis in heart tissue of infected WT and GPR15−deficient mice. Only infected mice were stained, n numbers are shown in Fig. 2a. (b) Gene expression of Gpr15 was determined in blood of WT mice. N numbers stated in Fig. 2a. Gene expression of specific immune cell markers for T cells (Cd3) and T cell sub-populations TH (Cd4), TC (Cd8) and Treg cells (Foxp3) was determined in blood of WT and GPR15-deficient mice. Ct-values were normalised to 18 S and the corresponding sham (sh) controls (ΔΔCt). 2¬ ΔΔCt values were plotted (geo-mean ± 95% CI). Significance was tested using an unpaired two-tailed t-test with Bonferroni correction. (c) Gene expression of Gpr15 was determined in lymph nodes of WT mice. N numbers stated in Fig. 2a. Gene expression of specific immune cell markers for T cells (Cd3) and T cell sub-populations TH (Cd4), TC (Cd8) and Treg cells (Foxp3) was determined in lymph nodes of WT and GPR15-deficient mice. Ct-values were normalised to Cdkn1b and the corresponding sham (sh) controls (ΔΔCt). 2¬ ΔΔCt values were plotted (geo-mean ± 95% CI). Unpaired two-tailed t-test with Bonferroni correction. Significant, compared * to sh of the same genotype, # between similarly treated groups of different genotypes. (*, **, ***, ****; p < 0.05, 0.01, 0.001, 0.0001)
Figure 3c summarizes the results of the gene expression measurements in terms of their temporal course as a heat map for each genotype individually. In WT mice, gene expression of cytokines and immune cell markers increased as early as day 5, remained elevated on day 6 and decreased significantly on day 7. In GPR15-deficient mice, the gene expression significantly increased from day 5 to day 6 p.i. and remained elevated until 7 days p.i. This summary suggests, again, a delayed and prolonged cardiac inflammation during acute myocarditis in GPR15-deficient mice.
In addition, we measured gene expression of the same T cell markers in blood and lymph nodes (Extended Data Fig. 1b,c). Five days p.i., the expression of all investigated markers (Cd3, Cd4, Foxp3 and Cd8) were significantly increased in blood of CVB3-infected mice compared to sham controls and tended to be even higher in blood of infected GPR15-deficient mice. In contrast to blood, in lymph nodes, the expression of Cd4 and Cd8 was significantly lower at day 5, which was in line with the observations in LV tissue. This opposite effect implies that the T cells remain in the circulation due to delayed tissue invasion.
More upregulated differentially expressed genes in Gpr15gfp/gfp mice at 7 days p.i
We aimed to identify the patho-mechanisms behind the adverse outcome observed at day 16 p.i. Given the decreasing body weight, higher virus load and more severe cardiac inflammation in GPR15-deficient mice, we assumed that the divergent disease progression might start at day 7. Thus, we next investigated this turning point by whole transcriptome analysis to determine the differences in gene expression between infected WT and infected GPR15-deficient mice from days 6 and 7. Therefore, 3′ mRNA sequencing was performed on a set of LV tissue samples (six sham controls and 3–6 infected mice per genotype). RNA sequencing revealed more than 30,000 annotated genes whose expression was subsequently compared between different groups.
The 242 genes assigned to the Gene Ontology (GO) term GO:0006915 ‘response to virus’ are highlighted in the volcano plot in Extended Data Fig. 2. At 6 days p.i., approximately 30% of those genes were significantly increased in both genotypes, confirming the response to virus infection. At 7 days p.i., however, the number of significantly increased genes was diminished in WT mice to 16% but remained similar in GPR15-deficient mice. Lastly, both sham groups were compared, and only two slightly downregulated genes of that GO term were found in Gpr15gfp/gfp mice. With respect to the TaqMan measurements in Figs. 3 and 4, we labeled the genes assigned to the highlighted GO term additionally with their gene symbol (Ccl5, Cxcl10, Cd8a, Ifnβ, Ifnγ and Foxp3). This demonstrates that the genes in the volcano plots revealed similar expression, as measured by TaqMan.
Extended Data Fig. 2. Volcano plots to visualise DEGs assigned to the GO term GO:0006915 “response to virus”.

(a) Myocarditis – 6 days: CVB3-infected WT (left) and Gpr15gfp/gfp (right) mice compared to their sham (sh) control 6 days p.i. (b) Myocarditis – 7 days: CVB3-infected WT (left) and Gpr15gfp/gfp (right) mice compared to their sh control 7 days p.i. (c) Comparison of sh WT and sh Gpr15gfp/gfp mice. Transcriptome analysis was performed by MACE-RNA seq of LV tissue. Fold change (FC) and p-value are displayed in the Volcano plot to visualise significant differences in gene expression. Genes assigned to the GO term GO:0006915 “response to virus” were highlighted as follows: (i) Not significantly regulated (black), (ii) significantly upregulated (FC > 1.5 & p-value < 0.05 (light red), p-value < 0.001 (dark red)) or (iii) significantly downregulated (FC < -1.5 & p-value < 0.05 (light blue), p-value < 0.001 (dark blue)). Genes labelled with their symbol are part of this GO term and were quantified by TaqMan analyses and plotted in Fig. 3. Genes not assigned to the GO term GO:0006915 are displayed in grey. DEGs were calculated with DESeq2 using Wald test.
Fig. 4. Identification of DEGs comparing cardiac tissue samples from Gpr15gfp/gfp and WT mice at 6 days and 7 days after CVB3 infection.

a, Transcriptome analysis was performed by bulk RNA sequencing of LV tissue from CVB3-infected Gpr15gfp/gfp mice compared to infected WT mice at 6 days (left) and 7 days (right) p.i. FC on x axis and P value on y axis are displayed in the volcano plot. Genes that reveal an FC of at least ±1.5 and P < 0.05 are highlighted. DEGs downregulated in GPR15-deficient mice are shown in blue (P < 0.05, light blue; P < 0.001, dark blue), whereas upregulated DEGs are shown in red (P < 0.05, light red; P < 0.001, dark red). The five most significant upregulated and downregulated DEGs are labeled. DEGs were calculated with DESeq2 using the Wald test. b, Heat map of all top DEGs (FC ± 1.5 and P < 0.001) on day 6 (left) and day 7 (right) p.i. Gene expression is normalized to the mean of the respective infected WT group and plotted as FC (log2) for each sample separately. DEGs were calculated with DESeq2 using the Wald test. c, To validate sequencing data, gene expression of three top DEGs on day 7 (Gbp6, Parp14 and Irgm1) was determined in all mice (n numbers stated in Fig. 2a). Ct values were normalized to the mean of 18S and Cdkn1b and to the corresponding sham controls (ΔΔCt). Unpaired two-tailed t-test with Bonferroni correction. 2−ΔΔCT values were plotted (geo-mean ± 95% CI). Significant, compared * to sham of the same genotype, # between similarly treated groups of different genotypes. (*, **, ***, ****; P < 0.05, 0.01, 0.001, 0.0001). d, days; Reg., regulated; sh, sham.
The comparison of infected genotypes is depicted as volcano plots in Fig. 4a for 6 days p.i. (left) and 7 days p.i. (right). Differentially expressed genes (DEGs) with a P value lower than 0.05 and a fold change (FC) higher or lower than 1.5 or −1.5 are highlighted. Comparing both volcano plots revealed more DEGs on day 7 (Fig. 4a, right). Although at 6 days p.i., 372 DEGs were detected (250 upregulated (red) and 122 downregulated (blue) in Gpr15gfp/gfp mice), 540 DEGs were identified (404 upregulated (red) and 136 downregulated (blue)) at 7 days p.i. The DEGs were further restricted by P < 0.001, resulting in top DEGs. The 30 top DEGs for 6 days p.i. and the 48 top DEGs for 7 days p.i are displayed as a heat map in Fig. 4b.
To confirm the RNA sequencing data, three of the top DEGs from day 7 were quantified, including all mice, using TaqMan. Therefore, we selected genes related to immune response. As shown in Fig. 4c, the gene expression of Gbp6, Parp14 and Irgm1 was similar between both infected genotypes at 6 days p.i. and revealed higher expression in GPR15-deficient mice at 7 days p.i.
Chemotaxis and T-cell-related GO terms are enriched at day 7
To go beyond single genes, RNA sequencing data were further processed using GO enrichment analyses. First, we focused on general differences between the infected genotypes on both days. Comparing the DEGs from days 6 and 7, we identified 69 DEGs that were similarly regulated on both days (Fig. 5a). Although 11 DEGs were downregulated, 58 were upregulated in GPR15-deficient mice (Fig. 5a and Extended Data Fig. 3). Based on the overlapping DEGs, we identified GO terms including at least three of these 69 similarly regulated DEGs. GO terms with more than 2,000 annotated genes and P > 0.01 were excluded, and closely related or redundant GO terms were removed using REVIGO25. The remaining 17 terms with the assigned DEGs are depicted in the GO chord graph (Fig. 5b). GPR15-deficent mice revealed upregulated DEGs that were assigned to ‘response to virus’ and ‘defense response’ (highlighted in bold). The 17 GO terms are plotted as a dot plot to visualize their regulation and significance (Fig. 5c). Of note, all GO terms were enriched with upregulated genes in infected Gpr15gfp/gfp mice (red z-scores). Besides general terms—for example, ‘defense response’ and ‘immune system process’—we also found more specific terms, such as ‘cell migration’ and ‘cellular response to interferon-alpha’. Again, it became obvious that the difference between both genotypes was more pronounced on day 7. These results indicate that the cardiac inflammation as response to virus infection seems to be prolonged in the GPR15-deficient mice.
