

Abstract
Epigenetics, a field that investigates alterations in gene function that can be inherited without changes in DNA sequence, encompasses molecular pathways such as histone variants, posttranslational modifications of amino acids, and covalent modifications of DNA bases. These pathways modulate the transformation of genotypes into specific phenotypes. Epigenetics plays a substantial role in cell growth, development, and differentiation by dynamically regulating gene transcription and ensuring genomic stability. This regulation is carried out by three key players: writers, readers, and erasers. In recent years, epigenetic proteins have played a crucial role in epigenetic regulation and have gradually become important targets in drug research and development. Targeted therapy is an essential strategy; however, the effectiveness of targeted drugs is often limited by drug resistance, posing a significant dilemma in clinical practice. Targeted protein degradation technologies, including proteolysis-targeting chimeras (PROTACs), have great potential in overcoming drug resistance and targeting undruggable targets. These areas of research are gaining increasing attention to various epigenetic related disease. In this review, we have provided a summary of the recently developed degraders targeting epigenetic readers, writers, and erasers. Additionally, we have outlined new applications for epigenetic protein degraders. Finally, we have addressed several unresolved challenges within the PROTAC field and offered potential solutions from our perspective. As the field continues to advance, the integration of these innovative methodologies holds great promise for addressing the challenges associated with PROTAC development.
1. Introduction
Epigenetics, which studies changes in gene function that are heritable without an alteration in DNA sequence, is first defined by Conrad Waddington in the early 1940s.1 Originally, epigenetics includes molecular pathways such as histone variants, posttranslational modifications of amino acids, and covalent modifications of DNA bases, that modulate the expression of genotypes into specific phenotypes (Scheme 1).2–6
Scheme 1.
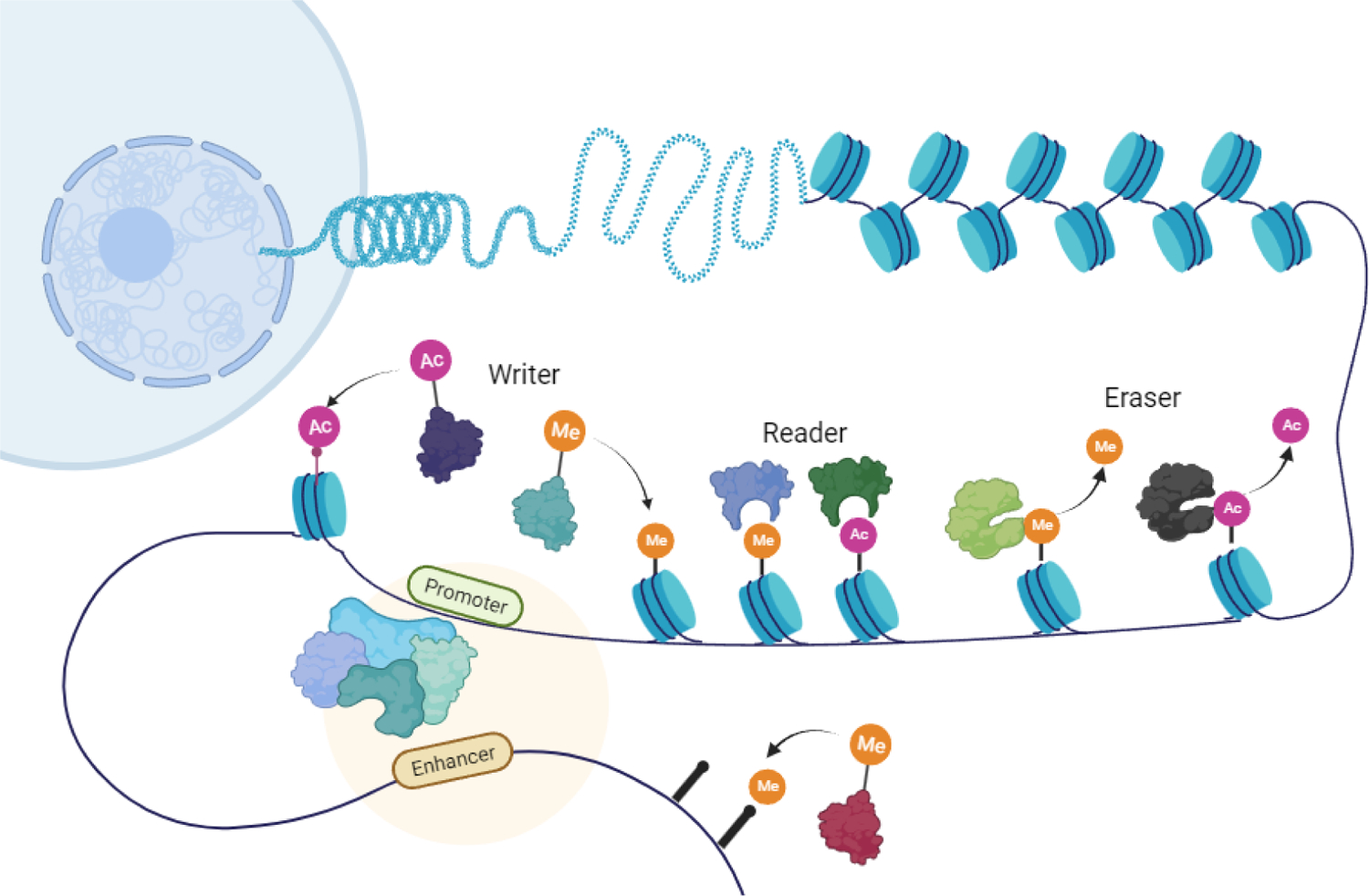
Role of epigenetics in human gene regulation.
Epigenetics contributes significantly to cell growth, development, and differentiation through dynamic regulation of gene transcription and genomic stability,7–13 which is carried out by writers (DNMTs, HATs, ubiquitin E3 ligases and HMTs), readers (bromodomains) and erasers (HDACs, KDMs and deubiquitinating enzymes). Writers could add epigenetic marks to DNA or histone tails, readers recognize epigenetic marks and erasers remove the epigenetic marks (Scheme 2).14–18
Scheme 2.
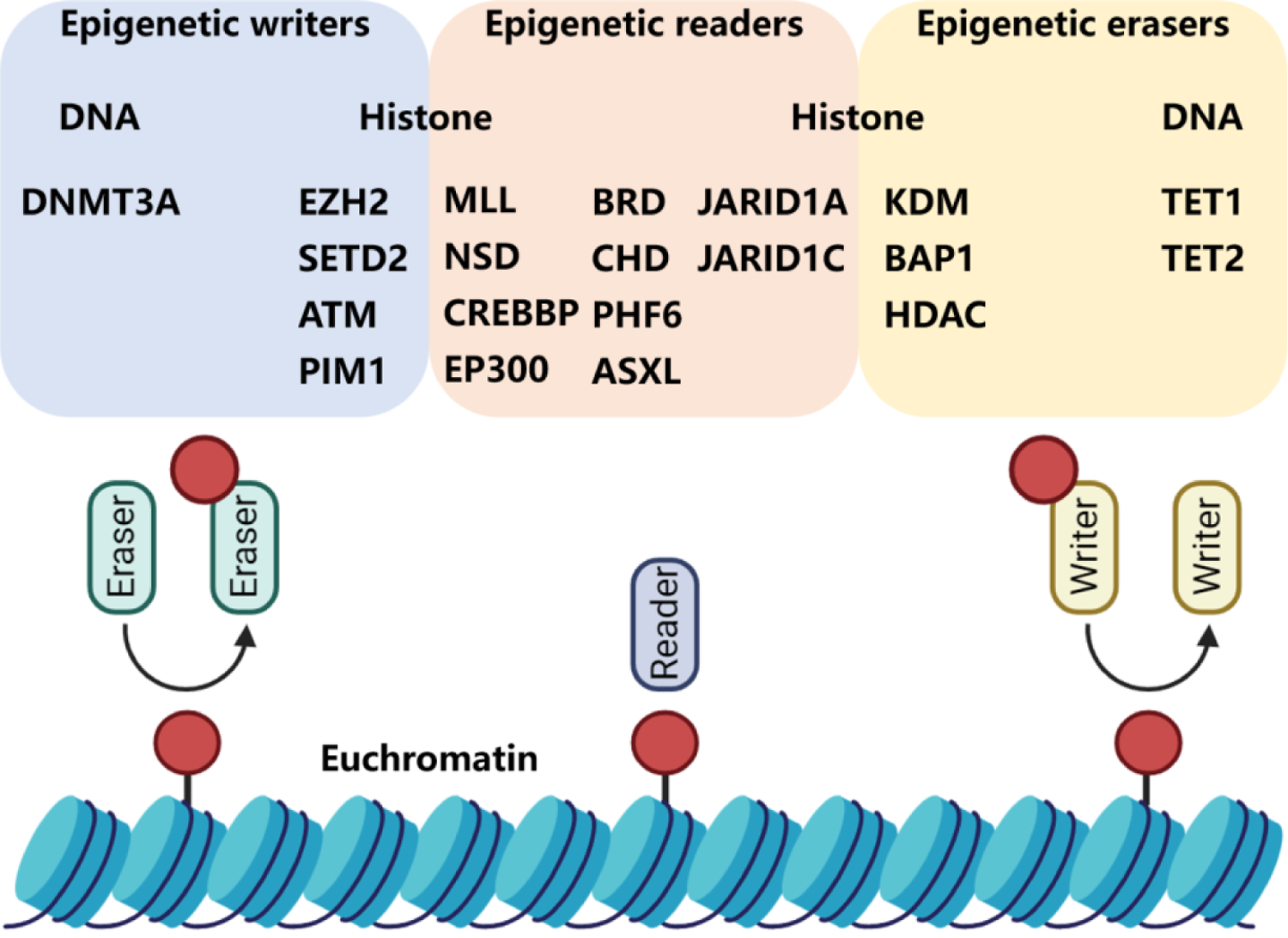
Epigenetic tools. Brown circles represent methylation, acetylation, or phosphorylation marks. DNMT3A, DNA (cytosine-5)-methyltransferase 3A; EZH2, enhancer of zeste homolog 2 protein; SETD2, SET domain containing 2 protein; ATM, ataxia-telangiectasia mutated protein; PIM1, proto-oncogene serine/threonine-protein kinase; MLL, mixed-lineage leukemia histone methyltransferase; NSD, nuclear receptor-binding SET domain protein; CREBBP, cAMP-responsive element-binding protein-binding protein; EP300, E1A Binding Protein P300; BRD, bromodomain-containing protein; CHD, congenital heart disease protein; PHF6, plant homeodomain factor 6 protein; ASXL, additional sex combs-like protein; JARID1A, JumonjiC and ARID domain-containing histone lysine demethylase 1A; JARID1C, JumonjiC and ARID domain-containing histone lysine demethylase 1C; KDM, histone lysine demethylase; BAP1, BRCA1-associated protein 1; HDAC, Histone deacetylase; TET1, ten-eleven translocation protein 1; TET2, ten-eleven translocation protein 2.
Since epigenetics is a key component of an organism’s normal development, it is not surprising that epigenetic dysregulation contributes to the origin and progression of human diseases such as cancer, metabolic diseases.19 For example, histone deacetylases (HDACs) are usually over-expressed in cancers.20 DNMT3A (R882) mutation is associated with acute myeloid leukaemia (AML).21 Vorinostat and romidepsin derived from phenotypic screens have been identified as HDAC inhibitors (Reddy SA. Romidepsin for the treatment of relapsed/refractory cutaneous T-cell lymphoma.22–23 Three other HDAC inhibitors, belinostat, panobinostat, and chidamide, have gained regulatory approval based on lead compound optimization.24 Tazemetostat, as an inhibitor of polycomb repressive subcomplex (PRC) protein enhancer of zeste homolog 2 (EZH2), has been approved by FDA for the treatment of relapsed or refractory follicular lymphoma.25 Up to date, the use of therapeutic epigenetic inhibitors in the clinic has made progress on a wide range of tumoral and non-tumoral disease, nevertheless, it is mainly limited to haematological malignancies.
PROteolysis TArgeting Chimeras (PROTACs) are hetero-bifunctional molecules, consisted of an E3 ligase ligand, a ligand of the target protein and a linker for connection. This kind of bifunctional molecules is designed to bring the target protein and the E3 ligase in proximity, causing ubiquitination of the target protein and its subsequent 26 S proteasomal degradation (Figure 1).26–33 Unlike traditional inhibitor approach of drug discovery, which is limited by the target protein’s “druggability” and asks for enzymatic activity that can be inhibited, PROTACs eliminate target protein through endogenous degradation pathways rather than merely inhibiting its function, thus, the whole protein could be depleted. As newly emerging techniques, PROTACs demonstrate huge advantage. Firstly, PROTACs require modest target protein binding affinity, they are less susceptible to mutation or over-expression. Secondly, PROTACs are event driven and initiate degradation process in a repeatable manner, allowing a lower dosage, administration frequency and toxicity. Due to this, PROTACs techniques have attracted widely research interest in academia as well as industry.34–37
Figure 1.
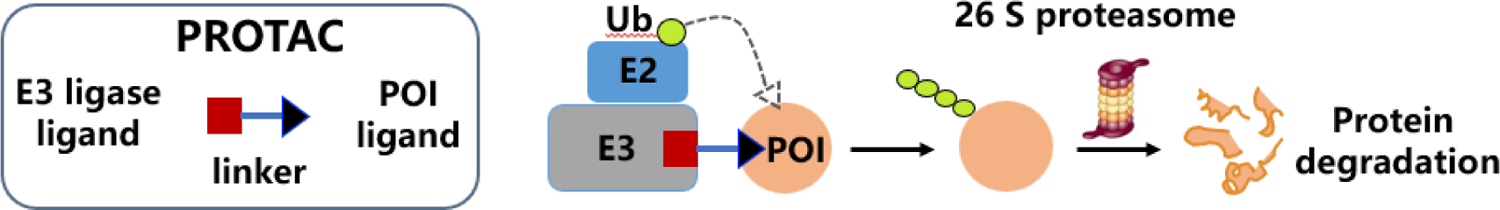
Introduction to PROTAC technology
Epigenetic proteins are always formed as complexes with multiple functions, thus, it’s difficult to prohibit their functions concurrently with occupied inhibitors. However, elimination of epigenetic proteins undoubtedly solves this issue.38 Based on the above, PROTACs has been successfully applied into degradation of epigenetic proteins (Figure 2). In this review, we aim to provide a comprehensive summary of reported degraders for epigenetic proteins categorized as readers, erasers, and writers.
Figure 2.

Targeting epigenetic protein using PROTAC degraders
2. Main text
2.1. Protein degraders for epigenetic readers
BET.
Bromodomian is highly conservative structure, it could recognize acetylated lysine in histone tails and regulate transcription and remodels chromatin.39 As bromodomain-containing proteins, Bromo and extral terminal domain family (BET) proteins, such as BRD2, BRD3, BRD4, BRD7 and BRD9 has attracted wide research interest. Recently, numerous degraders targeting BET proteins have been reported, which, demonstrated potent tumor growth suppression.40–42
In recent years, PROTACs targeting BET proteins has attracted wide interest and numerous degraders have been reported.43–47 In 2016, Crews group developed a pan-BET protein degrader, named as ARV-771 (Figure 3). This molecule was a von Hippel–Landau (VHL) E3 ligase-based PROTAC and demonstrated potent degradative activity in enzalutamide-resistant prostate carcinoma cells, 22Rv1 (DC50 < 5 nM), which resulted in down-regulation of cMyc levels and cell apoptosis. Interestingly, ARV-766, a diastereomer of ARV-771, showed little c-MYC suppression effect. This meant, for cellular degradative activity, ARV-771 worked through a catalytic event. Unlike the secondary resistance caused by BET inhibitors, ARV-771 showed potent efficacy in castration-resistant prostate cancer cells. Furthermore, a similar effect is reflected in the VCaP tumor model, leading to a 60% tumor growth inhibition with weight maintenance. No apparent effect was found when the group was treated with enzalutamide.48
Figure 3.
