

Summary
Prostate cancer (PCa) is the most common malignant tumor in men. Currently, there are few prognosis indicators for predicting PCa outcomes and guiding treatments. Here, we perform comprehensive proteomic profiling of 918 tissue specimens from 306 Chinese patients with PCa using data-independent acquisition mass spectrometry (DIA-MS). We identify over 10,000 proteins and define three molecular subtypes of PCa with significant clinical and proteomic differences. We develop a 16-protein panel that effectively predicts biochemical recurrence (BCR) for patients with PCa, which is validated in six published datasets and one additional 99-biopsy-sample cohort by targeted proteomics. Interestingly, this 16-protein panel effectively predicts BCR across different International Society of Urological Pathology (ISUP) grades and pathological stages and outperforms the D’Amico risk classification system in BCR prediction. Furthermore, double knockout of NUDT5 and SEPTIN8, two components from the 16-protein panel, significantly suppresses the PCa cells to proliferate, invade, and migrate, suggesting the combination of NUDT5 and SEPTIN8 may provide new approaches for PCa treatment.
Keywords: prostate cancer, proteomics, DIA-MS, BCR-free survival, NUDT5 and SEPTIN8, prognosis prediction
Graphical abstract
Highlights
-
•
Three molecular subtypes of PCa revealed by comprehensive proteomic analysis
-
•
A 16-protein panel effectively predicts BCR for patients with PCa across different stages
-
•
Inhibiting NUDT5 and SEPTIN8 suppresses the malignant features of PCa
Sun et al. perform a comprehensive proteomic profiling of 306 Chinese patients with prostate cancer (PCa) and develop a 16-protein panel that effectively predicts BCR for patients with PCa across different PCa stages. This study provides an effective prognosis prediction technique and potential therapeutic strategies for patients with PCa.
Introduction
Prostate cancer (PCa) is the most common type of cancer and the second leading cause of cancer-related deaths among males worldwide.1 In medically advanced regions, the proportion of limited or locally progressive PCa at the time of initial diagnosis is gradually increasing and tends to dominate the mainstream diagnosed population. The major curative treatment for this population is radical prostatectomy with adjuvant/neoadjuvant therapy.2 The incidence of biochemical recurrence (BCR) following radical prostatectomy can be as high as 40%,3,4,5 and it is strongly linked with later clinical recurrence, metastasis, and cancer-specific death.6,7 As a result, the prediction of BCR risk using a variety of clinical indicators to direct clinical work-up is now a widely used approach in the PCa treatment environment. These indicators typically involve the International Society of Urological Pathology (ISUP) grouping, prostate-specific antigen (PSA) at diagnosis, clinical stage, etc. The most classic D'Amico risk categorization method, which integrates the ISUP group and PSA value at diagnosis, has consistently been endorsed by a large number of experts across ethnic groupings. According to the D’Amico risk classification method, patients with PCa can be grouped into low-, intermediate-, and high-risk groups, with estimated risks of 5-year BCR of <25%, 25%–50%, and >50%, respectively.8 The effectiveness of combining radiological and clinical parameters to measure the BCR risk was also evaluated, and it was shown to increase the predictive accuracy of the risk stratification method.9 However, because of the widespread PSA screening of PCa, the distribution of patients gradually shifted over time from the high-risk groups into the low- and intermediate-risk groups; the clinical relevance of this classification scheme may be limited and diminishing at present.10 In addition, the current stratification methods fail at explaining the mechanisms of PCa progression and do not provide novel therapeutic targets for treating PCa.
Extensive genomic, epigenomic, and transcriptomic investigations have proposed novel classifications for PCa based on specific genomic abnormalities, such as specific gene mutations (SPOP, FOXA1, IDH1, etc.) and fusions (ERG, ETV1/4, FLI1, etc.).11,12,13 However, proteins are the major executors of cellular processes, and alterations in genetic and transcriptional profiles are not always consistent with alterations in protein profiles or activities. Therefore, a protein-based risk prediction and classification strategy might provide better insights into the pathological processes, therapeutic instruction, and prognosis prediction for cancer research.14 While thousands of PCa genomes and transcriptomes have been profiled to date,15 the published proteomic profiles of PCa were limited to small numbers of PCa samples. A proteogenomic study analyzed 39 PCa samples and identified a nine-gene sub-network that positively correlated with more aggressive tumor phenotypes and predicted recurrence-free survival.16 Sinha et al. showed that the proteomic-based prognostic biomarkers identified in a cohort of 76 patients with PCa were superior to the genomic and transcriptomic-based ones.17 The crucial involvement of the tricarboxylic acid (TCA) cycle in PCa progression was evidenced by proteomics rather than transcriptomics through a multi-omics study of 28 patients with PCa.18 A proteomic study of 28 patients with PCa found that pro-NPY might be a potential prognostic biomarker.19 A phosphoproteomic profiling, together with genomic and transcriptomic analysis, revealed clinically relevant pathway information potentially suitable for patient stratification and targeted therapies in late-stage PCa using 27 samples.20 A large-scale proteomic study of 278 Western patients with PCa derived a risk classification strategy for intermediate PCa risk.21 However, PCa is highly heterogeneous, and there are known differences in genetic alterations in PCa among different ethnic populations.12,22,23 Most proteomic-based studies were conducted on relatively small cohorts from different Western countries. Our knowledge of the molecular mechanisms behind PCa progression and BCR is still insufficient, especially in the Chinese population. Therefore, a comprehensive proteomic analysis of a large cohort of patients with PCa is urgent: it could provide a far more accurate classification of patients with PCa and new insights in its molecular mechanisms.
Data-independent acquisition mass spectrometry (DIA-MS)-based proteomics analysis provides a wide range of applications to explore novel biomarkers and therapeutic targets in various medical fields due to its high throughput and accurate quantification advantages.21,24,25,26,27,28,29 In particular, parallel accumulation-serial fragmentation combined with data-independent acquisition technology is a state-of-the-art proteomics technology that achieves higher depth of proteome identification through reducing noise interference and increasing signal sensitivity.30
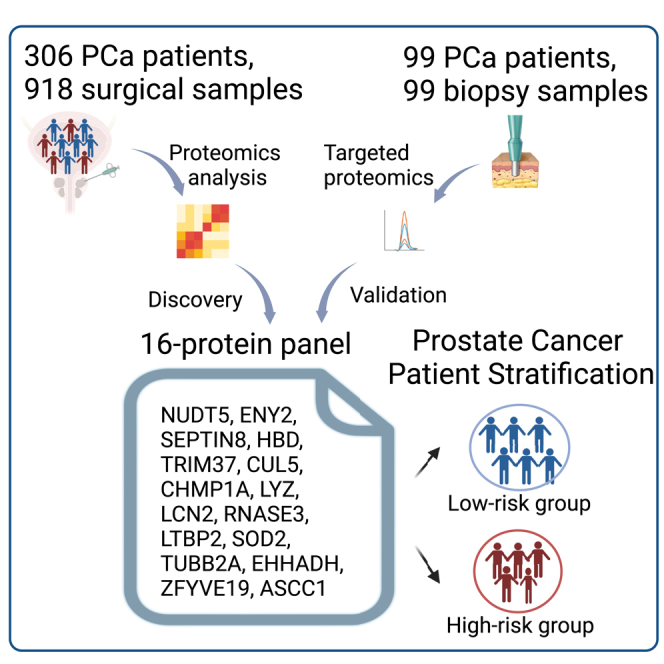
Here, we used DIA-MS technology to characterize the proteome landscape of PCa using 918 samples from 306 Chinese patients with PCa who underwent radical prostatectomy surgery (PCSHA). We measured over 10,000 proteins, mapped the proteomic characteristics associated with different ISUP groups, and identified three PCa subtypes with significant clinical and molecular differences. Notably, we identified a panel of 16 proteins that efficiently predict BCR, which outperforms the strategies currently used in clinical practice. This model was effectively validated in six external datasets. Furthermore, it was validated in an independent Chinese cohort comprising 99 biopsy samples quantified by a targeted proteomic technology. Among these 16 proteins, we highlighted NUDT5 and SEPTIN8 as promising candidates for synthetic targets in PCa treatment. Our comprehensive proteomic dataset fills the gap in the field of Chinese PCa proteomic studies by revealing new molecular mechanisms, prognostic prediction methods, and potential therapeutic strategies for PCa.
Results
Generation of a comprehensive proteomic landscape of PCa
To characterize the proteome of Chinese patients with PCa, we collected the surgical formalin-fixed paraffin-embedded (FFPE) blocks of 306 patients with PCa who underwent radical prostatectomy for localized PCa (henceforth named PCSHA cohort). The basic information of our study cohort, including age, PSA level at diagnosis, Gleason score, and pathological stage, is shown in Tables S1 and S7. Three punches were taken from each FFPE block: two from the primary and secondary Gleason score grading areas and one from the adjacent non-cancerous area (Figure 1A; Figure S1A), for a total of 918 FFPE punches. Each punch location was selected based on the hematoxylin and eosin staining of the FFPE block, which was evaluated by at least two pathologists (Figure S1B; Table S1). The diameter for each punched tissue is 1 mm, ensuring that the percentage of tumor cells is over 90% in the sample for proteomic analysis.
Figure 1.

Overview of the PCSHA cohort analysis
(A) The graphic illustrates the workflow encompassing tissue sample punching, sample information annotation, batch design, and subsequent proteomic data analysis. GS1 denoted a more aggressive tumor region, whereas GS2 represented a less malignant region.
(B) Heatmap displaying the relative protein expression levels of 10,071 proteins from 918 prostate tissue samples (N = 918). The gray bricks represented benign tissue. BE, preoperative endocrine therapy, PE, postoperative endocrine therapy; PR, postoperative radiotherapy. The patients were graded following the ISUP guidance.
(C) Boxplots representing the intra- and inter-individual correlation comparisons based on proteomic data. In the boxplot, the middle bar represents the average value, and the box represents the interquartile range.
(D) Volcano plot showing 1,784 differentially expressed proteins between tumor and benign samples (Welch’s t test, B-H adjusted p value < 0.05, fold change > 1.5).
(E) Pathways enriched (p value < 0.05) by these differentially expressed proteins in Figure 1D using Metascape. Blue and orange pathways were enriched by 61 downregulated proteins and by 2,009 upregulated proteins in the tumor samples, respectively.
See also Table 1, Figure S1, and Table S1. Patient information, related to Figure 1, Table S2. Protein quantification matrix, related to Figure 1, Table S3. Pathway enrichment from differentially expressed proteins between tumor and benign samples, related to Figure 1.
