

Abstract
Polycystic kidney disease (PKD), a disease characterized by the enlargement of the kidney through cystic growth is the fourth leading cause of end-stage kidney disease world-wide. Transient receptor potential Vanilloid 4 (TRPV4), a calcium-permeable TRP, channel participates in kidney cell physiology and since TRPV4 forms complexes with another channel whose malfunction is associated to PKD, TRPP2 (or PKD2), we sought to determine whether patients with PKD, exhibit previously unknown mutations in TRPV4. Here, we report the presence of mutations in the TRPV4 gene in patients diagnosed with PKD and determine that they produce gain-of-function (GOF). Mutations in the sequence of the TRPV4 gene have been associated to a broad spectrum of neuropathies and skeletal dysplasias but not PKD, and their biophysical effects on channel function have not been elucidated. We identified and examined the functional behavior of a novel E6K mutant and of the previously known S94L and A217S mutant TRVP4 channels. The A217S mutation has been associated to mixed neuropathy and/or skeletal dysplasia phenotypes, however, the PKD carriers of these variants had not been diagnosed with these reported clinical manifestations. The presence of certain mutations in TRPV4 may influence the progression and severity of PKD through GOF mechanisms. PKD patients carrying TRVP4 mutations are putatively more likely to require dialysis or renal transplant as compared to those without these mutations.
Keywords: polycystic kidney disease, TRPV4, ion channels, PKD, kidney disease, renal function, gain-of-function, channelopathies
Graphical Abstract
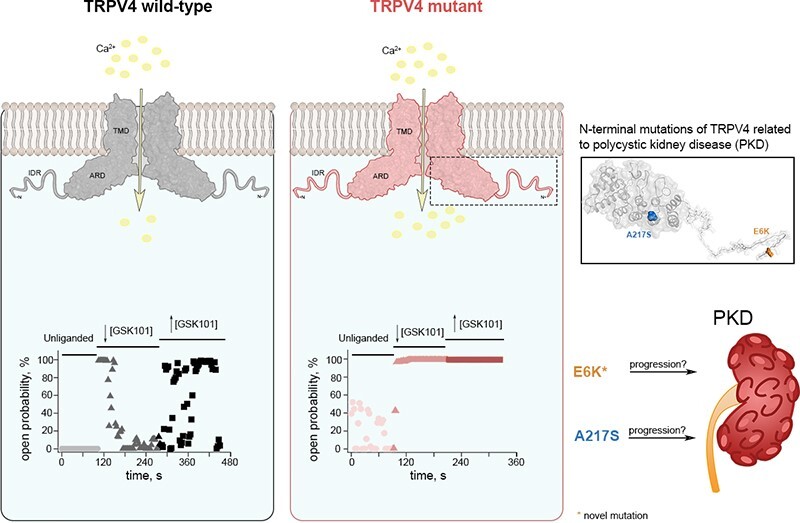
Graphical Abstract.
Introduction
Polycystic kidney disease (PKD) is the most frequent genetic cause of chronic kidney disease.1 PKD is genetically heterogeneous.2 Most patients carry pathogenic variants in either the polycystin-1 (PKD1) and polycystin-2 (PKD2) genes and show autosomal dominant inheritance. It has also been suggested that the TRPP1 and TRPP2 channels are altered by mutations, affecting Ca2+ homeostasis and co-contributing to the severity of PKD. Although a prominent role for mechanosensation in primary cilia of kidney cells and changes in Ca2+ influx into these structures had been suggested for PKD,3 recent studies have shown that PKD due to TRPP1 and TRPP2 dysfunction seems to rely on a deficit of Ca2+ signals in response to mechanical effects on the cilia.4,5
The transient receptor potential Vanilloid 4 (TRPV4) calcium-permeable channel6,7 has been shown to function as an osmo-, mechano-, and chemosensory protein,8–11 responding to several other stimuli,12−15 including the synthetic agonist GSK1016790A.16 This channel is expressed in different tissues and regulates the function of kidneys, the lung barrier,17–22 epithelial tissues (ie, skin),23–26 and of endothelial microvasculature (ie, brain).27–31
TRPV4 is essential for neuritogenesis in developing neurons, skeletal homeostasis, and remodeling of bone in response to physical activity (mechanical loading).32–34 Hence, it comes as no surprise that mutations in TRPV4, result in several human pathologies, being prominently associated with motor neuropathies and inborn motor neuron diseases, although overlapping phenotypes are possible.35–41 Some TRPV4 mutations mainly affect endochondral ossification, with shorter, wider bones and cartilage, this is consistent with high TRPV4 expression in chondrocytes.42–44
Disease-causing mutations of TRPV4 can be widespread throughout all channel domains.45–48 The human TRPV4 channel homotetramer contains subunits with a 6-helix transmembrane region, where the first four α-helices (S1-S4) form a voltage sensor-like domain, and the S5 and S6 α-helices form the pore domain.49 The cytoplasmic N- and C-termini contain the phosphoinositide binding domain (PBD), the ankyrin repeat domain (ARD), the proline-rich domain (PRD), the coupling domain, the TRP box, the calmodulin-binding domain (CAM), and the PDZ-like domain.49 Additionally, TRPV4 contains a highly flexible N-terminal intrinsically disordered region (IDR), which regulates TRPV4 function in response to other molecules, such as lipids.50 Skeletal dysplasia- and neuropathy-associated mutations are widely distributed and affect residues located in several regions, including the N-terminus of the channel, as reviewed in.45
TRPV4 has been shown to form complexes with the TRPP2 and TRPC1 channels,51–54 both of which are also relevant for PKD and some studies have proposed links between the activity of TRPV4 and PKD.55–57 Here, we searched for mutations in the TRPV4 gene of PKD patients and identified four heterozygote missense variants: E6K, A217S, P19S (previously characterized58,59), and E29A, all located at the N-terminus, two of were functionally characterized.
We also studied the previously uncharacterized gene variants S94L38 and R315W,60 which are associated to skeletomuscular dysplasias and atrophies. Our data show that these TRPV4 mutants exhibit a GOF phenotype, with increased sensitivity to the agonist, GSK1016790A and increased unliganded open probability. Importantly, these TRPV4 mutant proteins were present in the patients with more severe phenotypes, which carried additional mutations in PKD-associated genes.
Methods
Patients and Sample Collection
The study received approval of the Ethics and Research Committees from the Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán (INCMNSZ), approval no. 3232. A random sample of the registered PKD patients diagnosed with PKD through ultrasound scans61 according to previously established criteria62 at the Nephrology Department of the INCMNSZ were contacted and invited to participate in the study. Of these, 27 unrelated Mexican adults, men and women (31-78 years old), were recruited. All participants signed a written consent, and the study was conducted in agreement with the Declaration of Helsinsky. Patient oral epithelial cells were collected for DNA extraction using swabs (FAB-SWAB, Puritan).63 DNA was used for Sanger Sequencing to screen for genetic variants in all exons and exon-intron junctions of TRPV4 gene. Whole exome-sequencing (WES) was performed in 5 patients exhibiting most severe PKD phenotypes. DNA in these patients was extracted using the QIAamp Blood Kit (Qiagen). WES and bioinformatic analyses were performed at DREAMGenics (Oviedo, Spain).
Cell Transfection and Electrophysiology
Human embryonic kidney cells (HEK AD-293) (Agilent, #240085) were cultured and transfected with DNA for WT and mutant TRPV4 channels, as previously described.64,65
TRPV4 currents from transiently transfected cells were recorded with the patch-clamp technique using excised membrane patches in the inside-out configuration.66–69 Recording solutions for both the bath and the pipette contained (in m m): 130 NaCl, 3 HEPES, and 1 EDTA (pH 7.2). GSK1016790A (GSK101, Sigma-Aldrich) was prepared, as previously reported.65,70 The Hill equation was fitted to the data to obtain the Hill coefficient (n) and the apparent dissociation constant (KD).71 All-point histograms were constructed from traces where closings and openings could be readily distinguished as described in,72 and the open probability (Po) was calculated as the sum of the total open time divided by the sweep duration, also as previously described.65,70 Time-dependent open probability was represented as a time plot of all recorded sweeps. Average data for single-channel current amplitude i and Po in all experiments in each condition was represented as a scatter plot.
