

Summary
Familial hypercholesterolemia (FH) is a genetic disorder that affects 1 in 300 people, leading to high cholesterol levels and significantly increased cardiovascular risk. The limitations of existing FH treatments underscore the need for innovative therapeutics, and gene therapy offers a promising alternative to address FH more effectively. In this review, we survey approved gene therapy drugs first and then delve into the landscape of gene addition, gene inactivation, and gene editing therapies for hypercholesterolemia, highlighting both approved interventions and those in various stages of development. We also discussed recent advancements in gene editing tools that are essential for their application in gene therapy. Safety considerations inherent to gene therapy are also discussed, emphasizing the importance of mitigating potential risks associated with such treatments. Overall, this review highlights the progress and prospects of gene therapies for FH treatments, underscoring their potential to revolutionize the management of this prevalent and challenging condition.
Subject areas: Clinical genetics, Endocrinology, Human metabolism
Graphical abstract
Clinical genetics; Endocrinology; Human metabolism
Introduction
Familial hypercholesterolemia (FH) is an autosomal dominant genetic disorder,1 with prevalence rates as high as ∼ 1:300 for heterozygous FH (HeFH) and approximately ∼1:300,000 for homozygous FH (HoFH).2 FH may be caused by mutations in several genes that control low-density lipoprotein cholesterol (LDL-C) removal in the liver, including the low-density lipoprotein cholesterol receptor (LDLR), apolipoprotein B (APOB), proprotein convertase subtilisin/kexin type 9 (PCSK9), and LDL receptor adaptor protein 1 (LDLRAP1). The vast majority of FH cases (80–90%) involve mutations in LDLR, which binds and endocytoses low-density lipoproteins (LDLs) in the blood.3 Individuals suffering from FH exhibit substantially elevated LDL-C blood levels from a young age, predisposing them to chronic hypercholesterolemia.1 As elevated cholesterol levels are directly associated with an increased risk of atherosclerosis and atherosclerotic cardiovascular diseases (ASCVDs),4 individuals with FH generally experience an elevated incidence and mortality rates of ASCVDs compared to the general population.3
Diagnosis of FH mainly relies on serum LDL-C levels, family medical history, clinical manifestations such as skin/tendon xanthomas, corneal arcus, and so forth, and genetic testing.5 Among these methods, genetic testing represents the most accurate diagnostic approach, and can also offer insights into treatment strategies and prognosis for patients.6 Nevertheless, the accessibility of genetic testing is low due to its high-cost.7 The primary objective of FH treatment is to reduce the LDL-C blood levels, thereby mitigating the risk or delaying the onset and progression of ASCVDs.8 Complemented by non-pharmacological interventions such as lifestyle adjustments9,10,11 and lipoprotein apheresis,12,13 pharmacological interventions stand as the current cornerstone of FH treatment.14,15
Among pharmacological interventions, statins, PCSK9 inhibitors, and ezetimibe are widely employed as first-line treatments. Statins, renowned for their extensive use worldwide, exert their effects by inhibiting 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase, which catalyzes the rate-limiting step in cholesterol biosynthesis (Figure 1).16 However, although statins can typically reduce blood LDL-C by 20–50%, concerns persist regarding their adverse effects, which happen at a frequency ranging from 0.1% to 10%.17,18 Among the predominant musculoskeletal issues caused by Statins, myalgia is the most common, myositis is relatively rare, and rhabdomyolysis, though very rare, is a serious concern as it can lead to kidney damage and other complications.19 Furthermore, certain statins carry a potential risk of inducing diabetes, limiting the tolerability of high-dose therapy.20 In clinical practice, stains are often administered concurrently with Ezetimibe or PCSK9 inhibitors.21 Ezetimibe impedes intestinal cholesterol absorption through its inhibition of Niemann-Pick C1-Like 1 (NPC1L1), thereby reducing the cholesterol levels (Figure 1).22 Nevertheless, it also poses several adverse effects, including upper respiratory tract infections (URTI), arthralgia, and extremity pain.23 On the other hand, PCSK9 reduces the number of LDLRs on the surface of hepatocytes by enhancing their degradation.24 Therefore, PCSK9 inhibitors can promote the recycling of LDLR and bolster the LDLR-mediated clearance of LDL-C from the bloodstream (Figure 1).25 However, though only on the market for a relatively short period, PCSK9 inhibitors have also been associated with allergic reactions,26 neurocognitive effects,27,28 and liver enzyme abnormalities.29
Figure 1.

The working mechanism of first-line drug statins, PCSK9 inhibitors, and ezetimibe
Statins inhibit HMG-CoA reductase, thereby reducing cholesterol biosynthesis and lowering plasma cholesterol levels. Ezetimibe inhibits the absorption of dietary cholesterol by NPC1L1 in the intestine. PCSK9 inhibitors improve the stability of LDLR, thereby enhancing the LDLR-mediated clearance of cholesterol from blood.
Besides the above mainstay FH medications, a few other treatments have been approved in recent years to help patients with severe FH restore normal LDL-C levels, including antisense oligonucleotide (ASO) therapy. An ASO inhibitor targeting APOB has been shown to reduce LDL-C levels by an additional 24.7% when combined with high-dose statin therapy.30 Lomitapide, an inhibitor of microsomal triglyceride transport protein (MTP), indirectly inhibits the assembly and secretion of apolipoprotein B (apoB) with lipoproteins in the liver and intestine, resulting in an additional 50% reduction in LDL-C levels when used alongside statins. However, it can also cause significant gastrointestinal side effects.31 Lipoprotein Apheresis can reduce LDL levels by up to 65%. However, it is a costly procedure and financially unfeasible for most patients.13,32 Currently, liver transplantation is the only method capable of rapidly normalizing cholesterol levels in patients with HoFH. Nevertheless, it remains an uncommon treatment due to immune rejection risks and the severe shortage of organ donors.33
In summary, the current treatment of FH is challenged by various factors, including low diagnosis rates, high drug costs, severe side effects, intricate drug interactions, and considerable variability in individual drug response. To this end, the burgeoning field of gene therapy offers a promising therapeutic alternative for patients with FH which may revolutionize the current FH treatment landscape. In this review, we provide an overview of the present landscape of gene therapies for FH, with a specific emphasis on several gene editing-based therapies currently under development.
The landscape of gene therapy
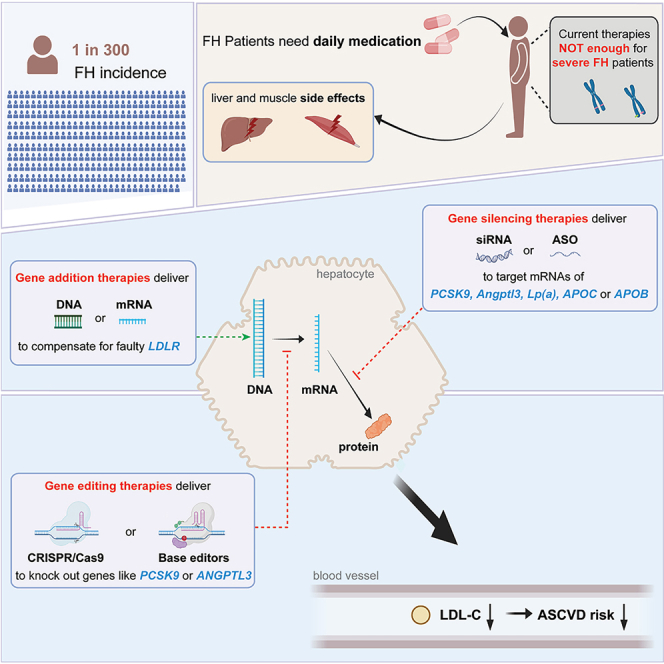
With the ongoing development of biotechnology, several innovative treatment options have emerged in the past decades. Among these, gene therapy stands out as one of the most promising approaches for addressing human hereditary diseases. In general, gene therapy treats genetic disorders by introducing genetic engineering materials into human cells, with the primary goal of modulating therapeutic gene expression at levels sufficient to alleviate or even cure the symptoms of the disorders.34 From a narrow perspective, two strategies are mainly employed in gene therapy, gene addition, and gene editing. While gene addition introduces a functional copy of a gene into cells to compensate for a faulty or missing gene, gene editing instills precise the modification of the cellular genome to correct or modify genetic defects associated with diseases and holds the potential of a “one-time editing, benefit forever” prospect.35,36
Approved gene addition and gene editing therapies
The past decade has witnessed an explosion in gene therapy research and application.37 Gene therapy can be given in two ways, in vivo and ex vivo. In vivo gene therapies deliver therapeutic genes or gene-modifying components directly to the target organ or tissue via delivery vectors such as adeno-associated virus vectors (AAVs) or lipid nanoparticles (LNPs).38,39,40,41 In contrast, ex vivo gene therapy involves the isolation of recipient cells from the patient, followed by the introduction of therapeutic genes or gene-modifying components into these cells. Subsequently, the modified cells are infused back into the patient.42,43,44,45 Currently, over 500 gene therapy clinical studies have been initiated globally, spanning cancers, hereditary genetic disorders, cardiovascular diseases, and other conditions. Among all the conditions, cancer and single-gene rare diseases stand out as the most extensively researched areas.46
As of 2023, dozens of gene therapy drugs have been approved for marketing worldwide, including 4 ex vivo gene therapies and 11 in vivo gene therapies (Table 1). The world’s first gene therapy to receive regulatory approval was Gendicine. Approved by the Chinese State Food and Drug Administration (SFDA) in 2003, Gendicine treats head and neck squamous cell carcinoma (HNSCC) by intratumorally delivering a functional copy of the tumor suppressor gene p53 on a recombinant adenovirus vector (rAd-p53).47 However, Gendicine has not been approved for use in the United States or other countries. The first gene therapy approved by the European Medicines Agency (EMA) is Glybera, which received conditional marketing approval in 2012. Glybera leverages an AAV vector to deliver a functional copy of the lipoprotein lipase (LPL) gene for the treatment of familial lipoprotein lipase deficiency through intravenous infusion.48 Another significant approval was granted to Strimvelis by the EMA in 2016, which treats rare genetic disorder called severe combined immunodeficiency due to adenosine deaminase deficiency (ADA-SCID).49,50 Using a gammaretrovirus vector to transfer a functional copy of the adenosine deaminase (ADA) gene into hematopoietic stem cells (HSCs) isolated from the patients, Strimvelis represents a notable example of ex vivo gene therapy.50 The first gene therapy approval in the United States was given to Luxturna by the Food and Drug Administration (FDA) in 2017, which delivers a functional RPE65 gene vectored on AAV2 through subretinal injection to treat Leber Congenital Amaurosis (LCA).51 Following these approvals, 10 other gene therapies that use either AAV, adenovirus (ADV), or retroviral vectors to deliver functional copies of therapeutic genes for the treatment of a range of genetic disorders were approved in 2019–2023 (Table 1). Most of these approvals are one-time in vivo gene therapies administered intravenously/intrathecally or ex vivo gene therapies given to autologous HSCs. However, VYJUVEK for the treatment of Dystrophic epidermolysis bullosa (DEB) is an exception. Administered topically, VYJUVEK represents the first redosable topical gene therapy in the world.52,53
Table 1.
Gene therapies approved by the SFDA, FDA or EMA
| Product | Indications | Approval | Delivery route | Delivery vector | Mechanism | References |
|---|---|---|---|---|---|---|
| Gendicine | head and neck cancer | 2003 | intratumoral injection | recombinant Adenovirus vector | deliver functional copies of the p53 gene | Peng et al.47 |
| Glybera | familial lipoprotein lipase deficiency | 2012 | intravenous infusion | AAV vector | deliver functional copies of the LPL gene | Gaudet et al.48 |
| Strimvelis | ADA-SCIDa | 2016 | ex vivo/HSCe | gamma-retroviral vector | deliver functional copies of the ADA gene | Cicalese et al.49 |
| Luxturna | LCAb or retinitis pigmentosa | 2017 | subretinal injection | AAV2 | deliver functional copies of the RPE65 gene | Russell et al.51 |
| Zolgensma | spinal muscular atrophy (SMA) | 2019 | intravenous infusion | AAV9 | deliver functional copies of the SMN1 gene | Mendell et al.54 |
| Zynteglo | transfusion-dependent β-thalassemia | 2019 | ex vivo/HSCe | gamma-retroviral vector | deliver functional copies of the β-globin gene | Locatelli et al.55 |
| ROCTAVIAN | hemophilia A | 2019 | intravenous infusion | AAV5 | deliver functional copies of FVIII gene | Ozelo et al.56 |
| Libmeldy | metachromatic leukodystrophy (MLD) | 2020 | intrathecal injection | lentiviral vector | deliver functional copies of the arylsulfatase A gene | Sessa et al.57 |
| ADSTILADRIN | high-grade, BCG-unresponsive NMIBCc | 2022 | intravesical | adenovirus vector | deliver functional copies of the IFNα2b gene | Boorjian et al.58 |
| HEMGENIX | hemophilia B | 2022 | intravenous infusion | AAV5 | deliver functional copies of the factor IX gene | Pipe et al.59 |
| SKYSONA | boys with early, active CALDd | 2022 | ex vivo/HSCe | lentiviral vector | deliver functional copies of the ABCD1 gene | Eichler et al.60 |
| ELEVIDYS | duchenne muscular dystrophy (DMD) | 2023 | intravenous infusion | AAV vector | deliver functional copies of an engineered dystrophin gene | Mendell et al.61 |
| VYJUVEK | dystrophic epidermolysis bullosa | 2023 | tropical | non-replicating Herpes simplex virus | deliver functional copies of the COL7A1 gene | Guide et al.52 |
| CASGEVY | sickle cell diseases | 2024 | ex vivo/HSCe | electroporation | deliver CRISPR/Cas9-sgRNA RNP to disrupt the BCL11A enhancer & restore γ-globin expression | Frangoul et al.62 |
| LYFGENIA | sickle cell diseases | 2024 | ex vivo/HSCe | lentiviral vector | deliver functional copies of the β-globin gene | Kanter et al.63 |
ADA-SCID: severe combined immunodeficiency due to adenosine deaminase deficiency.
