


Abstract
Multiplex quantitative PCR based on novel design of fluorescent primers is described. Fluorogenic primers are labeled with a single fluorophore on a base close to the 3′ end with no quencher required. A tail of 5–7 nt is added to the 5′ end of the primer to form a blunt-end hairpin when the primer is not incorporated into a PCR product. This design provides a low initial fluorescence of the primers that increases up to 8-fold upon formation of the PCR product. The hairpin oligonucleotides (ΔG from –1.6 to –5.8 kcal/mol) may be as efficient as linear primers and provide additional specificity to the PCR by preventing primer-dimers and mispriming. Multiple fluorogenic primers were designed by specialized software and used for real-time quantitation of c-myc and IL-4 cDNAs in the presence of reference genes such as β-actin, GAPDH and 18S rRNA. Targets of 10–107 copies were detected with precision in PCR using FAM-labeled primers for variable genes and JOE-labeled primers for the reference genes. This method was also used to detect single nucleotide polymorphism of the human retinal degeneration gene by allele-specific PCR with end-point detection using a fluorescent plate reader or a UV-transilluminator. We conclude that fluorogenic mono-labeled primers are an efficient and cost-effective alternative to FRET-labeled oligonucleotides.
INTRODUCTION
Nucleic acid detection and quantification methods play an increasing role in medical diagnostics and drug discovery, thus the development of reliable, fast and inexpensive detection methods is important. Quantitative, reverse transcription– polymerase chain reaction (RT–PCR) is used for determining the amount of specific mRNA transcripts in biological samples (1–3). The method has evolved from a low-throughput format with gel-based analysis to the use of fluorescence techniques that do not require the separation of the reaction product (‘closed tube’ format) (1–11). The amount of cDNA amplified by a PCR correlates with an increase in the fluorescent signal that results from an interaction between a fluorescent reporter and the other PCR reactants. The amount of starting DNA is then estimated by analyzing the fluorescence at each cycle of PCR (real-time), or after the PCR (end-point). These fluorescent techniques are faster and can be less expensive since they do not require post-PCR manipulations and the assays may be facilitated by high-throughput robotics. These methods also reduce contamination of the laboratory with PCR amplicon molecules that may interfere with subsequent assays.
The fluorescent techniques used in PCR-based detection may be of various kinds. DNA-binding dyes, such as SYBR Green™, that fluoresce brightly when bound to double-stranded DNA have been used for real-time detection during PCR (4,7). Other methods incorporate the use of an oligonucleotide labeled with a fluorophore and a quencher moiety. The quencher reduces the fluorescence of the fluorophore by fluorescence resonance energy transfer (FRET) when the two moieties are separated by <100 Å (12). During PCR, the fluorophore and quencher are separated causing an increase in fluorescence. The separation occurs either by cleavage of the oligonucleotide (5,6,11,13), or by a change in secondary structure of the oligonucleotide probe when it anneals to target DNA, as occurs with molecular beacons (8). An alternative approach for nucleic acid detection and quantitation employs reporter and quencher moieties attached directly to the PCR primers instead of the hybridization probe (9,10). By excluding the probe from the reaction, this technique simplifies PCR kinetics and decreases the cost. However, production of oligonucleotides with dual-modifications is still relatively expensive.
The properties of many fluorophores may change through their interaction with nucleobases (14–20). A significant effect on fluorescence was demonstrated for guanosine, an efficient electron donor (16,18–20). Some oligonucleotides labeled with a single fluorophore at the 5′ end decrease fluorescence upon hybridization (21,22), which may provide a basis for a cost-effective DNA detection method (23–25). This decrease in fluorescence was attributed to the proximity of guanosine in the complementary strand upon hybridization. However, the amount of signal generated by this decrease is small in comparison to the signal generated by FRET-labeled probes, which might affect the sensitivity of the detection. In another report, we describe the effects that primary and secondary oligo_deoxynucleotide structure has on the fluorescent properties of an internally conjugated fluorophore (26). Up to a 10-fold change in fluorescence was demonstrated for oligonucleotides with various structures. This phenomenon was exploited here to devise a novel method for detection and quantitation of PCR products. The method is unique because a several-fold gain in fluorescent signal arises during PCR by using a fluorogenic primer labeled with only a single fluorescent dye.
We performed multiplex quantitative real-time PCR and single nucleotide polymorphism (SNP) end-point detection using these fluorogenic primers. Various primers, templates and dye combinations were tested to demonstrate the utility of this method. The results indicate that fluorogenic primers may be used to successfully quantify at least two genes in a sample with high sensitivity and broad dynamic range. We conclude that this new method is an efficient, reliable and cost-effective alternative to present methods for high-throughput, quantitative real-time PCR and allele-specific PCR.