Fig. 5. Upregulated GO terms related to T-cell-mediated immune response and chemotaxis in infected Gpr15gfp/gfp mice on day 7 p.i.

a, Venn diagram visualizes the overlap of upregulated and downregulated DEGs between the infected genotypes on both days p.i. Sixty-nine DEGs—58 upregulated and 11 downregulated—showed similar regulation on days 6 and 7 in GPR15-deficient mice. b, GO terms with at least three of the 69 DEGs were selected. Only GO terms with fewer than 2,000 genes and P < 0.01 were considered, further narrowed down by REVIGO and presented in the GO chord graph. The genes on the left side are connected by lines to the appertaining GO term on the right side. DEGs assigned to ‘response to virus’ and ‘defense response’ are highlighted in bold. c, GO terms from the GO chord (b) are depicted as a dot plot for days 6 and 7. GO terms are sorted according to the q value on day 7. d, Based on the differential expression between both infected groups, 957 GO terms of the domain BP were significantly regulated on day 6 and/or day 7. Using semantic similarity, GO terms were grouped into eight clusters. Within one cluster, the number of GO terms based on their P value is displayed as stacked bar charts to compare the distribution of significantly regulated GO terms between days 6 and 7. e, The number of GO terms of clusters 2 and 6 are shown in detail using alluvial plots. This visualizes changes of significantly regulated GO terms from day 6 to day 7. f, Dot plot of selected GO terms of all three domains between both infected groups on day 7. Selection is based on gene ratio (DEGs/annotated genes) greater than 0.1 and q < 0.001. Redundant and closely related GO terms were removed by REVIGO. GO terms assigned to clusters 2 and 6 are highlighted in bold. c,f, Size of the squares represents the number of annotated genes, and color represents the z-score. Red z-scores indicate more upregulated than downregulated DEGs in GPR15-deficient mice. Terms are sorted according to the –log10 of q value, which is displayed on the x axis. b–f, Significance of GO term enrichment was calculated by Fisher’s exact test based on DEGs (P < 0.05 and FC > +1.5 or FC < −1.5). Adjusted P values (q values) of GO terms were determined using the Benjamini–Hochberg correction. d, days; NS, not significant.
Extended Data Fig. 3. 69 similar regulated DEGs found by comparing DEGs from day 6 and day 7.

Heat maps of the 69 DEGs (58 up- and 11 downregulated in GPR15-deficient mice, FC + /-1.5 and a p-value < 0.001) that were similar regulated on day 6 (left) and 7 (right) p.i. Gene expression is normalized to the mean of the respective CVB3-infected WT group and plotted as fold change (FC, log2) for each sample separately. DEGs were calculated with DESeq2 using Wald test.
Next, we aimed to investigate the changes from day 6 to day 7 in more detail. Therefore, we selected all GO terms from the domain biological process (BP) that were significantly enriched with DEGs between both infected genotypes on day 6 (358 GO terms), on day 7 (782 GO terms) or on both days (183 GO terms). These GO terms were clustered by semantic similarity, as shown in Extended Data Fig. 4. The GO terms of each cluster were sorted based on their P value as follows: (1) not significant (NS), (2) significant (P < 0.05, gray) and (3) highly significant (P < 0.01, dark gray). As shown in Fig. 5d, the number of significantly and highly significantly enriched GO terms noticeably increased from day 6 to day 7. This can be observed in almost all clusters but is particularly clear for cluster 2 (‘chemotaxis/migration immune cells’) and cluster 6 (‘immune response/alphabeta T cells’). These changes of significance are displayed as alluvial plots in Fig. 5e. In cluster 2 (‘chemotaxis/migration immune cells’), only eight of 48 GO terms were significantly enriched on day 6. In contrast, 47 GO terms were significantly or highly significantly enriched with DEGs on day 7. Similarly, cluster 6 (‘immune response/alphabeta T cells’), containing 179 GO terms, revealed 62 significantly enriched GO terms on day 6, which was increased to 158 significantly enriched GO terms on day 7. In summary, significantly enriched GO terms regarding chemotaxis and T cell immune response became evident for day 7.
Extended Data Fig. 4. Similarity matrix of significant GO terms of the domain biological process.

957 GO terms that were significantly different on day 6 and, or on day 7 between both infected genotypes were clustered based on semantic similarity using the R package simplify enrichment. “Lin” method was used to calculate the similarity matrix for these GO terms. 15 semantic similarity clusters were determined (right) and were further summarized into 8 main clusters due to strong similarities (left, C1-C8).
Due to these results, we examined the 7-day timepoint in more detail to identify important GO terms out of all three domains (BP, cellular component (CC) and molecular function (MF)). To identify the most prominently enriched GO terms, the 939 significantly enriched GO terms from day 7 were restricted by gene ratio > 0.1 and q < 0.001 and narrowed down using REVIGO to finally 25 GO terms, which were plotted in Fig. 5f. These 25 GO terms are almost exclusively related to the immune response to a viral infection and include genes significantly upregulated in infected Gpr15gfp/gfp mice. Of note, many GO terms are included in clusters 2 and 6 (highlighted in bold). Particularly noteworthy are GO terms linked to chemotaxis and to T cells. So far, the GO analyses of RNA sequencing data showed marked upregulation of genes in GPR15-deficient mice on day 7 assigned to GO terms associated with chemotaxis of immune cells and T-cell-mediated immune response.
GPR15 deficiency abolished chemotaxis of T cells
So far, the previous results point toward alterations in chemotaxis and cardiac inflammation, particularly associated with T cells, in GPR15-deficient mice during myocarditis. Based on those findings, we investigated the interactions of GPR15 and its two known ligands, GPR15L and the EGF-like domain 5 of thrombomodulin (TME5), in vitro.
Regarding the results of the GO analysis, which particularly highlighted chemotaxis, we hypothesized that GPR15 may function as a chemokine receptor mediating T cell homing during myocarditis. In addition to the receptor expression, we, therefore, reconsidered the blood samples of infected mice to evaluate the gene expression of the ligand Gpr15l in the acute phase of myocarditis. The Gpr15l expression was significantly increased in blood of GPR15-deficient mice at 4 days p.i. as well as 5 days p.i. (Fig. 6a). Next, we performed in vitro stimulation experiments on resident cardiac cells using the pro-inflammatory cytokine TNFα to investigate the regulation of Gpr15l. As shown in Fig. 6b, endothelial heart cells (MHEC-5T) and primary cardiac fibroblasts (cFBs), but not HL1 cells (Extended Data Fig. 5a), revealed an increased gene expression of Gpr15l after TNFα stimulation.
Fig. 6. GPR15-expressing splenocytes and their interactions with the ligand GPR15L.

a, Gpr15l gene expression in blood (n numbers stated in Fig. 2a). Ct values were normalized to 18S and the corresponding sham controls (ΔΔCt). Significance was tested using an unpaired two-tailed t-test with Bonferroni correction. b, Gpr15l gene expression in MHEC-5T (25 ng ml−1, 5 h, n = 4 biological replicates) and cFBs (10 ng ml−1, 6 h, n = 13 biological replicates in three independent experiments) after TNFα stimulation. Ct values were normalized to 18S and the corresponding untreated controls (ΔΔCt). Unpaired two-tailed t-test. c, Left y axis: splenocytes, pooled from 3–5 WT mice, were sorted into four CD45+ groups—CD4+CD25− TH, CD4+CD25+ Treg, CD8+ TC and DN cells—using FACS. Gpr15 gene expression was determined in three independent experiments. Ct values were normalized to the mean of Cdkn1b and 18S and to the DN cells (ΔΔCt). Repeated-measures one-way ANOVA. Right y axis: splenocytes from Gpr15gfp/gfp mice (n = 8) were analyzed by flow cytometry. Percentage of GFP+ cells was determined in the same four groups (mean ± 95% CI). Repeated-measures one-way ANOVA. d, Evaluation of actin polymerization mediated by GPR15L. Isolated T cells (pooled from 2–3 WT mice) were incubated with 500 nM GPR15L for 180 s, immediately fixed and subsequently labeled and analyzed by flow cytometry. Actin polymerization was determined in two independent experiments. Median fluorescence intensity of six biological replicates was normalized to non-stimulated cells (mean ± 95% CI). Unpaired two-tailed t-test. e, GPR15L-mediated migration of primary splenocytes by Boyden chamber assays (n = 12) in three independent experiments. After 3 h, migrated cells were analyzed by flow cytometry. Results were normalized to cells passively migrated to media without GPR15L (mean ± 95% CI). Unpaired two-tailed t-test with Bonferroni correction. Gene expression data were plotted as 2−ΔΔCt (geo-mean ± 95% CI). Significant, compared * to control or control of the same genotype, # between similarly treated groups of different genotypes. (*, **, ***, ****; P < 0.05, 0.01, 0.001, 0.0001). d, days; sh, sham.
Extended Data Fig. 5. Adhesion assay and analysis of GPR15-mediated T cell functionality.