Utilizing the data-independent acquisition (DIA) technology, we identified 116,631 peptides belonging to 10,071 proteins, with a false discovery rate of 1% (Table S2A). Over 6,500 detected proteins were found in over 50% of the samples. In general, more proteins were identified in tumor samples than in adjacent non-cancerous samples (Figure S1C), 10,023 proteins were identified in all tumor samples and 285 specific proteins in tumor samples, and 9,768 proteins in all benign samples and only 30 specific proteins in benign samples. Our proteomic data were highly reproducible, as the median Pearson correlation of the quality control samples was 0.92 (Figures S1D and S1E). No batch effect (different batches or mass spectrometry machines) was observed in the quality control samples or in all samples. These quality control analyses demonstrate that the proteomics data are of high quality. The proteomics profile of tumor samples is clearly different from normal samples (Figures 1B and S1F). However, the distinction between different ISUP grades (Figure S1G), pathology stages (Figure S1H), and BCR prognosis (Figure S1I) was less significant. The expression of KLK3 (also known as PSA), an abundant protein expressed in prostate tissues, was detected in our study as expected; however, the protein expression of KLK3 in prostate tissue did not exhibit any correlation with the serum PSA level (Figure S1J).
It is widely recognized that PCa is highly heterogeneous, as evidenced by the various Gleason scores and genomic alterations among various regions of PCa within a single patient.22,31 But when we evaluated the differences in proteome profiles between two Gleason score regions within the same patient (intra-patient) or the same Gleason score among different patients (inter-patient), we discovered that inter-patient heterogeneity significantly dominates intra-patient heterogeneity in PCa (Figure 1C). Furthermore, when we treated the two tumor samples from the same patient separately, in combination or by taking the mean of them, and then compared differentially expressed proteins (DEPs) between the tumor samples and the benign samples (Figure S1K), we discovered that by taking the mean of the two tumor samples, we identified the most common DEPs and the least unique DEPs. Therefore, for the subsequent study, we employed the averaged intra-patient protein expression data for latter analysis. We first evaluated the differential expressed proteins between tumor and benign samples and identified 1,784 DEPs (Figure 1D; Table S3A), including proteins such as AMACR that have been shown to be associated with PCa. We then analyzed these DEPs using Metascape.32 The proteins upregulated in tumor samples were enriched in various metabolism pathways, such as mitochondrial translation initiation, organic acid catabolic process, amino acid metabolic process, ribosome biogenesis, fatty acid metabolism, and RNA metabolism, whereas downregulated proteins in the tumor samples were enriched in actin-associated pathways, prostaglandin synthesis and regulation, and long-chain fatty acid transport pathways (Table S3; Figure 1E). These findings are consistent with previous studies.17,18,19,33
Activated cell cycle and inhibited cell adhesion in high-grade PCa
In general, patients with a higher Gleason score at diagnosis have a more aggressive disease course than those with a lower Gleason score.34,35 To explore the underlying mechanisms of PCa progression, we analyzed those differently expressed proteins within tumor samples across different ISUP groups. We discovered that dysregulated proteins could be categorized into 12 distinct clusters spanning five ISUP grades using mFuzz analysis (Figure 2A; Table S2C). Dysregulated proteins from clusters 1 (n = 679) and 2 (n = 644) were enriched in DNA damage repair pathways and cell cycle, and the expression of these proteins consistently increased with the increase in ISUP grades (Figures 2B–2D). Furthermore, the most significantly changed proteins (ANOVA p < 0.01) in cluster 1 were associated with RNA metabolism and the cell population proliferation (Figure 2E). In contrast, the expression of the dysregulated proteins from clusters 3 (n = 1,008) and 4 (n = 1,330) was continuously downregulated with the increase in ISUP grade. And these proteins were associated with cell adhesion and cytoskeleton-related pathways (Figure 2D), indicating that a higher PCa grade had a greater capacity for migration. The most significantly changed proteins (ANOVA p < 0.01) in cluster 3 were abundant in numerous metabolic pathways and the immunological route neutrophil degranulation (Figure 2F). Therefore, the progression of PCa may be influenced by metabolism and the microenvironment. Specifically, higher cell cycle activity and lower cell adhesion point to tumor cells that are more stem-like, less differentiated, and more malignant.
Figure 2.

Dysregulated proteins across different ISUP grades
(A) 12 different clusters identified by mFuzz clustering across different ISUP grades in the tumor samples.
(B) Normalized protein expression across five ISUP grades in clusters 1, 2, 3, and 4.
(C) Normalized average protein expression of all the proteins in each cluster across five ISUP grades.
(D) Pathway enrichment from all the proteins in each cluster across five ISUP grades (p < 0.05, Metascape). BE, preoperative endocrine therapy; PE, postoperative endocrine therapy; PR, postoperative radiotherapy.
(E and F) Networks based on the enriched pathways obtained using the most significantly dysregulated proteins from each cluster (one-way ANOVA p < 0.01). Orange points (E) show the proteins from cluster 1 and the blue ones (F) show the proteins from cluster 4.
Molecular subtyping of PCa based on proteomic profiles
To understand the proteomic patterns of primary PCa, we then performed an unsupervised clustering of DEPs, non-negative matrix factorization (NMF) analysis (Figure S2), and identified three patient subgroups: CPC1, CPC2, and CPC3 (Figure 3A). We then examined how clinical parameters varied in these patient subgroups (Figures 3A–3C). We discovered that the distribution of clinical variables, such as age, ISUP grade, pathological stage, and treatment, was similar in all subgroups (Figure 3C); however, the BCR-free survival varied significantly (Figure 3B), suggesting that specific components of the proteomes of the three subgroups may predict the BCR-free survival of patients with PCa. We thus looked at which biological functions were enriched in each subgroup. CPC1 and CPC2 show similar proteomic characteristics, which are featured by activation of metabolism-related pathways, including amino acid, sugar, and fatty acid metabolism, and downregulation in the cell cycle, immune-related signaling pathways, and genetic information processing pathways (Figure 3A). Of note, CPC3, which included patients with the poorest prognosis (Figure 3B), was characterized by the downregulation of metabolism-related processes and cellular processes involved in cell-cell interaction and by the upregulation in genetic information processing and immune cell receptor signaling pathways (Figure 3A). Comparing with mFuzz clustering, we identified similar pathways, such as DNA damage repair pathways and cell cycle, and the expression of these proteins consistently increased with the increase in ISUP grades in clusters 1 and 2 (Figures 2B–2D). In contrast, immune-associated pathways were decreased in clusters 3 and 4 (Figures 2B–2D).
Figure 3.

Proteomic pathway-based stratification of the patients with PCa associated with their prognosis
(A) The heatmap shows the normalized enrichment scores of the three patient subtypes (CPC1, N = 126; CPC2, N = 57; and CPC3, N = 43) using NMF analysis. The associations of proteomic subtypes with clinical characteristics (pathological stages, PSA, biochemical recurrence [BCR], and ISUP grade) are annotated.
(B) Kaplan-Meier curves of the BCR-free survival of each patient subtype from our cohort (log rank p = 0.0012).
(C) Distribution of the clinical indexes in the three patient subtypes.
(D) Relative expression of the dysregulated druggable proteins in the three patient subtypes. One-way ANOVA, B-H adjusted p value: ∗ < 0.05; ∗∗ < 0.01; ∗∗∗ < 0.001; ∗∗∗∗ < 0.0001. BE, preoperative endocrine therapy; PE, postoperative endocrine therapy; PR, postoperative radiotherapy.
See also Figure S2.
To identify new potential therapeutic targets for PCa treatment, we intersected our 1,784 DEPs with a database of 744 proteins that can be targeted in the Human Protein Atlas portal (www.proteinatlas.org). We found that 19 proteins of our 1,784 DEPs can potentially be drug targets for PCa (see STAR Methods). Indeed, the expression of these 19 molecules was significantly different in the three subgroups (Figure 3D). However, all these 19 proteins were downregulated in the CPC3 patient subgroup, which had the poorest prognosis, indicating that these proteins may not directly be suitable targets for PCa treatment. As previous studies suggested, the lack of known effective therapeutic targets for PCa treatment requires a different study approach.12
Identification and validation of a panel of 16 protein biomarkers for prognostic prediction
ISUP grading is associated with BCR disease-free survival in the PCSHA cohort (Figure S3A). To define a protein-based signature for predicting the prognosis of patients with PCa, we screened for proteins most significantly associated with the prognosis after excluding the proteins with missing values > 50% (Figure 4A). In our data, BCR mostly occurred within 1,000 days after surgery (Figure S3B), which is probably due to the fact that most patients with PCa were in the middle-to-late stages when diagnosed; therefore, we focused on BCR follow-up of 1–3 years in our study. A prognostic prediction model based on 16 proteins was built using Lasso regression on the PCSHA dataset to predict BCR in patients with PCa (Figure 4A; Table S4A, for details of the modeling see STAR Methods). The C-index for the 16-protein panel was determined to be 0.71, obtained through 1,000 bootstrapping iterations. The expression of the 16 proteins is shown in Figure 4B and Figure S4.
Figure 4.

Identification and validation of a panel of 16 proteins for BCR prediction
(A) Workflow for the development and validation of the 16-protein panel.
(B) Heatmap showing the relative abundance of 16 proteins in two different risk groups. The associations of the two risk groups with clinical characteristics were tested by Fisher’s exact test. The differential expression of each protein between the high- and low-risk groups was tested by Welch’s t test, B-H adjusted p value: ∗ < 0.05; ∗∗ < 0.01; ∗∗∗ < 0.001; ∗∗∗∗ < 0.0001.
(C–F) Kaplan-Meier plot for BCR-free survival (left panel) and ROC curves (right panel) of the 16-protein model in the PCSHA validation dataset (C), TCGA dataset (D), the MSKCC dataset (E), and the CPGEA dataset (F) for BCR-free survival based on our 16-protein prediction model, with a 1-year predictive power (yellow), a 2-year predictive power (blue), and a 3-year predictive power (red).
(G–I) Kaplan-Meier plots and ROC curves of BCR-free survival based on the 16-protein prediction model in different ISUP grades using the PCSHA validation set (G), TCGA dataset (H), and the MSKCC dataset (I).
See also Figures S3 and S4 and Table S4.
Based on the expression of these 16 proteins, we clustered patients with PCa into high- and low-risk groups (Figure 4B). Patients in the high-risk group were significantly associated with poor BCR-free survival, while those in the low-risk group were associated with better BCR-free survival (log rank p < 0.0001) (Figures 4C, S3C, and S3D). The area under the curve values (AUCs) based on the PCSHA discovery set for BCR prediction until 1, 2, and 3 years was 0.850, 0.856, and 0.899, respectively. Similar results were observed in the PCSHA validation cohort (Figures 4C and 4D). These results show that the prediction model based on the 16-protein panel performs well in predicting BCR-free survival in patients with PCa.