MATERIALS AND METHODS
Primers
Unlabeled and fluorophore-labeled primers were supplied by Synthetic Genetics or by Invitrogen. Primer design was performed with the help of specialized software (http://pf.lifetech.com/primerf/pages/Test_PrimerF_Launch.cfm) (Invitrogen). The synthesis of FAM-labeled oligonucleotides was performed either through direct incorporation of the fluorescein–dT phosphoramidite or by a two-step process, which included the coupling of the amino modifier C6 T phosphoramidite (Glen Research) followed by post-synthetic modification using carboxyfluorescein NHS ester (mixture of 5- and 6-isomers; Molecular Probes). The two-step process was used to synthesize all JOE-labeled oligonucleotides, using 5-carboxy JOE NHS ester (Molecular Probes). Reverse-phase purification of the modified primers was performed by absorption on polystyrene resin.
Templates
All the nucleotides, enzymes and other reagents used are Invitrogen products. Total RNA was isolated from HeLa cells and human blood lymphocytes using the Trizol reagent according to the manufacturer’s protocol. The samples of RNA isolated from stimulated or aspirin-treated peripheral-blood T cells (PBTs) were a generous gift from Dr Vincenzo Casolaro (Johns Hopkins University, Baltimore, MD). First-strand cDNAs were synthesized from total RNA by RT using the Superscript II kit with oligo-d(T12–18) or random hexamer primers as described by the manufacturer. The cloned cDNA template of interleukin-4 (IL-4) was generated by RT–PCR of total RNA from stimulated lymphocytes. Standard ‘hot-start’ PCR was performed using gene-specific primers (primer sequences not shown) and Taq polymerase with Taq polymerase antibodies (Platinum Quantitative PCR Supermix-UDG). The PCRs amplified the appropriate cDNAs, nearly full-length, which were then cloned into bacteria using the TOPO™ T/A cloning kit. Finally, plasmid DNAs from clones were purified by alkaline lysis followed by centrifugation, then linearized and further purified by phenol extraction and ethanol precipitation before use as PCR templates. Copy number of plasmids was calculated using absorbance measurements at 260 nm excitation wavelength. The other cloned cDNA templates, c-myc oncogene, β-actin, glyceraldehyde-6-phosphate dehydrogenase (GAPDH) and 18S rRNA, were generated from HeLa cell RNA using similar methods.
Fluorogenic PCR
Each 50 µl reaction contained the relevant template cDNA, 80–200 nM of each gene-specific primer, 1× Platinum Quantitative PCR Supermix-UDG, including 200 µM each dATP, dGTP, dCTP and 400 µM dUTP, 1 U uracil DNA glycosylase, 3 mM MgCl2, 20 mM Tris–HCl, pH 8.4, 50 mM KCl, DNA polymerase antibodies, stabilizers and 1.5 U Taq polymerase and 1× ROX reference dye. For three-step cycling, reactions were incubated at 25°C for 2 min, 95°C for 2 min, then cycled using 95°C for 15 s, 55°C for 30 s and 72°C for 30 s, followed by incubation at 25°C for 2 min. Exceptions were made for amplicons >500 bp. These included an extension time of 120 s and a post-cycling hold of 4 min at 72°C. Two-step cycling consisted of 95°C for 15 s and 65°C for 30 s. Reactions were conducted in a 96-well spectrofluorometric thermal cycler (ABI PRISM 7700) or i-Cycler (BioRad). Fluorescence was monitored during every PCR cycle at the annealing step.
RESULTS
Design and properties of fluorogenic PCR primers
Fluorogenic primers were designed to increase their fluorescence intensity when incorporated into the double-stranded PCR product. This design is based on a previous study that demonstrated the effects of the primary and secondary structure of oligonucleotides on the emission properties of the conjugated fluorophores (26). Briefly, the design factors are the presence of either a C or G as the terminal 3′ nucleotide of the primer, the fluorophore being attached to the second or third base (T) from the 3′ end, the presence of one or more Gs within the 3 nt flanking the labeled nucleotide and, for hairpin primers, the existence of a 5′ tail that is complementary to the 3′ end of the primer. The 5′ tail forms a blunt-end hairpin at temperatures below its melting point. The stem of the hairpin primers have a ΔG ranging from –1.6 to –5.8 kcal/mol (Table 1). The above characteristics and other standard characteristics of the primers, such as length and Tm, were included in the primer design by the specially developed software. These design rules enabled the software to output numerous primer pairs that were located throughout the target sequence. For example, the software output 94 usable primer pairs for the GAPDH sequence (1310 bp).
Table 1. Characteristics of primer sets used in PCR.

