

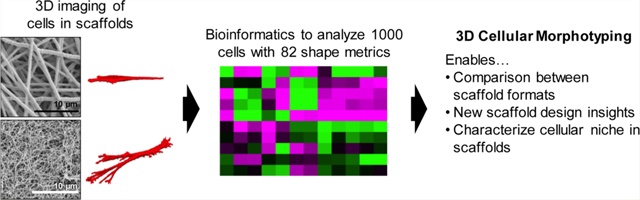
Abstract
Many biomaterial scaffolds have been advanced to provide synthetic cell niches for tissue engineering and drug screening applications; however, current methods for comparing scaffold niches focus on cell functional outcomes or attempt to normalize materials properties between different scaffold formats. We demonstrate a three-dimensional (3D) cellular morphotyping strategy for comparing biomaterial scaffold cell niches between different biomaterial scaffold formats. Primary human bone marrow stromal cells (hBMSCs) were cultured on 8 different biomaterial scaffolds, including fibrous scaffolds, hydrogels, and porous sponges, in 10 treatment groups to compare a variety of biomaterial scaffolds and cell morphologies. A bioinformatics approach was used to determine the 3D cellular morphotype for each treatment group by using 82 shape metrics to analyze approximately 1000 cells. We found that hBMSCs cultured on planar substrates yielded planar cell morphotypes, while those cultured in 3D scaffolds had elongated or equiaxial cellular morphotypes with greater height. Multivariate analysis was effective at distinguishing mean shapes of cells in flat substrates from cells in scaffolds, as was the metric L1-depth (the cell height along its shortest axis after aligning cells with a characteristic ellipsoid). The 3D cellular morphotyping technique enables direct comparison of cellular microenvironments between widely different types of scaffolds and design of scaffolds based on cell structure−function relationships.
Keywords: mesenchymal stem cells, biomaterials, tissue engineering, cell shape, regenerative medicine
Graphical Abstract
INTRODUCTION
Cells in vivo reside within extracellular matrix (ECM), a three-dimensional (3D) network of proteins and polysaccharides, in structures that are organized for evolutionarily designed functions.1 The ECM that surrounds the cell comprises its cell niche or microenvironment, which influences the cell morphology and in turn, influences the cell function. This interplay between the cell and its niche influences cell fate and is a critical factor in many biological processes, including development and malignant progression in cancer. 3D biomaterial scaffolds create a synthetic niche for in vitro cell culture that promotes cell behavior that better mimics the cell behavior observed in vivo than cell culture on two-dimensional (2D) tissue culture polystyrene surfaces. The advancement of a variety of 3D biomaterial scaffolds including fibrous scaffolds,2 porous sponges,3 and hydrogels,4,5 among others, has enabled researchers to provide a variety of synthetic cell niches. Current approaches design scaffold niches to promote desired cell functions, such as proliferation or differentiation to target lineages.6–8 While useful, these approaches do not enable a reasoned approach to synthetic niche design, where the scaffold is designed to elicit a desired 3D cell shape and thereby controls cell function.
There is significant evidence linking cell shape and function9–13 that provides a basis for engineering the biomaterial scaffold microenvironment to direct cell fate or guide cells into more natural morphologies. Cells that compose tissues in vivo have characteristic shapes based on their function, as noted with epithelial cells that have elongated (columnar epithelium), planar (squamous epithelium), or equiaxial (cuboidal epithelium) shapes.14 Consequently, it follows that biomaterial scaffold niches that drive cells into elongated, planar, or equiaxial cell morphologies may better promote the phenotypes of columnar, squamous, or cuboidal epithelia, respectively. Cells respond to forces and structures in their microenvironment through mechanotransduction using the actomyosin cytoskeleton complex to alter their shape, which affects signaling, differentiation, nuclear shape, and gene expression.10,15–17 Cell shape has also been shown to affect the sensitivity of the cell to extracellular and intracellular signaling through biophysical effects on signaling cascades by altering the spatial distribution of signal transduction events.18–20 Nanofiber scaffolds have been shown to promote osteogenic differentiation of bone marrow stromal cells and embryonic stem cells through effects on cell and nuclear shape,7,21–23 which demonstrates that biomaterial scaffolds can direct cell fate by altering cell shape through the cellular structure−function relationship.
Herein, we present a method for characterizing the biomaterial scaffold niche by measuring the 3D cell shape that results from culture of the same cells in a variety of biomaterial scaffolds, called 3D cellular morphotyping. In the context of this paper, morphotyping refers to characterization of the biomaterial scaffold cellular niche by measurements of cell morphology and the morphological variation within a cell population during culture in a biomaterial scaffold. The cellular morphotype provides a proxy measurement for characterizing the biomaterial scaffold niche since cell shape results from the cellular interaction with the scaffold microenvironment and its physical properties, enabling direct comparison of biomaterial scaffold niches. A key aspect of 3D cellular morphotyping is fitting a characteristic ellipsoid (gyration tensor) to each cell and assessing the cell dimensionality through ratios of the ellipsoid principal moments. Primary human bone marrow stromal cells (hBMSCs) were used for this study because of their clinical relevance for tissue engineering and regenerative medicine24 and the intense interest in guiding their behavior through environmental cues.2–5,7,8,10,11,21–23 hBMSCs were cultured in 8 biomaterial scaffolds in 10 treatment groups. The 3D shape of hBMSCs was determined by staining and imaging the actin cytoskeleton and nucleus with confocal laser scanning microscopy (CLSM) to generate 3D image volumes (Z-stacks). The Z-stacks were quantitatively evaluated with a bioinformatics approach using 82 shape metrics to assess 3D cellular morphotype for the 10 treatment groups that included approximately 1000 cells in total. Multivariate analysis methods were used to identify the key shape metrics for 3D cellular morphotyping of the niches. Additionally, 2D and 3D image analysis was compared to assess the differences between the two analysis methods.
METHODS
Data Deployed Online.
All image data acquired and analyzed in this manuscript is publicly accesssible online at: https://isg.nist.gov/deepzoomweb/zstackDownload. A web-based interactive 3D cell viewer has been built that enables browsing and comparison of the 3D cell renderings: https://isg.nist.gov/deepzoomweb/stemcells3d/index.html.
Materials.
Poly(ε-caprolactone) (PCL) (relative molecular mass 80000 g/mol), fibrinogen from human plasma (catalog no. F3879), thrombin from human plasma (catalog no. T6884), bovine serum albumin (BSA), dexamethasone, β-glycerophosphate, L-ascorbic acid, 10× Dulbecco’s modified Eagle’s medium (DMEM), chloroform, and methanol were purchased from Sigma-Aldrich (St. Louis, MO). Tissue culture polystyrene (TCPS) 100 mm diameter Petri dishes were purchased from FisherScientific (Pittsburgh, PA). Dulbecco’s phosphate buffered saline (D-PBS), α-minimum essential medium (α-MEM), L-glutamine, trypsin (0.25% by mass containing 1 mmol/L ethylenediaminetetraacetate (EDTA)), penicillin/streptomycin (P/S), AlexaFluor 546 phalloidin, and 4′,6-diamidino-2-phenylindole, dihydrochloride (DAPI) were purchased from Life Technologies (Carlsbad, CA). Fetal bovine serum (FBS) was purchased from Atlanta Biologicals (Atlanta, GA). PureCol bovine type I collagen (catalog # 5005-B) was purchased from Advanced BioMatrix (San Diego, CA). Reduced growth factor Matrigel (catalog # 354230) was purchased from BD Biosciences (San Jose, CA). Alvetex scaffolds were purchased from Reinnervate (Durham, United Kingdom). Primary human bone marrow stromal cells (hBMSCs) were obtained from Tulane Center for Gene Therapy (donor 7038, healthy 29 year old female, isolated from iliac crest).