LCA: Leber Congenital Amaurosis.
BCG-unresponsive NMIBC: Bacillus Calmette-Guerin (BCG)-unresponsive non-muscle invasive bladder cancer (NMIBC).
CALD: cerebral adrenoleukodystrophy.
HSC: hematopoietic stem cell.
In 2023, another milestone of gene therapy was reached by the groundbreaking approval of CASGEVY, which is the world’s first gene editing-based gene therapy.64 Through delivering the gene editing tool CRISPR/Cas9 to disrupt the enhancer region of BCL11A gene in autologous HSCs, CASGEVY restores the expression of γ-globin to replace the mutated β-globin for the treatment of sickle cell disease (SCD) in patients aged 12 years and older.62,64 Notably, CASGEVY does not use viral vectors for gene delivery. Instead, it is administered to patients-isolated HSCs through electroporation.62
Approved gene silencing therapies
From a broader perspective, gene silencing (at the RNA level) drugs can also be considered as gene therapies since they also introduce genetic engineering materials, such as small interfering RNA (siRNA) or ASOs, into human cells for the treatment of genetic disorders.65 siRNA is a 20–25 nt long double-stranded RNA, capable of eliciting gene silencing through RNA interference (RNAi). Different from siRNAs, ASOs and Splice switching oligonucleotides (SSOs) are short, single-stranded nucleic acid sequences, typically 16–53 nt in length. ASOs and SSOs specifically bind to target mRNAs or pre-mRNAs and control gene expression through translation blockage, splicing modulation, RNase H-mediated RNA degradation, and so forth.66 In general, these synthetic small nucleic acid molecules all modulate the expression of a disease-causing gene in some way to treat corresponding diseases. To date, more than 10 siRNA, ASO, and SSO drugs have been approved by EMA or FDA. Two recent reviews have made excellent summaries of these approved oligonucleotide drugs.65,67 Thus, they will not be discussed in detail here.
Gene therapy for hypercholesterolemia
Since the beginning of the 21st century, gene therapy has emerged as an important research area in modern biomedical engineering and clinical practice. In the realm of FH treatment, researchers also explored many new gene therapy possibilities. These endeavors can be grouped into three categories.
Gene addition therapy for hypercholesterolemia
This one-time therapy involves viral vectors- or exosomes-mediated in vivo delivery of functional gene copies to compensate for the body’s inability to produce normal proteins for cholesterol metabolism. The earliest clinical trial of gene addition therapies for FH can be traced back to the early 1990s, wherein retroviral vector containing LDLR gene was transduced into autologous hepatocytes obtained through partial hepatectomy before they were infused back into patients (NCT00004809) (Table 2). However, researchers only observed low levels of LDLR expression plus a variable and moderate decrease in plasma LDL-C levels in studied patients, both of which prevented the further progression of this trial.68 In 2015, Wang et al. employed AAV8 to deliver human LDLR gain-of-function mutants into an LDLR-deficient mouse model (Ldlr−/−, Apobec1−/− double knockout). Remarkably, they observed a 98% reduction in cholesterol levels and no significant side effects in the mice, suggesting promise for this strategy in FH treatment.69 Although a phase I clinical trial using AAV for functional LDLR delivery was registered in 2016, interim efficacy analysis was not released due to the early termination of this trial (Table 2). The use of AAV for therapeutic LDLR gene delivery remains controversial due to the potential of AAV to trigger an immune response. Furthermore, the transduction efficiency of AAV in human hepatocytes is lower than that in mouse hepatocytes, suggesting that higher doses of AAV may be required for human applications and posing the risk of suboptimal cholesterol reduction in humans.70,71,72
Table 2.
Functional gene addition therapy for FH in clinical trials
| Target | Delivery route | Delivery vector | Clinical Status | Indications | Ref. ID |
|---|---|---|---|---|---|
| LDL-R | ex vivo | retrovirus vector | phase 1 | HoFH | NCT00004809 |
| intravenous infusion | AAV8 | phase 1/2 | HoFH | NCT02651675 | |
| intravenous infusion | Exosome | phase 1 | HoFH | NCT05043181 |
Meanwhile, exosomes are disk-shaped vesicles that originate from the endosomes of the cell nucleus. As “natural nanoparticles,” they can carry various types of nucleic acid cargoes, such as DNA and RNA, and transmit them across cellular membranes. By altering the targeting proteins, exosomes can also be directed to target specific organs or tissues.73 In 2021, Li et al. delivered the mRNA encoding functional LDLR via exosomes into LDLR knockout mice to alleviate atherosclerosis induced by a high-fat diet.74 This study observed an increase in LDLR levels and a concurrent decrease in serum LDL-C levels and mice liver lipid deposition, without the detection of exosome-associated toxicity.74 Encouraged by the positive preclinical results, a phase-I clinical trial employing the same strategy was registered under the ID NCT05043181 in the same year, although no interim data was released yet (Table 2). Currently, exosome-based therapies are still in the early stages of development. Enhancement in their purity and targeting specificity, alongside validations of their ability to carry lengthy mRNAs are expected steps before the approval and clinical applications of exosome-based therapies.75,76
Gene inactivation therapy for hypercholesterolemia
To date, two gene inactivation therapies, namely Mipomersen and Inclisiran, have been approved for treating HoFH and HeFH respectively, although the marketing of Mipomersen has been discontinued due to concerns about its adverse effects.77,78,79 As an ASO, Mipomerson targets the mRNAs of apolipoprotein B-100 (apoB-100) to prevent its translation, thereby reducing the production of LDL particles for the treatment of HoFH (Table 3).30 Meanwhile, Inclisiran is a siRNA that inhibits the expression of PCSK9, thereby increasing LDLR levels in the liver and improving the LDLR-mediated clearance of LDL-C from the blood.80 The long-lasting durability and safety of Inclisiran were well demonstrated in the ORION-3 clinical study.78 Over a period of four years, patients who received Inclisiran twice annually experienced an up to 78% reduction in PCSK9 expression levels and an average decrease in LDL-C levels of 42.2% (Table 3), albeit nearly all participants reported drug-related adverse effects, albeit mild, which primarily present as nasopharyngitis and injection site reactions (ISRs).78 Of note, both Inclisiran and Mipomersen need to be used as an adjunct to other lipid-lowering medications such as maximally tolerated statin therapy, in order to achieve greater reduction in LDL-C and better control of cardiovascular risks.
Table 3.
Lipid-lowering gene inactivation therapies approved and in clinical trials
| Type | Target | Name | Clinical Status | Indications | Lipid-Lowering Effect (-%) | References |
|---|---|---|---|---|---|---|
| siRNA | PCSK9 | Inclisiran | approved | HeFH or ASCVD | 44% (LDL-C) | Ray et al.78 |
| Angptl3 | ARO-ANG3 | phase 2b | HoFH/Mixed Dyslipidemia | up to 32% (non-HDL-C) | Watts et al.81 | |
| APOC3 | ARO-APOC3 | Phase 2b | Mixed Dyslipidemia/Hypertriglyceridemia | up to 27% (non-HDL-C) | Vasas et al.82 | |
| Lp(a) | Olpasiran | Phase 2 | Elevated Lp(a) | up to 25% (LDL-C) | O'Donoghue et al.83 | |
| Lp(a) | SLN360 | phase 1 | Elevated Lp(a) | up to 26% (LDL-C) | Nissen et al.84 | |
| ASO | apoB-100 | Mipomerson | Discontinued | HoFH | 25% (LDL-C) | Raal et al.30 |
| PCSK9 | AZD8233 | phase 2b | Hyperlipidemia/Dyslipidemia | up to 79% (LDL-C) | Hofherr et al.85 | |
| Angptl3 | Vupanorsen | phase 2b | Dyslipidemia/FCSa | up to 28% (non-HDL-C) | Bergmark et al.86 | |
| APOC3 | Volanesorsen | phase 3 | Hypertriglyceridemia/FCSa | 46% (non-HDL-C) | Witztum et al.87 | |
| APOC3 | Olezarsen | Phase 2 | Hypertriglyceridemia | up to 24% (non-HDL-C) | Tardif et al.88 | |
| Lp(a) | Pelacarsen | phase 2 | Elevated Lp(a) and CVDb | / | Tsimikas et al.89 |
FCS: Familial Chylomicronemia Syndrome.
CVD: cardiovascular disease.
Besides PCSK9, ANGPTL3 is another intervention target for FH as its inhibition would promote the activity of LPL and endothelial lipase (EL), which hydrolyze triglycerides (TGs) and phospholipids respectively within very low-density lipoproteins (VLDLs) to prevent the conversion of VLDLs into LDLs.90,91 Xu et al. delivered ARO-ANG3, an siRNA targeting ANGPTL3, to five different mouse models and human hepatoma cells. During the experiment, an approximately 97% reduction in ANGPTL3 expression was observed on day 3, accompanied by a 73% reduction in LDL-C.92 Moreover, preliminary findings from a phase 1 clinical trial (NCT03747224) evaluating ARO-ANG3 demonstrated a ∼50% reduction in LDL-C levels among participants.93 Additionally, a phase 2b clinical trial (NCT04832971) further revealed that ARO-ANG3 led to up to 56% reduction in circulating TG levels and up to 32% reduction in non-high-density lipoprotein cholesterol (non-HDL-C) levels 24 weeks post-dosing (Table 3).81 It is important to note, however, that the drop in LDL-C level was not as prominent as that in TG levels.81
Besides siRNAs, ASO drugs targeting PCSK9 and ANGPTL3 have also advanced into phase 2 clinical trials. Of them, PCSK-targeting AZD8233 showed up to 79% reduction in LDL-C levels from baseline at 90 mg dosage (Table 3).85 Meanwhile, although the ANGPTL3-targeting Vupanorsen exhibited up to 28% and 57% reduction in non-HDL-C and TG levels respectively, the decrease in LDL-C levels only achieved statistical significance at high dose (Table 3).86 Accordingly, the indication of Vupanorsen has changed into dyslipidemia instead of HoFH. Notably, both ASOs exhibited signs of mild liver damage at high doses, suggesting more efforts in promoting their efficacy at low doses. However, as negatively charged and hydrophilic molecules, ASOs face challenges in efficiently crossing the cell membrane, which presents a significant hurdle in their application for gene therapy and may compromise their efficacy, especially at low doses. To boost their delivery efficiency, the LNP system has been employed to administer PCSK9-targeting ASO to mice, resulting in a 75% decrease in PCSK9 mRNA expression.94 Alternatively, ASOs may also be conjugated with target ligands to enhance their stability, cellular uptake, and organ specificity. Indeed, Vupanorsen is an N-acetyl galactosamine (GalNAc)-conjugated ASO targeting liver.95
Another target, APOC3, has also been the focus of lipid-lowering gene inactivation therapy development as well. APOC3 is a small apolipoprotein primarily found in triglyceride-rich lipoproteins (TRLs) and LDLs.96 It inhibits the activity of LPL and reduces the clearance of triglyceride-rich lipoproteins (TRLs) from the bloodstream.97,98 Though not considered as a direct target for FH, the inhibition of APOC3 can produce an LDL-lowering effect and reduce the risk of cardiovascular diseases. Volanesorsen, an APOC3-targeting ASO, has entered a phase 3 clinical trial and led to a ∼46% decrease in non-HDL-C levels in participants (Table 3).87 However, thrombocytopenia was observed in about half of the participants, therefore volanesorsen is approved with restrictions in Europe but not in the US.99 Olezarsen and ARO-APOC3, GalNAc-conjugated ASO and siRNA respectively, are designed to preserve the lipid-lowering effect of volanesorsen while minimizing its side effects.99 They have both finished phase 2 clinical trials and shown positive interim results (Table 3).82,88
Lipoprotein (a), or Lp(a) for short, is a unique lipoprotein particle composed of an LDL-like core plus an additional glycoprotein called apolipoprotein (a).100,101 Elevated levels of Lp(a) are recognized as an independent risk factor for ASCVD.102 As the inhibition of Lp(a) holds promise for reducing blood cholesterol levels and cardiovascular diseases risks, several gene-inactivation therapies targeting Lp(a) are also reviewed here, including two siRNAs and one ASO. Olpasiran and SLN360 are both GalNAc-conjugated siRNAs targeting Lp(a) mRNAs. In a phase 2 clinical trial conducted in patients with established ASCVD and elevated Lp(a) levels, Olpasiran effectively reduces the Lp(a) concentration by more than 95% and achieves up to 25% reduction in LDL-C level in the high-dose groups (Table 3).83 Meanwhile, SLN360, also known as Zerlasiran, reduces LDL-C levels by up to 26%, when administered at a single dose of 600 mg in a phase 1 clinical trial (Table 3).84 Pelacarsen, on the other hand, is an ASO against Lp(a). When administered at a weekly dose of 20 mg, Pelacarsen showed a reduction in Lp(a) levels of up to 79.5% and a modest decrease in LDL-C levels (Table 3).89 Notably, approximately 30% of the enrolled patients in the Pelacarsen clinical trial were diagnosed with FH, suggesting the potential of Pelacarsen in controlling LDL-C for FH treatment.89
Gene editing therapy for hypercholesterolemia
Following the emergence of CRISPR-Cas9 technology, the rapid development of the CRISPR/Cas9 system and the subsequent Base editing (BE) system have spurred widespread interest in gene editing therapy based on these systems. Recently, the first CRISPR/Cas9-based gene editing therapy, Casgevy, was even approved for the treatment of Sickle Cell Disease (SCD).103 The promise of gene editing therapy in treating genetic disorders also stimulated the exploration of gene editing-based FH treatment. In this section, we will review the rapid development of gene editing toolkit and gene editing-based FH therapies in development, targeting either PCSK9 or ANGPTL3. We will also briefly discuss the emerging new target ASGR1.