The dye is conjugated to the bold ‘T’; underlined bases denote non-specific sequence built onto the primer to allow the hairpin conformation. The ΔG of the hairpin was calculated using Oligo 6.0 software (Molecular Biology Insights, Inc). Two versions of the sequence of the forward primer for RDS gene allele-specific PCR (primer pair 15) are denoted by bases in parentheses.
A schematic of PCR with these fluorogenic primers is presented in Figure 1. The primer maintains a hairpin structure with a blunt end that has a relatively low fluorescence at temperatures below the Tm. When the hairpin melts, the fluorescence increases several-fold, and reaches its maximum when the 3′ end of the primer is extended into the double-stranded PCR product. We demonstrated that a hairpin primer generates a stronger signal during real-time PCR than a linear primer because it is more quenched when not extended (Fig. 2). Two labeled reverse primers were prepared, one hairpin and one linear, with identical sequence-specific portions (primer sets 1 and 2, Table 1). They were used with the same forward unlabeled linear primer to amplify IL-4 cDNA. The fluorescence of the PCR mixture was measured during a 25°C pre-reaction step, a 94°C denaturation step, the annealing step (55°C) of each PCR cycle and at 25°C after completion of PCR using an ABI PRISM 7700 (Fig. 2). The fluorescence of the linear primer at the 25°C pre-incubation step is higher than at the 94°C denaturation step because of the known effect of temperature and pH on fluorescence (27). During the PCR with the linear labeled primer, the fluorescence rises to a plateau with a gain of 2000 relative fluorescence units. The hairpin primer at 25°C is quenched 3-fold more than the linear primer, so the resulting gain in fluorescence at the PCR end-point is much greater for the hairpin primer than for the linear one, and equal to 5700 fluorescence units.
Figure 1.

Signal generation by fluorogenic primers. The oligonucleotide in blunt-end hairpin conformation emits the lowest fluorescent signal. The signal increases when the primer is linear, and reaches its maximum when the primer is incorporated into the double-stranded DNA.
Figure 2.

Fluorescence readings during real-time PCR with either a linear or a hairpin fluorogenic primer. Fluorescence was measured at 25°C before the PCR, during a preheating step at 94°C, at 55°C during each cycle of PCR, and finally at 25°C after the cycling. (A) PCR using a FAM-labeled linear reverse primer (primer set 1, Table 1). (B) PCR using a FAM-labeled hairpin reverse primer (primer set 2, Table 1). PCRs were performed on an ABI 7700 with 106 copies of cloned IL-4 cDNA using similar conditions and with the same unlabeled forward primer. (C) Plot of temperature versus detector readings.
To investigate the effect of the hairpin structure on PCR efficiency and specificity, we compared the threshold cycle (CT) and the slopes of the amplification curves for the real-time PCRs performed with either a linear or hairpin primer. No significant differences between linear and hairpin primers were observed (Figs 2 and 3). Furthermore, we demonstrated that the use of blunt-ended hairpin primers improves the specificity of PCR by reducing the formation of primer-dimer artifacts in the absence of target. We compared primer-dimer formation in separate reactions incorporating a linear labeled primer with an unlabeled counterpart primer that was either linear or hairpin (Fig. 3; primer sets 3 and 4, Table 1). Reactions included 10-fold serial dilutions of genomic DNA and no-DNA controls. When both primers were linear, primer-dimers were amplified at the 45th cycle of PCR in the control with no added template. Conversely, a PCR containing a hairpin primer did not result in primer-dimer amplification even after 55 cycles (Fig. 3).
Figure 3.

Effect of hairpin structure on PCR specificity and efficiency. The RDS gene was amplified using either a linear (dotted line), or a hairpin (solid line) unlabeled reverse primer with the same fluorogenic linear forward primer (primer sets 3 and 4, Table 1). PCRs for both primer sets were performed using 100, 2, 0.08 and 0 ng of genomic DNA.
Quantitative real-time PCR
Samples with templates comprising a 10-fold serial dilution of cloned c-myc cDNA ranging from 10 to 107 copies are discriminated by two-step real-time PCR (Fig. 4; primer set 3, Table 1). A linear relationship (r2 = 0.999) exists between the CT and starting copy numbers. Comparable results were obtained using 10-fold and 3-fold serial dilutions of IL-4 cloned cDNA (primer set 2, Table 1) with correlation coefficients of 0.992 and 0.999 respectively.
Figure 4.

Sensitivity, precision and dynamic range of fluorogenic real-time PCR. Serial dilutions (10-fold) of c-myc cDNA were amplified and detected using a FAM-labeled fluorogenic primer in two-step PCR on an ABI 7700 as described in Materials and Methods (primer set 5, Table 1). (A) Amplification plot. (B) Initial cDNA concentrations versus CT, standard deviations are shown as error bars (12 replicates per dilution).