Mean current values were measured at steady state (∼1.5 min) and for recordings of macroscopic currents of unliganded channels, the membrane potential was held at 0 mV and the voltage was stepped from −120 to 120 mV in increments of 20 mV and then back to 0 mV.
Dose-responses for GSK101 agonist were obtained for WT and mutant TRPV4 channels, as previously described.65 Single-channel recordings were performed by acquiring several sweeps at +60 mV for 3 s, in 10-50 repetitions using previously reported protocols.65,73 To study whether the hTRPV4 mutants were constitutively open, traces were obtained in the absence of the ligand and in the presence of a given concentration of GSK1016790A.
Mutagenesis
Human TRPV4-channel mutants were constructed by a 2-step PCR method 66,67 and the DNA was sequenced to confirm the insertion of a given mutation.
Statistical Analysis
Group data are reported as the mean ± standard error of the mean (s.e.m.). The 2-tailed t-test and one-way analysis of variance, followed by Tukey’s post hoc test, was used for group comparison, and calculated with Prism software (Graphstats Technologies). Significant differences between means were considered to exist when the P-value was less than 0.05.
Results
Twenty-seven patients diagnosed with PKD underwent screening for mutations within the TRPV4 gene. Clinical characterization of the patients is shown in Table 1.74 Tables 2 and S1 show the presence of the TRPV4 gene variants identified. Four heterozygote missense variants were identified at the N-terminus of the channel: E6K (7 patients), A217S (3 patients), P19S (2 patients), and E29A (1 patient). In addition, one patient carrying both E6K + P19S, and a second one carrying the P19S + A217S variants were identified (Table 2). A total of 15 out of 27 patients were carriers of TRPV4 gene variants. Five of the most severe PKD cases (stage G5) were selected for WES analysis to identify the underlying PKD gene mutations (Table 2). Interestingly, all have additional mutations in genes associated to PKD such as HNF1B + ATP6V1B; SEC63; PKD1 + CFTR; PKD1 + PKHD1 + TSC2, and PKD2 + HNF1B + DZIP1L, the latter presented without a TRPV4 mutation (Table 2).
Table 1.
Clinical Features of Patient Cohort
| Patient | Gender | Current Age | Current CKD Stage | Progression Time | Age at Which Reached Dialysis or Transplant | Delta 2 yr (Age/KDIGO eGFR CKD-EPI) | Skeletal or Neuromuscular Manifestations | Other Clinical Manifestations | Family History |
|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 56 | G5T | 5 | 51 | −1 | no | Systemic high blood pressure, Liver and Pancreas cysts and Intracraneal aneurysm | yes |
| 2 | M | 54 | G2 | NA | NA | −4 | no | Nephrolithiasi and Diabetes type 2 | no |
| 3 | F | 78 | G4 | NA | NA | −12 | Osteoporosis | Systemic high blood pressure | no |
| 4 | F | 38 | G2 | NA | NA | −3 | no | Systemic high blood pressure and Liver cysts | yes |
| 5 | M | 57 | G4 | NA | NA | −8 | no | Systemic high blood pressure | no |
| 6 | M | 67 | G5D | 7 | 60 | 0 | no | no | no |
| 7 | F | 55 | G4 | – | NA | 0 | Occasional low back pain | Systemic high blood pressure | yes |
| 8 | M | 54 | G3aT | 9 | 45 | −17 | no | Liver cysts | no |
| 9 | F | 66 | G3bT | 15 | 51 | −2 | no | no | no |
| 10 | F | 49 | G3aT | 5 | 44 | −2 | no | no | yes |
| 11 | F | 64 | G5T | 6 | 58 | 2 | Osteoporosis | Systemic high blood pressure | no |
| 12 | M | 45 | G5D | 2 | 43 | −4 | no | Systemic high blood pressure | yes |
| 13 | M | 31 | G2 | NA | NA | −12 | no | no | no |
| 14 | F | 64 | G3a | NA | NA | −5 | Osteopenia | Nephrolithiasis and Systemic high blood pressure | no |
| 15 | F | 54 | G2 | NA | NA | −17 | no | Nephrolithiasis and Systemic high blood pressure | yes |
| 16 | M | 57 | G3bT | 11 | 46 | 2 | Myopathy (associated with steroid treatment?) | Systemic high blood pressure | yes |
| 17 | M | 72 | G3aT | 11 | 61 | −12 | Osteoporosis | no | no |
| 18 | M | 53 | G3bT | 9 | 44 | −13 | no | Systemic high blood pressure | no |
| 19 | M | 60 | G1T | 14 | 46 | −1 | no | Systemic high blood pressure | yes |
| 20 | M | 56 | G3b | NA | NA | −25 | no | no | no |
| 21 | M | 62 | G5T | 2 | 60 | −5 | no | no | yes |
| 22 | M | 59 | G3bT | 5 | 54 | −13 | no | Nephrolithiasis and Systemic high blood pressure | yes |
| 23 | F | 53 | G2 | NA | NA | −17 | no | Systemic high blood pressure and Liver cysts | no |
| 24 | F | 35 | G1 | NA | NA | −1 | no | Systemic high blood pressure | no |
| 25 | M | 51 | G3aT | 6 | 45 | −13 | no | no | no |
| 26 | F | 68 | G3aT | 7 | 61 | −3 | no | no | yes |
| 27 | F | 69 | G1T | 7 | 62 | 21 | Osteoporosis | no | no |
Current CKD stage = chronic kidney disease stage based on “Age/KDIGO eGFR CKD-EPI (mL/min, calculated by CKD-EPI creatinine equation, 2021)”: G1= ≥90 (Normal to high kidney function), G2 = 60-89 (Mildly decreased kidney function), G3a = 45-59 (Mildly to moderately decrease kidney function), G3b = 30-44 (Moderately to severely decreased kidney function) G4 = 15-29 (Severely decreased kidney function), and G5 = <15 (Kidney failure) (G1-G5). Kidney transplant recipients are T = patient with kidney transplant and D = patient on dialysis. Delta 2 yr (Age/KDIGO eGFR CKD-EPI) is: Delta of “Age/KDIGO eGFR CKD-EPI” between 2 yr creatinine measurements. Delta eGFR is an intuitive method of assessing progression and regression of chronic kidney disease (CKD). For kidney transplant recipients a « T » letter is added at the end of the stage nomenclature: CKD G1T–G5T. In the case of patients on dialysis, a « D » letter is added: CKD G1D–G5D. NA = Not applicable
Table 2.
Severity of Disease in PKD Patients
| Cases With Stage G5, Dialysis or Transplant | *Age/Stage G5, Dialysis or Transplant (Mean ± SD) | Cases Not Reaching Stage G5, Dialysis or Transplant | Total Cases | |
|---|---|---|---|---|
| Total cases | 16 | 11 | 27 | |
| Total cases with TRPV4 mutation | 12 | 50.58 ± 7.11 | 3 | 15 |
| Total cases without TRPV4 mutation | 4 | 56 ± 7.44 | 8 | 12 |
| Single mutation carriers | ||||
| E6K carriers | 6 | 52.4 ± 7.63 | 1 | 7 |
| A217S carriers | 1 | 51 | 2 | 3 |
| P19S carriers | 2 | 43,46 | 0 | 2 |
| E29A carriers | 1 | 54 | 0 | 1 |
| Double mutation carriers (not included in single mutations carriers account) | ||||
| E6K + P19S carriers | 1 | 44 | 0 | 1 |
| P19S + A217S carriers | 1 | 62 | 0 | 1 |
| Cases with found mutations in PKD genes and the TRPV4 variants present in these cases | ||||
| HNF1B (Q521Pfs*30; P) and ATP6V1B (I386Hfs*56; P) + TRPV4 (A217S) | 1 | 51 | 1 | |
| SEC63 (R239*; P) + TRPV4 (E6K) | 1 | 45 | 1 | |
| PKD1 (Q2606*; P) and CFTR (L997F; P) + TRPV4 (P19S) | 1 | 43 | 1 | |
| PKD1 (R3039S; LP), PKHD1 (D3795N; VUS) and TSC2 (G1579S; LP) + TRPV4 (E6K + P19S) | 1 | 44 | 1 | |
| PKD2 (R496C; VUS), HNF1B (Q521Pfs*30; P) and DZIP1L (A591T; VUS) without TRPV4 mutation | 1 | 45 | 1 | |
Age of patients carrying TRPV4 gene variants at which they reached stage G5 (CKD stage = chronic kidney disease stage based on Age/KDIGO eGFR CKD-EPI), dialysis or transplant. The first column presents total cases with TRPV4 mutations, total cases without TRPV4 mutations, carriers of different TRPV4 variants (E6K, A217S, P19S, E29A), and the carriers of two TRPV4 variants (E6K + P19S and P19S + A217S). Also, cases with mutations in PKD genes are presented, with TRPV4 variants if they were detected. The 5 cases with mutations in PKD-associated genes with or without mutations in TRPV4. Variants were identified on genome assembly GRCh38 (hg38). PKD-associated genes: ATP6V1B (ATPase H+ transporting V1 subunit B1), CFTR (cystic fibrosis transmembrane conductance regulator), DZIP1L (DAZ interacting zinc finger protein 1 like), HNF1B (hepatocyte nuclear factor1-beta), PKD1 (polycystin 1), PKD2 (polycystin 2), PKHD1 (polycystic kidney and hepatic disease 1, ciliary IPT domain containing fibrocystin/polyductin), SEC63 (SEC63 homolog, protein translocation regulator), and TSC2 (tuberous sclerosis complex subunit 2). The PKD gene mutations are shown followed by their clinical significance: Pathogenic (P), Likely Pathogenic (LP), and Variant of Uncertain Significance (VUS).