Development of gene editing toolkit
In recent years, a set of gene editing techniques capable of precisely modifying an organism’s genome and its transcripts at specific sites to knock out or modify disease-causing genes has been developed. Among the existing gene editing tools, the CRISPR-Cas9 system and its derivatives represent the latest advance in gene editing technology.104 Currently, different types of genome editors have derived from the CRISPR-Cas9 system, including nucleases, base editors (BEs), and prime editors (PEs), further expanding the toolkit for gene therapy.105
Of these tools, the CRISPR-Cas9 nucleases are more adept at gene inactivation and are unavoidably related to detrimental large DNA fragment deletions, cell-cycle arrest and p53-mediated apoptosis or even chromosomal translocations in edited cells given that they work by introducing DNA double-strand breakage (DSBs) into the genome (Figure 2).106,107,108 In contrast, BEs are capable of introducing precise and highly efficient single-point mutations in a target DNA sequence without causing DSBs or requiring a donor DNA template, thus offering significant advantages for gene therapy applications compared to traditional CRISPR-Cas9 nucleases (Figure 2).109 BEs generally comprise a catalytically dead Cas9 (dCas9) or a nickase Cas9 (nCas9) to act as the “locator,” and a fused single-stranded DNA deaminase such as APOBEC3A to act as the “effector.”104 Two main classes of BEs were developed first: cytosine base editors (CBEs), proficient at converting C-G base pairs into T-A,110 and adenine base editors (ABEs), adept at converting A-T into G-C.111 Together, CBEs and ABEs can efficiently mediate four types of transition mutations (C→T, A→G, T→C, G→A). These BEs offer remarkable efficiency and precision and have been widely applied across various contexts. More recently, the adenine base inversion editor (AYBE)112 and the glycosylase base editor (CGBE)113 are developed to catalyze A-to-T/C and C-to-G/A inversions respectively, further expanding the BEs repertoire.
Figure 2.

The pros and cons of the CRISPR/Cas9-derived nuclease, base editor, and prime editor systems
Though free of DSBs-related safety concerns, BEs do carry the risk of off-target editing due that its “locator” moiety may bind to off-target sites and its “effector” moiety may induce non-sgRNA-dependent off-target mutations in single-strand DNA (ssDNA) or single-strand RNA (ssRNA).104,109 To further enhance the precision and safety profile of BEs, Wang et al. developed a transformer base editor (tBE) system, which induces efficient on-target editing with only background levels of genome-wide and transcriptome-wide OT mutations (Figure 2).114 By fusing a cleavable deoxycytidine deaminase inhibitor (dCDI) element (i.e., mA3CDI) to the “effector” mA3CDA1, tBE remains inactive at OT sites, thereby eliminating unintended mutations. However, when located at the on-target sites, tBE transforms to cleave off the dCDI element and thus catalyzes intended deamination efficiently. The high precision and safety profile makes tBE a reliable and efficient tool for gene therapy.114
While BEs have shown remarkable capabilities, they do have limitations when it comes to executing certain types of gene editing, such as simultaneous base substitutions of mixed types or small insertions/deletions at precise locations.115 To address these limitations, the Prime Editor (PE) was developed. Different from BEs, PEs use a wild-type reverse transcriptase (RT) derived from the Loni murine leukemia virus (MMLV) to work as the “effector,” which can reverse-transcribe a template to replace the target sequence.116 In PE-meditated editing, prime editing guide RNA (pegRNA) first guides the nCas9-RT fusion protein to the target site, nCas9 then makes a nick in the non-target strand (NTS) near the target site, and RT uses the template provided in pegRNA for reverse transcription, thereby effectively inserting the desired modifications into the NTS and getting the modifications fixed during subsequent DNA repair.117 The design of PE enables it to introduce all 12 types of point mutations as well as small insertions and deletions without the generation of DSBs (Figure 2). However, it is important to note that PEs also have their limitations, particularly in terms of editing efficiency, and still require further optimization to maximize their potential in gene therapy applications (Figure 2).105
Gene editing therapy targeting proprotein convertase subtilisin/kexin type 9
PCSK9, originally identified as neuroapoptosis-regulated convertase 1 (NARC-1), is the ninth proprotein convertase discovered to date.118,119 It is primarily expressed in the liver and intestine, with lower expression observed in the kidneys, skin, and brain.120,121 PCSK9 binds to LDLR and promotes its degradation in the lysosome, leading to decreased LDLR density on the cell membrane and elevated LDL-C levels in the blood.24 Inhibiting PCSK9 can thus increase the number of LDLR on hepatocyte surfaces and enhance LDL-C clearance from the blood, thereby preventing or treating ASCVDs.122,123,124 Indeed, although gain-of-function mutations in PCSK9 can lead to FH,125 individuals with naturally occurring loss-of-function mutations in PCSK9 exhibit LDL-C levels approximately 40% lower than those who do not carry these mutations.124 They also demonstrate a lower risk of atherosclerosis126 and do not experience adverse health consequences.127
To further emphasize the significance of PCSK9 as a therapeutic target for FH, several medications targeting it have been approved and marketed in recent years. One such agent is Evolocumab, a monoclonal antibody against PCSK9, which has been shown to have a favorable long-term efficacy and safety profile.128 Likewise, Alirocumab is another monoclonal antibody targeting PCSK9129 and is used as a second-line drug in the treatment of hypercholesterolemia, often in conjunction with first-line drugs such as statins.130 Clinical studies have demonstrated that alirocumab is highly effective for the treatment of hypercholesterolemia, and can be used to treat patients with acute myocardial infarction and reduce the size of their atherosclerosis.131 Besides, semi-annual injections of Inclisiran, a siRNA that inhibits the translation of PCSK9, was able to produce a ∼42% decrease in LDL-C levels without significant adverse effects.78
The optimistic prospects of targeting PCSK9 for FH treatment motivated gene editing exploration toward it. To date, various gene editing tools have been employed to induce mutations at specific loci of PCSK9 for sustained gene regulation. The earliest attempt to edit PCSK9 was conducted in 2014 by Ding et al. in mice. Using an adenovirus-delivered CRISPR/Cas9 system, they achieved >50% PCSK9 editing in the mouse liver and decreased plasma cholesterol levels by 35–40% (Table 4).132 In 2021, Rothgangl et al. utilized LNPs to deliver mRNAs encoding ABEs to edit PCSK9 in mice. They achieved 61% editing efficiency and observed a 95% reduction in plasma PCSK9 levels and a 58% reduction in LDL-C levels on average (Table 4).133 The same study achieved a 26% editing efficiency and a 14% reduction in plasma LDL-C levels in macaques (Table 4).133 In the same year, another research team also utilized an LNP-delivered ABE system to modify PCSK9 in the livers of cynomolgus monkeys, achieving ∼60% blood LDL-C reduction (Table 4).134
Table 4.
Gene editing therapy for FH in development
| Target | Delivery Strategy | Model | Editing tools | Editing efficiency | Lipid-Lowering Effect | References |
|---|---|---|---|---|---|---|
| PCSK9 | LNP | NHPsa | ABE8.8 | ∼79.4% | 60% | Musunuru et al.134 |
| LNP | Mice | ABE | 61% | 58% | Rothgangl et al.133 | |
| LNP | NHPsa | ABE | 26% | 14% | Rothgangl et al.133 | |
| ADV | Mice | CRISPR-Cas9 | >50% | 35-40% | Ding et al.132 | |
| ANGPTL3 | ADV | LDLR−/− Mice | CBE3 | ∼35% (ANGPTL3) | 51% | Chadwick et al.135 |
| LNP | Mice | CRISPR-Cas9 | 38.5% | 56.8% | Qiu et al.136 | |
| AAV9 | Mice | ABE4max | 63.30% | 61% | Zuo et al.137 | |
| GalNAc-LNP | Mice | ABE8.8 | 64% | / | Lee et al.138 | |
| GalNAc-LNP | NHPs1 | ABE8.8 | ∼61% | 35% | Kasiewicz et al.139 |
NHPs: non-human primates.
The positive feedback from preclinical research also spurred the advancement of PCSK-9 editing clinical trials. Recently, Verve Therapeutics released interim data from its ongoing Phase 1b clinical trial of VERVE-101, which uses LNP-delivered ABE to switch off PCSK9 expression for the treatment of HeFH.140 The data showed a dose-dependent reduction in blood LDL-C levels among participants, with the highest dosage demonstrating a 55% decrease in blood LDL-C levels after a single treatment lasting up to 180 days or more.140 However, severe adverse events potentially related to the treatment were also observed in two patients from the high-dose group, raising safety concerns about VERVE-101. Notably, one patient with underlying ASCVD experienced a Grade 3 myocardial infarction, while the other exhibited transiently elevated liver enzymes and a drop in platelet levels.141 Besides, it is worth noting that the effectiveness of this PCSK9 targeting strategy for FH treatment is contingent upon the functionality of LDLR. For instance, in HoFH cases where LDLR is absent (LDLR−/−), targeting PCSK9 may not achieve the desired lipid-lowering effect.142,143 Thus, exploring other lipid-lowering targets for FH treatment is necessary.
Gene editing therapy targeting angiopoietin-like 3
ANGPTL3 acts as a natural inhibitor of LPL and EL, which hydrolyze TGs and phospholipids within chylomicrons and VLDLs into free fatty acids (FFA) and glycerol monoester.91,144 Reduction in ANGPTL3 expression would enhance LPL and EL activity, thereby accelerating the clearance of VLDLs and reducing the conversion of VLDLs into LDLs (Figure 3).91 In 2009, Romeo et al. discovered a significant association between mutations in ANGPTL3 and TG levels in humans.145 Subsequently, researchers from Harvard Medical School and other institutions jointly identified four patients with loss-of-function ANGPTL3 mutations. All four patients manifested broad-spectrum hypolipidemia (low LDL-C, HDL-C, and triacylglycerols), were in good health, and demonstrated normal fertility.146 In May 2017, a genetics study involving over 90,000 individuals was published in the New England Journal of Medicine, revealing that individuals with loss-of-function mutations in ANGPTL3 exhibit significantly lower LDL-C levels and a reduced risk of cardiovascular disease compared to the general population.147 Together, these discoveries reinforce that ANGPTL3 could represent a safe target for gene editing-based hypercholesterolemia treatment. Notably, ANGPTL3’s regulation of LDL-C is independent of the LDL receptor and other established mechanisms,148 thus suggesting a potential therapeutic role of ANGPTL3 in HoFH treatment.
Figure 3.

ANGPTL3 as gene therapy targets
ANGPTL3 inhibits the activities of LPL and EL, which hydrolyze TGs and phospholipids respectively within VLDLs. Reduction in ANGPTL3 expression enhances LPL and EL activity, thereby increasing the conversion of VLDL into VLDL remnants with accelerated clearance and reducing VLDL conversion into LDL. As elevated LDL levels in blood vessels contribute to atherosclerosis, targeting ANGPTL3 expression through gene editing (e.g., BE or CRISPR/Cas9) or gene inactivation tools (siRNA or ASO) can potentially reduce the risk of ASCVD.
Several studies have explored the use of base editors for ANGPTL3 editing. In 2018, Chadwick et al. utilized an adenovirus vector to deliver the CBE3 to hyperlipidemic LDLR−/− mice, achieving an average editing efficiency of ∼35% and 51% reduction in cholesterol levels after 14 days. Meanwhile, the researchers compared the therapeutic effects of editing ANGPTL3 with those of editing PCSK9, finding that targeting ANGPTL3 yielded superior lipid-lowering effects. They also edited both targets simultaneously but observed neither additivity nor synergism between the two.135 In 2021, instead of using the FDA-approved MC-3 LNP, Qiu et al. developed a 306-O12B LNP delivery platform and used it to deliver the CRISPR/Cas9 system for liver-targeted ANGPTL3 editing. They observed a median editing efficiency of 38. 5% in mouse liver and a significant reduction in blood ANGPTL3, LDL-C, and TG levels (65.2, 56.8, and 29.4%).136 In 2023, Zuo et al. delivered an optimized CBE, AncBE4max, via AAV to edit ANGPTL3 and observed an editing efficiency of 63.3 ± 2.3% at the target site. Four weeks after administration, the blood TG and total cholesterol levels decreased by 58% and 61%, respectively.137 To bypass the dependency of LNP-mediated hepatic delivery on the functionality of LDLR, Verve Therapeutics, in collaboration with the University of Pennsylvania, USA, designed a GalNAc-LNP system. Utilizing this delivery platform, they achieved an ANGPTL3 editing efficiency of 64% in mice liver and a 98% reduction in plasma ANGPTL3 protein on average.138 Moreover, they observed an average hepatic ANGPTL3 editing efficiency of 61% and a significant (89% and 35%) and durable (up to three months) reduction in plasma ANGPTL3 and LDL-C levels in cynomolgus monkeys.139
Overall, targeting ANGPTL3 represents an attractive target for treating patients with severe FH, particularly those with LDLR-deficient phenotypes. The significance of ANGPTL3 is supported by the up to 50% reduction in LDL-C levels observed even in patients with HoFH during a phase 3 clinical trial of Evinacumab, a monoclonal antibody against ANGPTL3.149 However, it is important to note that the safety and efficacy of ANGPTL3-targeted therapies still require long-term follow-up to determine their impact on ASCVD risks and any potential adverse effects or complications.