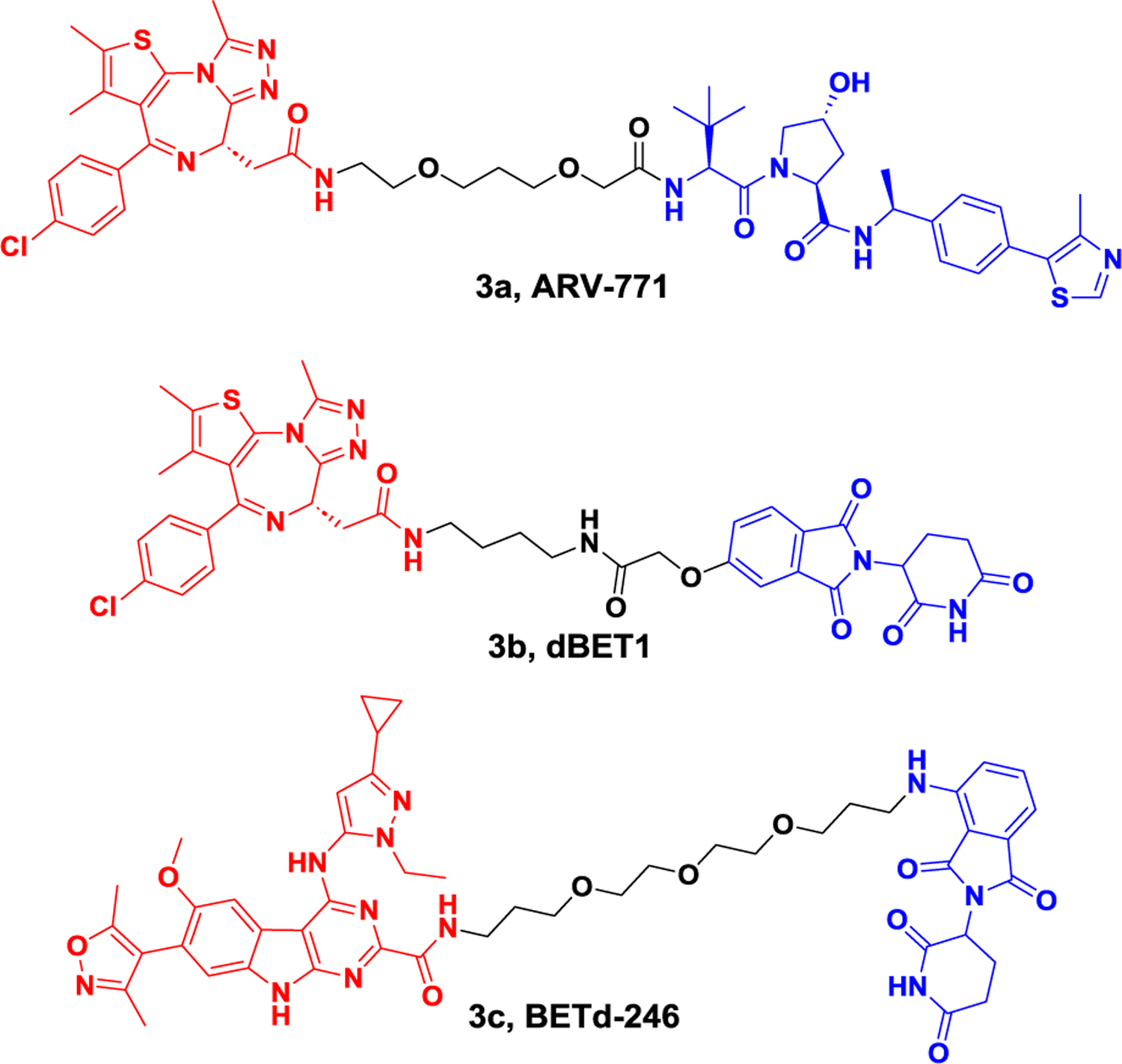
Representative PROTACs targeting drug-resistant BET.
dBET1, a molecule conjugating JQ1 and pomalidomide, was developed for BET protein degradation by the Bradner group in 2015 (Figure 3).49 The treatment of MV4–11 cells with dBET1 led to >85% reduction of BRD4 even at a low concentration (100 nM, 18 h). dBET1 was also found to be more effective than JQ1 in inhibiting the proliferation of human MV4–11 leukemia cells. In vivo studies using a murine hind-limb xenograft model with MV4–11 cells demonstrated that dBET1 was able to degrade BRD4 and inhibit tumor growth without affecting animal weight or normal blood counts. Furthermore, excised tumors showed a significant downregulation of MYC compared to the vehicle group.
Triple-negative breast cancer (TNBC) patients typically respond well to chemotherapy.50 However, high rates of metastatic disease often occur due to the amplification of MCL1 loci, which represents frequent genetic changes in chemo-refractory tumors. In TNBC patients, it’s discovered that MCL1 is both an intrinsic and acquired resistance factor, which has limited the application of numerous anticancer agents. In 2018, BETd-246, deriving from BETi-211, was made by the Wang group (Figure 3). As a degrader for BET proteins in TNBC, BETd-246 degraded BRD2, BRD3, and BRD4 in a dose-dependent manner (30–100 nmol/L, 1 or 3 hours), which led to a nearly complete depletion for the mentioned proteins. At a concentration of 10 nmol/L, when BETd-246 was tested in TNBC cell lines, cell growth was inhibited, a rapid downregulation of MCL1 protein was observed as well. BETd-246 (5 mpk, i.v., triweekly, 3 weeks) demonstrated comparative anti-tumor activity with BETi-211 (50 mpk, p.o., daily, 3 weeks), providing an alternative approach for TNBC to overcome drug resistance.51
Subsequently, numerous BET protein degraders were reported. BI2536, as a dual inhibitor, targeted two important therapeutic targets for AML, Polo-like kinase 1 (PLK1) and BRD4. In 2020, Lu group reported a dual degrader (HBL-4, Figure 4A) targeting BRD4 and PLK1 by connecting BI2536 with pomalidomide.52 HBL-4 led to induced rapid protein degradation in human leukemia cells (e.g., MV4–11, MOLM-13, and KG1). Compared with BI2536, more potent anti-proliferation as well as c-Myc level suppression efficacy was observed in MV4–11 tumor xenograft model, providing possible threptic option for acute myeloid leukemia. In 2019, MacPROTAC-1, derived from BET degrader MZ1, was reported by Ciulli group, in which a macrocycle was introduced into the molecule (Figure 4A).53 This type of conformational constrained strategy helped the molecule to maintain a bioactive configuration thus reducing the energetic penalty, affording an alternative approach to drug development. Compared with BET degrader MZ1, MacPROTAC-1 resulted in a more pronounced difference in binding affinity between BD1 and BD2. Furthermore, MacPROTAC-1 exhibited comparable degradation activity and cell proliferation inhibition in BET-sensitive 22RV1 cells.
Figure 4A.
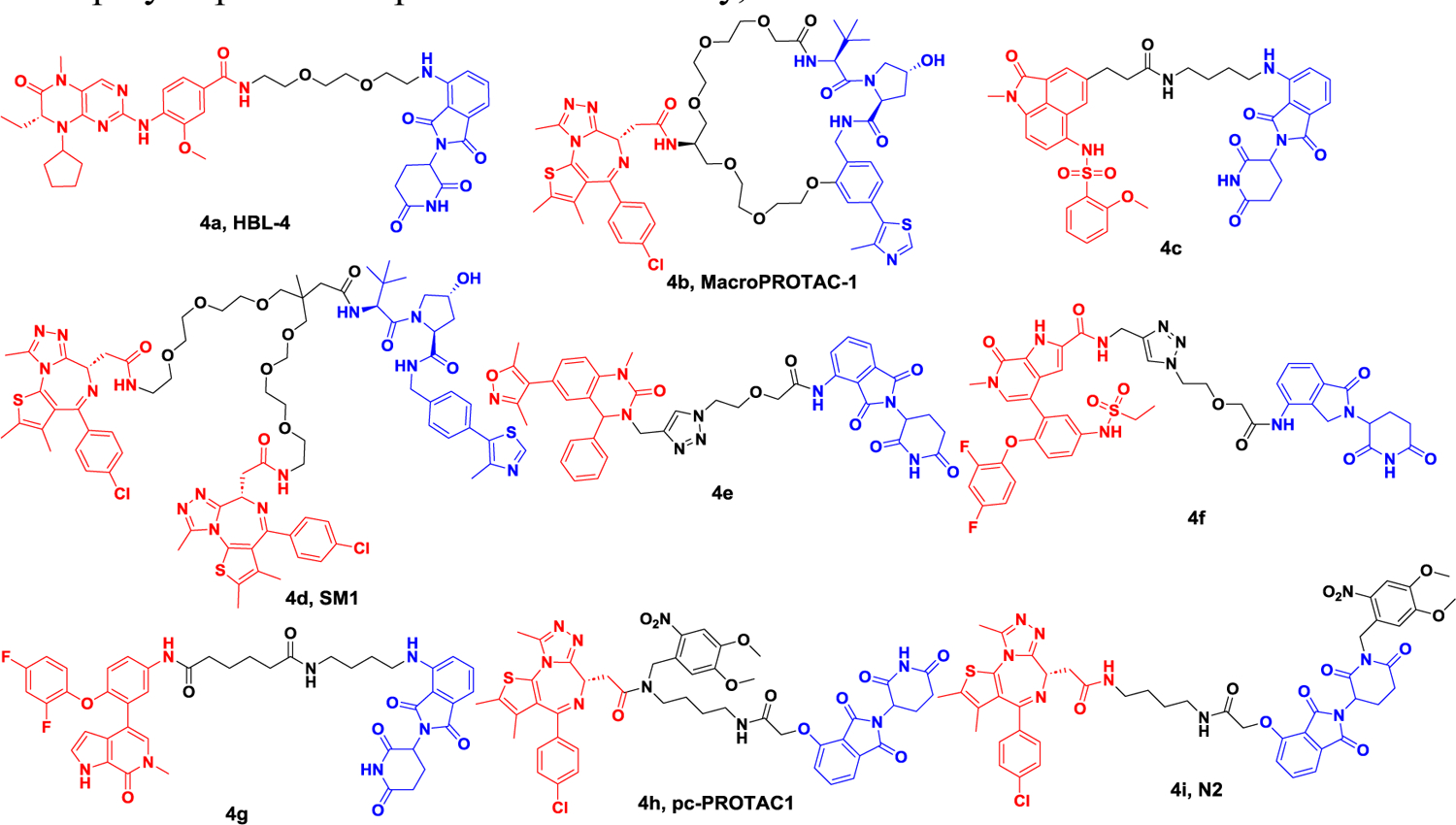
Representative PROTACs targeting BRD.
Although great progress has made in the development of BET protein degraders, selective subtype BET protein degrader avoiding off-target, is still in high demand. In 2020, one degrader 4c combined BD1 selective inhibitor with thalidomide was reported by Wang group, demonstrating high BRD2 and BRD4 selectivity over other subtypes (Figure 4A).54 BRD4 was completely degraded after 8 h treatment with degrader 4c at a concentration at 1 μM in either hematoma or solid tumor cells, demonstrating potent cell growth suppression effect. Besides, no obvious cytotoxicity was observed.
In 2021, Ciulli group developed trivalent PROTACs by connecting bivalent BET inhibitor with E3 ligand.55 Compared with bivalent PROTACs, the trivalent degrader 4d with a VHL ligand moiety (SM1, Figure 4A), showed higher degradation potency and resulted in stronger anticancer activity. Mechanistically, the degrader 4d (SM1) formed a 1:1:1 ternary complex with VHL, BD1 and BD2, leading to a longer residence time. Despite an increase in the molecular weight of the trivalent degrader, degrader 4d (SM1) demonstrated improved cell permeability and a highly favorable PK profile. In addition to common structures like JQ1, an increasing number of BET inhibitors have been utilized in the development and production of PROTACs.
In the year 2019, a novel type of degrader was created by Zhang’s team utilizing a highly effective dihydroquinazolinone-based BRD4 inhibitor. Within this group, degrader 4e (Figure 4A), demonstrated complete BRD4 degradation at a concentration of 1 μM following a 3-hour treatment. Additionally, the degrader displayed a remarkable ability to inhibit cell growth, with an IC50 of 0.81 μM in human leukemia monocytic cells, THP-1, surpassing the potency of the inhibitor by four-fold in the antiproliferative assay. In subsequent studies, ABBV-075 derivatives were utilized by the Zhang group56 to develop PROTACs. In 2020, the Zhang group incorporated linkers at the pyrrole ring position, resulting in the creation of degrader 4f (Figure 4A). This degrader effectively induced degradation of BRD4, cell cycle arrest, and apoptosis in the human pancreatic cancer cell line BxPC-3. Compared to ABBV-075, the antiproliferative activity of degrader 4f against the BxPC-3 cell line (IC50 = 0.165 μM) was improved by approximately seven-fold. In 2021, the Yu group reported the development of another BRD4 degrader, 4g (Figure 4A), which was created by connecting an ABBV-075 derivative with an E3 ligand.57 This molecule demonstrated a DC50 of 0.25 nM in MV4–11 cells. In RS4–11 cells, the DC50 was 3.15 nM. Additionally, it caused a suppression of cell proliferation in human leukemia cells (MV4–11 and RS4–11), with an IC50 of 0.5 nM and 4.8 nM, respectively.
The crucial function of BET protein in cells implies that PROTACs targeting it may lead to harmful effects on healthy cells, thereby limiting their clinical use. To address this challenge, several strategies have been employed to regulate PROTACs in space and time. One popular approach involves incorporating a photocaged group into the PROTACs. In 2019, the Pan research group modified dBET1 at the nitrogen site of JQ1 moiety with a bulky 4,5-dimethoxy-2-nitrobenzyl group, affording the degrader 4h (pcPROTAC1, as shown in Figure 4A). Upon light irradiation, this degrader could induce degradation of BRD4 in live cells. In the zebrafish model, treatment with the degrader led to a reduction in BRD4 levels and corresponding changes in phenotype.58 The Li research group developed a similar type of degrader (N2, Figure 4A) by incorporating the 4,5-dimethoxy-2-nitrobenzyl group onto the glutarimide nitrogen of dBET1. Upon exposure to UV light, the degrader induced the degradation of BRD4 in HEK293T cells, while in a zebrafish xenograft model, it suppressed the growth of tumors derived from tongue squamous cell carcinoma (TSCC) cells, HN-6.59
In 2020, Xing-sheng Lu et al. discovered that the BRD4-degrading PROTAC A1874 (Figure 4B) induces the degradation of BRD4 protein, leading to the down-regulation of BRD-dependent genes such as c-Myc, Bcl-2, and cyclin D1 in colon cancer. Furthermore, it was observed that A1874 exhibited greater efficacy against colon cancer compared to BRD4 inhibitors like JQ 1, CPI203, and I-BET151. Additionally, in BRD4-knockdown colon cancer, A1874 maintained its cytotoxicity, suggesting a BRD4-independent mechanism. This alternative mechanism may be mediated by A1874’s capacity to enhance p53 stability and ROS production in a dose-dependent manner. This study provides confirmation of the outstanding anticancer activity of A1874 against colon cancer cells, laying the groundwork for potential clinical translation.60
Figure 4B.
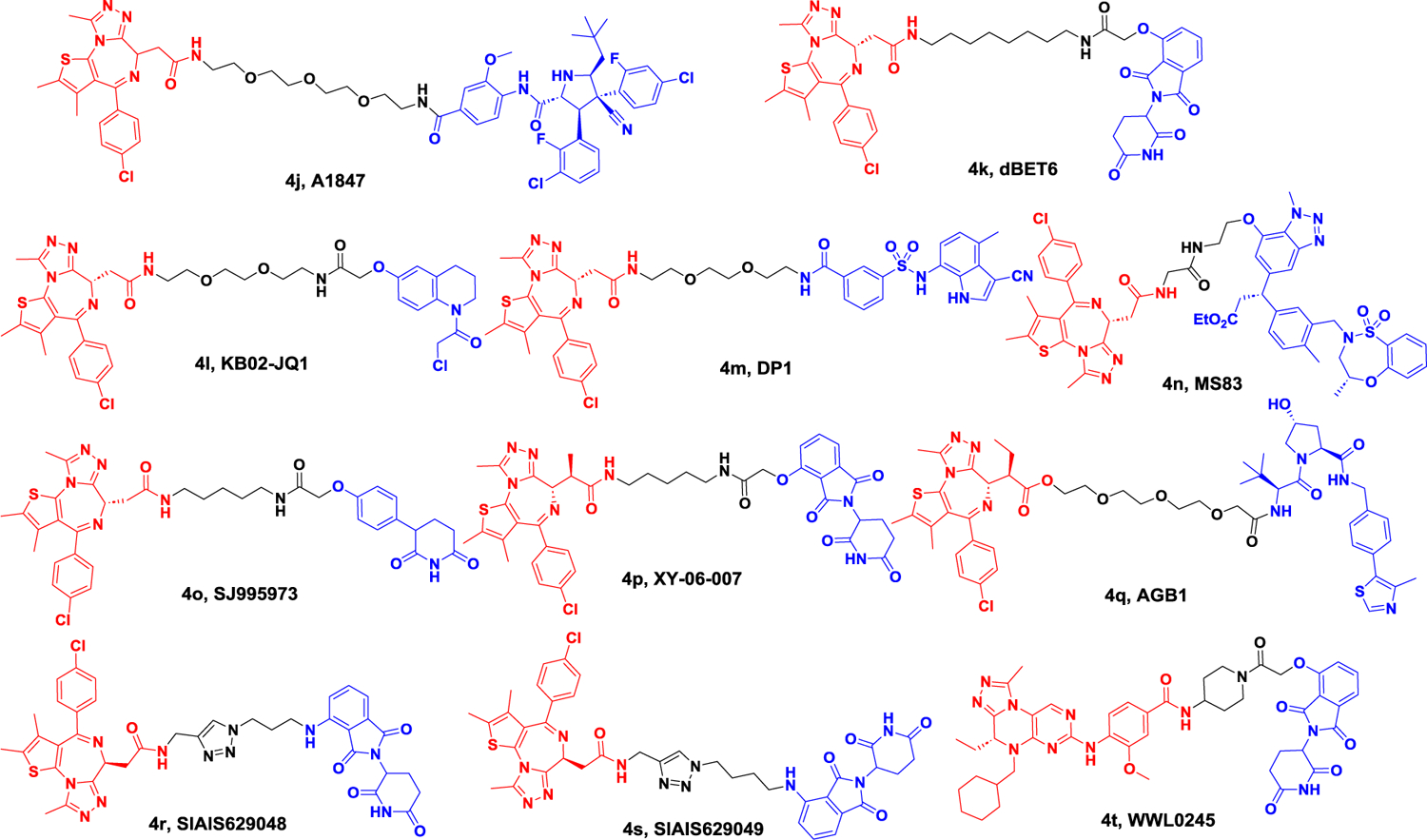
Representative PROTACs targeting BRD.
In 2021, the Dashwood group investigated the anti-tumor activity of BET degrader (dBET6) and HDAC3-specific inhibitor (BG45). They found that BG45 combined dBET6 (Figure 4B) showed significant anti-tumor activity in metastatic colon cancer, whereas individual agents were less effective, which provides further supports for combination HDAC3 with BRD461.
Furthermore, novel E3 ligases that are appropriate for use in PROTACs have been evaluated for their potential to develop BRD4 degrading agents. The Cravatt research group confirmed DCAF16 as a newly discovered E3 ligase in 2019. As a result of this discovery, they designed a BRD4 degrader, called KB02-JQ1 (Figure 4B), which utilized reversible ligands to target DCAF16 along with JQ1. However, to achieve degradation, a concentration of 20 μM was required.62 In 2020, the Chen group developed a novel type of BRD4 degrader 4m by utilizing E7820, a DCAF15 ligand, in combination with JQ1 (Figure 4B). This degrader was tested in SUDHL-4 cells, and its DC50 value was measured at 10.84 μM. Furthermore, this degrader demonstrated an impressive 98% maximum degradation rate.63 The Jin group also developed a novel BET protein degrader 4n by conjugating JQ1 with KI696, a KEAP1 ligand (Figure 4B). Notably, in human breast cancer cells, MDAMB-231, this degrader was found to decrease protein levels of both BRD4 and BRD3 but had no impact on BRD2 protein levels. Additionally, the degrader selectively degraded the short isoform of BRD4, while leaving the long isoform unaffected.64 The Rankovic group recently discovered that phenyl glutarimide could bind to CRBN, leading to the development of a new BRD4 degrader, SJ995973 (Figure 4B). This degrader was found to have similar binding affinity to previously reported degraders that utilize CRBN as the E3 ligase, but with improved stability. In MV4–11 cells, SJ995973 was able to induce half of BRD4 degradation at a concentration of only 0.87 nM.65
Based on a bump-and-hole strategy, Fischer group designed a CRBN-based degrader (XY-06–007) that specifically identified BRD4BD1L94V in 2021 (Figure 4B).66 Proteomics analysis showed this degrader had excellent BRD4BD1L94V selectivity over wild-type or other BET family of bromodomains. Besides, XY-06–007 demonstrated good pharmacokinetics in vivo studies.