The E6K variant, although not reported in the literature as a causal mutation for any known disease, was found in population databases with a very low allele frequency (rs769769057, gnomAD = 0.000007126) and has been reported as a Variant of Uncertain Significance in the ClinVar database. Our data shows the presence of TRPV4 mutants E6K and A217S in PKD patients harboring different underlying PKD gene mutations (Table 2). None of these patients had been diagnosed with skeletal dysplasia or neuropathic manifestations, although these mutations have been associated to these disease.37,38,41,60 PKD patients carrying TRPV4 mutations are more likely to require dialysis or renal transplant (12 out of 15), as compared to those without TRPV4 mutations (4 out of 12) (Tables 2 and S1).
Mutant TRPV4 Channels in the IDR Present Constitutive Activity and Increased Agonist Sensitivity
Mutations in the N-terminal IDR of TRPV4 are present in patients with peripheral neuropathy or are mixed with skeletal dysplasia38,75 with affected residues located adjacent to critical regulatory sites, such as phosphorylation sites,76,77 PBD78 and the PRD.79,80
Activation of the E6K and S94L mutants in the IDR was evaluated under several conditions, by recording currents in HEK293 cells expressing WT or mutant channels. Control experiments using cells transfected only with plasmid with green fluorescent protein showed no channel openings in the presence of GSK1016790A (Figure S1), indicating that these cells do not express TRPV4 and constitute an adequate experimental model. The novel E6K mutant located at the beginning of the N-terminus (Figure 1A), was found in several PKD patients. The S94L mutation, located near the IDR regulatory regions (Figure 1A) has been described in patients with congenital spinal muscular atrophy and arthrogryposis (CSMAA) and severe skeletal abnormalities38 and patients with CSMAA but not with skeletal dysplasia, highlighting the wide phenotypic variability within the clinical spectrum of the same TRPV4 mutation.75 This mutation was not present in our PKD patient cohort, but had not been previously characterized, hence it was studied here for comparison.
Figure 1.

Mutations of two residues in the N-terminal intrinsically disordered region (IDR) result in constitutive activity of TRPV4. (A) Structure of the human TRPV4 channel (PDB: 8FC8),70 where one subunit is highlighted for visual clarity. The N-terminal IDR was modelled with the SWISS-MODEL server (Guex and Peitsch, 1997; Guex et al., 2009; Waterhouse et al., 2018) for representative purposes. Residues E6 and S94 are depicted. (B-D) Representative currents were obtained at −120 mV and +120 mV for 100 ms from a holding potential of 0 mV in the inside-out configuration in wild-type (WT, B) and mutant E6K (C) and S94L (D) channels. Leak or initial currents and currents in the presence of 10 n m or 1 µm GSK1016790A (GSK) are shown, as indicated by labels. (E) Dose-response curves for activation by GSK measured at +120 mV in inside-out patches of HEK293 cells expressing WT or mutant E6K or S94L channels. The smooth curves are fits with the Hill equation [KD = 95 ± 5 n m for WT (n = 8-17), 65 ± 5 n m for E6K (n = 4-8) and 93 ± 6 n m for S94L (n = 5-11)]. Hill coefficients were 1.9 for WT, 2.6 for E6K, and 2.4 for S94L. A single GSK concentration was tested per membrane patch and normalized to the current at saturating 1 µm GSK in the same patch. Group data are reported as the mean ± s.e.m. Statistical significance was of *P < 0.01 between WT versus E6K and *P < 0.01 versus S94L in the presence of 10 n m GSK; *P < 0.01 between WT versus E6K and *P < 0.01 versus S94L in the presence of 20 n m GSK; *P < 0.05 between WT versus S94L in the presence of 50 n m GSK and *P < 0.01 versus E6K in the presence of 100 n m GSK. No statistical differences were observed starting from 200 n m to higher GSK concentrations between activation of the WT and mutant channels. A parametric unpaired t-test was performed. (F) Representative traces of single-channel recordings from WT TRPV4 channels in the unliganded state (Po = 0; i = 0 pA, n = 7) and with 10 n m (Po = 0.34 ± 0.06; i = 6.3 ± 0.5 pA, n = 7) or 100 n m (Po = 0.8 ± 0.07; i = 6.3 ± 0.3 pA, n = 6) GSK. (G) Representative single-channel recordings for the mutant TRPV4- E6K channel in the unliganded state (Po = 0.18 ± 0.003; i = 6.7 ± 0.5 pA, n = 4) and with 10 n m (Po = 0.94 ± 0.03; i = 5.8 ± 0.2 pA, n = 5) or 100 n m (Po = 0.91 ± 0.08; i = 6.15 ± 0.2 pA, n = 2) GSK. (H) Representative traces for single-channel recordings from the mutant TRPV4-S94L channel in the unliganded state (Po = 0.51 ± 0.08; i = 5.96 ± 0.4 pA, n = 11) and with 10 n m (Po = 0.82 ± 0.08; i = 5.98 ± 0.4 pA, n = 6) or 100 n m (Po = 0.94 ± 0.03; i = 5.97 ± 0.09 pA, n = 3) GSK. Closed and open states are marked with letters C and O, respectively. The histograms below each trace plot the average single-current amplitude elicited in the unliganded state or in the presence of different GSK concentrations. All single-channel recordings were performed in the inside-out configuration of the patch-clamp technique. Representative open probability (Po) of WT (I), mutant E6K (J) and S94L (K) channels through time. Values for the Po are shown at +60 mV from 0 (3 s) to up to 160 sweeps (480 s) in the unliganded state and in the presence of 10 and 100 n m GSK. Po was determined for the 3 s duration for each sweep at +60 mV.
Figure 1B-D shows representative macroscopic current traces of WT and E6K and S94L-TRPV4 channels activated with two concentrations of the agonist, GSK1016790A (GSK). The current-voltage relationships (Figure S2A-C) show the increased sensitivity of the E6K and S94L mutants to lower GSK concentrations at all voltages tested, as compared to WT channels.
To substantiate the observation that the E6K and S94L mutant TRPV4 channels displayed an apparent increase in sensitivity to GSK, we performed dose-response curves to GSK and compared them to the WT TRPV4 response. Figure 1E shows that activation of E6K (orange symbols) and S94L (purple symbols) mutants was left-shifted, as compared to the WT (black symbols) and activation curves in response to GSK yielded reduced apparent dissociation constants (KD).
The shift to the left in the dose-reponse to GSK for E6K and S94L mutants suggested a GOF mechanism, as previously proposed for S94L using cytotoxicity assays.38 Examination of the leak or unliganded macroscopic currents showed spontaneous openings in both mutants, that were not observed in WT channels, suggesting constitutive activity of the mutant channels (Figure S2D-F).
Single-channel recordings were performed to examine the biophysical details of the activation of the WT and E6K and S94L mutants, in unliganded conditions and with different concentrations of GSK. WT channels did not open in the absence of ligand (Figure 1F, light gray), however, in the presence of 10 n m GSK, opening bursts were observed (dark grey, Figure 1F) and the average single-channel current amplitude was 6.33 ± 0.5 pA (right, middle, Figure 1F). With 100 n m GSK, the channel exhibited an increase in opening events (black, Figure 1F) without changes in the single-channel current amplitude (right, bottom, Figure 1F).
