

Graphical abstract
Keywords: BRD4, Senescence, Inflammation, Autophagy, Aging-related diseases
Highlights
-
•
Brd4 is closely related with a variety of aging related diseases;
-
•
The role of Brd4 in regulating aging-related diseases is complex and multifaceted;
-
•
Cell cycle, apoptosis, cell senescence, SASP, senolytic effect, autophagy, and mitochondria dysfunction may be involved in Brd4 regulating aging.
Abstract
Background
Aging, a complex and profound journey, leads us through a labyrinth of physiological and pathological transformations, rendering us increasingly susceptible to aging-related diseases. Emerging investigations have unveiled the function of bromodomain containing protein 4 (BRD4) in manipulating the aging process and driving the emergence and progression of aging-related diseases.
Aim of review
This review aims to offer a comprehensive outline of BRD4′s functions involved in the aging process, and potential mechanisms through which BRD4 governs the initiation and progression of various aging-related diseases.
Key scientific concepts of review
BRD4 has a fundamental role in regulating the cell cycle, apoptosis, cellular senescence, the senescence-associated secretory phenotype (SASP), senolysis, autophagy, and mitochondrial function, which are involved in the aging process. Several studies have indicated that BRD4 governs the initiation and progression of various aging-related diseases, including Alzheimer's disease, ischemic cerebrovascular diseases, hypertension, atherosclerosis, heart failure, aging-related pulmonary fibrosis, and intervertebral disc degeneration (IVDD). Thus, the evidence from this review supports that BRD4 could be a promising target for managing various aging-related diseases, while further investigation is warranted to gain a thorough understanding of BRD4′s role in these diseases.
Introduction
Aging is a multifaceted process marked by a gradual deterioration of an organism's physiological functions over time. This leads to an escalating vulnerability to aging-related diseases, including neurodegenerative disorders, cardiovascular diseases, ischemic cerebrovascular diseases, and more [1]. The incidence of such aging-related diseases has seen a steady rise, largely attributed to demographic shifts resulting in aging populations. While the biological mechanisms that underpin aging remain incompletely understood, a growing perspective suggests that alterations of epigenetic information regulate the aging process [2]. Several studies have shed light on this phenomenon. Through targeting epigenetic changes, such as the overexpression of sirtuin2, or the deletion of the histone methyltransferase gene Set2, lifespan extension has been shown in yeast [3], [4], [5], [6], [7]. Furthermore, recent breakthroughs by Dr. Sinclair and his team have established that epigenetic information deficiency causes aging in mammals and highlighted the potential of epigenetic reprogramming in reversing the aging process [8]. Among the various epigenetic changes observed during aging, acetylation modification has received substantial attention. Adding (histone acetyltransferases, HATs) and removing (histone deacetylases, HDACs) acetyl groups have emerged as therapeutic strategies for a range of aging-related diseases [9], [10], [11]. As epigenetic readers for acetyl groups, bromodomain (BD)-containing proteins (BRDs) have been shown to be closely associated with various diseases in the past two decades [12]. However, the involvement of BRD4 in the aging process requires further investigation.
BRD2, BRD3, BRD4, and BRDT are the four bromodomain and extra-terminal (BET) domain proteins, which are broadly expressed in various species [13]. Structurally, a bromodomain is formed by four alpha-helices (alpha Z, alpha A, alpha B, and alpha C) and two loops (ZA loop and BC loop), and is responsible for recognizing acetylated lysine residues [14]. Each BET protein has two N-terminal bromodomains (BD1 and BD2) and an extra-terminal (ET) domain, and exhibit a highly similar structure [15]. BRD4 and BRDT also include a C-terminal domain (CTD), which helps facilitate histones to interact with other proteins [16], which is crucial for the regulation of gene expression and the control of chromatin structure. Fig. 1 depicts the detailed structure of the four BET family proteins.
Fig. 1.

Structure of four BET proteins. Two isoforms of BRD4 are presented. BD: bromodomain. ET: extra-terminal domain. CTD: C-terminal domain. GPA: glycine-proline-alanine.
BRD4, the most thoroughly studied member of the BET family, has two known mammalian splice variants including the BRD4 long variant (BRD4-L) and BRD4 short variant (BRD4-S) [17]. BRD4-L has been the focus of most studies, distinguished by a unique CTD that enables interactions with the positive transcription elongation factor b (P-TEFb), which facilitates the transcriptional machinery [18], [19]. Despite the lack of a CTD, BRD-S was also found to have its own unique biological functions [20]. Besides transcriptional regulation, studies have also proposed that BRD4 may function as a HAT [21], [22], [23].
This review aims to deliver a current synopsis of the potential mechanisms and roles of BRD4 in the aging process. Specifically, the study will delve into the regulatory function of BRD4 in cellular processes such as the cell cycle, apoptosis, senescence, autophagy, and mitochondrial dysfunction within the context of aging. Additionally, we will scrutinize the implications of BRD4 in the development of aging-related diseases, such as neurodegenerative disorders, ischemic cerebrovascular diseases, cardiovascular diseases, and other aging-related ailments. By consolidating the present understanding of BRD4′s functions and mechanisms, this review intends to offer novel insights into potential therapeutic targets for delaying the aging process and alleviating aging-related diseases.
The cellular mechanisms of BRD4 in aging
Cell cycle regulation by BRD4
The cell cycle is an intricately coordinated process crucial for cell growth and proliferation. During the G1 phase, cells ready themselves for DNA synthesis. The S phase is marked by DNA replication, while the G2 phase prepares cells for division. Lastly, the M phase encompasses the stage of cell division. Importantly, cell cycle arrest is a primary characteristic of cellular senescence [24].
BRD4 was initially found to be correlated with cell growth [25], [26]. In 2002, Dr. Houzelstein and his colleagues first reported that BRD4 nullizygous embryos cannot survive after implantation [27]. Additionally, BRD4 heterozygotes exhibit prenatal and postnatal growth defects associated with decreased proliferation rates, and a similar phenomenon is observed in cultured primary heterozygous cells [27]. These findings underscore the critical role of BRD4 in mammalian growth. Specifically, BRD4 is essential in cerebellar development [28]. Mechanistically, a study indicated that BRD4 stimulates G1 phase gene transcription and promotes cell cycle progression through recruiting P-TEFb and RNA polymerase II to promoters in HeLa cells [29]. Similarly, in NIH3T3 cells and mouse embryonic fibroblasts, BRD4 knockout inhibits gene expression related to G1 phase (Ccnd1, Ccnd2, Mcm2, Ranbp1, Nid1, Orcl2, Pop1, etc.), consequently arresting cells in the G0/G1 phase [30]. Furthermore, promoters of late and early postmitotic genes are first selectively marked by BRD4, and BRD4 directs transcription of these genes in daughter cells [31].Besides directing G1 gene expression, BRD4 has also been suggested to promote cell transit from the G2 to the M phase [26]. Intriguingly, akin to BRD4 knockout, an earlier study found that ectopic expression of BRD4 in NIH3T3 and HeLa cells impedes cell cycle progression from the G1 to the S phase [32]. In this scenario, BRD4 interacts with replication factor C (RFC) through BD2 instead of BD1, thus interfering with RFC-dependent DNA replication [32]. Collectively, these studies suggest a complex regulatory role for BRD4 in cell growth. Both the knockdown and ectopic expression of BRD4 seem to inhibit the progression of the cell cycle, albeit through distinct mechanisms.
BRD4 in apoptosis
Apoptosis is crucial for maintaining tissue homeostasis and eliminating damaged or abnormal cells. Numerous studies have extensively explored the relationship between BRD4 and apoptosis. However, similar to BRD4′s role in cell cycle regulation, its effect on apoptosis also diverges into two directions. Studies involving oncogenic cells, including colorectal cancer [33], renal carcinoma [34], retinoblastoma [35], gallbladder cancer [36], [37], [38], myeloma [39], non-small cell lung cancer [40], acute myeloid leukemia [41], and cholangiocarcinoma [42], indicate that pharmacological inhibition or genetic knockout of BRD4 triggers cell apoptosis, thus highlighting its potential as an anti-cancer or anti-tumor target. Several reviews have well summarized the apoptosis-inducing effect of BRD4 inhibition in oncogenic cells [43], [44]. The commonly mentioned mechanism underlying the anti-cancer effect is downregulation of the oncogene c-Myc [45]. Moreover, dislodging of BRD4-NUT (nuclear protein in testis) from chromatin, which trigger terminal squamous cell differentiation and apoptosis, was reported in midline carcinoma [46], [47]. RNA:DNA hybrids (R-loops) caused by BRD4 loss is another involved mechanism, which finally leads to increased replication stress and DNA damage [48]. Specifically, through triggering transcriptional downregulation of TopBP1 (DNA topoisomerase 2 binding protein 1), which is a DNA damage response protein, BRD4 inhibition fails to promote ATR-Chk1 pathway activation culminating in apoptotic cell death despite increased replication stress [48]. These findings underscore that BRD4 inhibition causes transcription-replication conflicts, DNA damage, and apoptosis in oncogenic cells [48].
However, with increasing research on BRD4 across various disease fields, several studies have suggested that BRD4 inhibition has an anti-apoptotic effect, thus playing a protective role in certain diseases. In the case of ischemia/reperfusion injury (I/R), decreased expression of endoplasmic reticulum stress-associated and pro-apoptotic proteins were observed after using the BET bromodomain inhibitor JQ1, or genetic knockdown of BRD4; a similar phenomenon was observed under hypoxia/reoxygenation (H/R) stimulation [49]. The researchers reported this protective effect was likely due to blocking FOXO4-dependent reactive oxygen species (ROS) generation during I/R or H/R through regulating the PI3K/AKT pathway [49]. Furthermore, BRD4 upregulation has been noted in chronic obstructive pulmonary disease (COPD), and BRD4 knockdown alleviated BEAS-2B cell apoptosis induced by cigarette smoke extract, a widely used COPD cell model [50]. In the intervertebral disc degeneration (IVDD) disease model, BRD4 inhibition reduces apoptosis in nucleus pulposus (NP) cells by inducing autophagy [51]. Although an anti-apoptotic effect was suggested in the above studies, the detailed mechanism needs further exploration and validation.
In conclusion, published studies showcase the contrasting roles of BRD4 in oncogenic and non-oncogenic cells, emphasizing its multifaceted function. Therefore, comprehending the role of BRD4 in apoptosis could have substantial implications for the development of therapies across various diseases.
BRD4 and senescence
Cellular senescence. Cellular senescence is characterized by an irreversible state of growth arrest, and alterations in gene expression and morphology [24]. Various stressors, including DNA damage, oxidative stress, and telomere dysfunction, deficiency of epigenetic information, can initiate senescence. Over time, senescent cells accumulate in tissues and are believed to initiate various aging-related diseases such as neurodegenerative disorders and cardiovascular diseases [52], [53], [54].