Other fluorogenic PCRs were performed for amplicons of various sizes. FAM-labeled primers were designed for amplicons of c-myc with sizes 66, 562 and 1107 bp and GAPDH with sizes 74, 570 and 956 bp (Table 1). The fluorogenic PCRs (three-step cycling) were performed using cDNA from first-strand synthesis reactions (20 µl) using HeLa total RNA (2.5 ng) as a template. PCRs were performed with 2, 0.2, 0.02 or 0 µl of these first-strand reactions. For all primer sets, cDNA dilutions were determined with evenly spaced CT values (data not shown), which confirms that PCR efficiency is comparable for the amplicons of various sizes.
Multiplex quantitative PCR
Quantitative, real-time, multiplex fluorogenic PCR with two sets of differently labeled gene-specific primers is useful because one primer set may be used to detect the amount of a gene that is variable and another to detect a gene that is relatively constant and used as a reference. We performed multiplex PCR using FAM-labeled primers for either c-myc or IL-4 and JOE-labeled primers for the reference gene. We discriminated between 3-fold serial dilutions of cloned IL-4 cDNA (primer set 2, Table 1) ranging from 22 to 3 × 105 copies, with each dilution containing 106 copies of cloned GAPDH cDNA (primer set 12, Table 1; Fig. 5A). The correlation coefficient of CT versus IL-4 copy number was 0.999 for the best two out of three replicates. We then performed a similar set of PCRs using a cloned cDNA for c-myc (primer set 5, Table 1) as the variable gene and GAPDH as the constant gene. The results in Figure 5B indicate that discrimination between dilutions of c-myc and IL-4 are similar. cDNAs other than GAPDH may be used as the reference gene. Serial dilutions (3-fold) of target concentration (IL-4) were discriminated by fluorogenic PCR when using either 106 copies of cloned cDNA β-actin (primer set 13, Table 1) or 18S rRNA (primer set 14, Table 1) as the reference gene. Standard curve plots yield r2 values of 0.995 and 0.998 for β-actin and 18S rRNA respectively.
Figure 5.

Multiplex fluorogenic PCR on ABI PRISM 7700. Amplification plots for PCR comprising a 3-fold serial dilution of cloned cDNA: (A) IL-4 cDNA (gray) from 303 030 to 22 copies; (B) c-myc cDNA (gray) from 106 to 22 copies; each dilution had 106 copies of cloned GAPDH cDNA (black). FAM-labeled fluorogenic primers were used to detect IL-4 (primer set 2, Table 1) and c-myc (primer set 5, Table 1) and a JOE-labeled primer was used to detect GAPDH (primer set 12, Table 1). Corresponding plots of initial cDNA concentrations (two duplicate reactions per concentration) versus CT are shown below the amplification plots.
Furthermore, first-strand cDNA from HeLa cell total RNA was used as a source of the reference gene in place of specific cloned cDNA. This was done to determine whether the PCRs would amplify their specific targets within a mixture of non-specific cDNAs. For these experiments, the variable template was cloned IL-4 cDNA (3-fold dilutions) and the constant template was a fixed amount of first-strand cDNA from the reverse transcription of HeLa total RNA (Fig. 6). Standard curves yield r2 values of 0.997 for β-actin (primer set 13, Table 1), 0.996 for GAPDH (primer set 12, Table 1) and 0.999 for 18S (primer set 14, Table 1). The fluorogenic PCR primers amplified only their appropriate target. Analysis of the PCR products by agarose gel electrophoresis revealed either insignificant or no non-specific PCR products or primer-dimers.
Figure 6.
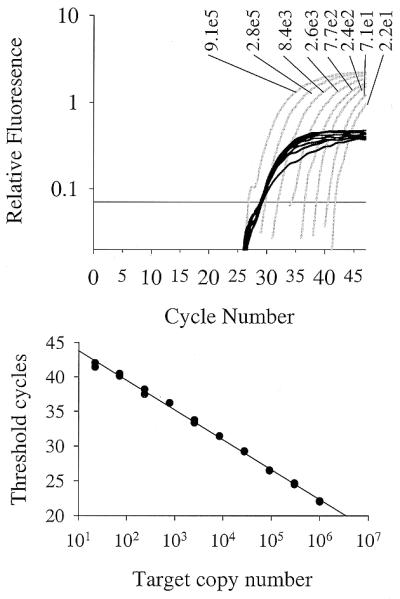
Multiplex real-time fluorogenic PCRs with a population of first-strand cDNAs from HeLa cells. Three-step PCRs were performed with templates comprising a serial dilution of cloned IL-4 cDNA from 91 000 to 22 copies in the presence of a constant amount of cDNA generated from total HeLa RNA. The variable IL-4 target was detected using a FAM-labeled hairpin primer (primer set 2, Table 1; gray) and the constant GAPDH target was detected with a JOE-labeled hairpin primer (primer set 12, Table 1; black).