In recently, Ciulli group also developed a novel VHL-based degrader 4q (AGB1, Figure 4B) based on the “bump-and-hole” approach.67 In an inducible BromoTag degron system, the degrader 4q (AGB1) not only formed a strong, cooperative ternary complex between VHL and the BromoTag-BRD2 but also completely induced the degradation of BromoTagged target proteins with low nanomolar potency. The degrader 4q (AGB1) exhibited exquisite selectivity over the native wild-type BET, which led it not cytotoxic in several cancer relevant cell lines. In summary, these two distinct methods provided a useful tool to study the effect and implications of rapid and highly selective degradation of a target protein.
In 2021, the Jiang group reported the synthesis of azide substrates based on IMiDs. These compounds were then linked to various POIs (Protein of Interest) through a ‘click reaction,’ enabling the screening of degraders that target BET family proteins. Among these compounds, degraders SIAIS629048 and SIAIS629049 exhibited potent activity in degrading BET proteins at a concentration of 50 nM. Additionally, these degraders demonstrated strong anti-proliferative activity against MV4–11 cells (Figure 4B).68
In 2022, the Wang group synthesized WWL0245, which demonstrated selective degradation of BRD4 (Figure 4B). This degrader exhibited potent antiproliferative effects in AR-positive prostate cancer cell lines. Additionally, WWL0245 induced the degradation of BRD4 with a sub-nanomolar DC50 and achieved >99% Dmax in the aforementioned cell line. In BETi-sensitive cancer cells, such as AR-positive prostate cancer cells, WWL0245 still displayed potent antiproliferative activity, with an IC50 of 3 nM in MV4–11 cells.69
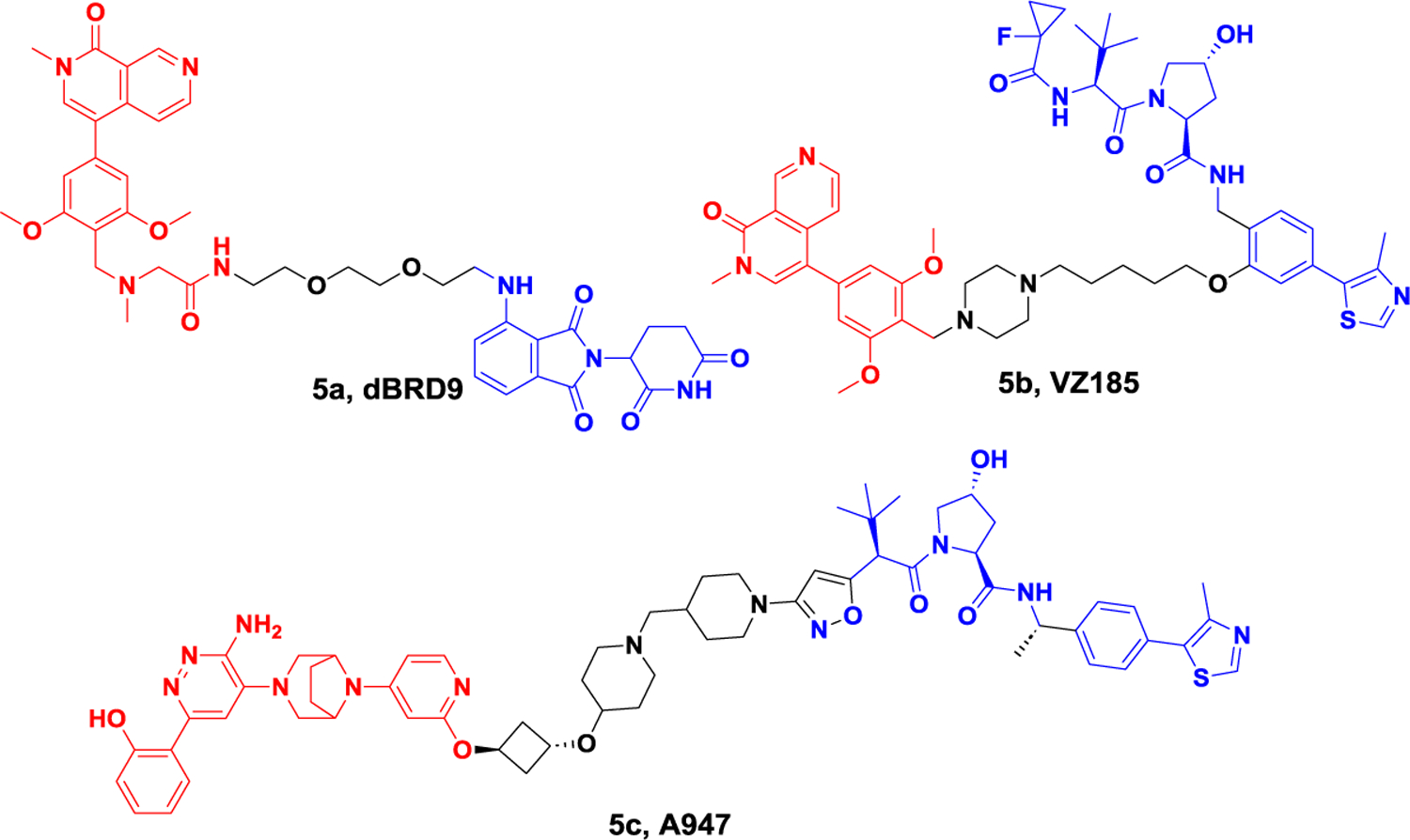
In 2017, the Bradner group first reported the degradation of BRD9, a subunit of the human BAF (SWI/SNF) nucleosome remodeling complex, by a molecule named dBRD9. This molecule exhibited potent degradation ability towards BRD9 at a concentration of 50 nM. Additionally, in the human acute myeloid leukemia MOLM13 cell line, dBRD9 demonstrated a superior anti-proliferative effect compared to the non-degrading probe, with a range of 10- to 100-fold excess (Figure 5).70
Figure 5.
Representative PROTACs targeting protein of BAF nucleosome complex.
In 2019, the Ciulli group developed VZ185 by conjugating ligands of VHL and BRD9 (Figure 5). In addition to degrading BRD9 with a DC50 value of 1.8 nM, VZ185 also demonstrated degradation of BRD7 with a DC50 value of 4.5 nM. In the EOL-1 (acute myeloid eosinophilic leukemia) and A-204 (malignant rhabdoid tumor) cell lines, VZ185 exhibited potent cytotoxic effects, with EC50 values of 3 and 40 nM, respectively.71
The catalytic function of BAF nucleosome complex is executed by one of ATP-dependent helicases, SMARCA2 and SMARCA4. These two proteins have a conserved bromodomain (BD) that can interact with acetylated chromatin. These two proteins share strong protein sequence homology. Preclinical genetic studies would indicate that achieving selective inhibition of SMARCA2 will likely be essential for a successful therapeutic. In 2022, the Yauch group discovered A947, a potent and highly selective PROTAC molecule for SMARCA2 (Figure 5). In SW1573 cells, A947 exhibited the potential to degrade SMARCA2 with a DC50 of 39 pM, achieving 96% maximal degradation at 10 nM. In contrast, it required a 28-fold higher concentration of A947 to achieve a DC50 (1.1 nM) on SMARCA4, achieving 92% maximal degradation at approximately 100 nM. Global ubiquitin profiling and proteomic analysis further confirmed the high specificity of A947 in degrading these target proteins at high concentrations.72
ENL.
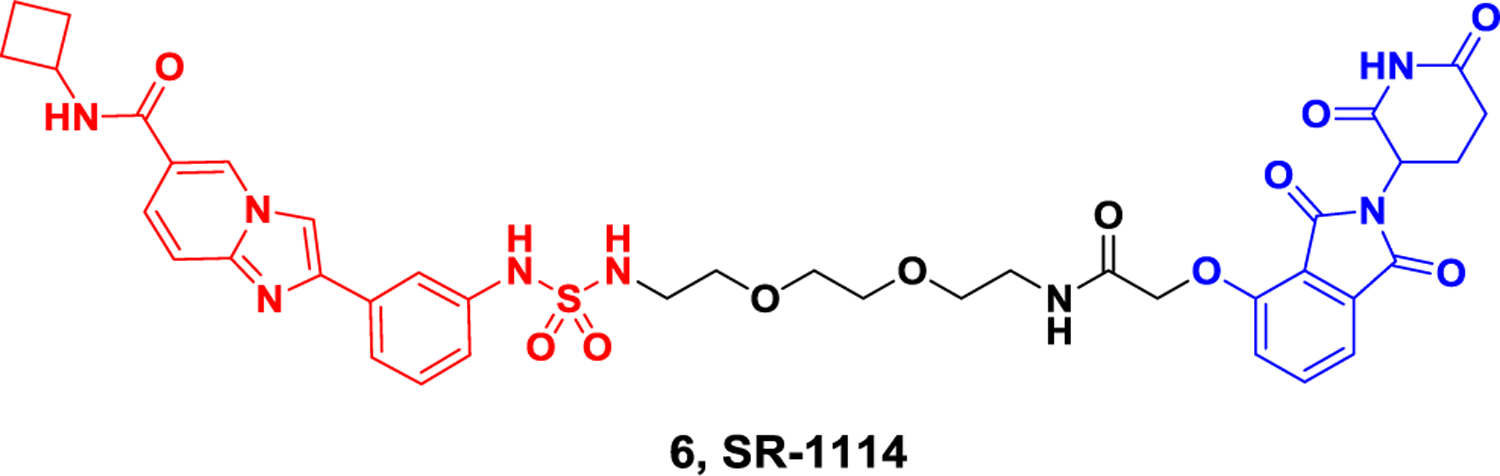
The YEATS domain is classified as a histone acetylation “reader”. ENL is one of the four human genome encoded YEATS domain containing proteins. ENL protein is essential for the survival of acute myeloid leukemia (AML). Anti-leukemia effect and leukemia growth inhibition of leukemia can be observed when ENL is knocked out. In 2021, the Erb group implemented a SuFEx-based strategy for high-throughput medicinal chemistry, leading to the discovery of a remarkably effective inhibitor called SR-0813 targeting the ENL YEATS domain. Building upon the identification of SR-0813, they subsequently developed SR-1114, a degrader that specifically targets ENL (Figure 6). In MV4;11 cells, treatment with SR-1114 resulted in the CRBN-dependent degradation of ENL, with a DC50 value of 150 nM. The maximum degradation of ENL was observed within 4 hours at a concentration of 10 μM, but ENL could be resynthesized within 24 hours.73
Figure 6.
Representative PROTAC targeting ENL.
2.2. Protein degraders for epigenetic writers
CBP.
The paralogous chromatin regulators CREB-binding protein (CBP) and p300.
CBP and p300 (also known as KAT3A and KAT3B) are key transcription factors that establish and activate enhancer mediations.74 p300/CBP can exert enzymatic function through a lysine acetyltransferase region, which can dynamically acetylate about 5000 lysines on more than 21000 proteins. They can also mediate protein-protein interactions on chromatin.75 In cancer, p300/CBP is considered to be an oncogene and tumor suppressor, and selective inhibitors targeting its KAT domain have proven to be a promising cancer treatment strategy.76 However, inhibition of a single domain is not sufficient to completely eliminate p300/CBP activity in cells. Therefore, it is necessary to develop inhibitors that can simultaneously inhibit multiple domains or even completely eliminate p300/CBP. In 2021, Ott et al. reported the first CRBN-based p300/CBP PROTAC, dCBP-1 (Figure 7).77 This degrader has high antiproliferative activity in multiple myeloma and can cause significant downregulation of the oncogene MYC. Treatment with dCBP-1 can also reduce the intensifier histone acetylation and chromatin accessibility and is more effective than KAT domain and bromine domain inhibitors alone or in combination. As a highly potent CBP/p300 degrader, dCBP-1 is a useful tool to investigate the mechanism by which these factors coordinate enhancer activity in cells.
Figure 7.
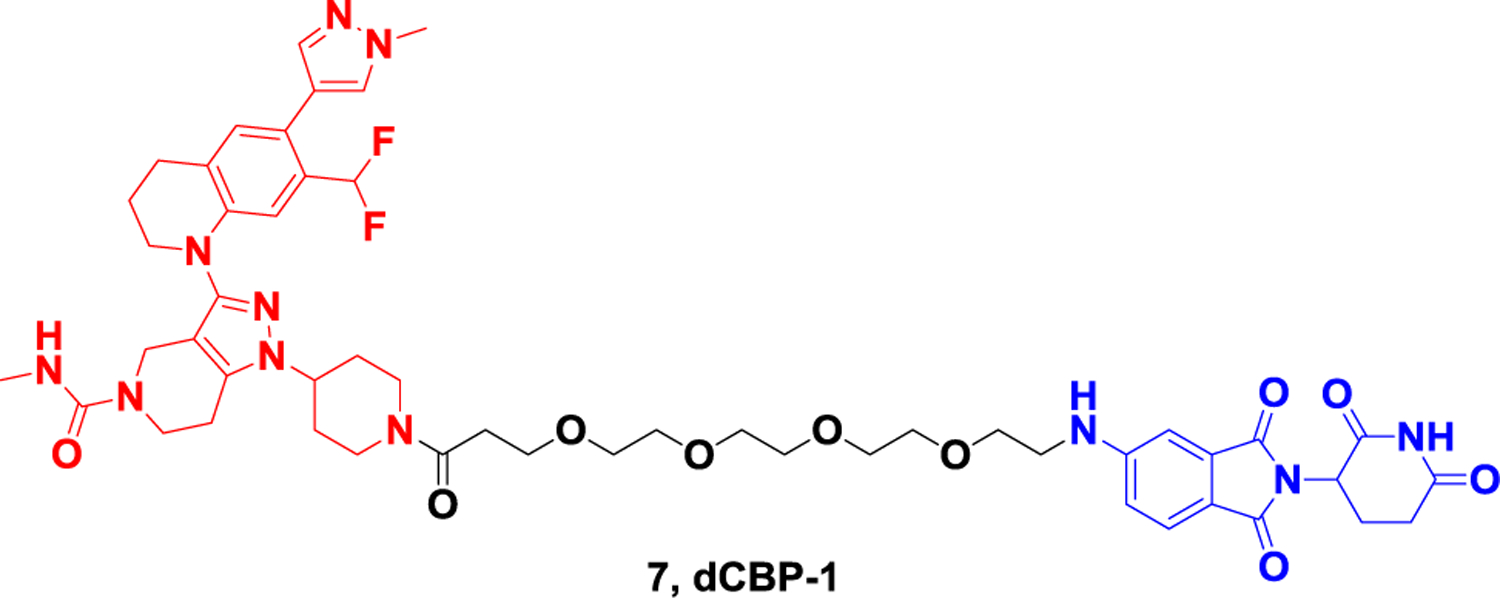
Representative PROTAC targeting CBP and p300.
PRC2.
The polycomb repressive complex 2 (PRC2) is an epigenetic regulator of transcription and consists of four subunits, EZH1/2, EED, SUZ12 and RbAp46/RbAp48. EZH2 is a key catalytic subunit in the PRC2 complex78. PRC2 has the activity of histone methyltransferase, which can methylate H3K27, and hyper-trimethylation of H3K27 can be observed in various types of tumors. PRC2 is both an oncogene and a suppressor of tumorigenesis in multiple cancer types, such as colorectal, breast, and prostate cancers79. Existing inhibitors targeting EZH2 and EED subunits can effectively inhibit the catalytic activity of the PRC2 complex and have effective anti-tumor activity. However, resistance mutations to this small-molecule inhibitor have been observed in clinical trials, and targeting PRC2 protein degradation could serve as an alternative strategy for this kind of competitive inhibition80.
Since 2019, a series of PROTACs targeting PRC2 subunits including EED and EZH2 have been developed, which can inhibit the activity of the PRC2 complex. Bloeche et al. developed the first PRC2 degrader 8a (Figure 8) that is capable of promoting ternary complex formation by VHL by combining the existing EED inhibitor MAK683 with a VHL ligand. EED-targeted PROTACs can simultaneously induce efficient and selective degradation of EED, EZH2, and SUZ12, effectively inhibit PRC2 enzymatic activity (pIC50~8.1) and reduce EZH2-dependent cancer cell proliferation (GI50= 49–58 nM)81. At the same time, James et al. also reported the PROTAC degrader UNC6852 (Figure 8) developed based on the EED ligand and VHL ligand. This degrader can selectively degrade EED (DC50 = 0.79 μM), EZH2 (DC50 = 0.3 μM), and SUZ12, resulting in loss of PRC2 catalytic activity and decreased H3K27me3 levels in HeLa cells. This degrader has antiproliferative effects in Diffuse Large B-Cell Lymphoma (DLBCL) cell lines with EZH2 activating mutations82. In 2020, Jin et al. generated MS1943 (Figure 8) based on a selective non-covalent inhibitor of EZH2 using a hydrophobic labeling method. This degrader is a first-in-class EZH2 selective degrader, which can effectively reduce the level of EZH2 in MDA-MB-468 cells at 5 μM. In addition, MS1943, the degrader targeting the EZH2 protein, significantly inhibited the growth and induced apoptosis of triple-negative breast cancer (TNBC) cells compared with inhibitors83. In 2021, Yu et al84. developed the degrader E7 (Figure 8) based on the CRBN ligand and the clinical EZH2 inhibitor EPZ6438, which induced degradation of all PRC2 subunits at 1 μM in WSU-DLCL-2 cells (EZH2 72%, SUZ12 81%, EED 75%, RbAp48 74%). In the same year, Wen et al85. reported a similar VHL-based degrader that induced 50% degradation of EZH2 protein levels at 1 mM. Degrader YM181 (Figure 8) induced robust cell viability inhibition in diffuse large B-cell lymphoma (DLBCL) and other subtypes of lymphomas. Overall, degrading PRC2 for cancer therapy has proven to be an effective strategy, and some degraders have equal or even better anticancer activity than inhibitors.
