

Abstract
Novel proteins can originate de novo from non-coding DNA and contribute to species-specific adaptations. It is challenging to conceive how de novo emerging proteins may integrate pre-existing cellular systems to bring about beneficial traits, given that their sequences are previously unseen by the cell. To address this apparent paradox, we investigated 26 de novo emerging proteins previously associated with growth benefits in yeast. Microscopy revealed that these beneficial emerging proteins preferentially localize to the endoplasmic reticulum (ER). Sequence and structure analyses uncovered a common protein organization among all ER-localizing beneficial emerging proteins, characterized by a short hydrophobic C-terminus immediately preceded by a transmembrane domain. Using genetic and biochemical approaches, we showed that ER localization of beneficial emerging proteins requires the GET and SND pathways, both of which are evolutionarily conserved and known to recognize transmembrane domains to promote post-translational ER insertion. The abundance of ER-localizing beneficial emerging proteins was regulated by conserved proteasome- and vacuole-dependent processes, through mechanisms that appear to be facilitated by the emerging proteins’ C-termini. Consequently, we propose that evolutionarily conserved pathways can convergently govern the cellular processing of de novo emerging proteins with unique sequences, likely owing to common underlying protein organization patterns.
Keywords: de novo gene birth, de novo proteins, ER, targeting, degradation, localization
Introduction
New protein-coding genes can evolve de novo from sequences that were previously non-genic (Fig 1A). Once considered rare, de novo gene birth has now been identified in many species and is gaining considerable attention as a mechanism of molecular innovation and species-specific adaptation (Van Oss and Carvunis 2019, Weisman 2022, Broeils, Ruiz-Orera et al. 2023, Zhao, Svetec et al. 2024). However, the mechanisms by which de novo proteins integrate into cellular systems to provide fitness benefits remain poorly understood (Parikh, Houghton et al. 2022, Zhao, Svetec et al. 2024). Ancient proteins have been coevolving for millions of years with the systems that help them fold and localize correctly, regulate their homeostasis, and enable their beneficial activities (Bohnsack and Schleiff 2010, Powers and Balch 2013, Gabaldon and Pittis 2015, Rebeaud, Mallik et al. 2021). How are de novo proteins, which are initially naïve to these systems, recognized and processed by the cell (Fig. 1A)? We sought to address this apparent paradox with a focus on identifying the systems regulating de novo protein homeostasis and localization.
Figure 1. Association between ER localization and beneficial phenotype.
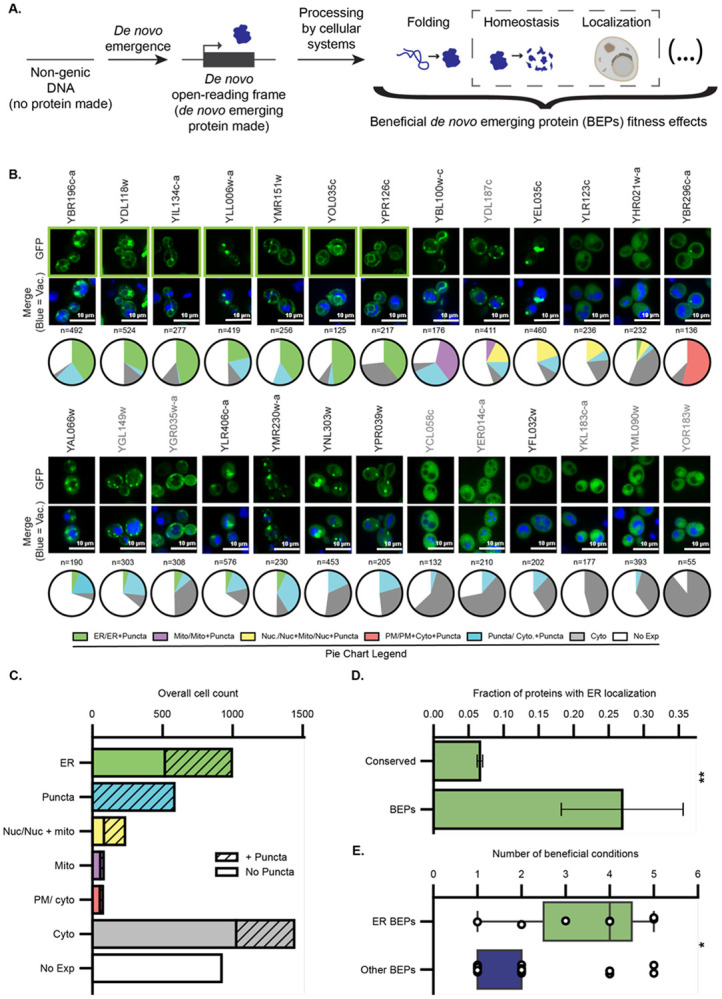
A. Schematic representation of the emergence of BEPs and the various processing by cellular systems that enables their beneficial fitness effects. These processes include, folding, homeostasis, and localization, among others (indicated by the ellipsis on far right). The culmination of these processes ultimately gives rise to the beneficial effects conferred by BEPs. The specific processes and mechanisms that BEPs use to integrate into the existing cellular systems remain unknown. The processes of homeostasis and localization (boxed by dashed outline) are the specific focus of the current study. B. Seven out of 26 BEPs localize to the ER. Fluorescence microscopy of β-estradiol induced expression of BEP-EGFP fusions from a 2μ plasmid. Images of ER-localized BEPs are outlined in green. Cells were stained with CMAC-blue to mark the vacuoles The distributions of localizations observed across individual cells are provided as pie-chart graphs below each image. The BEPs highlighted in grey correspond to those not presenting a band on immunoblotting analysis as shown on Figure S1. C. The ER is the predominant discernable localization for BEPs. The total cell counts from panel B are summed here to display the overall frequency of individual localizations in the BEP-EGFPs. D. BEPs are enriched in the ER relative to conserved proteins. The fraction of all BEP-EGFP fusions in this study (n=26) that localize to the ER upon induced expression, vs the fraction of the whole S. cerevisiae proteome fused C-terminally to GFP and natively expressed (n=4065, GFP collection; Chong et al 2015) that localize to the ER. Error bars indicate the standard error of the proportion. **:Fisher exact test, odds ratio= 5.2, p= 1.3 x 10−3. E. ER-localized BEPs are beneficial across more conditions than other BEPs. Distribution of the number of experimental conditions ER-localized and other BEPs were found to be beneficial when overexpressed by Vakirlis and colleagues (Vakirlis, Acar et al. 2020). *:Mann Whitney U test, p= 0.025, n= 26.
Several lines of evidence suggest that specialized systems capable of regulating de novo protein homeostasis and localization exist. First, several de novo proteins have been shown to localize at discrete subcellular compartments, such as the endoplasmic reticulum (ER), mitochondria, or nucleus (Verster, Styles et al. 2017, van Heesch, Witte et al. 2019, Vakirlis, Acar et al. 2020, Dong, Zhang et al. 2022, Sandmann, Schulz et al. 2023, Wacholder, Parikh et al. 2023). Therefore, systems that allow de novo proteins to attain these specific locations must exist. It is unknown, however, if the same systems that regulate targeting of the ancient proteome also operate with de novo proteins or if novel processes are used by these emerging proteins. Second, recent studies have suggested that the products of non-genic translation can carry molecular features recognized by degradation pathways that regulate the homeostasis of ancient proteins (Kesner, Chen et al. 2023, Casola, Owoyemi et al. 2024). For ancient proteins, recognition by targeting and degradation pathways often relies on biophysical characteristics, e.g. presence of a transmembrane domain (TMD) or degenerate amino acid targeting sequence (Chen, Shanmugam et al. 2019, Mehlhorn, Asseck et al. 2021). Even random sequences can be recognized by select targeting and degradation pathways (Kaiser, Preuss et al. 1987, Lemire, Fankhauser et al. 1989, Hayashishita, Kawahara et al. 2019, Hasenjager, Bologna et al. 2023). Given these potentially permissive requirements, recently emerged de novo proteins might possess characteristics that enable processing by conserved systems despite having had little time to adapt to the cellular machinery. To our knowledge, this hypothesis has not been experimentally tested in any species and the pathways that regulate homeostasis and localization of de novo proteins remain undefined.
In this study, we demonstrate for the first time that the localization and homeostasis of de novo proteins can be regulated by conserved targeting and degradation pathways. We focused on a suite of 28 beneficial de novo emerging proteins (BEPs) that we previously identified as beneficial for growth in systematic overexpression screens across related nutrient stress conditions (Vakirlis, Acar et al. 2020). Our results show that BEPs preferentially localize at the ER and require the conserved post-translational targeting machinery (GET and SND pathways) to attain localization at this organelle. We further show that conserved degradation pathways regulate the homeostasis of ER-localized BEPs. Strikingly, all ER-localized BEPs are predicted to encode a C-terminal TMD followed by a short hydrophobic C-terminus. We propose that these common protein characteristics allow BEPs to engage with the specialized conserved pathways that regulate their localization and homeostasis. These specialized pathways may constrain the evolutionary trajectories of de novo emerging proteins, thus, shaping molecular innovation.
Results
Beneficial de novo Emerging Proteins (BEPs) preferentially localize to the Endoplasmic Reticulum (ER)
We began our investigation by systematically assessing the subcellular localization of BEPs. To this aim, we made C terminal fusions with eGFP and expressed them on plasmids under the control of the β-estradiol-inducible GEV system (Veatch, McMurray et al. 2009, McIsaac, Silverman et al. 2011, McIsaac, Oakes et al. 2013). Microscopy revealed that while some cells displayed diffuse or punctate cytosolic signals or no measurable fluorescence, for others there were discrete localization patterns (Fig 1B, C). The lack of expression in some cells could be due to plasmid loss, reduced transcript stability, reduced translation efficiency, or poor protein stability. In support of the microscopy, immunoblotting revealed a broad distribution of protein abundances and breakdown products, suggesting that BEPs are highly susceptible to degradation (Fig S1). Strikingly, among expressing cells with discrete localization patterns, the ER was by far the dominant subcellular localization (Fig 1B, C). Seven out of twenty-six (27%) successfully cloned BEP-expressing strains exhibited robust ER localization (Fig 1B). To put this figure in perspective, we analyzed a genome-wide localization survey of the ancient yeast proteome generated by high-content microscopy of chromosomally-integrated C-terminal GFP fusions (Chong, Koh et al. 2015). These analyses suggested that BEPs exhibit a significantly increased ER prevalence relative to the ancient proteome (Fisher exact test, odds ratio= 5.2, p= 1.3 x 10−3; Fig 1D). We then compared the phenotypic impacts of overexpressing ER-localized and other BEPs as measured in our overexpression screens (Vakirlis, Acar et al. 2020). ER-localized BEPs were found to provide growth benefits across a broader array of growth conditions than other BEPs (Fig 1E, Mann Whitney U test, p= 0.025, n= 26; see methods). These results reveal a strong association between ER localization and growth benefits among de novo emerging proteins in yeast.
ER-localized BEPs all contain Trans-Membrane Domains (TMDs) followed by short hydrophobic C-termini
After finding that BEPs preferentially localize to the ER, we sought to identify any molecular determinants within their protein sequences that may be responsible for their targeting. To do so, we investigated whether the ER-localized BEPs share common sequence or structure patterns that distinguish them from other BEPs. We previously showed that the sequences of most BEPs are predicted to encode TMDs (Vakirlis, Acar et al. 2020) (Fig S2). Structural predictions by Alpha-Fold2 (Jumper, Evans et al. 2021) were consistent with these TMD predictions (Fig 2), though some have low confidence owing to BEPs’ lack of homology with known proteins (Peng, Svetec et al. 2024, Terwilliger, Liebschner et al. 2024). Considering TMD predictions and structural modeling together, all the ER-localized BEPs displayed the biophysical potential to integrate into membranes, including the ER membrane. However, this potential was shared with many of the other BEPs. Thus, we asked whether the sequences of ER-localized BEPs differ from those of other BEPs in additional ways.