All the above results were obtained using the ABI PRISM 7700 system. We also performed fluorogenic multiplex PCR of variable amounts of cloned IL-4 mixed with a constant amount of cloned GAPDH on the BioRad i-Cycler IQ system. The BioRad i-Cycler IQ system detected the cDNAs with similar sensitivity and dynamic range as the ABI PRISM 7700 (data not shown).
We demonstrated that the results of quantitative PCR using fluorogenic primers might be analyzed by the comparative CT method (User Bulletin 2, ABI PRISM 7700 Sequence Detection System, P/N 4303859). This method of analysis does not require plotting of a standard curve of CT versus starting copy number. Instead, the amount of target is calculated based on the difference between the CT of the target and an endogenous reference gene. We obtained a subset of RNA samples that were used in a published study where the comparative CT method was used in conjunction with the 5′ nuclease assay to show that IL-4 expression is reduced in lymphocytes treated with aspirin (28). We analyzed the amount of IL-4 in these same samples using fluorogenic, multiplex PCR. Total RNA (1 ng) from stimulated PBTs that were treated, or untreated, with aspirin were reversed transcribed into cDNA and 2 µl samples of these reactions were used for real-time PCR (three replicates). The three-step protocol was performed using a FAM-labeled primer for IL-4 and a JOE-labeled primer for GAPDH (primer sets 2 and 12, Table 1). The CT values for the target gene (IL-4) and reference gene (GAPDH) were obtained using the ABI 7700. We determined that the level of IL-4 mRNA in an aspirin-treated PBT sample was 33% lower that that found in the untreated PBT sample, as calculated by the comparative CT method. This result is comparable with the 43% decrease in IL-4 expression after aspirin treatment that was reported in the previous study using TaqMan™ probes (28).
End-point detection of allele-specific PCR
To demonstrate end-point detection capability, allele-specific PCRs were performed using human genomic DNA as a template. These PCRs detected the presence of a C/T polymorphism at position 558 of the human retinal degeneration (RDS) gene (29). Two unlabeled, linear allele-specific forward primers with either a dC or a T at the 3′ end were designed to detect either the dC or T polymorphism (primer set 15, Table 1). Discrimination of the alleles is based on the ability of DNA polymerase to extend 3′ mismatches much less efficiently than correct matches (30). A common fluorogenic hairpin reverse primer was used to generate the signal. Two allele-specific PCRs were performed on each of two genomic DNA samples bearing different SNPs (Fig. 7). Following PCR the fluorescence was determined directly in the PCR tubes using either a fluorescence plate reader (Polarion, TECAN) or a UV-transilluminator. The results show that both alleles can be identified correctly and there is no signal increase in the absence of a target.
Figure 7.
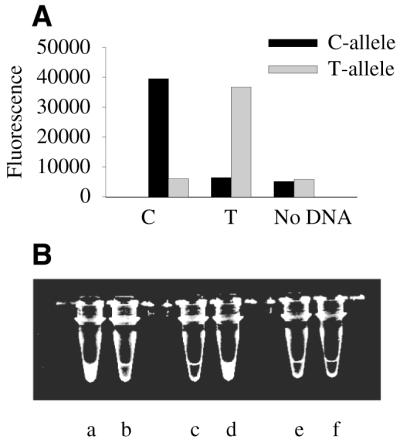
End-point detection of the fluorescent PCR product. PCR products specific for C558/T558 polymorphism in the RDS gene were generated using two different linear forward primers specific for the C allele or T allele and a common hairpin reverse primer labeled with FAM (primer set 15; Table 1). Three-step PCRs were performed as described in Materials and Methods through 40 cycles. (A) Fluorescence was determined on a plate reader (Polarion, TECAN) with 490 nm excitation, 525 nm emission, 20 nm bandwidth. (B) A photograph of the tubes was taken on a UV-transilluminator using a Kodak imaging system equipped with a green filter (520 nm, 40 nm bandwidth).