Evidence confirms a crucial role of BRD4 in regulating cellular senescence. BRD4 has been identified as a regulator of telomere length [55]. Telomere length is crucial for sustaining long-term cell division, and when telomeres become excessively short, cells cannot divide successfully and may undergo senescence [56]. Studies have found that four different BRD4 inhibitors, IBET151, JQ1, MS436, and OTX015, block telomere elongation, rather than influencing telomerase activity, in both mouse and human cells [55]. In this way, BRD4 inhibition could potentially expedite cell senescence. In esophageal cancer cells, both JQ1 and BRD4 knockdown via shRNA trigger cell senescence [57]. Mechanistically, BRD4 inhibition elevates p21 and decreases cyclin D1 protein expression, causing G1 phase cell cycle arrest. Independent of apoptosis, this inhibitory effect is caused by aurora kinase suppression [57]. Similarly, JQ1 inhibition and BRD4 depletion induce gastric cancer cellular senescence by downregulating miR-106b-5p, thus promoting p21 gene expression [58]. The same phenomenon was observed in head and neck oncogene cells in that JQ1 caused accumulation of DNA double-strand breaks and senescence-associated beta-galactosidase (SA-β-gal), and a decrease in phosphorylated Sirt1ser4, thus elevating p21 and p16 expression [59]. It is worth pointing out that, in these circumstances, loss of BRD4 transcriptionally activates p21 and increases cellular senescence independent of p53. P21 is generally thought to be activated by p53 to induce permanent cell cycle arrest [60], [61]. However, in p53 mutated and inactive gastric cancer cells [58], JQ1 and BRD4 depletion substantially increase p21 expression. Moreover, JQ1 enhances p21 protein levels and senescence but reduces p53 and Ac-p53 protein levels [59]. Collectively, these findings indicate that BRD4 inhibition induces oncogenic cell senescence in a p53 independent way.
In contrast to the aforementioned studies, BRD4 inhibition offers a protective role in IVDD through activating the AMPK/mTOR/ULK1 pathway and alleviate NP cell senescence through activating autophagy [51]. Altogether, the multifaceted functions of BRD4 indicate its capability to both counteract and induce cellular senescence, albeit through distinct mechanisms.
BRD4 and senescence-associated secretory phenotype (SASP). SASP is another distinct feature of cellular senescence, characterized by the release of many cytokines, chemokines, and growth factors [62]. Depending on the context, SASP can have both beneficial and detrimental impacts on neighboring cells. On the one hand, SASP can bolster tissue repair and regeneration by recruiting immune cells and facilitating tissue remodeling. And conversely, SASP can also contribute to chronic inflammation and increased cellular senescence, thereby leading to aging-related diseases [63].
In 2016, Tasdemir et al. reported that during senescence development, extensive remodeling happens in super-enhancers (SEs), which are enhancers enriched with H3K27Ac. SE remodeling results in the emergence of new SEs adjacent to key SASP-related genes, thus promoting SASP [64]. Furthermore, this study highlighted the necessity of BRD4 binding to SEs for SASP-related gene expression and subsequent paracrine signaling in senescent instead of quiescent cells; genetic deletion or pharmacological inhibition of BRD4 collapses SASP-related gene expression [64]. Thus, the available evidence underscores the importance of BRD4 in facilitating the SASP and downstream paracrine signaling, which are crucial for senescence immune surveillance and execution of the tumor-suppressive program. Other studies also point out the pivotal role of BRD4 in SASP. For instance, BRD4 inhibition prevents lipopolysaccharide (LPS)-induced senescence in macrophages [65]. Specifically, LPS triggers the activation of NF-kB and leads to a redistribution of BRD4 on chromosomes, enhancing the expression of SASP-related genes, and reinforcing the macrophage senescent phenotype through paracrine pathways, a process termed inflammaging [66]. Studies conducted on lung epithelial cells [67] and islet cells [68] further substantiate the pivotal role of BET proteins in initiating the SASP transcriptional program. Taken together, these studies suggest that BRD4 is essential for SE formation to promote SASP gene expression.
BRD4 and senolysis. As mentioned above, cell senescence has been detected in most organs, and accumulation of senescent cells has numerous detrimental effects [52], [53], [54]. Selective elimination of senescent cells, called senolysis, can prolong health span and alleviate a wide range of aging-related diseases. In 2015, Dr. Kirkland and his colleagues first discovered that a drug combination of dasatinib and quercetin could selectively kill senescent cells [69]. Over the past few years, senolytic pharmacotherapy has been recognized as a new therapeutic modality. In 2020, BET family proteins were identified as a novel senolytic target by Wakita et al. through high-throughput screening [70]. They discovered that both JQ1 and ARV-825 could selectively induce death in senescent cells, rather than proliferating and quiescent cells. Mechanistically, ARV-825 instigated senolysis through inhibiting non-homologous end joining (NHEJ) and promoting autophagic gene transcription [70]. Intriguingly, a recent study revealed that senescent cells can also promote normal healing by activating stem cell repair [71]. While senolysis therapy may have anti-aging effects by removing senescent cells, it may also negatively impact normal cell repair. Indiscriminate destruction of senescent cells poses risks, hence there is a significant journey ahead to confirm the senolytic effects of these BET family protein inhibitors and evaluate their safety.
Based on the studies in cellular senescence, SASP, and senolysis, the role of BRD4 in the regulation of senescence seems paradoxical. First, the loss of BRD4 can induce a senescent phenotype, potentially limiting tumor development while promoting aging-related diseases. Second, BRD4 is critical for the expression of SASP-related genes, which suggests it may facilitate aging-related diseases. Third, SASP is crucial for immune surveillance and the clearance of senescent cells; a deficiency in immune surveillance could potentially lead to the manifestation of latent tumors or the accumulation of senescent cells. Lastly, the potential senolytic effect of BRD4 inhibition may help clear senescent cells and rejuvenate tissues or organs. One of the reasons is that BRD4 usually functions as a scaffold connecting transcription factors at promoters with SEs [72], [73], which determines cell fate by controlling gene expression in a cell type-specific pattern [74], [75]. This means that the function of BRD4 appears to depend on the cellular context and the genetic makeup of the target cells. The complex function of BRD4 makes it even more difficult to demonstrate its role in senescence studies. Additional studies are necessary to fully elucidate its role in regulating senescence and the process of various aging-related diseases.
BRD4 and autophagy
Autophagy is an effective catabolic process that directs excess or damaged cytoplasmic components to lysosomes for degradation [76]. The relationship between autophagy and aging has been extensively studied, spanning from cellular senescence to lifespan extension [77], [78]. Autophagy can be regulated at both the transcriptional and posttranscriptional level [79].
Abundant evidence supports an inhibitory effect of BRD4 on autophagy. For instance, JQ1 promotes autophagosome formation in acute myelogenous leukemia (AML) stem cells, implicating that BET proteins may participate in autophagy regulation [80]. In 2017, Sakamaki et al. reported that BRD4 inhibits transcription of autophagy and lysosomal genes [81], [82], representing the first direct evidence of BRD4′s role in autophagy regulation. By abolishing interaction between autophagy gene promoters and methyltransferase G9a, loss of BRD4 promotes autophagy and lysosomal biosynthesis not only in nutrient deprivation-induced autophagy, but also autophagy induced by rapamycin, glucose starvation, hypoxia, trehalose, etc. In contrast, overexpressing BRD4 blocks the formation of LC3-II induced by rapamycin [82], [83]. Furthermore, BRD4 inhibition promotes autophagy in a TFEB (transcription factor EB)-TFE3 (transcription factor E3)-MITF (microphthalmia transcription factor) independent way [82]. Follow-up studies demonstrated that BRD4 inhibition has a protective role in intervertebral disc NP cells [51], [84], [85], [86], cadmium-induced acute kidney injury [87], and pancreatitis [88] through autophagy activation. Moreover, JQ1 activates PTEN-induced putative protein kinase 1 (PINK1) and the E3 ubiquitin ligase Parkin-mediated mitophagy [89], which specifically degrades damaged mitochondria [90].
These studies underscore the significance of BRD4-regulated autophagy in the pathophysiological processes across various cells and tissues. A recent investigation by Li et al. revealed that curcumin promotes autophagy, effectively diminishing ROS [91]. The reduction in ROS subsequently inhibits activities of acetyltransferase P300, leading to a decrease in BRD4 recruitment to the promoters of inflammation-related genes (IL-1β, IL-6, IL-8, TNF-α, and MCP-1), thus mitigating inflammation [91]. This research demonstrates a reciprocal regulation between autophagy and BRD4. Collectively, BRD4 inhibition can directly activate autophagy and lysosome-related gene transcription. Conversely, the activation of autophagy can limit BRD4 recruitment to the promoters of corresponding genes.
In contrast, certain studies propose that BRD4 plays a positive role in activating autophagy. For instance, in AML cells, BRD4 inhibitors such as JQ1 and I-BET-151 have been found to impede autophagy [92]. While the activation of autophagy promotes the degradation of nucleophosmin 1 (NPM1) and hexamethylene bisacetamide-inducible protein 1 (HEXIM1), in turn, stimulating the BET pathway [92]. It is unclear why they got these conflicting results in different studies. Thus, more studies are still warranted to investigate the role of BRD4 in autophagy, especially in different cell contexts.
BRD4 and mitochondrial function
Mitochondrial dysfunction, characterized by diminished respiratory capacity and mitochondrial membrane potential, can arise from various factors, including DNA mutations, mitochondrial mass homeostasis, mTOR signaling, low NAD+/NADH ratios, and Ca2+ overload [93]. Among these factors, mitochondrial mass homeostasis is maintained by mitochondrial biogenesis, mitophagy to clear damaged mitochondria, and balance of mitochondrial fission and fusion. Mitochondria dysfunction is a defining characteristic of aging [93]. Interestingly, studies suggested that BRD4 is indispensable for mitochondrial function regulation.
First, it is well documented that BRD4 deficiency enhances the levels of oxidative phosphorylation (OXPHOS) and its activities, leading to a remodeling of the mitochondrial proteome [94]. Dysfunctions in OXPHOS have been observed in various models of cellular senescence, suggesting a cause-effect relationship between OXPHOS and senescence [95], [96]. Additionally, an SE for peroxisome proliferator-activated receptor-gamma coactivator-1α (PGC-1α), which is a transcriptional cofactor essential to control mitochondrial biogenesis, is highly occupied by BRD4; BET inhibitors significantly reduce PGC-1α expression [97]. In cardiomyocytes, BRD4 controls nucleus-encoded mitochondrial transcription, and acute depletion of BRD4 leads to the loss of mitochondrial function in myocytes [98], [99]. Moreover, either the pharmacological inhibition (OTX015) or knockout of BRD4 suppresses the gene expression of mitochondrial fission factor (MFF), blocking fission and leading to mitochondrial hyperfusion in prostate cancer cells [100]. Mitochondrial hyperfusion, characterized by mitochondrial elongation, has been found to be associated with senescence-associated phenotypes [101], [102]. However, another study discovered that JQ1 increases fission-related gene expression and decreases fusion-related gene expression, resulting in apoptosis in melanoma cells [39], which contrasts the former result in prostate cancer [100]. It is uncertain that the contrasting results were caused by the different BET inhibitors or the different cell context. In conclusion, these findings suggest that BRD4 has a functional role in the regulation of mitochondrial function and the aging process.
The potential mechanisms of BRD4 in regulating the aging process are summarized in Table 1.
Table 1.
