

Significance
Chromosome arm 8q is the most frequently gained chromosome arm in aneuploid prostate cancer and is associated with worse prognosis. Using population science and laboratory approaches, we identified increased RAD21 expression in prostate cancer as a key adverse prognostic factor related to 8q gains that enables tumors to sustain proliferation despite oncogenic stress and DNA damage from oncogenic mutations. These data pinpoint one mechanism through which aneuploidy promotes tumor progression and suggests a potential therapeutic target for aneuploid prostate cancer.
Keywords: prostate cancer, RAD21, DNA damage, organoid, clinical outcomes
Abstract
Higher levels of aneuploidy, characterized by imbalanced chromosome numbers, are associated with lethal progression in prostate cancer. However, how aneuploidy contributes to prostate cancer aggressiveness remains poorly understood. In this study, we assessed in patients which genes on chromosome 8q, one of the most frequently gained chromosome arms in prostate tumors, were most strongly associated with long-term risk of cancer progression to metastases and death from prostate cancer (lethal disease) in 403 patients and found the strongest candidate was cohesin subunit gene, RAD21, with an odds ratio of 3.7 (95% CI 1.8, 7.6) comparing the highest vs. lowest tertiles of mRNA expression and adjusting for overall aneuploidy burden and Gleason score, both strong prognostic factors in primary prostate cancer. Studying prostate cancer driven by the TMPRSS2-ERG oncogenic fusion, found in about half of all prostate tumors, we found that increased RAD21 alleviated toxic oncogenic stress and DNA damage caused by oncogene expression. Data from both organoids and patients indicate that increased RAD21 thereby enables aggressive tumors to sustain tumor proliferation, and more broadly suggests one path through which tumors benefit from aneuploidy.
Aneuploidy, characterized by chromosomal gains and losses that cause an imbalanced genome, is a hallmark of cancer (1, 2). Prostate cancer has a relatively lower average burden of aneuploidy compared to many other cancer types (3), but wide variation in aneuploidy burden is still found in prostate cancers from different patients (4). Chromosomal gains and losses are strongly associated with progression to metastases and lethal disease (4), yet how aneuploidy or aneuploidy-associated mechanisms promote prostate cancer aggressiveness is unknown.
Chromosome arm 8q (chr8q) is the most commonly gained chromosome arm in aneuploid prostate cancer (3, 4), and is a strong adverse prognostic factor in diverse cohorts of patients with prostate cancer (4–7). Intriguingly, numerous germline genetic risk variants for prostate cancer are also located on chr8q (8). The mechanism(s) through which chr8q gains, or other aneuploidies, affect prostate cancer tumorigenesis and progression, and which, if any, individual genes are responsible, is incompletely understood. Gene-dosage change caused by copy number alteration has been implicated in driving specific chromosomal gains and losses in cancers by conferring certain proliferative advantages (9, 10), including losses of tumor suppressor genes and gains of oncogenes (3, 11). Thus, advantage resulting from such copy number alterations is one potential explanation for the association between greater aneuploidy and lethal prostate cancer.
Here, we investigate what drives chr8q gains and which genes might affect prostate cancer development and progression. To consider possibilities in addition to an obvious candidate, the MYC oncogene located on chr8q, we undertook two complementary approaches: in patients, we systematically ranked associations of per-gene mRNA expression levels of genes on chr8q and lethal prostate cancer in prospective epidemiology studies with long-term follow-up that provides lethal outcomes. In parallel, we used experimental models and prostate organoids for mechanistic studies. Both approaches converged on the cohesin subunit gene, RAD21 as a potential benefit of chr8q gain. RAD21 plays a role in a wide array of cellular functions including DNA replication/repair, mitosis, apoptosis, and transcription regulation (12), and is frequently amplified and modified in many cancers including prostate cancer (1, 13–15). While few studies have investigated gain of RAD21 in prostate cancer, we recently implicated RAD21 in driving chr8q gain in pediatric Ewing sarcoma (16). Strikingly, both Ewing sarcoma and prostate cancer can be driven by gene fusions involving structurally and functionally similar oncogenes from the ETS (E26 transformation-specific) family transcription factors, such as EWS-FLI1 in Ewing sarcoma and TMPRSS2-ERG in prostate cancer (17–19). To address whether increased RAD21 promotes prostate cancer in the same manner as it does in Ewing sarcoma, we utilized a primary mouse organoid model enabling an inducible TMPRSS2-ERG fusion oncogene (hereafter, referred T-ERG) which approximately half of all prostate cancers harbor. We investigated cellular consequences following the induction of T-ERG in these organoids. Through these organoids, we elucidated the role of increased RAD21 expression in mitigating toxic oncogenic stress and reducing DNA damage, which may represent a fundamental mechanism for promoting prostate cancer development. Gain of RAD21, in conjunction with other relevant chromosome 8q genes, acts synergistically to promote the aggressiveness of prostate cancer.
Results
A Systematic Approach in Patients Identifies Top Chromosome Arm 8q Genes Related to Lethal Progression in Prostate cancer.
To systematically prioritize genes that underlie the relation between increased chr8q gene expression and lethal prostate cancer, we assessed whole-transcriptome profiling (mRNA) data from 403 patients with prostate cancer who were participants of the prospective Health Professionals Follow-up Study (HPFS) and Physicians’ Health Study (PHS) cohorts and had up to 32 y of clinical follow-up (Table 1). Patients were selected for this study if they either developed lethal disease (metastases/death from prostate cancer, n = 120) or had nonlethal disease (without metastases >8 y after diagnosis, n = 283), overrepresenting the two extremes of prostate cancer prognosis and thereby increasing precision for quantifying biological differences between tumors. Among all 400 chr8q genes profiled, seven had unadjusted odds ratios (OR) of >2 for lethal prostate cancer per one SD higher gene expression (Fig. 1): YWHAZ, RAD21, MTDH, UBR5, SQLE, POLR2K, and PABPC1. MYC DNA copy number alterations have been shown to be prognostic in other studies (20, 21), unlike MYC mRNA expression in our present study population, as previously reported (22) (Fig. 1). These data suggest that multiple genes may be strongly associated with lethality, and thus one or more of these genes might, in principle, be responsible for driving chr8q gains in prostate cancer.