Cell Culture.
Primary hBMSCs were expanded in complete medium (α-MEM containing 16.5% by volume FBS, 4 mmol/L L-glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin) to 70% confluency. hBMSCs were detached from culture flask with trypsin and seeded onto scaffolds at passage 5 at a density of 2500 cells/cm2 as detailed below. Two sample groups were cultured with complete medium plus osteogenic supplements (OS), which consisted of 10 nmol/L dexamethasone, 20 mmol/L β-glycerophosphate, and 0.05 mmol/L L-ascorbic acid. The cells on scaffolds were cultured (humidified incubator, 37 °C, 5% CO2 by volume) for 24 h prior to preparation for confocal imaging.
PCL Substrate Preparation.
PCL spun coat (SC), nanofiber (NF), and microfiber (MF) substrates were prepared for the study. PCL SC substrates were prepared by spin coating 0.8 mL PCL solution (10% by mass in glacial acetic acid) on to TCPS dishes at 1000 rpm for 30 s. The PCL-coated TCPS dishes were dried overnight at room temperature and annealed by warming with hot air (60 °C) for several minutes to crystallize the PCL films. The dish was hot-punched into 10 mm diameter discs with PCL SC films for cell culture.
PCL NF and MF scaffolds were prepared by electrospinning PCL solution onto TCPS discs (10 mm dia., hot-punched from TCPS dishes). PCL solutions were used to prepare NF scaffolds (15% PCL by mass in 5:1 volume ratio chloroform:methanol) and MF scaffolds (20% PCL by mass in 9:1 volume ratio chloroform:methanol). A home-built electrospinning device was used to make NF (0.5 mL/h syringe pump rate, 21 gauge flat tip needle, 13.5 kV, 20.4 cm needle to target, 6 h spinning) and MF (18 gauge flat tip needle, 2.0 mL/h syringe pump rate, 13 kV, 1.5 h spinning) by electrospinning onto TCPS discs placed on an aluminum foil target.
SC, NF, and MF samples were placed in 48-well TCPS plates and affixed to the bottom of the well with silicone vacuum grease. The 48-well plates with samples were sterilized with ethylene oxide (Andersen Products, Haw River, NC) for 12 h and then degassed for 2 d under house vacuum. Prior to cell seeding, the scaffolds were incubated in complete medium (0.5 mL/well) in a humidified incubator at 37 °C with 5% CO2 for 2 days. For cell seeding of SC, SC+OS, NF, NF+OS, and MF samples, medium was removed and replaced with 0.5 mL of cells in solution (5000 cells/mL) to yield a seeding density of 2500 cells/cm2.
Porous Polystyrene Scaffold.
Porous polystyrene scaffolds (PPS, Alvetex, 6-well insert) were used according to manufacturer’s instructions. Scaffold inserts were placed in 6-well plates and soaked in 70% by volume ethanol, then washed twice with excess complete medium. PPS in 6-well plates were incubated overnight in complete medium (10 mL) prior to cell seeding. For cell seeding of PPS, medium was removed and 175 μL of cell suspension (141 250 cells/mL, ≈25 000 cells) was pipetted onto the scaffolds and allowed to adhere for 30 min in the incubator at 37 °C prior to adding 10 mL of medium.
Matrigel.
Reduced growth factor Matrigel (MG) was used as received. The samples were prepared according to a previously published protocol.25 Briefly, a 50 μL MG coating layer was applied to sterile 12 mm diameter glass coverslips in a 24-well plate and incubated at 37 °C for 30 min to allow MG to gel. Next, 300 μL MG containing 5000 cells was applied on top of the coated surface. The samples were incubated at 37 °C for 30 min to allow MG to gel and then 1 mL of complete medium was added to each well.
Fibrin Gel.
Fibrin gel (FG) was prepared by polymerizing filter-sterilized fibrinogen (6 mg/mL in D-PBS) and thrombin (25 U/mL in 2% by mass CaCl2 solution in deionized water) solutions. Fibrinogen-cell suspension (10,000 cells in 500 μL of fibrinogen solution) was mixed with 500 μL of thrombin in a 1.5 mL tube and then 500 μL was rapidly seeded onto a sterile 12 mm diameter glass coverslip in a 24-well plate. After allowing the gel to set for 10 min at room temperature, 1 mL of complete medium was added to each well.
Collagen Gel.
Collagen gel (CG) was prepared from type I collagen (3 mg/mL) using 8 parts collagen and 1 part 10× DMEM and was neutralized to pH 7.2 with 0.1 mol/L NaOH. Cells were suspended in the collagen solution (10,000 cells/mL) and 500 μL of the collagen-cell suspension was pipetted onto sterile 12 mm diameter glass coverslips in a 24-well plate. The samples were incubated at 37 °C for 2 h to allow collagen to gel and then 1 mL of complete medium was added to each well.
Collagen Fibrils.
Collagen fibrils (CF) were prepared from type I collagen (3 mg/mL) according to a previously developed protocol.26 Specifically, a 300 μg/mL collagen solution was prepared in D-PBS, neutralized with 0.1 mol/L NaOH and added to a 35 mm diameter untreated polystyrene culture dish. The samples were incubated for 24 h at 37 °C in a humidified incubator with 5% by volume CO2. After incubation, the samples were washed once with D-PBS and once with deionized water (18.2 MΩ). The samples were dried gently with a nitrogen stream (35 psi) for 20 s, immersed in D-PBS and stored in PBS at 4 °C until use (less than 3 d). For cell seeding, D-PBS was removed and each 35 mm diameter dish was seeded with 25 000 cells in 2 mL of medium.
Substrate Characterization.
Scanning electron microscopy (SEM) was used to image substrates and for determining fiber diameter for NF and MF. Substrates were sputter-coated with gold and imaged with SEM (S-4700eII FE-SEM, Hitachi, Tokyo, Japan). SEM images (>9 images per sample type; >3 images taken from each of 3 samples for each sample type) were used to determine the mean diameter of the NFs (589 ± 116 nm, n = 151 fibers) and the MFs (4.378 ± 0.419 μm, n = 54 fibers). The line tool in ImageJ (NIH, Bethesda, MD) was used to measure several fiber diameters in each SEM image.
Hydrogel (MG, FG, and CG) and CF samples were prepared for SEM imaging by fixing samples with 3.7% by volume formaldehyde for 1 h at 37 °C followed by three D-PBS washes. Samples were dehydrated in a series of ethanol washes (0, 30, 50, 70, 85, 95, and 100%) with each wash conducted 3 times for a total of 15 min. The samples were then critical point dried, mounted, and sputter-coated with gold, then imaged with SEM (S-4700eII FE-SEM).
Surface roughness of the SC films (SC and SC+OS treatment groups) was measured using atomic force microscopy (AFM, Dimension Icon, Bruker, Billerica, MA). Three SC film samples were analyzed with 3 spots per sample for a total of 9 spots measured. The spot size was 50 μm × 50 μm and the scans were acquired with 256 samples per line. The images were analyzed with Nanoscope Analysis (Bruker) and the root-mean-square (RMS) roughness was reported for each analyzed spot and averaged to produce an average value for the SC film.
Fluorescent Imaging.
Cells were cultured on substrates for 24 h, which enabled them to achieve a stable cell morphology. Samples were fixed with 3.7% by volume formaldehyde in D-PBS for 1 h at 37 °C, washed three times with D-PBS, permeabilized with 0.1% by volume Triton X-100 for 10 min, washed three times with D-PBS, blocked with 5% by mass BSA for 30 min at room temperature, stained with AlexaFluor 546-phalloidin (F-actin stain, 330 nmol/L in 5% by mass BSA) for 30 min at room temperature, washed two times with 1% by mass BSA, washed with D-PBS, stained with DAPI (nucleus stain, 0.03 mmol/L in D-PBS) for 5 min, washed three times with D-PBS and stored in D-PBS at 4 °C until imaged.