Single-channel currents of the E6K mutant revealed the presence of brief opening bursts in the unliganded state (light orange, Figure 1G). Single-channel current amplitudes for the unliganded state and with 10 or 100 n m GSK, remained similar among them (Figure 1G, right, top, middle and bottom), as compared to WT. Single-channel current amplitude for the S94L mutant channel was measured and remained constant at different concentrations of GSK (Figure 1H).
The open probability (Po) as a function of time for these single-residue mutations in TRPV4 was evaluated. Representative time courses for changes in Po for unliganded and after exposure to two GSK concentrations are shown for WT, E6K-, and S94L-TRPV4 channels (Figure 1I-K). As for the WT channel (Figure 1I), the unliganded state displayed very low Po values throughout the duration of the experiments (circles, Figure 1I). Addition of 10 and 100 n m GSK led to opening bursts and an increase in the Po (triangles and squares, Figures 1I). In the unliganded state, the E6K-TRPV4 displayed increased Po as compared to the WT. Addition of 10 and 100 n m GSK stabilized the open state and the Po readily increased (Figures 1J, triangles and squares, respectively), hence the open state of the E6K mutant channel is favored by lower concentrations of the agonist, as compared to WT channels. A similar behavior was observed for the S94L (circles, triangles and squares, Figure 1K).
A summary of the data obtained both for single-channel current amplitudes and for Po values for unliganded and liganded WT, E6K-, and S94L are shown in (Figure S3A-B). These experiments allowed us to conclude that the presence of E6K and S94L mutations in the IDR of the N-terminus of TRPV4 lead to an increase in activity even in the absence of an agonist.
Gain of Function of ARD TRPV4 Mutants
Most TRPV4 channel mutations identified in patients with mixed skeletal and neuromuscular disease phenotypes are localized to the ARD in the N-terminus of the channel.41 We decided to study the functional behavior of the A217S TRPV4-mutant channel, identified in 4/27 of the studied PKD patients and previously highlighted as a possible cause for spondylometaphyseal dysplasias Kozlowski type (SMDK) and scapuloperoneal spinal muscular atrophy (SPSMA).37 For comparison, we also performed electrophysiological experiments with the R315W mutant, previously shown to increase calcium influx and cytotoxicity as a GOF mutant.38,81 The R315W mutant has been identified in patients with neuropathies35,82,83 and patients with a mixed phenotype with SMDK and Charcot-Marie-Tooth disease type 2C neuropathy,41,84 but was not present in our patient cohort.
A217 and R315 residues, are located at the N-terminal region in the ARD’s (Figure 2A). A217 resides within the α-helix of the second ankyrin repeat (AR2), while R315 residue protrudes from the fourth ankyrin repeat (AR4) and is exposed toward the membrane-facing surface. Our experiments show that these mutants exhibit increased activation at a lower agonist concentration (10 n m), as compared to WT channels (Figure 2B-D). The activation of the A217S and R315W mutant TRPV4 channels in response to different concentrations of GSK was also shifted to the left, yielding slightly lower apparent KD values for both mutants, as compared to the WT channels (Figure 2E). Finally, the increase in the response of the A217S and R315W mutants to 10 nm GSK (triangles), as compared to WT channels, was evident at all voltages (Figure S4A-C).
Figure 2.

Mutations of two residues of ARD of the N-terminus of TRPV4 result in constitutively activated macroscopic currents and a shift in the response to the agonist. (A) Depiction of two subunits of the cryo-EM structure (PDB: 8FC8) [82] of the human TRPV4 channel. Residues A217 and R315W are depicted. Representative currents at −120 mV and +120 from a holding potential of 0 mV of wild-type (WT, B) and mutant A217S (C) or R315W (D) channels. Inside-out membrane patches were exposed to bath solution without agonist (initial), 10 n m and 1 µm GSK1016790A (GSK). (E) Dose-response curves for activation by GSK measured at +120 mV in inside-out patches of HEK293 cells expressing WT or mutant A217S or R315W channels. The smooth curves are fits with the Hill equation (KD = 95 ± 5 n m for WT [n = 8-17, same data as in Figure 1E), 66 ± 6 n m for A217S (n = 5-11) and 83 ± 4 n m for R315W (n = 4-11)]. Hill coefficients were 1.9 for WT, 2.1 for A217S, and 2.7 for R315W. A single GSK concentration was tested per membrane patch and normalized to the current at saturating 1 µm GSK in the same patch. Group data are reported as the mean ± s.e.m. Statistical significance was of *P < 0.01 between WT versus A217S and *P < 0.001 versus R315W in the presence of 10 n m GSK; *P < 0.01 between WT versus A217S and *P < 0.0001 versus R315W in the presence of 20 n m GSK; *P < 0.01 between WT versus A217S and *P < 0.01 versus R315W in the presence of 50 n m GSK and *P < 0.001 versus A217S and *P < 0.01 versus R315W in the presence of 100 n m GSK. No statistical differences started from 200 n m GSK between the WT and mutant channels. A parametric unpaired t-test was performed. Representative single-channel recordings for the A217S TRPV4 mutant in the (F) unliganded state and in the presence of (G)10 n m or (H) 100 n m GSK at +60 mV. The average single-current amplitude elicited in the unliganded state or in the presence of different GSK concentrations is plotted in the histograms below each trace. (I) Changes in open probability (Po) through time in the unliganded state and in the presence of 10 and 100 n m GSK, as indicated in the figure. Values for the Po are shown at +60 mV from 0 (3 s) to up to 150 sweeps (450 s). Po was determined for the 3 s duration for each sweep at +60 mV. The Hill equation was fitted to the data to obtain the Hill coefficient (n) and the apparent dissociation constant (KD).71 All-point histograms were constructed, and Po was calculated and graphed as described in Figure 1.
Similarly to the IDR mutants, macroscopic currents from A217S mutant channels showed constitutive activity in the unliganded state (Figure S4D-F). Single-channel analysis of the A217S channel determined the presence of unliganded opening events (Figure 2F). Application of 10 and 100 n m GSK resulted in longer-lived openings (Figure 2G-H). Single-channel current-amplitudes were like the WT (Figure 2F-H, lower panels).
The A217S mutant TRPV4 exhibited increased Po in the unliganded state (Figure 2I, circles) and exposure to 10 or 100 n m GSK also stabilized the open state (Figures 2I, triangles and squares). No significant statistical difference in the magnitude of the unitary current among WT and A217S mutant channels (Figure S5A) was observed but the Po in the unliganded state was increased, as compared to WT channels (Figure S5B). These data suggest that the A217S mutant presents GOF mechanisms due to the increased open probability in the unliganded state.
Discussion
To date, over 70 disease-causing mutations have been described in different domains of the hTRPV4 channel structure. Functional studies show that most mutations yield GOF phenotypes and are associated with increased Ca2+ entry into the cells where they are expressed.38,41,42,81,85–93 Several of the mutations reported for TRPV4 have not been electrophysiologically studied, hence, a detailed biophysical characterization was pivotal to understand the GOF mechanisms in mutant channels, especially for some which were identified in patients with PKD. Here, we describe and characterize the novel E6K TRPV4 mutation, which is also present in patients diagnosed with PKD. We also identified and characterized other TRPV4 mutants localized in the N-terminal region of the channel within different domains. While E6K and S94L reside within the IDR, A217S locates to the ARD. Here, we show that TRPV4 mutants present in the PKD patients studied, exhibit constitutive activity in the absence of ligand, with rather high open probabilities, and an apparent increase in the affinity for a selective agonist of the channel.
Our recordings were performed using excised cell-membrane patches, hence, an increase in the number of channels, as previously reported,38 does not explain the GOF phenomenon we observed but an increase in unliganded Po does.