Table 1.
Characteristics of men with primary prostate cancer from the Health Professionals Follow-up Study and Physicians’ Health Study (n = 403, diagnosed 1982 to 2005), by RAD21 tumor tissue expression*
| RAD21 mRNA | Tertile 1 (lowest) | Tertile 2 | Tertile 3 (highest) |
|---|---|---|---|
| N | 135 | 134 | 134 |
| Age at diagnosis [years] | 65.6 (61.6, 69.0) | 65.4 (61.6, 69.0) | 66.0 (61.0, 70.0) |
| White race | 132 (98%) | 126 (94%) | 128 (96%) |
| Gleason score | |||
| <7 | 26 (19%) | 16 (12%) | 17 (13%) |
| 3 + 4 | 59 (44%) | 51 (38%) | 29 (22%) |
| 4 + 3 | 28 (21%) | 38 (28%) | 36 (27%) |
| 8 | 11 (8%) | 6 (4%) | 23 (17%) |
| 9 to 10 | 11 (8%) | 23 (17%) | 29 (22%) |
| Predicted altered chromosome arms | 1.0 (0.0, 3.0) | 2.0 (0.0, 4.0) | 3.5 (1.2, 6.0) |
| Predicted altered chromosome arms | |||
| 0 | 44 (33%) | 35 (26%) | 16 (12%) |
| 1 to 2 | 48 (36%) | 38 (28%) | 32 (24%) |
| 3 to 4 | 24 (18%) | 36 (27%) | 36 (27%) |
| 4+ | 19 (14%) | 25 (19%) | 50 (37%) |
| ERG positive | 69 (53%) | 62 (53%) | 51 (42%) |
| Unknown | 5 | 17 | 13 |
| Ki-67 > 1% positive nuclei | 6 (6%) | 13 (12%) | 30 (29%) |
| Unknown | 29 | 30 | 31 |
| PTEN loss | 14 (17%) | 23 (26%) | 25 (29%) |
| Unknown | 54 | 45 | 47 |
| p53 positive | 4 (5%) | 2 (2%) | 8 (10%) |
| Unknown | 55 | 46 | 55 |
| BRCA1 positive | 8 (7%) | 11 (10%) | 22 (20%) |
| Unknown | 24 | 24 | 24 |
*Values are median (quartile 1, quartile 3) or count (percent).
Fig. 1.

mRNA expression analysis identifies chr8q genes associated with lethal prostate cancer. Shown are all chr8q genes (n = 400, normalized to units of SD for comparability) and odds ratios for lethal disease (metastases/death from prostate cancer), compared to nonlethal disease (no metastases for >8 y after diagnosis) in a whole-transcriptome profiling study among men with prostate cancer from the Health Professionals Follow-up Study and Physicians’ Health Study (n = 403, 1982 to 2019). Genes are sorted by odds ratio, and genes with odds ratio >2 as well as MYC are highlighted. Bars and estimates in parentheses are 95% CI.
Increased RAD21 Tumor Expression Is Strongly Prognostic for Lethal Prostate Cancer.
RAD21, located on cytoband chr8q24, was a surprising top-ranked gene, given that its role in promoting prostate cancer has received little attention thus far. Although correlated with the six other top-ranked genes, RAD21 tumor expression was not perfectly predicted by mRNA levels of the other genes, with Pearson correlations with the other genes ranging from r = 0.46 to r = 0.62 (SI Appendix, Fig. S1). Tumors with higher RAD21 expression also tended to have other features of aggressiveness, including higher Gleason score, greater aneuploidy burden, and other biomarkers, compared to tumors with lower expression (Table 1).
When ranking genes according to their average difference in expression between tumor and tumor-adjacent histologically normal prostate tissue, RAD21 expression ranked 98th, far from the top among the 400 chr8q genes (SI Appendix, Table S1). MYC, in contrast, had the 5th greatest expression difference between tumor and adjacent normal tissue of all chr8q genes, but neither its mRNA levels (Fig. 1) nor protein expression by immunohistochemistry were prognostic for lethality (22). Nevertheless, we established that RAD21 tumor expression and the difference between tumor and normal tissue was strongly associated with lethal prostate cancer (Table 2). As expected, RAD21 expression in adjacent normal tissue was not prognostic (Table 2). In sum, RAD21 was not noticeably overexpressed in tumors compared to their adjacent normal tissue in all tumors, but when overexpression was present, it was associated with a much worse prognosis.
Table 2.