For imaging, the samples were immersed in D-PBS and were imaged with a CLSM (Leica SP5 II confocal microscope, Leica Microsystems, Buffalo Grove, IL) using a 63× water immersion objective (0.9 numerical aperture). Z stacks of images (voxel dimensions of 240 nm × 240 nm × 710 nm, 1 Airy unit, line average 3, 400 Hz) of hBMSCs were captured for each substrate type. Z-stacks were collected on 2 channels for each cell: 1) orange fluorescence, actin, Alexa Fluor 546 phalloidin (excitation 543 nm, emission range 564 to 663 nm); and 2) blue fluorescence, nucleus, DAPI, (excitation 405 nm, emission range 434 to 517 nm). As determined from calculations based on the lateral and axial Rayleigh criterion equations,27 the limits of resolution are 255 nm (blue) and 308 nm (orange) for lateral (XY) and 1026 nm (blue) and 1376 nm (orange) for axial (Z) resolution.
Cells near the middle of substrates and away from substrate edges were selected for imaging to reduce edge effects and ensure that imaged cells were only in contact with the substrate material and not touching the TCPS or glass coverslips. Effort was made to image only individual hBMSCs that were not touching other cells (one nucleus per object). During image captures, cells were centered in the field of view so that cells were not touching the edges of the images to enable accurate analysis of cell shape. Over one hundred cells were imaged for each treatment group (n > 100 cells).
Image Segmentation.
The 3D imaged cells were analyzed by exporting all optical cross sections acquired by a CLSM as a stack of Z-frames Z-stack) in a TIFF file format. The Z-stacks were automatically segmented to isolate the cell (foreground) from the background. The 3D segmentation consisted of (1) mapping imaging and geometrical assumptions into algorithmic steps, (2) evaluating segmentation accuracy, precision, and efficiency, and (3) verifying segmentation results visually to guarantee segmentation quality, as described previously.28 The 3D segmentation was applied to a data set of 1253 hBMSCs (or 128,460 image frames with on average 125 cells/substrate × 10 substrate types × 2 stains × 51 frames/cell). 3D segmentation achieved an average precision of 0.82, an average accuracy of 0.84 (as measured by the Dice similarity index29) and a probability of segmentation success of 0.85, based on comparisons to human visual verification. A computation time of 42.3 h was required to process all 1253 Z-stacks. Over 110 cells per substrate group were imaged (1253 total cells), but several cells were not segmented correctly in either actin or nuclear channel during the automated image segmentation and were removed from the data set and not analyzed. There were between 87 and 102 cells analyzed per group (969 total cells, Table 1). For each segmented Z-stack, 39 shape metrics were computed for both actin and nucleus channels, 4 shape metrics were ratios of nucleus to actin values, resulting in a total of 82 shape metrics (Figure S1).
Table 1.
Scaffold Descriptions and Characteristics
| abbrev. | description | properties ((mean (S.D.)) | no. of cells analyzed |
|---|---|---|---|
| SC | flat PCLa spun-coat films | surface roughness = 92.76 nm (10.69 nm) | 99 |
| SC+OS | flat PCL spun-coat films with OSb | surface roughness = 92.76 nm (10.69 nm) | 96 |
| NF | electrospun PCL nanofibers | fiber dia. 589 nm (116 nm) | 101 |
| NF+OS | electrospun PCL nanofibers with OS | fiber dia. 589 nm (116 nm) | 95 |
| MF | electrospun PCL microfibers | fiber dia. 4.38 μm (0.42 μm) | 87 |
| PPS | porous polystyrene scaffold (Alvetex) | pore size 36−40 μm | 98 |
| MG | matrigel | mouse tumor extract, rich in Type IV collagen | 98 |
| FG | fibrin gel | polymerized fibrinogen (6 mg/mL) | 92 |
| CG | collagen gel | type I collagen (2.4 mg/mL) | 101 |
| CF | collagen fibrils | type I collagen (300 μg/mL), fibril diameter ∼200 nm | 102 |
PCL = poly(ε-caprolactone).
OS = osteogenic supplements.
Hierarchical Cluster Analysis.
For analyses of all 969 cells (ALL), data were first log2-transformed, then median-centered for each shape metric. For each treatment group, the 10% trimmed mean of all cells within a treatment group was used to produce an averaged data set (AVG). These values were then log2-transformed and median-centered for each shape metric. For both the ALL and AVG data sets, the shape metrics were divided into actin, nucleus, or actin and nucleus data sets for further analysis. Principal component analysis (PCA) and hierarchical cluster analysis (HCA) were performed for each of these subclasses (R Stats Package version 3.1.3 and the package dendextend version 0.17.5). HCA was based on Euclidian distance and average linkage to produce a dendrogram to demonstrate the relationship among the treatments (Figure 5 and Figure S6a, b), and visually organize the shape metrics for the heatmap in Figure 5.
Figure 5.

Hierarchical cluster analysis (HCA) of actin shape metrics. The scales for the dendrograms are Euclidian distance in multidimensional space. The top dendrogram shows the relationships between the twenty-eight (28) shape metrics based upon the response across treatment groups. The 28 shape metrics used for clustering were selected from the full list of 43 metrics (39 actin metrics and 4 nucleus-actin metrics) in Figure S1 by removing metrics calculated from cells that were not oriented by gyration tensor moments. The dendrogram to the right shows the relationships between the treatment groups based upon the response across shape metrics. The input data for the dendrograms are shown in the heatmap to visualize these response patterns. The data points represent the log2-transformed average (10% trimmed mean) of each shape metric for all cells within a particular treatment group. A color scale based upon the shape metric percentile is used to indicate whether a value is near the high end (magenta) or low end (green) of the range for each shape metric.
Statistical Analysis.
Cells (n > 100 cells) were imaged and analyzed (n > 87 cells) for each of the ten treatment groups. Data is presented as mean ±2 standard deviations of the mean. Statistical significance was defined as p < 0.05 using one-way analysis of variance (ANOVA) with Tukey’s HSD test for multiple comparisons.
RESULTS
Biomaterial Scaffolds Promote Characteristic Cell Shapes.
To evaluate our 3D cellular morphotyping strategy for characterization of biomaterial scaffold niches, the cell niches of a variety of biomaterial scaffolds that were produced from natural and synthetic polymers were investigated (Table 1, Figure 1), including flat surfaces (SC, SC+OS, CF), fibrous scaffolds (NF, NF+OS, MF), porous sponges (PPS), and hydrogels (MG, FG, CG). Within this array of substrates, there were subsets with systematically varied morphological and biochemical properties that included: (1) spun coat films (SC), nanofibers (NF), and microfibers (MF) were made from the same polymer (poly(ε-caprolactone), PCL) that was processed into different morphologies; (2) Matrigel (MG), fibrin gel (FG) and collagen gel (CG) hydrogels were prepared from three different natural materials; (3) collagen gel (CG) and collagen fibrils (CF) were prepared from collagen that was processed into different morphologies; (4) osteogenic supplements (OS) were added to NF and SC cultures and compared to nonsupplemented cultures (SC vs SC+OS and NF vs NF +OS) to assess the influence of OS on 3D cellular morphotype; and (5) SC was included as a flat surface control for validation of the morphotyping approach.
Figure 1.

Scaffold morphology. Top: Photographs of the scaffold macrostructure. Bottom: Scanning electron micrographs of scaffold microstructure.