BRD4 implicated in aging.
| Mechanisms | Functions | Molecular target | Ref |
|---|---|---|---|
| Cell cycle | BRD4 inhibition blocks G0/G1 phase | Inhibits G1 gene expression (Ccnd1, Ccnd2, Mcm2, Ranbp1, Nid1, Orc2, Pop1, et al.) | [29], [30], [31] |
| G2/M phase | Unknown | [26] | |
| BRD4 ectopic expression inhibits G0/G1 phase | Inhibits RFC-dependent DNA elongation | [32] | |
| Apoptosis | BRD4 inhibition triggers apoptosis | Downregulates c-Myc | [45] |
| Inhibits BRD4-NUT oncogene | [46], [47] | ||
| Accumulates RNA-DNA hybrids | [48] | ||
| BRD4 inhibition alleviates apoptosis | Inhibits FOXO4-dependent ROS generation through PI3K/AKT pathway | [49] | |
| Induces autophagy through MAPK/mTOR/ULK1 pathway | [51] | ||
| Cell senescence | BRD4 inhibition induces cell senescence | Blocks telomere elongation | [55] |
| Upregulates p21 expression | [57], [58], [59] | ||
| BRD4 inhibition alleviates cell senescence | Downregulates SASP-related genes | [67], [68], [64], [65] | |
| Induces autophagy via AMPK/mTOR/ULK1 pathway | [51] | ||
| SASP | BRD4 inhibition alleviates SASP | Inhibits SASP via SE remodeling | [64] |
| Senolysis | ARV-825 and JQ1 provoke senolysis | Attenuates NHEJ and upregulates autophagic gene expression | [70] |
| Autophagy | BRD4 inhibition inhibits autophagy | Unknown | [92] |
| BRD4 inhibition promotes autophagy | Promotes autophagy and lysosomal biosynthesis through methyltransferase G9a | [82] | |
| BRD4 inhibition promotes mitophagy | Activates PINK1/Parkin pathway | [89] | |
| Mitochondria dysfunction | BRD4 inhibition promotes mitochondria dysfunction | Increases the levels and activity of OXPHOS protein complexes | [94] |
| BRD4 inhibits mitochondria biogenesis | Downregulates PGC-1α gene expression | [97] | |
| BRD4 inhibition blocks mitochondria fission | Inhibits MFF expression | [100] | |
| BRD4 inhibition increases mitochondria fission | Inhibits c‐Myc and alters mitochondrial dynamics | [39] |
RFC: replication factor C; ROS: reactive oxygen species; SASP: senescence-associated secretory phenotype; NHEJ: non-homologous end joining; OXPHOS: oxidative phosphorylation; MFF: mitochondrial fission factor.
BRD4′s role in aging-related diseases
BRD4 has emerged as a key player in the development and progression of various aging-related diseases. Based on the previous sections, dysregulation of BRD4 has been linked to impaired cellular processes associated with the senescence and aging process. Thus, BRD4 may be a promising target for treating aging-related diseases. A summary of BRD4 in aging-related diseases is shown in Table 2.
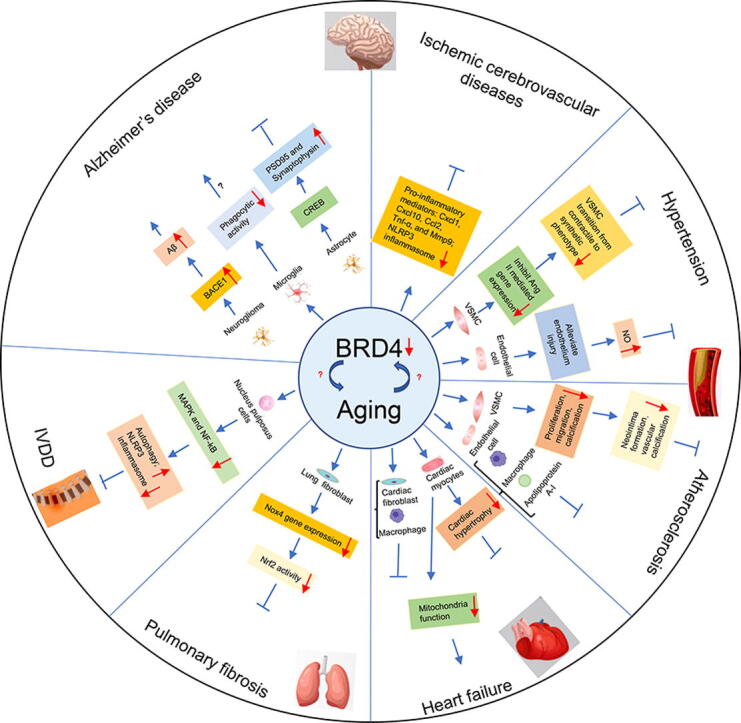
Fig. 2.

Mechanisms of BRD4 regulation in the aging process. Aβ, amyloid-beta; BACE1, beta-site APP-cleaving enzyme 1; CREB, cAMP response element binding protein; IVDD, intervertebral disc degeneration; VSMC, vascular smooth muscle cell; PSD95, post synaptic density protein 95; NO, nitric oxide.
Table 2.
Mechanisms of BRD4 implicated in aging-related diseases.
| Diseases | Functions (BRD4 inhibition) | Study tools | Potential mechanisms | Ref |
|---|---|---|---|---|
| Alzheimer’s disease | Alleviates Alzheimer’s disease | JQ1 | Improves spatial memory via CREB signaling and upregulating synaptic proteins of PSD95 and synaptophysin | [104] |
| JQ1, Genetic (siRNA) inhibition of BET proteins | Decreases the expression of a subset of phagocytosis-related genes and reduced the expression of Cd33, Trem2, and Zyx | [105] | ||
| JQ1 | Partly via downregulation of TNF-α and activation of CREB signaling | [106] | ||
| JQ1 | Elicits gene expression associated with ion channel activity, transcription and DNA repair | [107] | ||
| Exacerbates Alzheimer’s disease | JQ1, ARV-825 | Increases the levels of BACE1 | [108] | |
| Ischemic cerebrovascular diseases | Alleviates ischemic cerebrovascular diseases | dBET1, JQ1 | Inhibits inflammation | [110], [113] |
| Hypertension | Decreases blood pressure | JQ1 | Inhibits VSMC phenotype transition from contractile to synthetic state | [118] |
| Reduces oxidative stress and inflammatory response, alleviates endothelial cell damage, ameliorates aortic injury | [120] | |||
| Neointima formation | Inhibits neointima formation | JQ1 | Inhibits VSMC proliferation | [126], [127], [128] |
| Atherosclerosis | Alleviates atherosclerosis | JQ1 | Inhibits VSMC proliferation, migration, and calcification | [133], [134] |
| JQ-1, I-BET762, RVX-208 |
Regulates macrophage function | [65], [73], [91], [134] | ||
| JQ1, RVX-208 | Alleviates endothelial inflammation | [73], [135], [136], [137] | ||
| RVX-208 | Inhibits synthesis of apolipoprotein A-I | [138] | ||
| Heart failure | Alleviates heart failure | JQ1 | Inhibits the hypertrophic response of cardiac myocytes triggered by extracellular growth cues | [139], [140] |
| JQ1 | Inhibits fibrosis and inflammation | [141], [142], [143], [144] | ||
| Exacerbates heart failure | IBET 151, BRD4-specific knockout in cardiomyocyte | Damages mitochondria function | [98], [99], [145] | |
| Pulmonary fibrosis | Alleviates pulmonary fibrosis | BET inhibitors (IBET, JQ1, CG223, ZL0591, ZL0420 and ZL0454, OTX015) |
Decreases profibrotic gene Nox4 expression | [157], [159] |
| IVDD | Alleviates IVDD | JQ1 | Suppresses MAPK and NF-kB signaling pathways | [86] |
| JQ1 | Induces autophagy Prevents NP cell senescence and apoptosis |
[51], [86] |
CREB: cAMP response element binding protein; PSD95: postsynaptic density protein 95; VSMC: vascular smooth muscle cell; IVDD: intervertebral disc degeneration; NP: nucleus pulposus.
BRD4 and Alzheimer’s disease
Alzheimer's disease (AD) is characterized by a progressive neurodegenerative disorder that mostly affects the elderly population, and there are no effective curable drugs available at present [103]. Accumulation of extracellular amyloid-beta (Aβ) plaques and intracellular neurofibrillary tangles in the brain is the primary feature of AD, which leads to synaptic dysfunction and neuronal loss [103]. While the underlying mechanisms of AD are still not fully understood, there is evidence linking BRD4 to AD pathogenesis.
A study utilizing rat models of AD revealed that chronic inhibition of BRD4 improved spatial memory through cAMP response element binding protein (CREB) signaling, and by upregulating expression of postsynaptic density protein 95 (PSD95) and synaptophysin [104]. The protective role of BRD4 inhibition is negated by inhibiting astrocytes, thereby providing new insights into the roles of astrocytes and BRD4 in AD [104]. Moreover, both pharmacological and genetic loss of BRD4 in a murine microglial cell line, BV2, curtailed its phagocytic activity, and reduced the transcription of genes implicated in the pathological mechanism of AD, such as Cd33, Trem2, and Zyx [105]. Furthermore, simultaneous treatment with MS-275, an HDAC inhibitor, and JQ1, enhanced cognitive functions in AD-afflicted rats [106]. Additionally, JQ1 treatment incited a hippocampal gene expression program associated with ion channel activity, transcription, and DNA repair, thereby improving cognitive performance and long-term potentiation in brain function [107]. In conclusion, these findings suggest that BRD4 could serve as a potential therapeutic target for treating neuropathology and cognitive deficits in AD through inhibiting transcriptional expression of AD-related genes.
Conversely, a recent study examined the impacts of BRD4 degradation or inhibition on an AD cell model [108]. The investigation revealed that both BRD4 degradation and inhibition elevated Aβ levels through the amplification of beta-site APP-cleaving enzyme 1 (BACE1) levels, which is responsible for cleaving the amyloid-beta protein precursor (APP), and generating Aβ protein. At the same time, the downregulation of BRD4 increased the levels of AD-related phosphorylated Tau (pTau) protein. Speculatively, BRD4 inhibition may worsen AD-related cognitive dysfunction. Consequently, additional research is necessary to fully comprehend the role of BRD4 and the BET family proteins in AD pathogenesis.