Figure 2. ER-localized BEPs are rich in alpha helices and predicted TMDs.
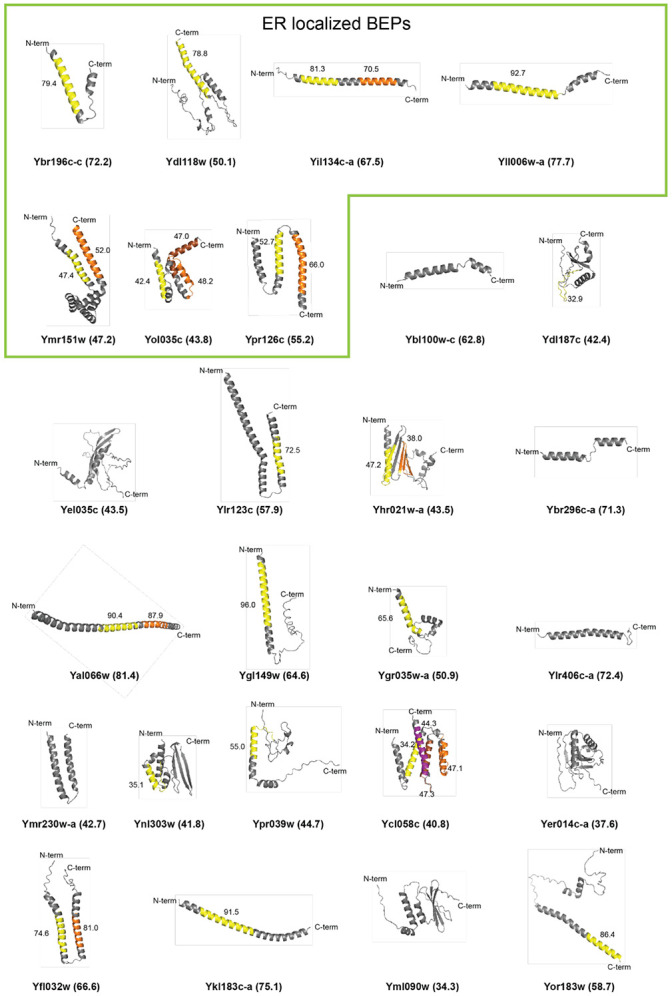
Alphafold 2.0 (Jumper, Evans et al. 2021, Varadi, Anyango et al. 2022) predicted tertiary structures for the ER-localized and other BEPs. The N- and C-termini for each BEP are indicated. The confidence of the alpha-fold prediction for each model is presented in parenthesis after the ORF name. TMDs predicted by Phobius (Kall, Krogh et al. 2004) are indicated by colors and the prediction confidence is presented next to each putative TMD.
Typically, proteins access the ER either co-translationally via the translocon pore or post-translationally by using the Guided Entry of Tail-anchored proteins (GET) and/or Srp-iNDependent targeting (SND) pathways (Aviram and Schuldiner 2017). Co-translationally inserted ER proteins often contain a signal sequence recognized by the signal-recognition particle (SRP) to help direct them to the translocon (Akopian, Shen et al. 2013). However, there were no signal sequences predicted in the ER-localized BEPs, neither by TargetP 2.0 (Almagro Armenteros, Salvatore et al. 2019) nor by SignalP 6.0 (Teufel, Almagro Armenteros et al. 2022). The GET and SND pathways do not require specific sequence motifs for post-translational tail-anchor insertion. Rather, these pathways recognize TMDs that are in the middle of the protein or close to the C-terminus (Shao and Hegde 2011, Shan 2016). Consistent with a tail-anchored insertion mechanism, the C-terminal regions following the last or only predicted TMD of ER-localized BEPs were significantly shorter than those of other TMD-containing BEPs (Fig 3A-B, S3A). In contrast, the overall lengths of ER-localized BEPs, as well as the lengths of their N-termini or TMDs, were statistically indistinguishable from those of other TMD-containing BEPs (Fig 3B, S3A). Therefore, rather than the TMD itself, it is likely that the context of the TMD, in close proximity to the C-terminus, enables ER-localized BEPs to be post-translationally inserted into the ER membrane. The longer C-termini of other TMD-containing BEPs may reduce the possibility for tail-anchored insertion.
Figure 3. ER-localized BEPs have TMDs followed by short and hydrophobic C-termini.
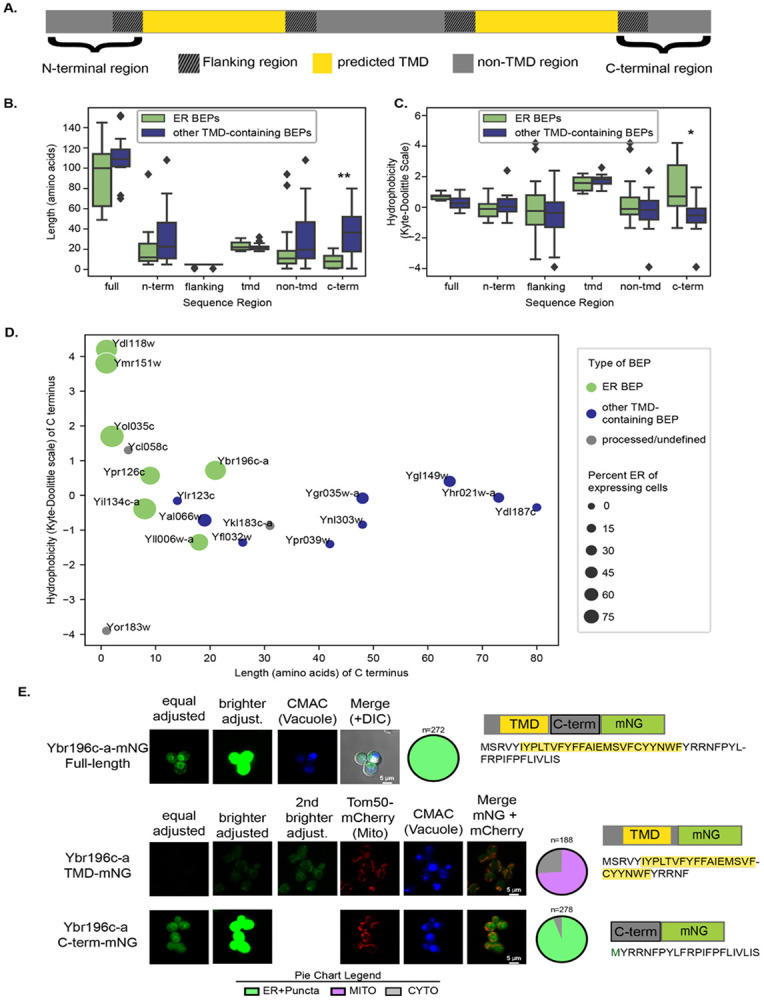
A. Cartoon diagram of a TMD-containing BEP sequence. Each sequence was separated into regions based on predicted TMDs. These regions included 1) the N terminus, all residues from the start of the sequence to the start of a predicted TMD; 2) TMD regions as predicted by Phobius (Kall, Krogh et al. 2004) ; 3) non-TMD regions including all residues not predicted to be in a TMD; 4) flanking regions, including up to 5 residues before and after a predicted TMD; and 5) the C terminus, all residues following the most distal TMD in the sequence. B. ER-localized BEPs have significantly shorter C-terminal ends than other TMD-containing BEPs. Length in amino acid residues for different sequence regions in ER-localized and other TMD-containing BEPs. C. ER-localized BEPs have more hydrophobic C-terminal ends than other TMD-containing BEPs. Hydrophobicity values (Kyte-Doolittle scale) for sequence regions in both ER and other TMD-containing BEPs. B-C. All P values calculated from Mann-Whitney U test. **: p <0.005, *:p<0.05 Full=Full sequence, n-term=N-terminal region, c-term=C-terminal region. For full, n-term, and c-term, n=19. For tmd, n=30. For flanking, n=60. For non-tmd n=49. D. Hydrophobicity versus length in amino acid residues for the C termini of all TMD containing BEPs. Processed/undefined: BEPs that showed some evidence of breakdown in western blots (see S1 Fig). Size of the dots represent percent of expressing cells that had ER localization. Corresponding systematic gene names from Saccharomyces Genome Database shown next to each dot. E. The C-terminus of Ybr196c-a drives localization to the ER. Fluorescent micrographs are shown of full length and two split versions of the Ybr196c-a protein fused with mNG, represented as TMD or C-term fragments (see cartoon next to images), corresponding to the predicted TMD and the C-terminal region of the sequence in S. cerevisiae. An mCherry-tagged Tom50 was integrated into these strains to allow visualization of the mitochondria and cells were stained with CMAC-blue to mark the vacuoles. The distributions of localizations observed for each construct are represented in the pie-chart graphs below to the right of each image. For simplicity, in the split versions the merge shows the co-localization between the mNG and the mCherry signal only.
In addition to being shorter, the C-terminal sequences after the TMDs of ER-localized BEPs were also significantly more hydrophobic than those of other TMD-containing BEPs (Fig 3C, S3B). This is notable because previous work has shown that hydrophobic C-termini can act as signals for protein degradation by the proteasome in humans (Kesner, Chen et al. 2023, Casola, Owoyemi et al. 2024, Yang, Li et al. 2024). Upon closer inspection of the predicted structures (Fig 2) however, it becomes apparent that while several BEPs have unstructured C-termini, all ER-localized BEPs end either with a TMD or with an alpha-helical motif. These motifs could reflect true TMDs that were missed by prediction algorithms. If so, the true C-termini would be even shorter than suggested by our calculations, and thus, a post-translational tail-anchored insertion of the ER-localized BEPs would be even more favorable. Nevertheless, based on current predictions, all ER-localized BEPs have C-termini between one and 26 amino acids, the estimated cutoff for post-translational targeting of TMDs into the ER (Fig 3D, S2, S3C; (Borgese, Coy-Vergara et al. 2019)).
The C-terminus of a model ER-localized BEP drives its localization
To investigate whether the common protein features of ER-localized BEPs play a role in their localization, we first focused on a model ER-localized BEP, Ybr196c-a. We previously showed that Ybr196c-a integrates into the ER membrane using a combination of biochemical approaches and microscopy (Vakirlis, Acar et al. 2020). Here, we again observed ER localization for Ybr196c-a when its plasmid-based expression is under the β-estradiol-inducible GEV system (Fig 1B). Plasmid-based expression systems in yeast yield higher incidents of cell-to-cell variability, and to mitigate these issues, we chromosomally integrated Ybr196c-a fused with mNG at the HIS3 locus and expressed it under a Z3EV β-estradiol-inducible system (McIsaac, Oakes et al. 2013). With this expression and tagging strategy, Ybr196c-a also displayed an ER localization, but now the prevalence of this localization was increased to be present in 100% of cells (Fig 3E compared to Fig 1B). Therefore, Ybr196c-a is a very robust example of an ER-localized BEP, making it a suitable candidate as a model for ER-localized BEPs.
To map the sequence determinants that dictate Ybr196c-a’s ER localization, we similarly expressed two split versions of the protein: the first from the N-terminus to the end of its predicted TMD, and the second from the end of the predicted TMD to the end of C-terminus. As expected from our analyses (Fig. 3A-D), the second split displayed clear localization at the ER (Fig 3E). The first split was intriguingly localized to the mitochondria. From this experiment, it was clear that the C-terminal region of Ybr196c-a dictates its targeting to the ER, as might be expected for a tail-anchored protein.