Confocal imaging of hBMSCs demonstrated that the biomaterial scaffolds influenced cell shape (Figure 2 and 3, Figures S2 and S3). The variety of biomaterial scaffolds (Table 1, Figure 1) resulted in a variety of representative cell morphologies, shown as both 2D maximum projections and 3D renderings in Figure 2. These cell morphologies were assessed with 82 shape metrics, consisting of 39 actin shape metrics, 39 nucleus shape metrics, and 4 nucleus-actin shape metrics (Figure S1), to quantify the 2D and 3D cell morphology (Figure 3, Figure S2a−h). A characteristic gyration tensor ellipsoid was determined for each cell (Figure 3a, inset) to assess dimensionality. The ratios of the gyration tensor ellipsoid principal moments provided a 3D measure of cell aspect ratio. These principal moment ratios were plotted to yield a dimensionality plot for actin (Figure 3a) and nucleus (Figure 3b) that evaluated the dimensionality characteristic of the cell niche for each treatment group. The dimensionality plots (Figure 3a, b) organize objects within a triangle whose hypotenuse is defined by the line x = y, so that the most equiaxial shapes (3D Sphere) are in the bottom left, elongated shapes (1D Rod) are in the top right, and planar shapes (2D Disk) are in the bottom right.
Figure 2.

Representative cells for each treatment group. (a) 2D maximum intensity projections and (b) 3D renderings of representative cells, with nuclei as insets.
Figure 3.

Analysis of cell shape dimensionality. (a) Dimensionality plot of the ratios of the square roots of the gyration tensor ellipsoid principal moments for actin. Upper right corner is more 1D-character (rod), bottom right is more 2D character (disk), and bottom left is more 3D character (sphere). Inset: Gyration tensor ellipsoid with principal moments labeled; √(L1) is the shortest semiaxis, √(L2) is the middle semiaxis, and √(L3) is the longest semiaxis. (b) Dimensionality plot of the ratios of the square roots of the gyration tensor ellipsoid principal moments for nucleus. Inset: Expanded view of the data. (c) L1-depth for actin. L1-depth is the depth of the cell parallel to the L1-axis (caliper distance) representing the thinnest cell dimension and is a gauge of cell height. (d) L1-depth for nucleus. (e) 3D plot of the √(L3/L1), √(L3/L2), and L1-depth for actin using data from panels a and c. (f) 3D plot of the √(L3/L1),√(L3/L2), and L1-depth for nucleus using data from panels b and d. Approximately 100 cells were imaged for each scaffold (Table 1) and error bars represent 2 standard deviations of the mean. In panels c and d, bars with an a, b, c, d, e, f, g, h, i or j are significantly different from SC, SCOS, NF, NFOS, MF, PPS, MG, FG, CG, or CF, respectively (P < 0.05, 1-way ANOVA with Tukey’s test, Figure S3).
The actin dimensionality plot (Figure 3a) demonstrated that substrates promoted cell shapes with different dimensionality. The error bars are 2 standard deviations of the mean which are nearly the same as 95% confidence intervals. Thus, data points whose error bars do not overlap are likely to have significantly different means. Cells from SC, SC+OS, and CF showed the most planar morphology and were closest to the bottom right “2D Disk” corner of the dimensionality plot (Figure 3a). MG was closest to the bottom left “3D Sphere” corner, indicating that it induced the most equiaxial morphology. The remaining substrate groups showed elongated cell morphology with NF being closest to the “1D Rod” corner. MF and CG groups overlapped, indicating that these substrates had cell niches with similar dimensionality.
The nucleus dimensionality plot showed that the nuclei in all of the different biomaterial scaffold niches had similar dimensionality with high degree of equiaxial morphology, causing the groups to overlap (Figure 3b). The expanded view (inset) showed that nuclear shape was influenced by cell niche but not as profoundly as the actin cytoskeleton. Similar to the actin results, the SC and SC+OS nuclei showed the most planar morphology while the MG nuclei showed the most equiaxial morphology, indicating a minor correlation between cell and nucleus shape.
Actin L1-depth is a measure of the thinnest dimension of the ellipsoid fit to the cell and is a gauge of cell dimensionality, particularly cell height (Figure 3c). The cells in the seven scaffold niches (NF, NF+OS, MF, PPS, MG, FG, CG) had a greater L1-depth than cells on the planar substrate niches (SC, SC+OS, CF). These results make sense intuitively, since cells in 3D scaffolds would be expected to take on morphologies with greater 3D character as compared to those cultured on 2D surfaces. Cells cultured on MF scaffolds had a greater L1-depth than those cultured on NF and NF+OS scaffolds, suggesting that the larger diameter fibers in MF promoted cell shapes with greater height. For nucleus L1-depth (Figure 3d), cells cultured in MG and FG had the greatest L1-depth, whereas those cultured on SC and SC+OS had the smallest L1-depth.
The relationship for L1-depth across treatment groups between actin and nucleus was found to be 0.78 using Pearson’s correlation, indicating a correlation between actin and nucleus dimensionality. The combination of the dimensionality plots and L1-depth data into a 3D plot provided a visualization of cell niche dimensionality in biomaterial scaffolds and enhanced the separation of 3D scaffold niches from 2D planar substrate niches (Figures 3e, f). The combination of metrics in the 3D plot for actin (Figure 3e) is able to differentiate between cells cultured in biomaterial scaffolds with greater height, like collagen gel or porous polystyrene scaffolds, from scaffolds with lesser height, like nanofiber scaffolds, whose fibers form a dense mat. Furthermore, this plot also separates between microfiber scaffolds and nanofiber scaffolds, demonstrating its sensitivity to the influence of fiber diameter on cell morphology.
Statistical analysis demonstrated many significant differences in cell shape between pairwise comparisons of substrates (Figure S3). A visual arrangement of pairwise comparisons between treatment groups and shape metrics was prepared. The shape metrics and treatment group pairings were sorted by the sum of the p-values from lowest to highest for the columns and rows. Therefore, the columns toward the left and rows near the top have greater significant differences (shaded boxes). The two shape metrics oriented to the gyration tensor ellipsoid with most significant differences between the treatment groups for actin were √(L1) and L1-depth, which are both measures of the thinnest dimension of the cells. This is a noteworthy finding because it indicates that measures of the thinnest cell dimension are the most effective of the evaluated shape metrics at separating biomaterial scaffold niches by cell shape and therefore are effective gauges of cell dimensionality.
All data in the current work has been deployed online for public access (https://isg.nist.gov/deepzoomweb/stemcells3d/index.html). The data can be downloaded for independent analysis or browsed online via a custom-built 3D cell viewer (Figure 4, Figures S4 and S5). Each of the 969 cells can be rendered into 3D representations, sorted by shape metrics, and rotated in 3D. The cell viewer enables results from quantitative cell shape measurements to be verified via visual examination. For instance, FG and CG had the greater L1-depth compared to SC and CF as determined by the algorithmic image analysis (Figure 3c) and this result was verified by visual examination of the cells in the 3D viewer, with the view down the L1-axis providing the visualization of the Z-area (cell spread area) and the view down the L3-axis providing the visualization of L1depth (Figure 4, Figure S5a, b). Likewise, the finding that nuclei had similar L1-depths under the different treatments (Figure 3d) was verified by browsing 3D viewer screen captures (Figure S5c, d).
Figure 4.

Novel web viewer enables side-by-side visual examination of hundreds of renderings at once. (a) Display of actin in all cells analyzed in the treatment groups SC, CF, FG, and CG were assembled from screen captures of the 3D cell viewer. Within each treatment group, the individual cells are ordered by L1-depth, with the largest L1-depth at the upper left of each treatment group. The blue spheres at the upper left of each treatment group are 100 μm in diameter and serve as the size scale. A guide to the gyration tensor axis alignments is given at the right of the panel. (b) Same as panel a, except cells have been rotated 90° on their L2-axis, so that L1-depth can be compared among treatments.
Hierarchical Clustering Separates 3D Scaffolds from 2D Substrates.