We further tested the prognostic prediction power of our 16-protein panel using other six public datasets. We first chose three transcriptomic datasets from The Cancer Genome Atlas (TCGA) (https://xenabrowser.net/) (432 Western patients with PCa), Memorial Sloan Kettering Cancer Center (MSKCC)36 (140 patients with PCa), and the Chinese Prostate Cancer Genome and Epigenome Atlas (CPGEA) (136 Chinese patients with PCa)12 (Table S7). In these three validation sets, the 16-protein panel was significantly correlated with the BCR-free survival (log rank p < 0.0001) (Figures 4D–4F). A similar prediction power (AUC values) was observed when predicting the prognosis of patients with PCa 1, 2, and 3 years after surgery (Figure 4; next, we validated the prognostic prediction performance of our model for PCa at different clinical stages using TCGA). Then, we tested the prediction power of our protein panel using three other published proteomic datasets (from Sinha et al.,17 Zhong et al.,21 and Charmpi et al.16) (Table S7). Once again, we found a significant association of our 16-protein panel with prognosis (log rank p < 0.05, Figures S3E–S3H). However, the AUCs were less than 0.7 in Charmpi et al.’s and the Sinha et al.’s datasets due to fewer proteins in the 16-protein panel detected in these datasets (12 unique proteins in Charmpi et al.’s dataset, seven unique proteins in Sinha et al.’s dataset, and 12 unique proteins in Zhong et al.’s dataset) (Figure S3H). Notably, our 16-protein panel did not perform well in Charmpi et al.’s dataset probably due to the small number of patients in these data (N = 2 in the low-risk group in Charmpi et al.’s dataset). In summary, the 16-protein panel demonstrated an excellent prediction power in predicting the prognosis of PCa.
The 16-protein prognosis prediction panel performed well across different ISUP grade and pathological stages
With the widespread of PSA screening both in western countries and in China, the spectrum of PCa at initial diagnosis gradually shifted from middle-to-late stages to early stage. As a result, risk classification based on clinical parameters such as pathological stages and ISUP grade may have limited values.10 Then, we assessed the effectiveness of this 16-protein prediction model in predicting PCa prognosis at various clinical stages. We found that this 16-protein panel performed well in predicting BCR-free survival across different ISUP grades and different pathological stages in both the PCSHA discovery set (Figures S5A and S5B) and the PCSHA validation dataset (Figure 4G).
Next, we validated the prognostic prediction performance of our model for PCa at different clinical stages using TCGA (https://xenabrowser.net/) and MSKCC36 datasets, which have more than ten patients with PCa with BCR in each clinical subgroup. Our 16-protein panel showed a significant association with BCR (log rank p < 0.05) and excellent BCR-free survival prediction power (AUCs >0.6) across different ISUP grades and different pathological stages in these two external datasets (Figures 4H–4I, S5D, and S5E). The 16-protein panel failed to predict prognosis only in one subgroup due to the small number of BCR occurrence (N = 5 in the T1-2 pathological subgroup of TCGA dataset (Figure S5D). Collectively, these results indicate that our 16-protein panel can effectively predict the BCR-free survival of patients at different levels of PCa aggressiveness, providing useful information for clinical decision-making and personalized treatment strategies after surgery.
The 16-protein prognosis prediction panel outperforms the prediction based on clinical parameters
We next compared the prognosis prediction performance of our 16-protein panel with commonly used prediction methods based on clinical parameters. We focused on the following clinical parameters: the PSA level at diagnosis (pre-PSA), the pathological staging, and the ISUP grade. Using a multivariate Cox regression analysis, these clinical parameters had a significantly high unfavorable risk score for BCR-free survival, indicating that PSA level at diagnosis, pathological staging, and ISUP grade are significantly associated with BCR (Figure 5A), in agreement with published results.8 Therefore, these three clinical parameters were further used in the comparison for prognostic prediction performance with the 16-protein panel. The hazard ratios (HRs) of pre-PSA, ISUP grade, and pathological T stage were between 1.07 and 2.07, while the HR of the 16-protein panel is 3.79, which is the highest (Figure 5A, labeled in red). We also found that the 16-protein panel exhibited a significant association with ISUP grade after being adjusted by multiple covariates (Table S4B). The AUCs of BCR-free prognosis prediction within three years using the 16-protein panel were significantly higher than using clinical parameters (Figures 5B and S5F), indicating that the 16-protein panel has a superior performance in prediction prognosis than the commonly employed methods based on clinical parameters. The decision curve analysis also supported that the 16-protein panel outperformed the clinical parameter-based methods (Figure S5G).
Figure 5.

Comparing the predictive performance of the 16-protein model and clinicopathological characteristics
(A) Forest plot showing the BCR-free prognostic score for each clinical parameter in a multivariate Cox regression analysis. The middle points indicate the hazard ratios. The endpoints represent the lower or upper 95% confidence intervals. BE, preoperative endocrine therapy; PE, postoperative endocrine therapy; PR, postoperative radiotherapy.
(B) ROC curves of the 16-protein panel and clinicopathological characteristics (PSA level, Gleason score, pathology stage, and D’Amico) at 1 (upper panel), 2 (middle panel), and 3 (lower panel) years in the PCSHA validation dataset.
(C–E) Sankey plots showing the patients with PCa overlapping among the 16-protein panel prediction system, the recurrence status, and the ISUP grade using the PCSHA validation dataset (C), TCGA dataset (D), and the MSKCC dataset (E).
Notably, Sankey plots showed that the risk classification based on our 16-protein panel overlaps with the observed BCR better than the risk classifications based on the ISUP grade in the PCSHA dataset (Figures 5C and S5H), TCGA dataset (Figure 5D), and the MSKCC dataset (Figure 5E), further demonstrating that the 16-protein panel outperforms the ISUP grade in predicting BCR-free survival.
The 16-protein prognosis prediction panel was validated in independent biopsy samples
In order to further validate the effectiveness and assess the clinical translation probability of the 16-protein panel in predicting BCR-free survival, a total of 99 FFPE biopsy samples were collected from 99 patients with PCa as an independent validation cohort, named as PCSHA-biopsy cohort (Table S5. Patient information for biopsy samples, related to Figure 6, Table S6. Protein quantification matrix detected by PRM, related to Figure 6, Table S7. Clinical characteristics of the seven PCa cohorts in this study, related to Figures 1 and 4–6). Upon examining the correlation between clinical characteristics and BCR-free survival, we discovered that there was no significant association between the prognosis of PCa and the D'Amico classification or pathological stages of this particular group (Figures 6A and 6B). Subsequently, the 16-protein panel’s prognostic prediction efficiency was assessed by measuring their expression level using parallel reaction monitoring (PRM)-targeted proteomics. The Pearson correlation of the quality control samples was greater than 0.99 (Figure S6A), indicating that the proteomic data are highly repeatable. Significant differences in the expression of 16 proteins from the 16-protein panel between high-risk and low-risk groups were observed (Figures 6C and S6B). This outcome is in line with the findings observed from the discovery cohort’s surgical samples, which are analyzed by DIA (Figure 4B).
Figure 6.

Validation of the 16-protein panel for BCR prediction in prostate cancer patients using biopsy samples
(A and B) Kaplan-Meier plots for BCR-free survival based on D’Amico (A) and pathological stage (B) in the PCSHA-biopsy dataset (N = 99).
(C) The peak groups of the unique peptides and the protein abundance between high- and low-risk patients.
(D) ROC curves (right panel) of the 16-protein model and clinicopathological characteristics (PSA level, ISUP grade based on biopsy and surgical samples, pathological stage, and D’Amico) in the PCSHA-biopsy test set.
(E) Kaplan-Meier plots of BCR-free survival based on the 16-protein prediction model in the PCSHA-biopsy set.
In this PCSHA-biopsy cohort, the AUC of the 16-protein panel for BCR prediction was 0.88 (Figures 6D and S6C), demonstrating superior performance of D'Amico, pre-PSA levels at diagnosis, the pathological staging, and the ISUP grade of both biopsy and surgical samples. Based on the expression of these 16 proteins, we were able to cluster patients with PCa into high- and low-risk groups. The risk groups are significantly linked to BCR-free survival (Figure 6E). These outcomes are in agreement with the findings from the DIA proteomics-analyzed discovery dataset, which further confirmed the effectiveness of this 16-protein panel in predicting BCR for patients with PCa. More importantly, the 16-protein panel was effectively verified in biopsy specimens, indicating that it is more practically applicable to evaluate the risk of BCR prior to radical prostatectomy, which could offer enhanced direction for prompt and vigorous interventions for patients with high-risk PCa.
NUDT5 and SEPTIN8 as the potential synthetic targets
Among the 16 proteins, half of them have a higher expression in tumor tissues than in adjacent non-tumor tissues (Figure S4). Five proteins, namely transcription and mRNA export factor ENY2 (ENY2), ADP-sugar pyrophosphatase (NUDT5), Septin-8 (SEPTIN8), latent transforming growth factor β (TGF-β)-binding protein 2 (LTBP2), and Cullin-5, were found to be positively associated with poor prognosis in patients with PCa (Table S4). The first three proteins were also found to be highly expressed in the high-risk group, the high Gleason score group (GS 8–10), and/or the advanced pathological stage (T3/T4) (Figure S4). ENY2 can regulate transcription widely and affect cell growth,37 which might not be suitable for therapeutic target. LTBP2 is a TGF-β-binding protein. The TGF-β signaling pathway can induce androgen receptor activation; therefore, LTBP2 could be one of the potential therapeutic targets for PCa.38 However, we found no significant correlation between the alteration of LTBP2 and the overall survival in TCGA database. NUDT5 is involved in the nucleic acid metabolism pathway and has been explored as a hormone-dependent protein regulating proliferation that promotes breast cancer cell growth. The inhibition of NUDT5 has been shown to effectively suppress the growth of breast cancer cells.39 SEPTIN8 is a cytoskeleton-related protein associated with intercellular communication, platelet secretion, and vesicle transport.40
Immunohistochemical staining was performed to explore the expression levels of NUDT5 and SEPTIN8 in 69 patients with PCa. The results showed that the expression of both NUDT5 and SEPTIN8 was significantly higher in the high-risk group compared to the low-risk group (Figures 7A and 7B; S7A), which was consistent with the findings shown in Figure 4B. We also examined the expression of NUDT5 and SEPTIN8 in several PCa cell lines by western blotting (Figure S7B). Interestingly, we observed a higher expression of NUDT5 in DU 145 and C4-2 cell lines compared to LnCaP, while there was no significant difference in the expression of SEPTIN8 among PCa cell lines. To further investigate the functional roles of NUDT5 and SEPTIN8 in PCa, we performed knockdown experiments targeting these genes in PC-3 cells (Figures 7C, S7C, and S7D). Interestingly, we found that knockdown of NUDT5 or SEPTIN8 alone did not significantly affect the proliferation (Figures 7E–7H), migration (Figures 7I–7K and S7E), or cell cycle (Figure S7F) of PC-3 cells. However, simultaneous knockdown of both NUDT5 and SEPTIN8 in PC-3 cells (Figure 7D) resulted in a significant inhibition of cell proliferation (Figures 7F–7H), migration (Figures 7I and 7J), and invasion (Figures 7L and 7M). These findings suggest a potential synergistic effect of NUDT5 and SEPTIN8 in promoting the malignant characteristics of PCa cells.