All ER-localized BEPs require the GET- and SND-dependent post-translational insertion machinery to localize to the ER
Since the C-terminal regions of the ER BEPs were important for their insertion into the ER, we next sought to determine whether conserved pathways are needed for their targeting. We began by defining the pathways that target the model ER-localized BEP Ybr196c-a to the ER using genetic approaches. Specifically, we evaluated whether Ybr196c-a localization was altered when members of the GET, SND, and SRP-dependent targeting pathways were deleted. We also evaluated the impact of deleting the mitochondrial protein quality control AAA-ATPase Msp1, as it can facilitate ER targeting of some single-pass transmembrane proteins after promiscuous insertion into the mitochondria (Wang and Walter 2020). The relevant gene deletions were engineered into strains expressing a chromosomally integrated Ybr196c-a-mNG fusion under a Z3EV3 β-estradiol-inducible system as in Fig. 3E. Upon disruption of Get1, Get2, and Get3, the core machinery of the GET complex, or Snd2 and Snd3, key pieces of the SND pathway, we found reduced Ybr196c-a targeting to the ER and increased mNG-tagged cytosolic puncta (Fig 4A-D). This dependence on the GET- and SND systems for Ybr196c-a’s ER targeting was observed regardless of whether the protein was N- or C-terminally tagged with mNG (Fig 4A-D). Interestingly, disruption of components that can act in concert with the GET pathway (e.g., Get4, Get5 and Sgt2) did not alter Ybr196c-a localization; thus, these accessory factors in the GET pathway are not required for Ybr196c-a targeting (Fig 4A). Loss of neither SRP targeting components nor Msp1 altered the ER localization of Ybr196c-a-mNG (Fig 4A). In addition, the loss of GET or SND targeting components had no impact on the localization of free mNG (S4A Fig). Together, these findings demonstrate that Ybr196c-a requires the post-translational insertion GET or SND machinery to access the ER, as might be expected for a tail-anchored protein with a TMD close to its C-terminus.
Figure 4. The GET and SND posttranslational ER-targeting pathways are needed for BEPs to access the ER.
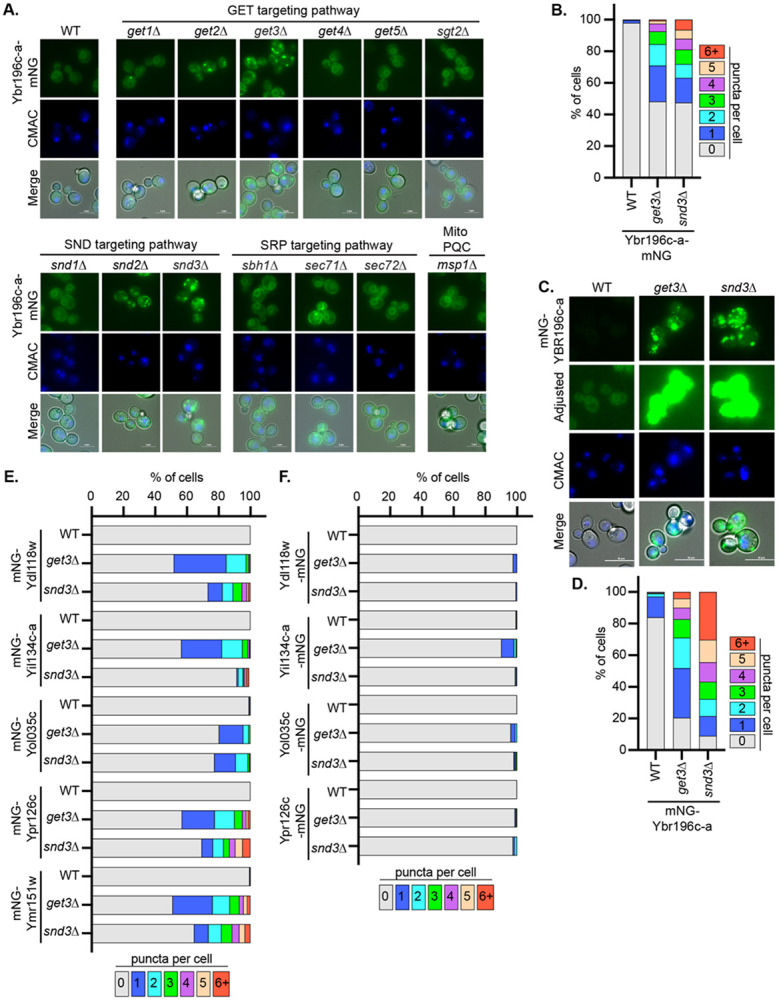
A and C. Fluorescence micrographs of A. Ybr196c-a-mNG or C. mNG-Ybr196c-a in the indicated deletion backgrounds are shown. B, D, E and F. The percentage of cells with the number of puncta per cell (based on images shown in panel A. and C. and Figure S4B and S4C, respectively), are indicated as a stacked distribution. Increasing numbers of puncta per cell, and concomitant loss of clear ER localization, are associated with disruption of the ER-insertion pathways needed for BEP targeting.
Expanding this approach beyond Ybr196c-a, we found that all the ER-localized BEPs that we successfully cloned in the relevant deletion contexts were also dependent upon the GET and SND pathways (Fig 4E-F and S4B-C Fig). Interestingly, this effect was more pronounced when the BEPs were N-terminally tagged and their C-termini were accessible. This is consistent with the notion that C-terminal tags would mask the terminal TMD needed for their post-translational ER insertion, as has been reported for other tail-anchored proteins (Weill, Krieger et al. 2019). Thus, for ER-localized BEPs to gain access to the ER the GET and SND pathways are needed. Given the structures of these ER BEPs, it is likely that they are anchored into the membrane by transmembrane domains that are close to their C-termini. The short C-termini may aid in preferentially targeting these proteins to the ER via the GET/SND pathways as opposed to insertion in the ER via alternative mechanisms (Aviram and Schuldiner 2017).
All ER-localized BEPs are degraded by conserved Protein Quality Control (PQC) pathways
Once we found that ER-localized BEPs are targeted to the ER via conserved pathways, we sought to determine if their degradation was regulated by established quality control mechanisms. To define the cellular pathways that control the homeostasis of ER-localized BEPs, we first evaluated whether the levels of Ybr196c-a depend on the major ER-resident E3 ubiquitin ligases, Doa10 and Hrd1(Swanson, Locher et al. 2001). Immunoblotting for Ybr196c-a, tagged either N- or C-terminally with mNG, revealed a strong increase in Ybr196c-a levels upon DOA10 deletion (Fig 5A-B). In contrast, free mNG was not stabilized by the loss of DOA10 (S5A-B Fig). Furthermore, this increase in abundance was independent of the tag, as Ybr196c-a fused to an HA tag was similarly dependent upon DOA10 (Fig 5C-D). We treated cells with the translation inhibitor cycloheximide (CHX) (Schneider-Poetsch, Ju et al. 2010), and examined the protein turnover rate of: 1) Ybr196c-a-mNG (Figs 5E-F), 2) free mNG (S5C-D Fig), 3) C-terminally HA-tagged Ybr196c-a (Fig 5G-H), 4) mNG-Ybr196c-a (S5E-F Fig). From these CHX-chase assays, we confirmed that all versions of Ybr196c-a (N- or C-terminally-tagged with either mNG or HA) were stabilized by loss of Doa10 whereas the mNG tag alone was not. Doa10 ubiquitination of ER-resident proteins often results in their retrotranslocation from the ER and subsequent proteasomal degradation (Nakatsukasa and Brodsky 2008). Accordingly, we found that in cells lacking PDR5 (which allows for improved retention of MG-132 in cells), Ybr196c-a was more stable when cells were treated with the proteasome inhibitor MG-132 than in vehicle control-treated cells (Fig. 5I-J), unlike free mNG (Fig. S5G) and irrespective of the positioning of the mNG tag (Fig. S5H). Together, these experiments demonstrate that Ybr196c-a abundance is regulated by Doa10- and proteasome-dependent degradation.
Figure 5. Ybr196c-a degradation is dependent upon Doa10 and the proteasome.

A. A representative immunoblot of protein extracts made for mNG N- or C-terminally tagged Ybr196c-a expressed in the indicated deletion backgrounds. B. The relative abundance of Ybr196c-a-mNG from six biological replicates of the immunoblot in A. for the strains indicated. A Student’s t-test comparing the WT to the PQC deletions was used to assess statistical differences. (not significant = ns; p-value < 0.05 = *). C. Similar analyses of C-terminally HA-tagged Ybr196c-a. D. The relative abundance of Ybr196c-a-HA from six biological replicates of the immunoblot in C. for the strains indicated. A Student’s t-test comparing the WT to the PQC deletions was used to assess statistical differences. (not significant = ns; p-value < 0.05 = *). E. A representative immunoblot of protein extracts expressing Ybr196c-a-mNG from WT or doa10Δ cells post cycloheximide (CHX) addition for the times indicated (in minutes) is shown. F. Quantification of the percent of Ybr196c-a-mNG remaining post-CHX addition is indicated. The dots represent the mean of the biological replicates (n) and the error bars show the standard error of the mean. A Student’s t-test comparing the WT to the doa10Δ cells was used to assess statistical differences (not significant = ns; p-value < 0.05 = *; p-value <0.0005 = ***). Similar analysis to those presented in E and F for an mNG control and the N-terminally mNG-tagged Ybr196c-a are presented in Figures S5C-D, S5E-F, respectively. G. A representative immunoblot of protein extracts expressing Ybr196c-a-HA from WT or doa10Δ cells post cycloheximide (CHX) addition for the times indicated (in minutes) is shown. H. Quantification of the percent of Ybr196c-a-HA remaining post-CHX addition is indicated. The dots represent the mean of the biological replicates (n=6) and the error bars show the standard error of the mean. A Student’s t-test comparing the WT to the doa10Δ cells was used to assess statistical differences (not significant = ns; p-value < 0.05 = *; p-value <0.0005 = ***). I. A representative immunoblot of protein extracts expressing Ybr196c-a-mNG in pdr5Δ cells that were untreated (+DMSO) or treated with the proteasomal inhibitor MG-132 post-CHX addition is shown. Similar analysis to those presented in I for an mNG control and the N-terminally mNG-tagged Ybr196c-a are presented in Figures S5G and S5H, respectively. J. Quantification of the percent of Ybr196c-a-mNG remaining post-CHX addition is indicated for the MG-132-treated or untreated (+DMSO control). The dots represent the mean of the biological replicates (n=3) and the error bars show the standard error of the mean. Student’s t-test comparing the DMSO- to the MG-132-treated cells at each time point was used to assess statistical differences. (not significant = ns; p-value < 0.05 = *; p-value <0.005 = **). For all immunoblots, REVERT total protein stain serves as the loading control.
To determine whether the homeostasis of other ER-localized BEPs is regulated by the same pathways, we performed additional immunoblotting assays of N- and C-terminal fusions of BEPs and mNG in cells lacking Doa10, Hrd1, or both. For many of the BEPs, we observed a faint or no band of the correct molecular weight in wild-type cells (S6A-E Fig). However, upon deletion of HRD1 and/or DOA10, distinct bands of the expected size were detected for each ER-localized BEP (S6A-E Fig). These findings raise the possibility that BEPs may attain distinct subcellular localizations, but their rapid turnover may preclude their detection at these locales. We explored this possibility by examining the localization and levels of each ER-localized BEP by fluorescence microscopy in the presence and absence of Doa10 and Hrd1. In wild-type cells, many BEPs displayed faint ER or predominantly cytosolic localization when expressed via the Z3EV as a chromosomal integration (Fig. 6A-C, S7, S8 Figs). Strikingly however, all exhibited clear ER localization and/or increased fluorescence intensities when DOA10 and/or HRD1 were deleted (Fig 6A-D and S7, S8 Figs) while our mNG control was unchanged (S4A Fig). These results indicate that homeostasis of ER-localized BEPs is regulated by ER resident ubiquitin ligases.
Figure 6. ER-localized BEPs are degraded via Hrd1 and Doa10.