The N-terminus is involved in protein-protein interactions that regulate channel function. Mutations in the ARD could lead to significant structural changes in the membrane-facing surface of the ARD’s, potentially affecting the interaction of TRPV4 with other proteins and destabilizing inhibitory complexes, leading to channel opening. Recently solved cryo-EM structures of TRPV4 in complex with the cytoskeletal remodeling GTPase RhoA, which inhibits channel activity, suggest that some residues in the ARD are essential for interaction between RhoA and TRPV4.49,94 Mutations in the ARD could disrupt this surface structure and lead to destabilization of the closed state. The A217 residue is not located within the contact interface with RhoA, but is adjacent to this region in the second ARD within its helical section, it is also surrounded by other non-polar residues with which the alanine interacts through hydrophobic bonding, possibly affecting folding of the domain.37
For the S94L mutant, mechanisms are possibly different to those hypothesized for the mutations in the ARD, and it remains difficult to explain the effect of these IDR mutations on the function of the channel. However, it is important to note that the S94 residue is located among phosphorylation sites, which could modulate the response of the channel to shear-stress77 and is located in the middle of the IDR, proposed to fine-tune the response of the channel to other stimuli.80 Therefore, a variation in some of these residues could promote modifications in the interaction with PIP2 that may cause a GOF effect, future studies will be required to address this.
The E6K mutation at the beginning of the N-terminal domain and outside the middle region of the IDR, was also constitutively open. The distal N-terminus of the IDR encompasses 30 negatively-charged amino acids, which modulate attractive and repulsive interactions with other regions of the IDR, which is enriched with positively charged residues.80 These two oppositely charged regions dynamically interact to modulate channel autoinhibition by competing for the binding of PIP2, hence, the E6K mutation could affect the autoinhibition mechanisms of the TRPV4 IDR.80
The mutant TRPV4 channels found in PKD patients studied in this report could be involved in the development of cystogenesis, as has been suggested.55 The finding of the E6K mutant in 30% of our studied PKD patients, suggests a role in the phenotypic expression of the disease, possibly modifying its severity, as 6 out of 7 patients carrying this variant required dialysis or renal transplant. As for the P19S mutant, this variant has been defined as a non-synonymous single nucleotide polymorphism with loss of function in cases of hyponatremia59 and GOF in chronic obstructive pulmonary disease.58 In respiratory epithelial cells, P19S leads to abnormal Ca2+ influx into human airway epithelia and the activation of the metalloproteinase MMP1.58 Since metalloproteinases are proteolytic proteins that cleave the extracellular matrix and regulate cell proliferation this mechanism could be relevant in PKD, where the activity of metalloproteins during the development and growth of cysts has been widely determined.95–98
Moreover, patients carrying any of the four identified TRPV4 mutations were more likely to require dialysis or renal transplant as opposed to those who did not carry any TRPV4 mutations (Table 2), supporting the involvement of TRPV4 in the etiology of PKD. In summary, our results show the presence of functionally altered TRPV4 channels in PKD patients and support its possible role as a modulator of the severity of the disease.
It is important to note that, although a reduction in the influx of Ca2+, due to mutations in the PKD1 and PKD2 (TRPP2 ion channel) genes, has been highlighted as a mechanism directly related to cyst growth in PKD animal and cell models,99,100 the effect of an abnormal and not regulated entry of Ca2+ due to GOF mutations, together with mutations in the PKD genes, remains unclear. Some experimental models have proposed that the formation of TRPP2-TRPV4 complexes and the stimulation of these channels to produce an excess of Ca2+ entry could influence PKD progression.51
As for the role of activation of TRPV4 in cell proliferation, there is contrasting information in the literature. While it has been shown that other GOF mutations are associated to the proliferation of some cell types,85,101 in some experimental models activation of TRPV4 by GSK1016790A can decrease cell proliferation102 or its inhibition can result in decreased proliferation.103 It has also been described that activation of TRPV4 in a rodent model with PKD and polycystic liver disease, produces a negative effect on cyst growth.56,57,104 While regulated activation/desensitization of TRPV4 may yield beneficial effects in disease progression in PKD cell and animal models with mutations in the PKD genes, it is not currently possible to predict the outcome of the presence of GOF mutations of TRPV4 can influence cystogenesis. For instance, in other cell types, such as the alveolo-capillary barrier, excess of Ca2+ entry promotes cell damage105 and some GOF mutations in TRPV4 promote breakdown of the blood-central nervous system barrier, leading to deterioration of motor neurons.106
As the field advances, the role of different molecular effectors that may influence PKD progression are being discovered. An example of this is TMEM16A (Anoctamin 1), which is upregulated upon knock-down of PKD1 and PKD2 and whose activation due to increases in Ca2+ leads to cyst formation through the secretion of Cl− in rodent experimental models.107,108 Thus, we propose that further studies considering other molecular drivers are required to advance our knowledge on this disease.
Limitations and Future Studies
In this study, we have determined the presence of mutations, one of them previously not described in the literature, in the TRPV4 ion channel in Mexican patients with PKD and focused in studying the biophysical properties of these mutant channels in a heterologous expression system. The limitations of our work and future studies and experiments include the following: (1) Studying the presence of these mutations in a larger patient cohort, although our study constitutes the foundation for broader scope genome-wide association studies in a larger and more diverse population; (2) Studying the biophysical properties of the mutant channels in complex (heterotetramers) with TRPP21 in heterologous expression systems to determine how the function of these channels is altered; (3) Producing PKD animal models that contain mutations in PKD genes, as well as the mutations here described to determine whether the presence of these amino acid substitutions in TRPV4 channels lead to differences in the development of PKD and studying the effects at the cellular, subcellular109–111 and organismal levels; (4) Clarifying how an abnormal Ca2+ entry to the cells and organelles may influence PKD onset/progression.
Supplementary Material
Acknowledgement
We thank Consejo Nacional de Humanidades, Ciencias y Tecnologías (CONAHCYT) for a scholarship to A.M. Hernández-Vega. Ana María Hernández-Vega is a doctoral student from Programa de Maestría y Doctorado en Ciencias Bioquímicas, Universidad Nacional Autónoma de México (UNAM) and has received CONAHCYT fellowship (CVU 928982). We also thank the following personnel from the Instituto de Fisiología Celular of the Universidad Nacional Autónoma de México: Laura Ongay Larios, Ana María Escalante Gonzalbo, Francisco Pérez Eugenio, Juan Manuel Barbosa Castillo, and Gerardo Coello Coutiño for technical assistance.
Contributor Information
Ana M Hernández-Vega, Departamento de Neurociencia Cognitiva, Instituto de Fisiología Celular, Universidad Nacional Autónoma de México, Ciudad de México 04510, Mexico.
Itzel Llorente, Departamento de Neurociencia Cognitiva, Instituto de Fisiología Celular, Universidad Nacional Autónoma de México, Ciudad de México 04510, Mexico.
Raúl Sánchez-Hernández, Departamento de Neurociencia Cognitiva, Instituto de Fisiología Celular, Universidad Nacional Autónoma de México, Ciudad de México 04510, Mexico.
Yayoi Segura, Unidad de Biología Molecular y Medicina Genómica, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, Ciudad de México14080, Mexico.
Teresa Tusié-Luna, Unidad de Biología Molecular y Medicina Genómica, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, Ciudad de México14080, Mexico; Instituto de Investigaciones Biomédicas, Universidad Nacional Autónoma de México, Ciudad de México 04510, Mexico.
Luis E Morales-Buenrostro, Departmento de Nefrología y Metabolismo Mineral, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, Ciudad de México 14080, México.
Refugio García-Villegas, Departamento de Fisiología, Biofísica y Neurociencias, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, Av. Instituto Politécnico Nacional 2508, Ciudad de México 07360, Mexico.
León D Islas, Departamento de Fisiología, Facultad de Medicina. Universidad Nacional Autónoma de México,Ciudad de México 04510, Mexico.
Tamara Rosenbaum, Departamento de Neurociencia Cognitiva, Instituto de Fisiología Celular, Universidad Nacional Autónoma de México, Ciudad de México 04510, Mexico.
Funding
This research was funded by the Secretaría de Educación, Ciencia, Tecnología e Innovación (SECTEI) del Gobierno de la Ciudad de México (grant number: SECTEI/208/2019) to T. R.
Conflict of Interest
None declared.
Data Availability
The data underlying this article are available in the article and in its online supplementary material. The clones of the mutant TRPV4 channels are available upon request.