Figure 7.

Validation of potential synthetic targets
(A and B) Immunohistochemical (IHC) staining showing the protein expression of SEPTIN8 and NUDT5 in benign and tumor samples of low-risk and high-risk PCa patients (A), and the statistics for the IHC intensity score of 69 patients with PCa were shown in (B).
(C and D) Western blot showing the efficacy of the single (C) or double (D) knockdown of SEPTIN8 or NUDT5 in PC-3 cells.
(E and F) Cell proliferation assay of the single (E) or double (F) knockout of SEPTIN8 or NUDT5 knockdown PC-3 cells using a ZenCell Owl live-cell imaging system.
(G and H) Cell proliferation assay of SEPTIN8-knockout (KO), NUDT5 KO, and double KO (dKO) (SEPTIN8/NUDT5-dKO) using EdU staining method, and the statistics for the percentage of EdU-positive cells were shown in (H).
(I and J) Wound healing assay of dKO in PC-3 cell line, and the statistics were shown in (J).
(K) Bar chart shows the statistics of the wound healing assay for NUDT5 or SEPTIN8 single knockout cells.
(L and M) Invasion assay of SEPTIN8/NUDT5-dKO in PC-3 cell line using transwell, and the statistics were shown in (M). Experiments were repeated in triplicates. Data are represented as mean ± SD. The p values are calculated by Welch’s t test, p value: ∗ < 0.05, ∗∗∗ < 0.001.
See also Figure S7.
Discussion
The comprehensive proteomic resource of the Chinese patients with PCa
In this study, using DIA-MS technology, we generated a highly valuable and comprehensive in-depth proteomic landscape of PCa. In total, 10,045 proteins were identified in 918 FFPE prostate tissues from 306 Chinese patients with PCa. With the use of AI technology, the highly reproducible proteomics data produced by DIA-based proteomics techniques may be investigated further to identify more proteins and learn additional important insights. Patients with PCa included in previous mass spectrometry-based proteome investigations are from western country.16,17,18,19,21 Therefore, to the best of our knowledge, this proteomic dataset—which includes the most patients with PCa and the most proteins that have been identified thus far—is the largest and most comprehensive one, thus serving as a valuable proteomic resource to the PCa field.
To account for the intra-tumor heterogeneity, we collected at least two tumor samples from each patient. However, we observed that intra-tumor heterogeneity was much less than inter-tumor one, highlighting the importance of enrolling a large number of patients to fully understand the underlying mechanisms of PCa.
Unlike other large-scale research on PCa proteins, our cohort is distinctive in that it includes a large number of Chinese patients with PCa. Except for the common dysregulation pathways, such as increased oxidative phosphorylation capacity, dysregulation of the TCA cycle, and mitochondrial dysfunction in tumor samples,12,18,19 we confirmed the different mutation convergence into the common pathway.12,16 Our investigation also discovered several distinct pathways enriched in tumors, including RNA metabolism, ribosome synthesis, mitochondrial translation, and metabolic digestion of amino acids.
The molecular taxonomy of patients with PCa
Given the extensive heterogeneity of PCa, molecular subtyping could help explore the divergent disease mechanisms and predict variable clinical outcomes and treatments. Genomics has been used to identify molecular subtypes of patients with PCa based on specific gene mutations.11 Building on this classical mutation-based genotype, more classifications have been proposed by incorporating transcriptomic data, clinical parameters, and other factors.41,42,43
We proposed a new subtyping of patients with PCa (CPC1, CPC2, and CPC3) based on the proteome of our cohort and showed that it agrees with the observed BCR prognosis of the patients. CPC3 had the poorest prognosis and was characterized by the downregulation of metabolism-related processes and cellular processes involved in cell-cell interaction and the upregulation of genetic information processing and immune cell receptor signaling pathways. Some of these signaling pathways were observed in previous studies.44,45,46 In the amino acid metabolism pathways, arginine, glycine, serine, and fatty acid metabolism have been reported to play important roles in PCa progression.47 FOXA1 mutation has been found in Chinese patients with PCa,12 which is a pioneer factor to regulate DNA binding by distinct transcription factors and RNA splicing.48 Thus, the genetic information processing might be more venerable in our PCSHA cohort, which hints novel insights for PCa treatment guidance and prognostic prediction.
In our work, the molecular clusters are linked to the prognosis of PCa (Figure 2B), which could inform clinicians the molecular features for PCa with low or high BCR risk, and provide guidance to clinicians to develop personalized treatment plan for PCa with different molecular features.
A 16-protein panel for BCR prediction
We developed a 16-protein panel capable of predicting BCR of patients with PCa. The model demonstrated superior BCR prediction performance over other commonly employed prediction system based on the PSA level, ISUP grade, pathology stage, or D’Amico risk classification (Figures 6 and 7). Importantly, the 16-protein model can effectively predict BCR across different ISUP grades and pathological stages, suggesting its potential application for predicting BCR even in the early stages of PCa.
PCSHA validation dataset, as well as other six published PCa datasets, including three transcriptomic datasets and three proteomic datasets were used to assess the efficacy and generalizability of the 16-protein model in BCR prediction. Similar to its BCR prediction power in the PCSHA discovery dataset, the 16-protein model effectively predicted BCR for patients with PCa both in the PCSHA validation dataset and in the three transcriptomic datasets (Figures 3C–3F). However, the prognosis prediction capacity of the 16-protein panel was less effective in the three proteomic dataset (with AUCs less than 0.7) (Figures S4D–S4F), which could potentially be attributed to the lower detection depth and higher missing rate observed in the published proteomic datasets, as well as the smaller cohort sizes compared to our cohort. It is important to emphasize that robust evaluation and validation necessitate an adequate number of samples.
Zhong et al. developed an 18-protein panel (HR = 2.72) for prognosis prediction using proteomic data.49 The HR is lower than ours (HR = 3.79). However, when their 18-protein model was applied to the PCSHA dataset for BCR prediction, its AUC was found to be 0.768, lower than that of our 16-protein panel. Given the significant variability in epidemiology and genetic background of PCa among different ethnic groups,12,50,51 the efficacy of the 18-protein BCR prediction panel for Chinese patients with PCa may be limited. Consequently, these two cohorts from different ethnicities complement each other, expanding the protein landscape of PCa and providing valuable resources for researchers to better understand PCa.
Additionally, we used PRM-targeted proteomics to confirm the efficacy of this 16-protein panel in BCR prediction in an independent biopsy cohort of 99 patients with PCa. The BCR prediction efficacy of the 16-protein model in biopsy samples was similar to that in radical surgery samples (Figure 6), demonstrating the robustness and effectiveness of this 16-protein panel in predicting BCR for patients with PCa at various stages. Furthermore, based on these findings, it may be possible to employ this 16-protein panel to assess the prognosis of PCa prior to radical prostatectomy. This would not only give patients with PCa more information to help them make decisions about their subsequent treatment plan, but it would also give clinicians better guidance on how to treat patients with high-risk PCa promptly and aggressively.
Notably, in both the radical surgical and biopsy samples, the predictive efficacy of this 16-protein panel for BCR far outperforms that of the most widely used D'Amico prediction method, suggesting that the 16-protein could potentially replace D'Amico for BCR prediction in the clinic. Nevertheless, this model still needs to be validated in larger clinical cohorts before it can be further translated into the clinic.
Although high-resolution mass spectrometry is currently not directly applicable to routine clinical testing, targeted mass spectrometry could be transferred into clinical testing in the near future.52 Meanwhile, measuring the expression of these 16-proteins by immunohistochemistry could be implemented in clinic. Hence, our 16-protein prediction panel, which is simple to implement, highly operational, and accessible, can be used to predict the prognosis of PCa in the clinical setting. It can be used to improve diagnostic effectiveness as well as in the decision-making process to find therapeutic targets and options.
Potential therapy targets of PCa
Our finding that many druggable proteins are downregulated in patients with poor prognosis not only explains to some extent the limited availability of targeted drugs for PCa but also suggests new ideas for novel drug mechanisms of action. In this study, we identified four upregulated proteins within the 16-protein panel and found that two proteins, NUDT5 and SEPTIN8, may serve as novel targets for synthetic lethality, which provides a new strategy for targeted therapy for PCa.