The emerging new target ASGR1
Lectins are cell-surface proteins that specifically recognize and bind to sugars on glycoproteins. They play a crucial role in coordinating intercellular functions across a variety of cell types.150 ASGR1 is a lectin involved in maintaining circulating glycoprotein homeostasis.151,152,153,154 It is the major subunit of the Asialoglycoprotein receptor (ASGPR), which is a heterotrimeric protein complex assembled from major and minor subunits. ASGPR maintains serum glycoprotein homeostasis by mediating the endocytosis and lysosomal degradation of serum glycoproteins bearing exposed terminal galactose or GalNAc residues (Figure 4).155,156,157 When asialoglycoproteins were injected into ASGPR-deficient mice, impaired clearance of these glycoproteins was observed.158 However, it is important to note that these glycoproteins do not accumulate in the serum, indicating that ASGPR is not the sole regulator of blood glycoprotein levels.158
Figure 4.

ASGR1 as gene therapy targets
ASGR1-mediated endocytosis and lysosomal degradation of serum glycoproteins lead to the mTORC1 pathway activation, AMPK pathway inhibition, as well as the translocation of transcription factor SREBPs from the ER to the nucleus, thereby leading to the upregulation of genes involved in cholesterol biosynthesis and PCSK9-mediated LDLR degradation. Importantly, ASGR1 also acts through the AMPK pathway to downregulate LXRα, which in turn downregulates ABC-family transporters ABCG5/G8 to inhibit cholesterol efflux into bile and intestine. In theory, ASGR1 knockout (ASGR1 KO) would inhibit intracellular cholesterol synthesis, promote ABCG5/G8-mediated cholesterol efflux, and promote LDLR recycling to enhance the LDLR-mediated clearance of blood cholesterol (dotted lines).
Specifically expressed in hepatocytes, ASGR1 serves as an ideal target for drug delivery to the liver.159 Indeed, the GalNAc-LNP took advantage of ASGR1 for its hepatic delivery.139 More importantly, ASGR1 seems to have a significant impact on lipid metabolism. A population genetics study conducted in 2016 among Icelanders revealed that individuals carrying a 12 bp deletion (Del12) in intron 4 of ASGR1 exhibit notably lower cholesterol levels (0.4 mmol per liter) and a 34% lower risk of coronary artery disease, as compared to the noncarriers.160 This Del12 mutation would activate a cryptic splice site, resulting in a frameshift and the synthesis of a truncated 89 aa protein (full-length ASGR1 is 291 aa long) that is susceptible to degradation, thereby representing an ASGR1 Loss-of-function phenotype. Besides, another loss-of-function ASGR1 variant, p.W158X, was also found to exhibit much lower LDL-C levels than non-carriers. p.W158X is caused by a 4 bp base insertion and would result in a premature stop codon at position 158. Hence, the loss-of-function variants of ASGR1 seem to be associated with lower cholesterol levels and reduced cardiovascular disease risk.160
In 2021, Xu et al. and Xie et al. employed CRISPR/Cas9 to disrupt the ASGR1 gene in mouse and pig embryos respectively.161,162 Using these genetically modified animals, the authors revealed that ASGR1 deficiency promotes the retention of sterol regulatory element binding proteins (SREBPs) in the endoplasmic reticulum (ER).161 SREBPs are transcription factors that are activated and translocated from the ER to the nucleus in response to low cellular cholesterol levels, thereby leading to the transcription of genes involved in lipogenesis biosynthesis and uptake.163,164 Therefore, ASGR1 deficiency inhibits cholesterol biosynthesis (Figure 4).161,162 Besides, as PCSK9 is also the downstream gene of SREBPs, the expression level of PCSK9 also decreased by 50% in ASGR1-deficient mice, hinting that ASGR1 deficiency might also enhance LDL-C clearance through PCSK9/LDLR axis (Figure 4).161 In 2022, the linkages between ASGR1 and SREBPs were further revealed to be mTORC1 and AMPK, which sense cellular nutrients and regulate various cellular metabolic pathways (Figure 4).165 ASGR1 deficiency impedes the glycoprotein endocytosis and decreases lysosomal amino acid levels, thereby inhibiting mTORC1 and activating AMPK pathways to interfere with the downstream lipogenesis pathways.165 More excitingly, the authors discovered that ASGR1 deficiency could also act through the AMPK pathway to upregulate LXRα, which in turn upregulates ABCA1 and ABCG5/G8 to facilitate cholesterol efflux (Figure 4).165 ABCG5/8 are ABC-family transporters responsible for cholesterol transport into bile, and their upregulation would increase biliary and fecal cholesterol excretion. Together, these findings suggest that ASGR1 may represent a highly promising new target for FH treatment, especially considering its unique role in regulating cholesterol efflux.
To date, studies exploring the potential of targeting ASGR1 for FH treatment are limited, only the work by Wang et al. has shown that an anti-ASGR1 neutralizing antibody could increase cholesterol excretion in mice and produce synergistic lipid-lowering effects when combined with statins or ezetimibe.165 More explorations into gene therapies targeting ASGR1 are expected in the future.
Overall, the rapid development of gene therapy holds promise for potentially curing patients with FH. As technologies develop, an imperative objective of research will be to provide cost-effective and enduring gene therapy options for all patients. However, existing gene therapy approaches are hampered by several limitations, including the complexity of delivery methods and safety concerns.
Safety considerations of gene therapy
Safety is the primary concern in the clinical application of any drug. Thus, ensuring the safety of gene therapy remains a critical consideration in both its basic research and clinical application. Unintended integration of therapeutic genes, potential antigenicity of viral vectors, and off-target editing are the primary safety concerns of gene therapy.166,167,168,169 Later in discussion, we discuss the key aspects of gene therapy safety concerns in detail.
Safety of vectors
Commonly used vectors for gene therapy delivery fall into two categories: viral and non-viral vectors. Viral vectors, such as AAVs, harness the infectivity of viruses to transfer exogenous genes, i.e., the payloads. After incorporating the payloads into the viral genome, the edited virus is then used to infect the patient’s cells and introduce target genes into these cells. Although viral vectors offer efficient gene delivery and long-term or even permanent gene expression, they may also lead to serious adverse consequences. For instance, certain gene therapies that use retroviral vectors may cause genomic instability and even trigger malignancies such as leukemia through unintended integration of exogenous genes.167 Meanwhile, AAV vectors, while lacking the ability to integrate, may still pose safety concerns such as hepatotoxicity, kidney damage, and neurological loss due to their ability to trigger immune responses and prolonged inflammation.170 Besides, viral vectors generally have limited cargo capacity, thus constraining the size of the payload. Therefore, careful consideration of the immunogenicity, payload size, and potential risk of insertional mutagenesis needs to be taken when using viral vectors for delivery.
Besides viral vectors, exogenous payloads can also be delivered through non-viral vectors such as liposomes, LNPs, nanoparticles, gene guns, and so forth.171 These vectors encapsulate or absorb the payloads before delivering them into cells. Non-viral vectors offer several advantages, including reduced immunogenicity, ease of production, and potential for carrying large cargo. They are also free of integration concerns associated with viral vectors.171 However, non-viral vectors generally exhibit low transfection efficiency and shorter duration of gene expression as compared to viral vectors. They also face challenges in achieving targeted delivery and pose risks of cytotoxicity and tissue damage.172 For instance, the adverse events observed in the high-dose group of the VERVE-101 trial are potentially attributable to the LNP delivery system, as similar liver toxicity was observed in animals dosed with empty LNPs or LNPs carrying inactive payloads.141 Consequently, Verve Therapeutics has temporarily paused the VERVE-101 trial and shifted its focus to the VERVE-102 trial, which delivers the same payload as VERVE-101 but utilizes a GalNAc-LNP delivery system instead.141
Safety of payloads
In gene therapy, the payloads could consist of proteins, mRNAs, ASOs, or siRNAs. Safety concerns associated with these payloads include immunogenicity, genotoxicity, off-target potentials, and so forth. For example, ASOs and siRNAs may bind to unintended mRNA sequences, leading to undesired gene silencing or modulation.173,174 Meanwhile, exogenous proteins, mRNAs, as well as ASOs and siRNAs can all provoke immune responses, potentially leading to the abnormal activation of the immune system and even the development of autoimmune disorders.174,175,176 Besides, excessive expression of the delivered gene may disrupt physiological homeostasis, causing toxicity or adverse effects. Therefore, careful consideration of the immunogenicity, stability, specificity, and controllability of payloads is crucial to maximize the efficacy of gene therapies while minimizing their adverse effects. It is also advisable to avoid selecting oncogenic genes or genes that may produce adverse effects as payloads. Moreover, attention should also be paid to whether the cells may undergo stress response or apoptosis due to the presence of foreign genes. Besides, the expression level, duration, and distribution of the gene in the body need to be continuously monitored to prevent the occurrence of other potential side effects.
Safety of gene editing tools
Recent research indicates potential safety concerns associated with the CRISPR/Cas9 system due to off-target effects.177 Moreover, the Cas9 cleavage-mediated DSBs can lead to deletions of large DNA segments, causing genomic instability and even chromosomal translocations.106,108 Furthermore, DSBs may also activate DNA damage response pathways to trigger cell-cycle arrest and p53-mediated apoptosis in edited cells.107,178,179 In contrast, BEs do not generate DSBs in general and thus are free of large DNA fragment deletions or chromosomal translocations. Nonetheless, they carry the risk of off-target editing at both the DNA and RNA levels.104,109 It is worth noting that mismatches in the sgRNA can lead to the incorrect binding of the base editor at off-target sites with sequence similarity to the target site, resulting in sgRNA-dependent off-target mutations. Meanwhile, given that the “effector” of BEs possesses inherent deamination activity toward ssDNA or ssRNA, the base editor may also induce non-sgRNA-dependent off-target mutations at a genome-wide and transcriptome-wide level.180,181 Notably, to further enhance the safety profile of BEs for clinical applications, a new BE system named tBE was recently developed, which could reduce the genome-wide and transcriptome-wide OT mutations to the background level, thus presenting an ideal tool for gene therapy.114
Conclusions
Patients with severe FH are at a high risk of developing cardiovascular disease and are often unresponsive to conventional therapies. Gene therapy targeting disease-causing factors such as LDLR, PCSK9, ANGPTL3, and APOB represents a promising new strategy for FH treatment. Several gene therapy drugs for FH treatment are currently undergoing clinical trials, with some showing encouraging interim results. With the continuous development of gene editing tools, mRNA, siRNA, and ASOs technology, and delivery systems, it is anticipated that patients with FH may benefit from these gene therapy drugs and achieve long-lasting relief or even cure from them in the future. Nevertheless, it is also worth noting that the development and manufacturing of gene therapies require substantial investments. Consequently, gene therapies often carry a high price tag and substantial implementation requirements, which may render them inaccessible to certain people or regions. In this context, any scientific or technical innovation that can reduce the cost of gene therapy is of great significance, as it will greatly improve the accessibility and practical application of gene therapy.
Limitations of the study
Due to space limitations, some important works may not be fully discussed and acknowledged.
Acknowledgments
This work is supported by grant 2019YFA0802804 to B.Y from the National Key Research and Development Program of China., grants 32371272 and 32070170 to B.Y. from the National Natural Science Foundation of China (NSFC), and grant 23ZR1442500 to B.Y. from the Shanghai Municipal Science and Technology Commission. This work was also supported in part by a Shanghai Municipal Education Commission (SMEC) grant to the Shanghai Frontiers Science Center for Biomacromolecules and Precision Medicine at ShanghaiTech University. The figures were first drafted with BioRender.com and then modified in Adobe Illustrator (Adobe).
Author contributions
B.Y. and Y.X.L. drafted the article, Y.X.L., B.Y., and Y.F.H. prepared the illustrations, all authors have reviewed the literature and edited the article. All authors have read the final article and approve its submission.
Declaration of interests
The authors declare no competing interests.