(a) Gpr15l gene expression in HL1 cardiomyocytes after TNFα stimulation (10 ng/ml, 6 h, n = 4 biological replicates). Ct-values were normalised to Cdnk1b and the corresponding untreated control (ΔΔCt). Unpaired two-tailed t-test. (b) Quiescent or activated T cells were incubated in the presence or absence of the receptor agonist GPR15L (500 nM). Percentage of positive cells for the intracellular markers (GranzymeB (GrB), IL10, IL17, IFNγ, TNF-α) was quantified for three different T cell populations: CD4+CD25-: T helper cells (TH); CD4+CD25+: regulatory T cells (Treg); CD8+: cytotoxic T cells (TC). (c) Thrombomodulin (Thbd) expression after TNFα stimulation in MHEC‐5 T (25 ng/mL, 5 h, n = 4 biological replicates). Ct‐values were normalised to 18 S and the untreated control (ΔΔCt). Gene expression data were plotted as 2-ΔΔCt (geo-mean ± 95% CI). Unpaired two‐tailed t‐test. (d) MHEC‐5 T monolayer were either cultured with growth medium alone or supplemented with 25 ng/mL TNFα for 24 h. Adhesion of primary splenocytes to MHEC‐5 T monolayer (n = 6-13 biological replicates in 3 independent experiments) was investigated during constant flow for 20 min. The adhesion strength of primary splenocytes was analysed after 5 min of flow pause to allow cells to adhere before flow was turned on to remove nonadherent splenocytes. Adherent splenocytes were counted via FIJI (mean ± 95% CI). Unpaired two‐tailed t‐test. Significant, compared * to control or control of the same genotype (*, **,****; p < 0.05, 0.01, 0.0001).
Subsequently, we investigated the impact of GPR15 on T cell chemotaxis. In a first approach, we treated WT T cells with the ligand GPR15L and quantified induced actin polymerization by phalloidin staining (Fig. 6d). Subsequent flow cytometry analysis revealed that TC and Treg cells significantly increased their actin polymerization in response to GPR15L treatment. Because actin polymerization is a key mechanism in the process of cell migration, we next tested the migration of lymphocytes in a Boyden chamber assay. Therefore, the chemotactic migration of WT or GPR15-deficient splenocytes toward the ligand GPR15L was examined. As depicted in Fig. 6e, the ligand GPR15L significantly increased the number of migrated T cells, with Treg and TC cells showing the highest effect. This chemotaxis was completely abolished in cells lacking the receptor GPR15.
Of note, both chemotaxis assays presented here indicate a T-cell-specific effect. Thus, we quantified the gene expression of Gpr15 in different T cell subsets (Fig. 6c, left y axis). Although the TH cells revealed only slightly higher Gpr15 gene expression compared to double-negative (DN) cells, the expression in both Treg and TC cells was remarkably higher. Additionally, we used Gpr15gfp/gfp splenocytes to quantify the number of GFP+ cells, here used as equivalent of GPR15 protein expression, within the different T cell subsets by flow cytometry (Fig. 6c, right y axis). In contrast to TH cell populations, TC and Treg cell populations revealed significant increased number of GFP+ cells compared to DN lymphocytes, with TC showing the highest number. This is in line with the Gpr15 gene expression data.
To address whether GPR15 alters the functionality of T cells, isolated T cells were activated in the absence or presence of the receptor agonist GPR15L. As shown in Extended Data Fig. 5a, GPR15L itself did not significantly change the protein expression of various T cell activation markers. TME5, a domain of the transmembrane protein thrombomodulin on endothelial cells, is described as another GPR15 ligand19. Pro-inflammatory stimulation of endothelial heart cells using TNFα led to an increased gene expression of thrombomodulin (Extended Data Fig. 5b). To investigate the influence of GPR15 on leukocyte adhesion on endothelial cells, a flow assay on an endothelial monolayer was performed. As depicted in Extended Data Fig. 5c, the number of adherent splenocytes increased on TNFα-activated endothelial cells but was not different between WT and GPR15-deficient splenocytes. In terms of adhesion strength, on average around 50% of the splenocytes remained firmly attached to the stimulated endothelial cells, regardless of the presence of GPR15 (Extended Data Fig. 5b). These results indicate that GPR15 has no relevant impact on cell adhesion to endothelial cells.
Taken together, GPR15 mediated the chemotactic migration of Treg and TC cells toward its chemoattractant ligand GPR15L rather than contributing to leukocyte attachment before transendothelial migration or influencing T cell function. Therefore, our experiments strengthen the hypothesis that GPR15 is essential for the recruitment of T cells to the site of inflammation in the acute phase of CVB3-induced myocarditis.
IFNγ reduced CVB3 replication in cardiomyocytes
Furthermore, we aimed to investigate the causes and subsequent effects of the lower IFNγ levels in the LV tissue of infected GPR15-deficient mice at day 5 p.i. To this end, we analyzed Ifnγ gene expression in and IFNγ secretion from activated T cell subtypes in vitro (Fig. 7a,b). Although the increase of Ifnγ gene expression after stimulation was significant only in Tc cells, IFNγ secretion was significantly increased in all T cell subtypes, with the increase being highest in TC cells. This indicates that activated TC cells are one of the main sources of IFNγ in the infected myocardium.
Fig. 7. IFNγ in different T cell subtypes and its influence on virus load and replication in HL1 cells.

a, Isolated T cells (pooled from four mice) were activated and sorted by FACS. Ifnγ gene expression was determined in three independent experiments. Ct values were normalized to Cdkn1b. Unpaired two-tailed t-test. Gene expression data were plotted as 2−ΔCt (mean ± s.e.m.). b, Isolated T cells (n = 4 biological replicates, each pooled from 2–3 mice) were activated to determine IFNγ secretion. The number of IFNγ+ cells was normalized to the number of IFNγ+ cells in the respective control group (mean ± 95% CI). Unpaired two-tailed t-test. c, HL1 cells were treated with 100 ng ml−1 IFNγ for 20 h before infection with 0.5 MOI CVB3 for 1 h. IFNγ treatment was maintained during and after infection (n = 12 biological replicates in three independent experiments). HL1 cells were lysed 6 h or 24 h after CVB3 infection. To determine CVB3 virus load, isolated RNA was reversely transcribed by random primer. Ct values were normalized to Hprt and untreated CVB3-infected cells (6 h p.i.) (ΔΔCt). Unpaired two-tailed t-test with Bonferroni correction. 2−ΔΔCt values were plotted (geo-mean ± 95% CI). To quantify virus replication, isolated RNA was reversely transcribed using the tagged (−) strand-specific primer. Copy number of the CVB3 minus strand was calculated based on a purified and quantified PCR product. The number of samples without detectable replication is shown below the dot plot. After 6 h, virus replication was not detectable in seven samples of the IFNγ-treated group (geo-mean ± 95% CI). Unpaired two-tailed t-test with Bonferroni correction. d, Virus replication was determined in LV tissue after 5 days, 6 days and 7 days p.i. (n numbers are shown in Fig. 2a). The number of samples without detectable replication is shown below the dot plot. Unpaired two-tailed t-test with Bonferroni correction (geo-mean ± 95% CI). Unpaired two-tailed t-test with Bonferroni correction. Significant, compared * to control, # between similarly treated groups of different genotypes. (*, **, ****; P < 0.05, 0.01, 0.0001). d, days; h, hours.
Next, we analyzed the influence of IFNγ on virus load and replication in infected HL1 cells (Fig. 7c). Therefore, IFNγ-treated HL1 cells were infected with 0.5 multiplicity of infection (MOI) CVB3. Although IFNγ-treated cells revealed no difference in virus load but less virus replication at 6 h p.i., at 24 h after infection both virus load and replication were decreased compared to untreated CVB3-infected cells.
Considering that Ifnγ gene expression was lower in GPR15-deficient mice 5 days p.i. (Fig. 2g), we returned to the animal experiment and examined virus replication in LV tissue after 5 days, 6 days and 7 days p.i. As shown in Fig. 7d, comparing infected WT and GPR15-deficient mice, virus replication in LV tissue was not different at 5 days and 6 days p.i. In contrast, virus replication was not detected at 7 days p.i. in any of the infected WT mice but was detected in all infected GPR15-deficient mice. This suggests a late consequence on virus replication of the lower Ifnγ expression in these mice at 5 days p.i.
Discussion
In this study, we examined the consequences of GPR15 deficiency on development, progression and recovery in experimental CVB3-induced viral myocarditis. Our main findings are as follows: (1) GPR15 deficiency led to insufficient virus elimination accompanied by further aggravated disease severity and impaired cardiac function in the subacute phase of myocarditis. (2) Although similar on day 5 and day 6, GPR15-deficient mice exhibited higher cardiac virus load at day 7 p.i. (3) GPR15-deficient mice exhibited a delayed recruitment of Treg cells and lower Ifny expression at day 5 p.i. but more pronounced inflammatory response at day 7 p.i. compared to WT mice. (4) Bulk RNA sequencing revealed that the response to virus did not decline from day 6 to day 7 in GPR15-deficient mice as observed in WT mice. Significant enrichment of upregulated DEGs in GO terms related to chemotaxis and TC cells in GPR15-deficient mice on day 7 was shown. (5) GPR15 was highly expressed on Treg and TC cells, and its deficiency abolished chemotaxis of T cells toward GPR15L in vitro.
GPR15 was discovered owing to structural homology to known chemokine receptors11. In humans, GPR15 is highest expressed in the intestine and lymphoid tissues and is found on T and B cells in various organs (Human Protein Atlas: GPR15; https://www.proteinatlas.org/ENSG00000154165-GPR15). In the murine intestine, GPR15 is preferentially expressed on distinct T cell subsets, particularly Treg and TC cells, whereas a very low number of TH cells express GPR15 (ref. 15).
GPR15 appears to be a counter-regulator of inflammation under patho-physiological conditions;26 its knockout exacerbates skin16 and colon15 inflammation in mice. During colitis, the receptor is described as a T cell homing receptor especially for Treg cells, but, in other organs, such as skin, it also regulates the migration of other T cell subsets during acute or chronic inflammatory states18. Furthermore, clinical cohort studies identified GPR15 as a major risk for cardiovascular diseases27–29. In various murine heart failure models, Gpr15 expression was highly increased during the inflammatory stage of myocardial infarction and even more during acute viral myocarditis. Therefore, GPR15-deficient mice were investigated in the context of CVB3-induced viral myocarditis.
