


Abstract
The proliferation of genome sequence data has led to the development of a number of tools and strategies that facilitate computational analysis. These methods include the identification of motif patterns, membership of the query sequences in family databases, metabolic pathway involvement and gene proximity. We re-examined the completely sequenced genome of Thermotoga maritima by employing the combined use of the above methods. By analyzing all 1877 proteins encoded in this genome, we identified 193 cases of conflicting annotations (10%), of which 164 are new function predictions and 29 are amendments of previously proposed assignments. These results suggest that the combined use of existing computational tools can resolve inconclusive sequence similarities and significantly improve the prediction of protein function from genome sequence.
INTRODUCTION
Since the seminal publication of the genome of Haemophilus influenzae (1), more than 20 complete genomes have been published (http://igweb.integratedgenomics.com/GOLD/) (2). While the quantity of data have dramatically increased, their interpretation remains a major bottleneck (3). Interpretation can be defined as genome annotation, including gene definition and function prediction, which leads to cellular reconstructions (4), and ultimately to general functional overviews of a given species. Therefore, the foundation of understanding the biology of any genome lies in gene annotation.
Following the original publications of a number of complete genomes, various research groups have reported significant improvements on annotation quality. Some examples include the genomes of H.influenzae (5), Mycoplasma genitalium (6) and Methanococcus jannaschii (7,8). These improvements consist of mostly false negative cases, and are usually accompanied by a small number of false positive descriptions, which reduce the overall accuracy of functional assignment. Cases of conflicting annotations are not easily reproducible (9–11) and further complicate the comparison of annotation quality (9,12).
This situation sharply contrasts with the experience accumulated over the past few years regarding strategies, methods and technology that improve genome sequence analysis and interpretation (extensively reviewed in 13). Function prediction has traditionally been based solely on sequence similarity to proteins of known function. The two most widely used tools that detect sequence similarity (i.e. BLAST and FASTA) usually provide functional predictions for 50–70% of genes for any sequenced genome (depending on the phylogenetic position of the organism), without compromising accuracy.
An additional 10–20% of genes have no homologs in the public databases while the remaining 10–30% have a marginal sequence matches making function assignment non-trivial. This gray area is frequently referred to as the ‘twilight zone’ of sequence similarity and it is here that most cases of conflicting annotations do occur.
Recently, a number of additional tools have been developed to assist sequence analysis, increasing the accuracy of function predictions in general. The advent of different algorithms for protein family clusters, profiles, fingerprints and Hidden Markov Models (HMMs) has led to the development of a number of pattern databases such as PROSITE (14), Pfam (15), PRINTS (16), ProDom (17), COGs (18) and the BioDictionary (19). Although most of these tools may have greater sensitivity in identifying remote similarities and critically conserved regions than either BLAST or FASTA, they also depend on sequence similarity.
Yet, over the last few years, we have witnessed a number of alternative approaches to identifying likely roles for gene products that depend mainly on genome context rather than on sequence similarity (20–22). Some of these tools have already been implemented in systems like GeneQuiz (http://columba.ebi.ac.uk:8765/ext-genequiz/) (23) and WIT (http://wit.integratedgenomics.com/IGwit) (24). This integration has been proven invaluable for the manual analysis of genome sequences, allowing the discovery of gene function in the twilight zone of sequence similarity.
Herein, we focus on the combined use of available tools to predict functions, and we re-evaluate the recently published genome of Thermotoga maritima (25). We report the identification of 16% additional functions over the original publication, the majority of which are corroborated by more than one method.
MATERIALS AND METHODS
We have compared the entire set of function predictions for the 1877 open reading frames (ORFs) of the completely sequenced genome of T.maritima (25) with the corresponding predictions available in WIT (24). After discarding all cases where the two independent analyses agreed (985 cases), we analyzed in detail cases of apparent disagreement (29 cases), as well as the remaining hypothetical proteins (863 cases).