RAD21 mRNA expression in tumor, tumor-adjacent histologically normal tissue, and the between-compartment difference in relation to lethal disease from the Health Professionals Follow-up Study and Physicians’ Health Study (follow-up 1982 to 2019, n = 403)
| mRNA | Tertile 1 (lowest) | Tertile 2 | Tertile 3 (highest) | Trend* |
|---|---|---|---|---|
| RAD21 in tumor tissue | ||||
| Lethal/nonlethal cases | 18/117 | 38/96 | 64/70 | |
| Odds ratio (95% CI), adjusted for | ||||
| – (Unadjusted) | 1 (reference) | 2.6 (1.40, 4.9) | 5.9 (3.3, 11) | 2.4 (1.87, 3.2) |
| – Extent of aneuploidy | 1 (reference) | 2.4 (1.30, 4.7) | 4.4 (2.4, 8.4) | 2.0 (1.55, 2.7) |
| – Extent of aneuploidy, Gleason | 1 (reference) | 2.1 (1.02, 4.3) | 3.7 (1.84, 7.6) | 1.82 (1.35, 2.5) |
| – Extent of aneuploidy, Gleason, PTEN† | 1 (reference) | 2.5 (0.92, 7.2) | 3.8 (1.42, 11) | 1.95 (1.28, 3.1) |
| – p53‡ | 1 (reference) | 2.4 (1.11, 5.6) | 5.0 (2.3, 12) | 2.1 (1.48, 3.1) |
| Odds ratio (95% CI), jointly by chr8q | ||||
| – 8q diploid | 1 (reference) | 1.97 (0.98, 4.1) | 3.0 (1.44, 6.3) | 1.62 (1.12, 2.4) |
| – 8q gain | 1.93 (0.40, 7.1) | 9.9 (3.8, 27) | 16 (7.8, 37) | 2.3 (1.48, 3.7) |
| Odds ratio (95% CI), by ERG§ | ||||
| – ERG-negative | 1 (reference) | 4.2 (1.49, 14) | 11 (4.1, 33) | 3.8 (2.4, 6.4) |
| – ERG-positive | 1 (reference) | 1.68 (0.71, 4.1) | 4.0 (1.74, 9.6) | 1.83 (1.28, 2.7) |
| RAD21 in normal tissue | ||||
| Lethal/nonlethal cases | 23/44 | 19/48 | 22/45 | |
| Odds ratio (95% CI) | 1 (reference) | 0.76 (0.36, 1.57) | 0.94 (0.45, 1.92) | 1.58 (0.73, 3.5) |
| RAD21 difference tumor–normal | ||||
| Lethal/nonlethal cases | 13/54 | 17/50 | 34/33 | |
| Odds ratio (95% CI) | 1 (reference) | 1.41 (0.63, 3.2) | 4.3 (2.0, 9.5) | 2.4 (1.71, 3.6) |
*Per one SD higher RAD21 expression.
†Among cases with measured PTEN protein, n = 257.
‡Among cases with measured p53 protein, indicating TP53 missense mutations, n = 247. Not otherwise adjusted.
§Among cases with measured ERG protein, indicating TMPRSS2-ERG fusions, n = 368. Not otherwise adjusted.
We next assessed to what extent RAD21 tumor expression was associated with lethal disease beyond known clinical and molecular prognostic factors in prostate cancer (Table 2). Adjusting for the overall aneuploidy burden somewhat attenuated associations between RAD21 mRNA and lethal disease, as expected given the strong association between RAD21 mRNA and the predicted count of chromosome arms altered by gains or losses (4). Associations between RAD21 mRNA expression and lethal disease tended to be stronger among tumors with predicted chr8q gain than among those with predicted diploid chr8q as well as among ERG-negative than ERG-positive tumors, but data were not conclusive about potential differences. Yet even after adjusting for both aneuploidy burden and Gleason score, strong prognostic factors in our cohorts (23), there remained an association between higher tumor RAD21 mRNA expression and lethal prostate cancer, with an odds ratio of 3.7 (95% CI 1.8, 7.6) comparing the 33% of tumors in the highest tertile of expression to the 33% of tumors in the lowest tertile of expression (Table 2). Further adjustment for PTEN loss, a strong single-gene adverse prognostic factor (24) that was associated with higher RAD21 mRNA (Table 2), did not attenuate this association. Additionally, we detected TP53 missense mutations, the single gene mutation most strongly associated with aneuploidy burden across cancers (3), using a genetically validated immunohistochemistry assay for p53 overexpression (25, 26). While p53 expression was associated with higher RAD21 tumor expression (Table 1), concomitant TP53 missense mutations did not explain the associations of high RAD21 expression with lethal disease (Table 2). Collectively, these results credentialed RAD21 mRNA expression in primary prostate cancer as a strong prognostic factor. This association was present beyond established prognostic factors in prostate cancer and potentially in synergy with additional chr8q genes.
To validate the association of RAD21 alterations with prognosis, we additionally assessed RAD21 DNA alterations across clinical disease states of prostate cancer among 2,592 patients in the Memorial Sloan Kettering (MSK) IMPACT Prostate Clinical-Genomic Database (SI Appendix, Table S2). Of the 88 patients (3%) with RAD21 tumor alterations annotated as oncogenic or likely oncogenic, 92% had amplifications of RAD21 DNA copy numbers, of which 93% co-occurred with MYC amplifications. RAD21-altered tumors had, on average, 4.8 more chromosome arms altered by aneuploidy (95% CI 3.5, 6.2) than RAD21-intact tumors (SI Appendix, Fig. S2). RAD21 alterations were associated with lower overall survival (hazard ratio 2.1, 95% CI 1.5, 3.0), similar to the 3% of tumors with MYC amplifications alone (SI Appendix, Fig. S3). These data underscore a worse prognosis with RAD21 alterations but pertain to presumed two-copy gains in RAD21 that co-occur with copy number alterations in MYC and are relatively rare. In contrast, the strong relation of RAD21 mRNA to prognosis was notable at expression levels present in large proportions of primary prostate tumors.
Acute Tmprss2-ERG Fusion Gene Expression Promotes Early Oncogenic Signatures but Impairs Proliferation in Primary Prostate Organoids.
We recently showed that increased RAD21 mitigates oncogenic stress in early oncogenesis in Ewing sarcoma driven by the EWS-FLI1 onco-fusion (16). Therefore, we hypothesized that RAD21 overexpression might also play a role in promoting ERG-fusion-positive prostate cancer at an early stage of cancer development. To study this, we utilized a mouse model carrying a copy of Cre recombinase-inducible Tmprss2-ERG (T-ERG) fusion (27) and derived organoids to model the early events induced by T-ERG expression. We isolated normal prostate tissues from the mice in which an N-terminal truncated human ERG gene was knocked into exon 2 of the mouse Tmprss2 locus, namely a T-fl/fl-ERG construct [SI Appendix, Fig. S4 A and B), (27)]. From the primary prostate tissues, we developed prostate organoids that are proliferative and express the androgen receptor (AR) (SI Appendix, Fig. S4C). To induce ERG expression from the Tmprss2 locus, we transduced these organoids with an adenoviral particle containing a Cre recombinase (AAV-Cre) at the early passage postisolation (SI Appendix, Fig. S4B and Methods). By visualizing coexpressed GFP, we were able to microscopically isolate single isogenic clones of organoids expressing the T-ERG fusion (Fig. 2A). To verify epithelial lineage, we detected both basal (p63) and luminal (CK8, cytokeratin 8) markers by immunohistochemistry (Fig. 2B).