A-B. Representative fluorescence microscopy of A. N-terminally- or B. C-terminally-mNG tagged Ybr196c-a is shown. The distribution of fluorescence observed across the cell populations is indicated in the pie chart below each diagram (n=number of cells assessed). Cells are stained with CMAC-blue to mark vacuoles and imaged via DIC, which is shown in the merge. Analogous imaging data for all ER BEPs is provided in Figures S7 and S8. C. Quantification of the whole cell fluorescence intensities determined using the NIS.ai and GA3 software (see methods), for the cells shown in panels A and B. A one-way ANOVA with Dunnett’s multiple comparisons test was performed relative to the fluorescence in WT cells to identify statistically significant changes (p-value < 0.0005 = ***). D. A heatmap indicating the fold change in the mNG fluorescent signal for ER-localized BEPs in the protein quality control deletion strains relative to the WT controls (based on data in Figures 6C, S7C, S7F, S7H, S8C, S8F) is shown.
When the impact of the mNG tag orientation was considered holistically, it was clear that the N-terminally tagged ER-localized BEPs had more robust increases in fluorescence intensity upon ubiquitin ligase deletion than those that were C-terminally tagged (Fig 6D). This is consistent with the notion that C-terminal tags would mask the BEP’s C-terminal TMD and hydrophobic tail, which might act as a degradation signal as has been reported for human and random sequences (Kesner, Chen et al. 2023, Casola, Owoyemi et al. 2024, Claudio Casola 2024). It is therefore possible that the same common feature of ER-localized BEPs, their short hydrophobic C-termini immediately following a TMD, might contribute to both their ER targeting (Fig. 4, S4 Fig), and their degradation (Fig. 6, S7-8). Alternatively, since the C-terminally tagged BEPs do not localize to the ER as robustly as those that are N-terminally tagged, there may simply be less access to these BEPs for the ER-resident Ubiquitin ligases, which could explain why we observe less of an effect with these constructs.
We considered the possibility that the ER-localized BEPs’ stability might be influenced by N-terminal degrons. N-degrons target proteins for degradation by the acetylation- or arginine-dependent N-end rule pathways (Varshavsky 2011) and are predicted to be abundant among proteins originating from non-genic translation in humans (Casola, Owoyemi et al. 2024). N-end rule substrates are ultimately ubiquitinated by E3 ligases Doa10, Ubr1 or Mot2/Not4 (Sherpa, Chrustowicz et al. 2022). Given that several ER-localized BEPs are dependent on Doa10, recognition via the N-end rule is a possibility. We identified the Nbox1 N-end degron motif in the sequence of two ER-localized BEPs, Ydl118w and Ypr126c. However, these BEPs were not stabilized by loss of the Ubr1 ubiquitin ligase (S9A-B Fig). Therefore, a Ubr1-mediated, N-end rule pathway does not seem to be a driver of ER-localized BEP degradation.
Finally, we investigated whether ER-localized BEPs can be degraded through proteasome-independent mechanisms in the vacuole. The vacuole is the yeast equivalent of the lysosome (Li and Kane 2009). ER-localized proteins can be degraded in the vacuole, if and only if they leave the ER, either through ER-phagy or through the secretory pathway (Knupp, Pletan et al. 2023). We saw little evidence of such transit via microscopy in wild-type cells, but still assessed the impact of inhibiting vacuolar degradation on ER-localized BEPs. Vacuolar proteases are matured by the master protease, Pep4. Thus, cells lacking Pep4 often accumulate vacuole-targeted proteins (Woolford, Daniels et al. 1986, Hecht, O'Donnell et al. 2014). Deletion of PEP4 caused a significant increase in the ratio of vacuolar to whole-cell fluorescence, indicating vacuolar accumulation of the ER-localized BEPs (Fig 7 and S10 Fig). These findings demonstrate that not only are ER-localized BEPs regulated by the ER PQC machinery and the proteasome, but they can also leave the ER and transit to the vacuole for degradation. It is therefore abundantly clear that conserved PQC pathways govern the stability and turnover of these evolutionarily novel ER-localized BEPs.
Figure 7. ER-localized BEPs can transit to the vacuole to be degraded.

A and D. Fluorescence micrographs of cells expressing A. mNG-Ybr196c-a or D. N-terminal mNG-tagged BEPs in WT or pep4Δ cells are shown. Vacuoles are stained with CMAC-blue and indicated on the green channel images using a white dashed line. B and E. The scatter plots indicate the whole-cell fluorescence intensities, as determined using NIS.ai and GA3 software, for mNG-tagged BEPs shown in A and D, respectively. The median fluorescence intensity and 95% confidence interval are represented by the horizontal lines. C and F. The ratio of the vacuolar fluorescence over the whole cell fluorescence for the cells imaged in A and D, respectively, is shown as a scatter plot. For graphs in B, C, E, and F, a Student’s t-test assessed statistical difference between the WT and pep4Δ cell populations (p < 0.0005 = ***). Similar analyses for C-terminal mNG-tagged BEPs is provided in Figure S10.
Discussion
In this study, we report the first experimental investigation of cellular processing of de novo proteins. Our results demonstrate that conserved pathways control the targeting and homeostasis of a group of yeast BEPs. We find it remarkable that the same systems that have regulated the ancient proteome for millions of years may also accept young de novo proteins as clients. For example, the GET and SND post-translational membrane insertion pathways that we find to be required for BEP ER targeting are conserved across fungi, plants, and animals (Mehlhorn, Asseck et al. 2021). ER localization is surprisingly prevalent among the yeast BEPs we investigated. Future studies are required to determine if this finding will generalize to other de novo proteins in yeast and beyond. We propose that the common localization observed here results from a common protein organization with a short C-terminal TMD context. Altogether, our findings suggest that de novo proteins can integrate cellular systems through molecular convergence: whether neutrally or under the action of natural selection (Stayton 2015), the yeast BEPs that we investigated possess common characteristics that seem to enable select pathways to regulate their localization and homeostasis at the ER.
In a previous study, we showed that the TMD of Ybr196c-a, a model ER-localized BEP, has arisen neutrally owing to the high thymine richness of its locus of origin (Vakirlis, Acar et al. 2020). Given that the non-genic sequences of yeasts are generally rich in TMD potential (Tassios, Nikolaou et al. 2023), TMD-containing de novo proteins may emerge frequently in this phylum. It follows that, since stop codons also appear frequently throughout yeast non-genic sequences (Dujon 1996), many de novo TMD-containing proteins might spontaneously be born with the characteristics required for recognition by the post-translational tail-anchor ER insertion machinery. In the case of Ybr196c-a, our previous experiments suggested that the efficiency of ER targeting may have increased over evolutionary time since its initial de novo emergence (Vakirlis, Acar et al. 2020). In the future, we plan to dissect the mechanisms by which the shared protein organization of ER-localized BEPs arose and evolved to establish whether it is the product of selection, neutral evolution, or a combination of both.
In general, this line of questioning represents an exciting direction of research at the intersection of cell biology and de novo gene birth. For example, we show that the homeostasis ER-localized BEPs depends both on the proteasome and the vacuole. This implies that a pool of BEPs is not maintained at the ER. Accordingly, the ER-localized BEPs lack the canonical KDEL retrieval sequence (Figure S2) (Newstead and Barr 2020) and therefore they will not be retrieved if they are non-selectively incorporated into vesicles leaving the ER. It will be interesting to define the molecular pathway that takes BEPs to the vacuole, such as the direct Golgi-to-endosome-to vacuole routes (Ihrke, Kyttala et al. 2004), the cytoplasmic-vacuole-transit (CVT) pathway of autophagy (Reggiori and Klionsky 2013), and/or ER-phagy (Bernales, Schuck et al. 2007). Depending on the mechanism by which BEPs depart the ER, they may sample different cellular compartments and this may ultimately facilitate the evolution of novel activities outside the ER.
As another example, when we dissected the coding sequence of Ybr196c-a, we found that the sequence fragment containing the C-terminus drives its ER localization whereas the sequence fragment containing the N-terminus and its single predicted TMD drives localization to the mitochondria. This may hint at two distinct targeting potentials within a single, small de novo protein. Previous studies have shown that the ER and mitochondrial targeting pathways can actively compete for clients and that the biochemical properties of amino acids immediately surrounding a TMD can profoundly influence which cellular membrane they are sorted to (Rao, Okreglak et al. 2016, Vitali, Sinzel et al. 2018, Mehlhorn, Asseck et al. 2021). Understanding the precise molecular mechanisms of Ybr196c-a targeting may prove a useful tool to help define the biochemical parameters that dictate cargo recognition between different pathways. We suspect that the second fragment, which contains the protein’s C-terminus possesses a TMD, that is not predicted by the computational methods we employed, but mediates insertion at the ER. Evolutionary analyses of the YBR196C-A locus across budding yeast species are also consistent with the possible presence of a second TMD (Vakirlis, Acar et al. 2020). Because different TMD prediction algorithms rely on criteria based on the structures of conserved proteins to identify potential transmembrane regions and have not been extensively evaluated on de novo emerged proteins, it is possible to obtain false negatives. Several other studies have also reported weaknesses when prediction algorithms trained on the ancient proteome are applied to de novo proteins (Aubel, Eicholt et al. 2023, Peng and Zhao 2023). Mechanistic research on recently-emerged de novo proteins, which are presumably still adapting to the conserved cellular machinery, may improve our understanding and predictive ability across diverse areas of molecular and cellular biology.
Materials and Methods
Yeast strains and growth conditions
The yeast strains used are described in Supplemental Table 1 and are all derived from S288c genetic backgrounds of S. cerevisiae. The methods for building gene deletions in this background are described in this table, but typically DNA cassettes targeted to the region of interest using primers containing sequences homologous to the genomic locus were employed. Yeast cells were grown in either synthetic complete medium (SC) lacking the appropriate amino acids for plasmid selection, prepared as described in (Amberg 2005) and using ammonium sulfate as a nitrogen source, or YPD medium where indicated. Liquid medium was filter-sterilized and solid plate medium had 2% agar (w/v) added before autoclaving. When necessary for selection, Hygromycin B or G-418 (H75020-1.0 and G64000-5.0, Research Products International) was added to the media to a final concentration of 200ug/ml. Yeast cells were grown at 30°C and where appropriate, 10-20μM of β-estradiol (E2758-1G, Millipore-Sigma) was added to cultures for 3 hours to induce GEVpr or Z3EVpr expression systems (McIsaac, Oakes et al. 2013).
The initial strain containing the ACT1pr-Z3EV-NatMX expression system (DBY12394) was generously provided by the Noyes lab (McIsaac, Oakes et al. 2013). The constructs containing the Z3EVpr followed by the coding sequences for individual BEPs tagged with mNeon-Green (mNG) at N’ or C’ terminus were incorporated at the HIS3 chromosomal locus. For a complete list of strains generated in this way see Supplemental Table 1.
Yeast Transformation
The constructs or DNA cassettes amplified from plasmids were integrated in the genome using the lithium acetate, polyethyleneglycol and salmon sperm DNA transformation protocol (Dunham MJ 2015) with an adaptation to be performed at high-throughput scale. The background strain was grown in 1ml of YPD media on 96-deep well plates and used for transformation performed with the liquid-handler EVO 150 (Tecan Group Ltd, Switzerland). Cells were washed in 750 μl of water, followed by a washing step in 1 ml of lithium acetate tris-EDTA buffer and finally resuspended in 500 μl of the same buffer. Transformations were carried out using 5 μl of ssDNA (2mg/ml) with 5 μl of purified PCR product or 500 ng of plasmid and 50μl of cells. 250μl of PEG-LiAC-TE mix was added to each cell mixture and incubated at 30°C for 15 min, followed by 60 min at 42°C.
Cells were then pelleted, resuspended in 200 μl of YPD or SC media for a recovery step at 30°C for 3h, prior to being used to seed 3 μl drops on selection plates. Plates were incubated at 30°C for 2 to 4 days until transformants grow. Agar plates were then used to pin into liquid media with the Singer RoToR (Singer Instruments, San Francisco, CA) and then used to make glycerol stocks.