Figure 8.
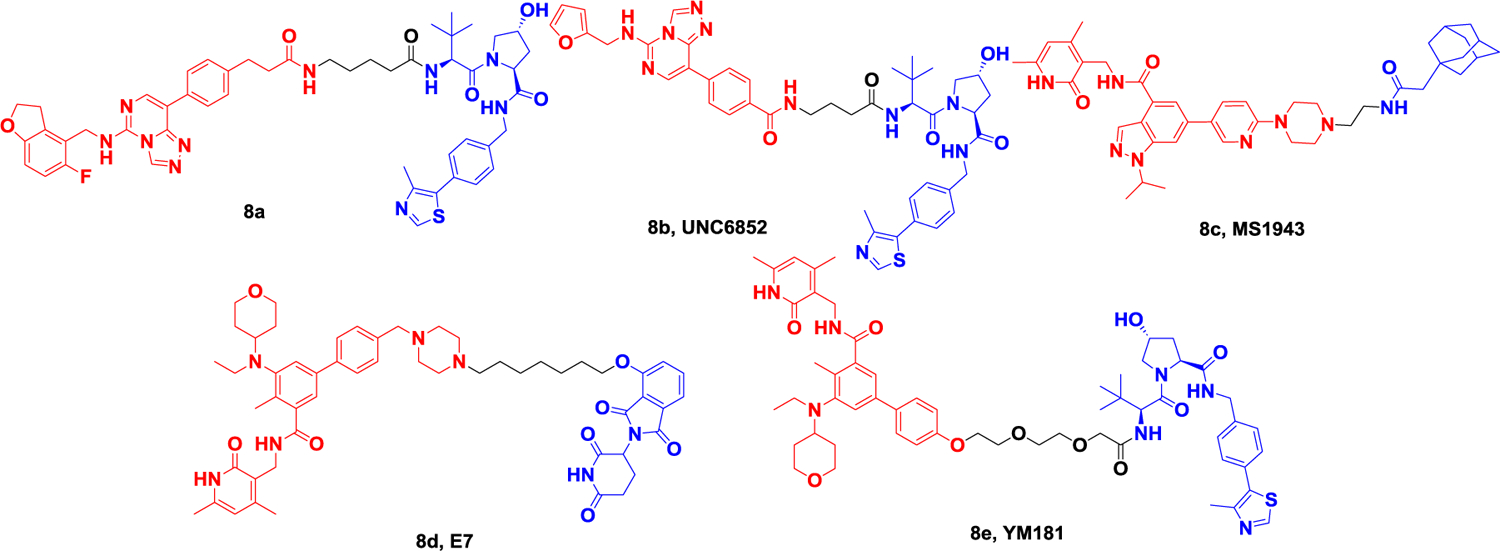
Representative PROTACs targeting PRC2 (EZH2, EED).
PRMT5.
Arginine methylation is a common post-translational modification, which is mainly regulated by protein arginine methyltransferase (PRMT). Nine PRMT species have been identified in mammals that catalyze the production of three different forms of methylated arginine86. PRMT is classified as type I, Type II, or Type III enzymes depending on the methylation products. The function of type I PRMTs is to catalyze the formation of monomethylarginine, which is subsequently further catalyzed to form asymmetric dimethylarginine87, including PRMT1,2,3,4,6 and 8. The function of type II PRMTs is to catalyze the formation of monomethylarginine intermediates, which are further catalyzed into symmetric dimethyl intermediates, including PRMT5 and 7. Type III PRMT enzymes only catalyze the substrate to form monomethylation products, mainly PRMT7.
PRMT5 (also known as Hsl7, Jbp1, Skb1) is often considered a strong transcriptional suppressor and was first identified as a JAK2-binding protein that methylates its H2A, H3, and H4. PRMT5 plays an important role in development and cancer. Overexpression of PRMT5 has been linked to heart disease, infectious diseases, and cancers such as breast, lung, and liver cancer, making it an important drug target88. In 2020, Jin et al89. reported the first PRMT5 degrader by linking the inhibitor EPZ01566619 and the VHL ligand through PEG chains to develop the PRMT5 selective degrader MS4322 (Figure 9). The degrader can effectively reduce PRMT5 protein level in human breast cancer cell line MCF-7 with DC50 of 1.1uM. MS4322 was also able to significantly reduce PRMT172 protein levels in other cancer lines, such as HeLa, A5, A549, and Jurkat cells, and inhibit the proliferation of these cells. This degrader is therefore a valuable chemical tool for exploring PRMT5’s function in health and disease.
Figure 9.
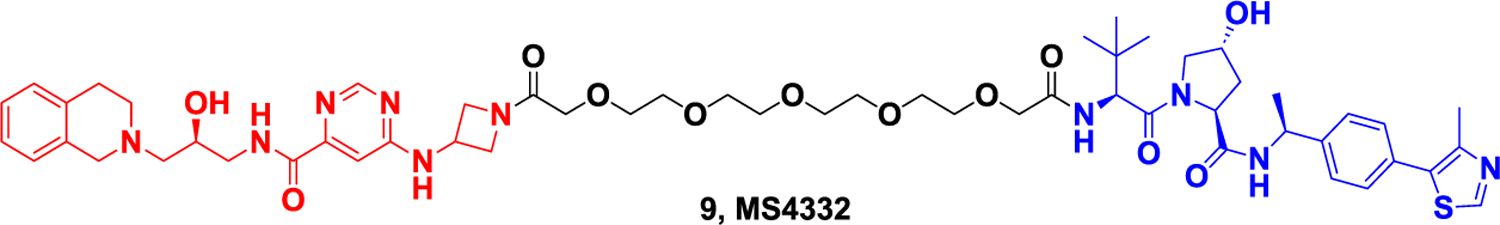
Representative PROTAC targeting PRMT5.
WDR5.
The chromatin associated WD50 repeat domain protein 5 (WDR5) is a functional subunit of the mixed lineage leukemia (MLL) histone methyltransferase complex (also known as the MLL complex), which is responsible for catalyzing H3K4 methylation and has significant catalytic activity only when the same series of regulatory proteins interact to form the complex90–92. WDR5, a major component of this complex, is essential for methylation on chromatin catalyzed by gene transcriptional regulation. WDR5 uses a doughnut-shaped propeller structure, and there are two main binding sites on the surface of its proteins, called WIN (W DR5-interact site) and WBM (WDR5-binding motif) sites93. The WIN position is required for WDR5 chromatin recruitment and interaction with the KMT2 enzyme, and MLL1 is particularly dependent on this interaction. WBM dots mediate protein-protein interactions (PPI) with a variety of non-MLL partners, such as c-MYC. WDR5 is overexpressed in many solid tumors and promotes tumorigenesis, including pancreatic ductal adenocarcinoma (PDAC)94–95. Many inhibitors that block the binding of WDR5 to its partner have been successfully developed by targeting WIN and WBM binding sites respectively; however, these inhibitors that block PPI between WDR5 and its binding partner generally have only relatively weak antitumor activity96–97. On the one hand, it may be due to its occupation-driven mode that it cannot permanently and completely block protein-protein interaction, and on the other hand, it may be due to the inhibitor only targeting part of WDR and not all carcinogenic functions. Therefore, a new treatment strategy needs to be developed. PROTACs have been shown to have a unique role relative to inhibitors, which enable the degradation of POI pharmacologically, thereby temporarily eliminating all functions of POI98.
In May 2021, Knapp et al99. reported two different families of WDR5 degraders based on two WIN site-binding stents, one based on the existing inhibitor OICR-9429 and one based on a modified pyrroimidazole scaffold. A variety of E3 ligands were connected to the modified OICR-9429 scaffold with linker of different lengths to obtain the degrader 10a (Figure 10). It induced the degradation of WDR5 of 58% with a DC50 value of 53 nM in MV4–11 cells. The 10b (Figure 10) molecule was developed based on the modified pyrrole imidazole scaffold and VHL ligand, which induced the degradation of WDR5 of 53% with a DC50 value of 1.24 μM. In September of the same year, Jin’s research group also reported MS67 (Figure 10) based on the inhibitor OICR-9429 and VHL ligand. Through the first round of design, MS33 degrader (Figure 10) (DC50 = 260 nM) was obtained, and the structure of the VHL-MS33-WDR5 ternary complex was successfully solved. Based on the crystal structure, they further optimized the inhibitor ligand and linker to obtain MS67 (DC50=3.7nM), and obtained the VHL-MS67-WDR5 ternary complex. They compared MS67 with other WDR5 inhibitors in a mouse model of AML PDX and showed that MS67 was able to significantly inhibit tumor growth.
Figure 10.
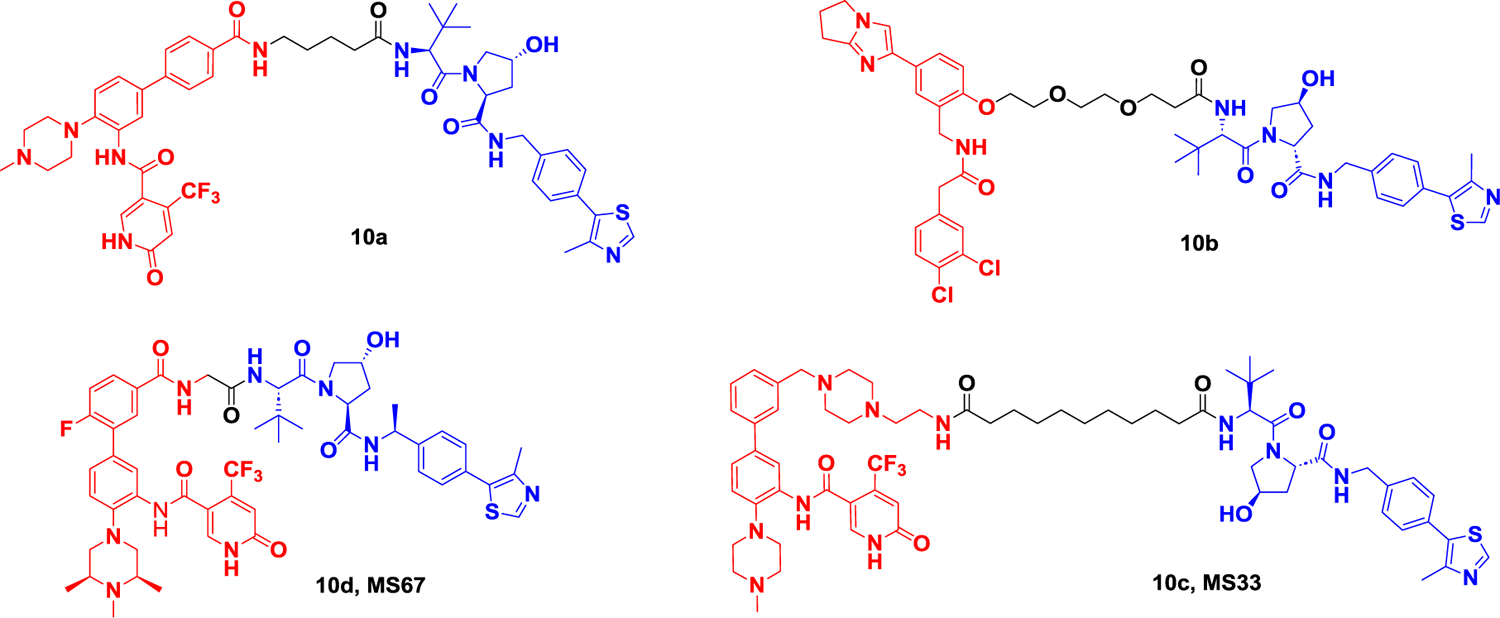
Representative PROTACs targeting WDR5.
NSD3.
Nuclear receptor binding SET domain protein 3 (NSD3; also known as KMT3F or WHSC1L1), is a lysine methyltransferase at position 36 of histone H3 (H3K36) that catalyzes the dimethylation of H3K36101. The NSD3 gene, localized within an amplicon locus of 8p11-p12 in breast and squamous lung cancers, encodes two splicing variants, NSD3-short (NSD3S) and NSD3-long (NSD3L) isoforms102. Overactivity of NSD3 protein is closely associated with many types of tumor development, including human acute myeloid leukemia (AML), breast tumors and lung cancer103–105. Therefore, NSD3 is considered a potential target for the development of novel anticancer drugs. Several inhibitors of NSD3 have been reported, among which BI-9321 blocks the NSD3 PWWP1 domain (a moda retained in both NSD3L and NSD3S), but this inhibitor does not have effective anti-cancer activity106. Using PROTAC strategy to develop NSD3-targeting degraders is expected to obtain more efficient antitumor compounds. In 2022, Wang et al107. reported the first selective degrader of NSD3, MS9715 (Figure 11) by linking the inhibitor BI-9321 to different E3 ligands via different lengths of linker. MS9715 showed the best NSD3 degradation activity in human acute myeloid leukemia cells (MOLM-13) with DC50 of 4.9 μM and Dmax greater than 80%. In addition, transcriptomic analysis showed that MS9715 can effectively inhibit the expression of NSD3 and cMyc-related genes and has better anti-tumor activity than inhibitors.
Figure 11.
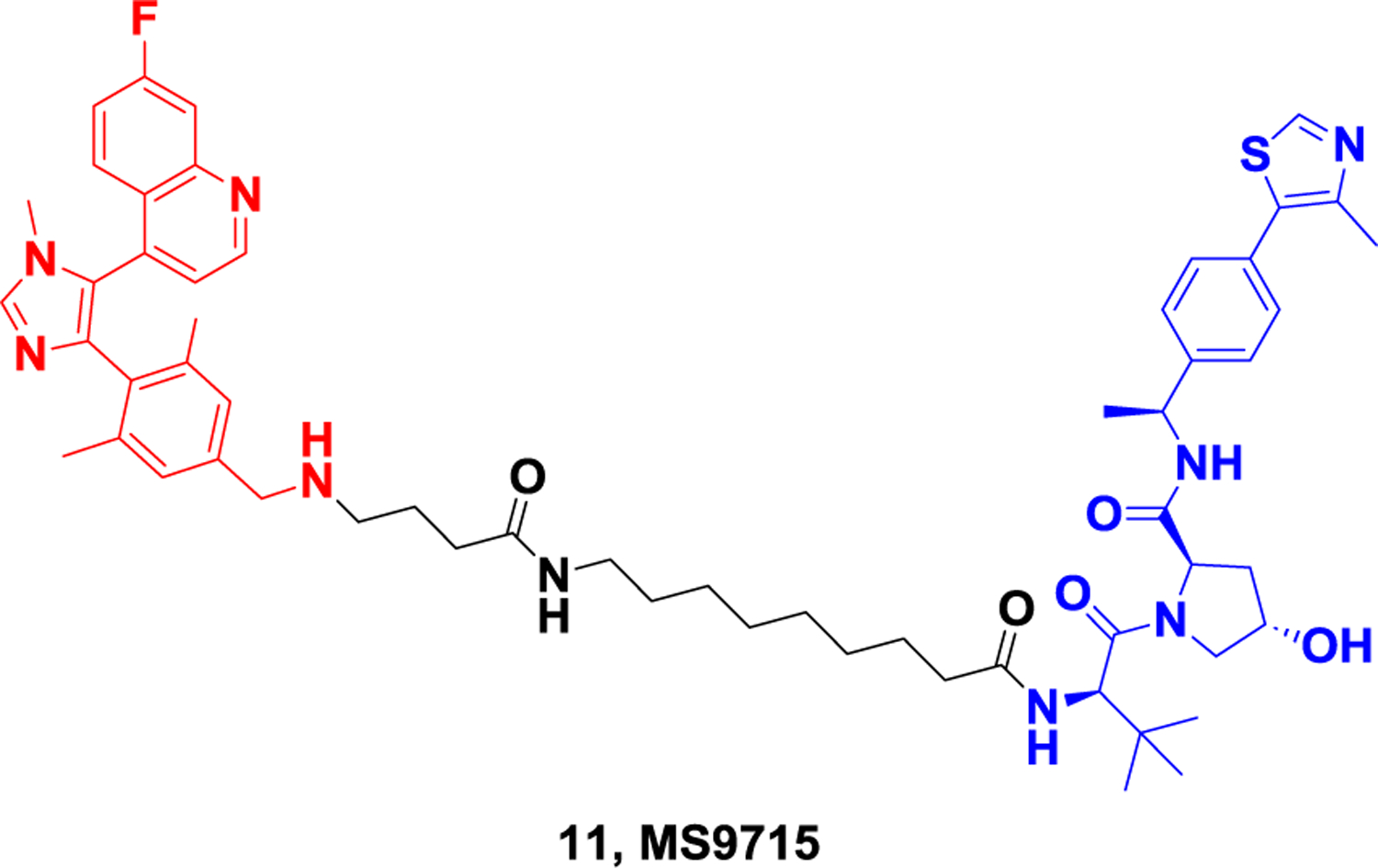
Representative PROTAC targeting NSD3.
2.3. Protein degraders for epigenetic erasers
HDAC.
Histone deacetylases (HDAC) are a class of important epigenetic regulatory factors, which are responsible for catalyzing the deacetylation of histones as “erasers”, thus making histones bind to negatively charged DNA tightly and inhibit gene transcription. Histone deacetylases (HDAC) play a key role in both gene expression regulation and regulation of cell signal transduction pathways and are considered attractive therapeutic targets108. There are currently 18 HDAC enzymes known in humans, which can be divided into four classes. Class 1 consists of HDACs 1, 2, 3, and 8; Class 2 is divided into Class 2a (HDACs 4, 5, 7, 9) and Class 2b (HDACs6 and 10); Class 3, also known as SIRT, consists of SIRT 1,2,3,4,5,6 and 7; Class 4 is represented by HDAC11109,110. Class I HDAC is expressed in a variety of tissues and is mainly located in the nucleus. HDACs1, 2, and 3 isoenzymes exist in large multiprotein complexes, and HDAC8 acts independently of the multiprotein complex111–113. Class II enzymes are more tissue-specific, responding to different cell signaling responses and shuttling between the nucleus and cytoplasm. HDAC6 is the only protein in the HDACs family with two functionally independent catalytic domains and a ubiquitin-bound zinc finger domain114,115. Class III is completely different from the atypical histone deacetylase family of other HDACs. HDAC11 type IV is expressed in the brain, heart, kidney, testis, and skeletal muscle and has nuclear localization characteristics. HDAC enzymes have also been shown to have varying degrees of catalytic reactivity to acetylated lysine, and only HDACs 1, 2, 3 and 6 have been shown to observe lysine deacetylase activity in vitro116.