Fig. 2.
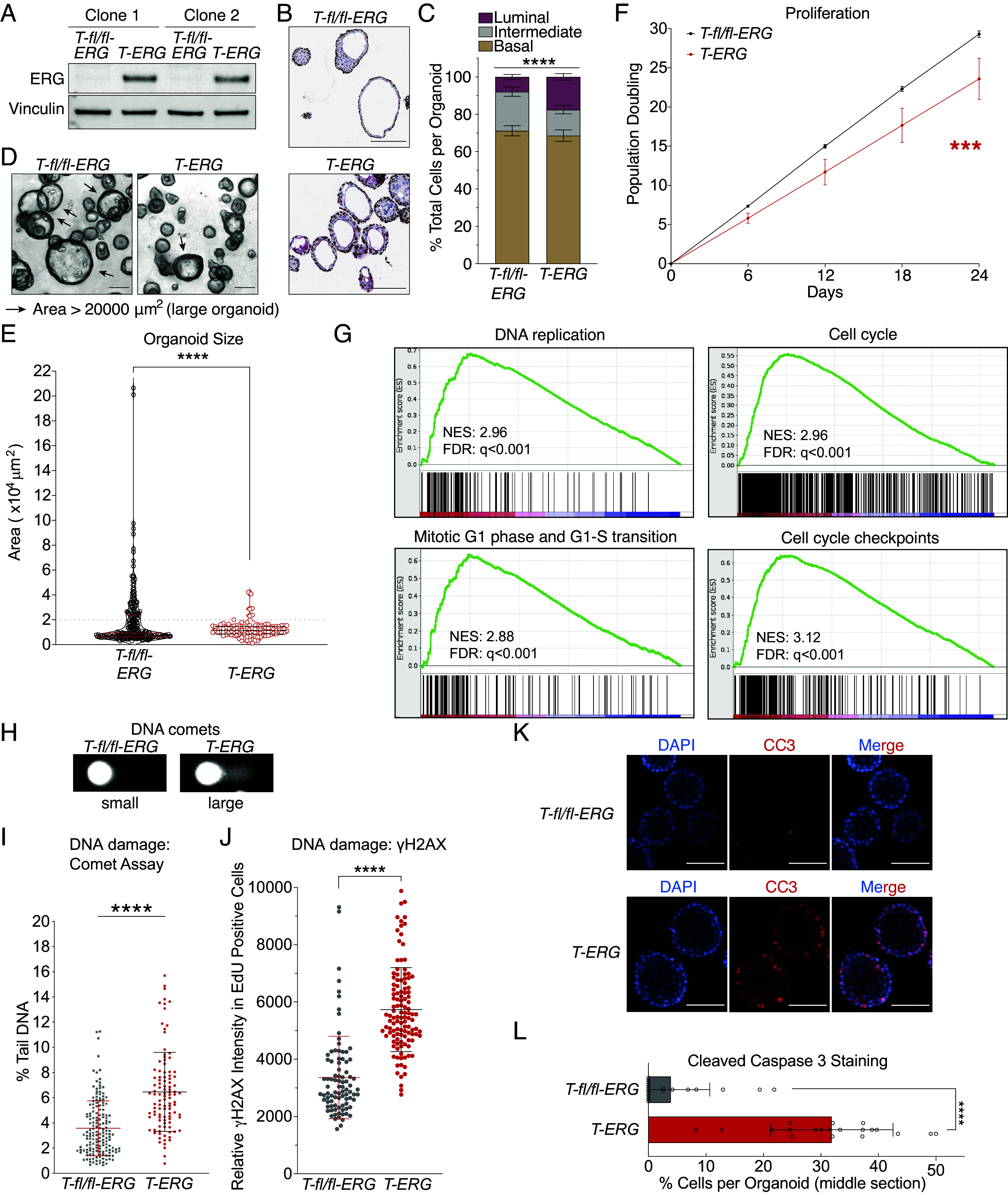
Induction of Tmprss2-ERG in prostate organoids causes oncogenic replication stress. (A) Expression of T-ERG by Western blot. Vinculin was used as a loading control. (B and C) T-ERG induction increases basal-to-luminal transition. Immunohistochemical analysis for basal marker, P63 (brown) and luminal marker, CK8 (red) (B) and quantification of basal-to-luminal transition (C). Error bars represent SEM of independent organoid clones, n = 11. (Scale bar: 150 µm.) ****P < 0.0001, two-way ANOVA. (D–F) Expression of T-ERG impairs organoid proliferation. Example and definition of large and small organoid in this study (D). (Scale bar: 150 µm.) Proliferation was measured by organoid size after 6-d growth (E) and by population doublings (F). Each circle represents an organoid, and lines represent median (Middle) and quartiles (Top and Bottom). Gray dash line: cut-off between large and small organoids. Each dot represents the mean of 2 independent replicates with SEM (error bars) ***P < 0.001, linear regression (G) Cell cycle and replication genes were enriched in the T-ERG organoids. GSEA was performed on RNA sequencing data of T-ERG organoids over T-fl/fl-ERG organoids; Top 20 pathways were listed in SI Appendix, Fig. S5A. NES: normalized enrichment score; FDR: false discovery rate. (H and I) T-ERG expression elevates single-cell total DNA damage level in prostate organoids. Total DNA damage levels were measured by alkaline comet assays: examples of small and large DNA comets in (H) and quantification in (I, each dot represents a single comet). Middle line: mean, error bar: SD. ****P < 0.0001, t test. (J) T-ERG expression increases DNA damage in S-phase cells. DNA damage was measured by γH2AX intensity in EdU-positive cells: examples of stained cells in (SI Appendix, Fig. S6B). Each dot represents an EdU positive cell. The middle line represents the average with SD (error bar). ****P < 0.0001, t test. (Scale bar: 10 µm.) (K and L) T-ERG expression induces cellular apoptosis. DNA (DAPI) is in blue and the apoptotic maker, cleaved caspase 3 (CC3) is in red (K). Quantification of CC3 is in (L). Each bar represents average with SD. ****P < 0.0001, t test.