Limitations of the study
RNA sequencing data have been used to validate the 16-protein panel’s predictive power in Western cultures for the most part; validation based on proteomics data will shed more light on the panel’s predictive power in other ethnic populations. Although the effectiveness of this 16-protein panel in predicting BCR has been confirmed in a sizable cohort, more clinical cohort validation is still required before this model can be further applied in the clinic. We also acknowledge that the functional significance of these 16 proteins in the initiation and advancement of PCa has not been fully confirmed by this investigation.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-NUDT5 | Abcam | Cat #ab129172; RRID:AB_11150622 |
| anti-Septin8 | Abcam | Cat #ab191404 |
| Anti-GAPDH | Cell signaling technology | Cat #2118; RRID:AB_561053 |
| Bacterial and virus strains | ||
| E. coli TOP10 competent cells | GB104 | Tiangen |
| Biological samples | ||
| FFPE samples | This paper | PCSHA (Table S1) |
| FFPE biopsy samples | This paper | PCSHA-biopsy (Table S5) |
| Chemicals, peptides, and recombinant proteins | ||
| Heptane | Sigma-Aldrich | Cat #246654-2L |
| Ethanol | Sinopharm Chemical Reagent limited corporation | Cat #10009218 |
| Urea | Sigma-Aldrich | Cat #U1250-1KG |
| Trizma Base | Sigma-Aldrich | Cat #T6791-500G |
| Formic acid (FA) | Fisher Scientific | Cat #A117-50 |
| HCl | Hualishi | Cat #HLS HCL001C |
| Thiourea | Sigma-Aldrich | Cat #T8656-500G |
| Ammonium bicarbonate (ABB) | General-Reagent | Cat #G12990A |
| Tris (2-carboxyethyl) phosphine (TCEP) | Adamas-beta | Cat #61820E |
| Iodoacetamide (IAA) | Sigma-Aldrich | Cat #I6125 |
| Trypsin | Hualishi Tech | Cat #HLS TRY001C |
| Lys-C | Hualishi | Cat #HLS LYS001C |
| Trifluoroacetic acid (TFA) | Fisher Chemical | Cat #85183 |
| Water | Fisher Chemical | Cat #W6-4 |
| Acetonitrile | Fisher Chemical | Cat #A955-4 |
| Methanol | Sigma-Aldrich | Cat #HLS TRY001C |
| Acetonitrile | General-Reagent | Cat #G80988B |
| Xylene | SINOPHARM | 10023418 |
| Neutral balsam | SINOPHARM | 10004160 |
| Blasticidin S HCl | Beyotime | ST018 |
| Puromycin | Yeasen | 60209ES59 |
| Phosphate Buffer Solution | Servicebio | G4202 |
| Matrigel® Matrix | Corning | 354234 |
| Crystal Violet Staining Solution | Beyotime | C0121-100mL |
| Critical commercial assays | ||
| BeyoClick™ EdU Proliferation Kit with Alexa Fluor 488 | Beyotiome | C0071S |
| Deposited data | ||
| Raw data | This paper | https://www.iprox.org/(IPX0005748000) |
| Experimental models: Cell lines | ||
| PC-3 | National Collection of Authenticated Cell Cultures | TCHu158 |
| DU 145 | National Collection of Authenticated Cell Cultures | TCHu222 |
| LNCaP | National Collection of Authenticated Cell Cultures | TCHu173 |
| HEK293T | National Collection of Authenticated Cell Cultures | GNHu17 |
| Oligonucleotides | ||
| CRISPR-SEPTIN8-gRNA1 | This paper | 5′- TGAGGAAGCCAGTCACCATG -3′ |
| CRISPR-NUDT5-gRNA1 | This paper | 5′- GAAACAGTTCCGACCACCAA -3′ |
| CRISPR-SEPTIN8-gRNA2 | This paper | 5′- ACTGGCTTCCTCAGTCTCGA -3′ |
| CRISPR-NUDT5-gRNA2 | This paper | 5′- TCACTATGAGTGTATCGTTC -3′ |
| CRISPR-Scramble-gRNA | This paper | 5′- ACGGAGGCTAAGCGTCGCAA -3′ |
| Recombinant DNA | ||
| Lenti-CRISPR-V2 Vector | Addgene | Addgene Plasmid # 52961 |
| Software and algorithms | ||
| Xcalibur | Thermo Fisher Scientific | OPTON-30965 |
| DIA-NN version 1.8.0-linux | Demichev et al.53 | https://github.com/vdemichev/DiaNN |
| R version 4.0.2 | R Project | https://www.r-project.org |
| Metascape | Zhou et al., 201932 | https://metascape.org/gp/index.html#/main/step1 |
| Other | ||
| SOLAμ | Thermo Fisher Scientific | Cat # 62209-001 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Chenghua Yang (Chenghua-yang@qq.com).
Materials availability
The study did not generate new unique reagents.
Data and code availability
-
•
All data are available in the manuscript or the supplementary material. The mass spectrometry proteomics raw data have been deposited to the ProteomeXchange Consortium (https://proteomecentral.proteomexchange.org) via the iProX partner repository54,55 with the dataset identifier PXD054025.
-
•
The study did not generate new code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Experimental model and subject details
Patients and tissue samples
With the ethical approval by the Translational Medicine Ethic Committee, Second Military Medical University (TMEC2014-001), this study examined the formalin-fixed paraffin-embedded (FFPE) PCa tissue samples from 306 PCa patients (Table S1) who underwent radical prostatectomy between 2015 and 2020. For each patient, three FFPE punches were obtained: two FFPE punches were from the tumor locations (tumor cell components are over 90%) with primary (G1) and secondary (G2) Gleason score grades, respectively; and one punch was from a benign region. Two experienced pathologists examined the tissue blocks from each patient, and regions with major and minor Gleason scores were marked for sample punching (Figure S1). An expert pathologist punctured the tissue samples (1 mm in diameter, 0.5–1 mm thick, and roughly 0.6–1.2 mg in weight, including wax) in blocks of FFPE prostate tissues for proteomics analysis (Figure S1).
For PRM targeted proteomic analysis, 99 FFPE biopsy samples were collected from 99 PCa patients at initial diagnosis before radical prostatectomy (Table S5). The biopsy for prostate cancer at diagnosis is performed as a combination of systemic biopsy and targeted biopsy. Each FFPE biopsy sample selected for the targeted proteomics analysis contains more than 70% tumors, which is evaluated by experienced pathologist.
All patients were staged according to the 4th edition of the WHO classification on male and genitourinary tumors from 2016. International Society of Urological Pathology (ISUP) system divides PCa into five grades: grade 1 (GS ≤ 6), grade 2 (GS = 7 with a primary and secondary pattern of 3 + 4), grade 3 (GS = 7 with a primary and secondary pattern of 4 + 3), grade 4 (GS = 8), and grade 5 (GS = 9–10).56
Cell culture
The PCa cell lines, including PC-3, DU 145, C4-2 and LNCaP cell lines, were purchased from National Collection of Authenticated Cell Cultures (https://www.cellbank.org.cn/). PC-3 cells were cultured in F-12K medium with 10% FBS, DU 145 cells in MEM with 10% FBS, C4-2 cells in DMEM medium with 10%FBS, and LNCaP cells in RPMI medium with 10%FBS. All cell culture media were completed with 2 mM glutamine, 10 mM HEPES, and penicillin G/streptomycin, and cultures at 37°C, with a humidified 5% CO2 environment. Short Tandem Repeats (STR) profiling was used to confirm the legitimacy of these prostate cancer cell line.
Method details
Study design and quality control
To minimize batch effects among different batches of samples, 918 samples were randomly distributed into 72 batches, each comprising 15 prostate samples and one fresh mouse liver sample for pressure cycling technology (PCT) quality control. To control for individual mouse heterogeneity and liver lobe heterogeneity, we divided the same liver lobe of a mouse into 72 parts. We evaluated the quality of these quality control (QC) samples (Figure S2). During mass spectrometry acquisition, it was necessary to include a duplicate sample of one randomly selected sample. The mass spectrometry accusation sample was a mixture of randomly selected prostate tissues from PCSHA, including benign and tumor samples with different Gleason scores. In order to ensure that the systematic error of the experiment was negligible, we assessed the reproducibility and batch effect of the QC samples and replicates. We found that the inter-patient heterogeneity was significantly higher than intra-individuals one (Figure S2D); thus, we took the average value of the two tumor samples protein intensity for each patient.
For prostate cancer subtyping analysis and the development of prognosis prediction model, we randomly divided all the patients, in a ratio of 3:1, into two groups, a discovery set (N = 226) and a validation set (N = 80), so that we can validate the analysis in an independent patient cohort.
Proteomics analysis
In brief, about 0.5 mg of FFPE PCa samples were processed into clean peptides through dewaxing, rehydration, protein denaturation assisted with pressure cycling technology (PCT), and digestion with lysC (enzyme to substrate ratio is 1:80)/trypsin (enzyme to substrate ratio is 1:20) assisted with PCT, as described previously.57 Peptide samples were separated with a nanoElute system at 300 nL/min flow rate and 217.5 bar. The mobile phase was mixed with buffer A (2% ACN, 0.1% formic acid) and buffer B (98% ACN, 0.1% formic acid). The buffer B (%) was linearly increased from 5 to 27%, followed by an increase to 40% within 10 min and a further boost to 80%. Peptides were scanned by a CaptiveSpray nanoelectrospray ion source on a hybrid trapped ion mobility spectrometer (TIMS) quadrupole time-of-flight mass spectrometer (timsTOF Pro, Bruker Daltonics, Germany). We performed data-dependent acquisition (DDA) in parallel cumulative continuous fragmentation mode with 10 PASEF scans per top-N acquisition cycle, which was used to generate a library of ion mobility-enhanced spectra.30 A total cycle time of 1.17 s was achieved with an accumulation and ramp time of 100 ms each for the dual TIMS analyzer. The ion mobility was scanned from 0.6 to 1.6 V/cm2. The MS1 and MS2 acquisitions were performed in the m/z range of 100 to 1,700 Th. Precursors reaching a target value of 20,000 arbitrary units were dynamically excluded for 0.4 min, and singly charged precursors were excluded at positions in the m/z plane of ion mobility. For DIA acquisition, the data were collected using a data independent collection of parallel cumulative sequential fragmentation.30 Ion mobility was scanned from 0.7 to 1.3 V/cm2. MS1 and MS2 acquisitions were performed in the m/z range of 100 to 1,700 Th. We set a window with a width of 25 Da, and the rest of the parameters were the same as for DDA.30
For the DIA analysis, we used DIANN (version 1.8.0-linux) for raw data parsing. Based on deep learning spectrograms and FASTA theoretical enzymatic cuts, we selected the options "unrelated sun", "use isotopologues", and "MBR". In addition, Trypsin was set as the digestive enzyme, carbamidomethylation was set as a fixed modification, and N-term methionine excision and methionine oxidation were set as variable modifications. Other parameters were left to their default values except for the protein inference, which was set to “protein name”. The background file was a human FASTA file downloaded from the UniProt website on January 26, 2020.
In total, 10,071 proteins were identified with 44.7% missing rate of the whole proteome. The batch effect across all samples and the reproducibility of the measurement were evaluated by TSNE plots and the correlation coefficient of 144 quality control samples (Figure S2).
PRM analysis
99 FFPE biopsy samples were collected and randomly divided into seven batches for targeted proteomic analysis, with each batch comprising 15 samples and a QC sample. In brief, about 0.5 mg of FFPE PCa samples were processed into clean peptides using the same process as in the FFPE surgical samples. The clean peptide samples were analyzed using parallel reaction monitoring (PRM) targeted proteomics, which was performed on Q Exactive HF MS (Thermo Fisher Scientific) system with UltiMate 3000 RSLCnano System (Thermo Fisher Scientific). The linear LC was 60 min 5%–30% buffer B (98% ACN, 0.1% formic acid), separating at a flow rate of 300 nL·min−1. The time-scheduled acquisition was in a ±5 min retention time window. Full scans were acquired with an m/z range of 300–2000 Th at a resolution of 60,000 FWHM. The AGC target was set at 3E6, and the maximum injection time was 55 ms. Target precursors were isolated using an m/z window of 1.6 Th and then fragmented at 27% normalized collision energy. The resulting product ions were scanned at a resolution of 30,000 FWHM, with an AGC target value of 2E5 charges and a maximum injection time of 100 ms. After evaluating the peak shape and the idotp score, 16 peptide precursors from 16 proteins and 20 CiRT peptide precursors were analyzed using skyline (Version 21.1). The Skyline-generated peptide quantitative result was log2 transformed and quantile normalized before transformed into a protein matrix. The quantification results of each peptide from each sample were detailed in Table S6.