BRD4 and ischemic cerebrovascular diseases
Ischemic cerebrovascular diseases, including stroke, rank among the principal causes of morbidity and mortality globally, particularly in elderly populations [109]. Recent research suggests that BRD4 may have a part to play in the development and progression of these diseases. In 2019, Dr. Candelario-Jalil's team first reported that dBET1 diminishes brain injury in ischemic stroke in aged mice [110]. In this study, they revealed that dBET1 degrades BRD4 in the brain without affecting BRD2 [110]. BRD3 was not detected due to its extremely low abundance [111]. Despite the fact that protein levels of BRD2 and BRD4 remained unchanged after the ischemic injury induced by middle cerebral artery occlusion (MCAO), dBET1 significantly reduced stroke volume and pro-inflammatory mediators (Cxcl1, Cxcl10, Ccl2, Tnf-α, and Mmp9) [110], which are crucial drivers of neurovascular injury [112]. Subsequently, another study published in the same year reported that BRD4 expression significantly increased at 12 h following reperfusion in a mouse MCAO model, and continued to rise over time [113]. While JQ1 decreases BRD4 expression in the MCAO model, it mitigates against the pyrin domain-containing protein 3 (NLRP3) inflammasome-mediated inflammatory response and pyroptosis, inhibits glial activation, and ultimately eased MCAO-induced brain injury. Although siRNA-specific knockdown of BRD4 could mimic the effect of JQ1, attributing all effects to BRD4 remains challenging, since the researchers did not comment on any changes in BRD2 and BRD3 [113]. Another research paper suggested that remote ischemic preconditioning protected against cerebral ischemic injury via the miR-204-5p-mediated BRD4/Pink1/Parkin pathway [114]. Furthermore, Dr. Candelario-Jalil's team recently updated their research, showing that dBET1 ameliorates neurological deficits and brain injury in the occurrence of transient ischemic stroke [115]. In conclusion, evidence implies that loss of BRD4 might be beneficial in treating ischemic cerebrovascular diseases. However, whether BRD4 can serve as a biomarker for early detection of cerebral ischemic injury, or if all the protective effects of BRD4 inhibition can be ascribed solely to BRD4, is yet to be determined.
Roles of BRD4 in hypertension and atherosclerosis
Angiotensin II (Ang II) serves as a significant pro-inflammatory and pro-atherogenic growth factor that activates vascular smooth muscle cells (VSMCs) in the progression of hypertension and atherosclerosis [116], [117], which are two of the most prevalent aging-related cardiovascular diseases (CVDs). In 2017, a report first indicated that BRD4 facilitates Ang II-mediated gene expression (Esm1, Spyr2, etc.) through mediating SE formation. This promotes a transition of the VSMC phenotype from a contractile to a synthetic state. Furthermore, it was noted that JQ1 mitigates Ang II-induced elevated blood pressure, vascular medial hypertrophy, and inflammation response in mice [118]. These findings suggest novel functional roles for BRD4 in Ang II responses and VSMC dysfunction, and are in agreement with previous discoveries that JQ1 is effective in animal models of aortic aneurysm [119]. Subsequently, Yang et al. reported that BRD4 has a higher expression level in patients with essential hypertension, and that JQ1 can reduce the oxidative, inflammatory response and endothelial cell (EC) damage, aortic injury, and blood pressure in spontaneously hypertensive rats [120]. Furthermore, Qiu et al. carried out a clinical study and genotyped single nucleotide polymorphisms in patients with hypertension and normotensive control patients [121]. They discovered that patients with genetic mutations within BRD4 (rs4808278) have increased susceptibility to high pulse pressure. Collectively, these findings imply that BRD4 may regulate blood pressure in a manner dependent on both VSMCs and ECs. Hence, BRD4 inhibition might represent a novel target for lowering blood pressure.
Atherosclerosis is a complex process [122]. Briefly, stimulation factors including hypercholesterolaemia, diabetes mellitus, hypertension, etc. promote circulating monocytes to transmigrate into the subendothelial space and differentiate into macrophages, followed by accumulation of oxidized LDL cholesterol and development of foam cells [123]. At the same time, VSMCs can transdifferentiate into the synthetic phenotype, migrate into the subendothelial layer and proliferate[124], or transdifferentiate into foam cells and accelerate plaque formation [125]. Numerous studies suggest that BRD4 promotes intimal hyperplasia, and that BRD4 deficiency impedes neointima lesion development [126], [127], [128], which is characterized by VSMC proliferation[129], [130], [131], [132]. Similarly, BRD4 inhibition may mitigate diabetic atherosclerosis by curbing the proliferation and migration of VSMCs [133]. Notably, apabetalone counteracts transdifferentiation and calcification of VSMCs, thereby preventing vascular calcification [134]. Beyond regulating VSMC function, BRD4 also has a role in the process of atherosclerosis through its regulation of macrophage function [65], [73], [91], [134], endothelial inflammation [73], [134], [135], [136], [137], and the synthesis of apolipoprotein A-I [138]. It is important to note, however, that BRD4 is widely expressed in all the vascular components. This broad expression makes it challenging to attribute the pharmacological effects of BET inhibitors observed in animal disease models to specific cell types or solely to BRD4.
BRD4 and heart failure
To date, research into the role of BRD4 in regulating cardiac function has yielded mixed results. Some early studies affirmed the therapeutic potential of BRD4 inhibition in heart failure. For instance, it was found that JQ1 could suppress pressure overload-induced cardiac hypertrophy in mice, and that siRNA knockdown of BRD4 in cultured neonatal cardiac myocytes could prevent the hypertrophic response triggered by external growth cues [139], [140]. These findings suggest that BRD4 inhibition could directly benefit hypertrophic cardiomyocytes therapeutically. Moreover, it was reported that administering JQ1 does not result in baseline cardiac dysfunction [139]. Other studies have indicated that inhibiting BET proteins might influence non-myocyte pathways, such as alleviating fibrosis and inflammation, in preclinical heart failure animal models [141], [142], [143], [144].
In contrast, exposure to the IBET-151 was found to cause cardiomyopathy in healthy male mice and rats, likely due to damaging cardiac mitochondria [145]. Subsequently, two studies simultaneously published in Circulation that structured a cardiomyocyte-specific BRD4 knockout mouse model, reported that acute deletion of BRD4 in developing and adult hearts rapidly led to contractile dysfunction and heart failure [98], [99]. RNA sequencing identified that loss of BRD4 transcriptionally represses gene expression related to the mitochondria electron transport chain and collapses mitochondria function [98], [99]. Furthermore, mice with a heterozygous deletion of BRD4 developed delayed heart failure, suggesting that even a reduced level of BRD4 protein is extremely harmful to heart function [98]. Collectively, these studies suggest that BRD4 is crucial for maintaining cardiac mitochondrial function and normal cardiac function.
As highlighted by the last study [98], there are several potential explanations for the stark contrast in results between JQ1 inhibition and genetic knockout of BRD4 in heart failure. First, pharmacological intervention may be not sufficient to induce cardiac functional changes. Changes in mitochondria-related gene transcription, akin to those seen in BRD4-specific knockout mice, was observed in JQ1 treated hearts [98]. Second, JQ1 does not exclusively target BRD4 [47]. Therefore, it is necessary to consider the potential involvement of BRD2 and BRD3 in the observed pharmacological effects in heart. Third, the absence of BRD4 might impact cells differently under varying conditions. For instance, in heart failure models, BRD4 knockout specifically in cardiomyocytes may yield therapeutic effects similar to those of JQ1 pharmacological inhibition. Finally, there could be off-target effects of JQ1, as previously reported [146].
BRD4 and aging-related pulmonary fibrosis
In 2013, Tang et al. first illustrated that BRD2 and BRD4 participate in initiating lung fibrosis induced by bleomycin exposure [147], [148]. IBET and JQ1 were found to impede bleomycin-induced lung fibrosis. This finding was substantiated by the specific knockout of BRD2, BRD3, and BRD4 using respective siRNAs, revealing that inhibition of BRD2 and BRD4 plays a role, whereas BRD3 does not. Subsequent research has demonstrated that BRD4 inhibition can mitigate bleomycin-induced lung fibrosis (JQ1, CG223, ZL0591) [149], [150], [151], radiation-induced lung fibrosis (JQ1) [152], innate inflammation-driven airway remodeling (ZL0420 and ZL0454, highly specific BRD4 inhibitors) [153], and allergen-induced inflammation and remodeling (ZL0454) [154], highlighting the therapeutic effect of BRD4 inhibition in lung fibrosis.
The main target of BRD4 in lung fibrosis is NADPH oxidase 4 (Nox4), an enzyme that generates ROS and is a pro-fibrotic gene involved in lung fibrosis [155], [156]. Research indicates that JQ1 reduces Nox4 gene expression by disrupting its promoter's association with BRD3 and BRD4, but not with BRD2. Reduced Nox4 enhances the activity of NFE2-related factor 2 (Nrf2) [157]. Furthermore, it has been shown that silencing Nox4 itself can trigger Nrf2 activation in lung fibroblasts [158]. Another study demonstrated that BRD4 inhibition (utilizing I-BET-762, JQ1, and OTX015 in vivo, and siRNA-BRD4 in vitro) counters aging-related lung fibrosis by disrupting the connection of BRD4, p300, and acetylated histone H4K16 with the Nox4 promoter, thereby downregulating Nox4 gene expression and its activities [159]. Collectively, these consistent findings suggest that BRD4 inhibition may potentially serve as an effective strategy for treating various types of lung fibrosis, including those related to aging.
BRD4 and intervertebral disc degeneration (IVDD)
IVDD is a prevalent chronic degenerative disease affecting the skeletal muscles, typically associated with increased age. In instances of diabetic IVDD, both BRD4 and matrix metalloproteinase 13 (MMP-13) levels are increased in NP cells [84]. Significantly, BRD4 inhibition has been demonstrated to suppress the MAPK and NF-kB signaling pathways and enhance autophagy, cause a reduction in MMPs and the prevention of diabetic IVDD. A further study confirmed that BRD4 inhibition can provide protection against IVDD by enhancing autophagy and repressing NLRP3 inflammasome activity via inhibiting NF-κB signaling in NP cells [86], or by inhibiting senescence and apoptosis of NP cells through the induction of autophagy [51]. Nevertheless, the limited evidence suggests that BRD4 inhibition might serve as a promising therapeutic method for IVDD but need further validation and exploration.
Conclusions
BRD4, an important epigenetic reader, plays a fundamental role in initiating gene expression and has a functional role in a variety of aging-related diseases. However, the detailed mechanisms through which BRD4 operates are far from being understood. The regulatory role of BRD4 in aging-related diseases is complex and multifaceted. In some instances, BRD4 inhibition has been demonstrated to have protective effects, while in others, it appears to produce detrimental effects. Several factors could explain these inconsistent findings. First, the function of BRD4 may be cell type- and disease-specific, which could account for the conflicting results observed in different cell types and disease models. Second, BRD4 knockout is lethal in embryos, so most in vitro studies rely on pharmacological inhibition, which may not specifically target BRD4. Third, some BET inhibitors, such as JQ1, may have off-target effects. Given the current complicated research results concerning BRD4, further investigation is necessary to completely comprehend the function of BRD4 in the aging process and to ascertain the most effective strategies for targeting it in the treatment of aging-related diseases.
Funding
This review was supported by funds from the Canadian Institutes of Health Research (CIHR, PJT-178010 to X.-L. Zheng, PJT-165941 to X.-L. Zheng) and from the Natural Sciences and Engineering Research Council of Canada (RGPIN-2020-04592 to X.-L. Zheng). Jiaxing Sun was supported by the China Scholarship Council (CSC).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Biographies

Xi-Long Zheng established his lab at the University of Calgary in 2001 after his Ph.D. and postdoctoral training in Calgary and New York (1993 - 2000). Dr. Zheng's research involves the pathogenesis and treatment of cardiovascular diseases, including hypertension, atherosclerosis, and coronary arterial heart disease.