The non-susceptible C57BL/6J mouse strain used in this study develops acute myocarditis to eliminate the virus, resolves cardiac inflammation thereafter and, finally, recovers from myocarditis21,22,30. Kim et al.15 unveiled GPR15 as a homing receptor for Treg cells during colitis and showed that GPR15 is not required for controlling the infection but, rather, for dampening the immune response. In the present study, we first analyzed the CVB3-induced myocarditis in the subacute phase at 16 days p.i. Starting with a similar virus load, the virus elimination was significantly impaired in GPR15-deficient mice. Although we determined virus persistence in most GPR15-deficient mice, they managed to dispel cardiac inflammation. However, cardiac function was impaired, accompanied by enhanced cardiac fibrosis, in GPR15-deficient mice.
Next, we extensively studied the key moment (5 days, 6 days and 7 days p.i.) when body weight development diverged between WT and GPR15-deficient mice, which may emphasize the deteriorated disease progression and severity in GPR15-deficent mice. WT and GPR15-deficient mice revealed equal virus loads in LV tissue at days 5 and 6 p.i., although, at day 5 p.i., the gene expression of Ifnγ, Cd3 and Foxp3 was significantly lower in GPR15-deficient mice. Lower Foxp3 expression suggests delayed recruitment of Treg cells. However, this conclusion is based only on gene expression data, as the barely abundant cell type of Treg cells is hardly detectable by histology and flow cytometry. At day 6 p.i., both genotypes showed similar inflammatory responses in the cardiac tissue. In line with the divergent weight development, virus load and inflammatory response started to decline in infected WT mice at day 7 but remained consistently high in GPR15-deficient mice. We, therefore, investigated this turning point between day 6 and day 7 by RNA sequencing. GO term analyses uncovered several chemotaxis-related terms to be significantly enriched with highly upregulated genes in GPR15-deficent mice at 7 days p.i. Because GPR15 is described as a chemokine receptor11,12, especially on T cells, we assume that GPR15 deficiency causes a primarily delayed immune cell recruitment and, afterwards, a prolonged inflammatory response, resulting in an impaired outcome. Similarly, deficiency of other chemokine receptors, CCR5 or CX3CR1, leads to aggravated pathogen-induced myocarditis described by increased mortality or impaired cardiac function31,32.
Two ligands are known for GPR15: GPR15L, a soluble chemoattractant protein, and TME5, a subunit of the integral membrane protein thrombomodulin18,19. In our study, Gpr15l expression was upregulated in cardiac residential cells after pro-inflammatory stimulation facilitating T cell recruitment to the site of injury, as shown in other models20. In vitro, GPR15 deficiency abolished chemotactic migration of Treg and TC cells toward GPR15L, supporting the hypothesis that GPR15 acts as a chemokine receptor. This finding was further strengthened by increased actin polymerization after GPR15L treatment in Treg and TC cells. In addition, we investigated the influence of GPR15 on the adhesion of splenocytes to endothelial cells via the ligand TME5 expressed on the cell surface of endothelial cells and on cytokine production of T cells, whereby no influence of GPR15 could be proven.
Previous studies showed that both TC and Treg cells have an impact on the outcome of CVB3-induced myocarditis. Adoptive Treg transfer during the inflammatory phase of viral myocarditis or before induction of myocarditis protects the heart against inflammatory damage33,34. Here, delayed recruitment of Treg cells is followed by prolonged inflammation and more severe cardiac damage. Henke et al.35 showed that depletion of TC cells led to improved survival in the early phase of CVB3-induced myocarditis but increased virus load, emphasizing their important role in virus elimination35. Here, we show that activated TC cells highly increase their IFNγ secretion and that IFNγ reduces virus load and virus replication in CVB3-infected cardiomyocytes in vitro. Therefore, we assume that TC cells are one of the main sources of the increased Ifnγ expression in vivo and that the lower Ifnγ expression in GPR15-deficient mice might cause the impaired virus elimination. However, the exact cause of the decreased Ifnγ expression 5 days p.i. in the LV tissue of GPR15-deficient mice cannot be elucidated here. It can be assumed that the recruitment not only of Treg cells but also that of TC cells is impaired by GPR15 deficiency. This would correspond to our in vitro results, according to which TC cells react strongly chemotactically to the ligand GPR15L. Furthermore, IFNγ has been shown not only to be an anti-viral agent in itself36 but also to reduce fibrosis and to prevent the development of severe chronic myocarditis37. The here-observed late sequela of myocarditis by GPR15 deficiency might be traced back to delayed recruitment of Treg cells, but possibly also TC cells, into the virus-infected myocardium.
In conclusion, we identified GPR15 as a chemokine receptor crucial for a better outcome after acute viral myocarditis. In vitro, GPR15 facilitates GPR15L-mediated recruitment of T cells, particularly TC cells essential for virus elimination and Treg cells essential for dampening cardiac inflammation. In myocarditis, GPR15 deficiency leads to delayed migration of Treg cells to the infected myocardium, resulting in inflammatory-induced cardiac injury from day 7 p.i. In addition, expression of Ifnγ was delayed in LV tissue of GPR15-deficient mice, which might lead to ongoing virus replication in the early phase of myocarditis and, thus, virus persistence later. In line with Kim et al.15, GPR15 is a homing receptor for Treg cells and important for dampening the cardiac inflammation. Moreover, the GPR15-mediated migration of TC cells is essential for virus elimination in viral myocarditis to prevent progression to heart failure.
Methods
Animal model
For the present study, we employed the previously described knock-in mouse strain (B6; 129P2-Gpr15tm1.1Litt/J) in which the endogenous Gpr15 gene was replaced by the sequence of the GFP15. Thus, these mice can be used as Gpr15 knockouts. Mice were bred on B6 background, and male littermates or offsprings from littermates identified as homozygous B6 WT or GPR15-deficient mice were used for subsequent experiments.
Additionally, cDNA from LV tissue obtained by previous studies was analyzed38. Three murine heart failure models were investigated: (1) chronic hypertension induced by continuous infusion of angiotensin II, (2) myocardial infarction induced by permanent occlusion of the left anterior descending artery and (3) CVB3-induced myocarditis21,39,40.
Experimental viral myocarditis
As depicted in Figs. 1b and 2a, male B6 WT (Gpr15+/+) or GPR15-deficient (Gpr15gfp/gfp) mice were used at the age of 7–9 weeks to induce experimental viral myocarditis21. Therefore, 5 × 105 plaque-forming units (PFU) of CVB3 (Nancy strain) were injected intraperitoneally under short-time CO2/O2 anesthesia. Sham controls were treated equally, but saline was used for injection. On day 4, blood was drawn via facial vein to prove infection. The body weight was monitored daily. If blood sampling did not yield sufficient RNA to prove viremia, but virus infection was proven in the LV tissue later, mice were included into the analysis. CVB3-infected mice that did not show virus load in blood at day 4 p.i. or in LV tissue later were excluded from the study. Timelines studying acute or subacute phase of myocarditis are depicted in Figs. 2a and 1b, respectively. The n numbers of included CVB3+ mice and the original n numbers are shown separately for each experiment and were calculated using G*Power (version 3.1.9.7).
To study the acute phase, mice were killed 5 days, 6 days or 7 days p.i. Blood was taken from the beating heart and snap frozen in liquid nitrogen. After heart explantation, atria were removed, and hearts were transversally cut to create a cross-section that was then fixed in 10% neutral-buffered formalin solution for 24 h. The other part of the LV tissue was immediately snap frozen in liquid nitrogen and stored at −80 °C. In addition, lymph nodes were harvested for the 5-day timepoint and snap frozen in liquid nitrogen. To study the subacute phase, 16 days p.i., cardiac function was recorded by hemodynamic measurements. Heart and blood were collected and processed as described above.
All mice were housed under pathogen-free conditions in the animal facility of the University Medical Centre Hamburg-Eppendorf at 22 °C with ad libitum access to water and standard laboratory chow diet. All animal experiments were approved by the local bioethics committee of Hamburg, Germany (G13/115, G15/060, N060/2020, ORG821 and ORG1068) and conform to the Guide for the Care and Use of Laboratory Animals, published by the US National Research Council (8th edition, revised 2011)41.
Hemodynamics
A PV loop system (ADV500, Transonic) was used for hemodynamic measurements in closed-chest approach39,42. Mice were anesthetized using urethane (0.8–1.2 g kg−1 body weight) accompanied by buprenorphine analgesia (0.1 mg kg−1 body weight). First, a tracheotomy was performed for artificial ventilation. Subsequently, a pressure–conductance catheter (1.2 F, Transonic) was inserted in the right carotid artery and carefully pushed forward into the LV. The catheter’s position inside the ventricle was optimized until rectangular-shaped loops were obtained. PV loops were recorded under short-time apnea. The inferior caval vein (ICV) was occluded by gentle compression during the PV loop measurements. Using the wash-in technique, a bolus of hypertonic saline (10%) was injected into the left jugular vein to estimate the volume43. Data were acquired using iox2 (version 2.9.5.73, Emka Technologies). Subsequent analyses of PV loops were performed in LabChart 7.3 Pro (ADInstruments). For baseline analysis, 5–10 consecutive loops during end-expiratory ventilation pause were selected to calculate preload-dependent parameters. Preload-independent parameters were analyzed by selecting loops during ICV occlusion43. Mice with too deep anesthesia or excessive bleeding during the measurement were excluded from the analysis.