Dozens of drugs with HDAC-inhibiting properties have been developed, mainly for the treatment of hematoma, and promising advances have been made in inflammatory diseases and neurodegenerative diseases. However, some HDACs enzymes have scaffold functions in addition to catalysis, such as HDAC3, which limits the efficacy of inhibitors117. In addition, many of the currently approved HDAC inhibitors non-selectively target various HDACs and exhibit significant toxicity. PROTAC technology, as a new protein degradation technology, is expected to overcome various problems existing in inhibitors.
In 2018, Tang et al. developed dHDAC6 (Figure 12A), the first degrader of the HDAC family, based on a non-selective HDAC inhibitor and the E3 ligand Pemadomide118.The concentration at which half-maximal degradation was achieved (DC50) and the maximum percentage of degradation (Dmax) are 34 nM and 70.5% respectively. In 2019, Rao et al. obtained A novel HDAC6 degrader, NP8 (Figure 12A), based on the selective HDAC6 inhibitor Nexturastat A (Nex A) and the CRBN ligand pomadomide. Subsequently, they changed the location of the inhibitor to linker, connected pomalidomide to the benzene ring of NexA, and obtained the HDAC6 degrader NH2 (Figure 12A), whose activity was significantly improved compared to that of NP8, with a DC50 of 3.2 nM in human multiple myeloma cells, MM.1S. This shows that the ternary compound has good flexibility119. In 2020, Tang Group obtained the first selective HDAC6 degrader 12d (Figure 12A) based on the NexA and VHL ligand. The DC50’s of the most potent degrader 12d are 7.1 nM and 4.3 nM in human MM1S and mouse 4935 cell lines, respectively. They then developed a competitive assay to evaluate the binding affinity of different E3 ligands in cells and screened for libraries of thalidomide analogs, including those with partial linkers. By combining the most active E3 ligand with the pan inhibitor SAHA, they found a selective HDAC6 degradation product, YZ167 (Figure 12A), with a DC50 of 1.94 nM in MM.1S cells. In addition, the degradation compound YZ268 (Figure 12A), which is designed based on the selective HDAC6 inhibitor Next-A, also has selective degradation activity on HDAC6, without affecting the new substrates IKZFs and GSPT1120–122. In 2021, He et al. coupled a selective HDAC6 inhibitor derived from the natural product indirubin with pemadomide to obtain a new degrader 12g (Figure 12B) with a DC50 of 108.9 nM and a Dmax of 88%. The application of this HDAC6 reducer in LPS-induced mice attenuated NLRP3 inflammasome activation, demonstrating for the first time that HDAC6 PROTAC may be a novel strategy for the treatment of NLRP3 inflammasome-associated diseases123.
Figure 12A.
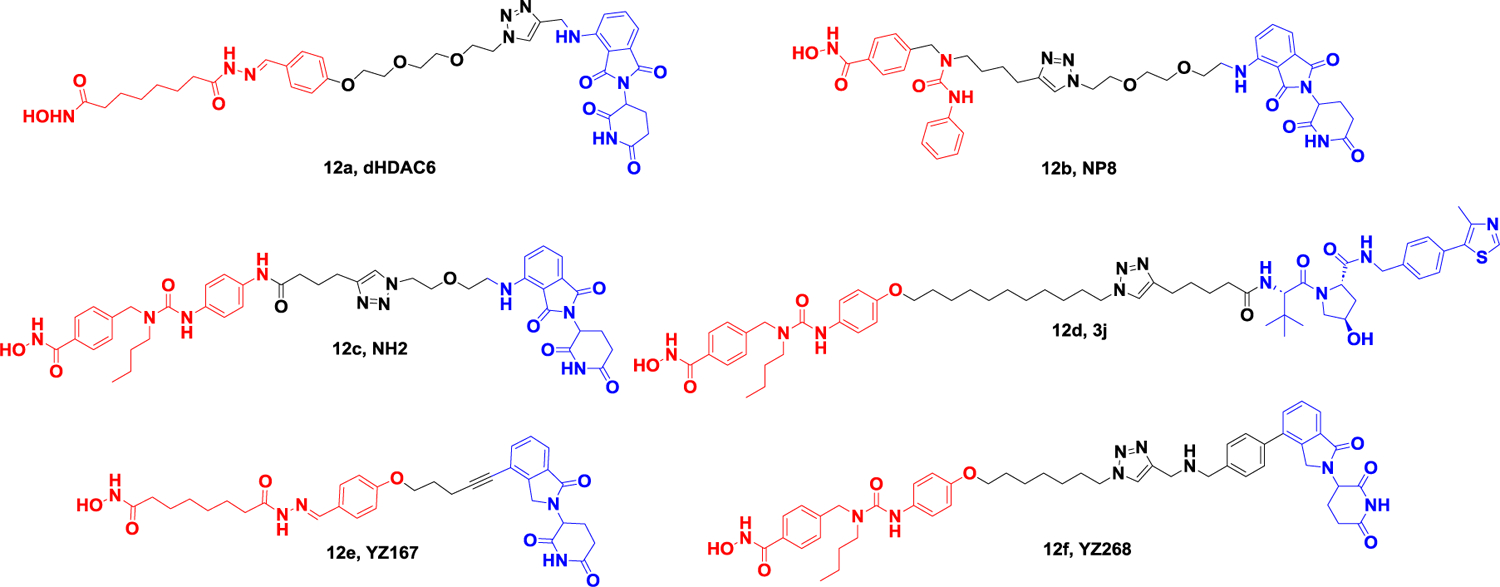
Representative PROTACs targeting HDAC.
Figure 12B.
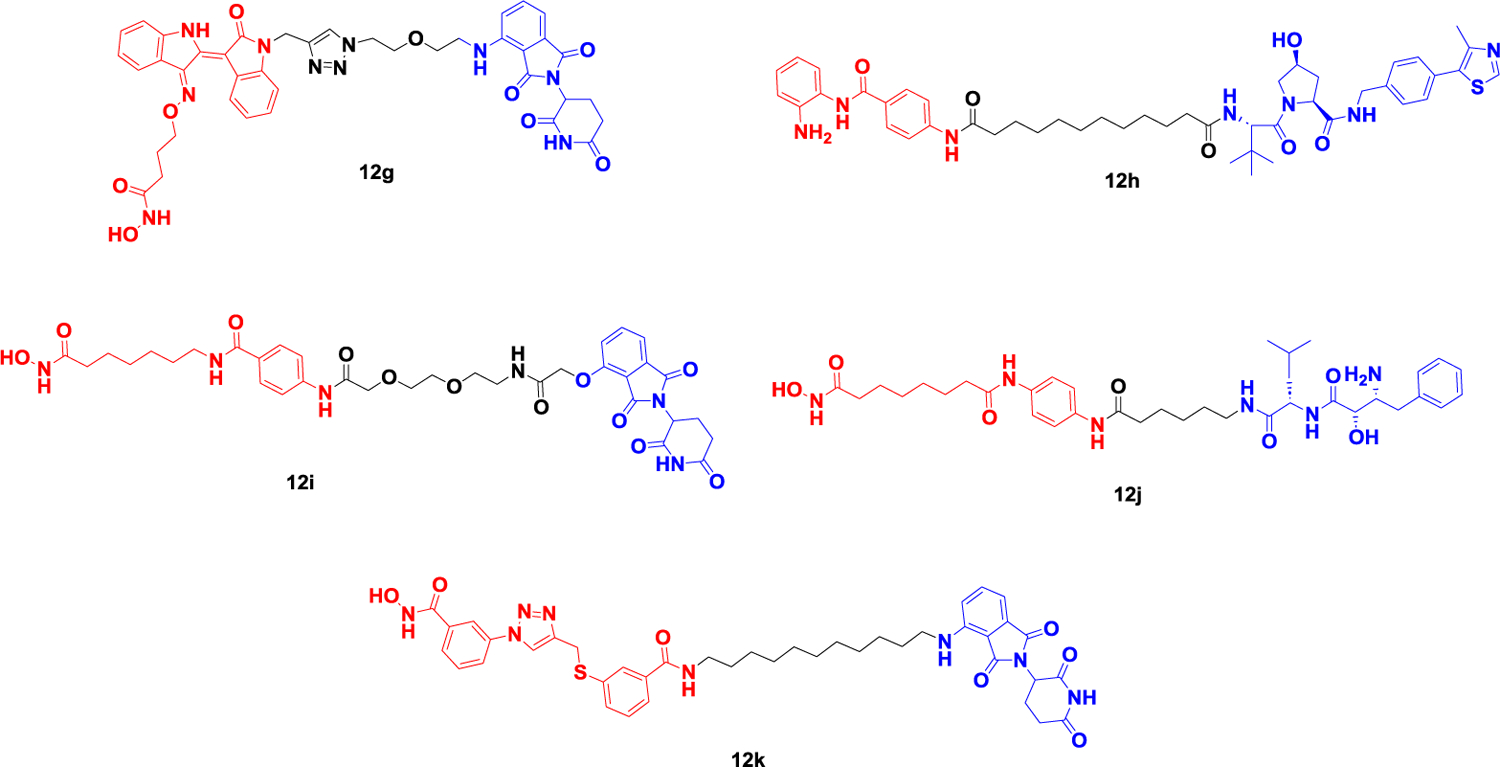
Representative PROTACs targeting HDAC.
In addition to the above-mentioned degradation agents that selectively target HDAC6, many degradation agents targeting other HDAC subtypes have also been reported in the past two years, including HDAC1, HDAC2, HDAC3, and HDAC8. Hodgkinson’s group reported a Class I HDAC (HDAC1/2/3) degrader 12h (Figure 12B) designed based on the VHL and HDAC inhibitor CI-994, which induced the degradation of HDAC1/2/3 in HCT116 cells. They then optimized the linker and VHL ligands on the basis of the developed Class I histone deacetylase PROTACs to obtain a selective degrader targeting HDAC1/2 by optimizing the linker of 12h and further demonstrated that HDAC1/2 is essential for inducing apoptosis and cell arrest in cancer cells124,125.
Hansen et al. discovered a series of alkylated HDACIs using pharmacophore linking strategies and applied HAIR technology to synthesize a proof-of-concept HDAC degrader 12i (Figure 12B) based on pomalidomide and SAHA. Degrader 12i has strong inhibitory activity on a variety of HDAC subtypes but only induces the degradation of HDAC1 and HDAC6, which indicates that the selectivity of the degrader is different from its affinity activity and provides a reference for the subsequent development of selective degraders126.
Based on Bestatin and SAHA, Zhang et al. recruited apoptosis protein 1 (cIAP1) E3 ubiquitin ligase to achieve targeted degradation of HDAC6. In fact, treatment with the degrader 12j (Figure 12B) in human multiple myeloma cells (RPMI-8226) for 24 h resulted in the effective degradation of HDAC1/6/8. In addition, the degrader also exhibited more potent aminopeptidyl N (APN, CD13) inhibitory activity and antiangiogenic activity than the approved APN inhibitor Bestatin, meaning that the compound is both an HDAC1/6/8 degrader and a dual inhibitor of both APN and HDAC127. In 2022, Suzuki et al. successfully developed a selective HDAC8 degrader 12k (Figure 12B) based on the HDAC8 selective inhibitor and CRBN ligand they reported previously. Compared with HDAC8 inhibitors, the deactivator can more effectively inhibit the growth of T-cell leukemia Jurkat cells128.
SIRT2.
Mammalian sirtuin protein is a niacinamide adenine (NAD+) -dependent histone deacetylase that uses NAD+ as a co-substrate to regulate the acetylation and ribosylation of a variety of proteins129. The human sirtuin family consists of seven subtypes, SIRT1-SIRT7, all of which have highly conserved NAD binding domains and catalytic functional domains. Of the seven sirtuins, SIRT2 is the only one that resides primarily in the cytoplasm and can remove acetyl and other acyl groups from protein lysine residues120,131. SIRT2 promotes tumor growth and regulates various biological pathways through lysine deacetylation and degreasing acylation, which makes SIRT2 an attractive target for cancer therapy.
In 2018, Manfred et al. reported the first PROTAC 13a (Figure 13) to degrade SIRT2132. They combined the structural characteristics of sirt2 selectivity and high-efficiency triazolyl SirReals with the ligand thalidomide to achieve chemically induced degradation of Sirt2. This is the first example of a PROTAC targeting an epigenetic erasure protein. Compound 12 showed significant SIRT2 protein selective degradation in human cervical cancer cells (HeLa) at 10 μM level for 2 hours. In 2020, Lin et al. reported a case of the PROTAC molecule TM-P4-Thal (Figure 13), combined with the thiomyristoyl lysine-based SIRT2 selective inhibitor TM and CRBN ligand through a PEG linker and can simultaneously inhibit the activity of SIRT2 protein deacetylase and ester acylase133.The degrader TM-P4-Thal can effectively and selectively degrade SIRT2 in MCF7 cells at the level of 0.5μM for 48 hours, and the deacetylation of α-tubulin, a downstream target of SIRT2, can be significantly inhibited at the level of 10 μM for 12 hours. TM-P4-Thal also effectively inhibited the defatting acylation of SIRT2 downstream target K-Ras4a in HEK 293T cells at the level of 1μM for 48 hours.
Figure 13.
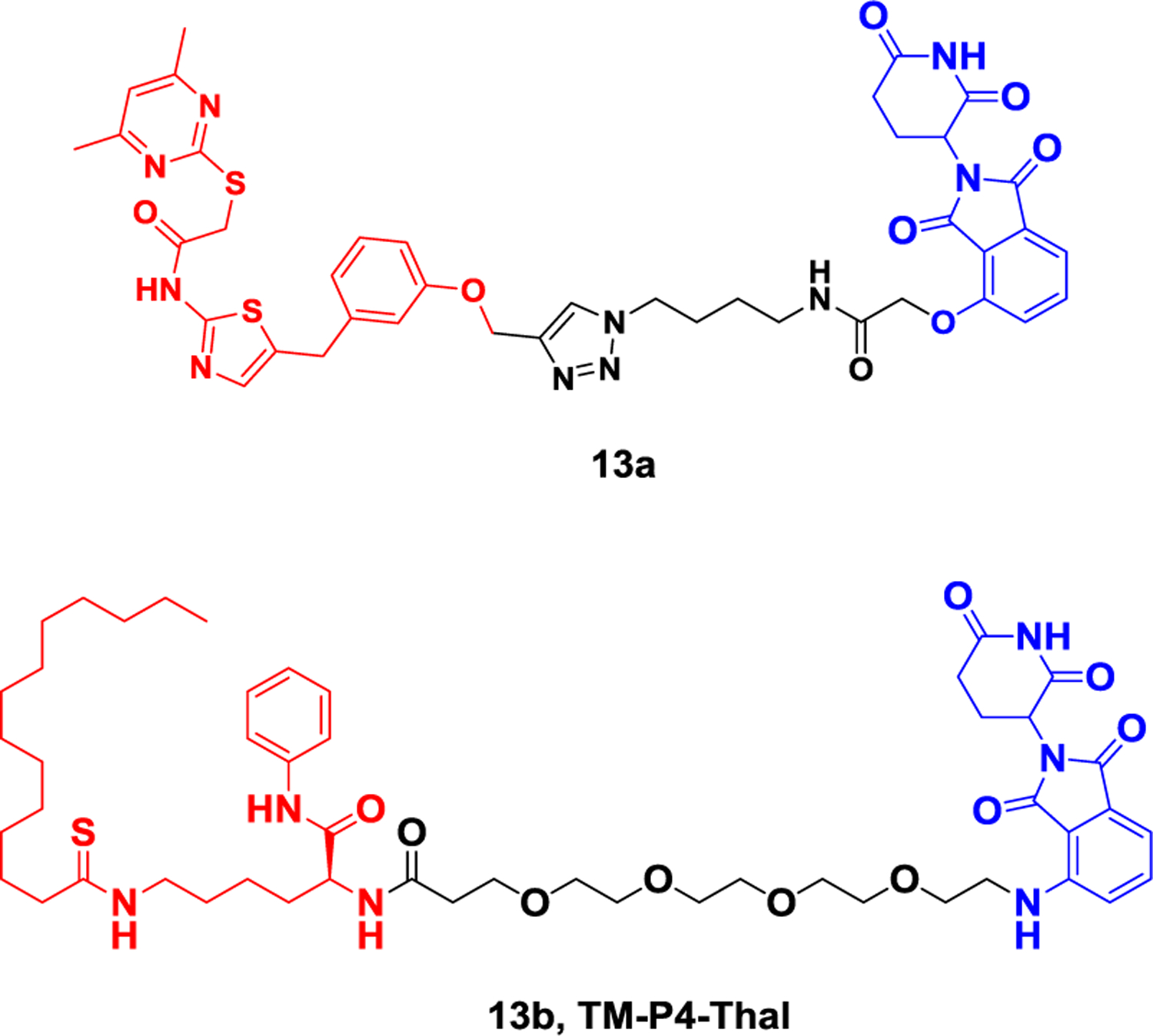
Representative PROTAC targeting SIRT2.
KDM5C.