DISCUSSION
Oligonucleotides labeled with a single reporter but no specific quencher can be used to efficiently detect nucleic acids by PCR in real time, or at end point, without opening the reaction vessel. The fluorogenic PCR primers are chemically synthesized oligodeoxynucleotides (22–29 nucleobases) with a fluorophore attached to the C5 position of thymidine that increases their fluorescence when incorporated into a double-stranded PCR product. This phenomenon results from having the fluorophore close to the 3′ end of an oligonucleotide with G or C at the 3′ end, the existence of a G within a few bases of the label, and the ability of the oligonucleotide to form a blunt-end hairpin at temperatures close to the annealing temperature of the primer. The fact that all primers are labeled at a thymidine may be significant and we are currently determining whether fluorogenic properties result from labeling on other bases. We believe that the mechanism for this change in fluorescence intensity is charge separation between the nucleobases, specifically guanosines, and the fluorophore (16,18–20), which decreases the fluorescence of unincorporated primers. The high quenching efficiency of the dG–dC and dC–dG base pair at the blunt end of the hairpin provides low background fluorescence. The formation of the double-stranded PCR product prevents charge separation and an increase of signal is observed (26). We report a 3-fold increase in signal at 55°C during PCR, and an 8-fold increase in signal between the initial and end-points of PCR at room temperature and show that the hairpin conformation of the primers (ΔG from –1.6 to –5.8 kcal/mol) does not adversely affect the efficiency of the PCR.
Fluorogenic PCR may be routinely used to quantify 100 or less copies of target in a background of non-specific templates over a broad dynamic range of 107–10 copies. The fluorogenic primer method discriminated between 3-fold dilutions of target in multiplex PCR. Here, we used FAM- and JOE-labeled primers for multiplex PCR, but other dyes, including TET, HEX, TAMRA, ROX and Alexa-594 may be used (26). The method may be highly specific as demonstrated by the lack of signal when no template is added to the PCR.
The relative quantification of cDNA using fluorogenic primers is comparable in sensitivity and dynamic range to other published methods of quantification, such as the 5′ nuclease assay (1) or SYBR Green (7). However, direct comparisons between methods of quantitation are difficult because these methods are functionally different and may be affected differently by various factors. All probe-based technologies have inherent complexities related to the kinetics of the hybridization and amplification. The fluorogenic primer method is not susceptible to this problem. The detection methods involving DNA-binding dyes, such as SYBR Green I, are limited in their ability to detect multiple targets in a single reaction. DNA-binding dyes may also increase the stability of the double-stranded structures (31) and, therefore, may facilitate the annealing of primers to non-specific targets. Fluorogenic primers in the hairpin conformation can actually enhance the specificity of the PCR by helping to prevent primer-dimers and mispriming. The use of blunt-ended hairpin PCR-primers has been previously shown to reduce primer-dimers and mispriming (32,33). This enhanced specificity of the hairpin primers might be very useful for mutation detection by allele-specific PCR.
The detection of potential mispriming products and primer-dimers that may amplify during PCR is a problem of the fluorogenic primer method, as in other PCR methods. In order to reduce the appearance of these artifacts, a ‘hot-start’ DNA polymerase and a relatively low magnesium concentration may be used. Performing multiplex PCR may also aid in reducing artifacts. The amplification of the reference gene may out-compete potential primer-dimer amplification for the limited amount of DNA polymerase in PCR. Despite efforts to optimize PCR and primer design, we find that some primer pairs still form artifacts. Probe-based methods can discriminate between the correct amplicon and PCR artifacts. However, PCR artifacts may affect even probe-based methods, because primer-dimer formation suppresses the formation of the specific amplicon causing false negative results or inaccurate quantitation. When artifacts do occur using the fluorogenic primer method, the problem may be easily overcome, because other functional primer pairs can be easily generated due to the flexibility of the design rules, availability of the software and low production cost.
The large gain in fluorescence intensity at room temperature after the PCR permits efficient detection of target at end-point using a fluorescence plate reader or even a UV-transilluminator. The gain in signal during PCR occurs with fluorogenic primers labeled with various fluorophores (26). This enables simultaneous detection of multiple targets in a wide variety of instruments with various detection capabilities.
PCR using fluorogenic primers may have great value as a tool in DNA detection, including quantitative real-time PCR and SNP detection, because of several important features. The synthesis of mono-labeled oligonucleotides is less expensive and the purification requirement is less rigorous compared to dual-labeled probes and primers. Fluorogenic primers may more easily detect targets with high frequency of mutations, such as HIV, or targets with alternate splice forms, which are problematic using hybridization probes. The incorporation of fluorescence into the PCR product also allows the separation of nucleic acids by size using electrophoretic techniques. Finally, fluorogenic primers allow a ‘universal format’ of detection. The same universal, labeled, primer can incorporate into different amplicons through the use of unlabeled primer pairs, where one unlabeled primer has an adaptor-tail. The universal format was successfully used with dual-labeled primers (34,35). In addition to the applications mentioned, the ability of fluorogenic oligonucleotides to generate a strong signal in response to the changes in their primary and secondary structure may be useful for studying enzymatic reactions as well as other interactions between proteins and nucleic acids.