RNA-based analyses
RNA isolation
Total RNA was isolated from snap-frozen tissue samples using QIAzol lysis reagent and further purified using an miRNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. Previously, frozen tissue was disrupted by stainless steel beads in a 2 -ml tube filled with QIAzol using a tissue lyser II (Qiagen). To obtain total RNA from blood and cells, an RNeasy Mini Kit (Qiagen) was used according to the manufacturer’s protocol. To avoid genomic DNA contamination within the isolated RNA, DNase-I (Qiagen) was applied directly on the column during the purification protocol. RNA concentration was determined using a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific). RNA was stored at −80 °C.
Reverse transcription and gene expression analysis
RNA was reversely transcribed into cDNA using a high-capacity cDNA kit (Life Technologies). Depending on the underlying experiment, we used 1 µg of RNA from tissue samples, 0.1 µg from blood and 0.25 µg from cell culture experiments for cDNA synthesis. Reverse transcription was carried out at 37 °C for 2 h, followed by an inactivation step of 5 min at 85 °C. The resulting cDNA was diluted to a final working concentration of 10 ng µl−1 for tissue samples or 1.25 ng µl−1 for blood and cell culture samples.
Quantitative real-time PCR was performed to assess gene expression of target genes using 2.5 µl of gene expression master mix (Thermo Fisher Scientific) and 0.25 µl of gene expression assay (Supplementary Table 1). CVB3 load was determined using primers and probe (Supplementary Table 2)24,44. A volume of 1 µl of cDNA was used as template in a final volume of 5 µl. Each sample was analyzed in duplicates. Real-time PCR was performed on a QuantStudio 7 Flex or QuantStudio 7 Pro system (Thermo Fisher Scientific) using QuantStudio software version 1.3 or Design and Analysis software 2.6.0, respectively. Gene expression of Cdkn1b and/or 18S or Hprt was determined as endogenous controls, and ΔCt values were calculated for the target genes. The formula 2−ΔCt was used to calculate absolute gene expression, and obtained values were plotted as x-fold to Cdkn1b or 18S or the mean of both or Hprt. For normalization, mean of ΔCt values of the respective control group was used to calculate ΔΔCt values. Relative gene expression data were determined using the formula 2−ΔΔCt and plotted as x-fold to the respective control45.
Detection of virus replication
Virus replication was determined by strand-specific reverse transcription of the intermediate minus RNA strand of CVB3, which is essential for production of the viral genome of viral progeny. Therefore, isolated total RNA was reversely transcribed into strand-specific cDNA using a high-capacity cDNA kit (Life Technologies) in the presence of 1 µM minus strand-specific primer (RT-CVB3_SE_tagged; 5′-TGAGATAATTGCCCTGAATGCGGCTAATCC-3′, TIB Molbiol). To avoid subsequent detection of non-specific cDNA synthesis, the minus strand-specific primer for reverse transcription was tagged by adding 11 nucleotides at the 5′ end (Supplementary Table 3). In addition, cDNA synthesis was performed in the absence of the minus strand-specific primer as an additive control of specificity. Subsequently, the tagged minus strand-specific cDNA was quantified with specific primers amplifying only tagged cDNA by real-time PCR as described above. Copy number was calculated using a purified and quantified PCR product, because endogenous control genes were not available.
MACE RNA sequencing analysis
Massive analysis of cDNA ends (MACE) is a 3′ mRNA sequencing method based on the analysis of Illumina reads derived from fragments that originate from 3′ mRNA ends46. Thirty-one RNA samples from the acute myocarditis model were used for RNA sequencing (WT sham (n = 6) / Gpr15gfp/gfp sham (n = 6) / WT CVB3 (6 days n = 6; 7 days n = 3) / Gpr15gfp/gfp CVB3 (6 days n = 5; 7 days n = 5)). RNA samples were processed by GenXPro using the MACE Kit (version 2) according to the manufacturerʼs manual. In brief, RNA was fragmented, and polyadenylated mRNA was enriched and amplified by competitive PCR after poly(A)-specific reverse transcription and template-switch-based second-strand syntheses. Duplicate reads as determined by the implemented unique molecular identifiers (TrueQuant IDs) were removed from the raw dataset. Low-quality sequence bases were removed by Cutadapt software (version 2.3)47, and poly(A) tails were clipped by an in-house Python script. The reads were mapped to the mouse genome (mm10), and transcripts were quantified by HTSeq. Differential gene expression was determined using DESeq2 (version 1.20)48 and plotted as volcano plots using Graph Pad Prism (GraphPad Software). For selected DEGs, gene expression of all replicates was visualized as a heat map. A pseudo-count was introduced for every not-expressed gene to allow logarithmic calculation after normalization. The normalized gene count was standardized to the mean of the normalized gene count of the respective sham group and plotted as log2 FC. For data visualization, the R statistical software (version 3.6.3, R Foundation for Statistical Computing) tool ComplexHeatmap was used49.
GO enrichment analysis
GO enrichment analysis was performed using the R package topGO (version 2.42.0)50. GO annotation data were based on ENSEMBL. Enrichment of GO terms was calculated by Fisher’s exact test based on DEGs (P < 0.05 and FC of at least ±1.5). The results were visualized using different R packages. Clustering of GO terms was performed with the R package simplifyEnrichment (version 1.0.0)51. First, the semantic similarity measurement was calculated based on the method of Lin52. The resulting similarity matrix was then clustered by the k-means method. Based on their P values, GO terms of particular clusters were split into three categories and plotted as alluvial plots created with ggforce (version 0.3.3)53 and ggplot2 (version 3.3.3)54. The latter was also used to generate dot plots. Color of the symbol is based on the GO term’s z-score. It is calculated for each GO term by taking the number of upregulated DEGs, subtracting the number of downregulated DEGs and dividing this number by the square root of the number of annotated genes. Whether most DEGs in this GO term are upregulated or downregulated in Gpr15gfp/gfp mice is indicated by the color gradient from red to blue, respectively. The chord plot was generated using GOChord from the R package GOplot (version 1.0.2)55. It represents the association of genes to GO terms. The chord plot also visualizes the direction of regulation for depicted genes, with red representing upregulation and blue representing downregulation in Gpr15gfp/gfp mice.
Histological analyses
Cross-sections of murine hearts were fixed in 10% neutral-buffered formalin solution, dehydrated and embedded in paraffin (formalin-fixed paraffin-embedded (FFPE)) afterwards. Then, 4-µm-thick cross-sections were cut using a microtome. For all staining methods, FFPE sections were deparaffinized and rehydrated in a descending ethanol series.
Picrosirius red staining
Picrosirius red (PSR) staining was used to assess fibrosis of the cardiac tissue. Rehydrated slides were incubated for 90 s in Mayer’s hemalum solution (Merck Millipore) diluted 1:1 with distilled water to stain acidic structures and then blued in tap water at approximately 60 °C for 10 min. The basic tissue structures were counterstained by incubation for 60 s in a 1% eosin solution (Merck Millipore) slightly acidified with glacial acetic acid (Roth). For collagen staining, a PSR stain kit (Polysciences) was used according to the manufacturer’s protocol. Then, sections were dehydrated using an ascending ethanol series, followed by incubation in xylene substitute. Finally, the sections were covered with EUKITT (ORSAtec).
Chromogenic immunohistochemistry
After dewaxing and inactivation of endogenous peroxidases (PBS/3% hydrogen peroxide), antigen retrieval was performed using the Ventana BenchMark XT machine. Sections were incubated with anti-CD3 or anti-CD8 antibody (Supplementary Table 4) for 32 min. Anti-Rabbit Histofine Simple Stain MAX PO conjugated with universal immunoperoxidase polymer (medac-diagnostika) was used as secondary antibody. Detection of secondary antibody was performed with an ultraView Universal DAB Detection Kit (Ventana). For subsequent counterstaining, Hematoxylin Ventana Roche and Bluing Reagent Ventana Roche were used. Regarding chromogenic immunohistological staining and subsequent quantification of CD3+ and CD8+ T cells in LV tissue: for one GPR15-deficient CVB3-infected mouse (5 days), FFPE tissue was not available. Furthermore, for one GPR15-deficient CVB3-infected mouse (5 days) and one WT CVB3-infected mouse (5 days), CD8 staining was not successful.
RNAscope
An RNAscope Multiplex Fluorescent v2 Reagent Kit (323100, ACD) or an RNAscope 2.5 High Definition Reagent Kit-RED (322350, ACD) was used to perform in situ hybridization to detect CVB3 plus strand RNA on FFPE sections of ventricular tissue56,57. In brief, sections were incubated for 1 h at 60 °C and deparaffinized by xylene and 100% ethanol. To quench internal peroxidase activity, sections were incubated with hydrogen peroxide incubation for 10 min, followed by target retrieval at 95 °C for 10 min. Sections were permeabilized with Protease Plus at 40 °C for 30 min. Probes were incubated for 2 h at 40 °C, followed by RNAscope amplification steps. For subsequent immunofluorescent staining, sections were blocked with 3% BSA/TBS at room temperature for 2 h. Primary anti-troponin T antibody was incubated overnight at 4 °C in 1% BSA/TBS. After washing, sections were incubated with Alexa Fluor 488–coupled secondary antibody and Alexa Fluor 633–coupled wheat germ agglutinin (WGA) for 2 h at room temperature. The slides were mounted in DAPI Fluoromount-G. For chromogenic signal detection, nuclei were counterstained using hematoxylin. More detailed information about antibodies, probes and reagents is provided in Supplementary Table 5.