Shenghua Zhou is the Director of Cardiovascular Department and a doctoral supervisor in the Second Xiangya Hospital, Central South University, China. Dr Zhou’s research focuses on the cardiovascular diseases and the pathological mechanisms investigation.

Jiaxing Sun is a M.D from Central South University, China and currently a visiting Ph.D. student (Biochemistry and Molecular Biology) in the University of Calgary, Canada. Her research interest is vascular aging and its regulation by epigenetic mechanisms.

Yu Gui holds a M.D. from Sun Yat-Sen University of Medical Sciences, China and a Ph.D. from University of Calgary, Canada. Her current research work mainly focuses on investigating the regulation of smooth muscle functions (such as contraction and proliferation) by hormones and cell metabolites in health and cardiovascular diseases.
Contributor Information
Shenghua Zhou, Email: zhoushenghua@csu.edu.cn.
Xi-Long Zheng, Email: xlzheng@ucalgary.ca.
References
- 1.Katzir I., Adler M., Karin O., Mendelsohn-Cohen N., Mayo A., Alon U. Senescent cells and the incidence of age-related diseases. Aging Cell. 2021;20:e13314. doi: 10.1111/acel.13314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Magalhães J.P. Ageing as a software design flaw. Genome Biol. 2023;24:51. doi: 10.1186/s13059-023-02888-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dang W., Steffen K.K., Perry R., Dorsey J.A., Johnson F.B., Shilatifard A., et al. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature. 2009;459:802–807. doi: 10.1038/nature08085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feser J., Truong D., Das C., Carson J.J., Kieft J., Harkness T., et al. Elevated histone expression promotes life span extension. Mol. Cell. 2010;39:724–735. doi: 10.1016/j.molcel.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu Z., Chen K., Xia Z., Chavez M., Pal S., Seol J.H., et al. Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes Dev. 2014;28:396–408. doi: 10.1101/gad.233221.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaeberlein M., McVey M., Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ryu H.Y., Rhie B.H., Ahn S.H. Loss of the Set2 histone methyltransferase increases cellular lifespan in yeast cells. Biochem. Biophys. Res. Commun. 2014;446:113–118. doi: 10.1016/j.bbrc.2014.02.061. [DOI] [PubMed] [Google Scholar]
- 8.Yang J.H., Hayano M., Griffin P.T., Amorim J.A., Bonkowski M.S., Apostolides J.K., et al. Loss of epigenetic information as a cause of mammalian aging. Cell. 2023;186:305–326.e27. doi: 10.1016/j.cell.2022.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Francois A., Canella A., Marcho L.M., Stratton M.S. Protein acetylation in cardiac aging. J. Mol. Cell. Cardiol. 2021;157:90–97. doi: 10.1016/j.yjmcc.2021.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh A.K. p300 in Cardiac Development and Accelerated Cardiac Aging. Aging Dis. 2020;11:916–926. doi: 10.14336/AD.2020.0401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maity S., Farrell K., Navabpour S., Narayanan S.N., Jarome T.J. Epigenetic mechanisms in memory and cognitive decline associated with aging and Alzheimer's disease. Int. J. Mol. Sci. 2021;22:12280. doi: 10.3390/ijms222212280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang N., Wu R., Tang D., Kang R. The BET family in immunity and disease. Signal Transduct. Target. Ther. 2021;6:23. doi: 10.1038/s41392-020-00384-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Filippakopoulos P., Picaud S., Mangos M., Keates T., Lambert J.P., Barsyte-Lovejoy D., et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeng L., Zhou M.M. Bromodomain: an acetyl-lysine binding domain. FEBS Lett. 2002;513:124–128. doi: 10.1016/s0014-5793(01)03309-9. [DOI] [PubMed] [Google Scholar]
- 15.Wu S.Y., Chiang C.M. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J. Biol. Chem. 2007;282:13141–13145. doi: 10.1074/jbc.R700001200. [DOI] [PubMed] [Google Scholar]
- 16.Spriano F., Stathis A., Bertoni F. Targeting BET bromodomain proteins in cancer: the example of lymphomas. Pharmacol. Ther. 2020;215 doi: 10.1016/j.pharmthera.2020.107631. [DOI] [PubMed] [Google Scholar]
- 17.Alsarraj J., Faraji F., Geiger T.R., Mattaini K.R., Williams M., Wu J., et al. BRD4 short isoform interacts with RRP1B, SIPA1 and components of the LINC complex at the inner face of the nuclear membrane. PLoS One. 2013;8:e80746. doi: 10.1371/journal.pone.0080746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schröder S., Cho S., Zeng L., Zhang Q., Kaehlcke K., Mak L., et al. Two-pronged binding with bromodomain-containing protein 4 liberates positive transcription elongation factor b from inactive ribonucleoprotein complexes. J. Biol. Chem. 2012;287:1090–1099. doi: 10.1074/jbc.M111.282855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.B.R. Sabari, A. Dall'Agnese, A. Boija, I.A. Klein, E.L. Coffey, K. Shrinivas, B.J. Abraham, N.M. Hannett, A.V. Zamudio, J.C. Manteiga, C.H. Li, Y.E. Guo, D.S. Day, J. Schuijers, E. Vasile, S. Malik, D. Hnisz, T.I. Lee, I.I. Cisse, R.G. Roeder, P.A. Sharp, A.K. Chakraborty, R.A. Young, Coactivator condensation at super-enhancers links phase separation and gene control, Science (New York, NY). 2018;361. [DOI] [PMC free article] [PubMed]
- 20.R.J. Conrad, P. Fozouni, S. Thomas, H. Sy, Q. Zhang, M.M. Zhou, M. Ott, The short isoform of BRD4 promotes HIV-1 latency by engaging repressive SWI/SNF chromatin-remodeling complexes, Molecular Cell 2017;67:1001-12.e6. [DOI] [PMC free article] [PubMed]
- 21.Devaiah B.N., Case-Borden C., Gegonne A., Hsu C.H., Chen Q., Meerzaman D., et al. BRD4 is a histone acetyltransferase that evicts nucleosomes from chromatin. Nat. Struct. Mol. Biol. 2016;23:540–548. doi: 10.1038/nsmb.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishiyama A., Dey A., Miyazaki J., Ozato K. Brd4 is required for recovery from antimicrotubule drug-induced mitotic arrest: preservation of acetylated chromatin. Mol. Biol. Cell. 2006;17:814–823. doi: 10.1091/mbc.E05-08-0729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nicodeme E., Jeffrey K.L., Schaefer U., Beinke S., Dewell S., Chung C.W., et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hernandez-Segura A., Nehme J., Demaria M. Hallmarks of cellular senescence. Trends Cell Biol. 2018;28:436–453. doi: 10.1016/j.tcb.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 25.Chua P., Roeder G.S. Bdf1, a yeast chromosomal protein required for sporulation. Mol. Cell Biol. 1995;15:3685–3696. doi: 10.1128/mcb.15.7.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dey A., Ellenberg J., Farina A., Coleman A.E., Maruyama T., Sciortino S., et al. A bromodomain protein, MCAP, associates with mitotic chromosomes and affects G(2)-to-M transition. Mol. Cell Biol. 2000;20:6537–6549. doi: 10.1128/mcb.20.17.6537-6549.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Houzelstein D., Bullock S.L., Lynch D.E., Grigorieva E.F., Wilson V.A., Beddington R.S. Growth and early postimplantation defects in mice deficient for the bromodomain-containing protein Brd4. Mol. Cell Biol. 2002;22:3794–3802. doi: 10.1128/MCB.22.11.3794-3802.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Penas C., Maloof M.E., Stathias V., Long J., Tan S.K., Mier J., et al. Time series modeling of cell cycle exit identifies Brd4 dependent regulation of cerebellar neurogenesis. Nat. Commun. 2019;10:3028. doi: 10.1038/s41467-019-10799-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang Z., He N., Zhou Q. Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression. Mol. Cell Biol. 2008;28:967–976. doi: 10.1128/MCB.01020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mochizuki K., Nishiyama A., Jang M.K., Dey A., Ghosh A., Tamura T., et al. The bromodomain protein Brd4 stimulates G1 gene transcription and promotes progression to S phase. J. Biol. Chem. 2008;283:9040–9048. doi: 10.1074/jbc.M707603200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dey A., Nishiyama A., Karpova T., McNally J., Ozato K. Brd4 marks select genes on mitotic chromatin and directs postmitotic transcription. Mol. Biol. Cell. 2009;20:4899–4909. doi: 10.1091/mbc.E09-05-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maruyama T., Farina A., Dey A., Cheong J., Bermudez V.P., Tamura T., et al. A Mammalian bromodomain protein, brd4, interacts with replication factor C and inhibits progression to S phase. Mol. Cell Biol. 2002;22:6509–6520. doi: 10.1128/MCB.22.18.6509-6520.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang P., Li R., Xiao H., Liu W., Zeng X., Xie G., et al. BRD4 inhibitor AZD5153 suppresses the proliferation of colorectal cancer cells and sensitizes the anticancer effect of PARP inhibitor. Int. J. Biol. Sci. 2019;15:1942–1954. doi: 10.7150/ijbs.34162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu X., Liu D., Gao X., Xie F., Tao D., Xiao X., et al. Inhibition of BRD4 suppresses cell proliferation and induces apoptosis in renal cell carcinoma. Cell. Physiol. Biochem. 2017;41:1947–1956. doi: 10.1159/000472407. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Y., Duan S., Jang A., Mao L., Liu X., Huang G. JQ1, a selective inhibitor of BRD4, suppresses retinoblastoma cell growth by inducing cell cycle arrest and apoptosis. Exp. Eye Res. 2021;202 doi: 10.1016/j.exer.2020.108304. [DOI] [PubMed] [Google Scholar]
- 36.Ba M., Long H., Yan Z., Wang S., Wu Y., Tu Y., et al. BRD4 promotes gastric cancer progression through the transcriptional and epigenetic regulation of c-MYC. J. Cell. Biochem. 2018;119:973–982. doi: 10.1002/jcb.26264. [DOI] [PubMed] [Google Scholar]
