

Abstract
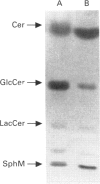
Glucosylceramide, a degradation product of complex glycosphingolipids, is hydrolysed in lysosomes by glucocerebrosidase (GlcCerase). Mutations in the human GlcCerase gene cause a reduction in GlcCerase activity and accumulation of glucosylceramide, which results in the onset of Gaucher disease, the most common lysosomal storage disease. Significant clinical heterogeneity is observed in Gaucher disease, with three main types known, but no clear correlation has been reported between the different types and levels of residual GlcCerase activity. We now demonstrate that a correlation exists by using a radioactive, short-acyl chain substrate, N-(1-[14C]hexanoyl)-D-erythro-glucosylsphingosine ([14C]hexanoyl-GlcCer). This substrate rapidly transferred into biological membranes in the absence of detergent [Futerman and Pagano (1991) Biochem. J. 280, 295-302] and was hydrolyzed to N-(1-[14C]hexanoyl)-D-erythro-sphingosine ([14C]hexanoyl-Cer) both in vitro and in situ, with an acid pH optimum. A strict correlation was observed between levels of [14C]hexanoyl-GlcCer hydrolysis and Gaucher type in human skin fibroblasts. The mean residual activity measured in vitro for 3 h incubation in type 1 Gaucher fibroblasts (the mild form of the disease) was 46.3 +/- 4.6 nmol of [14C]hexanoyl-Cer formed per mg protein (n = 9), and in type 2 and 3 fibroblasts (the neuronopathic forms of the disease) was 19.6 +/- 6.5 (n = 9). A similar correlation was observed when activity was measured in situ, suggesting that the clinical severity of a lysosomal storage disease is related to levels of residual enzyme activity.
Full text
PDF


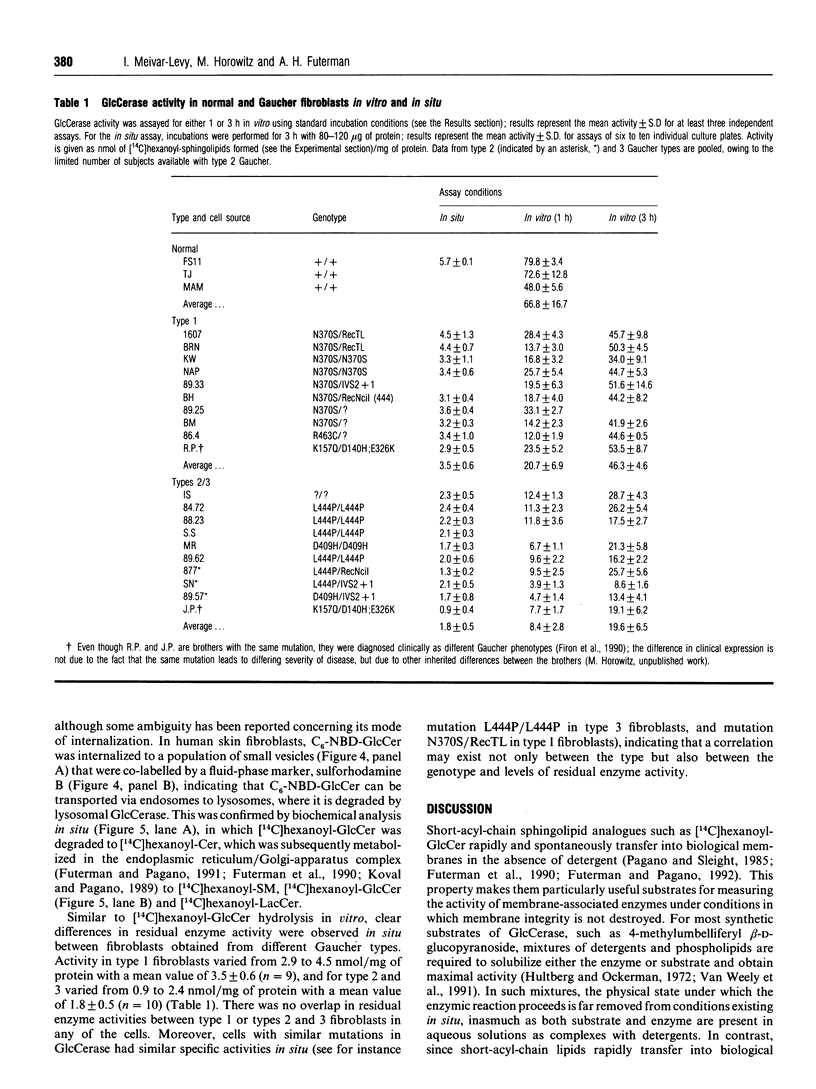

Images in this article
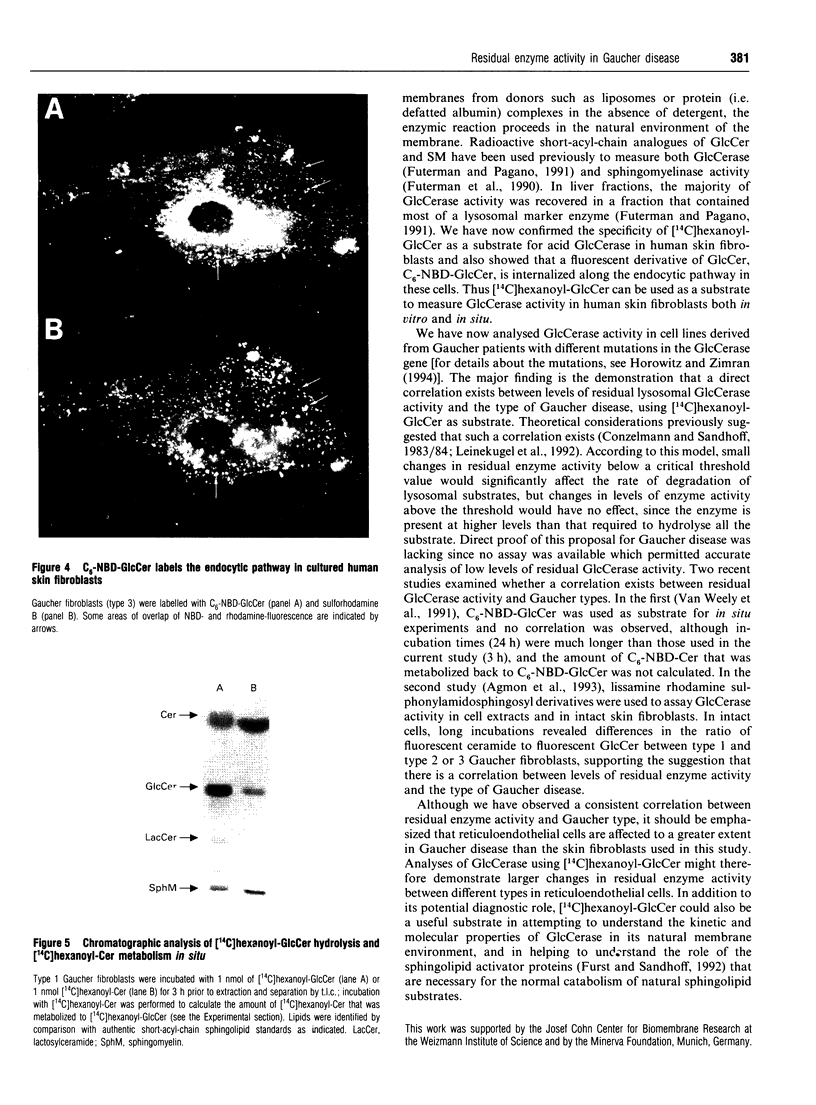
Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Agmon V., Cherbu S., Dagan A., Grace M., Grabowski G. A., Gatt S. Synthesis and use of novel fluorescent glycosphingolipids for estimating beta-glucosidase activity in vitro in the absence of detergents and subtyping Gaucher disease variants following administration into intact cells. Biochim Biophys Acta. 1993 Sep 29;1170(1):72–79. doi: 10.1016/0005-2760(93)90177-b. [DOI] [PubMed] [Google Scholar]