In total, we analyzed 892 T.maritima ORFs, using a variety of function prediction tools. Our analysis included: (i) up to five iterations of the PSI-BLAST algorithm (http://www.ncbi.nlm.nih.gov/blast/psiblast.cgi) (26); (ii) presence of PROSITE patterns (http://www.expasy.ch/prosite/) (14); (iii) membership of the query sequences in the family databases Pfam (http://pfam.wustl.edu/) (15) and COGs (http://www.ncbi.nlm.nih.gov/COG/) (18); (iv) metabolic pathway involvement (http://wit.mcs.anl.gov/) (27–29); and (v) chromosomal neighborhood of the corresponding genes (http://igweb.integratedgenomics.com/IGwit/CGI/operons.cgi) (20). In addition, the continuous annotation of released genomes and extensive literature support contributed towards a more reliable definition of functional roles. The domain organization of a number of T.maritima ORFs was performed with PRODOM (http://www.toulouse.inra.fr/prodom.html) (17). All BLAST searches were performed against the non-redundant database of NCBI until August 1999 to ensure the similar composition of the target database with the one of the original analysis.
Our results are available on the WWW at http://wit.integratedgenomics.com/TM.html with pointers to the combination of methods used, ensuring reproducibility.
RESULTS AND DISCUSSION
Our analysis demonstrates that there is 90% agreement in function assignments with the original report of the T.maritima genome (21). In total, we have identified 193 cases of conflicting annotations (10.3% of the entire genome) (Fig. 1), of which 164 are new function identifications (table 1, http://wit.integratedgenomics.com/TM.html) and the remaining 29 cases are amendments to previously proposed functions (table 2, http://wit.integratedgenomics.com/TM.html). The number of function assignments thus increases from 1014 (54%) to 1178 (63%), a 16% increase over the original publication (Fig. 1). The annotation pattern of the T.maritima genome is therefore similar to that of previous genome publications (5–8).
Figure 1.
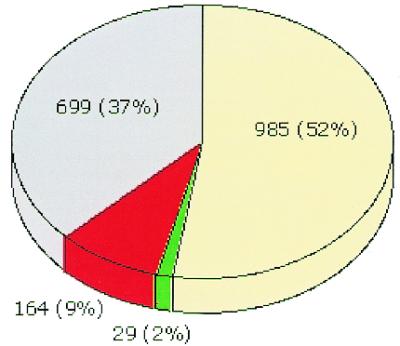
Representation of the annotation quality for the genome of T.maritima, as a pie chart. The cases of agreement are shown in pale yellow (known function) or gray (unknown function). The amended cases are shown in green (we suggest change of function) or red (we identify new function).
The combined use of traditional and novel tools and strategies in sequence analysis allows researchers to significantly increase the efficiency of annotations. In particular, virtually all cases have either readily detectable sequence similarity to proteins of known function, or marginal similarity (based on BLAST) that is further confirmed by other methods. The fact that the majority of our predictions are supported by BLAST merely suggests that this tool may be sufficient. To the contrary, it is the combination of methods, when available, that either support an unclear similarity, or distinguish between equally probable alternative predictions. At the time of our analysis, only three cases were detected exclusively on the basis of an experimental observation.
A large number of the functions we report here are identified for the first time in T.maritima, and greatly assists towards a more reliable metabolic reconstruction for this species. For instance: TM0385 is the ribonucleoside-triphosphate reductase (EC 1.17.4.2), an important enzyme in nucleotide biosynthesis; TM0474 is the nicotinate phosphoribosyltransferase (EC 2.4.2.11), which catalyzes the first step in the Preiss–Handler pathway leading to the synthesis of NAD; TM0790 is the phosphomethylpyrimidine kinase (thiD) (EC 2.7.4.7) in thiamine biosynthesis; and TM1621 is the glycerophosphoryl diester phosphodiesterase (EC 3.1.4.46), which hydrolyzes the deacetylated phospholipids to glycerol 3-phosphate and the corresponding alcohols. Although all the above mentioned assignments can be derived by careful inspection of similarity searches using BLAST, the additional supporting evidence from other resources enhances our ability to detect function more efficiently and to partially automate the process.
When the family of the query sequence contains multiple function descriptions, the prediction may be assisted by further constraints such as pathway or operon involvement. One such case in T.maritima is represented by a gene cluster for the enzymes involved in rhamnulose metabolism. This potential operon includes TM1073 [predicted here to be a rhamnulokinase (EC 2.7.1.5)], TM1072 [rhamnulose-1-phosphate aldolase (EC 4.1.2.19)] and TM1071 [xylose isomerase (EC 5.3.1.5)]. The latter is known to have wide substrate specificity and accepts rhamnulose as a substrate (30). All the enzymes of this gene cluster are involved in rhamnulose metabolism. Of interest is the hypothetical protein (Fig. 2) that belongs to the same gene cluster with the other three enzymes of the rhamnulose metabolism, suggesting potential functional coupling to that pathway. Functional coupling between these genes is preserved in a number of species as shown in Figure 2. These data provide additional confirmation for functional assignments for the participating genes.