Microscopy
Images of PSR or chromogenic immunohistochemistry staining were captured using a BZ 9000 microscope (Keyence) with ×10 CFI PL APO Lbd. (NA = 0.45), ×20 CFI PL APO Lbd. (NA = 0.75) or ×60 CFI PL APO Lbd. H (NA = 1.4) objective and processed with BZ II Analyzer software. Fibrotic areas were quantified using FIJI (version 2.14.0). To quantify positive immune cells, tissue area of 2–3 cross-sections per mouse was measured with QuPath (version 0.4.3)58, whereas positive cells were counted manually. To capture confocal images, a Leica TCS SP5 confocal microscope (Leica Microsystems) with ×40 HCX PL APO CS (NA = 1.3) and ×63 HCX PL APO Lbd. Bl. oil (NA = 1.4–0.6) objectives was used. A maximum projection of three-dimensional images was created using Leica LAS AF software over the full range of the signal.
Cell-based analyses
Murine cardiac fibroblasts
Primary murine cardiac fibroblasts were obtained by multiple repeated digestions of minced LV tissue from male WT B6 mice (10–12 weeks old) using 0.1 mg ml−1 Liberase (Roche)59,60. Obtained cardiac fibroblasts were resuspended in DMEM (Pan) containing 20% FCS (Thermo Fisher Scientific) and 100 U ml−1 penicillin–streptomycin (Thermo Fisher Scientific) and rapidly attached to cell culture flasks. Cells were kept in a humidified atmosphere at 37 °C, with 5% CO2 and 95% air.
Isolation of splenocytes and T cells
Primary lymphocytes from spleen were isolated from male WT and GPR15-deficient B6 mice at 8–16 weeks of age39. Spleen was removed, kept in ice-cold DPBS and mashed through a 70-µm cell strainer. Cells in the flow-through were collected, washed with DPBS and passed through a 40-µm cell strainer. For purification, cell suspension was carefully layered over 3 ml of Histopaque-1077 (Sigma-Aldrich) and centrifuged at 400g for 30 min. The splenocytes, contained in the interface, were aspirated, washed with DPBS and finally resuspended in RPMI VLE 1640 media (Pan) supplemented with 0.5% BSA. Before subsequent experiments, splenocytes were rested for 2 h in a humidified atmosphere at 37 °C, with 5% CO2 and 95% air. In case of subsequent T cell isolation by magnetic cell separation, Pan T Cell Biotin-Antibody Kit II (Milteny Biotec) was used according to the manufacturer’s protocol.
Chemotaxis assay
Freshly isolated primary splenocytes were used for a Boyden chamber assay. Cell culture inserts with 3 µm pore size (353492, Falcon) were coated with 50 µl of fibronectin (10 µg ml−1) at 37 °C for 1 h. Fibronectin solution was removed, and inserts were washed once with DPBS and dried at room temperature. Next, 1 × 106 splenocytes in 250 µl of medium were added to the inserts and settled for 30 min. Wells of suited companion plate (Falcon) were filled with 700 µl of media as negative control or media supplemented with 500 nM GPR15L (25-78, Phoenix Pharmaceuticals). Inserts containing splenocytes were applied to the wells of the companion plate. Cell migration was allowed for 3 h under cell culture conditions. Thereafter, inserts were removed to access the lower chamber containing the migrated cells. To determine the initial population, splenocytes were added directly to 700 µl of medium without an interjacent insert and used to calculate the percentage of migration.
Migrated splenocytes were analyzed using flow cytometry analyses (Supplementary Fig. 1). First, splenocytes were blocked with 2% rat serum (STEMCELL Technologies) and 500 ng ml−1 anti-CD16/CD32 monoclonal antibody (clone 2.4G2, Bio X Cell) for 5 min at room temperature. Next, splenocytes were incubated with the staining master mix (Supplementary Table 6). Pacific Orange was used for live/dead discrimination. After washing once with DPBS, splenocytes were resuspended in 500 µl of DPBS for flow cytometry analysis (LSRFortessa, BD Biosciences, using FACSDiva software (version 9.0.1)). Single-stained controls were used for compensation. Flow cytometry data were analyzed using FCSalyzer software (version 0.9.22-alpha). The gating strategy for this analysis is shown in Supplementary Fig. 1.
Analysis of GPR15-expressing T cells
Using splenocytes isolated from WT mice, Gpr15 expression of T cell populations was examined on transcriptomic level. WT splenocytes were sorted by fluorescence-activated cell sorting (FACS) (Aria IIIu, BD Biosciences, using FACSDiva software (version 9.0.1)) and stained with the master mix (Supplementary Table 6) as described in section 5.4.3. Cells were sorted into tubes filled with 1 ml of RLT buffer containing 1% β-mercaptoethanol for immediate cell lysis. RNA isolation and gene expression analyses were performed as described in section 5.2. GFP fluorescence as equivalent for GPR15 expression of T cell populations was examined using flow cytometry. Therefore, Gpr15gfp/gfp splenocytes were stained as described above. GFP+ cells were counted during the flow cytometry analysis. The gating strategy is shown in Supplementary Fig. 2.
Actin polymerization assay
Isolated T cells were incubated with or without 500 nM GPR15L for 180 s and immediately fixed in 4% paraformaldehyde at room temperature for 20 min. Extracellular staining master mix (Supplementary Table 7) was added and incubated for 30 min at 4 °C in the dark. Cells were washed in DPBS supplemented with 10% FCS and subsequently fixed with IC Fixation Buffer (eBioscience) for 30 min at 4 °C. Afterwards, cells were washed in permeabilization buffer (eBioscience) and incubated in 1× Phalloidin-iFluor 488 (Abcam) diluted in permeabilization buffer for 30 min at 4 °C in the dark. Cells were washed again in permeabilization buffer, fixed with CellFIX (BD Biosciences) and acquired in a FACSCanto (BD Biosciences, using FACSDiva software (version 9.0.1)). Phalloidin median fluorescence intensity of different T cell populations was analyzed with FlowJo software (version 10.8.1). The respective gating strategy is shown in Supplementary Fig. 3.
IFNγ secretion assay
Isolated T cells were activated with Dynabeads Mouse T-Activator CD3/CD28 (Thermo Fisher Scientific). In brief, Dynabeads were incubated with T cells in a 1:1 ratio for 12 h in a 24-well plate under cell culture conditions. After removal of Dynabeads, IFNγ secretion was detected using a Mouse IFNγ Secretion Assay (Milteny Biotec) as described in the user manual. For subsequent flow cytometric analysis, cells were stained with staining master mix (Supplementary Table 8) and acquired in a FACSCanto II (BD Biosciences). IFNγ secretion was analyzed for different cell populations using FlowJo software (version 10.8.1). The respective gating strategy is shown in Supplementary Fig. 4.
Ifnγ gene expression in T cell subsets
Isolated T cells were activated with Dynabeads Mouse T-Activator CD3/CD28 as described in the previous section. After removal of Dynabeads, cells were stained with the staining master mix (Supplementary Table 8) and sorted by FACS (Aria III, BD Biosciences, using FACSDiva software (version 9.0.1)). Cells were sorted into tubes filled with 2–3 ml of RLT buffer containing 1% β-mercaptoethanol for immediate cell lysis. RNA isolation and subsequent TaqMan gene expression analyses were performed as described in section 5.2.
IFNγ treatment of CVB3-infected HL1 cells
HL1 cells (Merck, SCC065) were plated on 24-well plates and grown until confluence. Then, cells were stimulated with 100 ng ml−1 IFNγ (PeproTech) in RPMI VLE 1640 medium (0.5 % FCS and 100 U ml−1 penicillin–streptomycin) for 20 h in the incubator (37 °C, 5% CO2 and 95% air). Subsequently, cells were infected with 0.5 MOI CVB3. After 1 h, virus suspension was removed, and cells were further incubated for 23 h. Cells were lysed with 350 µl of RLT buffer containing 1% β-mercaptoethanol. RNA isolation and subsequent gene expression analyses were performed as described in section 5.2.
T cell activation assay
Isolated T cells were activated with Dynabeads Mouse T-Activator CD3/CD28 (Thermo Fisher Scientific) and/or 500 nM GPR15L (25-78, Phoenix Pharmaceuticals) as described in previous sections. For subsequent FACS analysis, beads were removed magnetically, and T cells were resupended in DPBS supplemented with 10% FCS. Next, extracellular staining master mix (Supplementary Table 9) was added and incubated at 4 °C for 30 min in the dark. Cells were washed in DPBS supplemented with 10% FCS and subsequently fixed with IC Fixation Buffer (eBioscience) at 4 °C for 30 min. Afterwards, cells were washed in permeabilization buffer (eBioscience), and the staining master mix for intracellular staining (Supplementary Table 10) was added and incubated at 4 °C for 45 min in the dark. Once again, cells were washed in permeabilization buffer, fixed with CellFIX (BD Biosciences) and acquired in an LSRFortessa (BD Biocencies). Positive cells for the intracellular markers (granzyme B, IL-17, IFNγ and TNFα) were counted for different T cell populations using FlowJo software (version 10.8.1). The respective gating strategy is shown in Supplementary Fig. 5.
Flow adhesion assay
In vitro flow adhesion assay was used to determine the attachment of splenocytes to endothelial cells. First, 100 µl of cell suspension containing 5 × 104 MHEC-5T cells (DSMZ, ACC 336) was injected into the channel of μ-slides (0.4 Luer ibiTreat, ibidi)61. All subsequent steps were performed in a humidified atmosphere at 37 °C, with 5% CO2 and 95% air. After allowing cells to adhere for 4 h, ports were filled with growth medium (DMEM (Pan), 10% FCS, 100 U ml−1 penicillin and 100 µg ml−1 streptomycin), and slides were placed on a shaker with slow speed to ensure constant medium exchange. Approximately 12 h later, growth medium was carefully replaced by starvation medium (DMEM and 0.5% FCS) for 6–8 h. To enhance thrombomodulin expression, cells were stimulated with 25 ng ml−1 recombinant murine TNFα (PeproTech) in starvation medium for 24 h. Splenocytes were stained in medium supplemented with 2 µM CellTracker Red CMTPX (Thermo Fisher Scientific) for 30 min and rested for 2 h. Then, 1 × 107 stained splenocytes were resuspended in 500 µl of medium and applied to the in vitro flow adhesion assay.