Differentially expressed analysis
In case the different missing bias in both tumor and benign, proteins that were missing in more than 80% of the samples in both the tumor group and the benign group were first deleted. Then, the remaining missing values were filled with zeros, and the mean value of duplicate samples was taken as the final quantitative value for that sample. A two-sided unpaired Welch’s t-test was used to compare the two groups. And p-values were adjusted using the Benjamini-Hochberg (B-H) method. The differentially expressed proteins between tumor and benign tissues were filtered with fold change greater than 1.5, p-value less than 0.05 using the B-H test.
Proteomic-based clustering analysis
The pathway enrichment analysis was performed using differentially expressed proteins between tumor and benign tissues. The activation degree score of the pathway is calculated in GSVA.58
The enrichment analysis of pathways was conducted using the "enricher" function from the "clusterProfiler" package, with default parameters. The analysis utilized 50 hallmark gene sets obtained from the Molecular Signature Database v7.4. For the proteomic data, enrichment was performed using the "gsva" method within the GSVA framework.
To determine the optimal number of stable PCa subtypes, we utilized K-means clustering (implemented through the "kmeans" function in R), consensus clustering (implemented through the "consensusClusterPlus" package in R), and NbClust testing (implemented through the "NbClust" function in R). In order to cluster the samples based on the constituent pattern of each pathway, we scaled each sample. Consensus clustering was then employed to evaluate the robustness of the K-means clustering, with 1000 iterations and 80% resampling. NbClust testing provided 50 different test methods to determine the optimal number of clusters. Finally, a silhouette analysis was performed to confirm the stability of the clustering.
mFuzz analysis
In case bias is induced from the benign samples, proteins that were missing in more than 80% of the tumor samples were deleted. The minimum value is suitable for the normalization in the mFuzz analysis. Therefore, we filled the missing values in the matrix of each GS group by multiplying the minimum value of the protein matrix in the group by 0.8. One-way analysis of variance (ANOVA) was used to determine differences between samples with different GS grades. P-values were adjusted using the B-H method. The average normalized protein quantities by Z score in each GS grade were used for fuzzy c-means clustering with the R (version 4.0.2) package Mfuzz (version 2.48.0). The number of clusters was set to ten and the fuzzifier coefficient, M, was set to 1.25.
Druggable protein screening
To enhance the reliability of our data, we implemented rigorous criteria that excluded proteins with a deletion rate exceeding 30% (i.e., proteins that were undetected in over 30% of the samples). Following this process, we were left with a subset of 174 proteins. Nineteen of the proteins in our subset of 174 were found to overlap with the molecules identified in NMF clustering.
Prognostic prediction model building
To identify more robust prognostic biomarkers and subsequently validate them using external validation datasets, we implemented stricter criteria. Specifically, samples with over 50% missing ratios were excluded, and patients with recurrence-free survival (RFS) of less than 1 month were omitted. Additionally, proteins with an expression level of “NA” (not detected) in >50% of patients were removed, resulting in an expression matrix comprising 6753 proteins for 266 PCa patients. Utilizing the protein matrix obtained, and guided by the assessment outcomes from "NAguideR",59 we employed the impseq method60 to impute missing values.
Next, all PCa patients were randomly divided into a discovery set (N = 186) and a validation set (N = 80). Univariate Cox regression analysis was used to analyze the correlation between protein expression levels and BCR in the discovery set, revealing 973 proteins significantly correlated with BCR (p < 0.05). The expression levels of all proteins were then ranked, and the top 50% of them were selected for subsequent analysis. LASSO regression was performed 100 times using the glmnet package (version 4.1.6) in R software, with the parameters set to family = "cox" and type.measure = "deviance". 10-fold cross-validation was used, and other parameters were set to default. Proteins with an occurrence frequency of ≥90 were selected for subsequent modeling analysis, including P80188, Q96K21, P61626, Q08426, O94972, P02042, P04179, P12724, Q13885, Q14767, Q8N9N2, Q93034, Q9HD42, Q9NPA8, Q9UKK9, and Q92599 (Table S4). The univariate Cox regression analysis demonstrated that patients with a higher expression level of Q14767, Q92599, Q93034, Q9NPA8, and Q9UKK9 significantly increased the risk of recurrence. For O94972, P02042, P04179, P12724, P61626, P80188, Q08426, Q13885, Q8N9N2, Q96K21, and Q9HD42, a higher expression level was associated with better prognosis in prostate cancer patients.
A logistic regression algorithm was applied to develop a predictive model for prostate cancer recurrence risk. The formula for the model is as follows:
The Risk score represents a patient’s risk score for BCR, K refers to the number of proteins, Wi represents the regression coefficients from the multiple Cox regression analysis, and Ei refers to the protein expression levels. Proteins with Wi > 0 are defined as risk factors, and those with Wi < 0 are defined as protective factors. Patients were divided into high/low-risk groups using the optimal cut-off value, determined through the "surv_cutpoint" function from the R package survminer (version 0.4.9). The predictive model was then evaluated in the PCSHA validation cohort and another six independent cohorts.
Immunohistochemistry staining
We collected 69 FFPE samples from the proteogenomic cohort to validate the expression of NUDT5 and SEPTIN8 by Immunohistochemistry (IHC). Immunostaining was carried out as reported previously.61 Sections were stained using anti-NUDT5 (ab129172, Abcam) and anti-SEPTIN8 (ab191404, Abcam) antibodies at 1:1200 dilution and detected by the DAB reagents provided in detection kit (Wuxi Origene). Immunostaining was quantified based on the staining intensity (intensity score). The staining intensity was scored as 0 for negative staining; 1 for weak staining; 2 for moderate staining; 3 for strong staining.
Generation of NUDT5 and SEPTIN8 knockout cells
The CRISPR-cas9 system was used to generate NUDT5 or SEPTIN8 knockout cells. The sequence of Guide RNAs used to knockdout NUDT5 or SEPTIN8 were shown in the Key resources table. Scramble gRNA were used as a control. Guide RNA were cloned into the Lenti-CRISPR-V2 Vector (Addgene Plasmid # 52961). Lentivirus were packaged in HEK293T cells and used to infect the PC-3 cell line for 24 h. Then, stable cell lines were established for the subsequent experiment by puromycin selection using 1 g/mL for three days. Western blot and the surveyor assay were used to evaluate the effectiveness of the gene knockdown.
EdU staining
BeyoClickTM EdU Cell Proliferation Kit with Alexa Fluor 488 was used to measure the proliferation of the PC-3 cell line (Beyotime, C0071S). To label the cells in logarithmic growth, PC-3 cells were seeded into 6-well plates at a density of 1×105 cells per well and then treated for 2 h with media containing 10 mM EdU. Following a single PBS wash, the cells were fixed in 4% paraformaldehyde for 15 min. After fixation, the cells were permeabilized for 15 min in PBS containing 0.3% Triton X-100. Each click reaction well received a preset dose of click reaction buffer. Click Response Buffer was applied to the cells for 30 min at room temperature. Hoechst 33342 was then used to stain the cell nuclei for 30 min at room temperature while it was dark. Fluorescence microscopy was used to examine the prepared samples.
Western blot analysis
Cells were lysed with RIPA buffer (50 mM Tris-HCl pH 7.4, 1% Triton X-100, 1 mM EDTA, 150 mM NaCl, 0.1% SDS, 2mM sodium pyrophosphate, 50 mM NaF, and a cocktail of protease inhibitor) on ice. The prostate tissues were homogenized and then lysed in RIPA buffer on ice. All lysates were centrifuged at 12,000 rpm for 15 min at 4°C. Proteins were first separated by SDS-PAGE and then transferred to polyvinylidene fluoride membranes. The membranes were blocked using 5% milk in PBS and blotted using the antibodies. The anti-NUDT5 (ab129172) and the anti-Septin8 (ab191404) antibodies were from Abcam. The GAPDH (#2118) antibody was purchased from Cell signaling technology. The HRP-labeled Goat Anti-Mouse/Rabbit IgG(H + L) antibodies were purchased from Epizyme.
Cell proliferation assay
To measure the cell proliferation rate, 5×103 exponentially developing PC-3 cells were seeded in 24-well plates (Corning, Cat. No. 3516). The images of the PC-3 cells were recorded every 2 h using a ZenCell Owl live cell imaging system (Innome, Germany). Cell numbers were analyzed at each time point. The cell proliferation curves were plotted utilizing GraphPad Prism.
Wound healing assay
One mL of 1x105 exponentially growing PC-3 cells was seeded and grown to a cell density of roughly 90% in 12-well plates (Corning, Cat. No. 3513). The medium was changed to serum-free RPMI 1640 media after drawing straight lines in the wells. The cells were cultured for additional 48 h and imaged every 24 h using an EVOSTM XL Core. Image Pro Plus was used to calculate the migration rate of the PC-3 cells.
Invasion assay
A total of 5x105 exponentially growing PC-3 cells that were starved for 12h in 200 μL RPMI 1640 medium without FBS were seeded into transwell chambers (Corning, Cat. No. 3422), which were pre-coated with 60μL Matrigel solution (12.5% Matreigel in RPMI 1640 medium) for 3h at 37°C. Next, we added 500 μL RPMI 1640 medium with 30% FBS below the chambers. PC-3 cells were grown in the transwell chambers for 24h. The chambers were gently dipped in PBS three times and the non-invading cells were scrubbed from the membrane’s upper surface using cotton-tipped swabs. The cells were then fixed with 4% paraformaldehyde for 30 min, washed three times with PBS, and stained with Crystal Violet Staining Solution for 15 min. Finally, cells in transwell chambers were imaged using EVOSTM XL Core.
Cell cycle analysis
For cell cycle analysis, NUDT5 or SEPTIN8 knocked out PC-3 cells were, collected, washed with PBS, re-suspended in DNA Staining Solution (Multi Sciences, CCS012) and stained with Permeabilization Solution (Multi Sciences, CCS012) at 1:100 dilution for 30 min at room temperature in the dark. Percentage of different cell cycle phases were analyzed by FlowJo.
Quantification and statistical analysis
A two-sided unpaired Welch’s t-test was used to compare the two groups. And p-values were adjusted using the Benjamini-Hochberg (B-H) method. One-way analysis of variance (ANOVA) was used to determine differences between samples with different GS grades. P-values were adjusted using the B-H method.