Hierarchical cluster analysis (HCA) of the average values for 28 actin shape metrics per treatment group was performed (Figure 5). The HCA demonstrated that cells on CF substrates clustered with SC and SC+OS substrates, which were also the three planar substrates and promoted the most planar cell morphologies (Figure 3a, c). Six of the seven 3D scaffold treatment groups (FG, NF, NF+OS, CG, MF, and PPS) clustered together, while MG was on its own branch separated from the other 9 treatments with a high Euclidean distance compared to the other treatment groups (Figure 5). The treatment group dendrogram corresponded with the dimensionality plot, indicating that dimensionality was as effective at separating the treatment groups as the total collection of shape metrics. Characterization of the data set with HCA allowed for comparisons of treatment groups to be made. A key point from the HCA was that the addition of OS did not shift the average actin shape metric response significantly, as NF and NF+OS clustered together and SC and SC+OS clustered together. These results indicated that the substrate affected cell shape more than OS. HCA of average nucleus shape metrics per treatment group did not align with average actin metric clustering (Figure S6a). However, the Euclidian distances between treatments for nucleus were much lower (1.5 or less) than for actin (10 or less), indicating greater differences in actin cell shape by treatment group. This was confirmed by HCA of actin and nucleus metrics (Figure S6a), which was dominated by actin, resulting in the same dendrogram that was generated for the actin metrics. HCA for 969 individual cells across all treatment groups demonstrated heterogeneity for actin and nucleus shape metrics with considerable intermixing of different treatment groups (Figure S6b). The exception to this intermixing of treatment groups was MG for actin shape metrics, which grouped together (yellow, Figure S6b), indicating that MG had cells with the most distinctive shape out of all treatment groups analyzed. Principal component analysis (PCA), which projects multiparametric data into lower dimensional space to reveal relationships between different treatments, was largely congruent with HCA (Figure S7). The results of HCA and PCA support the use of cell dimensionality and other shape metrics for 3D cellular morphotyping of the cellular niche provided by biomaterial scaffolds.
Niche precision, defined as the dispersion of cell shapes on a particular substrate, identified the substrates that yield greater control over cell shape. Coefficient of variation (CV) was used to assess niche precision, with variation assessed per shape metric, then averaging the variations per shape metric to yield a CV per treatment group. According to the CV metric, MG had the lowest mean CV for all 28 actin shape metrics analyzed with HCA (Figure S8a). These results are congruent with the HCA and indicate that MG exerted the greatest control over cell shape. The actin mean CV values were greater than the nucleus mean CV values (Figure S8a, b), which indicated that actin shape measurements had greater variability than nucleus shape measurements. Shape metric precision, defined as the dispersion of cell shapes across all treatment groups for a particular shape metric, was assessed and compared to the average CV across all shape metrics for actin and nucleus. L1-based metrics, such as L1-depth and √(L1), had the lowest variation for actin shape metrics (Figure S8h). Taken together with the statistical analysis (Figure S3) and the CV results per treatment group (Figure S8e), these results indicate that L1-based metrics are the most informative for 3D cellular morphotyping of cell niches (Figure 3c).
Power analysis was performed to assess the number of cells required to detect significant differences in the cell shape measurements.30 The L1-depth for a subset of the treatment groups consisting of SC (12.3 μm, SD = 3.61, n = 99), NF (15.2 μm, SD = 3.38, n = 101) and MF (20.6 μm, SD = 5.67, n = 87), was selected since these three substrates have the same composition but different morphologies. Fifty cells would be needed to detect a significant difference between the three groups based on a one-way ANOVA (p = 0.05, power 0.8). For a pairwise comparison of SC and MF using a t test (p = 0.05, power 0.8), 13 cells would be needed to detect a significant difference. Collecting images for a large number (>87) of cells per treatment group provides sufficient power to the results presented in this work and enhances the quality of the 3D cellular morphotyping analysis.
2D vs 3D Shape Metric Analysis.
Because 3D imaging is more costly in terms of acquisition time, processing time, and data storage than 2D imaging, an investigation of the results from 3D shape metrics were compared to 2D shape metrics to assess the value added by 3D imaging and analysis. Cell spread area (Z-area) is a commonly measured 2D shape metric that is used to assess cell size and was compared to the corresponding 3D shape metric, cell volume (Figure 6). Z-area is the cell spread area obtained from the maximum projection of a Z-stack, which would be similar to a 2D image acquired by an epifluorescence microscope without 3D confocal optics. Comparison of actin Z-area and cell volume reveals similar trends (0.71 Pearson’s correlation) with some differences. For instance, FG had the largest cells by volume, whereas SC+OS had the largest cells by Z-area. Additionally, NF had the smallest cells by volume, whereas MG had the smallest cells by Z-area. These results indicate that use of the 2D metric of Z-area to estimate cell size will be more reliable when the cells have a smaller Z-depth. 2D imaging modalities cannot detect the cell volume along the Z-axis during a Z-area calculation. For cells that are randomly oriented in 3D, a spherical shape has the lowest possible surface area to volume ratio, has the largest possible relative Z-depth for a given volume, and will have the most highly underestimated size as determined by a Z-area calculation. Thus, MG cells, which have the most spherical shape, have the largest change from the Z-area plot to the volume plot (Figure 6a, b).
Figure 6.

Comparison of 2D and 3D shape metrics. Actin (a) Z-area and (b) volume. Nucleus (c) Z-area and (d) volume. Approximately 100 cells were imaged for each scaffold (see Table 1) and error bars represent 2 standard deviations of the mean. Statistical analysis is presented in Figure S3. The strength of the relationship between the four plots was evaluated with Pearson correlation coefficients: actin for Z-area vs volume = 0.71, Z-area for actin vs nucleus = 0.93, nucleus for Z-area vs volume = 0.20, and volume for actin vs nucleus = 0.85. Bars with an a, b, c, d, e, f, g, h, i, or j are significantly different from SC, SCOS, NF, NFOS, MF, PPS, MG, FG, CG, or CF, respectively (P < 0.05, 1-way ANOVA with Tukey’s test).
Nucleus Z-area and volume did not exhibit similar trends to one another (0.20 Pearson’s correlation), where SC and SC+OS had the largest nucleus Z-area while MG and FG had the largest nucleus volume. Comparison of Z-area for actin and nucleus and volume for actin and nucleus indicated correlation, with Pearson correlation coefficients of 0.93 and 0.85, respectively.
It is important to note that Z-area measurements underestimated the relative size of cells in 3D scaffolds. For instance, 2D Z-area measurements showed that cells in scaffolds (NF, NF+OS, MF, PPS, MG, FG, CG) were smaller than cells on flat substrates (SC, SC+OS, CF), whereas 3D volume measurements showed that cells in scaffolds were closer in size to the cells on flat substrates. These results indicate that the cell spread area, a 2D shape metric, underestimates the size of cells with greater height. Indeed, MG, which had the most equiaxial morphology, had the smallest Z-area even though it had a median volume. These findings demonstrate that 3D imaging and 3D shape metrics are necessary for accurate cell shape analysis, particularly for cell shapes with greater height, which would be commonly found in 3D scaffolds.
DISCUSSION
The scientific contributions of this work include (1) development of a novel measurement technique to analyze cell shape to assess 3D cell niches in biomaterial scaffolds, (2) a sizable data set of approximately 1000 3D confocal image volumes (Z-stacks) from ten treatment groups that is adequate for performing meaningful statistical analyses, (3) public deployment of the data set on the web to enable additional analyses to be conducted by other groups, and (4) the analysis of trade-offs between cell shape analysis with 2D and 3D shape metrics. 3D cell shape was analyzed with a series of shape metrics to generate a 3D cellular morphotype for the cell niche in a variety of biomaterial scaffolds. This process provided a means to assess the cell niche characteristic to various biomaterial scaffolds independent of materials properties by measuring the cell shape since the cell shape is influenced by the microenvironment. Using cell structure to measure scaffolds niches is analogous to cytotoxicity testing, where cells are used to evaluate drug toxicity by characterizing cell viability to measure drug IC50.31,32 In both cases, the cell response to the factor being tested, either biomaterial scaffold or drug, is evaluated instead of directly evaluating the scaffold or drug. A large number of 3D cell shape metrics (39 actin, 39 nucleus, 4 actin-nucleus ratios, 82 total) were tabulated which enabled identification of metrics that distinguished cells on flat substrates from cells in scaffolds and distinguished between scaffold formats.