Histone demethylase (KDMs) plays an important role in the epigenetic modification of histone methylation and demethylation. KDM family proteins can be divided into two types according to their demethylation mechanism. One type is composed of FAD-dependent enzymes, including KDM1A and KDM1B. The other type is composed of Fe(II)/α-ketoglutarate-dependent enzymes, including KDM2–7134. The KDM5 family (KDMA-D) interacts with the chromatin remodeling NuRD complex and histone deacetylase complex, catalyzing the demethylation of lysine 4 histone H3 (H3K4me3/2) that is dimethylated or trimethylated135. KDM5 enzymes are associated with cancer occurrence and neurodegenerative diseases regulated by epigenetic mechanisms136. Several KDM5 inhibitors have been successfully developed, which have good inhibitory activity in vitro but have not shown effective anticancer effects. KDM proteins regulate genetic gene expression through both enzymatic and scaffold functions, where the “catalytic function” oxidizes methyl from lysine residues in histones and removes them, and the “scaffold function” interacts with transcription factors to form protein complexes137. Traditional KDM5 inhibitors only inhibit their enzymatic function without interfering with their scaffold function, which may be one of the reasons for the poor clinical efficacy of inhibitors. Developing KDM5 degraders based on protein degradation technology is expected to overcome the limitations of inhibitors. In 2021138, Suzuki et al. reported the first case of a histone demethylase KDMs degrader 14 (Figure 14) based on a KDM5C inhibitor, which can achieve significant protein degradation at a level of 5 μM in prostate cancer PC-3 cells. The experiment also proved that the degrader 14 has significantly better anti-proliferative activity than the inhibitor. This is the first report of a histone demethylase KDMs degrader and lays the foundation for the development of related target degraders.
Figure 14.
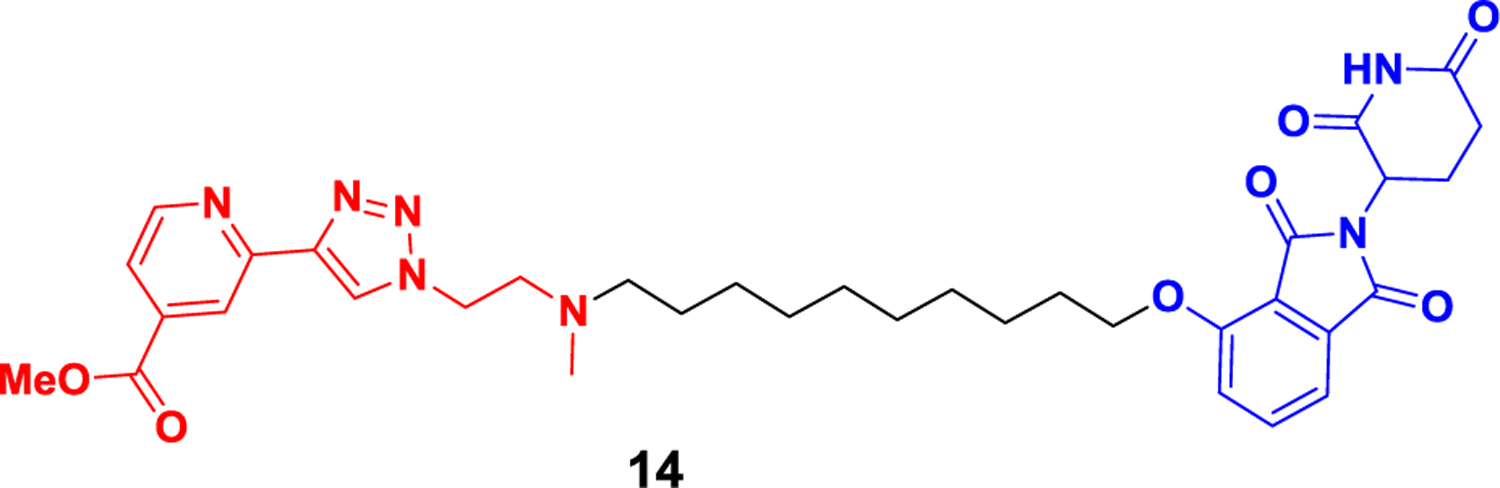
Representative PROTAC targeting KDM5C.
2.4. Representative ternary complex crystal structures
In 2017, the Ciulli group successfully solved the first ternary crystal structure of the VHL-MZ1-BRD4 complex (Figure 15a, PDB: 5T35). This groundbreaking structure revealed the presence of a novel protein-protein interaction between VHL and the second bromodomain of BRD4 (BD2 domain). This structural insight could provide valuable clues as to why MZ1 selectively binds to BRD4 and not to other isoforms.139 For another commonly used E3 ligase, cereblon (CRBN), the Fischer group successfully solved the ternary complex structure of CRBN-dBET23-BRD4 (Figure 15b, PDB: 6BN7). In this complex, dBET23 induced the second bromodomain of BRD4 (BD1 domain) to form de novo protein-protein interactions with both the thalidomide-binding domain (TBD) and the LON N domain of CRBN.140 These ternary complex crystal structures provide framework for the development of selective PROTACs for epigenetic proteins.
Figure 15.
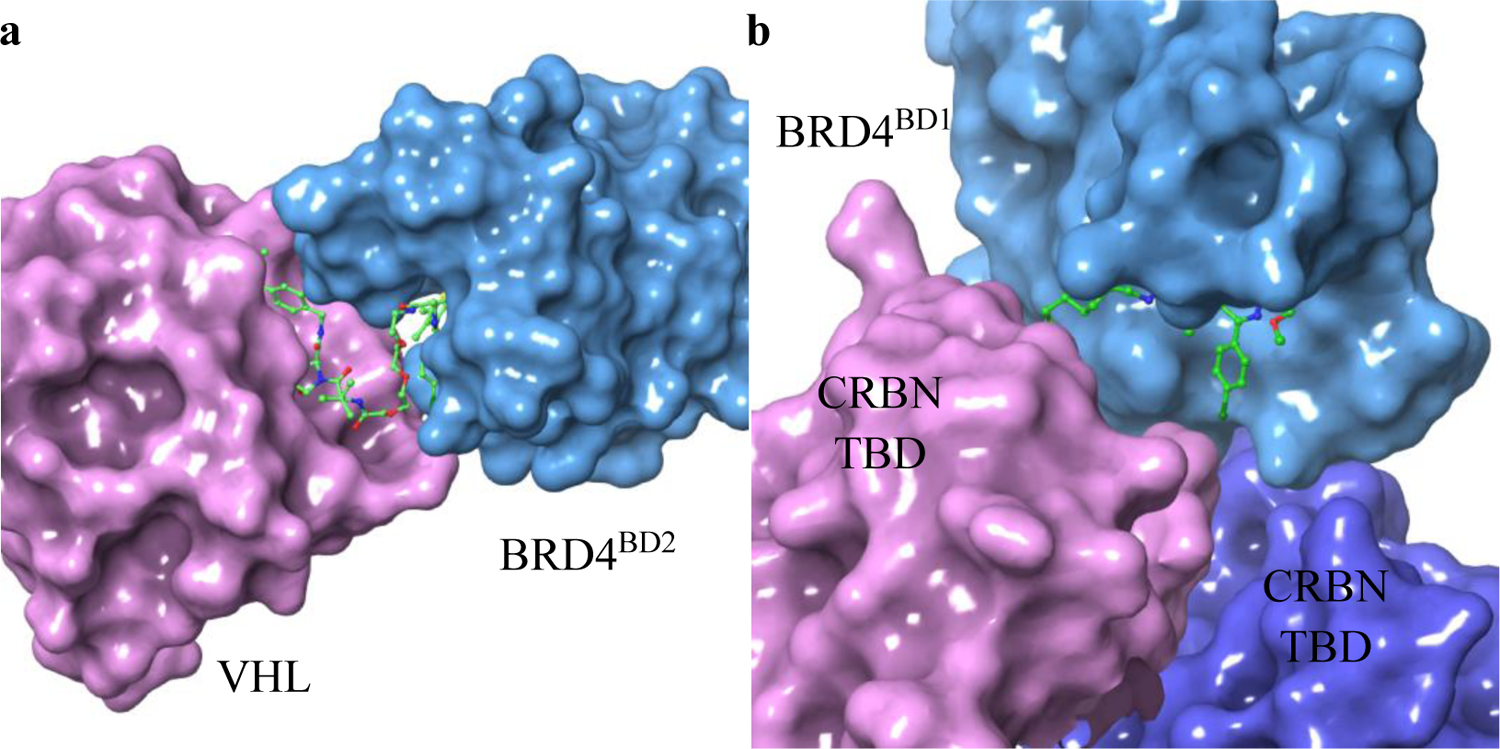
Representative ternary complex of E3 ligase, PROTAC and epigenetic protein. a, VHL-MZ1-BRD4 complex (PDB: 5T35); b, CRBN-dBET23-BRD4 complex (PDB: 6BN7).
2.5. New applications of epigenetic protein degraders
Antibody-BRD 4 degrader conjugate
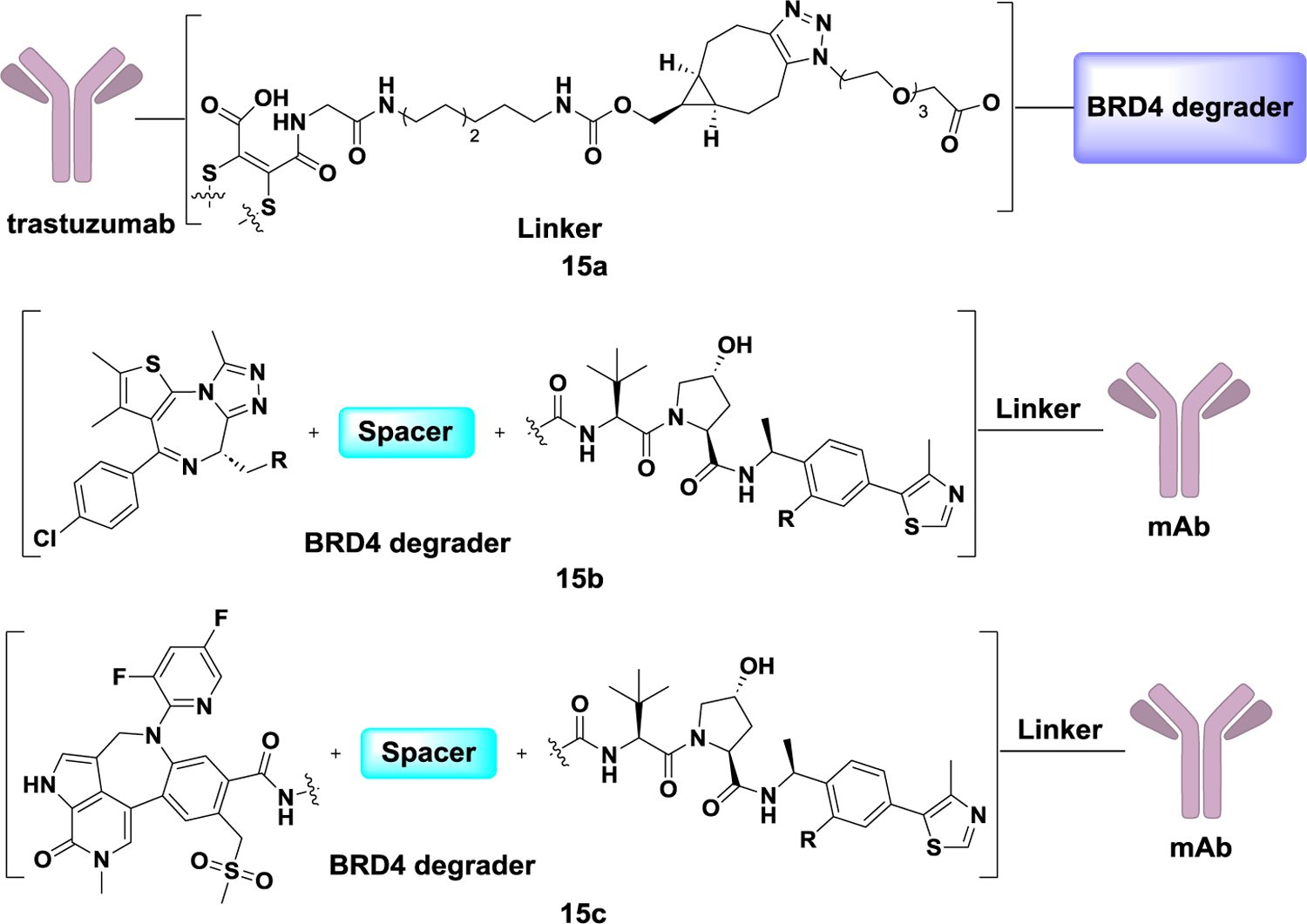
Although PROTAC technology has demonstrated significant advantages, most PROTAC molecules exhibit poor tissue selectivity and cannot distinguish between different cell types. The drawbacks have hindered the further application of this technology. In 2020, the research group led by Tate developed the AbPROTAC technology, which conjugates trastuzumab with a BRD4 degrader molecule 15a (Figure 16)141. This enables selective targeting of HER2-positive cells. Using confocal microscopy, the authors demonstrated that internalization and lysosomal transport occurred specifically in HER2-positive cells. The release of the PROTAC molecule inside the cells led to the degradation of BRD4.
Figure 16.
Representative PROTAC-antibody conjugate.
To address the issues of oral bioavailability and solubility of PROTAC molecules, in 2021, Peter S. Dragovich and colleagues connected MZ1 to an antibody that specifically recognizes the cell surface antigen STEAP1. In prostate cancer cells, the antibody-drug conjugate exhibited potent antigen-dependent anti-proliferative activity 15b (Figure 16)142. Subsequently, this research team utilized novel BRD4 degrader molecules to synthesize antibody-drug conjugates, which displayed even more potent antigen-dependent anti-tumor activity in HL-60 (human prostate cancer cell line) mouse xenograft 15c (Figure 16)143.
BRD 4 degrader in phase separation
Biomolecular condensates play crucial roles in various biological processes. However, specific regulators for these condensates are currently lacking. PROTACs can dynamically modulate biomolecular condensates by degrading key molecules within them. Recently, the Rao group discovered that BRD4 degrader ZXH-3–26 significantly reduce BRD4 condensates. They also identified that BRD4 condensates form preferentially, serving specific functions in the regulation of biological processes. The PROTAC technology offers an effective and targeted approach for studying biomolecular condensates (Figure 17).144
Figure 17.
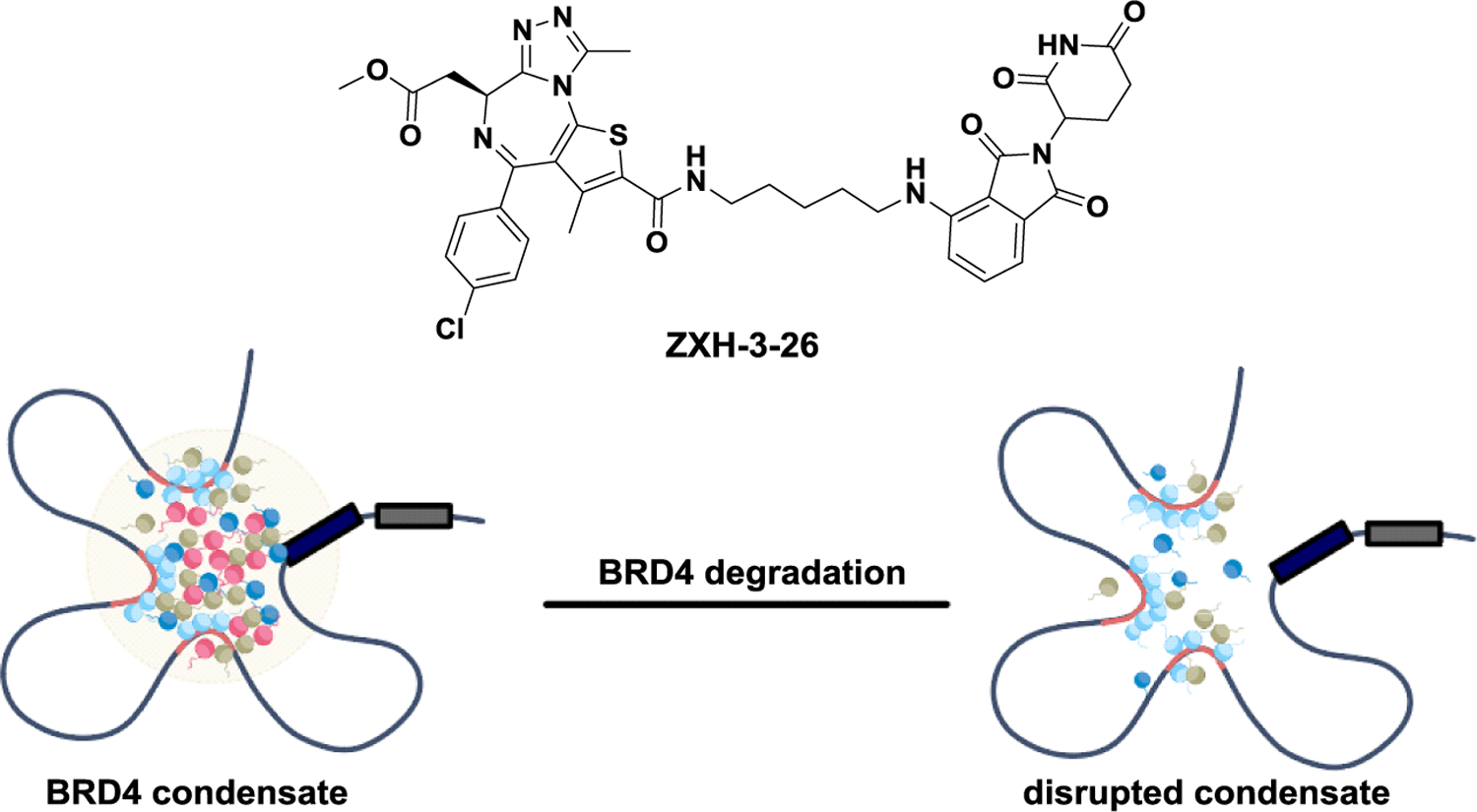
Modulating BRD4 condensate through PROTAC.
Delivery Systems for BRD4 degrader
In 2023, Xiang Gao team developed a tumor-targeting copolymer designed for the co-delivery of DOX (doxorubicin) and the BRD4 degrader ARV-825, referred to as ARV-DOX/cRGD-P, as a potential treatment approach for colorectal cancer (CRC). Their study identified BRD4 as a promising therapeutic target for adriamycin-resistant CRC, and the use of ARV-825 as a PROTAC degrader demonstrated the potential to enhance the sensitivity of CRC to adriamycin. The ARV-DOX/cRGD-P copolymer exhibited significant anti-tumor effects, suggesting its potential application in clinical CRC treatment.145
2.6. PROTACs available that target epigenetic proteins
As illustrated in Table 1, our review comprehensively outlines the existing PROTACs designed to target epigenetic proteins. This burgeoning class of therapeutic agents holds immense promise in modulating the activity of proteins associated with epigenetic regulation. In this context, the field of epigenetic-targeted PROTACs is dynamic, with continuous advancements propelling these compounds toward therapeutic breakthroughs.