- BLIGH E. G., DYER W. J. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959 Aug;37(8):911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Beutler E. Gaucher disease as a paradigm of current issues regarding single gene mutations of humans. Proc Natl Acad Sci U S A. 1993 Jun 15;90(12):5384–5390. doi: 10.1073/pnas.90.12.5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudker O., Futerman A. H. Detection and characterization of ceramide-1-phosphate phosphatase activity in rat liver plasma membrane. J Biol Chem. 1993 Oct 15;268(29):22150–22155. [PubMed] [Google Scholar]
- Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976 May 7;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Conzelmann E., Sandhoff K. Partial enzyme deficiencies: residual activities and the development of neurological disorders. Dev Neurosci. 1983;6(1):58–71. doi: 10.1159/000112332. [DOI] [PubMed] [Google Scholar]
- Dinur T., Grabowski G. A., Desnick R. J., Gatt S. Synthesis of a fluorescent derivative of glucosyl ceramide for the sensitive determination of glucocerebrosidase activity. Anal Biochem. 1984 Jan;136(1):223–234. doi: 10.1016/0003-2697(84)90329-4. [DOI] [PubMed] [Google Scholar]
- Firon N., Eyal N., Kolodny E. H., Horowitz M. Genotype assignment in Gaucher disease by selective amplification of the active glucocerebrosidase gene. Am J Hum Genet. 1990 Mar;46(3):527–532. [PMC free article] [PubMed] [Google Scholar]
- Futerman A. H., Pagano R. E. Determination of the intracellular sites and topology of glucosylceramide synthesis in rat liver. Biochem J. 1991 Dec 1;280(Pt 2):295–302. doi: 10.1042/bj2800295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futerman A. H., Pagano R. E. Use of N-([1-14C]hexanoyl)-D-erythro-sphingolipids to assay sphingolipid metabolism. Methods Enzymol. 1992;209:437–446. doi: 10.1016/0076-6879(92)09054-7. [DOI] [PubMed] [Google Scholar]