Although expression of T-ERG is not sufficient to initiate tumorigenesis without a secondary event, such as PTEN loss (27–30), expression of T-ERG alone significantly increased the portion of prostatic luminal cells (Fig. 2 B and C), an indication of increased basal-to-luminal transition, which has been reported as an early oncogenic event in prostate cancer (29, 31). Consistently, T-ERG induction showed no morphologic evidence of neoplasia during early passages (Fig. 2D). However, expression of T-ERG had a dramatic impact on organoid growth (Fig. 2 D–F). Organoid size was much smaller, and proliferation was slower in the T-ERG-expressing organoids than in controls (Fig. 2 D–F and SI Appendix, Fig. S4 D and E). However, compared to the expression of EWS-FLI1, which often caused senescence and a stall in growth in Ewing sarcoma (16, 32, 33), T-ERG expression did not stall organoid growth (Fig. 2F and SI Appendix, Fig. S4E). Taken together, our data argue proliferation of prostate organoids is impaired upon acute induction of T-ERG expression.
Tmprss2-ERG Induces Replication-Associated DNA Damage and Apoptosis in Primary Prostate Organoids.
To determine how acute induction of T-ERG impacts prostate cells, we conducted RNA sequencing on organoids within three passages of induction (Methods). With gene set enrichment analysis [GSEA (34, 35)] comparing the T-ERG with parental T-fl/fl-ERG organoids of the same passage number, we found that expression of T-ERG enriched for GSEA signatures related to the cell cycle and replication (Fig. 2G and SI Appendix, Fig. S5A), suggesting induction of replication stress by T-ERG. By comet assay, we observed a significantly elevated overall DNA damage level in the T-ERG cells compared to the uninduced cells within these isogenic organoids (Fig. 2 H and I). We further assessed the occurrence of DNA damage in the S-phase cells by using immunofluorescence staining and confocal microscopy (SI Appendix, Fig. S5B and Methods). EdU incorporation and staining methods specifically marked the cells in S/G2 phase with no overlap with cells in G1 phase (SI Appendix, Fig. S5C and Methods). Our findings revealed a significant increase in γH2AX levels in T-ERG-expressing cells with ongoing DNA replication (EdU-positive cells) (Fig. 2J), suggesting that acute T-ERG induction leads to elevated DNA damage likely stemmed from replication stress.
DNA damage caused by oncogenic replication stress can induce both senescence and apoptosis (36). T-ERG expression did not stop growth in our primary organoid (Fig. 2F), unlike what we previously observed in Ewing sarcoma (16), suggesting that oncogene-induced senescence is not a direct consequence of T-ERG expression in the cells tested. Through immunofluorescence staining of the apoptosis marker cleaved caspase 3 (CC3), we discerned a visually increased proportion of CC3-positive cells in the T-ERG organoids (Fig. 2 K and L). To further corroborate this observation, we conducted an apoptosis profiling assay, which revealed apoptotic signatures in the T-ERG-expressing organoids (SI Appendix, Fig. S5D). In summary, these observations collectively suggest that T-ERG promotes cell cycle progression but concurrently induces toxic replication stress that can lead to DNA damage and apoptosis in primary prostate cells. Interestingly, these cells remained euploid over the course of these analyses (SI Appendix, Fig. S5E), arguing that these T-ERG-driven adverse effects might suppress oncogenic genomic events in primary prostate epithelium.
Increased RAD21 Reduces DNA Damage and Apoptosis in Primary Tmprss2-ERG-Expressing Organoids.
To investigate whether increased RAD21 can ameliorate replication-associated DNA damage and apoptosis in the T-ERG-expressing organoids, we overexpressed the mouse Rad21 gene under control of a CMV promoter, using lentiviral transduction of isogenic T-ERG prostate organoid models (Methods). This intervention resulted in a roughly 20% increase in Rad21 protein in the control organoids and a 70% increase in the T-ERG organoids (Fig. 3A). The overexpression of Rad21 had a notable impact on the fitness of T-ERG-expressing organoids, leading to larger and more mature organoids (compared T-ERG, Vector vs. T-ERG, RAD21; Fig. 3 B and C). Furthermore, Rad21 overexpression effectively rescued the impaired proliferation caused by T-ERG, restoring it to the level observed in the cells carrying uninduced T-fl/fl-ERG construct (Fig. 3D). However, overexpression of Rad21 did not induce neoplastic changes in morphology, such as the invasive “finger-shape extrusion” associated with neoplastic invasion. These data suggest that Rad21 overexpression promotes proliferation and improves cell fitness in the setting of T-ERG-induced stress but does not per se cause transformation.
Fig. 3.

Increased Rad21 mitigates oncogenic stress and promotes growth in the Tmprss2-ERG-expressing prostate organoids. (A) Mild overexpression of Rad21, measured by Western blot. GAPDH is used as a loading control. Number on top indicates the extent of overexpression relative to vector expressing organoids. (B) Example of the organoids carrying indicated constructs 6 d post passage. (Scale bar: 150 µm.) (C and D) Rad21 overexpression promotes proliferation of T-ERG organoids. Proliferation was measured by organoid size after 6-day growth after passage (C) and by population doublings. Each circle is a single organoid, and lines represent median (Middle) and quartiles (Top and Bottom) in (C). ****P < 0.0001, t-test, compared between T-ERG, Vector and T-ERG, Rad21 in (C), and ****P < 0.0001, linear regression in (D). (E and F) Increased Rad21 reduces whole cell DNA damage by alkaline comet assays (E, each dot represents a single comet) and DNA damage in S-phase cells by γH2AX intensity in EdU positive cells (F, each dot represents a single EdU positive cell. Middle line: mean, error bar: SD). ****P < 0.0001 t-test, compared between T-ERG, Vector and T-ERG, Rad21. (G) Rad21 overexpression decreases apoptosis in T-ERG-expressing organoids. Error bar: SD. ****P < 0.0001, compared to T-ERG cells expressing vector; ####P < 0.0001, compared to T-fl/fl-ERG cells overexpressing Rad21; ^^^^P < 0.0001, ^P < 0.05, compared to T-fl/fl-ERG cells expressing the Vector; no symbol is shown if no significance is detected. Each data bar or population in (C–G) represents combined data from 2 independent clones with either Vector or Rad21.