Table 1.
The summary of currently available PROTACs targeting epigenetic proteins
| NO. | Name | Target | NO. | Name | Target |
|---|---|---|---|---|---|
| 1 | 3a, ARV-771 | BET | 19 | 8b, UNC6852 | PRC2 |
| 2 | 3b, dBET1 | BET | 20 | 8c, MS1943 | EZH2 |
| 3 | 3c, BETd-246 | BET | 21 | 8d, E7 | PRC2 |
| 4 | 4a, HBL-4 | BRD4 | 22 | 9, MS4332 | PRMT5 |
| 5 | 4b, MacroPROTAC-1 | BET | 23 | 10a | WDR5 |
| 6 | 4d, SM1 | BET | 24 | 10d, MS67 | WDR5 |
| 7 | 4f | BRD4 | 25 | 11, MS9715 | NSD3 |
| 8 | 4j, A1847 | BRD4 | 26 | 12a, dHDAC6 | HDAC6 |
| 9 | 4l, KB02-JQ1 | BRD4 | 27 | 12b, NP8 | HDAC6 |
| 10 | 4n | BRD3/4 | 28 | 12c, NH2 | HDAC6 |
| 11 | 4p, XY-06–007 | BRD4 | 29 | 12e, YZ167 | HDAC6 |
| 12 | 4q, AGB | BRD2 | 30 | 12h | HDAC1/2/3 |
| 13 | 5a, dBRD9 | BRD9 | 31 | 12i | HDAC1/6 |
| 14 | 5b, VZ185 | BRD7/9 | 32 | 12j | HDAC1/6/8 |
| 15 | 5c, A547 | SMARCA2 | 33 | 12k | HDAC8 |
| 16 | 6, SR1114 | ENL | 34 | 13a | SIRT2 |
| 17 | 7, dCBP-1 | CBP | 35 | 13b, TM-P4-Thal | SIRT2 |
| 18 | 8a | PRC2 | 36 | 14 | KDM5C |
3. Conclusion and outlook
In this review, we summarized the degraders reported for epigenetics targets, including writes, readers, and erasers. These epigenetic regulators have been implicated in various diseases, making them attractive therapeutic targets. Notably, researchers have developed innovative strategies to target epigenetic targets, which play crucial roles in modulating chromatin structure and gene expression. As the field of epigenetic degradation continues to evolve, it holds immense promise for the development of novel therapeutics across various diseases, ranging from cancer to neurodegenerative disorders. The successful clinical investigation of BRD9 degraders, such as FHD-609 and CFT8634, underscores the translational potential of this approach and paves the way for further advancements in epigenetic target degradation strategies.146 With ongoing research and innovative techniques, the future of epigenetics-based therapies appears bright, offering new avenues for precision medicine and improved patient outcomes.
While there is promise in the PROTAC field, several challenges remain unresolved. For instance: 1) Discovery of PROTAC molecules is currently confined to known ligands and established binding pockets. Prolonged treatment could lead to drug resistance; 2) Some undruggable epigenetic targets lack suitable ligands for degrader development; 3) The availability of E3 ligases is limited, with only CRBN and VHL being utilized in degrader development.
Addressing the challenges in the PROTAC field has spurred the development of innovative methodologies aimed at overcoming existing limitations and broadening the scope of targetable proteins. These emerging strategies hold the potential to revolutionize the field of targeted protein degradation:
1. Expanding Ligand Discovery and Binding Pockets: To overcome the limitation of relying solely on known ligands and binding pockets, researchers are actively exploring new approaches. This includes the use of computational methods to predict potential binding sites on target proteins, enabling the design of PROTACs that can engage previously unexplored regions. Additionally, advancements in chemical synthesis and high-throughput screening techniques have facilitated the identification of novel ligands, enabling the development of PROTACs against a wider range of protein targets. These efforts not only enhance target diversity but also reduce the risk of drug resistance due to prolonged treatment.
2. Exploration of new E3 Ligases: While CRBN and VHL have been the primary E3 ligases utilized in PROTAC development, efforts are underway to identify new E3 ligases. This diversification offers the possibility of targeting different cellular compartments, optimizing degradation kinetics, and minimizing off-target effects. By harnessing a wider array of E3 ligases, researchers can enhance the precision and versatility of PROTAC-based therapies.
3. Advanced Delivery Systems: Developing effective delivery systems is critical for the clinical translation of PROTACs. Researchers are actively exploring nanoparticle-based delivery platforms, cell-penetrating peptides, and other innovative techniques to enhance the intracellular uptake and stability of PROTAC molecules. These advancements aim to improve target engagement and tissue specificity while minimizing off-target effects.
As the field continues to evolve, the combination of these novel methodologies holds great promise in addressing the challenges posed by PROTAC development. With a concerted effort towards innovation and collaboration, researchers are working towards realizing the full therapeutic potential of targeted protein degradation across a wide range of diseases and previously challenging target proteins.
ACKNOWLEDGEMENTS
This work was supported by National Key R&D Program of China (#2021YFA1300200, #2021YFA1302100 and #2020YFE0202200), National Natural Science Foundation of China (#82125034, 82330115), and NIH grant R01CA177910.
Footnotes
CONFLICTS OF INTEREST
The authors declare no competing financial interest.
Reference
- 1.Waddington CH. The epigenotype. Endeavour 1942;1:18–20. [Google Scholar]
- 2.Cathérine Dupont D Armant Randall, Brenner Carol A. Epigenetics: definition, mechanisms and clinical perspective. Semin Reprod Med. 2009;27:351–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bird A. Perceptions of epigenetics. Nature. 2007;447:396–8. [DOI] [PubMed] [Google Scholar]
- 4.Hake SB, Allis CD. Histone H3 variants and their potential role in indexing mammalian genomes: the “H3 barcode hypothesis.” PNAS. 2006;103:6428–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Donnell KJ, Meaney MJ. Epigenetics, development, and psychopathology. Ann Rev Clin Psychol. 2020;16:327–50. [DOI] [PubMed] [Google Scholar]
- 6.Narlikar GJ, Sundaramoorthy R, Owen-Hughes T. Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell. 2013;154:490–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nepali Kunal, Liou Jing-Ping. Recent developments in epigenetic cancer therapeutics: clinical advancement and emerging trends. J Biomed Sci. 2021;28:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, et al. Identifcation of the major Abeta1-042-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med. 2000;6:143–50. [DOI] [PubMed] [Google Scholar]
- 9.Stilling RM, Fischer A. The role of histone acetylation in age-associated memory impairment and Alzheimer’s disease. Neurobiol Learn Mem. 2011;96:19–26. [DOI] [PubMed] [Google Scholar]
- 10.Caccamo A, Maldonado MA, Bokov AF, Majumder S, Oddo S. CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2010;107:22687–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kontopoulos E, Parvin JD, Feany MB. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet. 2006;15:3012–23. [DOI] [PubMed] [Google Scholar]
- 12.Liu R, Lei JX, Luo C, Lan X, Chi L, Deng P, et al. Increased EID1 nuclear translocation impairs synaptic plasticity and memory function associated with pathogenesis of Alzheimer’s disease. Neurobiol Dis. 2012;45:902–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chapuis J, Hansmannel F, Gistelinck M, Mounier A, Van Cauwen-berghe C, Kolen KV, et al. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol Psychiatry. 2013;18:1225–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Z, Schluesener YHJ. Oral administration of histone deacetylase inhibitor MS-275 ameliorates neuroinfammation and cerebral amyloidosis and improves behavior in a mouse model. J Neuropathol Exp Neurol. 2013;72:178–85. [DOI] [PubMed] [Google Scholar]
- 15.Miller AA, Kurschel E, Osieka R, Schmidt CG. Clinical pharmacology of sodium butyrate in patients with acute leukemia. Eur J Cancer Clin Oncol. 1987;23:1283–7. [DOI] [PubMed] [Google Scholar]
- 16.Chen KL, Wang SS, Yang YY, Yuang RY, Chen RM, Hu CJ. The epigenetic effects of amyloidbeta (1–40) on global DNA and neprilysin genes in murine cerebral endothelial cells. Biochem Biophys Res Comm. 2009;378:57–61. [DOI] [PubMed] [Google Scholar]
- 17.Zhang K, Schrag M, Crofton A, Trivedi R, Vinters H, Kirsch W. Targeted proteomics for quantifcation of histone acetylation in Alzheimer’s disease. Proteomics. 2012;12:1261–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Francis YI, Fa M, Ashraf H, Zhang H, Staniszewski A, Latchman DS, et al. Dysregulation of histone acetylation in the APP/PS1 mouse model of Alzheimer’s disease. J Alzheimer Dis. 2009;18:131–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Webb Thomas, Craigon Conner, Ciulli Alessio. Targeting epigenetic modulators using PROTAC degraders: Current status and future perspective. Bioorg. Med. Chem. Lett 2022, 63, 128653. [DOI] [PubMed] [Google Scholar]
- 20.Mithraprabhu S, Kalff A, Chow A, Khong T, Spencer A. Dysregulated Class I histone deacetylases are indicators of poor prognosis in multiple myeloma. Epigenetics. 2014;9(11):1511–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang L, Rau R, Goodell MA. DNMT3A in haematological malignancies. Nat Rev Cancer. 2015;15(3):152–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mycosis fungoides/Sezary syndrome): Use in a community setting. Crit Rev Oncol Hematol. 2016;106:99–107. [DOI] [PubMed] [Google Scholar]
- 23.Nakajima HK, Young Bae, Terano HY, Minoru Horinouchi Sueharu. FR901228, a Potent Antitumor Antibiotic. Is a Novel Histone Deacetylase Inhibitor. 1998;241: 126–133. [DOI] [PubMed] [Google Scholar]
- 24.Webb Thomas, Craigon Conner, Ciulli Alessio. Targeting epigenetic modulators using PROTAC degraders: Current status and future perspective. Bioorg. Med. Chem. Lett 2022, 63, 128653. [DOI] [PubMed] [Google Scholar]
- 25.U.S. Food and Drug Administration: FDA granted accelerated approval to tazemetostat for follicular lymphoma.
- 26.Walczak MJ, Petzold G & Thoma NH Targeted protein degradation. You can glue it too! Nat. Chem. Biol 13, 452–453 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Békés M, Langley DR & Crews CM PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov 21, 181–200 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Konstantinidou M et al. PROTACs- a game-changing technology. Expert Opin. Drug Discov 14, 1255–1268 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chirnomas D, Hornberger KR & Crews CM Protein degraders enter the clinic-a new approach to cancer therapy. Nat Rev Clin Oncol (2023). 10.1038/s41571-023-00736-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schreiber SL, Cell, 2021, 184, 3–9. [DOI] [PubMed] [Google Scholar]
- 31.Scholes NS, Mayor-Ruiz C & Winter GE Identification and selectivity profiling of small-molecule degraders via multi-omics approaches. Cell Chem. Biol 28, 1048–1060 (2021). [DOI] [PubMed] [Google Scholar]
- 32.Dale B et al. Advancing targeted protein degradation for cancer therapy. Nat. Rev. Cancer 21, 638–654 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He SP, Dong GQ, Cheng JF, Wu Y & Sheng CQ Strategies for designing proteolysis targeting chimaeras (PROTACs). Med. Res. Rev 42, 1280–1342 (2022).). [DOI] [PubMed] [Google Scholar]
- 34.Deshaies RJ Protein degradation. Prime time for PROTACs. Nat. Chem. Biol 11, 634–635 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Sun X, Gao H, Yang Y et al. PROTACs: great opportunities for academia and industry. Sig. Transduct. Target. Ther 4, 64 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He M, Cao C, Ni Z et al. PROTACs: great opportunities for academia and industry (an update from 2020 to 2021). Sig. Transduct. Target. Ther 7, 181 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cao C, He M, Wang L, He Y & Yu Rao. Chem. Soc. Rev 51, 7066–7114 (2022). [DOI] [PubMed] [Google Scholar]
- 38.Webb T, Craigon C & Ciulli A Bioorg. Med. Chem. Lett 63, 128653 (2022). [DOI] [PubMed] [Google Scholar]
- 39.Dhalluin C et al. Structure and ligand of a histone acetyltransferase bromodomain. Nature 399, 491–496 (1999). [DOI] [PubMed] [Google Scholar]
- 40.Georg EW et al. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kanak R et al. PROTAC-induced BET protein degradation as a therapy for castration resistant prostate cancer. Proc. Natl Acad. Sci. USA 113, 7124–7129 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qin C et al. Discovery of QCA570 as an exceptionally potent and efficacious proteolysis targeting chimera (PROTAC) degrader of the bromodomain and extra-terminal (BET) proteins capable of inducing complete and durable tumor regression. J. Med. Chem 61, 6685–6704 (2018).). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Winter GE et al. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raina K et al. PROTAC-induced BET protein degradation as a therapy for castration resistant prostate cancer. Proc. Natl Acad. Sci. USA 113, 7124–7129 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bai L et al. Targeted degradation of BET proteins in triple-negative breast cancer. Cancer Res. 77, 2476–2487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zengerle M, Chan K-H & Ciulli A Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem. Biol 10, 1770–1777 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou B et al. Discovery of a small-molecule degrader of bromodomain and extra-terminal (BET) proteins with picomolar cellular potencies and capable of achieving tumor regression. J. Med. Chem 61, 462–481 (2017).). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raina K et al. PROTAC-induced BET protein degradation as a therapy for castration resistant prostate cancer. Proc. Natl Acad. Sci. USA 113, 7124–7129 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Winter GE et al. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381 (2015).). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bai L et al. Targeted degradation of BET proteins in triple-negative breast cancer. Cancer Res. 77, 2476–2487 (2017).). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qin C et al. Discovery of QCA570 as an exceptionally potent and efficacious proteolysis targeting chimera (PROTAC) degrader of the bromodomain and extra-terminal (BET) proteins capable of inducing complete and durable tumor regression. J. Med. Chem 61, 6685–6704 (2018).) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mu XP, Bai LT, Xu YJ, Wang JY & Lu HB Protein targeting chimeric molecules specific for dual bromodomain 4(BRD4) and Polo-like kinase 1(PLK1)proteins in acute myeloid leukemia cells. Biochem. Biophys. Res. Commun 521, 833–839 (2020). [DOI] [PubMed] [Google Scholar]
- 53.Testa A et al. Structure-based design of a macrocyclic PROTAC. Angew. Chem. Int. Ed. Engl 59, 1727–1734 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiang F et al. Discovery of novel small molecule induced selective degradation of the bromodomain and extra-terminal(BET) bromodomain protein BRD4 and BRD2 with cellular potencies. Bioorg. Med. Chem 28, 115181 (2020). [DOI] [PubMed] [Google Scholar]
- 55.Imaide S et al. Trivalent PROTACs enhance protein degradation via combined avidity and cooperativity. Nat. Chem. Biol 17, 1157–1167 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang J et al. Development of small-molecule BRD4 degraders based on pyrrolopyridone derivative. Bioorg. Chem 99, 103817 (2020). [DOI] [PubMed] [Google Scholar]
- 57.Xiang W et al. Structure-guided discovery of novel potent and efficacious proteolysis targeting chimera (PROTAC) degrader of BRD4. Bioorg. Chem 115, 105238 (2021). [DOI] [PubMed] [Google Scholar]
- 58.Xue G, Wang K, Zhou DL, Zhong HB & Pan ZY Light-induced protein degradation with photocaged PROTACs. J. Am. Chem. Soc 141, 18370–18374 (2019). [DOI] [PubMed] [Google Scholar]
- 59.Li ZZ et al. Development of photocontrolled BRD4 PROTACs for tongue squamous cell carcinoma (TSCC). Eur. J. Med. Chem 222, 113608 (2021). [DOI] [PubMed] [Google Scholar]
- 60.Qin AC, Jin H, Song Y, Gao Y, Chen YF, Zhou LN, Wang SS, Lu XS. The therapeutic effect of the BRD4-degrading PROTAC A1874 in human colon cancer cells. Cell death & disease 2020, 11, 805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kapoor S, Gustafson T, Zhang M, Chen YS, Li J, Nguyen N, Perez JET, Dashwood WM, Rajendran P, Dashwood RH. Deacetylase Plus Bromodomain Inhibition Downregulates ERCC2 and Suppresses the Growth of Metastatic Colon Cancer Cells. Cancers 2021, 13, 1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang XY et al. Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat. Chem. Biol 15, 737–746 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li L et al. In vivo target protein degradation induced by PROTACs based on E3 ligase DCAF15. Signal Transduct. Target Ther 5, 129–131 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wei JL et al. Harnessing the E3 ligase KEAP1 for targeted protein degradation. J. Am. Chem. Soc 143, 15073–15083 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rankovic Z et al. Phenyl-Glutarimides: alternative cereblon binders for the design of PROTACs. Angew. Chem. Int. Ed. Engl 60, 26663–26670 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nowak RP et al. Structure-guided design of a “Bump-and-Hole” bromodomain-based degradation tag. J. Med. Chem 64, 11637–11650 (2021). [DOI] [PubMed] [Google Scholar]