L1-depth was also effective at distinguishing mean cell shapes on flat substrates from cells in scaffolds (Figure 3). The combination of L1-depth with the ratios of gyration tensor ellipsoid principle moments (Figure 3e) allowed for separation between 3D scaffold niches that had greater height, like collagen gel or porous polystyrene scaffolds, from scaffolds with lesser height, like nanofiber scaffolds, which form a dense mat. The large number of shape metrics analyzed enabled classification of mean cell shape by HCA and PCA, which also showed that flat substrates elicited cell shapes that were distinct from the cell shapes found in the scaffold treatments (Figure 5). In a single cell analysis, the HCA and PCA were not able to distinguish cells by scaffold (Figures S6 and S7). There may be too much heterogeneity in the cell shape, there may not be enough shape metrics or it may be that different, more effective metrics are needed. A useful goal for future work may be to identify a set metrics that can effectively sort the single cells by scaffold.
In addition to presenting a new 3D cellular morphotyping technique to characterize biomaterial scaffold cell niches, the publicly available data set allows other researchers to explore the data set and the results. This enables researchers to develop new hypotheses for cell shape-driven behavior in scaffold niches. For example, the current data indicates that L1-depth is greater on MF than on SC. According to the literature, this increase in cell height will decrease the efficiency of membrane-based signal transduction,18 and could inspire hypotheses regarding the effect of membrane-based signaling on cell functional outcomes. Public availability of the data set also enables researchers to identify characteristic shape metrics for each scaffold type. This would be informative for conducting studies of biological variables, such as hBMSCs from different donors cultured in a particular scaffold type or cellular response to a drug, where a set of characteristic shape metrics could be assessed between treatment groups to enhance understanding of the observed biological variation. Additionally, 3D cellular morphotyping could be applied to study cell response to scaffolds with varied properties.
The comparison of 2D and 3D cell shape metrics highlighted the advantages of 3D cell shape analysis compared to 2D cell shape analysis. When cells have a 3D shape with considerable height, the use of 2D shape metrics underestimates the results for the cell measurements. 2D imaging and 2D cell shape analysis can be effective in certain cases, particularly when cells have a planar morphology, allowing cells to be imaged and analyzed more rapidly. This study also quantitatively demonstrated the variability in cell shape within different treatment groups (Figure S6b). The HCA of individual cell shape results showed a high degree of intermixing between treatment groups, highlighting the need to image many cells to improve confidence in results.
To identify target cell morphologies for synthetic niche designs, knowledge of the in vivo 3D cell shape for cells of interest is required. Traditional understanding of in vivo 3D cell shapes comes from qualitative histological assessments.14 The use of 3D cellular morphotyping analysis enables a cellular structure−function strategy to be applied to scaffold design, where the 3D cell morphology in a tissue of interest can be quantified and matched through controlled design of a synthetic microenvironment that elicits a similar cellular morphotype. For example, squamous epithelia have a planar morphology that would yield a low L1-depth and would fall near SC on the actin dimensionality plot (Figure 3a). In contrast, chondrocytes have a spherical morphology, which would have a high L1-depth and would fall near MG on the actin dimensionality plot (Figure 3a). Therefore, based on the current cell shape measurements, flat surfaces could be used to culture squamous epithelia (low L1-depth, planar morphology) and promote their characteristic morphology and phenotype, whereas hydrogels could be used to culture chondrocytes (higher L1-depth, equiaxial morphology) to induce their characteristic morphology. The chemistry and mechanics of the microenvironment, as well as the cell type, are also important to cellular function and behavior and should be optimized to successfully direct tissue generation.
Six actions were performed to improve confidence in this data set. First, more than 100 cells were imaged for each treatment group, yielding a total of 1253 Z-stacks of hBMSCs (128 460 images, 128 GB). Second, significant effort was devoted to developing and optimizing automated image segmentation protocols, described elsewhere.28 Different algorithms and the sequence for thresholding, smoothing, cavity-filling, and small-size particle removal were tested. The protocols were assessed by comparison with “ground truth” segmentations, which were defined by manual segmentation of Z-stacks by 4 different experts, and the most accurate segmentation protocol was used for analyses. Third, each segmentation was verified by visual examination to eliminate poorly segmented cells.28 Fourth, the accuracy of the algorithms used to calculate the shape metrics was verified by comparing results from analysis of a synthetic in silico 3D sphere Z-stack to ideal values computed using mathematical formulas (Figure S9a). Fifth, shape metric results were verified by visual examination of 3D renderings of all cells that were analyzed using a custom, interactive 3D cell viewer (Figure 4, Figures S4 and S5). All of the data for the segmented Z-stacks has been deployed online for public access, allowing other investigators to explore the data and analyze the data with new algorithms and/or cell shape metrics. Sixth, fluorescent microspheres with known diameter distributions were imaged to assess confocal Z-aberrations that may contribute to errors in the shape metric analyses (Figure S9). These actions provide an assessment of the data quality that enables users to be informed as they use the data in planning or decision making.
hBMSCs were stained with phalloidin (actin stain), since phalloidin is stable and specific, which enables high-contrast fluorescence imaging of the cytoskeleton. Actin-staining is useful for imaging individual cells that are not in contact with other cells. The current work focused on the cell response to the scaffold, in the absence of contact from other cells. Since cells in vivo are often in contact with other cells, future work may assess the effect of cell seeding density and cell−cell contacts on cell morphology in 3D scaffolds in. A cell membrane stain that demarcates the cell boundaries may be useful for this type of study. A new experimental design and a new data set would be required to determine if the analytical tools developed herein would be effective for analyzing cells that are in contact with other cells.
Discussions of cell shape often trigger contemplation of whether cell volume is conserved during shape change. Herein, cell volume was not measured before and after cell seeding. However, there are several studies on this topic in the literature. Cells can change their volume during growth,33 during development,34 and in response to changes in osmolality.35 During adhesion, cells can accommodate shape changes by increasing the surface area of their plasma membrane by exocytosing intracellular vesicles or by unfolding the plasma membrane, which can alter plasma membrane composition and properties to influence cell behavior.36–38 These changes in surface area and volume can occur in a time scale of minutes or hours.
To develop synthetic biomaterial niches that better mimic the in vivo microenvironment, imaging of cells in vivo is required. Recent efforts have utilized advanced imaging techniques to image cells in vivo and measure their 3D cell shapes. These techniques have assessed the 3D shapes of bone marrow cells in live mice39 and the 3D shapes of mouse osteocytes in tissue sections using confocal microscopy.40 X-ray techniques have provided quantitative measurements of osteocyte lacunae and, presumably, osteocyte morphology in whole bones.41–43 Cells with elongated, planar, and equiaxial morphology have been observed in live zebrafish embryos during development.44 New imaging techniques, such as whole tissue clearing45 and light sheet microscopy,46 may also provide measurements of in vivo 3D cell shapes for synthetic niche designs. As more quantitative 3D cell shape data sets are obtained, the opportunity to directly compare cell shape data from the same cell type in both synthetic biomaterial scaffold niches and natural in vivo niches will be informative.