Plasmids and DNA Manipulations
Gateway Entry Clones
We attempted to tag each BEP chosen to be included in the study with mNeonGreen (mNG) both at N’ and C’ terminus. The first step was to create a collection of Entry Clones to be used for the cloning Gateway System (ThermoFisher, Waltham, MA). Primers were designed containing the attB1 and attB2 sequences to amplify each ORF with a stop codon for the N’ terminal tagging and without, for the C’ terminal tagging. The primers synthesized by Integrated DNA Technologies (IDT, Coralville, IA) were used to amplify each BEP using Q5 High-Fidelity 2x Master Mix (M0492L, New England BioLabs, Ipswich, MA) using genomic DNA extracted from the strain FY4 with the kit Yeast DNA Extraction (78870, ThermoFisher) as a template. The PCR conditions were as follows: 98°C for 30 sec, followed by 25 cycles of 98°C for 10sec, 55°C for 15 sec and 72°C for 30 sec and a final elongation step of 2 min at 72°C. The PCR products were purified with NucleoFast 96 PCR Plates (743100.10, Macherey Nagel, Allentown, PA) and quantified on a Nanodrop (Thermo Fisher, Waltham, MA) prior to use for recombination with the donor plasmid pDONR221, using BP Clonase II Enzyme mix (11789100, ThermoFisher, Waltham, MA). The recombination reactions were used to transform DH5alpha competent cells (C2987U, New England BioLabs, Ipswich, MA) and positive clones were grown and selected in Luria broth media supplemented with 50 μg/ml of Spectinomycin (158993, MP Biomedicals, Santa Ana, CA). The NucleoSpin 96 Plasmid kit (740625.4, Macherey Nagel, Allentown, PA) was used to extract the plasmids and quantification was done using the plate reader SpectraMax M4 (Molecular Devices, San Jose, CA).
Destination Plasmids
The destination plasmids were made by modification of the pAG415-GAL-ccdB-EGFP and pAG415-GAL-EGFP-ccdB from the Yeast Gateway kit (1000000011, Addgene, (Alberti, Gitler et al. 2007)) to create two new plasmids (pARC0031 and pARC0152) with the GAL promoter swapped to Z3EV promoter and the ccdB-EGFP replaced by ccdB-mNeonGreen-Tadh1 and hygromycin cassette (for the C terminally tag) or mNeonGreen-ccdB-Tadh1and hygromycin cassette (for the N terminally tag).
Expression plasmids
Expression plasmids were made by LR recombination between the Entry clones and the Destination plasmids previously prepared, using the LR Clonase II enzyme mix (11791020, ThermoFisher, Waltham, MA). The recombination reactions were used to transform DH5alpha competent cells (C2987U, New England BioLabs, Ipswich, MA) and positive clones were grown and selected in Luria broth media supplemented with 100ug/ml of Ampicillin (J60977, Alfa Aesar, Haverfill, MA). The NucleoSpin 96 Plasmid kit (740625.4, Macherey Nagel, Allentown, PA) was used to extract the plasmids and quantification was done using the plate reader SpectraMax M4 (Molecular Devices, San Jose, CA). Plasmids used in this work are described in Supplemental Table 2. The GALpr-BEP-eGFP plasmids, used in Figure 1, were made by LR recombination using the entry clones containing the BEP sequence without stop codon and the destination plasmid pAG415-GAL-ccdB-EGFP (Vakirlis, Acar et al. 2020).
For all the remaining figures in this work, using the expression plasmids as a template, we amplified the fragment containing the Z3EVpr -BEP-mNG, Z3EVpr-mNG-BEP or Z3EVpr -BEP-HA with the hygromycin cassette for chromosomal integration at the HIS3 locus. Q5 High-Fidelity 2x Master Mix (M0492L, New England BioLabs, Ipswich, MA) and the pair of primers ARC338 (TCTATATTTTTTTATGCCTCGGTAATGATTTTCATTTTTTTTTTTCCACCTAGCGG ATGACTCTTTTTTTTTCTTAGCGATTGGCATTATCACATAATGAATTATACATTAT ATAAAGTAATGTGATTTCTTCGAAGAATATACTAAAAAATGAGCAGGCAAGATAA ACGAAGGCAAAGacaaaagctggagctctagta) and ARC339 (AAAGAAAAAGCAAAAAGAAAAAAGGAAAGCGCGCCTCGTTCAGAATGACACGT ATAGAATGATGCATTACCTTGTCATCTTCAGTATCATACTGTTCGTATACATACTT ACTGACATTCATAGGTATACATATATACACATGTATATATATCGTATGCTGCAGC TTTAAATAATCGGTGTCAgcgaattgggtaccggcc) were used to amplify the fragment in a 50ul reaction. The PCR conditions were as follows: 98°C for 30 sec, followed by 25 cycles of 98°C for 10 sec, 56°C for 15sec and 72°C for 2 min and a final elongation step of 5min at 72°C. The PCR products were incubated at 37°C with 1ml of DpnI (R0176S, New England BioLabs, Ipswich, MA) and then purified with NucleoFast 96 PCR Plates (743100.10, Macherey Nagel, Allentown, PA) and quantified on a Nanodrop (Thermo Fisher, Waltham, MA) prior to being used for yeast transformation. For a complete list of plasmids generated in this section see Supplemental Table 2.
Yeast Protein Extraction and Immunoblot Analyses
Whole cell extracts of yeast proteins were generated using trichloroacetic acid (TCA) method as described in (Hager, Krasowski et al. 2018) and modified from (Volland, Urban-Grimal et al. 1994). In brief, cells were grown in SC medium to mid-exponential log phase at 30°C (A600 = 0.6-1.0) and an equal density of cells was harvested by centrifugation. Cell pellets were frozen in liquid nitrogen and stored at −80°C until processing. Cells were lysed using sodium hydroxide, precipitated with 50% TCA, and solubilized in SDS/Urea buffer [8 M Urea, 200 mM Tris-HCl (pH 6.8), 0.1 mM EDTA (pH 8.0), 100 mM DTT, 100 mM Tris (not pH adjusted)] and heated to 37°C for 15 min prior to loading on an SDS-PAGE gel. Immunoblotted proteins were detected using mouse anti-green fluorescent protein (GFP) antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), mNeon Green antibody (Cell Signaling Technology, Danvers, MA, USA) or HA antibody (Roche, Indianapolis, IN). Anti-mouse secondary antibodies conjugated to IRDye-800 or IRDye −680 were used to detect primary GFP or mNG antibodies on an Odyssey CLx infrared imaging system (LI-COR Biosciences, Lincoln, NE, USA). The HRP-conjugated anti-HA antibody was detected on a ChemiDoc (Bio-Rad, Hercules, CA). As a loading and transfer control, membranes were stained with Revert (LI-COR Biosciences, Lincoln, NE, USA) and detected using the Odyssey CLx.
Extracts containing HA-tagged BEPs were loaded on 16.5% Tris-tricine gels and run in the cold using 1X Tricine running buffer (Bio-Rad, Hercules, CA). Proteins were blotted to Immobilon-PSQ (0.2 micron) PVDF membrane using the Criterion system (Bio-Rad). Membranes were stained with REVERT total protein stain (LI-COR Biosciences, Lincoln, NE, USA), followed by blocking with TBST with 3% BSA and overnight incubation at 4°C on a platform rocker with an HRP-conjugated anti-HA antibody in TBST with 1 % BSA (1:5000, Roche). Membranes were then washed 3 times with TBST and detected using the SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo, Waltham, MA) on the Bio-Rad Chemidoc XRS+ Imaging System (Bio-Rad).
Protein Stability Assays
The stability of mNG- or HA-tagged YBR196c-a (as an N-terminal or C-terminal fusion, as indicated) or mNG alone expressed as a chromosomal integration from the Z3EVpr was assessed by cycloheximide (CHX) chase assay as described in (Schneider-Poetsch, Ju et al. 2010). Cells were grown to mid-exponential log phase and induced to express the tagged YBR196c-a or mNG using b-estradiol. Cells were next treated with 0.15 mg/ml CHX (Gold Bio, St. Louis, MD, USA) and equal densities of cells were harvested at the indicated times. Cell pellets were subjected to protein extraction, SDS-PAGE, and immunoblotting, as described above.
For assays that employed the proteasome inhibitor MG-132 (Fisher, Waltham, MA), cells were incubated with 10 μM of MG-132 (stock 10 mM in DMSO) or an equivalent volume of DMSO (vehicle control) for 1 h prior to CHX addition. When CHX was added to block new protein synthesis the t=0 timepoint was harvested and the time course initiated.
Fluorescence microscopy
For imaging experiments, cells were grown and induced to express as indicated above. Fluorescent proteins were localized using: 1) epifluorescence microscopy, 2) confocal microscopy in low-throughput, or 3) confocal microscopy in high-throughput. For epifluorescence microscopy and low-throughput confocal microscopy, cells were stained with 250 μM Cell Tracker Blue CMAC dye (Life Technologies, Carlsbad, CA) and 10 μM trypan blue (Gibco, Dublin, Ireland) and plated onto 35 mm glass bottom microwell dishes that were concanavalin A (MP Biomedicals, Solon, OH, USA) or poly-D-lysine coated (MatTek Corporation, Ashland, MA). For epifluorescence microscopy, cells were imaged using a Nikon Ti2 inverted microscope (Nikon, Chiyoda, Tokyo, Japan) outfitted with an Orca Flask 4.0 cMOS camera (Hammamatsu, Bridgewater, NJ) and a 100x objective (NA 1.49). For low-throughput confocal microscopy, cells were imaged using a Nikon Ti inverted microscope (Nikon) outfitted with a swept-field confocal scan head, EMCCD detection (iXon3; Andor, Belfast, UK) and a 100x objective (NA 1.49). For high-throughput confocal microscopy, imaging was done as described in Bowman et al. (Bowman, Jordahl et al. 2022).
In all cases, image acquisition was controlled using NIS-Elements software (Nikon) and all images within an experiment were captured using identical settings. Images were cropped and adjusted equivalently using NIS-Elements (Nikon), and where added adjusted images are needed to capture the range of fluorescence intensities in an experiment, additional images are presented that are also evenly adjusted and the change in this adjustment is indicated in the figure panel.
Fluorescence microscopy image analysis and statistical tests
To define the localizations represented for both EGFP- and mNG-tagged BEPs, all images were analyzed using the Image J (National Institutes of Health, Bethesda, MD) with the Cell Counter plugin to categorize the localization for every cell in a field. These manually defined localization patterns are summarized in the pie charts presented in Figures 1B, 3E, 6A-B, S7, S8, and S9.
To quantify the whole cell fluorescence intensity for mNG-tagged BEPs, we used Nikon NIS-Elements .ai (Artificial Intelligence) and Nikon General Analysis 3 (GA3) software packages. We trained imaging on a ground-truth set of samples, where cells were segmented using images acquired with the trypan blue-stained cell surfaces or the DIC images. Next, the NIS.ai software performed iterative training until a training loss threshold of <0.2 was obtained, which is indicative of a high degree of agreement between the initial ground truth and the output generated by NIS.ai. Fields of images were then processed, using the automated segmentation, and the mean fluorescence in the green channel (480nm) was measured for each cell. Any partial cells at the edges of images were removed. Fluorescence intensities are plotted as scatter plot using Prism (GraphPad Software, San Diego, CA, USA). We performed Kruskal-Wallis statistical tests with Dunn’s post hoc correction for multiple comparisons or Students t-tests where only two samples are compared to one another. In all cases, significant p-values from these tests are represented as: * <0.05; ** <0.0005, *** <0.0005; ns >0.05.
Biochemical properties, transmembrane domain prediction, and statistics
Amino acid sequences of the BEPs were analyzed for their biochemical properties using python. A custom script that used the packages Biopython and ‘peptides’ was used to calculate hydrophobicity and length in different sequence regions. Transmembrane domain prediction was conducted by accessing the Phobius (https://phobius.sbc.su.se) and TMHMM 2.0 (https://services.healthtech.dtu.dk/services/TMHMM-2.0/) online servers (Krogh, Larsson et al. 2001) . A fasta file containing all the amino acid sequences was uploaded and both analyses were run using default parameters. All statistical tests were performed in python using scipy.stats.