We used ibidi perfusion sets (WHITE: length 50 cm, ID 0.8 mm, 10-ml reservoirs) connected to a fluidic unit (ibidi, 10902), which was attached to a pump building up a continuous flow. First, the perfusion set was filled with pre-warmed 5 ml of RPMI VLE 1640 medium containing 0.5% BSA and attached to the μ-slide containing the endothelial monolayer, and air bubbles were removed. Next, the μ-slide was placed in an incubation unit (Tokai Hit, 5% CO2 and 37 °C) inside a fluorescence microscope (BZ-9000, Keyence). The fluidic unit was placed in an incubator (37 °C and 5% CO2). To start the flow adhesion assay, unidirectional flow with a shear stress of 1 dyne per cm2 was applied, and splenocytes were added to the perfusion set. To record the adhesion of splenocytes to the MHEC-5T monolayer, images were taken every 30 s for 20 min using bright-field and red fluorescence channel (tetramethylrhodamine (TRITC)). To investigate the adhesion strength, flow was switched off for 5 min. Subsequently, flow was switched on to remove not-adhered splenocytes, and images were captured. Adhered splenocytes were counted with FIJI software (version 2.14.0). The ratio between the total number of splenocytes and adherent splenocytes was calculated to analyze adhesion strength.
Statistics
Gene expression data (2−ΔCt and 2−ΔΔCt values) are displayed with geometric mean ± 95% confidence interval (CI). All other data are presented as mean ± 95% CI, unless otherwise stated. All statistical analyses were performed using Gaussian normally distributed data. Hence, the statistics for the gene expression analyses were calculated with ΔCt or ΔΔCt values. In general, statistical comparison of two groups was performed using the unpaired two-tailed t-test with P < 0.05 considered as statistically significant. If more than two groups from both genotypes, WT and Gpr15gfp/gfp, were compared, Bonferroni correction was additionally used to adjust for multiple comparisons. If more than two groups from one genotype, either WT or Gpr15gfp/gfp, were compared, ordinary one-way ANOVA or repeated-measures one-way ANOVA (matched data) was used, each corrected with the Holm–Sidak method. To test for significance of daily weight measurements, multiple t-tests with Holm–Sidak correction were used. Fisher’s exact test was used for the analysis of contingency tables. Significance of GO term enrichment was calculated by Fisher’s exact test based on DEGs (P < 0.05 and FC > +1.5 or FC < −1.5). Adjusted P values (q values) of GO terms were determined using the Benjamini–Hochberg correction. All data were analyzed using GraphPad Prism 6 (versions 6.07 and 9.5.1) or R studio (version 4.0.2).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary Information
Supplementary Figs. 1–5 and Supplementary Tables 1–10
Source data
Source data and exact P value for comparisons
Acknowledgements
We thank the core unit for cytometry and cell sorting; the UKE Microscopy Imaging Facility at the University Hospital Centre Hamburg-Eppendorf for providing flow cytometers, microscopes and support; and GenXPro for performing MACE analyses and bioinformatics. Furthermore, the authors thank O. E. Meyfarth and R. Scholz for technical assistance. We greatly appreciate the assistance of K. Hartmann and S. Krasemann (HEXT Mouse Pathology Core Facility, UKE Hamburg) in processing histological samples. We thank the Lighthouse Core Facility (LCF) for support with cell sorting and flow cytometry. LCF is funded, in part, by the Medical Faculty, University of Freiburg (project nos. 2021/A2-Fol and 2021/B3-Fol) and the German Research Foundation (DFG) (project no. 450392965). The transgenic DsRed mice were kindly provided by R. Kesselring (Department of General and Visceral Surgery, Medical Center, University of Freiburg). H.W., T.V., T.M., D. Wolf, I.H., D. Westermann and D.L. are members of SFB1425, funded by the DFG (project no. 422681845). This work was supported by the German Centre for Cardiovascular Research (DZHK) (FKZ 81Z0710108 to D. Westermann and D.L.).
Extended Data
Author contributions
Conceived and designed the experiments: B.S., H.W., D. Wolf, I.H., D. Westermann and D.L. Performed the experiments: B.S., H.W., L.B., S.K., T.V., T.M., I.Y., A.A. and D.L. Analyzed the data: B.S., H.W., L.B., T.V., T.M., F.E. and D.L. Contributed materials/analysis tools: B.S., H.W., L.B., T.V., T.M., M.A.B., P.M.B., F.E., S.V.K., K.K., P.K., S.B., T.Z., D. Wolf, I.H., D. Westermann and D.L. Wrote the manuscript: B.S., H.W., L.B., D. Westermann and D.L.
Peer review
Peer review information
Nature Cardiovascular Research thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Data availability
RNA sequencing data are available under GSE248521. Source data of all main figures are provided.
Code availability
The code used for processing of RNA sequencing data (poly(A) tail clipping) is available at https://github.com/AGLindner/GPR15-RNA-Sequencing.
Competing interests
All authors declare no competing financial interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Bastian Stoffers, Hanna Wolf.
Change history
8/23/2024
A Correction to this paper has been published: 10.1038/s44161-024-00540-x
Extended data
is available for this paper at 10.1038/s44161-023-00401-z.
Supplementary information
The online version contains supplementary material available at 10.1038/s44161-023-00401-z.
References
- 1.Fung, G., Luo, H., Qiu, Y., Yang, D. & McManus, B. Myocarditis. Circ. Res.118, 496–514 (2016). 10.1161/CIRCRESAHA.115.306573 [DOI] [PubMed] [Google Scholar]
- 2.Blauwet, L. A. & Cooper, L. T. Myocarditis. Prog. Cardiovasc. Dis.52, 274–288 (2010). 10.1016/j.pcad.2009.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cooper, L. T. Jr Myocarditis. N. Engl. J. Med.360, 1526–1538 (2009). 10.1056/NEJMra0800028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schultz, J. C., Hilliard, A. A., Cooper, L. T. Jr. & Rihal, C. S. Diagnosis and treatment of viral myocarditis. Mayo Clin. Proc.84, 1001–1009 (2009). 10.1016/S0025-6196(11)60670-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fabre, A. & Sheppard, M. N. Sudden adult death syndrome and other non-ischaemic causes of sudden cardiac death. Heart92, 316–320 (2006). 10.1136/hrt.2004.045518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuhl, U. et al. High prevalence of viral genomes and multiple viral infections in the myocardium of adults with ‘idiopathic’ left ventricular dysfunction. Circulation111, 887–893 (2005). 10.1161/01.CIR.0000155616.07901.35 [DOI] [PubMed] [Google Scholar]
- 7.Kandolf, R. et al. Mechanisms and consequences of enterovirus persistence in cardiac myocytes and cells of the immune system. Virus Res.62, 149–158 (1999). 10.1016/S0168-1702(99)00041-6 [DOI] [PubMed] [Google Scholar]
- 8.Bouin, A. et al. Enterovirus persistence in cardiac cells of patients with idiopathic dilated cardiomyopathy is linked to 5′ terminal genomic RNA-deleted viral populations with viral-encoded proteinase activities. Circulation139, 2326–2338 (2019). 10.1161/CIRCULATIONAHA.118.035966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lammermann, T. & Kastenmuller, W. Concepts of GPCR-controlled navigation in the immune system. Immunol. Rev.289, 205–231 (2019). 10.1111/imr.12752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dubyak, G. R. GPCRs in innate and adaptive immune responses. In GPCRs: Structure, Function, and Drug Discovery (eds Jastrzebska, B. & Park. P.S.-H.) 429–461 (Elsevier, 2020).