GSEA of RNAseq data comparing T-ERG-expressing organoids with Rad21 overexpression construct with those that do not overexpress Rad21 showed an increase in cohesin resolution (SI Appendix, Fig. S6A), suggesting that RAD21 overexpression increases the cohesin complex establishment. Similar to the data from T-ERG expression alone (SI Appendix, Fig. S5A), we also observed an enrichment of cell cycle and DNA replication gene signatures when overexpressing Rad21 in the T-ERG organoids (SI Appendix, Fig. S6A), suggesting that Rad21 overexpression promotes growth of the T-ERG cells. We next ascertained whether increased RAD21 exacerbates replication stress, or conversely mitigates DNA damage, when T-ERG is induced. Using a comet assay, we determined that Rad21 overexpression reduced the elevated DNA damage found in the T-ERG-expressing cells to levels similar to cells without T-ERG expression (Fig. 3E). We further detected reduced γH2AX signal in S-phase T-ERG-expressing cells when Rad21 was overexpressed (Fig. 3F; compared T-ERG, Rad21 to T-ERG, Vector). Intriguingly, this reduction was evident in uninduced T-fl/fl-ERG cells (T-fl/fl-ERG, Vector vs. T-fl/fl-ERG, Rad21) (Fig. 3F), suggesting that RAD21 may promote repair of lesions arising from replication abnormality upon oncogene induction as well as some baseline stress during normal DNA replication. Consistent with this model, apoptosis induced by T-ERG was significantly decreased upon Rad21 overexpression, with levels reduced to those observed in the T-fl/fl-ERG organoids carrying either Vector or Rad21 (Fig. 3G and SI Appendix, S6B).
Increased RAD21 Expression Promotes Proliferation in Models of Advanced Prostate Cancer Models.
To assess the relevance of RAD21 overexpression in more aggressive prostate cancer, we utilized the T-ERG-expressing mouse prostate organoid with a homozygous loss of PTEN (PtenL/L, T-ERG). This model exhibits high prostatic intraepithelial neoplasia lesions in vivo and invasive “finger-shape” structures in ex vivo organoids (27, 37). We found that Rad21 overexpression also increased proliferation of organoids derived from PtenL/L, T-ERG prostates, while it concurrently reduced replication stress-induced DNA damage (Fig. 4 A and B). However, Rad21 overexpression had a limited impact on apoptosis when compared to organoids without additional Rad21 (Fig. 4C). This observation may be attributed to activation of the PI3K pathway activation from PTEN loss, which has an independent role in suppressing apoptosis (38). These results highlight the fundamental role of increased RAD21 in reducing toxic DNA damage during replication, which is maintained in this prostate cancer model and is independent of PTEN status. Furthermore, Rad21 overexpression was associated with a lower level of γH2AX (lower DNA damage) and higher levels of phosphorylated AKT (p-AKT) in the PtenL/L, T-ERG organoid model (Fig. 4D), consistent with increased RAD21 promoting prostate cancer aggressiveness by reducing toxic DNA damage. Additionally, Rad21 overexpression significantly increased the invasiveness of PtenL/L, T-ERG organoids, with a higher percentage of “finger-shape” neoplasia structures into Matrigel than observed in the organoids carrying the Vector control (Fig. 4 E and F). Consistent with these observations, in the patient samples described earlier, higher RAD21 expression was positively correlated with the Ki-67-staining derived proliferative index in prostate tissue from HPFS/PHS studies (Table 1 and Fig. 4G). Collectively, these data support a model where increased RAD21 promotes prostate cancer progression by reducing replication stress-induced DNA damage.
Fig. 4.

Rad21 overexpression promotes prostate cancer development. (A) Rad21 overexpression promotes proliferation of the PtenL/L, T-ERG organoids. Proliferation was measured by population doublings. Each dot represents the mean of 2 independent replicates with SEM (error bars). **P < 0.01, linear regression. (B and C) Increased Rad21 reduces whole cell DNA damage by γH2AX intensity in EdU positive cells (B, each dot represents a single EdU positive cell. Middle line: mean, error bar: SD; ****P < 0.0001, t test.), but does not affect apoptosis (C, each circle: each organoid. Error bar: SD. ns: not significant, t test) in the PtenL/L, T-ERG organoids. Each data bar or population in (B and C) represents combined data from 2 independent clones with either Vector or Rad21. (D–F) PtenL/L, T-ERG organoids exhibit more advanced phenotypes when overexpressed Rad21. Overexpression of Rad21 upregulated phosphorylated AKT protein levels, measured by Western blot; tubulin is used as a loading control (D). Example of “finger-shape extrusion” structure indicating invasiveness of the organoids in (E) and the quantification of the extrusion structure in (F, each dot represents an independent culture composed of over 100 organoids. Error bar: SD; *P < 0.05, t test). (G) RAD21 mRNA expression and the proliferative index Ki-67 in primary tumors from participants of the two population-based studies (n = 313). Pearson correlations r and 95% CI are given. Note the log-scaled y axis. Dots at the bottom of the plot are tumors with 0% Ki-67-positive nuclei.
Discussion
In this study, we leveraged observational and experimental approaches to identify genes on chr8q that may promote cancer-specific aneuploidy and thereby promote lethality in prostate cancer. We first prioritized genes associated with the risk of lethal progression among patients with prostate cancer, finding increased RAD21 expression predicted which tumors would develop a lethal phenotype. The prognostic association of RAD21 with lethal prostate cancer was informative beyond overall aneuploidy, tumor grade, loss of PTEN, and TP53. Moreover, both RAD21 overexpression and chr8q gains appeared to drive lethal progression, suggesting that, while RAD21 is not the sole driver, it likely contributes to the selection benefits conferred by recurrent chr8q gains in prostate cancer. This finding is intriguing given that men who identify as Black or are of African ancestry face the highest burden of prostate cancer mortality. In some studies (25), but not others (39), Black men were substantially more likely to have tumors with chr8q gains, a difference that did not appear to be fully explained by issues related to healthcare access (25).