Acknowledgments
ACKNOWLEDGEMENTS
We thank the Invitrogen Inc. staff members Joe Hayes, Gulilat Gebeyehu, Rick Pires and Malek Masoud for expertise in producing fluorogenic primers, Joe Solus for expertise in SNP detection and Dave Saile for writing the primer design software.
REFERENCES
- 1.Heid C.A., Stevens,J., Livak,K.J. and Williams,P.M. (1996) Real time quantitative PCR. Genome Res., 6, 986–994. [DOI] [PubMed] [Google Scholar]
- 2.Freeman W.M., Walker,S.J. and Vrana,K.E. (1999) Quantitative RT–PCR: pitfalls and potential. Biotechniques, 26, 112–125. [DOI] [PubMed] [Google Scholar]
- 3.Bustin S.A. (2000) Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol., 25, 169–193. [DOI] [PubMed] [Google Scholar]
- 4.Higuchi R., Fockler,C., Dollinger,G. and Watson,R. (1993) Kinetic PCR analysis: real-time monitoring of DNA amplification reactions. Biotechnology, 11, 1026–1030. [DOI] [PubMed] [Google Scholar]
- 5.Lee L.G., Connell,C.R. and Bloch,W. (1993) Allelic discrimination by nick-translation PCR with fluorogenic probes. Nucleic Acids Res., 21, 3761–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Livak K.J., Flood,S.J.A., Marmaro,J., Giusti,W. and Deetz,K. (1995) Oligonucleotides with fluorescent dyes at opposite ends provide a quenched probe system useful for detecting PCR product and nucleic acid hybridization. PCR Methods Appl., 4, 357–362. [DOI] [PubMed] [Google Scholar]
- 7.Wittwer C.T., Herrmann,M.G., Moss,A.A. and Rasmussen,R.P. (1997) Continuous fluorescence monitoring of rapid cycle DNA amplification. Biotechniques, 22, 130–138. [DOI] [PubMed] [Google Scholar]
- 8.Tyagi S. and Kramer,F.R. (1996) Molecular beacons: probes that fluoresce upon hybridization. Nat. Biotechnol., 14, 303–308. [DOI] [PubMed] [Google Scholar]
- 9.Nazarenko I.A., Bhatnagar,S.K. and Hohman,R.J. (1997) A closed tube format for amplification and detection of DNA based on energy transfer. Nucleic Acids Res., 25, 2516–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thelwell N., Millington,S., Solinas,A., Booth,J. and Brown,T. (2000) Mode of action and application of Scorpion primers to mutation detection. Nucleic Acids Res., 28, 3752–3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Todd A.V., Fuery,C.J., Impey,H.L., Applegate,T.L. and Haughton,M.A. (2000) DzyNA–PCR: use of DNAzymes to detect and quantify nucleic acid sequences in a real-time fluorescent format. Clin. Chem., 46, 625–630. [PubMed] [Google Scholar]
- 12.Clegg R.M. (1992) Fluorescence resonance energy transfer and nucleic acids. Methods Enzymol., 211, 353–388. [DOI] [PubMed] [Google Scholar]
- 13.Lyamichev V., Brow,M.A., Varvel,V.E. and Dahlberg,J.E. (1999) Comparison of the 5′ nuclease activities of taq DNA polymerase and its isolated nuclease domain. Proc. Natl Acad. Sci. USA, 96, 6143–6148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lianos P. and Georghiou,S. (1979) Complex formation between pyrene and the nucleotides GMP, CMP, TMP and AMP. Photochem. Photobiol., 29, 13–21. [Google Scholar]
- 15.Shafirovich V.Y., Courtney,S.H., Ya,N. and Geacintov,N.E. (1995) Proton-coupled photoinduced electron transfer, deuterium isotope effects and fluorescence quenching in noncovalent benzo[α]pyrenetetraol-nucleoside complexes in aqueous solutions. J. Am. Chem. Soc., 117, 4920–4929. [Google Scholar]
- 16.Seidel C.A.M., Schulz,A. and Sauer,M.H.M. (1996) Nucleobase-specific quenching of fluorescent dyes. 1. Nucleobase one-electron redox potentials and their correlation with static and dynamic quenching efficiencies. J. Phys. Chem., 100, 5541–5553. [Google Scholar]
- 17.Widengren J., Dapprich,J. and Rigler,R. (1997) Fast interactions between Rh6G and dGTP in water studied by fluorescence correlation spectroscopy. Chem. Phys., 216, 417–426. [Google Scholar]
- 18.Walter N.G. and Burke,J.M. (1997) Real-time monitoring of hairpin ribozyme kinetics through base-specific quenching of fluorescein-labeled substrates. RNA, 3, 392–404. [PMC free article] [PubMed] [Google Scholar]
- 19.Sauer M., Drexhage,K.H., Lieberwirth,U., Muller,R., Nord,S. and Zander,C. (1998) Dynamics of the electron transfer reaction between an oxazine dye and DNA oligonucleotides monitored on the single-molecule level. Chem. Phys. Lett., 284, 153–163.