Acknowledgments
This work was supported by grants from the National Key R&D Program of China (no. 2022YF0608403), the National Natural Science Foundation of China (81972492, 81772695, and 82373011), the Shanghai Science and Technology Commission Outstanding Academic Leaders Program (22XD1405000), the Zhejiang Provincial Natural Science Foundation of China (LQ24C050002) and the “Leading Goose” R&D Program of Zhejiang (2024SSYS0035). We thank Westlake University Supercomputer Center for assistance in data generation and storage and the Mass Spectrometry & Metabolomics Core Facility at the Center for Biomedical Research Core Facilities of Westlake University for sample analysis. We thank Mr. Hang Wu, Ms. Jianfang Yu, and Jiayi Liu for assistance for the sample preparation and Mr. Youqi Liu for assistance with the data analysis of this manuscript.
Author contributions
C.Y., X.G., T.G., and R.S. designed and supervised the project. H.Y., Yan Wang, M.Q., J.Z., Z.D., Y. Zhang, B.Y., J.H., L.W., Z.L., W.Z., and C.X. collected the samples and clinical data. M.H. performed the pathological evaluation and punching of the FFPE samples. R.S. performed proteomic experiments. J.A., L.T., Yingrui Wang, Y. Zhu, M.L., L.H., W.G., H.C., X.D., K.M., and R.S. conducted proteomic data analysis. H.Y. and X.S. performed biological function validation. R.S., C.Y., and T.G. wrote the manuscript with inputs from co-authors.
Declaration of interests
T.G. is a shareholder of Westlake Omics, Inc. L.T., W.G., and L.H. are employees of Westlake Omics, Inc.
Published: August 8, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101679.
Contributor Information
Tiannan Guo, Email: guotiannan@westlake.edu.cn.
Xu Gao, Email: gaoxu.changhai@foxmail.com.
Chenghua Yang, Email: Chenghua-yang@qq.com.
Supplemental information
For each sample, “_1” represents the lower Gleason score tumor sample; “_2” represents the higher Gleason score tumor samples; and “_
References
- 1.Siegel R.L., Miller K.D., Wagle N.S., Jemal A. Cancer statistics, 2023. CA A Cancer J. Clin. 2023;73:17–48. doi: 10.3322/caac.21763. [DOI] [PubMed] [Google Scholar]
- 2.Rebello R.J., Oing C., Knudsen K.E., Loeb S., Johnson D.C., Reiter R.E., Gillessen S., Van der Kwast T., Bristow R.G. Prostate cancer. Nat. Rev. Dis. Prim. 2021;7:9. doi: 10.1038/s41572-020-00243-0. [DOI] [PubMed] [Google Scholar]
- 3.Van den Broeck T., van den Bergh R.C.N., Arfi N., Gross T., Moris L., Briers E., Cumberbatch M., De Santis M., Tilki D., Fanti S., et al. Prognostic Value of Biochemical Recurrence Following Treatment with Curative Intent for Prostate Cancer: A Systematic Review. Eur. Urol. 2019;75:967–987. doi: 10.1016/j.eururo.2018.10.011. [DOI] [PubMed] [Google Scholar]
- 4.Stephenson A.J., Scardino P.T., Eastham J.A., Bianco F.J., Jr., Dotan Z.A., Fearn P.A., Kattan M.W. Preoperative nomogram predicting the 10-year probability of prostate cancer recurrence after radical prostatectomy. J. Natl. Cancer Inst. 2006;98:715–717. doi: 10.1093/jnci/djj190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hull G.W., Rabbani F., Abbas F., Wheeler T.M., Kattan M.W., Scardino P.T. Cancer control with radical prostatectomy alone in 1,000 consecutive patients. J. Urol. 2002;167:528–534. doi: 10.1016/S0022-5347(01)69079-7. [DOI] [PubMed] [Google Scholar]
- 6.Freedland S.J., Humphreys E.B., Mangold L.A., Eisenberger M., Dorey F.J., Walsh P.C., Partin A.W. Risk of prostate cancer-specific mortality following biochemical recurrence after radical prostatectomy. JAMA. 2005;294:433–439. doi: 10.1001/jama.294.4.433. [DOI] [PubMed] [Google Scholar]
- 7.Pound C.R., Partin A.W., Eisenberger M.A., Chan D.W., Pearson J.D., Walsh P.C. Natural history of progression after PSA elevation following radical prostatectomy. JAMA. 1999;281:1591–1597. doi: 10.1001/jama.281.17.1591. [DOI] [PubMed] [Google Scholar]
- 8.D'Amico A.V., Whittington R., Malkowicz S.B., Schultz D., Blank K., Broderick G.A., Tomaszewski J.E., Renshaw A.A., Kaplan I., Beard C.J., Wein A. Biochemical outcome after radical prostatectomy, external beam radiation therapy, or interstitial radiation therapy for clinically localized prostate cancer. JAMA. 1998;280:969–974. doi: 10.1001/jama.280.11.969. [DOI] [PubMed] [Google Scholar]
- 9.Mazzone E., Gandaglia G., Ploussard G., Marra G., Valerio M., Campi R., Mari A., Minervini A., Serni S., Moschini M., et al. Risk Stratification of Patients Candidate to Radical Prostatectomy Based on Clinical and Multiparametric Magnetic Resonance Imaging Parameters: Development and External Validation of Novel Risk Groups. Eur. Urol. 2022;81:193–203. doi: 10.1016/j.eururo.2021.07.027. [DOI] [PubMed] [Google Scholar]
- 10.Hernandez D.J., Nielsen M.E., Han M., Partin A.W. Contemporary evaluation of the D'amico risk classification of prostate cancer. Urology. 2007;70:931–935. doi: 10.1016/j.urology.2007.08.055. [DOI] [PubMed] [Google Scholar]
- 11.Cancer Genome Atlas Research N. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163:1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J., Xu C., Lee H.J., Ren S., Zi X., Zhang Z., Wang H., Yu Y., Yang C., Gao X., et al. A genomic and epigenomic atlas of prostate cancer in Asian populations. Nature. 2020;580:93–99. doi: 10.1038/s41586-020-2135-x. [DOI] [PubMed] [Google Scholar]
- 13.Fraser M., Sabelnykova V.Y., Yamaguchi T.N., Heisler L.E., Livingstone J., Huang V., Shiah Y.J., Yousif F., Lin X., Masella A.P., et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature. 2017;541:359–364. doi: 10.1038/nature20788. [DOI] [PubMed] [Google Scholar]
- 14.Wang J., Ma Z., Carr S.A., Mertins P., Zhang H., Zhang Z., Chan D.W., Ellis M.J., Townsend R.R., Smith R.D., et al. Proteome Profiling Outperforms Transcriptome Profiling for Coexpression Based Gene Function Prediction. Mol. Cell. Proteomics. 2017;16:121–134. doi: 10.1074/mcp.M116.060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Attard G., Parker C., Eeles R.A., Schroder F., Tomlins S.A., Tannock I., Drake C.G., de Bono J.S. Prostate cancer. Lancet. 2016;387:70–82. doi: 10.1016/S0140-6736(14)61947-4. [DOI] [PubMed] [Google Scholar]
- 16.Charmpi K., Guo T., Zhong Q., Wagner U., Sun R., Toussaint N.C., Fritz C.E., Yuan C., Chen H., Rupp N.J., et al. Convergent network effects along the axis of gene expression during prostate cancer progression. Genome Biol. 2020;21:302. doi: 10.1186/s13059-020-02188-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sinha A., Huang V., Livingstone J., Wang J., Fox N.S., Kurganovs N., Ignatchenko V., Fritsch K., Donmez N., Heisler L.E., et al. The Proteogenomic Landscape of Curable Prostate Cancer. Cancer Cell. 2019;35:414–427.e6. doi: 10.1016/j.ccell.2019.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Latonen L., Afyounian E., Jylha A., Nattinen J., Aapola U., Annala M., Kivinummi K.K., Tammela T.T.L., Beuerman R.W., Uusitalo H., et al. Integrative proteomics in prostate cancer uncovers robustness against genomic and transcriptomic aberrations during disease progression. Nat. Commun. 2018;9:1176. doi: 10.1038/s41467-018-03573-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iglesias-Gato D., Wikstrom P., Tyanova S., Lavallee C., Thysell E., Carlsson J., Hagglof C., Cox J., Andren O., Stattin P., et al. The Proteome of Primary Prostate Cancer. Eur. Urol. 2016;69:942–952. doi: 10.1016/j.eururo.2015.10.053. [DOI] [PubMed] [Google Scholar]
- 20.Drake J.M., Paull E.O., Graham N.A., Lee J.K., Smith B.A., Titz B., Stoyanova T., Faltermeier C.M., Uzunangelov V., Carlin D.E., et al. Phosphoproteome Integration Reveals Patient-Specific Networks in Prostate Cancer. Cell. 2016;166:1041–1054. doi: 10.1016/j.cell.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhong Q., Rui S., Aref A.T., Noor Z., Anees A., Zhu Y., Lucas N., Poulos R.C., Lyu M., Zhu T., et al. Proteomic-based stratification of intermediate-risk prostate cancer patients. Life Sci. Alliance. 2023;7 doi: 10.26508/lsa.202302146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haffner M.C., Zwart W., Roudier M.P., True L.D., Nelson W.G., Epstein J.I., De Marzo A.M., Nelson P.S., Yegnasubramanian S. Genomic and phenotypic heterogeneity in prostate cancer. Nat. Rev. Urol. 2021;18:79–92. doi: 10.1038/s41585-020-00400-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jaratlerdsiri W., Jiang J., Gong T., Patrick S.M., Willet C., Chew T., Lyons R.J., Haynes A.M., Pasqualim G., Louw M., et al. African-specific molecular taxonomy of prostate cancer. Nature. 2022;609:552–559. doi: 10.1038/s41586-022-05154-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun Y., Selvarajan S., Zang Z., Liu W., Zhu Y., Zhang H., Chen W., Chen H., Li L., Cai X., et al. Artificial intelligence defines protein-based classification of thyroid nodules. Cell Discov. 2022;8:85. doi: 10.1038/s41421-022-00442-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Demichev V., Tober-Lau P., Lemke O., Nazarenko T., Thibeault C., Whitwell H., Rohl A., Freiwald A., Szyrwiel L., Ludwig D., et al. A time-resolved proteomic and prognostic map of COVID-19. Cell Syst. 2021;12:780–794.e7. doi: 10.1016/j.cels.2021.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Messner C.B., Demichev V., Wendisch D., Michalick L., White M., Freiwald A., Textoris-Taube K., Vernardis S.I., Egger A.S., Kreidl M., et al. Ultra-High-Throughput Clinical Proteomics Reveals Classifiers of COVID-19 Infection. Cell Syst. 2020;11:11–24.e4. doi: 10.1016/j.cels.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goncalves E., Poulos R.C., Cai Z., Barthorpe S., Manda S.S., Lucas N., Beck A., Bucio-Noble D., Dausmann M., Hall C., et al. Pan-cancer proteomic map of 949 human cell lines. Cancer Cell. 2022;40:835–849. doi: 10.1016/j.ccell.2022.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo T., Kouvonen P., Koh C.C., Gillet L.C., Wolski W.E., Rost H.L., Rosenberger G., Collins B.C., Blum L.C., Gillessen S., et al. Rapid mass spectrometric conversion of tissue biopsy samples into permanent quantitative digital proteome maps. Nat. Med. 2015;21:407–413. doi: 10.1038/nm.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ludwig C., Gillet L., Rosenberger G., Amon S., Collins B.C., Aebersold R. Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial. Mol. Syst. Biol. 2018;14 doi: 10.15252/msb.20178126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meier F., Brunner A.D., Frank M., Ha A., Bludau I., Voytik E., Kaspar-Schoenefeld S., Lubeck M., Raether O., Bache N., et al. diaPASEF: parallel accumulation-serial fragmentation combined with data-independent acquisition. Nat. Methods. 2020;17:1229–1236. doi: 10.1038/s41592-020-00998-0. [DOI] [PubMed] [Google Scholar]