- 37.Hao J., Yang Z., Wang L., Zhang Y., Shu Y., Jiang L., et al. Downregulation of BRD4 inhibits gallbladder cancer proliferation and metastasis and induces apoptosis via PI3K/AKT pathway. Int. J. Oncol. 2017;51:823–831. doi: 10.3892/ijo.2017.4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu S., Li F., Pan L., Yang Z., Shu Y., Lv W., et al. BRD4 inhibitor and histone deacetylase inhibitor synergistically inhibit the proliferation of gallbladder cancer in vitro and in vivo. Cancer Sci. 2019;110:2493–2506. doi: 10.1111/cas.14102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li L., Meng Y., Wu X., Li J., Sun Y. Bromodomain-containing protein 4 inhibitor JQ1 promotes melanoma cell apoptosis by regulating mitochondrial dynamics. Cancer Sci. 2021;112:4013–4025. doi: 10.1111/cas.15061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi L., Xiong Y., Hu X., Wang Z., Xie C. BRD4 inhibition promotes TRAIL-induced apoptosis by suppressing the transcriptional activity of NF-κB in NSCLC. Int. J. Med. Sci. 2021;18:3090–3096. doi: 10.7150/ijms.60776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pericole F.V., Lazarini M., de Paiva L.B., Duarte A., Vieira Ferro K.P., Niemann F.S., et al. BRD4 inhibition enhances azacitidine efficacy in acute myeloid leukemia and myelodysplastic syndromes. Front. Oncol. 2019;9:16. doi: 10.3389/fonc.2019.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu Q., Ding X., Huang T., Zhang S., Li Y., Xu L., et al. BRD4 degrader ARV-825 produces long-lasting loss of BRD4 protein and exhibits potent efficacy against cholangiocarcinoma cells. Am. J. Transl. Res. 2019;11:5728–5739. [PMC free article] [PubMed] [Google Scholar]
- 43.Duan Y., Guan Y., Qin W., Zhai X., Yu B., Liu H. Targeting Brd4 for cancer therapy: inhibitors and degraders. MedChemComm. 2018;9:1779–1802. doi: 10.1039/c8md00198g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.White M.E., Fenger J.M., Carson W.E., 3rd. Emerging roles of and therapeutic strategies targeting BRD4 in cancer. Cell. Immunol. 2019;337:48–53. doi: 10.1016/j.cellimm.2019.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi J., Vakoc C.R. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell. 2014;54:728–736. doi: 10.1016/j.molcel.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.French C.A. Pathogenesis of NUT midline carcinoma. Annu. Rev. Pathol. 2012;7:247–265. doi: 10.1146/annurev-pathol-011811-132438. [DOI] [PubMed] [Google Scholar]
- 47.Filippakopoulos P., Qi J., Picaud S., Shen Y., Smith W.B., Fedorov O., et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lam F.C., Kong Y.W., Huang Q., Vu Han T.L., Maffa A.D., Kasper E.M., et al. BRD4 prevents the accumulation of R-loops and protects against transcription-replication collision events and DNA damage. Nat. Commun. 2020;11:4083. doi: 10.1038/s41467-020-17503-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu H., Wang L., Weng X., Chen H., Du Y., Diao C., et al. Inhibition of Brd4 alleviates renal ischemia/reperfusion injury-induced apoptosis and endoplasmic reticulum stress by blocking FoxO4-mediated oxidative stress. Redox Biol. 2019;24 doi: 10.1016/j.redox.2019.101195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu X., Wang J., Luo H., Xu C., Chen X., Zhang R. MiR-218 Inhibits CSE-Induced Apoptosis and Inflammation in BEAS-2B by Targeting BRD4. Int. J. Chron. Obstruct. Pulmon. Dis. 2020;15:3407–3416. doi: 10.2147/COPD.S278553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang G.Z., Chen H.W., Deng Y.J., Liu M.Q., Wu Z.L., Ma Z.J., et al. BRD4 inhibition suppresses senescence and apoptosis of nucleus pulposus cells by inducing autophagy during intervertebral disc degeneration: an in vitro and in vivo study. Oxid. Med. Cell. Longev. 2022;2022:9181412. doi: 10.1155/2022/9181412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kida Y., Goligorsky M.S. Sirtuins, cell senescence, and vascular aging. Can. J. Cardiol. 2016;32:634–641. doi: 10.1016/j.cjca.2015.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McHugh D., Gil J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018;217:65–77. doi: 10.1083/jcb.201708092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu R.M. Aging, cellular senescence, and Alzheimer's disease. Int. J. Mol. Sci. 2022;23:1989. doi: 10.3390/ijms23041989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang S., Pike A.M., Lee S.S., Strong M.A., Connelly C.J., Greider C.W. BRD4 inhibitors block telomere elongation. Nucleic Acids Res. 2017;45:8403–8410. doi: 10.1093/nar/gkx561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.d'Adda di Fagagna F., Reaper P.M., Clay-Farrace L., Fiegler H., Carr P., Von Zglinicki T., et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 57.Xu J.L., Yuan Y.J., Lv J., Qi D., Wu M.D., Lan J., et al. Inhibition of BRD4 triggers cellular senescence through suppressing aurora kinases in oesophageal cancer cells. J. Cell Mol. Med. 2020;24:13036–13045. doi: 10.1111/jcmm.15901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dong X., Hu X., Chen J., Hu D., Chen L.F. BRD4 regulates cellular senescence in gastric cancer cells via E2F/miR-106b/p21 axis. Cell Death Dis. 2018;9:203. doi: 10.1038/s41419-017-0181-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Webber L.P., Yujra V.Q., Vargas P.A., Martins M.D., Squarize C.H., Castilho R.M. Interference with the bromodomain epigenome readers drives p21 expression and tumor senescence. Cancer Lett. 2019;461:10–20. doi: 10.1016/j.canlet.2019.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abbas T., Dutta A. p21 in cancer: intricate networks and multiple activities. Nat. Rev. Cancer. 2009;9:400–414. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Macleod K.F., Sherry N., Hannon G., Beach D., Tokino T., Kinzler K., et al. p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes Dev. 1995;9:935–944. doi: 10.1101/gad.9.8.935. [DOI] [PubMed] [Google Scholar]
- 62.Young A.R., Narita M. SASP reflects senescence. EMBO Rep. 2009;10:228–230. doi: 10.1038/embor.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yin Y., Chen H., Wang Y., Zhang L., Wang X. Roles of extracellular vesicles in the aging microenvironment and age-related diseases. J. Extracellular Vesicles. 2021;10:e12154. doi: 10.1002/jev2.12154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tasdemir N., Banito A., Roe J.S., Alonso-Curbelo D., Camiolo M., Tschaharganeh D.F., et al. BRD4 connects enhancer remodeling to senescence immune surveillance. Cancer Discov. 2016;6:612–629. doi: 10.1158/2159-8290.CD-16-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang H., Fu H., Zhu R., Wu X., Ji X., Li X., et al. BRD4 contributes to LPS-induced macrophage senescence and promotes progression of atherosclerosis-associated lipid uptake. Aging. 2020;12:9240–9259. doi: 10.18632/aging.103200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Franceschi C., Bonafè M., Valensin S., Olivieri F., De Luca M., Ottaviani E., et al. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000;908:244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- 67.K. Meyer, T. Patra, Vijayamahantesh, R. Ray, SARS-CoV-2 spike protein induces paracrine senescence and leukocyte adhesion in endothelial cells, J. Virol. 2021;95:e0079421. [DOI] [PMC free article] [PubMed]
- 68.Thompson P.J., Shah A., Apostolopolou H., Bhushan A. BET proteins are required for transcriptional activation of the senescent islet cell secretome in type 1 diabetes. Int. J. Mol. Sci. 2019:20. doi: 10.3390/ijms20194776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhu Y., Tchkonia T., Pirtskhalava T., Gower A.C., Ding H., Giorgadze N., et al. The Achilles' heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015;14:644–658. doi: 10.1111/acel.12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wakita M., Takahashi A., Sano O., Loo T.M., Imai Y., Narukawa M., et al. A BET family protein degrader provokes senolysis by targeting NHEJ and autophagy in senescent cells. Nat. Commun. 2020;11:1935. doi: 10.1038/s41467-020-15719-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reyes N.S., Krasilnikov M., Allen N.C., Lee J.Y., Hyams B., Zhou M., et al. Sentinel p16(INK4a+) cells in the basement membrane form a reparative niche in the lung. Science (New York, N.Y.) 2022;378:192–201. doi: 10.1126/science.abf3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lovén J., Hoke H.A., Lin C.Y., Lau A., Orlando D.A., Vakoc C.R., et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brown J.D., Lin C.Y., Duan Q., Griffin G., Federation A., Paranal R.M., et al. NF-κB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol. Cell. 2014;56:219–231. doi: 10.1016/j.molcel.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Whyte W.A., Orlando D.A., Hnisz D., Abraham B.J., Lin C.Y., Kagey M.H., et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-André V., Sigova A.A., et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mizushima N., Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 77.Rubinsztein D.C., Mariño G., Kroemer G. Autophagy and aging. Cell. 2011;146:682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 78.Wong S.Q., Kumar A.V., Mills J., Lapierre L.R. Autophagy in aging and longevity. Hum. Genet. 2020;139:277–290. doi: 10.1007/s00439-019-02031-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hu L.F. Epigenetic regulation of autophagy. Adv. Exp. Med. Biol. 2019;1206:221–236. doi: 10.1007/978-981-15-0602-4_11. [DOI] [PubMed] [Google Scholar]
- 80.Jang J.E., Eom J.I., Jeung H.K., Cheong J.W., Lee J.Y., Kim J.S., et al. AMPK-ULK1-mediated autophagy AMPK–ULK1-mediated autophagy confers resistance to BET inhibitor JQ1 in acute myeloid leukemia stem cells. Clin. Cancer Res. 2017;23:2781–2794. doi: 10.1158/1078-0432.CCR-16-1903. [DOI] [PubMed] [Google Scholar]
- 81.Sakamaki J.I., Ryan K.M. Transcriptional regulation of autophagy and lysosomal function by bromodomain protein BRD4. Autophagy. 2017;13:2006–2007. doi: 10.1080/15548627.2017.1364822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.J.I. Sakamaki, S. Wilkinson, M. Hahn, N. Tasdemir, J. O'Prey, W. Clark, A. Hedley, C. Nixon, J.S. Long, M. New, T. Van Acker, S.A. Tooze, S.W. Lowe, I. Dikic, K.M. Ryan, Bromodomain protein BRD4 is a transcriptional repressor of autophagy and lysosomal function. Mol. Cell. 2017;66:517-32.e9. [DOI] [PMC free article] [PubMed]
- 83.Wen X., Klionsky D.J. BRD4 is a newly characterized transcriptional regulator that represses autophagy and lysosomal function. Autophagy. 2017;13:1801–1803. doi: 10.1080/15548627.2017.1364334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang J., Hu J., Chen X., Huang C., Lin J., Shao Z., et al. BRD4 inhibition regulates MAPK, NF-κB signals, and autophagy to suppress MMP-13 expression in diabetic intervertebral disc degeneration. FASEB J. 2019;33:11555–11566. doi: 10.1096/fj.201900703R. [DOI] [PubMed] [Google Scholar]
- 85.Li Y., Xiang J., Zhang J., Lin J., Wu Y., Wang X. Inhibition of Brd4 by JQ1 promotes functional recovery from spinal cord injury by activating autophagy. Front. Cell. Neurosci. 2020;14 doi: 10.3389/fncel.2020.555591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hong J., Li S., Markova D.Z., Liang A., Kepler C.K., Huang Y., et al. Bromodomain-containing protein 4 inhibition alleviates matrix degradation by enhancing autophagy and suppressing NLRP3 inflammasome activity in NP cells. J. Cell. Physiol. 2020;235:5736–5749. doi: 10.1002/jcp.29508. [DOI] [PubMed] [Google Scholar]