The longitudinal follow-up of patients for the clinically relevant outcomes of metastasis and prostate cancer enabled identification of RAD21 as a potential driver of prostate cancer progression. In cross-sectional comparisons of tumors and nontumor tissue at diagnosis, RAD21 would have been missed, because it is not overexpressed, on average, in tumor tissue relative to normal tissue. Leveraging human data to prioritize potential genes was also important given the lack of aneuploid prostate cancer mouse models. Nevertheless, using a mouse prostate organoid model harboring T-ERG as an oncogenic inducer, we observed that acute induction of T-ERG at the early tumor initiation stage mainly drove cell cycle progression, led to accumulation of DNA damage likely during DNA replication, which caused apoptosis and impaired proliferation in the absence of increased Rad21 expression. It is possible that T-ERG increases replication stress–induced DNA damage by suppressing Chk1 expression in a manner similar to a previous study (40). These DNA lesions are still toxic to the cells and impair proliferation especially during early oncogenesis with low mutational burden, as we observed in our primary organoid models. Indeed, expression of TMPRSS2-ERG (or TMPRSS2-ETV1) by itself is not sufficient to initiate invasive carcinomas (27–30). Increasing Rad21 expression significantly reduced DNA replication stress-induced damage and promoted proliferation despite expression of T-ERG. Interestingly, Rad21 played an antiapoptotic role in T-ERG-expressing prostate cells, distinct from the antisenescence role reported in EWS-FLI1-driven Ewing sarcoma (16). However, in both cases, RAD21 mitigation of DNA replication stress–induced DNA damage provided a growth advantage for oncogene-expressing cells. These observations suggest an important role of increased RAD21 in mechanistically sustaining oncogenic potential until the late stage of prostate cancers, and thus promote tumor progression.
RAD21 is overexpressed at the transcript level in various cancers but is rarely mutated (TCGA, cBioPortal). RAD21 or its yeast homolog SCC1 has been reported to be important in regulating homologous recombination (HR)-mediated repair, resolution of stalled replication forks, and mediating loop extrusion of induced double-strand breaks (41–47). How RAD21 plays a role in a dosage-dependent manner in mitigating replication-associated DNA damage and promoting replication fork progression is yet to be further elucidated. However, a recent study showed that increased RAD21 promotes immune evasion in ovarian cancer by interacting with TAP/TEAD4 and recruiting the NuRD complex (48). Prostate cancer represents a suppressive environment for autoreactive T cells, and antitumor immune responses are rare in this disease (49). Nevertheless, mitigating toxic oncogenic stress provides new mechanistic perspectives for increased RAD21 in promoting prostate cancer aggressiveness. Moreover, increased RAD21 expression could also potentially mitigate the effects of therapeutic radiation by promoting DNA repair. Higher RAD21 expression was positively associated with overexpression of the BRCA1 protein (Table 1) that is involved in HR repair and that tends to be overexpressed in proliferating tumors (50). While RAD21 inhibitors are not available at present, understanding the detailed mechanism of RAD21 in mitigating oncogenic stress and promoting DNA HR repair may suggest novel therapeutic strategies to treat prostate tumors with high levels of RAD21 expression.
Our data identified several other 8q genes besides RAD21 as associated with lethal progression. The MYC proto-oncogene mRNA and protein levels did not predict lethal progression as reported in our previous study (22), but was prognostic (in a later disease state) if measured by DNA copy numbers (20). MYC itself induces strong DNA replication stress, which can promote oncogene-induced senescence and apoptosis (36, 51). Interestingly, depletion of RAD21 failed to sustain MYC-induced replication stress and led to synthetic lethality in some studies (52, 53). Therefore, increased MYC expression may depend on elevated RAD21 expression. This may explain why gain of 8q is observed rather than amplification of only MYC, as gain of other relevant 8q genes may be needed to allow tumor progression.
Limitations of the current study include our inability to assess RAD21 in human populations on multiple levels, notably DNA copy numbers, mRNA expression, and protein expression, in the same individuals with long-term follow-up for clinical outcomes. Doing so would be informative to delineate at which level RAD21 gets dysregulated to inform therapy development, which other somatic alterations RAD21 alterations on the different levels co-occur with, and which level is most suitable for prognostication. In addition, despite some suggestions that findings related to 8q gains may be particularly relevant to the higher burden of prostate cancer mortality in Black men (25), the racial diversity in the current study was limited, and findings should be replicated in other settings. Further, we chose a T-ERG model because of our previous work in ETS-driven Ewing sarcoma (16), not because of any strong association between TMPRSS2-ERG status and RAD21 mRNA expression in our human prostate cancer samples, and thus it is not clear whether our findings would be restricted to prostate cancer with a TMPRSS2-ERG fusion. Additional model systems would be useful as part of future directions of this line of research.
Taken together, we identified increased RAD21 as a potential driver of lethal progression, positioning it among the top candidates on 8q, which underscores the critical role of high RAD21 in promoting prostate cancer development by mitigating toxic replication-associated DNA damage. These findings open up promising avenues for developing therapeutic strategies targeting RAD21-related vulnerabilities, given the relation of RAD21 to oncogenic replication stress, which could potentially influence responses to PARP or ATM inhibitors and other therapeutics in development.
Methods
Cohort Studies, Gene Expression Profiling, and Analysis.