- 31.Zhang W., Wang T., Wang Y., Zhu F., Shi H., Zhang J., Wang Z., Qu M., Zhang H., Wang T., et al. Intratumor heterogeneity and clonal evolution revealed in castration-resistant prostate cancer by longitudinal genomic analysis. Transl. Oncol. 2022;16 doi: 10.1016/j.tranon.2021.101311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A.H., Tanaseichuk O., Benner C., Chanda S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019;10:1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwon O.K., Ha Y.S., Na A.Y., Chun S.Y., Kwon T.G., Lee J.N., Lee S. Identification of Novel Prognosis and Prediction Markers in Advanced Prostate Cancer Tissues Based on Quantitative Proteomics. Cancer Genomics Proteomics. 2020;17:195–208. doi: 10.21873/cgp.20180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stark J.R., Perner S., Stampfer M.J., Sinnott J.A., Finn S., Eisenstein A.S., Ma J., Fiorentino M., Kurth T., Loda M., et al. Gleason score and lethal prostate cancer: does 3 + 4 = 4 + 3? J. Clin. Oncol. 2009;27:3459–3464. doi: 10.1200/JCO.2008.20.4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Egevad L., Granfors T., Karlberg L., Bergh A., Stattin P. Percent Gleason grade 4/5 as prognostic factor in prostate cancer diagnosed at transurethral resection. J. Urol. 2002;168:509–513. [PubMed] [Google Scholar]
- 36.Taylor B.S., Schultz N., Hieronymus H., Gopalan A., Xiao Y., Carver B.S., Arora V.K., Kaushik P., Cerami E., Reva B., et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Atanassov B.S., Mohan R.D., Lan X., Kuang X., Lu Y., Lin K., McIvor E., Li W., Zhang Y., Florens L., et al. ATXN7L3 and ENY2 Coordinate Activity of Multiple H2B Deubiquitinases Important for Cellular Proliferation and Tumor Growth. Mol. Cell. 2016;62:558–571. doi: 10.1016/j.molcel.2016.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang F., Chen Y., Shen T., Guo D., Dakhova O., Ittmann M.M., Creighton C.J., Zhang Y., Dang T.D., Rowley D.R. Stromal TGF-beta signaling induces AR activation in prostate cancer. Oncotarget. 2014;5:10854–10869. doi: 10.18632/oncotarget.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Page B.D.G., Valerie N.C.K., Wright R.H.G., Wallner O., Isaksson R., Carter M., Rudd S.G., Loseva O., Jemth A.S., Almlof I., et al. Targeted NUDT5 inhibitors block hormone signaling in breast cancer cells. Nat. Commun. 2018;9:250. doi: 10.1038/s41467-017-02293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shiryaev A., Kostenko S., Dumitriu G., Moens U. Septin 8 is an interaction partner and in vitro substrate of MK5. World J. Biol. Chem. 2012;3:98–109. doi: 10.4331/wjbc.v3.i5.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tomlins S.A., Alshalalfa M., Davicioni E., Erho N., Yousefi K., Zhao S., Haddad Z., Den R.B., Dicker A.P., Trock B.J., et al. Characterization of 1577 primary prostate cancers reveals novel biological and clinicopathologic insights into molecular subtypes. Eur. Urol. 2015;68:555–567. doi: 10.1016/j.eururo.2015.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao S.G., Chang S.L., Erho N., Yu M., Lehrer J., Alshalalfa M., Speers C., Cooperberg M.R., Kim W., Ryan C.J., et al. Associations of Luminal and Basal Subtyping of Prostate Cancer With Prognosis and Response to Androgen Deprivation Therapy. JAMA Oncol. 2017;3:1663–1672. doi: 10.1001/jamaoncol.2017.0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen S., Huang V., Xu X., Livingstone J., Soares F., Jeon J., Zeng Y., Hua J.T., Petricca J., Guo H., et al. Widespread and Functional RNA Circularization in Localized Prostate Cancer. Cell. 2019;176:831–843.e22. doi: 10.1016/j.cell.2019.01.025. [DOI] [PubMed] [Google Scholar]
- 44.Zhao S.G., Chen W.S., Li H., Foye A., Zhang M., Sjostrom M., Aggarwal R., Playdle D., Liao A., Alumkal J.J., et al. The DNA methylation landscape of advanced prostate cancer. Nat. Genet. 2020;52:778–789. doi: 10.1038/s41588-020-0648-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meng J., Lu X., Jin C., Zhou Y., Ge Q., Zhou J., Hao Z., Yan F., Zhang M., Liang C. Integrated multi-omics data reveals the molecular subtypes and guides the androgen receptor signalling inhibitor treatment of prostate cancer. Clin. Transl. Med. 2021;11 doi: 10.1002/ctm2.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stelloo S., Nevedomskaya E., Kim Y., Schuurman K., Valle-Encinas E., Lobo J., Krijgsman O., Peeper D.S., Chang S.L., Feng F.Y., et al. Integrative epigenetic taxonomy of primary prostate cancer. Nat. Commun. 2018;9:4900. doi: 10.1038/s41467-018-07270-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strmiska V., Michalek P., Eckschlager T., Stiborova M., Adam V., Krizkova S., Heger Z. Prostate cancer-specific hallmarks of amino acids metabolism: Towards a paradigm of precision medicine. Biochim. Biophys. Acta Rev. Canc. 2019;1871:248–258. doi: 10.1016/j.bbcan.2019.01.001. [DOI] [PubMed] [Google Scholar]
- 48.Lupien M., Eeckhoute J., Meyer C.A., Wang Q., Zhang Y., Li W., Carroll J.S., Liu X.S., Brown M. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell. 2008;132:958–970. doi: 10.1016/j.cell.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhong Q., Sun R., Aref A.T., Noor Z., Anees A., Zhu Y., Lucas N., Poulos R.C., Lyu M., Zhu T., et al. Proteomic-based stratification of intermediate-risk prostate cancer patients. Life Sci. Alliance. 2024;7 doi: 10.26508/lsa.202302146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ren S., Wei G.H., Liu D., Wang L., Hou Y., Zhu S., Peng L., Zhang Q., Cheng Y., Su H., et al. Whole-genome and Transcriptome Sequencing of Prostate Cancer Identify New Genetic Alterations Driving Disease Progression. Eur. Urol. 2018;73:322–339. doi: 10.1016/j.eururo.2017.08.027. [DOI] [PubMed] [Google Scholar]
- 51.Kimura T. East meets West: ethnic differences in prostate cancer epidemiology between East Asians and Caucasians. Chin. J. Cancer. 2012;31:421–429. doi: 10.5732/cjc.011.10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodriguez H., Zenklusen J.C., Staudt L.M., Doroshow J.H., Lowy D.R. The next horizon in precision oncology: Proteogenomics to inform cancer diagnosis and treatment. Cell. 2021;184:1661–1670. doi: 10.1016/j.cell.2021.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Demichev V., Messner C.B., Vernardis S.I., Lilley K.S., Ralser M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods. 2020;17:41–44. doi: 10.1038/s41592-019-0638-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ma J., et al. iProX: an integrated proteome resource. Nucleic Acids Res. 2019;47:D1211–D1217. doi: 10.1093/nar/gky869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen T., et al. iProX in 2021: connecting proteomics data sharing with big data. Nucleic Acids Res. 2021;50:D1522–D1527. doi: 10.1093/nar/gkab1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Epstein J.I., Egevad L., Amin M.B., Delahunt B., Srigley J.R., Humphrey P.A., Grading C. The 2014 International Society of Urological Pathology (ISUP) Consensus Conference on Gleason Grading of Prostatic Carcinoma: Definition of Grading Patterns and Proposal for a New Grading System. Am. J. Surg. Pathol. 2016;40:244–252. doi: 10.1097/PAS.0000000000000530. [DOI] [PubMed] [Google Scholar]
- 57.Cai X., Xue Z., Wu C., Sun R., Qian L., Yue L., Ge W., Yi X., Liu W., Chen C., et al. High-throughput proteomic sample preparation using pressure cycling technology. Nat. Protoc. 2022;17:2307–2325. doi: 10.1038/s41596-022-00727-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hanzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 2013;14:1–15. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang S., Li W., Hu L., Cheng J., Yang H., Liu Y. NAguideR: performing and prioritizing missing value imputations for consistent bottom-up proteomic analyses. Nucleic Acids Res. 2020;48 doi: 10.1093/nar/gkaa498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Verboven S., Branden K.V., Goos P. Sequential imputation for missing values. Comput. Biol. Chem. 2007;31:320–327. doi: 10.1016/j.compbiolchem.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 61.Qu Y., Dai B., Ye D., Kong Y., Chang K., Jia Z., Yang X., Zhang H., Zhu Y., Shi G. Constitutively active AR-V7 plays an essential role in the development and progression of castration-resistant prostate cancer. Sci. Rep. 2015;5:7654. doi: 10.1038/srep07654. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
For each sample, “_1” represents the lower Gleason score tumor sample; “_2” represents the higher Gleason score tumor samples; and “_
Data Availability Statement
-
•
All data are available in the manuscript or the supplementary material. The mass spectrometry proteomics raw data have been deposited to the ProteomeXchange Consortium (https://proteomecentral.proteomexchange.org) via the iProX partner repository54,55 with the dataset identifier PXD054025.
-
•
The study did not generate new code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.