Description and re-analysis of published datasets
Localization annotations for ancient proteins (shown in Fig 1D) were obtained from a published study using the yeast GFP collection (Chong, Koh et al. 2015). The localizations assignments from replicate “WT1” were parsed and proteins were grouped into “ER” or “other” categories. For proteins that exhibited more than one localization, if one was ER, than that protein was counted as “ER”, otherwise it was placed in the “other”.
Fitness measurements for BEPs were obtained and reanalyzed from (Vakirlis, Acar et al. 2020). For each BEP we counted the number of conditions that it was beneficial relative to a control, with the minimum and maximum number being 1 and 5, respectively.
Supplementary Material
Figure S1. Immunoblot analyses of BEPs. (accompanies Figure 1)
A representative immunoblot of protein extracts made for EGFP C-terminally tagged BEPs expressed from plasmids as shown in Figure 1B. Labeled in green are the ER localized BEPs based on the microscopy shown in Figure 1B. The BEPs in gray are the cases identified as potentially being break down/processed products. REVERT total protein stain serves as the loading control.
Figure S2. ER BEPs amino acid sequences and predicted TMDs. (accompanies Figure 2)
Amino acid sequence of 21 of the 26 BEPs that have at least one predicted TMD region determined by Phobius (Kall, Krogh et al. 2004) highlighted in yellow, or underlined by TMHMM (Krogh, Larsson et al. 2001). The first seven sequences boxed in green correspond to the ER-localized BEPs identified in Figure 1B.
Figure S3. Sequence analysis of BEP protein sequences using TMHMM reveals ER-localized BEPs have short and hydrophobic C-termini.
(accompanies Figure 3).
A. ER-localized BEPs have significantly shorter C-terminal ends than other TMD-containing BEPs. Comparison of the distributions of the length in amino acid residues for different sequence regions in ER-localized and other TMD-containing BEPs as predicted by TMHMM (refer to Figure 3A for the definition of different sequence regions)). B. ER-localized BEPs have more hydrophobic C-terminal ends than other TMD-containing BEPs. Hydrophobicity values (Kyte-Doolittle scale) for sequence regions in both ER and other TMD-containing BEPs as predicted by TMHMM. A-B. All P values calculated from Mann-Whitney U test. **: p <0.005, *:p<0.05 Full=Full sequence, n-term=N-terminal region, c-term=C-terminal region. For full, n-term, and c-term, n=18. For tmd, n=28. For flanking, n=56. For non-tmd n=46. C. Hydrophobicity versus length in amino acid residues for the C termini of all TMD containing BEPs as predicted by TMHMM. Processed/undefined: BEPs that showed some evidence of breakdown in western blots (see Figure S1). Size of the dots represent percent of expressing cells that had ER localization. Corresponding systematic gene names from Saccharomyces Genome Database shown next to each dot.
Figure S4. ER BEPs in cells lacking functional GET or SND pathways. (accompanies Figure 4)
Fluorescence micrographs of A. mNG or B. ER-BEPs N-terminally tagged with mNG and C. ER-BEPs C-terminally tagged with mNG, in the indicated deletion backgrounds are shown. These micrographs represent the same analysis done for Ybr196c-a on Figure 4A and 4C. The percentage of cells with the number of puncta per cell are indicated as a stacked distribution, based on these images and are shown in Figure 4E and 4F, respectively. Increasing numbers of puncta per cell, and concomitant loss of clear ER localization, are associated with disruption of the ER-insertion pathways needed for BEP targeting.
Figure S5. Figure. mNG stability is not dependent upon Doa10 or the proteasome, but N-terminally tagged mNG-Ybr196c-a stability remains dependent on both these factors. (accompanies Figure 5)
A. A representative immunoblot of protein extracts made for free mNG in the indicated deletion backgrounds. B. The relative abundance of free mNG from four biological replicates of the immunoblot in A. for the strains indicated. A Student’s t-test comparing the WT to the PQC deletions was used to assess statistical differences. (not significant = ns; p-value < 0.05 = *). C. A representative immunoblot of protein extracts expressing free mNG from WT or doa10Δ cells post cycloheximide (CHX) addition for the times indicated (in minutes) is shown. D. Quantification of the percent of mNG remaining post-CHX addition is indicated. The dots represent the mean of the biological replicates (n=3) and the error bars show the standard error of the mean. A Student’s t-test comparing the WT to the doa10Δ cells was used to assess statistical differences (not significant = ns; p-value < 0.05 = *; p-value <0.0005 = ***). E. A representative immunoblot of protein extracts expressing mNG-Ybr196c-a from WT or doa10Δ cells post cycloheximide (CHX) addition for the times indicated (in minutes) is shown. F. Quantification of the percent of mNG-Ybr196c-a remaining post-CHX addition is indicated. The dots represent the mean of the biological replicates (n=3) and the error bars show the standard error of the mean. A Student’s t-test comparing the WT to the doa10Δ cells was used to assess statistical differences (not significant = ns; p-value < 0.05 = *; p-value <0.0005 = ***). G. A representative immunoblot of protein extracts expressing free mNG in pdr5Δ cells that were untreated (+DMSO) or treated with the proteasomal inhibitor MG-132 post-CHX addition is shown. H. A representative immunoblot of protein extracts expressing Ybr196c-a-mNG in pdr5Δ cells that were untreated (+DMSO) or treated with the proteasomal inhibitor MG-132 post-CHX addition is shown.
Figure S6. ER BEPs are stabilized by loss of ER PQC factors Doa10 and/or Hrd1. (accompanies Figure 5)
A-E. Representative immunoblot of protein extracts made for each ER-localized BEP expressing the BEP fused N or C-terminally with mNG in the indicated deletion backgrounds. Panels A-E show the different ER-localized BEPs as was done for Ybr196c-a in Figure 5A.
Figure S7. ER BEPs increase in abundance when ER PQC factors are lost. (accompanies Figure 6)
Representative fluorescence microscopy of N-terminally or C-terminally-mNG tagged for A-B, Ydl118w; D-E, Yil134c-a and G, Ymr151w, in the background deletions listed in the figure, as shown for Ybr196c-a in Figure 6A and 6B. The distribution of fluorescence observed across the cell populations is indicated in the pie chart below each diagram (n=number of cells assessed). Cells are stained with CMAC-blue to mark vacuoles and imaged via DIC, which is shown in the merge. C, F and H. Quantification of the whole cell fluorescence intensities determined using the NIS.ai and GA3 software (see methods), for the cells shown in panels A-B, D-E and G, respectively. A one-way ANOVA with Dunnett’s multiple comparisons test was performed relative to the fluorescence in WT cells to identify statistically significant changes (p-value < 0.0005 = ***).
Figure S8. ER BEPs increase in abundance when ER PQC factors are lost. (accompanies Figure 6)
Representative fluorescence microscopy of N-terminally or C-terminally-mNG tagged for A-B, Yol035c and D-E, Ypr126c, in the background deletions listed in the figure, as shown for Ybr196c-a in Figure 6A and 6B. The distribution of fluorescence observed across the cell populations is indicated in the pie chart below each diagram (n=number of cells assessed). Cells are stained with CMAC-blue to mark vacuoles and imaged via DIC, which is shown in the merge. C and F. Quantification of the whole cell fluorescence intensities determined using the NIS.ai and GA3 software (see methods), for the cells shown in panels A-B and D-E, respectively. A one-way ANOVA with Dunnett’s multiple comparisons test was performed relative to the fluorescence in WT cells to identify statistically significant changes (p-value < 0.0005 = ***).
Figure S9. Loss of Ubr1 does not improve ER localization or abundance of BEPs. (accompanies Figure 6)
A. Fluorescence micrographs of cells expressing Ydl118w-mNG or Ypr126c-mNG in WT and ubr1Δ backgrounds. Vacuoles are stained with CMAC-blue. B. Immunoblotting for detection of BEP-mNG. REVERT total protein stain serves as the loading control.
Figure S10. BEPs are stabilized by loss of vacuolar proteases. (accompanies Figure 7)
A and D. Fluorescence micrographs of cells expressing A. Ybr196c-a-mNG or D. C-terminal mNG-tagged ER-localized BEPs in WT or pep4Δ cells are shown. Vacuoles are stained with CMAC-blue and indicated on the green channel images using a white dashed line. B and E. The scatter plots indicate the whole-cell fluorescence intensities, as determined using NIS.ai and GA3 software, for mNG-tagged BEPs shown in A and D, respectively. The median fluorescence intensity and 95% confidence interval are represented by the horizontal lines. C and F. The ratio of the vacuolar fluorescence over the whole cell fluorescence for the cells imaged in A and D, respectively, is shown as a scatter plot. For graphs in B, C, E, and F, a Student’s t-test assessed statistical difference between the WT and pep4Δ cell populations (p < 0.0005 = ***). Similar analyses for N-terminal mNG-tagged BEPs are provided in Figure 7.
Acknowledgments
We thank the Dr. Jeff Brodsky and his lab (Univ. of Pittsburgh) for their insightful discussions on this work. We further acknowledge the technical assistance of Dr. Simon Watkins (Director of the Center for Biologic Imaging, Dept of Cell Biology, Univ. of Pittsburgh), Christina Goldbach (Nikon Instruments Inc.) and Dr. Chaowei Shang (Director of Microscopy and Imaging, Dietrich School of Arts and Sciences, Univ. of Pittsburgh) to establish the high-content imaging approaches used in this work. Additionally, we would like to thank all the members of the Carvunis and O’Donnell labs, as well as, the Pittsburgh Area Yeast Meeting group (PAYM) and Molecular Evolution Laboratory Discussion group (MELD), for their feedback and suggestions on various stages of this work. This work was supported by funds provided by the National Institute of General Medical Sciences of the National Institutes of Health grant DP2GM137422 awarded to A.-R.C. and the National Science Foundation grant MCB-2144349 awarded to A.-R.C and start-up funds provided by the Dept. of Biological Sciences, Univ of Pittsburgh to A.F.O.
Abbreviations:
- BEP
Beneficial Emerging Proteins
- ER
Endoplasmic Reticulum
- PQC
Protein Quality Control
- SND
Srp-iNDependent targeting
- GET
Guided Entry of Tail-anchored proteins
- CHX
Cycloheximide
- mNG
m-Neon Green
- TMD
Transmembrane Domain
- Ub
Ubiquitination
Footnotes
Declaration of interests
A.-R.C. is a member of the Scientific Advisory Board for Flagship Labs 69, Inc. (ProFound Therapeutics).