- Futerman A. H., Stieger B., Hubbard A. L., Pagano R. E. Sphingomyelin synthesis in rat liver occurs predominantly at the cis and medial cisternae of the Golgi apparatus. J Biol Chem. 1990 May 25;265(15):8650–8657. [PubMed] [Google Scholar]
- Fürst W., Sandhoff K. Activator proteins and topology of lysosomal sphingolipid catabolism. Biochim Biophys Acta. 1992 Jun 5;1126(1):1–16. doi: 10.1016/0005-2760(92)90210-m. [DOI] [PubMed] [Google Scholar]
- Grabowski G. A., Gatt S., Horowitz M. Acid beta-glucosidase: enzymology and molecular biology of Gaucher disease. Crit Rev Biochem Mol Biol. 1990;25(6):385–414. doi: 10.3109/10409239009090616. [DOI] [PubMed] [Google Scholar]
- Horowitz M., Zimran A. Mutations causing Gaucher disease. Hum Mutat. 1994;3(1):1–11. doi: 10.1002/humu.1380030102. [DOI] [PubMed] [Google Scholar]
- Hultbery B., Ockerman P. A. Artificial substrates in the assay of acid glycosidases. Clin Chim Acta. 1972 Jun;39(1):49–58. doi: 10.1016/0009-8981(72)90298-7. [DOI] [PubMed] [Google Scholar]
- Kok J. W., Babia T., Hoekstra D. Sorting of sphingolipids in the endocytic pathway of HT29 cells. J Cell Biol. 1991 Jul;114(2):231–239. doi: 10.1083/jcb.114.2.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kok J. W., Eskelinen S., Hoekstra K., Hoekstra D. Salvage of glucosylceramide by recycling after internalization along the pathway of receptor-mediated endocytosis. Proc Natl Acad Sci U S A. 1989 Dec;86(24):9896–9900. doi: 10.1073/pnas.86.24.9896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kok J. W., Hoekstra K., Eskelinen S., Hoekstra D. Recycling pathways of glucosylceramide in BHK cells: distinct involvement of early and late endosomes. J Cell Sci. 1992 Dec;103(Pt 4):1139–1152. doi: 10.1242/jcs.103.4.1139. [DOI] [PubMed] [Google Scholar]
- Kornfeld S., Mellman I. The biogenesis of lysosomes. Annu Rev Cell Biol. 1989;5:483–525. doi: 10.1146/annurev.cb.05.110189.002411. [DOI] [PubMed] [Google Scholar]
- Koval M., Pagano R. E. Lipid recycling between the plasma membrane and intracellular compartments: transport and metabolism of fluorescent sphingomyelin analogues in cultured fibroblasts. J Cell Biol. 1989 Jun;108(6):2169–2181. doi: 10.1083/jcb.108.6.2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legler G. Glucosidases. Methods Enzymol. 1977;46:368–381. doi: 10.1016/s0076-6879(77)46044-0. [DOI] [PubMed] [Google Scholar]