References
- 1. Cornec-Le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet North Am Ed. 2019;393(10174):919–935. [DOI] [PubMed] [Google Scholar]
- 2. Mantovani V, Bin S, Graziano C et al. Gene panel analysis in a large cohort of patients with autosomal dominant polycystic kidney disease allows the identification of 80 potentially causative novel variants and the characterization of a complex genetic architecture in a subset of families. Front Genet. 2020;11:464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nauli SM, Alenghat FJ, Luo Y et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33(2):129–137. [DOI] [PubMed] [Google Scholar]
- 4. Delling M, Indzhykulian AA, Liu X et al. Primary cilia are not calcium-responsive mechanosensors. Nature. 2016;531(7596):656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu X, Vien T, Duan J, Sheu SH, DeCaen PG, Clapham DE. Polycystin-2 is an essential ion channel subunit in the primary cilium of the renal collecting duct epithelium. eLife. 2018;7:e33183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Strotmann R, Harteneck C, Nunnenmacher K, Schultz G, Plant TD. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat Cell Biol. 2000;2(10):695–702. [DOI] [PubMed] [Google Scholar]
- 7. Liedtke W, Choe Y, Martí-Renom MA et al. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell. 2000;103(3):525–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Matthews BD, Thodeti CK, Tytell JD, Mammoto A, Overby DR, Ingber DE. Ultra-rapid activation of TRPV4 ion channels by mechanical forces applied to cell surface beta1 integrins. Integr Biol. 2010;2(9):435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Servin-Vences MR, Richardson J, Lewin GR, Poole K. Mechanoelectrical transduction in chondrocytes. Clin Exp Pharma Physio. 2018;45(5):481–488. [DOI] [PubMed] [Google Scholar]
- 10. Servin-Vences MR, Moroni M, Lewin GR, Poole K. Direct measurement of TRPV4 and PIEZO1 activity reveals multiple mechanotransduction pathways in chondrocytes. eLife. 2017;6:e21074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. O'Conor CJ, Leddy HA, Benefield HC, Liedtke WB, Guilak F. TRPV4-mediated mechanotransduction regulates the metabolic response of chondrocytes to dynamic loading. Proc Natl Acad Sci USA. 2014;111(4):1316–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ma X, He D, Ru X et al. Apigenin, a plant-derived flavone, activates transient receptor potential vanilloid 4 cation channel. British J Pharmacol. 2012;166(1):349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Smith PL, Maloney KN, Pothen RG, Clardy J, Clapham DE. Bisandrographolide from andrographis paniculata activates TRPV4 channels. J Biol Chem. 2006;281(40):29897–29904. [DOI] [PubMed] [Google Scholar]
- 14. Güler AD, Lee H, Iida T, Shimizu I, Tominaga M, Caterina M. Heat-evoked activation of the ion channel, TRPV4. J Neurosci. 2002;22(15):6408–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shibasaki K. TRPV4 ion channel as important cell sensors. J Anesth. 2016;30(6):1014–1019. [DOI] [PubMed] [Google Scholar]
- 16. Jin M, Wu Z, Chen L et al. Determinants of TRPV4 activity following selective activation by small molecule agonist GSK1016790A. PLoS One. 2011;6(2):e16713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liedtke W, Friedman JM. Abnormal osmotic regulation in trpv4-/- mice. Proc Natl Acad Sci USA. 2003;100(23):13698–13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Berrout J, Jin M, Mamenko M, Zaika O, Pochynyuk O, O'Neil RG. Function of transient receptor potential cation channel subfamily V member 4 (TRPV4) as a mechanical transducer in flow-sensitive segments of renal collecting duct system. J Biol Chem. 2012;287(12):8782–8791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jia Y, Wang X, Varty L et al. Functional TRPV4 channels are expressed in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2004;287(2):L272–L278. [DOI] [PubMed] [Google Scholar]
- 20. Hamanaka K, Jian MY, Weber DS et al. TRPV4 initiates the acute calcium-dependent permeability increase during ventilator-induced lung injury in isolated mouse lungs. Am J Physiol Lung Cell Mol Physiol. 2007;293(4):L923–L932. [DOI] [PubMed] [Google Scholar]
- 21. Lorenzo IM, Liedtke W, Sanderson MJ, Valverde MA. TRPV4 channel participates in receptor-operated calcium entry and ciliary beat frequency regulation in mouse airway epithelial cells. Proc Natl Acad Sci USA. 2008;105(34):12611–12616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alenmyr L, Uller L, Greiff L, Högestätt ED, Zygmunt PM TRPV4-mediated calcium influx and ciliary activity in human native airway epithelial cells. Basic Clin Pharmacol Toxicol. 2014;114(2):210–216. [DOI] [PubMed] [Google Scholar]
- 23. Sokabe T, Fukumi-Tominaga T, Yonemura S, Mizuno A, Tominaga M The TRPV4 channel contributes to intercellular junction formation in keratinocytes. J Biol Chem. 2010;285(24):18749–18758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Oláh A, Tóth BI, Borbíró I et al. Cannabidiol exerts sebostatic and antiinflammatory effects on human sebocytes. J Clin Invest. 2014;124(9):3713–3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mergler S, Valtink M, Taetz K et al. Characterization of transient receptor potential vanilloid channel 4 (TRPV4) in human corneal endothelial cells. Exp Eye Res. 2011;93(5):710–719. [DOI] [PubMed] [Google Scholar]
- 26. Khalifa Ahmed M, Takumida M, Ishibashi T, Hamamoto T, Hirakawa K. Expression of transient receptor potential vanilloid (TRPV) families 1, 2, 3 and 4 in the mouse olfactory epithelium. Rhin. 2009;47(3):242–247. [DOI] [PubMed] [Google Scholar]
- 27. Hatano N, Suzuki H, Itoh Y, Muraki K. TRPV4 partially participates in proliferation of human brain capillary endothelial cells. Life Sci. 2013;92(4-5):317–324. [DOI] [PubMed] [Google Scholar]
- 28. Marrelli SP, O'neil RG, Brown RC, Bryan RMJ. PLA2 and TRPV4 channels regulate endothelial calcium in cerebral arteries. Am J Physiol Heart Circ Physiol. 2007;292(3):H1390–H1397. [DOI] [PubMed] [Google Scholar]
- 29. Schierling W, Troidl K, Apfelbeck H et al. Cerebral arteriogenesis is enhanced by pharmacological as well as fluid-shear-stress activation of the Trpv4 calcium channel. Eur J Vasc Endovasc Surg. 2011;41(5):589–596. [DOI] [PubMed] [Google Scholar]
- 30. Earley S, Pauyo T, Drapp R, Tavares MJ, Liedtke W, Brayden JE. TRPV4-dependent dilation of peripheral resistance arteries influences arterial pressure. Am J Physiol Heart Circ Physiol. 2009;297(3):H1096–H1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Saifeddine M, El-Daly M, Mihara K et al. GPCR-mediated EGF receptor transactivation regulates TRPV4 action in the vasculature. British J Pharmacol. 2015;172(10):2493–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Du G, Li L, Zhang X et al. Roles of TRPV4 and piezo channels in stretch-evoked Ca(2+) response in chondrocytes. Exp Biol Med (Maywood). 2020;245(3):180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang L, You X, Zhang L, Zhang C, Zou W. Mechanical regulation of bone remodeling. Bone Res. 2022;10(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ma Q, Miri Z, Haugen HJ, Moghanian A, Loca D. Significance of mechanical loading in bone fracture healing, bone regeneration, and vascularization. J Tissue Eng. 2023;14:204173142311725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen DH, Sul Y, Weiss M et al. CMT2C with vocal cord paresis associated with short stature and mutations in the TRPV4 gene. Neurology. 2010;75(22):1968–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Unger S, Lausch E, Stanzial F et al. Fetal akinesia in metatropic dysplasia: the combined phenotype of chondrodysplasia and neuropathy?. Am J Med Genetics Pt A. 2011;155(11):2860–2864. [DOI] [PubMed] [Google Scholar]
- 37. Cho TJ, Matsumoto K, Fano V et al. TRPV4-pathy manifesting both skeletal dysplasia and peripheral neuropathy: a report of three patients. Am J Med Genetics Pt A. 2012;158A(4):795–802. [DOI] [PubMed] [Google Scholar]