References
- 1.Defesche J.C., Gidding S.S., Harada-Shiba M., Hegele R.A., Santos R.D., Wierzbicki A.S. Familial hypercholesterolaemia. Nat. Rev. Dis. Prim. 2017;3 doi: 10.1038/nrdp.2017.93. [DOI] [PubMed] [Google Scholar]
- 2.Santos R.D., Gidding S.S., Hegele R.A., Cuchel M.A., Barter P.J., Watts G.F., Baum S.J., Catapano A.L., Chapman M.J., Defesche J.C., et al. Defining severe familial hypercholesterolaemia and the implications for clinical management: a consensus statement from the International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel. Lancet Diabetes Endocrinol. 2016;4:850–861. doi: 10.1016/s2213-8587(16)30041-9. [DOI] [PubMed] [Google Scholar]
- 3.Brandts J., Ray K.K. Familial Hypercholesterolemia: JACC Focus Seminar 4/4. J. Am. Coll. Cardiol. 2021;78:1831–1843. doi: 10.1016/j.jacc.2021.09.004. [DOI] [PubMed] [Google Scholar]
- 4.Gofman J.W., Lindgren F., Elliott H., Mantz W., Hewitt J., Strisower B., Herring V., Lyon T.P. The role of lipids and lipoproteins in atherosclerosis. Science. 1950;111:166–186. doi: 10.1126/science.111.2877.166. [DOI] [PubMed] [Google Scholar]
- 5.Watts G.F., Gidding S.S., Mata P., Pang J., Sullivan D.R., Yamashita S., Raal F.J., Santos R.D., Ray K.K. Familial hypercholesterolaemia: evolving knowledge for designing adaptive models of care. Nat. Rev. Cardiol. 2020;17:360–377. doi: 10.1038/s41569-019-0325-8. [DOI] [PubMed] [Google Scholar]
- 6.Sturm A.C., Knowles J.W., Gidding S.S., Ahmad Z.S., Ahmed C.D., Ballantyne C.M., Baum S.J., Bourbon M., Carrié A., Cuchel M., et al. Clinical Genetic Testing for Familial Hypercholesterolemia: JACC Scientific Expert Panel. J. Am. Coll. Cardiol. 2018;72:662–680. doi: 10.1016/j.jacc.2018.05.044. [DOI] [PubMed] [Google Scholar]
- 7.Watts G.F., Gidding S.S., Hegele R.A., Raal F.J., Sturm A.C., Jones L.K., Sarkies M.N., Al-Rasadi K., Blom D.J., Daccord M., et al. International Atherosclerosis Society guidance for implementing best practice in the care of familial hypercholesterolaemia. Nat. Rev. Cardiol. 2023;20:845–869. doi: 10.1038/s41569-023-00892-0. [DOI] [PubMed] [Google Scholar]
- 8.Lui D.T.W., Lee A.C.H., Tan K.C.B. Management of Familial Hypercholesterolemia: Current Status and Future Perspectives. J. Endocr. Soc. 2021;5 doi: 10.1210/jendso/bvaa122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Broekhuizen K., van Poppel M.N.M., Koppes L.L.J., Brug J., van Mechelen W. A tailored lifestyle intervention to reduce the cardiovascular disease risk of individuals with Familial Hypercholesterolemia (FH): design of the PRO-FIT randomised controlled trial. BMC Publ. Health. 2010;10:69. doi: 10.1186/1471-2458-10-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hegele R.A. Environmental modulation of atherosclerosis end points in familial hypercholesterolemia. Atherosclerosis Suppl. 2002;2:5–7. doi: 10.1016/S1567-5688(01)00013-7. [DOI] [PubMed] [Google Scholar]
- 11.Austin M.A., Hutter C.M., Zimmern R.L., Humphries S.E. Familial Hypercholesterolemia and Coronary Heart Disease: A HuGE Association Review. Am. J. Epidemiol. 2004;160:421–429. doi: 10.1093/aje/kwh237. [DOI] [PubMed] [Google Scholar]
- 12.Moriarty P.M. Lipoprotein apheresis: present and future uses. Curr. Opin. Lipidol. 2015;26:544–552. doi: 10.1097/mol.0000000000000234. [DOI] [PubMed] [Google Scholar]
- 13.Safarova M.S., Moriarty P.M. Lipoprotein Apheresis: Current Recommendations for Treating Familial Hypercholesterolemia and Elevated Lipoprotein(a) Curr. Atherosclerosis Rep. 2023;25:391–404. doi: 10.1007/s11883-023-01113-2. [DOI] [PubMed] [Google Scholar]
- 14.Wierzbicki A.S., Humphries S.E., Minhas R., Guideline Development Group Familial hypercholesterolaemia: summary of NICE guidance. Br. Med. J. 2008;337 doi: 10.1136/bmj.a1095. [DOI] [PubMed] [Google Scholar]
- 15.Kayikcioglu M., Tokgozoglu L. Current Treatment Options in Homozygous Familial Hypercholesterolemia. Pharmaceuticals. 2022;16:64. doi: 10.3390/ph16010064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taylor F., Huffman M.D., Macedo A.F., Moore T.H.M., Burke M., Davey Smith G., Ward K., Ebrahim S. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst. Rev. 2013;2013 doi: 10.1002/14651858.CD004816.pub5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ward N.C., Watts G.F., Eckel R.H. Statin Toxicity. Circ. Res. 2019;124:328–350. doi: 10.1161/circresaha.118.312782. [DOI] [PubMed] [Google Scholar]
- 18.Howard J.P., Wood F.A., Finegold J.A., Nowbar A.N., Thompson D.M., Arnold A.D., Rajkumar C.A., Connolly S., Cegla J., Stride C., et al. Side Effect Patterns in a Crossover Trial of Statin, Placebo, and No Treatment. J. Am. Coll. Cardiol. 2021;78:1210–1222. doi: 10.1016/j.jacc.2021.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pedro-Botet J., Núñez-Cortés J.M., Flores J., Rius J. Muscle symptoms related with statin therapy in general practice. Atherosclerosis. 2015;241:e197. [Google Scholar]
- 20.Chogtu B., Magazine R., Bairy K.L. Statin use and risk of diabetes mellitus. World J. Diabetes. 2015;6:352–357. doi: 10.4239/wjd.v6.i2.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramkumar S., Raghunath A., Raghunath S. Statin Therapy: Review of Safety and Potential Side Effects. Acta Cardiol. Sin. 2016;32:631–639. doi: 10.6515/acs20160611a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Phan B.A.P., Dayspring T.D., Toth P.P. Ezetimibe therapy: mechanism of action and clinical update. Vasc. Health Risk Manag. 2012;8:415–427. doi: 10.2147/vhrm.S33664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim B.K., Hong S.J., Lee Y.J., Hong S.J., Yun K.H., Hong B.K., Heo J.H., Rha S.W., Cho Y.H., Lee S.J., et al. Long-term efficacy and safety of moderate-intensity statin with ezetimibe combination therapy versus high-intensity statin monotherapy in patients with atherosclerotic cardiovascular disease (RACING): a randomised, open-label, non-inferiority trial. Lancet. 2022;400:380–390. doi: 10.1016/s0140-6736(22)00916-3. [DOI] [PubMed] [Google Scholar]
- 24.Lagace T.A. PCSK9 and LDLR degradation: regulatory mechanisms in circulation and in cells. Curr. Opin. Lipidol. 2014;25:387–393. doi: 10.1097/mol.0000000000000114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Handelsman Y., Lepor N.E. PCSK9 Inhibitors in Lipid Management of Patients With Diabetes Mellitus and High Cardiovascular Risk: A Review. J. Am. Heart Assoc. 2018;7 doi: 10.1161/JAHA.118.008953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gürgöze M.T., Muller-Hansma A.H.G., Schreuder M.M., Galema-Boers A.M.H., Boersma E., Roeters van Lennep J.E. Adverse Events Associated With PCSK9 Inhibitors: A Real-World Experience. Clin. Pharmacol. Ther. 2019;105:496–504. doi: 10.1002/cpt.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.di Mauro G., Zinzi A., Scavone C., Mascolo A., Gaio M., Sportiello L., Ferrajolo C., Rafaniello C., Rossi F., Capuano A. PCSK9 Inhibitors and Neurocognitive Adverse Drug Reactions: Analysis of Individual Case Safety Reports from the Eudravigilance Database. Drug Saf. 2021;44:337–349. doi: 10.1007/s40264-020-01021-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mefford M.T., Rosenson R.S., Cushman M., Farkouh M.E., McClure L.A., Wadley V.G., Irvin M.R., Bittner V., Safford M.M., Somaratne R., et al. PCSK9 Variants, Low-Density Lipoprotein Cholesterol, and Neurocognitive Impairment. Circulation. 2018;137:1260–1269. doi: 10.1161/CIRCULATIONAHA.117.029785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zafar Y., Sattar Y., Ullah W., Roomi S., Rashid M.U., Khan M.S., Schmidt L. Proprotein convertase subtilisin/Kexin type-9 (PCSK-9) inhibitors induced liver injury - a retrospective analysis. J. Community Hosp. Intern. Med. Perspect. 2020;10:32–37. doi: 10.1080/20009666.2019.1710952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raal F.J., Santos R.D., Blom D.J., Marais A.D., Charng M.J., Cromwell W.C., Lachmann R.H., Gaudet D., Tan J.L., Chasan-Taber S., et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375:998–1006. doi: 10.1016/s0140-6736(10)60284-x. [DOI] [PubMed] [Google Scholar]
- 31.Wetterau J.R., Lin M.C., Jamil H. Microsomal triglyceride transfer protein. Biochim. Biophys. Acta. 1997;1345:136–150. doi: 10.1016/s0005-2760(96)00168-3. [DOI] [PubMed] [Google Scholar]
- 32.Gidding S.S., Champagne M.A., de Ferranti S.D., Defesche J., Ito M.K., Knowles J.W., McCrindle B., Raal F., Rader D., Santos R.D., et al. The Agenda for Familial Hypercholesterolemia: A Scientific Statement From the American Heart Association. Circulation. 2015;132:2167–2192. doi: 10.1161/cir.0000000000000297. [DOI] [PubMed] [Google Scholar]
- 33.Moyle M., Tate B. Homozygous familial hypercholesterolaemia presenting with cutaneous xanthomas: response to liver transplantation. Australas. J. Dermatol. 2004;45:226–228. doi: 10.1111/j.1440-0960.2004.00103.x. [DOI] [PubMed] [Google Scholar]
- 34.Scheller E.L., Krebsbach P.H. Gene therapy: design and prospects for craniofacial regeneration. J. Dent. Res. 2009;88:585–596. doi: 10.1177/0022034509337480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Razi Soofiyani S., Baradaran B., Lotfipour F., Kazemi T., Mohammadnejad L. Gene therapy, early promises, subsequent problems, and recent breakthroughs. Adv. Pharmaceut. Bull. 2013;3:249–255. doi: 10.5681/apb.2013.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pathak S. Gene therapy – Principles and Applications. Glob. J. Transfus. Med. 2022;7:3–6. doi: 10.4103/gjtm.gjtm_87_21. [DOI] [Google Scholar]
- 37.Anguela X.M., High K.A. Entering the Modern Era of Gene Therapy. Annu. Rev. Med. 2019;70:273–288. doi: 10.1146/annurev-med-012017-043332. [DOI] [PubMed] [Google Scholar]
- 38.Ling Q., Herstine J.A., Bradbury A., Gray S.J. AAV-based in vivo gene therapy for neurological disorders. Nat. Rev. Drug Discov. 2023;22:789–806. doi: 10.1038/s41573-023-00766-7. [DOI] [PubMed] [Google Scholar]
- 39.Li C., Georgakopoulou A., Newby G.A., Chen P.J., Everette K.A., Paschoudi K., Vlachaki E., Gil S., Anderson A.K., Koob T., et al. In vivo HSC prime editing rescues sickle cell disease in a mouse model. Blood. 2023;141:2085–2099. doi: 10.1182/blood.2022018252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi D., Toyonaga S., Anderson D.G. In Vivo RNA Delivery to Hematopoietic Stem and Progenitor Cells via Targeted Lipid Nanoparticles. Nano Lett. 2023;23:2938–2944. doi: 10.1021/acs.nanolett.3c00304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li X., La Salvia S., Liang Y., Adamiak M., Kohlbrenner E., Jeong D., Chepurko E., Ceholski D., Lopez-Gordo E., Yoon S., et al. Extracellular Vesicle–Encapsulated Adeno-Associated Viruses for Therapeutic Gene Delivery to the Heart. Circulation. 2023;148:405–425. doi: 10.1161/CIRCULATIONAHA.122.063759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu X., He X., Liu F., Jiang X., Wang P., Zhang J., Jiang J. Development and clinical translation of ex vivo gene therapy. Comput. Struct. Biotechnol. J. 2022;20:2986–3003. doi: 10.1016/j.csbj.2022.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vavassori V., Ferrari S., Beretta S., Asperti C., Albano L., Annoni A., Gaddoni C., Varesi A., Soldi M., Cuomo A., et al. Lipid nanoparticles allow efficient and harmless ex vivo gene editing of human hematopoietic cells. Blood. 2023;142:812–826. doi: 10.1182/blood.2022019333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fischer A. Gene therapy for inborn errors of immunity: past, present and future. Nat. Rev. Immunol. 2023;23:397–408. doi: 10.1038/s41577-022-00800-6. [DOI] [PubMed] [Google Scholar]
- 45.Everette K.A., Newby G.A., Levine R.M., Mayberry K., Jang Y., Mayuranathan T., Nimmagadda N., Dempsey E., Li Y., Bhoopalan S.V., et al. Ex vivo prime editing of patient haematopoietic stem cells rescues sickle-cell disease phenotypes after engraftment in mice. Nat. Biomed. Eng. 2023;7:616–628. doi: 10.1038/s41551-023-01026-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singh G. Resveratrol Delivery via Gene Therapy: Entering the Modern Era. Turk. J. Pharm. Sci. 2022;19:104–109. doi: 10.4274/tjps.galenos.2020.89577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peng Z. Current status of gendicine in China: recombinant human Ad-p53 agent for treatment of cancers. Hum. Gene Ther. 2005;16:1016–1027. doi: 10.1089/hum.2005.16.1016. [DOI] [PubMed] [Google Scholar]