Data Availability
Supplementary table 1 is available for download: https://github.com/cjhough/ER_BEPS
References
- Akopian D., Shen K., Zhang X. and Shan S. O. (2013). "Signal recognition particle: an essential protein-targeting machine." Annu Rev Biochem 82: 693–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberti S., Gitler A. D. and Lindquist S. (2007). "A suite of Gateway cloning vectors for high-throughput genetic analysis in Saccharomyces cerevisiae." Yeast 24(10): 913–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almagro Armenteros J. J., Salvatore M., Emanuelsson O., Winther O., von Heijne G., Elofsson A. and Nielsen H. (2019). "Detecting sequence signals in targeting peptides using deep learning." Life Sci Alliance 2(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberg D. C. (2005). "Methods in yeast genetics: a Cold Spring Harbor Laboratory course manual." (No Title). [Google Scholar]
- Aubel M., Eicholt L. and Bornberg-Bauer E. (2023). "Assessing structure and disorder prediction tools for ovo emerged proteins in the age of machine learning." F1000Res 12: 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviram N. and Schuldiner M. (2017). "Targeting and translocation of proteins to the endoplasmic reticulum at a glance." J Cell Sci 130(24): 4079–4085. [DOI] [PubMed] [Google Scholar]
- Bernales S., Schuck S. and Walter P. (2007). "ER-phagy: selective autophagy of the endoplasmic reticulum." Autophagy 3(3): 285–287. [DOI] [PubMed] [Google Scholar]
- Bohnsack M. T. and Schleiff E. (2010). "The evolution of protein targeting and translocation systems." Biochim Biophys Acta 1803(10): 1115–1130. [DOI] [PubMed] [Google Scholar]
- Borgese N., Coy-Vergara J., Colombo S. F. and Schwappach B. (2019). "The Ways of Tails: the GET Pathway and more." Protein J 38(3): 289–305. [DOI] [PubMed] [Google Scholar]
- Bowman R. W. 2nd, Jordahl E. M., Davis S., Hedayati S., Barsouk H., Ozbaki-Yagan N., Chiang A., Li Y. and O'Donnell A. F. (2022). "TORC1 Signaling Controls the Stability and Function of alpha-Arrestins Aly1 and Aly2." Biomolecules 12(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broeils L. A., Ruiz-Orera J., Snel B., Hubner N. and van Heesch S. (2023). "Evolution and implications of de novo genes in humans." Nat Ecol Evol 7(6): 804–815. [DOI] [PubMed] [Google Scholar]
- Casola C., Owoyemi A. and Vakirlis N. (2024). "Degradation determinants are abundant in human noncanonical proteins." bioRxiv: 2024.2005.2001.592071. [Google Scholar]
- Chen Y., Shanmugam S. K. and Dalbey R. E. (2019). "The Principles of Protein Targeting and Transport Across Cell Membranes." Protein J 38(3): 236–248. [DOI] [PubMed] [Google Scholar]
- Chong Y. T., Koh J. L., Friesen H., Duffy S. K., Cox M. J., Moses A., Moffat J., Boone C. and Andrews B. J. (2015). "Yeast Proteome Dynamics from Single Cell Imaging and Automated Analysis." Cell 161(6): 1413–1424. [DOI] [PubMed] [Google Scholar]
- Casola Claudio, O. A., Vakirlis Nikolaos (2024). "Degradation determinants are abundant in human 2 noncanonical proteins." bioRxiv. [Google Scholar]
- Dong C., Zhang L., Xia S., Sosa D., Arsala D. and Long M. (2022). "New gene evolution with subcellular expression patterns detected in PacBio-sequenced genomes of Drosophila genus." bioRxiv: 2022.2011.2030.518489. [Google Scholar]
- Dujon B. (1996). "The yeast genome project: what did we learn?" Trends Genet 12(7): 263–270. [DOI] [PubMed] [Google Scholar]
- Dunham MJ, D. M., Gartenberg MR, Brown GW (2015). "Methods in yeast genetics and genomics : a Cold Spring Harbor Laboratory course manual." [Google Scholar]
- Gabaldon T. and Pittis A. A. (2015). "Origin and evolution of metabolic sub-cellular compartmentalization in eukaryotes." Biochimie 119: 262–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager N. A., Krasowski C. J., Mackie T. D., Kolb A. R., Needham P. G., Augustine A. A., Dempsey A., Szent-Gyorgyi C., Bruchez M. P., Bain D. J., Kwiatkowski A. V., O'Donnell A. F. and Brodsky J. L. (2018). "Select alpha-arrestins control cell-surface abundance of the mammalian Kir2.1 potassium channel in a yeast model." J Biol Chem 293(28): 11006–11021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasenjager S., Bologna A., Essen L. O., Spadaccini R. and Taxis C. (2023). "C-terminal sequence stability profiling in Saccharomyces cerevisiae reveals protective protein quality control pathways." J Biol Chem 299(9): 105166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashishita M., Kawahara H. and Yokota N. (2019). "BAG6 deficiency induces mis-distribution of mitochondrial clusters under depolarization." FEBS Open Bio 9(7): 1281–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht K. A., O'Donnell A. F. and Brodsky J. L. (2014). "The proteolytic landscape of the yeast vacuole." Cell Logist 4(1): e28023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihrke G., Kyttala A., Russell M. R., Rous B. A. and Luzio J. P. (2004). "Differential use of two AP-3-mediated pathways by lysosomal membrane proteins." Traffic 5(12): 946–962. [DOI] [PubMed] [Google Scholar]
- Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Zidek A., Potapenko A., Bridgland A., Meyer C., Kohl S. A. A., Ballard A. J., Cowie A., Romera-Paredes B., Nikolov S., Jain R., Adler J., Back T., Petersen S., Reiman D., Clancy E., Zielinski M., Steinegger M., Pacholska M., Berghammer T., Bodenstein S., Silver D., Vinyals O., Senior A. W., Kavukcuoglu K., Kohli P. and Hassabis D. (2021). "Highly accurate protein structure prediction with AlphaFold." Nature 596(7873): 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser C. A., Preuss D., Grisafi P. and Botstein D. (1987). "Many random sequences functionally replace the secretion signal sequence of yeast invertase." Science 235(4786): 312–317. [DOI] [PubMed] [Google Scholar]
- Kall L., Krogh A. and Sonnhammer E. L. (2004). "A combined transmembrane topology and signal peptide prediction method." J Mol Biol 338(5): 1027–1036. [DOI] [PubMed] [Google Scholar]
- Kesner J. S., Chen Z., Shi P., Aparicio A. O., Murphy M. R., Guo Y., Trehan A., Lipponen J. E., Recinos Y., Myeku N. and Wu X. (2023). "Noncoding translation mitigation." Nature 617(7960): 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knupp J., Pletan M. L., Arvan P. and Tsai B. (2023). "Autophagy of the ER: the secretome finds the lysosome." FEBS J 290(24): 5656–5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A., Larsson B., von Heijne G. and Sonnhammer E. L. (2001). "Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes." J Mol Biol 305(3): 567–580. [DOI] [PubMed] [Google Scholar]
- Lemire B. D., Fankhauser C., Baker A. and Schatz G. (1989). "The mitochondrial targeting function of randomly generated peptide sequences correlates with predicted helical amphiphilicity." J Biol Chem 264(34): 20206–20215. [PubMed] [Google Scholar]
- Li S. C. and Kane P. M. (2009). "The yeast lysosome-like vacuole: endpoint and crossroads." Biochim Biophys Acta 1793(4): 650–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIsaac R. S., Oakes B. L., Wang X., Dummit K. A., Botstein D. and Noyes M. B. (2013). "Synthetic gene expression perturbation systems with rapid, tunable, single-gene specificity in yeast." Nucleic Acids Res 41(4):e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIsaac R. S., Silverman S. J., McClean M. N., Gibney P. A., Macinskas J., Hickman M. J., Petti A. A. and Botstein D. (2011). "Fast-acting and nearly gratuitous induction of gene expression and protein depletion in Saccharomyces cerevisiae." Mol Biol Cell 22(22): 4447–4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehlhorn D. G., Asseck L. Y. and Grefen C. (2021). "Looking for a safe haven: tail-anchored proteins and their membrane insertion pathways." Plant Physiol 187(4): 1916–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsukasa K. and Brodsky J. L. (2008). "The recognition and retrotranslocation of misfolded proteins from the endoplasmic reticulum." Traffic 9(6): 861–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newstead S. and Barr F. (2020). "Molecular basis for KDEL-mediated retrieval of escaped ER-resident proteins - SWEET talking the COPs." J Cell Sci 133(19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh S. B., Houghton C., Van Oss S. B., Wacholder A. and Carvunis A. R. (2022). "Origins, evolution, and physiological implications of de novo genes in yeast." Yeast 39(9): 471–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J., Svetec N., Molina H. and Zhao L. (2024). "The Origin and Evolution of Sex Peptide and Sex Peptide Receptor Interactions." Mol Biol Evol 41(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J. and Zhao L. (2023). "The origin and structural evolution of de novo genes in Drosophila." bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers E. T. and Balch W. E. (2013). "Diversity in the origins of proteostasis networks--a driver for protein function in evolution." Nat Rev Mol Cell Biol 14(4): 237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao M., Okreglak V., Chio U. S., Cho H., Walter P. and Shan S. O. (2016). "Multiple selection filters ensure accurate tail-anchored membrane protein targeting." Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebeaud M. E., Mallik S., Goloubinoff P. and Tawfik D. S. (2021). "On the evolution of chaperones and cochaperones and the expansion of proteomes across the Tree of Life." Proc Natl Acad Sci U S A 118(21). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reggiori F. and Klionsky D. J. (2013). "Autophagic processes in yeast: mechanism, machinery and regulation." Genetics 194(2): 341–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandmann C. L., Schulz J. F., Ruiz-Orera J., Kirchner M., Ziehm M., Adami E., Marczenke M., Christ A., Liebe N., Greiner J., Schoenenberger A., Muecke M. B., Liang N., Moritz R. L., Sun Z., Deutsch E. W., Gotthardt M., Mudge J. M., Prensner J. R., Willnow T. E., Mertins P., van Heesch S. and Hubner N. (2023). "Evolutionary origins and interactomes of human, young microproteins and small peptides translated from short open reading frames." Mol Cell 83(6): 994–1011 e1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider-Poetsch T., Ju J., Eyler D. E., Dang Y., Bhat S., Merrick W. C., Green R., Shen B. and Liu J. O. (2010). "Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin." Nat Chem Biol 6(3): 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan S. O. (2016). "ATPase and GTPase Tangos Drive Intracellular Protein Transport." Trends Biochem Sci 41(12): 1050–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S. and Hegde R. S. (2011). "Membrane protein insertion at the endoplasmic reticulum." Annu Rev Cell Dev Biol 27: 25–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherpa D., Chrustowicz J. and Schulman B. A. (2022). "How the ends signal the end: Regulation by E3 ubiquitin ligases recognizing protein termini." Mol Cell 82(8): 1424–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stayton C. T. (2015). "What does convergent evolution mean? The interpretation of convergence and its implications in the search for limits to evolution." Interface Focus 5(6): 20150039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson R., Locher M. and Hochstrasser M. (2001). "A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matalpha2 repressor degradation." Genes Dev 15(20): 2660–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassios E., Nikolaou C. and Vakirlis N. (2023). "Intergenic Regions of Saccharomycotina Yeasts are Enriched in Potential to Encode Transmembrane Domains." Mol Biol Evol 40(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger T. C., Liebschner D., Croll T. I., Williams C. J., McCoy A. J., Poon B. K., Afonine P. V., Oeffner R. D., Richardson J. S., Read R. J. and Adams P. D. (2024). "AlphaFold predictions are valuable hypotheses and accelerate but do not replace experimental structure determination." Nat Methods 21(1): 110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teufel F., Almagro Armenteros J. J., Johansen A. R., Gislason M. H., Pihl S. I., Tsirigos K. D., Winther O., Brunak S., von Heijne G. and Nielsen H. (2022). "SignalP 6.0 predicts all five types of signal peptides using protein language models." Nat Biotechnol 40(7): 1023–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakirlis N., Acar O., Hsu B., Castilho Coelho N., Van Oss S. B., Wacholder A., Medetgul-Ernar K., Bowman R. W. 2nd, Hines C. P., Iannotta J., Parikh S. B., McLysaght A., Camacho C. J., O'Donnell A. F., Ideker T. and Carvunis A. R. (2020). "De novo emergence of adaptive membrane proteins from thymine-rich genomic sequences." Nat Commun 11(1): 781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heesch S., Witte F., Schneider-Lunitz V., Schulz J. F., Adami E., Faber A. B., Kirchner M., Maatz H., Blachut S., Sandmann C. L., Kanda M., Worth C. L., Schafer S., Calviello L., Merriott R., Patone G., Hummel O., Wyler E., Obermayer B., Mucke M. B., Lindberg E. L., Trnka F., Memczak S., Schilling M., Felkin L. E., Barton P. J. R., Quaife N. M., Vanezis K., Diecke S., Mukai M., Mah N., Oh S. J., Kurtz A., Schramm C., Schwinge D., Sebode M., Harakalova M., Asselbergs F. W., Vink A., de Weger R. A., Viswanathan S., Widjaja A. A., Gartner-Rommel A., Milting H., Dos Remedios C., Knosalla C., Mertins P., Landthaler M., Vingron M., Linke W. A., Seidman J. G., Seidman C. E., Rajewsky N., Ohler U., Cook S. A. and Hubner N. (2019). "The Translational Landscape of the Human Heart." Cell 178(1): 242–260 e229. [DOI] [PubMed] [Google Scholar]