- 38. Velilla J, Marchetti MM, Toth-Petroczy A et al. Homozygous TRPV4 mutation causes congenital distal spinal muscular atrophy and arthrogryposis. Neurol Genet. 2019;5(2):e312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu Y, Yan X, Chen Y, He Z, Ouyang Y.Novel TRPV4 mutation in a large Chinese family with congenital distal spinal muscular atrophy, skeletal dysplasia and scaly skin. J Neurol Sci. 2020;419:117153. [DOI] [PubMed] [Google Scholar]
- 40. Ragamin A, Gomes CC, Bindels-de Heus K et al. De novo TRPV4 Leu619Pro variant causes a new channelopathy characterised by giant cell lesions of the jaws and skull, skeletal abnormalities and polyneuropathy. J Med Genet. 2022;59(3):305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Taga A, Peyton MA, Goretzki B et al. TRPV4 mutations causing mixed neuropathy and skeletal phenotypes result in severe gain of function. Ann Clin Transl Neurol. 2022;9(3):375–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rock MJ, Prenen J, Funari VA et al. Gain-of-function mutations in TRPV4 cause autosomal dominant brachyolmia. Nat Genet. 2008;40(8):999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Phan MN, Leddy HA, Votta BJ et al. Functional characterization of TRPV4 as an osmotically sensitive ion channel in porcine articular chondrocytes. Arthritis Rheum. 2009;60(10):3028–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Leddy HA, McNulty AL, Lee SH et al. Follistatin in chondrocytes: the link between TRPV4 channelopathies and skeletal malformations. FASEB J. 2014;28(6):2525–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rosenbaum T, Benítez-Angeles M, Sánchez-Hernández R et al. TRPV4: a Physio and Pathophysiologically Significant Ion Channel. Int J Mol Sci. 2020;21(11):3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nilius B, Voets T. The puzzle of TRPV4 channelopathies. EMBO Rep. 2013;14(2):152–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nilius B, Owsianik G. Channelopathies converge on TRPV4. Nat Genet. 2010;42(2):98–100. [DOI] [PubMed] [Google Scholar]
- 48. White JPM, Cibelli M, Urban L, Nilius B, McGeown JG, Nagy I. TRPV4: molecular conductor of a diverse orchestra. Physiol Rev. 2016;96(3):911–973. [DOI] [PubMed] [Google Scholar]
- 49. Nadezhdin KD, Talyzina IA, Parthasarathy A, Neuberger A, Zhang DX, Sobolevsky AI. Structure of human TRPV4 in complex with GTPase RhoA. Nat Commun. 2023;14(1):3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Goretzki B, Guhl C, Tebbe F, Harder JM, Hellmich UA. Unstructural biology of TRP ion channels: the role of intrinsically disordered regions in channel function and regulation. J Mol Biol. 2021;433(17):166931. [DOI] [PubMed] [Google Scholar]
- 51. Zhang ZR, Chu WF, Song B et al. TRPP2 and TRPV4 form an EGF-activated calcium permeable channel at the apical membrane of renal collecting duct cells. PLoS One. 2013;8(8):e73424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Köttgen M, Buchholz B, Garcia-Gonzalez MA et al. TRPP2 and TRPV4 form a polymodal sensory channel complex. J Cell Biol. 2008;182(3):437–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Du J, Ma X, Shen B, Huang Y, Birnbaumer L, Yao X. TRPV4, TRPC1, and TRPP2 assemble to form a flow-sensitive heteromeric channel. FASEB J. 2014;28(11):4677–4685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tsiokas L, Arnould T, Zhu C, Kim E, Walz G, Sukhatme VP. Specific association of the gene product of PKD2 with the TRPC1 channel. Proc Natl Acad Sci USA. 1999;96(7):3934–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pyrshev K, Stavniichuk A, Tomilin VN et al. TRPV4 functional status in cystic cells regulates cystogenesis in autosomal recessive polycystic kidney disease during variations in dietary potassium. Physiol Rep. 2023;11(6):e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gradilone SA, Masyuk TV, Huang BQ et al. Activation of Trpv4 reduces the hyperproliferative phenotype of cystic cholangiocytes from an animal model of ARPKD. Gastroenterology. 2010;139(1):304–314.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zaika O, Mamenko M, Berrout J, Boukelmoune N, O'Neil RG, Pochynyuk O. TRPV4 dysfunction promotes renal cystogenesis in autosomal recessive polycystic kidney disease. J Am Soc Nephrol. 2013;24(4):604–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Li J, Kanju P, Patterson M et al. TRPV4-mediated calcium influx into human bronchial epithelia upon exposure to diesel exhaust particles. Environ Health Perspect. 2011;119(6):784–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tian W, Fu Y, Garcia-Elias A et al. A loss-of-function nonsynonymous polymorphism in the osmoregulatory TRPV4 gene is associated with human hyponatremia. Proc Natl Acad Sci USA. 2009;106(33):14034–14039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kang SS, Shin SH, Auh CK, Chun J. Human skeletal dysplasia caused by a constitutive activated transient receptor potential vanilloid 4 (TRPV4) cation channel mutation. Exp Mol Med. 2012;44(12):707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Belibi FA, Edelstein CL. Unified ultrasonographic diagnostic criteria for polycystic kidney disease. J Am Soc Nephrol. 2009;20(1):6–8. [DOI] [PubMed] [Google Scholar]
- 62. Ravine D, Sheffield L, Danks DM, Gibson RN, Walker RG, Kincaid-Smith P. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet North Am Ed. 1994;343(8901):824–827. [DOI] [PubMed] [Google Scholar]
- 63. Richards B, Skoletsky J, Shuber AP et al. Multiplex PCR amplification from the CFTR gene using DNA prepared from buccal brushes/swabs. Hum Mol Genet. 1993;2(2):159–163. [DOI] [PubMed] [Google Scholar]
- 64. Chen Y, Wang ZL, Yeo M et al. Epithelia-Sensory Neuron Cross Talk Underlies Cholestatic Itch Induced by Lysophosphatidylcholine. Gastroenterology. 2021;161(1):301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Benítez-Angeles M, Romero AEL, Llorente I et al. Modes of action of lysophospholipids as endogenous activators of the TRPV4 ion channel. J Physio. 2023;l601(9):1655–1673. [DOI] [PubMed] [Google Scholar]
- 66. Salazar H, Jara-Oseguera A, Hernandez-Garcia E et al. Structural determinants of gating in the TRPV1 channel. Nat Struct Mol Biol. 2009;16(7):704. [DOI] [PubMed] [Google Scholar]
- 67. Salazar H, Llorente I, Jara-Oseguera A et al. A single N-terminal cysteine in TRPV1 determines activation by pungent compounds from onion and garlic. Nat Neurosci. 2008;11(3):255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Morales-Lázaro SL, Llorente I, Sierra-Ramírez F et al. Inhibition of TRPV1 channels by a naturally occurring omega-9 fatty acid reduces pain and itch. Nat Commun. 2016;7:(1):13092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch Eur J Physiol. 1981;391(2):85–100. [DOI] [PubMed] [Google Scholar]
- 70. Benítez-Angeles M, Juárez-González E, Vergara-Jaque A et al. Unconventional interactions of the TRPV4 ion channel with beta-adrenergic receptor ligands. Life Sci Alliance. 2023;6(3):e202201704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Oseguera AJ, Islas LD, García-Villegas R, Rosenbaum T. On the mechanism of TBA block of the TRPV1 channel. Biophys J. 2007;92(11):3901–3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Islas LD. Patch clamping and single-channel analysis. In: Zheng J, Trudeau MC, eds. Handbook of Ion Channels. 1sted. Boca Raton: CRC Press, 2015. [Google Scholar]
- 73. Canul-Sánchez JA, Hernández-Araiza I, Hernández-García E et al. Different agonists induce distinct single-channel conductance states in TRPV1 channels. J Gen Physiol. 2018;150(12):1735–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Inker LA, Eneanya ND, Coresh J et al. New creatinine- and cystatin C–based equations to estimate GFR without race. N Engl J Med. 2021;385(19):1737–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lugo E, Graulau E, Ramos Cortes E, Carlo S, Ramírez N. Homozygous TRPV4 mutation broadens the phenotypic spectrum of congenital spinal muscular atrophy and arthrogryposis: a case report. Cureus. 2023;15(8):e43413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gao X, Wu L, O'Neil RG. Temperature-modulated diversity of TRPV4 channel gating: activation by physical stresses and phorbol ester derivatives through protein kinase C-dependent and -independent pathways. J Biol Chem. 2003;278(29):27129–27137. [DOI] [PubMed] [Google Scholar]