- 48.Gaudet D., Méthot J., Déry S., Brisson D., Essiembre C., Tremblay G., Tremblay K., de Wal J., Twisk J., van den Bulk N., et al. Efficacy and long-term safety of alipogene tiparvovec (AAV1-LPLS447X) gene therapy for lipoprotein lipase deficiency: an open-label trial. Gene Ther. 2013;20:361–369. doi: 10.1038/gt.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cicalese M.P., Ferrua F., Castagnaro L., Pajno R., Barzaghi F., Giannelli S., Dionisio F., Brigida I., Bonopane M., Casiraghi M., et al. Update on the safety and efficacy of retroviral gene therapy for immunodeficiency due to adenosine deaminase deficiency. Blood. 2016;128:45–54. doi: 10.1182/blood-2016-01-688226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.South E., Cox E., Meader N., Woolacott N., Griffin S. Strimvelis(®) for Treating Severe Combined Immunodeficiency Caused by Adenosine Deaminase Deficiency: An Evidence Review Group Perspective of a NICE Highly Specialised Technology Evaluation. Pharmacoecon. Open. 2019;3:151–161. doi: 10.1007/s41669-018-0102-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Russell S., Bennett J., Wellman J.A., Chung D.C., Yu Z.F., Tillman A., Wittes J., Pappas J., Elci O., McCague S., et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390:849–860. doi: 10.1016/s0140-6736(17)31868-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guide S.V., Gonzalez M.E., Bağcı I.S., Agostini B., Chen H., Feeney G., Steimer M., Kapadia B., Sridhar K., Quesada Sanchez L., et al. Trial of Beremagene Geperpavec (B-VEC) for Dystrophic Epidermolysis Bullosa. N. Engl. J. Med. 2022;387:2211–2219. doi: 10.1056/NEJMoa2206663. [DOI] [PubMed] [Google Scholar]
- 53.Khan A., Riaz R., Ashraf S., Akilimali A. Revolutionary breakthrough: FDA approves Vyjuvek, the first topical gene therapy for dystrophic epidermolysis bullosa. Ann. Med. Surg. 2023;85:6298–6301. doi: 10.1097/ms9.0000000000001422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mendell J.R., Al-Zaidy S., Shell R., Arnold W.D., Rodino-Klapac L.R., Prior T.W., Lowes L., Alfano L., Berry K., Church K., et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017;377:1713–1722. doi: 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- 55.Locatelli F., Thompson A.A., Kwiatkowski J.L., Porter J.B., Thrasher A.J., Hongeng S., Sauer M.G., Thuret I., Lal A., Algeri M., et al. Betibeglogene Autotemcel Gene Therapy for Non-β(0)/β(0) Genotype β-Thalassemia. N. Engl. J. Med. 2022;386:415–427. doi: 10.1056/NEJMoa2113206. [DOI] [PubMed] [Google Scholar]
- 56.Ozelo M.C., Mahlangu J., Pasi K.J., Giermasz A., Leavitt A.D., Laffan M., Symington E., Quon D.V., Wang J.D., Peerlinck K., et al. Valoctocogene Roxaparvovec Gene Therapy for Hemophilia A. N. Engl. J. Med. 2022;386:1013–1025. doi: 10.1056/NEJMoa2113708. [DOI] [PubMed] [Google Scholar]
- 57.Sessa M., Lorioli L., Fumagalli F., Acquati S., Redaelli D., Baldoli C., Canale S., Lopez I.D., Morena F., Calabria A., et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: an ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet. 2016;388:476–487. doi: 10.1016/S0140-6736(16)30374-9. [DOI] [PubMed] [Google Scholar]
- 58.Boorjian S.A., Alemozaffar M., Konety B.R., Shore N.D., Gomella L.G., Kamat A.M., Bivalacqua T.J., Montgomery J.S., Lerner S.P., Busby J.E., et al. Intravesical nadofaragene firadenovec gene therapy for BCG-unresponsive non-muscle-invasive bladder cancer: a single-arm, open-label, repeat-dose clinical trial. Lancet Oncol. 2021;22:107–117. doi: 10.1016/s1470-2045(20)30540-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pipe S.W., Leebeek F.W.G., Recht M., Key N.S., Castaman G., Miesbach W., Lattimore S., Peerlinck K., Van der Valk P., Coppens M., et al. Gene Therapy with Etranacogene Dezaparvovec for Hemophilia B. N. Engl. J. Med. 2023;388:706–718. doi: 10.1056/NEJMoa2211644. [DOI] [PubMed] [Google Scholar]
- 60.Eichler F., Duncan C., Musolino P.L., Orchard P.J., De Oliveira S., Thrasher A.J., Armant M., Dansereau C., Lund T.C., Miller W.P., et al. Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. N. Engl. J. Med. 2017;377:1630–1638. doi: 10.1056/NEJMoa1700554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mendell J.R., Sahenk Z., Lehman K.J., Lowes L.P., Reash N.F., Iammarino M.A., Alfano L.N., Lewis S., Church K., Shell R., et al. Long-term safety and functional outcomes of delandistrogene moxeparvovec gene therapy in patients with Duchenne muscular dystrophy: A phase 1/2a nonrandomized trial. Muscle Nerve. 2024;69:93–98. doi: 10.1002/mus.27955. [DOI] [PubMed] [Google Scholar]
- 62.Frangoul H., Altshuler D., Cappellini M.D., Chen Y.-S., Domm J., Eustace B.K., Foell J., de la Fuente J., Grupp S., Handgretinger R., et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 2021;384:252–260. doi: 10.1056/NEJMoa2031054. [DOI] [PubMed] [Google Scholar]
- 63.Kanter J., Walters M.C., Krishnamurti L., Mapara M.Y., Kwiatkowski J.L., Rifkin-Zenenberg S., Aygun B., Kasow K.A., Pierciey F.J., Bonner M., et al. Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease. N. Engl. J. Med. 2022;386:617–628. doi: 10.1056/NEJMoa2117175. [DOI] [PubMed] [Google Scholar]
- 64.Hoy S.M. Exagamglogene Autotemcel: First Approval. Mol. Diagn. Ther. 2024;28:133–139. doi: 10.1007/s40291-024-00696-z. [DOI] [PubMed] [Google Scholar]
- 65.Yamada Y. Nucleic Acid Drugs-Current Status, Issues, and Expectations for Exosomes. Cancers. 2021;13 doi: 10.3390/cancers13195002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dhuri K., Bechtold C., Quijano E., Pham H., Gupta A., Vikram A., Bahal R. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J. Clin. Med. 2020;9 doi: 10.3390/jcm9062004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kulkarni J.A., Witzigmann D., Thomson S.B., Chen S., Leavitt B.R., Cullis P.R., van der Meel R. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 2021;16:630–643. doi: 10.1038/s41565-021-00898-0. [DOI] [PubMed] [Google Scholar]
- 68.Grossman M., Rader D.J., Muller D.W., Kolansky D.M., Kozarsky K., Clark B.J., 3rd, Stein E.A., Lupien P.J., Brewer H.B., Jr., Raper S.E., et al. A pilot study of ex vivo gene therapy for homozygous familial hypercholesterolaemia. Nat. Med. 1995;1:1148–1154. doi: 10.1038/nm1195-1148. [DOI] [PubMed] [Google Scholar]
- 69.Wang L., Muthuramu I., Somanathan S., Zhang H., Bell P., He Z., Yu H., Zhu Y., Tretiakova A.P., Wilson J.M. Developing a second-generation clinical candidate AAV vector for gene therapy of familial hypercholesterolemia. Mol. Ther. Methods Clin. Dev. 2021;22:1–10. doi: 10.1016/j.omtm.2021.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bissig-Choisat B., Wang L., Legras X., Saha P.K., Chen L., Bell P., Pankowicz F.P., Hill M.C., Barzi M., Leyton C.K., et al. Development and rescue of human familial hypercholesterolaemia in a xenograft mouse model. Nat. Commun. 2015;6:7339. doi: 10.1038/ncomms8339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lisowski L., Dane A.P., Chu K., Zhang Y., Cunningham S.C., Wilson E.M., Nygaard S., Grompe M., Alexander I.E., Kay M.A. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature. 2014;506:382–386. doi: 10.1038/nature12875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Barzi M., Chen T., Gonzalez T.J., Pankowicz F.P., Oh S.H., Streff H.L., Rosales A., Ma Y., Collias S., Woodfield S.E., et al. A humanized mouse model for adeno-associated viral gene therapy. Nat. Commun. 2024;15:1955. doi: 10.1038/s41467-024-46017-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baek G., Choi H., Kim Y., Lee H.C., Choi C. Mesenchymal Stem Cell-Derived Extracellular Vesicles as Therapeutics and as a Drug Delivery Platform. Stem Cells Transl. Med. 2019;8:880–886. doi: 10.1002/sctm.18-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li Z., Zhao P., Zhang Y., Wang J., Wang C., Liu Y., Yang G., Yuan L. Exosome-based Ldlr gene therapy for familial hypercholesterolemia in a mouse model. Theranostics. 2021;11:2953–2965. doi: 10.7150/thno.49874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sun L., Xu R., Sun X., Duan Y., Han Y., Zhao Y., Qian H., Zhu W., Xu W. Safety evaluation of exosomes derived from human umbilical cord mesenchymal stromal cell. Cytotherapy. 2016;18:413–422. doi: 10.1016/j.jcyt.2015.11.018. [DOI] [PubMed] [Google Scholar]
- 76.Rezaie J., Feghhi M., Etemadi T. A review on exosomes application in clinical trials: perspective, questions, and challenges. Cell Commun. Signal. 2022;20:145. doi: 10.1186/s12964-022-00959-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ray K.K., Wright R.S., Kallend D., Koenig W., Leiter L.A., Raal F.J., Bisch J.A., Richardson T., Jaros M., Wijngaard P.L.J., et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020;382:1507–1519. doi: 10.1056/NEJMoa1912387. [DOI] [PubMed] [Google Scholar]
- 78.Ray K.K., Troquay R.P.T., Visseren F.L.J., Leiter L.A., Scott Wright R., Vikarunnessa S., Talloczy Z., Zang X., Maheux P., Lesogor A., Landmesser U. Long-term efficacy and safety of inclisiran in patients with high cardiovascular risk and elevated LDL cholesterol (ORION-3): results from the 4-year open-label extension of the ORION-1 trial. Lancet Diabetes Endocrinol. 2023;11:109–119. doi: 10.1016/S2213-8587(22)00353-9. [DOI] [PubMed] [Google Scholar]
- 79.Hair P., Cameron F., McKeage K. Mipomersen sodium: first global approval. Drugs. 2013;73:487–493. doi: 10.1007/s40265-013-0042-2. [DOI] [PubMed] [Google Scholar]
- 80.Frank-Kamenetsky M., Grefhorst A., Anderson N.N., Racie T.S., Bramlage B., Akinc A., Butler D., Charisse K., Dorkin R., Fan Y., et al. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc. Natl. Acad. Sci. USA. 2008;105:11915–11920. doi: 10.1073/pnas.0805434105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Watts G.F., Gaudet D., Altamirano D., Hegele R., Ballantyne C.M., Nicholls S., Chang T., Alagarsamy S., Fu R., San Martin J., Rosenson R.S. ARO-ANG3, an Investigational RNAi Therapeutic, Silences the Expression of ANGPTL3 and Decreases Atherogenic Lipoproteins in Patients With Mixed Dyslipidemia: ARCHES-2 Study Results. Circulation. 2023;148 doi: 10.1161/circ.148.suppl_1.17120. [DOI] [Google Scholar]
- 82.Vasas S., Azizad M., Clifton P., Gaudet D., Goldenberg R., Modesto K., Chang T., Melquist S., Fu R., San Martin J., Ballantyne C.M. ARO-APOC3, an Investigational RNAi Therapeutic, Silences APOC3 and Reduces Atherosclerosis-Associated Lipoproteins in Patients With Mixed Dyslipidemia: MUIR Study Results. Circulation. 2023;148 doi: 10.1161/circ.148.suppl_1.17091. [DOI] [Google Scholar]
- 83.O'Donoghue M.L., Rosenson R.S., Gencer B., López J.A.G., Lepor N.E., Baum S.J., Stout E., Gaudet D., Knusel B., Kuder J.F., et al. Small Interfering RNA to Reduce Lipoprotein(a) in Cardiovascular Disease. N. Engl. J. Med. 2022;387:1855–1864. doi: 10.1056/NEJMoa2211023. [DOI] [PubMed] [Google Scholar]
- 84.Nissen S.E., Wolski K., Balog C., Swerdlow D.I., Scrimgeour A.C., Rambaran C., Wilson R.J., Boyce M., Ray K.K., Cho L., et al. Single Ascending Dose Study of a Short Interfering RNA Targeting Lipoprotein(a) Production in Individuals With Elevated Plasma Lipoprotein(a) Levels. JAMA. 2022;327:1679–1687. doi: 10.1001/jama.2022.5050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hofherr A., Schumi J., Rudvik A., Vega R., Ryden-Bergsten T., Hurt-Camejo E., Johanson P., Carlsson B.C. The PCSK9-Targeted Antisense Oligonucleotide AZD8233 Reduces LDL-C, ApoB, and Lp(a) in Patients With Dyslipidemia on Statin Treatment - Data From the Phase 2b ETESIAN Study. Circulation. 2022;146 doi: 10.1161/circ.146.suppl_1.11482. [DOI] [Google Scholar]