- 67.Bond AG et al. Development of BromoTag: a “Bump-and-Hole”-PROTAC system to induce potent, rapid, and selective degradation of tagged target proteins. J. Med. Chem 64, 15477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu HX et al. Construction of an IMiD-based azide library as a kit for PROTAC research. Org. Biomol. Chem 19, 166–170 (2021). [DOI] [PubMed] [Google Scholar]
- 69.Hu R et al. Identification of a selective BRD4 PROTAC with potent antiproliferative effects in AR-positive prostate cancer based on a dual BET/PLK1 inhibitor. E. J. Med. Chem 227, 113922 (2022). [DOI] [PubMed] [Google Scholar]
- 70.Remillard D et al. Degradation of the BAF Complex Factor BRD9 by Heterobifunctional Ligands. Angewandte Chemie International Edition. 56, 5738–5743, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zoppi V et al. Iterative Design and Optimization of Initially Inactive Proteolysis Targeting Chimeras (PROTACs) Identify VZ185 as a Potent, Fast, and Selective von Hippel-Lindau (VHL) Based Dual Degrader Probe of BRD9 and BRD7. J Med Chem. 62, 699–726, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cantley J, Ye X, Rousseau E et al. Selective PROTAC-mediated degradation of SMARCA2 is efficacious in SMARCA4 mutant cancers. Nat Commun 13, 6814 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Garnar-Wortzel L et al. Chemical inhibition of ENL/AF9 YEATS domains in acute leukemia. ACS Cent. Sci 7, 815–830 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Merika M, Williams AJ, Chen G, Collins T, Thanos D. Recruitment of CBP/p300 by the IFNβ enhanceosome is required for synergistic activation of transcription. Mol. Cell, 1, 277–287 (1998). [DOI] [PubMed] [Google Scholar]
- 75.Weinert BT, Narita T, Satpathy S, Srinivasan B, Hansen BK, Schölz C, Hamilton WB, Zucconi BE, Wang WW, Liu WR, et al. Time-resolved analysis reveals rapid dynamics and broad scope of the CBP/p300 acetylome. Cell, 174, 1–27 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Attar N, Kurdistani SK. Exploitation of EP300 and CREBBP lysine acetyltransferases by cancer CSH Perspect. Med., 7 (2016), p. a026534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vannam R et al. Targeted degradation of the enhancer lysine acetyltransferases CBP and p300. Cell Chem. Biol 28, 503–514 (2021). [DOI] [PubMed] [Google Scholar]
- 78.Dimou A et al. Epigenetics during EMT in lung cancer: EZH2 as a potential therapeutic target. Cancer Treat. Res. Commun 12, 40–48 (2017). [Google Scholar]
- 79.Kim KH & Roberts CW Targeting EZH2 in cancer. Nat. Med 22, 128–134 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Potjewyd F, et al. Degradation of Polycomb Repressive Complex 2 with an EED-Targeted Bivalent Chemical Degrader. Cell Chem Biol, 2020, 27, 47–56 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hsu JH et al. EED-targeted PROTACs degrade EED, EZH2, and SUZ12 in the PRC2 complex. Cell Chem. Biol 27, 41–46 (2020). [DOI] [PubMed] [Google Scholar]
- 82.Potjewyd F et al. Degradation of polycomb repressive complex 2 with an EED-targeted bivalent chemical degrader. Cell Chem. Biol 27, 47–56 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ma AQ et al. Discovery of a first-in-class EZH2 selective degrader. Nat Chem Biol. 16, 214–222 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu ZH et al. Design and synthesis of EZH2-based PROTACs to degrade the PRC2 complex for targeting the noncatalytic activity of EZH2. J. Med. Chem 2021, 64, 2829–2848 (2021). [DOI] [PubMed] [Google Scholar]
- 85.Tu YL et al. Design, synthesis, and evaluation of VHL-based EZH2 degraders to enhance therapeutic activity against lymphoma. J. Med. Chem 64, 10167–10184 (2021). [DOI] [PubMed] [Google Scholar]
- 86.Lorenzo AD & Bedford MT Histone arginine methylation. FEBS Lett. 585, 2024–2031 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yang YZ & Bedford MT Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 13, 37–50 (2013). [DOI] [PubMed] [Google Scholar]
- 88.Stopa N, Krebs JE & Shechter D The PRMT5 arginine methyltransferase: many roles in development, cancer and beyond. Cell Mol. Life Sci 72, 2041–2059 (2015).; [DOI] [PMC free article] [PubMed] [Google Scholar]; Richters A. Targeting protein arginine methyltransferase 5 in disease. Future Med. Chem 9, 2081–2098 (2017). [DOI] [PubMed] [Google Scholar]
- 89.Shen YD et al. Discovery of first-in-class protein arginine methyltransferase 5 (PRMT5) degraders. J. Med. Chem 63, 9977–9989 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Miller T, Krogan NJ, Dover J, Erdjument-Bromage H, Tempst P, Johnston M, Greenblatt JF, Shilatifard A, COMPASS: A complex of proteins associated with a trithorax-related SET domain protein. Proc. Natl. Acad. Sci. U.S.A 98, 12902–12907 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Roguev A, Schaft D, Shevchenko A, Pijnappel WW, Wilm M, Aasland R, Stewart AF, The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4. EMBO J. 20, 7137–7148 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Trievel RC, Shilatifard A, WDR5, a complexed protein. Nat. Struct. Mol. Biol 16, 678–680 (2009). [DOI] [PubMed] [Google Scholar]
- 93.Schapira M, Tyers M, Torrent M, Arrowsmith CH, WD40 repeat domain proteins: A novel target class? Nat. Rev. Drug Discov 16, 773–786 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Thomas LR, Adams CM, Wang J, Weissmiller AM, Creighton J, Lorey SL, Liu Q, Fesik SW, Eischen CM, Tansey WP, Interaction of the oncoprotein transcription factor MYC with its chromatin cofactor WDR5 is essential for tumor maintenance. Proc. Natl. Acad. Sci. U.S.A 116, 25260–25268 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Thomas LR, Wang Q, Grieb BC, Phan J, Foshage AM, Sun Q, Olejniczak ET, Clark T, Dey S, Lorey S, Alicie B, Howard GC, Cawthon B, Ess KC, Eischen CM, Zhao Z, Fesik SW, Tansey WP, Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC. Mol. Cell 58, 440–452 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Senisterra G, Wu H, Allali-Hassani A, Wasney GA, Barsyte-Lovejoy D, Dombrovski L, Dong A, Nguyen KT, Smil D, Bolshan Y, Hajian T, He H, Seitova A, Chau I, Li F, Poda G, Couture JF, Brown PJ, Al-Awar R, Schapira M, Arrowsmith CH, Vedadi M, Small-molecule inhibition of MLL activity by disruption of its interaction with WDR5. Biochem. J 449, 151–159 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bolshan Y, Getlik M, Kuznetsova E, Wasney GA, Hajian T, Poda G, Nguyen KT, Wu H, Dombrovski L, Dong A, Senisterra G, Schapira M, Arrowsmith CH, Brown PJ, Al-Awar R, Vedadi M, Smil D, Synthesis, optimization, and evaluation of novel small molecules as antagonists of WDR5-MLL interaction. ACS Med. Chem. Lett 4, 353–357 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lai AC, Crews CM, Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov 16, 101–114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dölle A et al. Design, synthesis, and evaluation of WD-repeat-containing protein 5(WDR5) degraders. J. Med. Chem 64, 10682–10710 (2021). [DOI] [PubMed] [Google Scholar]
- 100.Yu XF et al. A selective WDR5 degrader inhibits acute myeloid leukemia in patient-derived mouse models. Sci. Transl. Med 13, eabj1578 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vougiouklakis T, Hamamoto R, Nakamura Y & Saloura V The NSD family of protein methyltransferases in human cancer. Epigenomics 7, 863–874 (2015). [DOI] [PubMed] [Google Scholar]
- 102.Shen C, Ipsaro JJ, Shi J, Milazzo JP, Wang E, Roe JS, Suzuki Y, Pappin DJ, Joshua-Tor L, Vakoc CR. NSD3-Short is an adaptor protein that couples BRD4 to the CHD8 chromatin remodeler Mol. Cell, 60 (2015), pp. 847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang GG, Cai L, Pasillas MP, Kamps MP. NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat. Cell Biol, 9 (2007), pp. 804–812. [DOI] [PubMed] [Google Scholar]
- 104.Turner-Ivey B, Smith EL, Rutkovsky AC, Spruill LS, Mills JN, Ethier SP. Development of mammary hyperplasia, dysplasia, and invasive ductal carcinoma in transgenic mice expressing the 8p11 amplicon oncogene NSD3. Breast Cancer Res. Treat, 164 (2017), pp. 349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chau NG, Ma C, Danga K, Al-Sayegh H, Nardi V, Barrette R, Lathan CS, DuBois SG, Haddad RI, Shapiro GI, et al. An anatomical site and genetic-based prognostic model for patients with nuclear protein in testis (NUT) midline carcinoma: analysis of 124 patients. JNCI Cancer Spectr, 4 (2020), p. pkz094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bottcher J, Dilworth D, Reiser U, Neumuller RA, Schleicher M, Petronczki M, Zeeb M, Mischerikow N, Allali-Hassani A, Szewczyk MM, et al. Fragment-based discovery of a chemical probe for the PWWP1 domain of NSD3. Nat. Chem. Biol, 15 (2019), pp. 822–829. [DOI] [PubMed] [Google Scholar]
- 107.Xu CX et al. A NSD3-targeted PROTAC suppresses NSD3 and cMyc oncogenic nodes in cancer cells. Cell Chem. Biol S2451–9456(21)00393–7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007; 26(37): 5310–5318. [DOI] [PubMed] [Google Scholar]
- 109.Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol. 2014; 6(4):a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004; 338(1): 17–31. [DOI] [PubMed] [Google Scholar]
- 111.De Ruijter AJ, Van Gennip AH, Caron HN, Kemp S, Van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003; 370(Pt 3): 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kelly RD, Cowley SM. The physiological roles of histone deacetylase (HDAC) 1 and 2: complex co-stars with multiple leading parts. Biochem Soc Trans. 2013; 41(3): 741–749. [DOI] [PubMed] [Google Scholar]
- 113.Chakrabarti A, Oehme I, Witt O, et al. HDAC8: a multifaceted target for therapeutic interventions. Trends Pharmacol Sci. 2015; 36(7): 481–492. [DOI] [PubMed] [Google Scholar]
- 114.Grozinger CM, Hassig CA, Schreiber SL. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc Natl Acad Sci USA. 1999; 96(9): 4868–4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li T, Zhang C, Hassan S, et al. Histone deacetylase 6 in cancer. J Hematol Oncol. 2018; 11(1): 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gao L, Cueto MA, Asselbergs F, Atadja P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J Biol Chem. 2002; 277(28): 25748–25755. [DOI] [PubMed] [Google Scholar]
- 117.Cao F, de Weerd S, Chen D, Zwinderman MRH, van der Wouden PE and Dekker FJ, Eur J Med Chem, 2020, 208, 112800. [DOI] [PubMed] [Google Scholar]
- 118.Yang Ka, Song Yanling, Xie Haibo, Wu Hao, Wu Yi-Ting, Leisten Eric D., Tang Weiping, Development of the first small molecule histone deacetylase 6 (HDAC6) degraders, Bioorganic & Medicinal Chemistry Letters, Volume 28, Issue 14,2018, Pages 2493–2497. [DOI] [PubMed] [Google Scholar]
- 119.An Zixuan and others, Developing potent PROTACs tools for selective degradation of HDAC6 protein, Protein & Cell, Volume 10, Issue 8, August 2019, Pages 606–609; [DOI] [PMC free article] [PubMed] [Google Scholar]; Yang Haiyan and others, Plasticity in designing PROTACs for selective and potent degradation of HDAC6. Chem. Commun, 2019,55, 14848. [DOI] [PubMed] [Google Scholar]
- 120.Development of Multifunctional Histone Deacetylase 6 Degraders with Potent Antimyeloma Activity. Wu Hao, Yang Ka, Zhang Zhongrui, Leisten Eric D., Li Ziyuan, Xie Haibo, Liu Jin, Smith Kerry A., Novakova Zora, Barinka Cyril, and Tang Weiping. Journal of Medicinal Chemistry 2019. 62 (15), 7042–7057. [DOI] [PubMed] [Google Scholar]
- 121.Development of Selective Histone Deacetylase 6 (HDAC6) Degraders Recruiting Von Hippel–Lindau (VHL) E3 Ubiquitin Ligase. Yang Ka, Wu Hao, Zhang Zhongrui, Leisten Eric D., Nie Xueqing, Liu Binkai, Wen Zhi, Zhang Jing, Cunningham Michael D., and Tang Weiping. ACS Medicinal Chemistry Letters 2020. 11 (4), 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yang Ka, Zhao Yu, Nie Xueqing, Wu Hao, Wang Bo, Almodovar-Rivera Chelsi M., Xie Haibo, Tang Weiping, A Cell-Based Target Engagement Assay for the Identification of Cereblon E3 Ubiquitin Ligase Ligands and Their Application in HDAC6 Degraders, Cell Chemical Biology, Volume 27, Issue 7,2020,Pages 866–876.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cao ZX et al. Attenuation of NLRP3 Inflammasome Activation by Indirubin-Derived PROTAC Targeting HDAC6. ACS Chem. Biol 16, 2746–2751 (2021). [DOI] [PubMed] [Google Scholar]
- 124.Smalley JP et al. PROTAC-mediated degradation of class I histone deacetylase enzymes in corepressor complexes. Chem. Commun 56, 4476–4479 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Optimization of Class I Histone Deacetylase PROTACs Reveals that HDAC1/2 Degradation is Critical to Induce Apoptosis and Cell Arrest in Cancer Cells. Smalley Joshua P., Baker India M., Pytel Wiktoria A., Lin Li-Ying, Bowman Karen J., Schwabe John W. R., Cowley Shaun M., and Hodgkinson James T.. Journal of Medicinal Chemistry 2022. 65 (7), 5642–5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sinatra L. et al. Hydroxamic acids immobilized on aesins(HAIRs): synthesis of dual-targeting HDAC inhibitors and HDAC degraders(PROTACs). Angew. Chem. Int. Ed. Engl 59, 22494–22499 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cao J, Zhao W, Zhao C, Liu Q, Li S, Zhang G, Chou CJ, Zhang Y. Development of a Bestatin-SAHA Hybrid with Dual Inhibitory Activity against APN and HDAC. Molecules. 2020; 25(21):4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Selective degradation of histone deacetylase 8 mediated by a proteolysis targeting chimera(PROTAC). Jiranan Chotitumnavee and others. Chem. Commun, 2022, 58, 4635. [DOI] [PubMed] [Google Scholar]
- 129.Imai S, Armstrong CM, Kaeberlein M & Guarente L et al. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800 (2000). [DOI] [PubMed] [Google Scholar]
- 130.Wang Y; Fung YME; Zhang W; He B; Chung MWH;Jin J; Hu J; Lin H; Hao Q Deacylation Mechanism by SIRT2 Revealed in the 1’-SH-2’-O-Myristoyl Intermediate Structure. Cell Chem. Biol 2017, 24 (3), 339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Feldman JL; Dittenhafer-Reed KE; Kudo N; Thelen JN; Ito A; Yoshida M; Denu JM Kinetic and Structural Basis for Acyl-Group Selectivity and NAD+ Dependence in Sirtuin-Catalyzed Deacylation. Biochemistry 2015, 54 (19), 3037–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Schiedel Matthias, Herp Daniel, Hammelmann Soren, Swyter Soren, Lehotzky Attila, Robaa Dina, Olah Judit, Ovadi Judit, Sippl Wolfgang, and Jung Manfred Journal of Medicinal Chemistry 2018. 61 (2), 482–491. [DOI] [PubMed] [Google Scholar]
- 133.Hong JY et al. Simultaneous inhibition of SIRT2 deacetylase and defatted-acylase activities via a PROTAC strategy. ACS Med. Chem. Lett 11, 2305–2311 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Suzuki T, Miyata N, Lysine Demethylases Inhibitors. J. Med. Chem 2012, 54, 8236–8250. [DOI] [PubMed] [Google Scholar]
- 135.Tu S, Teng Y-C, Yuan C, Wu Y-T, Chan M-Y, Cheng A-N, Lin P-H, Juan L-J, Tsai M-D, Nat. Struct. Mol. Biol 2008, 15, 419–421; [DOI] [PubMed] [Google Scholar]; Harmeyer KM, Facompre ND, Herlyn M, Basu D, Trends Cancer 2017, 3, 713–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Teng Y-C, Lee C-F, Li Y-S, Chen Y-R, Hsiao P-W, Chan M-Y, Lin F-M, Huang H-D, Chen Y-T, Jeng Y-M, Hsu C-H, Yan Q, Tsai M-D, Juan L-J, Cancer Res. 2013, 73, 4711–4721. [DOI] [PubMed] [Google Scholar]
- 137.Raiymbek G, An S, Khurana N, Gopinath S, Larkin A, Biswas S, Trievel RC, Cho U-S, Ragunathan K, eLife 2020, 9, e53155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Iida T et al. Design, synthesis, and biological evaluation of lysine demethylase 5C degraders. ChemMedChem. 16, 1609–1618 (2021). [DOI] [PubMed] [Google Scholar]
- 139.Gadd M, Testa A, Lucas X et al. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol 13, 514–521 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Nowak RP, DeAngelo SL, Buckley D et al. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat Chem Biol 14, 706–714 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Maneiro M; Forte N; Shchepinova MM; Kounde CS; Chudasama V; Baker JR; Tate EW Antibody–PROTAC Conjugates Enable HER2-Dependent Targeted Protein Degradation of BRD4. ACS Chem. Biol 2020, 15, 1306–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Dragovich PS; et al. Antibody-Mediated Delivery of Chimeric BRD4 Degraders. Part 1: Exploration of Antibody Linker, Payload Loading, and Payload Molecular Properties. J. Med. Chem 2021, 64, 2534–2575. [DOI] [PubMed] [Google Scholar]
- 143.Dragovich PS; et al. Antibody-Mediated Delivery of Chimeric BRD4 Degraders. Part 2: Improvement of In Vitro Antiproliferation Activity and In Vivo Antitumor Efficacy. J. Med. Chem 2021, 64, 2576–2607. [DOI] [PubMed] [Google Scholar]
- 144.Shi Y; Liao Y; Liu Q; Ni Z; Zhang Z; Shi M; Li P; Li H; Rao Y BRD4-targeting PROTAC as a unique tool to study biomolecular condensates. Cell Discovery 2023, 9, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.He Y, Ju Y, Hu Y, Wang B, Che S, Jian Y, Zhuo W, Fu X, Cheng Y, Zheng S et al. Brd4 proteolysis-targeting chimera nanoparticles sensitized colorectal cancer chemotherapy. Journal of controlled release 2023, 354, 155–166. [DOI] [PubMed] [Google Scholar]
- 146.Békés M, Langley DR and Crews CM. Nat. Rev. Drug Discovery, 2022, 21, 181–200. [DOI] [PMC free article] [PubMed] [Google Scholar]