BMSCs are effective at generating bone, cartilage and fat tissue. Although there is not a quantitative assessment of 3D cell shapes in vivo, histological sections of bone provide a 2D view of the morphologies of the cells involved in bone formation.14 Osteoprogenitor cells reside on the surface of bone and may have an elongated or flattened morphology. Osteoblasts can range in shape from cuboidal to a flattened ellipsoid morphology. Mature osteocytes are surrounded by mature bone and reside in lacunae, which are shaped like an elongated, flattened, ellipsoid.42,43 Osteocytes extend cell projections radially into canaliculi, which are tunnels that connect the lacuna to one another. The main cells in cartilage are chondrocytes and they have a rounded, ellipsoidal to spherical morphology. Early stage fat cells are lipoblasts, which are elongated and have many extensions.14 These become rounded and spherical as they develop into mature adipocytes. NF, CG, and MF generated cell morphologies that were elongated, flattened ellipsoids that may reflect osteoprogenitors, osteoblasts, osteocytes and lipoblasts. MG generated the most rounded, ellipsoidal or spherical morphologies, that best reflected some osteoblasts, chondrocytes and adipoctyes. In vitro studies of BMSC differentiation have observed that cell elongation (minor axis length) correlates with osteogenic differentiation, which would be best reflected by NF.7,9,47
CONCLUSIONS
In summary, the current work presents 3D cellular morphotyping, a technique for characterizing cell niches in biomaterial scaffolds across widely different types of biomaterial scaffolds by measuring cell shape. The technique was able to detect many significant differences between the 8 different biomaterial scaffolds and 10 treatment groups that were tested. Flat surfaces yielded planar cellular morphotypes whereas 3D scaffolds yielded cellular morphotypes with greater cell height (L1-depth), validating that the morphotyping technique was suitable for its intended use. The novelty of this work is the development of a method that enables direct comparison of the cellular niche between different biomaterial scaffolds. Additionally, the sizable data set of acquired image volumes is publicly accessible for researchers to test new hypotheses. Finally, 3D cellular morphotyping informs a strategy for designing niches that optimize cell structure−function relationships and that elicit the desired 3D cell morphologies observed in target tissues in vivo.
Supplementary Material
ACKNOWLEDGMENTS
S.J.F. and S.S. were supported by NIST-NRC Research Associateships. D.C. acknowledges support from National Science Foundation award PHY1205965. The authors acknowledge the assistance of Charles H. Camp, Jr., with calculating the chromatic aberrations and discussion of image distortion in confocal microscopy, Nathan A. Hotaling with power analysis, Young Jong Lee with 3D plots, and Elissa H. Williams with atomic force microscopy. The authors also acknowledge the NIST SHIP and SURF program students, Andrew Wang and Jacob Siegel, who worked on the 3D web-based visualization during the summer of 2014. The hBMSCs employed in this work were obtained from the Tulane Center for Gene Therapy (NCRR-NIH P40RR017447). The “standard deviation” (S.D.) is the same as the “combined standard uncertainty of the mean” for the purposes of this work. This article, a contribution of NIST, is not subject to US copyright. Certain equipment and instruments or materials are identified in the paper to adequately specify the experimental details. Such identification does not imply recommendation by NIST, nor does it imply the materials are necessarily the best available for the purpose.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACSPublicationswebsite at DOI: 10.1021/acsbiomaterials.7b00473.
Supplemental methods for shape metric verification and performance assessment of the imaging pipeline; supplemental figures for data that were not included in the main text, including a detailed list of shape metrics (Figure S1), shape metric plots (Figure S2), statistical analysis (Figure S3), screen captures of 3D cell viewer (Figures S4 and S5), dendrogram and principal component analysis (Figures S6 and S7), niche precision results (Figure S8), and error assessment (Figure S9) (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Cukierman E; Pankov R; Stevens DR; Yamada KM Taking cell-matrix adhesions to the third dimension. Science 2001, 294 (5547), 1708–1712. [DOI] [PubMed] [Google Scholar]
- (2).Liao S; Nguyen LTH; Ngiam M; Wang C; Cheng Z; Chan CK; Ramakrishna S. Biomimetic nanocomposites to control osteogenic differentiation of human mesenchymal stem cells. Adv. Healthcare Mater. 2014, 3 (5), 737–751. [DOI] [PubMed] [Google Scholar]
- (3).Florczyk SJ; Leung M; Li Z; Huang JI; Hopper RA; Zhang M. Evaluation of three-dimensional porous chitosan-alginate scaffolds in rat calvarial defects for bone regeneration applications. J. Biomed. Mater. Res, Part A 2013, 101 (10), 2974–2983. [DOI] [PubMed] [Google Scholar]
- (4).Chatterjee K; Lin-Gibson S; Wallace WE; Parekh SH; Lee YJ; Cicerone MT; Young MF; Simon CG Jr The effect of 3D hydrogel scaffold modulus on osteoblast differentiation and mineralization revealed by combinatorial screening. Biomaterials 2010, 31 (19), 5051–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Khetan S; Guvendiren M; Legant WR; Cohen DM; Chen CS; Burdick JA Degradation-mediated cellular traction directs stem cell fate in covalently crosslinked three-dimensional hydrogels. Nat. Mater. 2013, 12 (5), 458–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ranga A; Gobaa S; Okawa Y; Mosiewicz K; Negro A; Lutolf MP 3D niche microarrays for systems-level analyses of cell fate. Nat. Commun. 2014, 5, 4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kumar G; Tison CK; Chatterjee K; Pine PS; McDaniel JH; Salit ML; Young MF; Simon CG, Jr The determination of stem cell fate by 3D scaffold structures through the control of cell shape. Biomaterials 2011, 32, 9188–9196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kumar G; Waters MS; Farooque TM; Young MF; Simon CG Jr Freeform fabricated scaffolds with roughened struts that enhance both stem cell proliferation and differentiation by controlling cell shape. Biomaterials 2012, 33 (16), 4022–4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Chen D; Sarkar S; Candia J; Florczyk SJ; Bodhak S; Driscoll MK; Simon CG Jr.; Dunkers JP; Losert W. Machine learning based methodology to identify cell shape phenotypes associated with microenvironmental cues. Biomaterials 2016, 104, 104–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).McBeath R; Pirone DM; Nelson CM; Bhadriraju K; Chen CS Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev. Cell 2004, 6 (4), 483–495. [DOI] [PubMed] [Google Scholar]
- (11).Kilian KA; Bugarija B; Lahn BT; Mrksich M. Geometric cues for directing the differentiation of mesenchymal stem cells. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (11), 4872–4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Chen CS; Mrksich M; Huang S; Whitesides GM; Ingber DE Geometric Control of Cell Life and Death. Science 1997, 276 (5317), 1425–1428. [DOI] [PubMed] [Google Scholar]
- (13).Sailem HZ; Sero JE; Bakal C. Visualizing cellular imaging data using PhenoPlot. Nat. Commun. 2015, 6, 5825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ross MH; Wojciech P. Histology: A Text and Atlas, 5th ed.; Lippincott Williams and Wilkins: Baltimore, MD, 2006. [Google Scholar]