- 87.Gong Z.G., Zhao Y., Wang Z.Y., Fan R.F., Liu Z.P., Wang L. Epigenetic regulator BRD4 is involved in cadmium-induced acute kidney injury via contributing to lysosomal dysfunction, autophagy blockade and oxidative stress. J. Hazard. Mater. 2022;423 doi: 10.1016/j.jhazmat.2021.127110. [DOI] [PubMed] [Google Scholar]
- 88.Shen S., Li B., Dai J., Wu Z., He Y., Wen L., et al. BRD4 inhibition protects against acute pancreatitis through restoring impaired autophagic flux. Front. Pharmacol. 2020;11:618. doi: 10.3389/fphar.2020.00618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mu J., Zhang D., Tian Y., Xie Z., Zou M.H. BRD4 inhibition by JQ1 prevents high-fat diet-induced diabetic cardiomyopathy by activating PINK1/Parkin-mediated mitophagy in vivo. J. Mol. Cell. Cardiol. 2020;149:1–14. doi: 10.1016/j.yjmcc.2020.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ashrafi G., Schwarz T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013;20:31–42. doi: 10.1038/cdd.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li X., Zhu R., Jiang H., Yin Q., Gu J., Chen J., et al. Autophagy enhanced by curcumin ameliorates inflammation in atherogenesis via the TFEB-P300-BRD4 axis. Acta Pharm. Sin. B. 2022;12:2280–2299. doi: 10.1016/j.apsb.2021.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huang M., Garcia J.S., Thomas D., Zhu L., Nguyen L.X., Chan S.M., et al. Autophagy mediates proteolysis of NPM1 and HEXIM1 and sensitivity to BET inhibition in AML cells. Oncotarget. 2016;7:74917–74930. doi: 10.18632/oncotarget.12493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Miwa S., Kashyap S., Chini E., von Zglinicki T. Mitochondrial dysfunction in cell senescence and aging. J. Clin. Invest. 2022;132 doi: 10.1172/JCI158447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Barrow JJ, Balsa E, Verdeguer F, Tavares CD, Soustek MS, Hollingsworth LRt, et al. Bromodomain inhibitors correct bioenergetic deficiency caused by mitochondrial disease complex I mutations. Mol. Cell. 2016;64:163-75. [DOI] [PMC free article] [PubMed]
- 95.Yoon Y.S., Lee J.H., Hwang S.C., Choi K.S., Yoon G. TGF beta1 induces prolonged mitochondrial ROS generation through decreased complex IV activity with senescent arrest in Mv1Lu cells. Oncogene. 2005;24:1895–1903. doi: 10.1038/sj.onc.1208262. [DOI] [PubMed] [Google Scholar]
- 96.Yoon G., Kim H.J., Yoon Y.S., Cho H., Lim I.K., Lee J.H. Iron chelation-induced senescence-like growth arrest in hepatocyte cell lines: association of transforming growth factor beta1 (TGF-beta1)-mediated p27Kip1 expression. Biochem. J. 2002;366:613–621. doi: 10.1042/BJ20011445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gelato K.A., Schöckel L., Klingbeil O., Rückert T., Lesche R., Toedling J., et al. Super-enhancers define a proliferative PGC-1α-expressing melanoma subgroup sensitive to BET inhibition. Oncogene. 2018;37:512–521. doi: 10.1038/onc.2017.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kim S.Y., Zhang X., Schiattarella G.G., Altamirano F., Ramos T.A.R., French K.M., et al. Epigenetic reader BRD4 (bromodomain-containing protein 4) governs nucleus-encoded mitochondrial transcriptome to regulate cardiac function. Circulation. 2020;142:2356–2370. doi: 10.1161/CIRCULATIONAHA.120.047239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Padmanabhan A., Alexanian M., Linares-Saldana R., González-Terán B., Andreoletti G., Huang Y., et al. BRD4 (bromodomain-containing protein 4) interacts with GATA4 (GATA binding protein 4) to govern mitochondrial homeostasis in adult cardiomyocytes. Circulation. 2020;142:2338–2355. doi: 10.1161/CIRCULATIONAHA.120.047753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Civenni G, Bosotti R, Timpanaro A, Vàzquez R, Merulla J, Pandit S, Rossi S, et al. Epigenetic control of mitochondrial fission enables self-renewal of stem-like tumor cells in human prostate cancer. Cell Metabol. 2019;30:303–18.e6. [DOI] [PubMed]
- 101.Yoon Y.S., Yoon D.S., Lim I.K., Yoon S.H., Chung H.Y., Rojo M., et al. Formation of elongated giant mitochondria in DFO-induced cellular senescence: involvement of enhanced fusion process through modulation of Fis1. J. Cell. Physiol. 2006;209:468–480. doi: 10.1002/jcp.20753. [DOI] [PubMed] [Google Scholar]
- 102.Lee S., Jeong S.Y., Lim W.C., Kim S., Park Y.Y., Sun X., et al. Mitochondrial fission and fusion mediators, hFis1 and OPA1, modulate cellular senescence. J. Biol. Chem. 2007;282:22977–22983. doi: 10.1074/jbc.M700679200. [DOI] [PubMed] [Google Scholar]
- 103.Congdon E.E., Ji C., Tetlow A.M., Jiang Y., Sigurdsson E.M. Tau-targeting therapies for Alzheimer disease: current status and future directions. Nat. Rev. Neurol. 2023 doi: 10.1038/s41582-023-00883-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nikkar R., Esmaeili-Bandboni A., Badrikoohi M., Babaei P. Effects of inhibiting astrocytes and BET/BRD4 chromatin reader on spatial memory and synaptic proteins in rats with Alzheimer's disease. Metab. Brain Dis. 2022;37:1119–1131. doi: 10.1007/s11011-022-00940-7. [DOI] [PubMed] [Google Scholar]
- 105.Matuszewska M., Cieślik M., Wilkaniec A., Strawski M., Czapski G.A. The role of bromodomain and extraterminal (BET) proteins in controlling the phagocytic activity of microglia in vitro: relevance to Alzheimer's disease. Int. J. Mol. Sci. 2022;24:13. doi: 10.3390/ijms24010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Badrikoohi M., Esmaeili-Bandboni A., Babaei P. Simultaneous administration of bromodomain and histone deacetylase I inhibitors alleviates cognition deficit in Alzheimer's model of rats. Brain Res. Bull. 2022;179:49–56. doi: 10.1016/j.brainresbull.2021.12.004. [DOI] [PubMed] [Google Scholar]
- 107.Benito E., Ramachandran B., Schroeder H., Schmidt G., Urbanke H., Burkhardt S., et al. The BET/BRD inhibitor JQ1 improves brain plasticity in WT and APP mice. Transl. Psychiatry. 2017;7:e1239. doi: 10.1038/tp.2017.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang S., Bai P., Lei D., Liang Y., Zhen S., Bakiasi G., et al. Degradation and inhibition of epigenetic regulatory protein BRD4 exacerbate Alzheimer's disease-related neuropathology in cell models. J. Biol. Chem. 2022;298 doi: 10.1016/j.jbc.2022.101794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Benjamin E.J., Virani S.S., Callaway C.W., Chamberlain A.M., Chang A.R., Cheng S., et al. Heart disease and stroke statistics-2018 update: a report from the American heart association. Circulation. 2018;137:e67–e492. doi: 10.1161/CIR.0000000000000558. [DOI] [PubMed] [Google Scholar]
- 110.DeMars K.M., Yang C., Candelario-Jalil E. Neuroprotective effects of targeting BET proteins for degradation with dBET1 in aged mice subjected to ischemic stroke. Neurochem. Int. 2019;127:94–102. doi: 10.1016/j.neuint.2019.03.004. [DOI] [PubMed] [Google Scholar]
- 111.Shang E., Salazar G., Crowley T.E., Wang X., Lopez R.A., Wang X., et al. Identification of unique, differentiation stage-specific patterns of expression of the bromodomain-containing genes Brd2, Brd3, Brd4, and Brdt in the mouse testis. Gene expression patterns : GEP. 2004;4:513–519. doi: 10.1016/j.modgep.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 112.Yang C., Hawkins K.E., Doré S., Candelario-Jalil E. Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. Am. J. Physiol. Cell Physiol. 2019;316:C135–C153. doi: 10.1152/ajpcell.00136.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhou Y., Gu Y., Liu J. BRD4 suppression alleviates cerebral ischemia-induced brain injury by blocking glial activation via the inhibition of inflammatory response and pyroptosis. Biochem. Biophys. Res. Commun. 2019;519:481–488. doi: 10.1016/j.bbrc.2019.07.097. [DOI] [PubMed] [Google Scholar]
- 114.Jiao Y., Wang J., Jia Y., Xue M. Remote ischemic preconditioning protects against cerebral ischemia injury in rats by upregulating miR-204-5p and activating the PINK1/Parkin signaling pathway. Metab. Brain Dis. 2022;37:945–959. doi: 10.1007/s11011-022-00910-z. [DOI] [PubMed] [Google Scholar]
- 115.Liu L., Yang C., Lavayen B.P., Tishko R.J., Larochelle J., Candelario-Jalil E. Targeted BRD4 protein degradation by dBET1 ameliorates acute ischemic brain injury and improves functional outcomes associated with reduced neuroinflammation and oxidative stress and preservation of blood-brain barrier integrity. J. Neuroinflammation. 2022;19:168. doi: 10.1186/s12974-022-02533-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Brasier A.R., Recinos A., 3rd, Eledrisi M.S. Vascular inflammation and the renin-angiotensin system. Arterioscler. Thromb. Vasc. Biol. 2002;22:1257–1266. doi: 10.1161/01.atv.0000021412.56621.a2. [DOI] [PubMed] [Google Scholar]
- 117.Mehta P.K., Griendling K.K. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007;292:C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 118.Das S., Senapati P., Chen Z., Reddy M.A., Ganguly R., Lanting L., et al. Regulation of angiotensin II actions by enhancers and super-enhancers in vascular smooth muscle cells. Nat. Commun. 2017;8:1467. doi: 10.1038/s41467-017-01629-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Duan Q., Mao X., Liao C., Zhou H., Sun Z., Deng X., et al. Inhibition of BET bromodomain attenuates angiotensin II induced abdominal aortic aneurysm in ApoE(-/-) mice. Int. J. Cardiol. 2016;223:428–432. doi: 10.1016/j.ijcard.2016.08.238. [DOI] [PubMed] [Google Scholar]
- 120.Yang Y.M., Shi R.H., Xu C.X., Li L. BRD4 expression in patients with essential hypertension and its effect on blood pressure in spontaneously hypertensive rats. J. Am. Soci. Hypertension: JASH. 2018;12:e107–e117. doi: 10.1016/j.jash.2018.11.004. [DOI] [PubMed] [Google Scholar]
- 121.Qiu J.J., Yang R.Z., Tang Y.J., Lin Y.Y., Xu H.J., Zhang N., et al. BRD4 and PIN1 gene polymorphisms are associated with high pulse pressure risk in a southeastern Chinese population. BMC Cardiovasc. Disord. 2020;20:475. doi: 10.1186/s12872-020-01757-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tamargo I.A., Baek K.I., Kim Y., Park C., Jo H. Flow-induced reprogramming of endothelial cells in atherosclerosis. Nat. Rev. Cardiol. 2023:1–16. doi: 10.1038/s41569-023-00883-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Davignon J., Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109:Iii27 -32. doi: 10.1161/01.CIR.0000131515.03336.f8. [DOI] [PubMed] [Google Scholar]