Figure 2.

Inferred functional coupling based on the clustering of genes involved in rhamnulose metabolism in a number of genomes. The colors label genes occurring in corresponding clusters in each species, with distinct colors indicating distinct clusters (for example RTY00493 is not part of the cluster formed by the other genes of rhamnose metabolsim). Gene identifiers are internal WIT numbers that are hyperlinked to the data. Dashes represent enzymes that are either absent from the genome or not part of the particular cluster shown. For T.maritima, RTM01813 corresponds to TM1073 and RTM1814 to TM1072; TM1071 is not shown (predicted as a sugar isomerase replacing the missing l-rhamnose isomerase present in other species). The hypothetical protein also missing from T.maritima but present in all other species may also be involved in rhamnulose metabolism. Species abbreviations: BS, Bacillus subtilis; EF, Enterococcus faecalis; EC, Escherichia coli; TY, Salmonella typhi; YP, Yersinia pestis; TM, T.maritima.
Another potential source of inconsistency lies in large protein families where some of the members are not assigned with any functional descriptions at all. For example, the ORF TM1404 is described as an ABC transporter, while its paralogs (TM0387, TM0388, TM0543, TM1100, TM1303, TM1304, TM1306 and TM1326) are characterized as ‘conserved hypothetical proteins’. Of particular interest is the fourth largest protein family in T.maritima, which includes the hypothetical proteins TM0037, TM0739, TM1145, TM1147, TM1467, TM1678, TM1682 and TM1699, the response regulators TM0186 and TM0842, and the ABC transporters TM0593 and TM1170 (Fig. 3). The above proteins are related through the process of distinct fusion events between common and unique components (15,16). As shown in Figure 3, most of them are related through a common domain (HD-GYP domain) (31) in the C-terminus of the proteins. This domain has been predicted as a signal transduction component (31) and most of these proteins may be members of the two-component signal transduction superfamily. In the case of TM0186 the HD-GYP domain is fused to the chemotaxis protein CheY, while in TM0842, the CheY domain is fused to another domain of unknown function.
Figure 3.

Schematic representation of the domain organization for a number of T.maritima ORFs. Domain analysis was performed in WIT using the PRODOM database (17). For details, please see text.
Another example where the presence of distinct functional domains in one protein was not taken into account is TM0343 which was reported as chorismate mutase, while it is actually a composite protein conserved across a number of species and comprised of phospho-2-dehydro-3-deoxyheptonate aldolase (EC 4.1.2.15) and chorismate mutase (EC 5.4.99.5), both catalyzing different steps in the aromatic amino acids biosynthesis. Thus, even though the existence of such fusion proteins can greatly assist in the functional characterization of their components (or vice versa) (21,22), they can also become a major source of error propagation if their composite nature is not properly analyzed.
Where no precise function can be confidently assigned to the entire protein, the detection of additional features, such as sequence motifs (15,16,19) or functional domains, is particularly important. These data may provide a more general characterization of the proteins and they can direct further experimental analysis. The original T.maritima analysis (25) reported the presence of only a few of these motifs (the GGDEF motif, for example) in a number of hypothetical proteins. We have identified a large number of proteins containing short domains such as zinc fingers, TPR repeats, CBS domains and various transmembrane domains that were omitted (data available on WIT). Examples of conserved pattern detection in PROSITE is the ORF TM0462, a ribosomal large subunit pseudouridine synthase D (the other large subunit C being TM0940), and the ORF TM0774, a L7AE ribosomal protein. Other examples of similarity detection using family models in Pfam is the ORF TM1573, a protein containing the bacterial PQQ domain, and the ORF TM1741, an RNA methyltransferase. Examples of detection using orthologous clusters in COGs are the ORFs TM0097, which is predicted to be a nucleotidyltransferase, and TM0916, a periplasmic serine protease (all the above cases are listed in table 1, http://wit.integratedgenomics.com/TM.html).
We also report cases where there is a direct biochemical characterization or indirect computational analysis in the recent literature for T.maritima genes or their homologs in other species. Examples of the former include the ORFs TM0473 (pyridoxine biosynthesis protein), TM0511 (uracil–DNA glycosylase) and TM1394 (Hsp33) (listed in table 1, http://wit.integratedgenomics.com/TM.html). Examples of the latter include the ORFs TM0911 and TM1440; these were specifically assigned with the function of the α subunit of eIF-2B, while it has been previously demonstrated that they clearly form a different subunit (32).