- 77. Wegierski T, Lewandrowski U, Müller B, Sickmann A, Walz G. Tyrosine phosphorylation modulates the activity of TRPV4 in response to defined stimuli. J Biol Chem. 2009;284(5):2923–2933. [DOI] [PubMed] [Google Scholar]
- 78. Garcia-Elias A, Mrkonjić S, Pardo-Pastor C et al. Phosphatidylinositol-4,5-biphosphate-dependent rearrangement of TRPV4 cytosolic tails enables channel activation by physiological stimuli. Proc Natl Acad Sci USA. 2013;110(23):9553–9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Goretzki B, Glogowski NA, Diehl E et al. Structural Basis of TRPV4 N Terminus Interaction with Syndapin/PACSIN1-3 and PIP(2). Structure. 2018;26(12):1583–1593.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Goretzki B, Wiedemann C, McCray BA et al. Crosstalk between regulatory elements in disordered TRPV4 N-terminus modulates lipid-dependent channel activity. Nat Commun. 2023;14(1):4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Fecto F, Shi Y, Huda R, Martina M, Siddique T, Deng HX. Mutant TRPV4-mediated Toxicity Is Linked to Increased Constitutive Function in Axonal Neuropathies. J Biol Chem. 2011;286(19):17281–17291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Auer-Grumbach M, Olschewski A, Papić L et al. Alterations in the ankyrin domain of TRPV4 cause congenital distal SMA, scapuloperoneal SMA and HMSN2C. Nat Genet. 2010;42(2):160–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Echaniz-Laguna A, Dubourg O, Carlier P et al. Phenotypic spectrum and incidence of TRPV4 mutations in patients with inherited axonal neuropathy.. Neurology. 2014;82(21):1919–1926. [DOI] [PubMed] [Google Scholar]
- 84. Faye E, Modaff P, Pauli R, Legare J. Combined phenotypes of spondylometaphyseal Dysplasia-Kozlowski type and Charcot-Marie-Tooth disease type 2C secondary to a TRPV4 pathogenic variant. Mol Syndromol. 2019;10(3):154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Krakow D, Vriens J, Camacho N et al. Mutations in the gene encoding the calcium-permeable ion channel TRPV4 produce spondylometaphyseal dysplasia, kozlowski type and metatropic dysplasia. Am Hum Genet. 2009;84(3):307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Camacho N, Krakow D, Johnykutty S et al. Dominant TRPV4 mutations in nonlethal and lethal metatropic dysplasia. Am J Med Genetics Pt A. 2010;152A(5):1169–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Loukin S, Zhou X, Su Z, Saimi Y, Kung C. Wild-type and Brachyolmia-causing Mutant TRPV4 Channels Respond Directly to Stretch Force. J Biol Chem. 2010;285(35):27176–27181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Deng HX, Klein CJ, Yan J et al. Scapuloperoneal spinal muscular atrophy and CMT2C are allelic disorders caused by alterations in TRPV4. Nat Genet. 2010;42(2):165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Landouré G, Zdebik AA, Martinez TL et al. Mutations in TRPV4 cause Charcot-Marie-Tooth disease type 2C. Nat Genet. 2010;42(2):170–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Klein CJ, Shi Y, Fecto F et al. TRPV4 mutations and cytotoxic hypercalcemia in axonal Charcot-Marie-Tooth neuropathies. Neurology. 2011;76(10):887–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Loukin S, Su Z, Kung C. Increased basal activity is a key determinant in the severity of human skeletal dysplasia caused by TRPV4 mutations. PLoS One. 2011;6(5):e19533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Fiorillo C, Moro F, Brisca G et al. TRPV4 mutations in children with congenital distal spinal muscular atrophy. Neurogenetics. 2012;13(3):195–203. [DOI] [PubMed] [Google Scholar]
- 93. Klausen TK, Janssens A, Prenen J et al. Single point mutations of aromatic residues in transmembrane helices 5 and -6 differentially affect TRPV4 activation by 4α-PDD and hypotonicity: implications for the role of the pore region in regulating TRPV4 activity. Cell Calcium. 2014;55(1):38–47. [DOI] [PubMed] [Google Scholar]
- 94. Kwon DH, Zhang F, McCray BA et al. TRPV4-Rho GTPase complex structures reveal mechanisms of gating and disease. Nat Commun. 2023;14(1):3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Nakamura T, Ushiyama C, Suzuki S, Ebihara I, Shimada N, Koide H. Elevation of serum levels of metalloproteinase-1, tissue inhibitor of metalloproteinase-1 and type IV collagen, and plasma levels of metalloproteinase-9 in polycystic kidney disease. Am J Nephrol. 2000;20(1):32–36. [DOI] [PubMed] [Google Scholar]
- 96. Liu B, Li C, Liu Z, Dai Z, Tao Y. Increasing extracellular matrix collagen level and MMP activity induces cyst development in polycystic kidney disease. BMC Nephrol. 2012;13(1):109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kashyap S, Hein KZ, Chini CC et al. Metalloproteinase PAPP-A regulation of IGF-1 contributes to polycystic kidney disease pathogenesis. JCI Insight. 2020;5(4):135700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zakiyanov O, Kalousová M, Zima T, Tesař V Chapter Four—Matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases in kidney disease. In: Makowski GS, ed. Advances in Clinical Chemistry. Vol 105. Elsevier, 2021:141–212. [DOI] [PubMed] [Google Scholar]
- 99. Cowley BD. Calcium, cyclic AMP, and MAP kinases: dysregulation in polycystic kidney disease. Kidney Int. 2008;73(3):251–253. [DOI] [PubMed] [Google Scholar]
- 100. Staruschenko A, Alexander RT, Caplan MJ, Ilatovskaya DV. Calcium signalling and transport in the kidney. Nat Rev Nephrol. Available at: 10.1038/s41581-024-00835-z. Accessed 19 April 2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Fujii S, Tajiri Y, Hasegawa K et al. The TRPV4-AKT axis promotes oral squamous cell carcinoma cell proliferation via CaMKII activation. Lab Invest. 2020;100(2):311–323. [DOI] [PubMed] [Google Scholar]
- 102. Thoppil RJ, Adapala RK, Cappelli HC et al. TRPV4 channel activation selectively inhibits tumor endothelial cell proliferation. Sci Rep. Sci Rep. 2015;5(1):14257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Liu J, Guo Y, Zhang R et al. Inhibition of TRPV4 remodels single cell polarity and suppresses the metastasis of hepatocellular carcinoma. Cell Death Dis. 2023;14(6):379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mamenko MV, Boukelmoune N, Tomilin VN et al. The renal TRPV4 channel is essential for adaptation to increased dietary potassium. Kidney Int. 2017;91(6):1398–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Kuebler WM, Jordt SE, Liedtke WB. Urgent reconsideration of lung edema as a preventable outcome in COVID-19: inhibition of TRPV4 represents a promising and feasible approach. Am J Physiol Lung Cell Mol Physiol. 2020;318(6):L1239–L1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Sullivan JM, Bagnell AM, Alevy J et al. Gain-of-function mutations of TRPV4 acting in endothelial cells drive blood-CNS barrier breakdown and motor neuron degeneration in mice. Sci Transl Med. 2024;16(748):eadk1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Cabrita I, Buchholz B, Schreiber R, Kunzelmann K. TMEM16A drives renal cyst growth by augmenting Ca2+ signaling in M1 cells. J Mol Med. 2020;98(5):659–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Xu T, Chen M, Xu Q et al. Anoctamin 1 Inhibition Suppresses Cystogenesis by Enhancing Ciliogenesis and the Ciliary Dosage of Polycystins. Front Biosci (Landmark Ed). ) 2022; 27(7):216. [DOI] [PubMed] [Google Scholar]
- 109. Padhy B, Xie J, Wang R, Lin F, Huang CL. Channel Function of Polycystin-2 in the Endoplasmic Reticulum Protects against Autosomal Dominant Polycystic Kidney Disease. JASN. 2022;33(8):1501–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Liu X, Tang J, Chen XZ Role of PKD2 in the endoplasmic reticulum calcium homeostasis. Front Physiol. 2022;13:962571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Caplan MJ Polycystin-2 in the Endoplasmic Reticulum: bending Ideas about the Role of the Cilium. JASN. 2022;33(8):1433–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material. The clones of the mutant TRPV4 channels are available upon request.