- 86.Bergmark B.A., Marston N.A., Bramson C.R., Curto M., Ramos V., Jevne A., Kuder J.F., Park J.G., Murphy S.A., Verma S., et al. Effect of Vupanorsen on Non-High-Density Lipoprotein Cholesterol Levels in Statin-Treated Patients With Elevated Cholesterol: TRANSLATE-TIMI 70. Circulation. 2022;145:1377–1386. doi: 10.1161/CIRCULATIONAHA.122.059266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Witztum J.L., Gaudet D., Freedman S.D., Alexander V.J., Digenio A., Williams K.R., Yang Q., Hughes S.G., Geary R.S., Arca M., et al. Volanesorsen and Triglyceride Levels in Familial Chylomicronemia Syndrome. N. Engl. J. Med. 2019;381:531–542. doi: 10.1056/NEJMoa1715944. [DOI] [PubMed] [Google Scholar]
- 88.Tardif J.C., Karwatowska-Prokopczuk E., Amour E.S., Ballantyne C.M., Shapiro M.D., Moriarty P.M., Baum S.J., Hurh E., Bartlett V.J., Kingsbury J., et al. Apolipoprotein C-III reduction in subjects with moderate hypertriglyceridaemia and at high cardiovascular risk. Eur. Heart J. 2022;43:1401–1412. doi: 10.1093/eurheartj/ehab820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tsimikas S., Karwatowska-Prokopczuk E., Gouni-Berthold I., Tardif J.C., Baum S.J., Steinhagen-Thiessen E., Shapiro M.D., Stroes E.S., Moriarty P.M., Nordestgaard B.G., et al. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N. Engl. J. Med. 2020;382:244–255. doi: 10.1056/NEJMoa1905239. [DOI] [PubMed] [Google Scholar]
- 90.Lim G.B. ANGPTL3 inhibition for hypercholesterolaemia. Nat. Rev. Cardiol. 2021;18:72. doi: 10.1038/s41569-020-00483-3. [DOI] [PubMed] [Google Scholar]
- 91.Adam R.C., Mintah I.J., Alexa-Braun C.A., Shihanian L.M., Lee J.S., Banerjee P., Hamon S.C., Kim H.I., Cohen J.C., Hobbs H.H., et al. Angiopoietin-like protein 3 governs LDL-cholesterol levels through endothelial lipase-dependent VLDL clearance. J. Lipid Res. 2020;61:1271–1286. doi: 10.1194/jlr.RA120000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xu Y.X., Redon V., Yu H., Querbes W., Pirruccello J., Liebow A., Deik A., Trindade K., Wang X., Musunuru K., et al. Role of angiopoietin-like 3 (ANGPTL3) in regulating plasma level of low-density lipoprotein cholesterol. Atherosclerosis. 2018;268:196–206. doi: 10.1016/j.atherosclerosis.2017.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ying Q., Chan D.C., Watts G.F. Angiopoietin-like protein 3 inhibitors and contemporary unmet needs in lipid management. Curr. Opin. Lipidol. 2021;32:210–212. doi: 10.1097/MOL.0000000000000747. [DOI] [PubMed] [Google Scholar]
- 94.Yang L., Ma F., Liu F., Chen J., Zhao X., Xu Q. Efficient Delivery of Antisense Oligonucleotides Using Bioreducible Lipid Nanoparticles In Vitro and In Vivo. Mol. Ther. Nucleic Acids. 2020;19:1357–1367. doi: 10.1016/j.omtn.2020.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gaudet D., Karwatowska-Prokopczuk E., Baum S.J., Hurh E., Kingsbury J., Bartlett V.J., Figueroa A.L., Piscitelli P., Singleton W., Witztum J.L., et al. Vupanorsen, an N-acetyl galactosamine-conjugated antisense drug to ANGPTL3 mRNA, lowers triglycerides and atherogenic lipoproteins in patients with diabetes, hepatic steatosis, and hypertriglyceridaemia. Eur. Heart J. 2020;41:3936–3945. doi: 10.1093/eurheartj/ehaa689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Huff M.W., Hegele R.A. Apolipoprotein C-III. Circ. Res. 2013;112:1405–1408. doi: 10.1161/CIRCRESAHA.113.301464. [DOI] [PubMed] [Google Scholar]
- 97.Tromp T.R., Stroes E.S.G., Hovingh G.K. Gene-based therapy in lipid management: the winding road from promise to practice. Expert Opin. Invest. Drugs. 2020;29:483–493. doi: 10.1080/13543784.2020.1757070. [DOI] [PubMed] [Google Scholar]
- 98.Khetarpal S.A., Zeng X., Millar J.S., Vitali C., Somasundara A.V.H., Zanoni P., Landro J.A., Barucci N., Zavadoski W.J., Sun Z., et al. A human APOC3 missense variant and monoclonal antibody accelerate apoC-III clearance and lower triglyceride-rich lipoprotein levels. Nat. Med. 2017;23:1086–1094. doi: 10.1038/nm.4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hegele R.A. APOC3 Interference for Familial Chylomicronaemia Syndrome. touchREV. Endocrinol. 2022;18:82–83. doi: 10.17925/ee.2022.18.2.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Utermann G., Weber W. Protein composition of Lp(a) lipoprotein from human plasma. FEBS Lett. 1983;154:357–361. doi: 10.1016/0014-5793(83)80182-3. [DOI] [PubMed] [Google Scholar]
- 101.Gaubatz J.W., Heideman C., Gotto A.M., Jr., Morrisett J.D., Dahlen G.H. Human plasma lipoprotein [a]. Structural properties. J. Biol. Chem. 1983;258:4582–4589. [PubMed] [Google Scholar]
- 102.Tsimikas S., Fazio S., Ferdinand K.C., Ginsberg H.N., Koschinsky M.L., Marcovina S.M., Moriarty P.M., Rader D.J., Remaley A.T., Reyes-Soffer G., et al. NHLBI Working Group Recommendations to Reduce Lipoprotein(a)-Mediated Risk of Cardiovascular Disease and Aortic Stenosis. J. Am. Coll. Cardiol. 2018;71:177–192. doi: 10.1016/j.jacc.2017.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Philippidis A. CASGEVY Makes History as FDA Approves First CRISPR/Cas9 Genome Edited Therapy. Hum. Gene Ther. 2024;35:1–4. doi: 10.1089/hum.2023.29263.bfs. [DOI] [PubMed] [Google Scholar]
- 104.Yang L., Chen J. A Tale of Two Moieties: Rapidly Evolving CRISPR/Cas-Based Genome Editing. Trends Biochem. Sci. 2020;45:874–888. doi: 10.1016/j.tibs.2020.06.003. [DOI] [PubMed] [Google Scholar]
- 105.Anzalone A.V., Koblan L.W., Liu D.R. Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime. Nat. Biotechnol. 2020;38:824–844. doi: 10.1038/s41587-020-0561-9. [DOI] [PubMed] [Google Scholar]
- 106.Zuccaro M.V., Xu J., Mitchell C., Marin D., Zimmerman R., Rana B., Weinstein E., King R.T., Palmerola K.L., Smith M.E., et al. Allele-Specific Chromosome Removal after Cas9 Cleavage in Human Embryos. Cell. 2020;183:1650–1664.e15. doi: 10.1016/j.cell.2020.10.025. [DOI] [PubMed] [Google Scholar]
- 107.Haapaniemi E., Botla S., Persson J., Schmierer B., Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018;24:927–930. doi: 10.1038/s41591-018-0049-z. [DOI] [PubMed] [Google Scholar]
- 108.Kosicki M., Tomberg K., Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yang B., Yang L., Chen J. Development and Application of Base Editors. CRISPR J. 2019;2:91–104. doi: 10.1089/crispr.2019.0001. [DOI] [PubMed] [Google Scholar]
- 110.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A⋅T to G⋅C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tong H., Wang X., Liu Y., Liu N., Li Y., Luo J., Ma Q., Wu D., Li J., Xu C., Yang H. Programmable A-to-Y base editing by fusing an adenine base editor with an N-methylpurine DNA glycosylase. Nat. Biotechnol. 2023;41:1080–1084. doi: 10.1038/s41587-022-01595-6. [DOI] [PubMed] [Google Scholar]
- 113.Ye L., Zhao D., Li J., Wang Y., Li B., Yang Y., Hou X., Wang H., Wei Z., Liu X., et al. Glycosylase-based base editors for efficient T-to-G and C-to-G editing in mammalian cells. Nat. Biotechnol. 2024:1–10. doi: 10.1038/s41587-023-02050-w. [DOI] [PubMed] [Google Scholar]
- 114.Wang L., Xue W., Zhang H., Gao R., Qiu H., Wei J., Zhou L., Lei Y.-N., Wu X., Li X., et al. Eliminating base-editor-induced genome-wide and transcriptome-wide off-target mutations. Nat. Cell Biol. 2021;23:552–563. doi: 10.1038/s41556-021-00671-4. [DOI] [PubMed] [Google Scholar]
- 115.Landrum M.J., Lee J.M., Benson M., Brown G., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Hoover J., et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–D868. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chen P.J., Liu D.R. Prime editing for precise and highly versatile genome manipulation. Nat. Rev. Genet. 2023;24:161–177. doi: 10.1038/s41576-022-00541-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A., Liu D.R. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Seidah N.G., Benjannet S., Wickham L., Marcinkiewicz J., Jasmin S.B., Stifani S., Basak A., Prat A., Chretien M. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. USA. 2003;100:928–933. doi: 10.1073/pnas.0335507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Benjannet S., Rhainds D., Essalmani R., Mayne J., Wickham L., Jin W., Asselin M.-C., Hamelin J., Varret M., Allard D., et al. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J. Biol. Chem. 2004;279:48865–48875. doi: 10.1074/jbc.M409699200. [DOI] [PubMed] [Google Scholar]
- 120.O'Connell E.M., Lohoff F.W. Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) in the Brain and Relevance for Neuropsychiatric Disorders. Front. Neurosci. 2020;14:609. doi: 10.3389/fnins.2020.00609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shapiro M.D., Tavori H., Fazio S. PCSK9: From Basic Science Discoveries to Clinical Trials. Circ. Res. 2018;122:1420–1438. doi: 10.1161/circresaha.118.311227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Maxwell K.N., Soccio R.E., Duncan E.M., Sehayek E., Breslow J.L. Novel putative SREBP and LXR target genes identified by microarray analysis in liver of cholesterol-fed mice. J. Lipid Res. 2003;44:2109–2119. doi: 10.1194/jlr.M300203-JLR200. [DOI] [PubMed] [Google Scholar]
- 123.Horton J.D., Cohen J.C., Hobbs H.H. Molecular biology of PCSK9: its role in LDL metabolism. Trends Biochem. Sci. 2007;32:71–77. doi: 10.1016/j.tibs.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cohen J., Pertsemlidis A., Kotowski I.K., Graham R., Garcia C.K., Hobbs H.H. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 2005;37:161–165. doi: 10.1038/ng1509. [DOI] [PubMed] [Google Scholar]
- 125.Abifadel M., Varret M., Rabès J.-P., Allard D., Ouguerram K., Devillers M., Cruaud C., Benjannet S., Wickham L., Erlich D., et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003;34:154–156. doi: 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- 126.Cohen J.C., Boerwinkle E., Mosley T.H., Jr., Hobbs H.H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 2006;354:1264–1272. doi: 10.1056/NEJMoa054013. [DOI] [PubMed] [Google Scholar]
- 127.Rao A.S., Lindholm D., Rivas M.A., Knowles J.W., Montgomery S.B., Ingelsson E. Large-scale phenome-wide association study of PCSK9 variants demonstrates protection against ischemic stroke. Circ. Genom. Precis. Med. 2018;11 doi: 10.1161/CIRCGEN.118.002162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Koren M.J., Sabatine M.S., Giugliano R.P., Langslet G., Wiviott S.D., Ruzza A., Ma Y., Hamer A.W., Wasserman S.M., Raal F.J. Long-Term Efficacy and Safety of Evolocumab in Patients With Hypercholesterolemia. J. Am. Coll. Cardiol. 2019;74:2132–2146. doi: 10.1016/j.jacc.2019.08.1024. [DOI] [PubMed] [Google Scholar]
- 129.Schwartz G.G., Steg P.G., Szarek M., Bhatt D.L., Bittner V.A., Diaz R., Edelberg J.M., Goodman S.G., Hanotin C., Harrington R.A., et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018;379:2097–2107. doi: 10.1056/NEJMoa1801174. [DOI] [PubMed] [Google Scholar]
- 130.Pérez de Isla L., Díaz-Díaz J.L., Romero M.J., Muñiz-Grijalvo O., Mediavilla J.D., Argüeso R., Sánchez Muñoz-Torrero J.F., Rubio P., Álvarez-Baños P., Ponte P., et al. Alirocumab and Coronary Atherosclerosis in Asymptomatic Patients with Familial Hypercholesterolemia: The ARCHITECT Study. Circulation. 2023;147:1436–1443. doi: 10.1161/circulationaha.122.062557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Räber L., Ueki Y., Otsuka T., Losdat S., Häner J.D., Lonborg J., Fahrni G., Iglesias J.F., van Geuns R.-J., Ondracek A.S., et al. Effect of Alirocumab Added to High-Intensity Statin Therapy on Coronary Atherosclerosis in Patients With Acute Myocardial Infarction: The PACMAN-AMI Randomized Clinical Trial. JAMA. 2022;327:1771–1781. doi: 10.1001/jama.2022.5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ding Q., Strong A., Patel K.M., Ng S.L., Gosis B.S., Regan S.N., Cowan C.A., Rader D.J., Musunuru K. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ. Res. 2014;115:488–492. doi: 10.1161/circresaha.115.304351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rothgangl T., Dennis M.K., Lin P.J.C., Oka R., Witzigmann D., Villiger L., Qi W., Hruzova M., Kissling L., Lenggenhager D., et al. In vivo adenine base editing of PCSK9 in macaques reduces LDL cholesterol levels. Nat. Biotechnol. 2021;39:949–957. doi: 10.1038/s41587-021-00933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Musunuru K., Chadwick A.C., Mizoguchi T., Garcia S.P., DeNizio J.E., Reiss C.W., Wang K., Iyer S., Dutta C., Clendaniel V., et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature. 2021;593:429–434. doi: 10.1038/s41586-021-03534-y. [DOI] [PubMed] [Google Scholar]