Finally, of importance remains the observation that a number of essential genes escape identification (33,34). A notable such example in T.maritima is the gene for ribosomal protein L34 (located at 1 476 262–1 476 394 bp positions of the T.maritima genome). Similarly, the ribosomal genes S20P and L30P were not detected in the genome of Treponema pallidum (35) (N.C.Kyrpides, unpublished results).
We conclude that with the efficient use of a variety of biological sequence analysis tools and resources, genome annotation can be significantly improved. At the same time, it should be pointed out that a conservative approach to genome sequence annotation (3) resulting in underprediction, as is apparently the case for T.maritima (25), is preferable to overpredicting functions with low accuracy, if these annotations are not actively curated before entering public databases such as GenBank (36) and EMBL (37). The combination of various powerful bioinformatics methods should pave the way for more automatic procedures that generate reliable and reproducible genome sequence annotations.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Natalia Maltsev (Argonne National Laboratory), Uy Ear and Wei Yang (Integrated Genomics Inc.), for helpful suggestions and comments. This work was supported in part by Integrated Genomics Inc., the European Molecular Biology Laboratory and the TMR Programme (grant no. ERBFMRXCT960019) from the European Commission DGXII (Science, Research and Development).
REFERENCES
- 1.Fleischmann R.D., Adams,M.D., White,O., Clayton,R.A., Kirkness,E.F., Kerlavage,A.R., Bult,C.J., Tomb,J.-F., Dougherty,B.A., Merrick,J.M. et al. (1995) Science, 269, 496–512. [DOI] [PubMed] [Google Scholar]
- 2.Kyrpides N.C. (1999) Bioinformatics, 15, 773–774. [DOI] [PubMed] [Google Scholar]
- 3.Venter J.C., Smith,H.O. and Fraser,C.M. (1999) ASM News, 65, 322–327. [Google Scholar]
- 4.Selkov E., Maltsev,N., Olsen,G.J., Overbeek,R. and Whitman,W.B. (1997) Gene, 197, 11–26. [DOI] [PubMed] [Google Scholar]
- 5.Casari G., Andrade,M.A., Bork,P., Boyle,J., Daruvar,A., Ouzounis,C., Schneider,R., Tamames,J., Valencia,A. and Sander,C. (1995) Nature, 376, 647–648. [DOI] [PubMed] [Google Scholar]
- 6.Ouzounis C., Casari,G., Valencia,A. and Sander,C. (1996) Mol. Microbiol., 20, 897–899. [DOI] [PubMed] [Google Scholar]
- 7.Koonin E.V., Mushegian,A.R., Galperin,M.Y. and Walker,D.R. (1997) Mol. Microbiol., 25, 619–637. [DOI] [PubMed] [Google Scholar]
- 8.Kyrpides N.C., Olsen,G.J., Klenk,H.-P., White,O., Woese,C.R. (1996) Microb. Compar. Genomics, 1, 329–338. [PubMed] [Google Scholar]
- 9.Brenner S.E. (1999) Trends Genet., 15, 132–133. [DOI] [PubMed] [Google Scholar]
- 10.Kyrpides N.C. and Ouzounis,C.A. (1999) Mol. Microbiol., 32, 886–887. [DOI] [PubMed] [Google Scholar]
- 11.Tsoka S., Promponas,V. and Ouzounis,C.A. (1999) FEBS Lett., 451, 354–355. [DOI] [PubMed] [Google Scholar]
- 12.Andrade M., Casari,G., de Daruvar,A., Sander,C., Schneider,R., Tamames,J., Valencia,A. and Ouzounis,C. (1997) Comput. Appl. Biosci., 13, 481–483. [DOI] [PubMed] [Google Scholar]
- 13.Bork P., Dandekar,T., Diaz-Lazcoz,Y., Eisenhaber,F., Huynen,M. and Yuan,Y. (1998) J. Mol. Biol., 283, 707–725. [DOI] [PubMed] [Google Scholar]
- 14.Hofmann K., Bucher,P., Falquet,L. and Bairoch,A. (1999) Nucleic Acids Res., 27, 15–19. [Google Scholar]