- (15).Wang N; Tytell JD; Ingber DE Mechanotransduction at a distance: mechanically coupling the extracellular matrix with the nucleus. Nat. Rev. Mol. Cell Biol. 2009, 10 (1), 75–82. [DOI] [PubMed] [Google Scholar]
- (16).Khatau SB; Hale CM; Stewart-Hutchinson PJ; Patel MS; Stewart CL; Searson PC; Hodzic D; Wirtz D. A perinuclear actin cap regulates nuclear shape. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (45), 19017–19022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Halder G; Dupont S; Piccolo S. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat. Rev. Mol. Cell Biol. 2012, 13 (9), 591–600. [DOI] [PubMed] [Google Scholar]
- (18).Meyers J; Craig J; Odde DJ Potential for control of signaling pathways via cell size and shape. Curr. Biol. 2006, 16 (17), 1685–1693. [DOI] [PubMed] [Google Scholar]
- (19).Rangamani P; Lipshtat A; Azeloglu EU; Calizo RC; Hu M; Ghassemi S; Hone J; Scarlata S; Neves SR; Iyengar R. Decoding information in cell shape. Cell 2013, 154 (6), 1356–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Chan Y-HM; Marshall WF Scaling properties of cell and organelle size. Organogenesis 2010, 6 (2), 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ruckh TT; Kumar K; Kipper MJ; Popat KC Osteogenic differentiation of bone marrow stromal cells on poly(epsilon-caprolactone) nanofiber scaffolds. Acta Biomater. 2010, 6 (8), 2949–2959. [DOI] [PubMed] [Google Scholar]
- (22).Smith LA; Liu X; Hu J; Ma PX The influence of three-dimensional nanofibrous scaffolds on the osteogenic differentiation of embryonic stem cells. Biomaterials 2009, 30 (13), 2516–2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Smith LA; Liu X; Hu J; Wang P; Ma PX Enhancing osteogenic differentiation of mouse embryonic stem cells by nanofibers. Tissue Eng., Part A 2009, 15 (7), 1855–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Mendicino M; Bailey AM; Wonnacott K; Puri RK; Bauer SR MSC-based product characterization for clinical trials: an FDA perspective. Cell Stem Cell 2014, 14 (2), 141–5. [DOI] [PubMed] [Google Scholar]
- (25).Lee GY; Kenny PA; Lee EH; Bissell MJ Three-dimensional culture models of normal and malignant breast epithelial cells. Nat. Methods 2007, 4 (4), 359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Elliott JT; Halter M; Plant AL; Woodward JT; Langenbach KJ; Tona A. Evaluating the performance of fibrillar collagen films formed at polystyrene surfaces as cell culture substrates. Biointerphases 2008, 3 (2), 19–28. [DOI] [PubMed] [Google Scholar]
- (27).Cole RW; Jinadasa T; Brown CM Measuring and interpreting point spread functions to determine confocal microscope resolution and ensure quality control. Nat. Protoc. 2011, 6 (12), 1929–1941. [DOI] [PubMed] [Google Scholar]
- (28).Bajcsy P; Simon M; Florczyk S; Simon CG; Juba D; Brady M. A Method for the Evaluation of Thousands of Automated 3D Stem Cell Segmentations. J. Microsc. 2015, 260 (3), 363–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zou KH; Warfield SK; Bharatha A; Tempany CMC; Kaus MR; Haker SJ; Wells WM III; Jolesz FA; Kikinis R. Statistical Validation of Image Segmentation Quality Based on a Spatial Overlap Index. Acad. Radiol. 2004, 11 (2), 178–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Faul F; Erdfelder E; Lang A-G; Buchner A. G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behavior Research Methods 2007, 39 (2), 175–191. [DOI] [PubMed] [Google Scholar]
- (31).Krausz E; de Hoogt R; Gustin E; Cornelissen F; Grand-Perret T; Janssen L; Vloemans N; Wuyts D; Frans S; Axel A; Peeters PJ; Hall B; Cik M. Translation of a tumor microenvironment mimicking 3D tumor growth co-culture assay platform to highcontent screening. J. Biomol. Screening 2013, 18 (1), 54–66. [DOI] [PubMed] [Google Scholar]
- (32).Vinci M; Gowan S; Boxall F; Patterson L; Zimmermann M; Court W; Lomas C; Mendiola M; Hardisson D; Eccles SA Advances in establishment and analysis of three-dimensional tumor spheroid-based functional assays for target validation and drug evaluation. BMC Biol. 2012, 10, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zucker RM; Whittington K; Price BJ Differentiation of HL-60 cells: cell volume and cell cycle changes. Cytometry 1983, 3 (6), 414–8. [DOI] [PubMed] [Google Scholar]
- (34).Saias L; Swoger J; D’Angelo A; Hayes P; Colombelli J; Sharpe J; Salbreux G; Solon J. Decrease in Cell Volume Generates Contractile Forces Driving Dorsal Closure. Dev. Cell 2015, 33 (5), 611–21. [DOI] [PubMed] [Google Scholar]
- (35).Pritchard S; Guilak F. The role of F-actin in hypo-osmotically induced cell volume change and calcium signaling in anulus fibrosus cells. Ann. Biomed. Eng. 2004, 32 (1), 103–11. [DOI] [PubMed] [Google Scholar]
- (36).Kehle T; Herzog V. A colloidal gold labeling technique for the direct determination of the surface area of eukaryotic cells. Eur. J. Cell Biol. 1989, 48 (1), 19–26. [PubMed] [Google Scholar]
- (37).Gauthier NC; Rossier OM; Mathur A; Hone JC; Sheetz MP Plasma membrane area increases with spread area by exocytosis of a GPI-anchored protein compartment. Mol. Biol. Cell 2009, 20 (14), 3261–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Figard L; Sokac AM A membrane reservoir at the cell surface: unfolding the plasma membrane to fuel cell shape change. Bioarchitecture 2014, 4 (2), 39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lo Celso C; Fleming HE; Wu JW; Zhao CX; Miake-Lye S; Fujisaki J; Cote D; Rowe DW; Lin CP; Scadden DT Liveanimal tracking of individual haematopoietic stem/progenitor cells in their niche. Nature 2009, 457 (7225), 92–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Himeno-Ando A; Izumi Y; Yamaguchi A; Iimura T. Structural differences in the osteocyte network between the calvaria and long bone revealed by three-dimensional fluorescence morphometry, possibly reflecting distinct mechano-adaptations and sensitivites. Biochem. Biophys. Res. Commun. 2012, 417 (2), 765–770. [DOI] [PubMed] [Google Scholar]
- (41).Kerschnitzki M; Kollmannsberger P; Burghammer M; Duda GN; Weinkamer R; Wagermaier W; Fratzl P. Architecture of the osteocyte network correlates with bone material quality. J. Bone Miner. Res. 2013, 28 (8), 1837–1845. [DOI] [PubMed] [Google Scholar]
- (42).Carter Y; Thomas CDL; Clement JG; Cooper DML Femoral osteocyte lacunar density, volume and morphology in women across the lifespan. J. Struct. Biol. 2013, 183 (3), 519–526. [DOI] [PubMed] [Google Scholar]
- (43).Carter Y; Thomas CDL; Clement JG; Peele AG; Hannah K; Cooper DML Variation in osteocyte lacunar morphology and density in the human femur - a synchrotron radiation micro-CT study. Bone 2013, 52 (1), 126–132. [DOI] [PubMed] [Google Scholar]
- (44).Xiong F; Ma W; Hiscock TW; Mosaliganti KR; Tentner AR; Brakke KA; Rannou N; Gelas A; Souhait L; Swinburne IA; Obholzer ND; Megason SG Interplay of cell shape and division orientation promotes robust morphogenesis of developing epithelia. Cell 2014, 159 (2), 415–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Yang B; Treweek JB; Kulkarni RP; Deverman BE; Chen C-K; Lubeck E; Shah S; Cai L; Gradinaru V. Single-cell phenotyping within transparent intact tissue through whole-body clearing. Cell 2014, 158 (4), 945–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Krzic U; Gunther S; Saunders TE; Streichan SJ; Hufnagel L. Multiview light-sheet microscope for rapid in toto imaging. Nat. Methods 2012, 9 (7), 730–733. [DOI] [PubMed] [Google Scholar]
- (47).Marklein RA; Lo Surdo JL; Bellayr IH; Godil SA; Puri RK; Bauer SR High Content Imaging of Early Morphological Signatures Predicts Long Term Mineralization Capacity of Human Mesenchymal Stem Cells upon Osteogenic Induction. Stem Cells 2016, 34 (4), 935–47. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.