- 135.Chadwick A.C., Evitt N.H., Lv W., Musunuru K. Reduced blood lipid levels with in vivo CRISPR-Cas9 base editing of ANGPTL3. Circulation. 2018;137:975–977. doi: 10.1161/CIRCULATIONAHA.117.031335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Qiu M., Glass Z., Chen J., Haas M., Jin X., Zhao X., Rui X., Ye Z., Li Y., Zhang F., Xu Q. Lipid nanoparticle-mediated codelivery of Cas9 mRNA and single-guide RNA achieves liver-specific in vivo genome editing of Angptl3. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2020401118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zuo Y., Zhang C., Zhou Y., Li H., Xiao W., Herzog R.W., Xu J., Zhang J., Chen Y.E., Han R. Liver-specific in vivo base editing of Angptl3 via AAV delivery efficiently lowers blood lipid levels in mice. Cell Biosci. 2023;13:109. doi: 10.1186/s13578-023-01036-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lee R., Denizio J., Mizoguchi T., Dutta C., Clendaniel V., Garrity R., Cox N., Glass Z., Chamarthi H., Braun M., et al. An investigational in vivo base editing medicine targeting ANGPTL3, VERVE-201, achieves potent and LDLR-independent liver editing in mouse models. Eur. Heart J. 2023;44 [Google Scholar]
- 139.Kasiewicz L.N., Biswas S., Beach A., Ren H., Dutta C., Mazzola A.M., Rohde E., Chadwick A., Cheng C., Garcia S.P., et al. GalNAc-Lipid nanoparticles enable non-LDLR dependent hepatic delivery of a CRISPR base editing therapy. Nat. Commun. 2023;14:2776. doi: 10.1038/s41467-023-37465-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Horie T., Ono K. VERVE-101: a promising CRISPR-based gene editing therapy that reduces LDL-C and PCSK9 levels in HeFH patients. Eur. Heart J. Cardiovasc. Pharmacother. 2024;10:89–90. doi: 10.1093/ehjcvp/pvad103. [DOI] [PubMed] [Google Scholar]
- 141.Grinstein J.D. Bittersweet Symphony: Verve’s Pause on VERVE-101 Narrows LNP-Delivery Strategy. 2024. https://www.genengnews.com/topics/genome-editing/bittersweet-symphony-verves-pause-on-verve-101-narrows-lnp-delivery-strategy/
- 142.Coppinger C., Movahed M.R., Azemawah V., Peyton L., Gregory J., Hashemzadeh M. A Comprehensive Review of PCSK9 Inhibitors. J. Cardiovasc. Pharmacol. Therapeut. 2022;27 doi: 10.1177/10742484221100107. [DOI] [PubMed] [Google Scholar]
- 143.Karatasakis A., Danek B.A., Karacsonyi J., Rangan B.V., Roesle M.K., Knickelbine T., Miedema M.D., Khalili H., Ahmad Z., Abdullah S., et al. Effect of PCSK9 Inhibitors on Clinical Outcomes in Patients With Hypercholesterolemia: A Meta-Analysis of 35 Randomized Controlled Trials. J. Am. Heart Assoc. 2017;6 doi: 10.1161/jaha.117.006910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Eckel R.H. Lipoprotein lipase. N. Engl. J. Med. 1989;320:1060–1068. doi: 10.1056/NEJM198904203201607. [DOI] [PubMed] [Google Scholar]
- 145.Romeo S., Yin W., Kozlitina J., Pennacchio L.A., Boerwinkle E., Hobbs H.H., Cohen J.C. Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J. Clin. Invest. 2009;119:70–79. doi: 10.1172/JCI37118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Musunuru K., Pirruccello J.P., Do R., Peloso G.M., Guiducci C., Sougnez C., Garimella K.V., Fisher S., Abreu J., Barry A.J., et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N. Engl. J. Med. 2010;363:2220–2227. doi: 10.1056/NEJMoa1002926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Dewey F.E., Gusarova V., Dunbar R.L., O’Dushlaine C., Schurmann C., Gottesman O., McCarthy S., Van Hout C.V., Bruse S., Dansky H.M., et al. Genetic and pharmacologic inactivation of ANGPTL3 and cardiovascular disease. N. Engl. J. Med. 2017;377:211–221. doi: 10.1056/NEJMoa1612790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ge H., Cha J.Y., Gopal H., Harp C., Yu X., Repa J.J., Li C. Differential regulation and properties of angiopoietin-like proteins 3 and 4. J. Lipid Res. 2005;46:1484–1490. doi: 10.1194/jlr.M500005-JLR200. [DOI] [PubMed] [Google Scholar]
- 149.Raal F.J., Rosenson R.S., Reeskamp L.F., Hovingh G.K., Kastelein J.J.P., Rubba P., Ali S., Banerjee P., Chan K.C., Gipe D.A., et al. Evinacumab for Homozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020;383:711–720. doi: 10.1056/NEJMoa2004215. [DOI] [PubMed] [Google Scholar]
- 150.Raposo C.D., Canelas A.B., Barros M.T. Human Lectins, Their Carbohydrate Affinities and Where to Find Them. Biomolecules. 2021;11 doi: 10.3390/biom11020188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Morell A.G., Gregoriadis G., Scheinberg I.H., Hickman J., Ashwell G. The role of sialic acid in determining the survival of glycoproteins in the circulation. J. Biol. Chem. 1971;246:1461–1467. [PubMed] [Google Scholar]
- 152.Pricer W.E., Jr., Ashwell G. The binding of desialylated glycoproteins by plasma membranes of rat liver. J. Biol. Chem. 1971;246:4825–4833. [PubMed] [Google Scholar]
- 153.Sarkar M., Liao J., Kabat E.A., Tanabe T., Ashwell G. The binding site of rabbit hepatic lectin. J. Biol. Chem. 1979;254:3170–3174. [PubMed] [Google Scholar]
- 154.Steer C.J., Ashwell G. Studies on a mammalian hepatic binding protein specific for asialoglycoproteins. Evidence for receptor recycling in isolated rat hepatocytes. J. Biol. Chem. 1980;255:3008–3013. [PubMed] [Google Scholar]
- 155.Spiess M. The asialoglycoprotein receptor: a model for endocytic transport receptors. Biochemistry. 1990;29:10009–10018. doi: 10.1021/bi00495a001. [DOI] [PubMed] [Google Scholar]
- 156.Stockert R.J. The asialoglycoprotein receptor: relationships between structure, function, and expression. Physiol. Rev. 1995;75:591–609. doi: 10.1152/physrev.1995.75.3.591. [DOI] [PubMed] [Google Scholar]
- 157.Pricer W.E., Jr., Hudgin R.L., Ashwell G., Stockert R.J., Morell A.G. A membrane receptor protein for asialoglycoproteins. Methods Enzymol. 1974;34:688–691. doi: 10.1016/s0076-6879(74)34090-6. [DOI] [PubMed] [Google Scholar]
- 158.Roggenbuck D., Mytilinaiou M.G., Lapin S.V., Reinhold D., Conrad K. Asialoglycoprotein receptor (ASGPR): a peculiar target of liver-specific autoimmunity. Auto. Immun. Highlights. 2012;3:119–125. doi: 10.1007/s13317-012-0041-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.D'Souza A.A., Devarajan P.V. Asialoglycoprotein receptor mediated hepatocyte targeting - strategies and applications. J. Contr. Release. 2015;203:126–139. doi: 10.1016/j.jconrel.2015.02.022. [DOI] [PubMed] [Google Scholar]
- 160.Nioi P., Sigurdsson A., Thorleifsson G., Helgason H., Agustsdottir A.B., Norddahl G.L., Helgadottir A., Magnusdottir A., Jonasdottir A., Gretarsdottir S., et al. Variant ASGR1 associated with a reduced risk of coronary artery disease. N. Engl. J. Med. 2016;374:2131–2141. doi: 10.1056/NEJMoa1508419. [DOI] [PubMed] [Google Scholar]
- 161.Xu Y., Tao J., Yu X., Wu Y., Chen Y., You K., Zhang J., Getachew A., Pan T., Zhuang Y., et al. Hypomorphic ASGR1 modulates lipid homeostasis via INSIG1-mediated SREBP signaling suppression. JCI Insight. 2021;6 doi: 10.1172/jci.insight.147038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Xie B., Shi X., Li Y., Xia B., Zhou J., Du M., Xing X., Bai L., Liu E., Alvarez F., et al. Deficiency of ASGR1 in pigs recapitulates reduced risk factor for cardiovascular disease in humans. PLoS Genet. 2021;17 doi: 10.1371/journal.pgen.1009891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Xiao X., Song B.-L. SREBP: a novel therapeutic target. Acta Biochim. Biophys. Sin. 2013;45:2–10. doi: 10.1093/abbs/gms112. [DOI] [PubMed] [Google Scholar]
- 164.Zhu Y., Lin X., Zhou X., Prochownik E.V., Wang F., Li Y. Posttranslational control of lipogenesis in the tumor microenvironment. J. Hematol. Oncol. 2022;15:120. doi: 10.1186/s13045-022-01340-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wang J.Q., Li L.L., Hu A., Deng G., Wei J., Li Y.F., Liu Y.B., Lu X.Y., Qiu Z.-P., Shi X.J., et al. Inhibition of ASGR1 decreases lipid levels by promoting cholesterol excretion. Nature. 2022;608:413–420. doi: 10.1038/s41586-022-05006-3. [DOI] [PubMed] [Google Scholar]
- 166.Braun C.J., Boztug K., Paruzynski A., Witzel M., Schwarzer A., Rothe M., Modlich U., Beier R., Göhring G., Steinemann D., et al. Gene Therapy for Wiskott-Aldrich Syndrome—Long-Term Efficacy and Genotoxicity. Sci. Transl. Med. 2014;6:227ra33. doi: 10.1126/scitranslmed.3007280. [DOI] [PubMed] [Google Scholar]
- 167.Hacein-Bey-Abina S., Von Kalle C., Schmidt M., McCormack M.P., Wulffraat N., Leboulch P., Lim A., Osborne C.S., Pawliuk R., Morillon E., et al. LMO2-Associated Clonal T Cell Proliferation in Two Patients after Gene Therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 168.Manno C.S., Pierce G.F., Arruda V.R., Glader B., Ragni M., Rasko J.J., Ozelo M.C., Hoots K., Blatt P., Konkle B., et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 169.Mingozzi F., Meulenberg J.J., Hui D.J., Basner-Tschakarjan E., Hasbrouck N.C., Edmonson S.A., Hutnick N.A., Betts M.R., Kastelein J.J., Stroes E.S., High K.A. AAV-1–mediated gene transfer to skeletal muscle in humans results in dose-dependent activation of capsid-specific T cells. Blood. 2009;114:2077–2086. doi: 10.1182/blood-2008-07-167510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Suoranta T., Laham-Karam N., Ylä-Herttuala S. Strategies to improve safety profile of AAV vectors. Front. Mol. Med. 2022;2 doi: 10.3389/fmmed.2022.1054069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Yin H., Kanasty R.L., Eltoukhy A.A., Vegas A.J., Dorkin J.R., Anderson D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014;15:541–555. doi: 10.1038/nrg3763. [DOI] [PubMed] [Google Scholar]
- 172.Wang C., Pan C., Yong H., Wang F., Bo T., Zhao Y., Ma B., He W., Li M. Emerging non-viral vectors for gene delivery. J. Nanobiotechnol. 2023;21:272. doi: 10.1186/s12951-023-02044-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Jackson A.L., Linsley P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010;9:57–67. doi: 10.1038/nrd3010. [DOI] [PubMed] [Google Scholar]
- 174.Crooke S.T. Molecular Mechanisms of Antisense Oligonucleotides. Nucleic Acid Therapeut. 2017;27:70–77. doi: 10.1089/nat.2016.0656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Judge A.D., Sood V., Shaw J.R., Fang D., McClintock K., MacLachlan I. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat. Biotechnol. 2005;23:457–462. doi: 10.1038/nbt1081. [DOI] [PubMed] [Google Scholar]
- 176.Han G., Noh D., Lee H., Lee S., Kim S., Yoon H.Y., Lee S.H. Advances in mRNA therapeutics for cancer immunotherapy: From modification to delivery. Adv. Drug Deliv. Rev. 2023;199 doi: 10.1016/j.addr.2023.114973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Enache O.M., Rendo V., Abdusamad M., Lam D., Davison D., Pal S., Currimjee N., Hess J., Pantel S., Nag A., et al. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat. Genet. 2020;52:662–668. doi: 10.1038/s41588-020-0623-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Ihry R.J., Worringer K.A., Salick M.R., Frias E., Ho D., Theriault K., Kommineni S., Chen J., Sondey M., Ye C., et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018;24:939–946. doi: 10.1038/s41591-018-0050-6. [DOI] [PubMed] [Google Scholar]
- 180.Grünewald J., Zhou R., Garcia S.P., Iyer S., Lareau C.A., Aryee M.J., Joung J.K. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature. 2019;569:433–437. doi: 10.1038/s41586-019-1161-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Zhou C., Sun Y., Yan R., Liu Y., Zuo E., Gu C., Han L., Wei Y., Hu X., Zeng R., et al. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature. 2019;571:275–278. doi: 10.1038/s41586-019-1314-0. [DOI] [PubMed] [Google Scholar]