- 15.Bateman A., Birney,E., Durbin,R., Eddy,S.R., Howe,K.L., Erik,L.L. and Sonnhammer,E.L.L. (2000) Nucleic Acids Res., 28, 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Atwood T.K., Flower,D.R., Lewis,A.P., Mabey,J.E., Morgan,S.R., Scordis,P., Selley,J.N. and Wright,W. (1999) Nucleic Acids Res., 27, 220–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Corpet F., Servant,F., Gouzy,J. and Kahn,D. (2000) Nucleic Acids Res., 28, 267–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tatusov R.L., Galperin,M.Y., Natale,D.A. and Koonin,E.V. (2000) Nucleic Acids Res., 28, 33–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rigoutsos I., Floratos,A., Ouzounis,C., Gao,Y. and Parida,L. (1999) Proteins, 37, 264–277. [DOI] [PubMed] [Google Scholar]
- 20.Overbeek R., Fonstein,M., D’Souza,M., Pusch,G.D. and Maltsev,N. (1999) Proc. Natl Acad. Sci. USA, 96, 2896–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marcotte E.M., Pellegrini,M., Ng,H.L., Rice,D.W., Yeates,T.O. and Eisenberg,D. (1999) Science, 285, 751–753. [DOI] [PubMed] [Google Scholar]
- 22.Enright A.J., Iliopoulos,I., Kyrpides,N.C., and Ouzounis,C.A. (1999) Nature, 402, 86–90. [DOI] [PubMed] [Google Scholar]
- 23.Andrade M.A., Brown,N.P., Leroy,C., Hoersch,S., de Daruvar,A., Reich,C., Franchini,A., Tamames,J., Valencia,A., Ouzounis,C. and Sander,C. (1999) Bioinformatics, 15, 391–412. [DOI] [PubMed]
- 24.Overbeek R., Larsen,N., Pusch,G.D., D’Souza,M., Selkov,E.,Jr, Kyrpides,N., Fonstein,M., Maltsev,N. and Selkov,E. (1999) Nucleic Acids Res., 28, 123–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nelson K.E., Clayton,R.A., Gill,S.R., Gwinn,M.L., Dodson,R.J., Haft,D.H., Hickey,E.K., Peterson,J.D., Nelson,W.C., Ketchum,K.A. et al. (1999) Nature, 399, 323–329. [DOI] [PubMed] [Google Scholar]
- 26.Altschul S.F., Madden,T.L., Schäffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bono H., Ogata,H., Goto,H. and Kanehisa,M. (1998) Genome Res., 8, 203–210. [DOI] [PubMed] [Google Scholar]
- 28.Selkov E. Jr, Grechkin,Y., Mikhailova,N. and Selkov,E. (1998) Nucleic Acids Res., 26, 43–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karp P.D., Krummenacker,M., Paley,S. and Wagg,J. (1999) Trends Biotechnol., 17, 275–281. [DOI] [PubMed] [Google Scholar]
- 30.Collyer C.A., Goldberg,J.D., Viehmann,H., Blow,D.M., Ramsden,N.G., Fleet,G.W., Montgomery,F.J. and Grice,P. (1992) Biochemistry, 31, 12211–12218. [DOI] [PubMed] [Google Scholar]
- 31.Galperin M.Y., Natale,D.A., Aravind,L. and Koonin,E.V. (1999) J. Mol. Microbiol. Biotechnol., 1, 303–305. [PMC free article] [PubMed] [Google Scholar]
- 32.Kyrpides N. and Woese,C.R. (1998) Proc. Natl Acad. Sci. USA, 95, 3726–3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raghavan S. and Ouzounis,C.A. (1999) Nucleic Acids Res., 27, 4405–4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Delcher A.L., Harmon,D., Kasif,S., White,O. and Salzberg,S.L. (1999) Nucleic Acids Res., 27, 4636–4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fraser C.M., Norris,S.J., Weinstock,G.M., White,O., Sutton,G.G., Dodson,R., Gwinn,M., Hickey,E.K., Clayton,R., Ketchum,K.A. et al. (1998) Science, 281, 375–388. [DOI] [PubMed] [Google Scholar]
- 36.Benson D.A., Karsch-Mizrachi,I., Lipman,D.J., Ostell,J., Rapp,B.A. and Wheeler,D.L. (2000) Nucleic Acids Res., 28, 15–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baker W., van den Broek,A., Camon,E., Hingamp,P., Sterk,P., Stoesser,G. and Tuli,M.A. (2000) Nucleic Acids Res., 28, 19–23. [DOI] [PMC free article] [PubMed] [Google Scholar]