- Van Oss S. B. and Carvunis A. R. (2019). "De novo gene birth." PLoS Genet 15(5): e1008160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi M., Anyango S., Deshpande M., Nair S., Natassia C., Yordanova G., Yuan D., Stroe O., Wood G., Laydon A., Zidek A., Green T., Tunyasuvunakool K., Petersen S., Jumper J., Clancy E., Green R., Vora A., Lutfi M., Figurnov M., Cowie A., Hobbs N., Kohli P., Kleywegt G., Birney E., Hassabis D. and Velankar S. (2022). "AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models." Nucleic Acids Res 50(D1): D439–D444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A. (2011). "The N-end rule pathway and regulation by proteolysis." Protein Sci 20(8): 1298–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch J. R., McMurray M. A., Nelson Z. W. and Gottschling D. E. (2009). "Mitochondrial dysfunction leads to nuclear genome instability via an iron-sulfur cluster defect." Cell 137(7): 1247–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verster A. J., Styles E. B., Mateo A., Derry W. B., Andrews B. J. and Fraser A. G. (2017). "Taxonomically Restricted Genes with Essential Functions Frequently Play Roles in Chromosome Segregation in Caenorhabditis elegans and Saccharomyces cerevisiae." G3 (Bethesda) 7(10): 3337–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitali D. G., Sinzel M., Bulthuis E. P., Kolb A., Zabel S., Mehlhorn D. G., Figueiredo Costa B., Farkas A., Clancy A., Schuldiner M., Grefen C., Schwappach B., Borgese N. and Rapaport D. (2018). "The GET pathway can increase the risk of mitochondrial outer membrane proteins to be mistargeted to the ER." J Cell Sci 131(10). [DOI] [PubMed] [Google Scholar]
- Volland C., Urban-Grimal D., Geraud G. and Haguenauer-Tsapis R. (1994). "Endocytosis and degradation of the yeast uracil permease under adverse conditions." J Biol Chem 269(13): 9833–9841. [PubMed] [Google Scholar]
- Wacholder A., Parikh S. B., Coelho N. C., Acar O., Houghton C., Chou L. and Carvunis A. R. (2023). "A vast evolutionarily transient translatome contributes to phenotype and fitness." Cell Syst 14(5): 363–381 e368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L. and Walter P. (2020). "Msp1/ATAD1 in Protein Quality Control and Regulation of Synaptic Activities." Annu Rev Cell Dev Biol 36: 141–164. [DOI] [PubMed] [Google Scholar]
- Weill U., Krieger G., Avihou Z., Milo R., Schuldiner M. and Davidi D. (2019). "Assessment of GFP Tag Position on Protein Localization and Growth Fitness in Yeast." J Mol Biol 431(3): 636–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisman C. M. (2022). "The Origins and Functions of De Novo Genes: Against All Odds?" J Mol Evol 90(3-4): 244–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolford C. A., Daniels L. B., Park F. J., Jones E. W., Van Arsdell J. N. and Innis M. A. (1986). "The PEP4 gene encodes an aspartyl protease implicated in the posttranslational regulation of Saccharomyces cerevisiae vacuolar hydrolases." Mol Cell Biol 6(7): 2500–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Li Q., Stroup E. K., Wang S. and Ji Z. (2024). "Widespread stable noncanonical peptides identified by integrated analyses of ribosome profiling and ORF features." Nat Commun 15(1): 1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L., Svetec N. and Begun D. J. (2024). "De Novo Genes." Annu Rev Genet. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Immunoblot analyses of BEPs. (accompanies Figure 1)
A representative immunoblot of protein extracts made for EGFP C-terminally tagged BEPs expressed from plasmids as shown in Figure 1B. Labeled in green are the ER localized BEPs based on the microscopy shown in Figure 1B. The BEPs in gray are the cases identified as potentially being break down/processed products. REVERT total protein stain serves as the loading control.
Figure S2. ER BEPs amino acid sequences and predicted TMDs. (accompanies Figure 2)
Amino acid sequence of 21 of the 26 BEPs that have at least one predicted TMD region determined by Phobius (Kall, Krogh et al. 2004) highlighted in yellow, or underlined by TMHMM (Krogh, Larsson et al. 2001). The first seven sequences boxed in green correspond to the ER-localized BEPs identified in Figure 1B.
Figure S3. Sequence analysis of BEP protein sequences using TMHMM reveals ER-localized BEPs have short and hydrophobic C-termini.
(accompanies Figure 3).
A. ER-localized BEPs have significantly shorter C-terminal ends than other TMD-containing BEPs. Comparison of the distributions of the length in amino acid residues for different sequence regions in ER-localized and other TMD-containing BEPs as predicted by TMHMM (refer to Figure 3A for the definition of different sequence regions)). B. ER-localized BEPs have more hydrophobic C-terminal ends than other TMD-containing BEPs. Hydrophobicity values (Kyte-Doolittle scale) for sequence regions in both ER and other TMD-containing BEPs as predicted by TMHMM. A-B. All P values calculated from Mann-Whitney U test. **: p <0.005, *:p<0.05 Full=Full sequence, n-term=N-terminal region, c-term=C-terminal region. For full, n-term, and c-term, n=18. For tmd, n=28. For flanking, n=56. For non-tmd n=46. C. Hydrophobicity versus length in amino acid residues for the C termini of all TMD containing BEPs as predicted by TMHMM. Processed/undefined: BEPs that showed some evidence of breakdown in western blots (see Figure S1). Size of the dots represent percent of expressing cells that had ER localization. Corresponding systematic gene names from Saccharomyces Genome Database shown next to each dot.
Figure S4. ER BEPs in cells lacking functional GET or SND pathways. (accompanies Figure 4)
Fluorescence micrographs of A. mNG or B. ER-BEPs N-terminally tagged with mNG and C. ER-BEPs C-terminally tagged with mNG, in the indicated deletion backgrounds are shown. These micrographs represent the same analysis done for Ybr196c-a on Figure 4A and 4C. The percentage of cells with the number of puncta per cell are indicated as a stacked distribution, based on these images and are shown in Figure 4E and 4F, respectively. Increasing numbers of puncta per cell, and concomitant loss of clear ER localization, are associated with disruption of the ER-insertion pathways needed for BEP targeting.
Figure S5. Figure. mNG stability is not dependent upon Doa10 or the proteasome, but N-terminally tagged mNG-Ybr196c-a stability remains dependent on both these factors. (accompanies Figure 5)
A. A representative immunoblot of protein extracts made for free mNG in the indicated deletion backgrounds. B. The relative abundance of free mNG from four biological replicates of the immunoblot in A. for the strains indicated. A Student’s t-test comparing the WT to the PQC deletions was used to assess statistical differences. (not significant = ns; p-value < 0.05 = *). C. A representative immunoblot of protein extracts expressing free mNG from WT or doa10Δ cells post cycloheximide (CHX) addition for the times indicated (in minutes) is shown. D. Quantification of the percent of mNG remaining post-CHX addition is indicated. The dots represent the mean of the biological replicates (n=3) and the error bars show the standard error of the mean. A Student’s t-test comparing the WT to the doa10Δ cells was used to assess statistical differences (not significant = ns; p-value < 0.05 = *; p-value <0.0005 = ***). E. A representative immunoblot of protein extracts expressing mNG-Ybr196c-a from WT or doa10Δ cells post cycloheximide (CHX) addition for the times indicated (in minutes) is shown. F. Quantification of the percent of mNG-Ybr196c-a remaining post-CHX addition is indicated. The dots represent the mean of the biological replicates (n=3) and the error bars show the standard error of the mean. A Student’s t-test comparing the WT to the doa10Δ cells was used to assess statistical differences (not significant = ns; p-value < 0.05 = *; p-value <0.0005 = ***). G. A representative immunoblot of protein extracts expressing free mNG in pdr5Δ cells that were untreated (+DMSO) or treated with the proteasomal inhibitor MG-132 post-CHX addition is shown. H. A representative immunoblot of protein extracts expressing Ybr196c-a-mNG in pdr5Δ cells that were untreated (+DMSO) or treated with the proteasomal inhibitor MG-132 post-CHX addition is shown.
Figure S6. ER BEPs are stabilized by loss of ER PQC factors Doa10 and/or Hrd1. (accompanies Figure 5)
A-E. Representative immunoblot of protein extracts made for each ER-localized BEP expressing the BEP fused N or C-terminally with mNG in the indicated deletion backgrounds. Panels A-E show the different ER-localized BEPs as was done for Ybr196c-a in Figure 5A.
Figure S7. ER BEPs increase in abundance when ER PQC factors are lost. (accompanies Figure 6)
Representative fluorescence microscopy of N-terminally or C-terminally-mNG tagged for A-B, Ydl118w; D-E, Yil134c-a and G, Ymr151w, in the background deletions listed in the figure, as shown for Ybr196c-a in Figure 6A and 6B. The distribution of fluorescence observed across the cell populations is indicated in the pie chart below each diagram (n=number of cells assessed). Cells are stained with CMAC-blue to mark vacuoles and imaged via DIC, which is shown in the merge. C, F and H. Quantification of the whole cell fluorescence intensities determined using the NIS.ai and GA3 software (see methods), for the cells shown in panels A-B, D-E and G, respectively. A one-way ANOVA with Dunnett’s multiple comparisons test was performed relative to the fluorescence in WT cells to identify statistically significant changes (p-value < 0.0005 = ***).
Figure S8. ER BEPs increase in abundance when ER PQC factors are lost. (accompanies Figure 6)
Representative fluorescence microscopy of N-terminally or C-terminally-mNG tagged for A-B, Yol035c and D-E, Ypr126c, in the background deletions listed in the figure, as shown for Ybr196c-a in Figure 6A and 6B. The distribution of fluorescence observed across the cell populations is indicated in the pie chart below each diagram (n=number of cells assessed). Cells are stained with CMAC-blue to mark vacuoles and imaged via DIC, which is shown in the merge. C and F. Quantification of the whole cell fluorescence intensities determined using the NIS.ai and GA3 software (see methods), for the cells shown in panels A-B and D-E, respectively. A one-way ANOVA with Dunnett’s multiple comparisons test was performed relative to the fluorescence in WT cells to identify statistically significant changes (p-value < 0.0005 = ***).
Figure S9. Loss of Ubr1 does not improve ER localization or abundance of BEPs. (accompanies Figure 6)
A. Fluorescence micrographs of cells expressing Ydl118w-mNG or Ypr126c-mNG in WT and ubr1Δ backgrounds. Vacuoles are stained with CMAC-blue. B. Immunoblotting for detection of BEP-mNG. REVERT total protein stain serves as the loading control.
Figure S10. BEPs are stabilized by loss of vacuolar proteases. (accompanies Figure 7)
A and D. Fluorescence micrographs of cells expressing A. Ybr196c-a-mNG or D. C-terminal mNG-tagged ER-localized BEPs in WT or pep4Δ cells are shown. Vacuoles are stained with CMAC-blue and indicated on the green channel images using a white dashed line. B and E. The scatter plots indicate the whole-cell fluorescence intensities, as determined using NIS.ai and GA3 software, for mNG-tagged BEPs shown in A and D, respectively. The median fluorescence intensity and 95% confidence interval are represented by the horizontal lines. C and F. The ratio of the vacuolar fluorescence over the whole cell fluorescence for the cells imaged in A and D, respectively, is shown as a scatter plot. For graphs in B, C, E, and F, a Student’s t-test assessed statistical difference between the WT and pep4Δ cell populations (p < 0.0005 = ***). Similar analyses for N-terminal mNG-tagged BEPs are provided in Figure 7.
Data Availability Statement
Supplementary table 1 is available for download: https://github.com/cjhough/ER_BEPS