- McIntyre J. C., Sleight R. G. Fluorescence assay for phospholipid membrane asymmetry. Biochemistry. 1991 Dec 24;30(51):11819–11827. doi: 10.1021/bi00115a012. [DOI] [PubMed] [Google Scholar]
- Mistry P. K., Cox T. M. The glucocerebrosidase locus in Gaucher's disease: molecular analysis of a lysosomal enzyme. J Med Genet. 1993 Nov;30(11):889–894. doi: 10.1136/jmg.30.11.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld E. F. Lysosomal storage diseases. Annu Rev Biochem. 1991;60:257–280. doi: 10.1146/annurev.bi.60.070191.001353. [DOI] [PubMed] [Google Scholar]
- Osiecki-Newman K., Fabbro D., Legler G., Desnick R. J., Grabowski G. A. Human acid beta-glucosidase: use of inhibitors, alternative substrates and amphiphiles to investigate the properties of the normal and Gaucher disease active sites. Biochim Biophys Acta. 1987 Sep 2;915(1):87–100. doi: 10.1016/0167-4838(87)90128-2. [DOI] [PubMed] [Google Scholar]
- Pagano R. E. A fluorescent derivative of ceramide: physical properties and use in studying the Golgi apparatus of animal cells. Methods Cell Biol. 1989;29:75–85. doi: 10.1016/s0091-679x(08)60188-0. [DOI] [PubMed] [Google Scholar]
- Pagano R. E., Sleight R. G. Defining lipid transport pathways in animal cells. Science. 1985 Sep 13;229(4718):1051–1057. doi: 10.1126/science.4035344. [DOI] [PubMed] [Google Scholar]
- Schwarzmann G., Sandhoff K. Lysogangliosides: synthesis and use in preparing labeled gangliosides. Methods Enzymol. 1987;138:319–341. doi: 10.1016/0076-6879(87)38028-0. [DOI] [PubMed] [Google Scholar]
- Van Weely S., Van Leeuwen M. B., Jansen I. D., De Bruijn M. A., Brouwer-Kelder E. M., Schram A. W., Sa Miranda M. C., Barranger J. A., Petersen E. M., Goldblatt J. Clinical phenotype of Gaucher disease in relation to properties of mutant glucocerebrosidase in cultured fibroblasts. Biochim Biophys Acta. 1991 Jun 5;1096(4):301–311. doi: 10.1016/0925-4439(91)90066-i. [DOI] [PubMed] [Google Scholar]