- 11.Heiber, M. et al. A novel human gene encoding a G-protein-coupled receptor (GPR15) is located on chromosome 3. Genomics32, 462–465 (1996). 10.1006/geno.1996.0143 [DOI] [PubMed] [Google Scholar]
- 12.Deng, H. K., Unutmaz, D., KewalRamani, V. N. & Littman, D. R. Expression cloning of new receptors used by simian and human immunodeficiency viruses. Nature388, 296–300 (1997). 10.1038/40894 [DOI] [PubMed] [Google Scholar]
- 13.Nguyen, L. P. et al. Role and species-specific expression of colon T cell homing receptor GPR15 in colitis. Nat. Immunol.16, 207–213 (2015). 10.1038/ni.3079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lahl, K., Sweere, J., Pan, J. & Butcher, E. Orphan chemoattractant receptor GPR15 mediates dendritic epidermal T-cell recruitment to the skin. Eur. J. Immunol.44, 2577–2581 (2014). 10.1002/eji.201444628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim, S. V. et al. GPR15-mediated homing controls immune homeostasis in the large intestine mucosa. Science340, 1456–1459 (2013). 10.1126/science.1237013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jegodzinski, L. et al. The G protein-coupled receptor (GPR) 15 counteracts antibody-mediated skin inflammation. Front. Immunol.11, 1858 (2020). 10.3389/fimmu.2020.01858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fischer, A. et al. Differential effects of α4β7 and GPR15 on homing of effector and regulatory T cells from patients with UC to the inflamed gut in vivo. Gut65, 1642–1664 (2016). 10.1136/gutjnl-2015-310022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ocon, B. et al. A mucosal and cutaneous chemokine ligand for the lymphocyte chemoattractant receptor GPR15. Front. Immunol.8, 1111 (2017). 10.3389/fimmu.2017.01111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pan, B. et al. The fifth epidermal growth factor like region of thrombomodulin alleviates LPS-induced sepsis through interacting with GPR15. Thromb. Haemost.117, 570–579 (2017). 10.1160/TH16-10-0762 [DOI] [PubMed] [Google Scholar]
- 20.Suply, T. et al. A natural ligand for the orphan receptor GPR15 modulates lymphocyte recruitment to epithelia. Sci. Signal.10, eaal0180 (2017). [DOI] [PubMed]
- 21.Becher, P. M. et al. Cardiac function remains impaired despite reversible cardiac remodeling after acute experimental viral myocarditis. J. Immunol. Res.2017, 6590609 (2017). 10.1155/2017/6590609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bacmeister, L. et al. Inflammation and fibrosis in murine models of heart failure. Basic Res. Cardiol.114, 19 (2019). 10.1007/s00395-019-0722-5 [DOI] [PubMed] [Google Scholar]
- 23.Weinzierl, A. O. et al. Effective chemokine secretion by dendritic cells and expansion of cross-presenting CD4−/CD8+ dendritic cells define a protective phenotype in the mouse model of coxsackievirus myocarditis. J. Virol.82, 8149–8160 (2008). 10.1128/JVI.00047-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindner, D. et al. Cardiac fibroblasts aggravate viral myocarditis: cell specific coxsackievirus B3 replication. Mediators Inflamm.2014, 519528 (2014). 10.1155/2014/519528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Supek, F., Bosnjak, M., Skunca, N. & Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE6, e21800 (2011). 10.1371/journal.pone.0021800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bauer, M. The role of GPR15 function in blood and vasculature. Int. J. Mol. Sci.22, 10824 (2021). [DOI] [PMC free article] [PubMed]
- 27.Huan, T. et al. A whole-blood transcriptome meta-analysis identifies gene expression signatures of cigarette smoking. Hum. Mol. Genet.25, 4611–4623 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haase, T. et al. Novel DNA methylation sites influence GPR15 expression in relation to smoking. Biomolecules8, 74 (2018). [DOI] [PMC free article] [PubMed]
- 29.Haase, T. et al. G protein-coupled receptor 15 expression is associated with myocardial infarction. Int. J. Mol. Sci.24, 180 (2022). [DOI] [PMC free article] [PubMed]
- 30.Fairweather, D., Kaya, Z., Shellam, G. R., Lawson, C. M. & Rose, N. R. From infection to autoimmunity. J. Autoimmun.16, 175–186 (2001). 10.1006/jaut.2000.0492 [DOI] [PubMed] [Google Scholar]
- 31.Machado, F. S. et al. CCR5 plays a critical role in the development of myocarditis and host protection in mice infected with Trypanosoma cruzi. J. Infect. Dis.191, 627–636 (2005). 10.1086/427515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muller, I. et al. CX3CR1 knockout aggravates Coxsackievirus B3-induced myocarditis. PLoS ONE12, e0182643 (2017). 10.1371/journal.pone.0182643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pappritz, K. et al. Immunomodulation by adoptive regulatory T-cell transfer improves Coxsackievirus B3-induced myocarditis. FASEB J.10.1096/fj.201701408R (2018). [DOI] [PubMed]
- 34.Shi, Y. et al. Regulatory T cells protect mice against coxsackievirus-induced myocarditis through the transforming growth factor β–coxsackie-adenovirus receptor pathway. Circulation121, 2624–2634 (2010). 10.1161/CIRCULATIONAHA.109.893248 [DOI] [PubMed] [Google Scholar]
- 35.Henke, A., Huber, S., Stelzner, A. & Whitton, J. L. The role of CD8+ T lymphocytes in coxsackievirus B3-induced myocarditis. J. Virol.69, 6720–6728 (1995). 10.1128/jvi.69.11.6720-6728.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang, S., Brown, H. M. & Hwang, S. Direct antiviral mechanisms of interferon-gamma. Immune Netw.18, e33 (2018). 10.4110/in.2018.18.e33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fairweather, D. et al. Interferon-γ protects against chronic viral myocarditis by reducing mast cell degranulation, fibrosis, and the profibrotic cytokines transforming growth factor-β1, interleukin-1β, and interleukin-4 in the heart. Am. J. Pathol.165, 1883–1894 (2004). 10.1016/S0002-9440(10)63241-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Becher, P. M. et al. Role of Toll-like receptors and interferon regulatory factors in different experimental heart failure models of diverse etiology: IRF7 as novel cardiovascular stress-inducible factor. PLoS ONE13, e0193844 (2018). 10.1371/journal.pone.0193844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hinrichs, S. et al. Precursor proadrenomedullin influences cardiomyocyte survival and local inflammation related to myocardial infarction. Proc. Natl Acad. Sci. USA115, E8727–E8736 (2018). 10.1073/pnas.1721635115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Westermann, D. et al. Selective PDE5A inhibition with sildenafil rescues left ventricular dysfunction, inflammatory immune response and cardiac remodeling in angiotensin II-induced heart failure in vivo. Basic Res. Cardiol.107, 308 (2012). 10.1007/s00395-012-0308-y [DOI] [PubMed] [Google Scholar]
- 41.National Research Council. Guide for the Care and Use of Laboratory Animals 8th edn (National Academies Press, 2011). [PubMed]
- 42.Pacher, P., Nagayama, T., Mukhopadhyay, P., Batkai, S. & Kass, D. A. Measurement of cardiac function using pressure–volume conductance catheter technique in mice and rats. Nat. Protoc.3, 1422–1434 (2008). 10.1038/nprot.2008.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bacmeister, L. et al. Assessment of PEEP-ventilation and the time point of parallel-conductance determination for pressure–volume analysis under β-adrenergic stimulation in mice. Front. Cardiovasc. Med.6, 36 (2019). 10.3389/fcvm.2019.00036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lindner, D. et al. Protective function of STAT3 in CVB3-induced myocarditis. Cardiol. Res. Pract.2012, 437623 (2012). 10.1155/2012/437623 [DOI] [PMC free article] [PubMed] [Google Scholar]
-
45.Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the
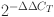 method. Methods 25, 402–408 (2001). [DOI] [PubMed]
method. Methods 25, 402–408 (2001). [DOI] [PubMed] - 46.Zawada, A. M. et al. Massive analysis of cDNA ends (MACE) and miRNA expression profiling identifies proatherogenic pathways in chronic kidney disease. Epigenetics9, 161–172 (2014). 10.4161/epi.26931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J.1710.14806/ej.17.1.200 (2011).
- 48.Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol.15, 550 (2014). 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics32, 2847–2849 (2016). 10.1093/bioinformatics/btw313 [DOI] [PubMed] [Google Scholar]
- 50.Alexa, A. & Rahnenfuhrer, J. Analysis for Gene Ontology. R package version 2.42.0; https://bioconductor.org/packages/topGO (2020).
- 51.Gu, Z. & Hübschmann, D. simplifyEnrichment: A Bioconductor Package for Clustering and Visualizing Functional Enrichment Results. Genomics Proteomics Bioinformatics21, 190–202 (2023). 10.1016/j.gpb.2022.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin, D. An information-theoretic definition of similarity. ICML ʼ98: Proc. of the Fifteenth International Conference on Machine Learning98, 296–304 (1998). [Google Scholar]
- 53.Pedersen T. ggforce: Accelerating ‘ggplot2’. version 3.3.3; https://ggforce.data-imaginist.com, https://github.com/thomasp85/ggforce (2021).
- 54.Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer, 2016).
- 55.Walter, W., Sanchez-Cabo, F. & Ricote, M. GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics31, 2912–2914 (2015). 10.1093/bioinformatics/btv300 [DOI] [PubMed] [Google Scholar]
- 56.Scherschel, K. et al. Characterization of the HCN interaction partner TRIP8b/PEX5R in the intracardiac nervous system of TRIP8b-deficient and wild-type mice. Int. J. Mol. Sci.22, 4772 (2021). [DOI] [PMC free article] [PubMed]
- 57.Brauninger, H. et al. Cardiac SARS-CoV-2 infection is associated with pro-inflammatory transcriptomic alterations within the heart. Cardiovasc. Res.118, 542–555 (2022). 10.1093/cvr/cvab322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bankhead, P. et al. QuPath: open source software for digital pathology image analysis. Sci. Rep.7, 16878 (2017). 10.1038/s41598-017-17204-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Voss, S. et al. Macrophage migration inhibitory factor (MIF) expression increases during myocardial infarction and supports pro-inflammatory signaling in cardiac fibroblasts. Biomolecules9, 38 (2019). [DOI] [PMC free article] [PubMed]
- 60.Lindner, D. et al. Cardiac fibroblasts support cardiac inflammation in heart failure. Basic Res. Cardiol.109, 428 (2014). [DOI] [PubMed]
- 61.Plendl, J., Sinowatz, F. & Auerbach, R. A transformed murine myocardial vascular endothelial cell clone: characterization of cells in vitro and of tumours derived from clone in situ. Virchows Arch426, 619–628 (1995). 10.1007/BF00192118 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figs. 1–5 and Supplementary Tables 1–10
Source data and exact P value for comparisons
Data Availability Statement
RNA sequencing data are available under GSE248521. Source data of all main figures are provided.
The code used for processing of RNA sequencing data (poly(A) tail clipping) is available at https://github.com/AGLindner/GPR15-RNA-Sequencing.