- 124.Bennett M.R., Sinha S., Owens G.K. Vascular smooth muscle cells in atherosclerosis. Circ. Res. 2016;118:692–702. doi: 10.1161/CIRCRESAHA.115.306361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wang Y., Dubland J.A., Allahverdian S., Asonye E., Sahin B., Jaw J.E., et al. Smooth muscle cells contribute the majority of foam cells in ApoE (apolipoprotein E)-deficient mouse atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2019;39:876–887. doi: 10.1161/ATVBAHA.119.312434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang B., Zhang M., Takayama T., Shi X., Roenneburg D.A., Kent K.C., et al. BET bromodomain blockade mitigates intimal hyperplasia in rat carotid arteries. EBioMedicine. 2015;2:1650–1661. doi: 10.1016/j.ebiom.2015.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dutzmann J., Haertlé M., Daniel J.M., Kloss F., Musmann R.J., Kalies K., et al. BET bromodomain-containing epigenetic reader proteins regulate vascular smooth muscle cell proliferation and neointima formation. Cardiovasc. Res. 2021;117:850–862. doi: 10.1093/cvr/cvaa121. [DOI] [PubMed] [Google Scholar]
- 128.Zhang M., Wang B., Urabe G., Huang Y., Plutzky J., Kent K.C., et al. The BD2 domain of BRD4 is a determinant in EndoMT and vein graft neointima formation. Cell. Signal. 2019;61:20–29. doi: 10.1016/j.cellsig.2019.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Matturri L., Cazzullo A., Turconi P., Lavezzi A.M. Cytogenetic aspects of cell proliferation in atherosclerotic plaques. Cardiologia (Rome, Italy) 1997;42:833–836. [PubMed] [Google Scholar]
- 130.Pan X., Wang B., Yuan T., Zhang M., Kent K.C., Guo L.W. Analysis of combined transcriptomes identifies gene modules that differentially respond to pathogenic stimulation of vascular smooth muscle and endothelial cells. Sci. Rep. 2018;8:395. doi: 10.1038/s41598-017-18675-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tennant M., Dilley R.J., McGeachie J.K., Prendergast F.J. Histogenesis of arterial intimal hyperplasia and atherosclerosis. Aust. N. Z. J. Surg. 1990;60:79–85. [PubMed] [Google Scholar]
- 132.Khurana R., Zhuang Z., Bhardwaj S., Murakami M., De Muinck E., Yla-Herttuala S., et al. Angiogenesis-dependent and independent phases of intimal hyperplasia. Circulation. 2004;110:2436–2443. doi: 10.1161/01.CIR.0000145138.25577.F1. [DOI] [PubMed] [Google Scholar]
- 133.Wu Y., Zhang M., Xu C., Chai D., Peng F., Lin J. Anti-diabetic atherosclerosis by inhibiting high glucose-induced vascular smooth muscle cell proliferation via Pin1/BRD4 pathway. Oxid. Med. Cell. Longev. 2020;2020:4196482. doi: 10.1155/2020/4196482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gilham D., Tsujikawa L.M., Sarsons C.D., Halliday C., Wasiak S., Stotz S.C., et al. Apabetalone downregulates factors and pathways associated with vascular calcification. Atherosclerosis. 2019;280:75–84. doi: 10.1016/j.atherosclerosis.2018.11.002. [DOI] [PubMed] [Google Scholar]
- 135.Shahid S, Pantakani M, Binder L, Fischer A, Pantakani K and Asif AR. Small Molecule BRD4 Inhibitors Apabetalone and JQ1 Rescues Endothelial Cells Dysfunction, Protects Monolayer Integrity and Reduces Midkine Expression. Molecules (Basel, Switzerland). 2022;27. [DOI] [PMC free article] [PubMed]
- 136.Jarausch J., Neuenroth L., Andag R., Leha A., Fischer A., Asif A.R., et al. Influence of shear stress, inflammation and BRD4 inhibition on human endothelial cells: a holistic proteomic approach. Cells. 2022;11:3086. doi: 10.3390/cells11193086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wasiak S., Tsujikawa L.M., Daze E., Gilham D., Stotz S.C., Rakai B.D., et al. Epigenetic BET reader inhibitor apabetalone (RVX-208) counters proinflammatory aortic gene expression in a diet induced obesity mouse model and in human endothelial cells. Atherosclerosis. 2022;364:10–19. doi: 10.1016/j.atherosclerosis.2022.11.015. [DOI] [PubMed] [Google Scholar]
- 138.McLure K.G., Gesner E.M., Tsujikawa L., Kharenko O.A., Attwell S., Campeau E., et al. RVX-208, an inducer of ApoA-I in humans, is a BET bromodomain antagonist. PLoS One. 2013;8:e83190. doi: 10.1371/journal.pone.0083190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Anand P., Brown J.D., Lin C.Y., Qi J., Zhang R., Artero P.C., et al. BET bromodomains mediate transcriptional pause release in heart failure. Cell. 2013;154:569–582. doi: 10.1016/j.cell.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Spiltoir J.I., Stratton M.S., Cavasin M.A., Demos-Davies K., Reid B.G., Qi J., et al. BET acetyl-lysine binding proteins control pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2013;63:175–179. doi: 10.1016/j.yjmcc.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Duan Q., McMahon S., Anand P., Shah H., Thomas S., Salunga H.T., et al. BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci. Transl. Med. 2017;9:eaah5084. doi: 10.1126/scitranslmed.aah5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Antolic A., Wakimoto H., Jiao Z., Gorham J.M., DePalma S.R., Lemieux M.E., et al. BET bromodomain proteins regulate transcriptional reprogramming in genetic dilated cardiomyopathy. JCI insight. 2020;5:e138687. doi: 10.1172/jci.insight.138687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Stratton M.S., Bagchi R.A., Felisbino M.B., Hirsch R.A., Smith H.E., Riching A.S., et al. Dynamic chromatin targeting of BRD4 stimulates cardiac fibroblast activation. Circ. Res. 2019;125:662–677. doi: 10.1161/CIRCRESAHA.119.315125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Song S., Liu L., Yu Y., Zhang R., Li Y., Cao W., et al. Inhibition of BRD4 attenuates transverse aortic constriction- and TGF-β-induced endothelial-mesenchymal transition and cardiac fibrosis. J. Mol. Cell. Cardiol. 2019;127:83–96. doi: 10.1016/j.yjmcc.2018.12.002. [DOI] [PubMed] [Google Scholar]
- 145.Piquereau J., Boet A., Péchoux C., Antigny F., Lambert M., Gressette M., et al. The BET bromodomain inhibitor I-BET-151 induces structural and functional alterations of the heart mitochondria in healthy male mice and rats. Int. J. Mol. Sci. 2019;20:1527. doi: 10.3390/ijms20071527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wang L., Xu M., Kao C.Y., Tsai S.Y., Tsai M.J. Small molecule JQ1 promotes prostate cancer invasion via BET-independent inactivation of FOXA1. J. Clin. Invest. 2020;130:1782–1792. doi: 10.1172/JCI126327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tang X., Peng R., Ren Y., Apparsundaram S., Deguzman J., Bauer C.M., et al. BET bromodomain proteins mediate downstream signaling events following growth factor stimulation in human lung fibroblasts and are involved in bleomycin-induced pulmonary fibrosis. Mol. Pharmacol. 2013;83:283–293. doi: 10.1124/mol.112.081661. [DOI] [PubMed] [Google Scholar]
- 148.Tang X., Peng R., Phillips J.E., Deguzman J., Ren Y., Apparsundaram S., et al. Assessment of Brd4 inhibition in idiopathic pulmonary fibrosis lung fibroblasts and in vivo models of lung fibrosis. Am. J. Pathol. 2013;183:470–479. doi: 10.1016/j.ajpath.2013.04.020. [DOI] [PubMed] [Google Scholar]
- 149.Tian B., Zhao Y., Sun H., Zhang Y., Yang J., Brasier A.R. BRD4 mediates NF-κB-dependent epithelial-mesenchymal transition and pulmonary fibrosis via transcriptional elongation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016;311:L1183–L1201. doi: 10.1152/ajplung.00224.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kaneshita S., Kida T., Yoshioka M., Nishioka K., Raje M., Sakashita A., et al. CG223, a novel BET inhibitor, exerts TGF-β1-mediated antifibrotic effects in a murine model of bleomycin-induced pulmonary fibrosis. Pulm. Pharmacol. Ther. 2021;70 doi: 10.1016/j.pupt.2021.102057. [DOI] [PubMed] [Google Scholar]
- 151.Bernau K., Skibba M., Leet J.P., Furey S., Gehl C., Li Y., et al. Selective inhibition of bromodomain-containing protein 4 reduces myofibroblast transdifferentiation and pulmonary fibrosis. Front. Mol. Med. 2022;2 doi: 10.3389/fmmed.2022.842558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wang J., Zhou F., Li Z., Mei H., Wang Y., Ma H., et al. Pharmacological targeting of BET proteins attenuates radiation-induced lung fibrosis. Sci. Rep. 2018;8:998. doi: 10.1038/s41598-018-19343-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tian B., Liu Z., Litvinov J., Maroto R., Jamaluddin M., Rytting E., et al. Efficacy of novel highly specific bromodomain-containing protein 4 inhibitors in innate inflammation-driven airway remodeling. Am. J. Respir. Cell Mol. Biol. 2019;60:68–83. doi: 10.1165/rcmb.2017-0445OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tian B, Hosoki K, Liu Z, Yang J, Zhao Y, Sun H, et al.. Mucosal bromodomain-containing protein 4 mediates aeroallergen-induced inflammation and remodeling. J. Allergy Clin. Immunol. 2019;143:1380-94.e9. [DOI] [PMC free article] [PubMed]
- 155.Hecker L., Vittal R., Jones T., Jagirdar R., Luckhardt T.R., Horowitz J.C., et al. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009;15:1077–1081. doi: 10.1038/nm.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hecker L., Logsdon N.J., Kurundkar D., Kurundkar A., Bernard K., Hock T., et al. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med. 2014;6:231ra47. doi: 10.1126/scitranslmed.3008182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Stock C.J.W., Michaeloudes C., Leoni P., Durham A.L., Mumby S., Wells A.U., et al. Bromodomain and extraterminal (BET) protein inhibition restores redox balance and inhibits myofibroblast activation. Biomed Res. Int. 2019;2019:1484736. doi: 10.1155/2019/1484736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bernard K., Logsdon N.J., Miguel V., Benavides G.A., Zhang J., Carter A.B., et al. NADPH oxidase 4 (Nox4) suppresses mitochondrial biogenesis and bioenergetics in lung fibroblasts via a nuclear factor erythroid-derived 2-like 2 (Nrf2)-dependent pathway. J. Biol. Chem. 2017;292:3029–3038. doi: 10.1074/jbc.M116.752261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Sanders Y.Y., Lyv X., Zhou Q.J., Xiang Z., Stanford D., Bodduluri S., et al. Brd4-p300 inhibition downregulates Nox4 and accelerates lung fibrosis resolution in aged mice. JCI insight. 2020;5:e137127. doi: 10.1172/jci.insight.137127. [DOI] [PMC free article] [PubMed] [Google Scholar]