- 20.Lewis F.D., Letzinger,R.L. and Wasielewski,M.R. (2001) Dynamics of photoinduced charge transfer and hole transport in synthetic DNA hairpins. Acc. Chem. Res., 34, 159–170. [DOI] [PubMed] [Google Scholar]
- 21.Cardullo R.A., Agrawal,S., Flores,C., Zamecnik,P.C. and Wolf,D.E. (1988) Detection of nucleic acid hybridization by nonradiative fluorescence resonance energy transfer. Proc. Natl Acad. Sci. USA, 85, 8790–8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee S.P., Porter,D., Chirikjian,J.G., Knutson,J.R. and Han,M.K. (1994) A fluorometric assay for DNA cleavage reactions characterized with BamHI restriction endonuclease. Anal. Biochem., 220, 377–383. [DOI] [PubMed] [Google Scholar]
- 23.Knemeyer J.P., Marme,N. and Sauer,M. (2000) Probes for detection of specific DNA sequences at the single-molecule level. Anal. Chem., 72, 3717–3724. [DOI] [PubMed] [Google Scholar]
- 24.Crockett A.O. and Wittwer,C.T. (2001) Fluorescein-labeled oligonucleotides for real-time PCR: using the inherent quenching of deoxyguanosine nucleotides. Anal. Biochem., 290, 89–97. [DOI] [PubMed] [Google Scholar]
- 25.Kurata S., Kanagawa,T., Yamada,K., Torimura,M., Yokomaku,T., Kamagata,Y. and Kurane,R. (2001) Fluorescent quenching-based quantitative detection of specific DNA/RNA using a BODIPY® FL-labeled probe or primer. Nucleic Acids Res., 29, e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nazarenko I., Pires,R., Lowe,B., Obaidy,M. and Rashtchian,A. (2002) Effect of primary and secondary structure of oligodeoxyribonucleotides on the fluorescent properties of conjugated dyes. Nucleic Acids Res., 30, 2089–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lakowicz J.R. (1999) Principles of Fluorescence Spectroscopy, 2nd Edn. Kluver Academic/Plenum Publishers, New York, pp. 185–210.
- 28.Cianferoni A., Schroeder,J.T., Kim,J., Schmidt,J.W., Lichtenstein,L.M., Georas,S.N. and Casolaro,V. (2001) Selective inhibition of interleukin-4 gene expression in human T cells by aspirin. Blood, 97, 1742–1749. [DOI] [PubMed] [Google Scholar]
- 29.Farrar G.J., Kenna,P., Jordan,S.A., Kumar-Singh,R. and Humphries,P. (1991) A sequence polymorphism in the human peripherin/RDS gene. Nucleic Acids Res., 19, 6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petruska J., Goodman,M.F., Boosalis,M.S., Sowers,L.S., Cheong,C. and Tinoco,I.,Jr (1988) Comparison between DNA melting thermodynamics and DNA polymerase fidelity. Proc. Natl Acad. Sci. USA, 85, 6252–6256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wiederholt K., Rajur,S.B. and McLaughlin,L.W. (1997) Oligonucleotides tethering Hoechst 33258 derivatives: effect of the conjugation site on duplex stabilization and fluorescence properties. Bioconjugate Chem., 8, 119–126. [DOI] [PubMed] [Google Scholar]
- 32.Ailenberg M. and Silverman,M. (2000) Controlled hot start and improved specificity in carrying out PCR utilizing touch-up and loop incorporated primers (TULIPS). Biotechniques, 29, 1018–1024. [DOI] [PubMed] [Google Scholar]
- 33.Kaboev O.K., Luchkina,L.A., Tret’iakov,A.N. and Bahrmand,A.R. (2000) PCR hot start using primers with the structure of molecular beacons (hairpin-like structure). Nucleic Acids Res., 28, e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nuovo G.J., Hohman,R.J., Nardone,G.A. and Nazarenko,I. (1999) J. Histochem. Cytochem., 47, 273–279. [DOI] [PubMed] [Google Scholar]
- 35.Myakishev M.V., Khripin,Y., Hu,S. and Hamer,D.H. (2001) High-throughput SNP genotyping by allele-specific PCR with universal energy-transfer-labeled primers. Genome Res., 11, 163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]