The HPFS (n = 51,529) and PHS (n = 29,071) are prospective cohort studies that enrolled men nationwide. HPFS participants have an educational and professional background in health professions, were 40 to 75 y of age and free from cancer and chronic disease at enrollment in 1986, and have been reporting detailed data on lifestyle, diet, and health via biennial questionnaires until this day. PHS participants were physicians who fulfilled similar criteria when enrolled at ages 40 to 84 y in 1982; they initially participated in a randomized-controlled trial of aspirin and multivitamins in the prevention of chronic disease and were then followed as a prospective cohort. Incident prostate cancer diagnoses in both cohorts were self-reported by the participating health professionals and then verified and further followed by the study team through detailed prostate cancer-specific questionnaires, contact with treating physicians, and reviews of medical records, including for metastases. Prostate cancer primary tissue from diagnosis was retrieved from treating hospitals and clinics and centrally rereviewed for histology and Gleason scoring (23). Causes of death were adjudicated by an endpoint committee of physicians. End of follow up for lethal disease was January 2015 in PHS and January 2019, in this analysis, in HPFS.
For gene expression profiling of the primary tumor, participants with available formalin-fixed paraffin-embedded surgical tissue from prostatectomy or transurethral resection of the prostate in 1982 to 2005 were included. Lethal cases consisting of participants who developed metastases or died from prostate cancer as well as controls consisting of participants with a prostate cancer diagnosis who remained free from metastases for at least 8 y after cancer diagnosis were selected via cumulative-incidence sampling (54). High-density tumor cores were selected for RNA extraction and whole-transcriptome amplification, followed by expression profiling using the GeneChip Human Gene 1.0 ST microarray (Affymetrix; Gene Expression Omnibus, GSE79021). For 201 participants, tumor-adjacent histologically normal prostate tissue was profiled as well. Aneuploidy burden was predicted from the tumor transcriptome as previously described (4).
Tumor protein biomarkers were evaluated using tissue microarrays constructed from highest-grade nodules of prostatectomy blocks and transurethral resections of the prostate. ERG overexpression (55), PTEN loss (24), and p53 overexpression (25) were evaluated using genetically validated assays. Ki-67 was quantified as the proportion of positive nuclei (56) and adjusted for between-tissue microarray batch effects using quantile normalization (57).
The association of gene expression levels with lethal disease was evaluated using logistic regression, based on trends (slope) per continuous levels scaled to SD 1. For RAD21 mRNA, models were additionally adjusted for aneuploidy burden (0, 1 to 2, 3 to 4, 4+ altered chromosome arms), Gleason score (<7, 3 + 4, 4 + 3, 8, 9 to 10), and PTEN loss (complete loss, intact), BRCA1 expression (50), as well as separately adjusted for p53 status (overexpressed/TP53 mismatch mutation, intact) and stratified by predicted chr8q status (predicted diploid, predicted gain).
The independent validation study was conducted among patients with prostate cancer, without restrictions by clinical disease state, who were seen at Memorial Sloan Kettering (MSK) Cancer Center and had paired tumor–normal panel-based sequencing between 2015 and 2020. Sequencing used the MSK-IMPACT panel, an FDA-cleared targeted hybridization capture-based gene panel that included 341 to 468 genes (depending on its version) and captured single-nucleotide variants, small insertions and deletions, copy-number alterations, and structural rearrangements (58). Clinical data were captured by research assistants and clinical fellows in a previously described and validated research database (59). The current study excluded patients missing basic clinical data (2%), low tumor cellularity (2%; due to tumor mutational burden < 1, fraction genome altered by copy number alterations < 1%, and purity by pathology <20%), missing data on RAD21 (5%), and patients without survival follow-up (2%; SI Appendix, Fig. S7). RAD21, MYC, and TP53 alterations were restricted to oncogenic or likely oncogenic alterations as per OncoKB version 2.8 (60), of which for RAD21 >90% were copy number gains. Chromosome arm-level aneuploidy was called using ASCETS version 1.0 with breadth of coverage >0.5 (61). For prognostic analyses, patients were followed from the date of tumor sequencing to death, last contact, or end of follow-up in 2021. Hazard ratios were estimated using Cox regression with the timescale starting at sequencing.
Participants provided written informed consent. The research was conducted in accordance with the U.S. Common Rule and was approved by institutional review boards at Harvard T.H. Chan School of Public Health, Partners Healthcare, and Memorial Sloan Kettering Cancer Center.
Isolation and Development of Mouse Prostate Organoids.
Animal husbandry and euthanasia was performed according to our animal protocol approved by the MIT Institutional Animal Care and Use Committee (Protocol # 0122-004-25). A 4-month-old mouse carrying the desired genotype was euthanized, and aseptic surgical equipment was used to isolate the entire prostate. Anterior prostate, dorsolateral prostate, and ventral prostate were separated under a dissection microscope. Each lobe of a prostate was developed into organoid culture and organoids from the ventral prostate were consistently used throughout this study. Procedures for developing and subculturing mouse prostate organoids were described in detail (37, 62). The brand for each culture medium component in this study is listed in SI Appendix, Table S3. Organoids were subcultured every 6-7 d, depending on the specific experiments. To break the organoids down into single cells, they were incubated in TrypLE (Gibco) supplemented with 10 µM Y-27632 for 20 min at 37 °C at 1000 rpm in the Eppendorf ThermoMixer F1.5. Cells were spun down and resuspended in ADMEM/F12 +/+/+ medium (62). The number of cells was determined by using a Nexcelom cellometer after organoids were broken up into single cells. The desired number of cells were spun down to remove the ADMEM/F12 +/+/+ medium. Then, cells were resuspended in 100% Matrigel and seeded on a 24-well plate in 30 uL per Matrigel dome. 3000 single cells were seeded per 30 uL Matrigel dome to develop into organoids throughout this study, unless otherwise noted.
Proliferation Analyses.
Proliferation analysis for population doublings was performed by counting the cells at the time of splitting during every passage using a Nexcelom cellometer. At least 2 independent clones were tested genotype for proliferation. Cell counts were conducted in two technical replicates and the average was used for analysis. For cell size measurement, organoids were grown for 6 d in Matrigel in the culture condition under the described conditions. Z-stack pictures were taken using the Nikon Eclipse Ti microscope under 4X/0.13 Plan Fluor objective with NIS-Elements BR5.02.01 software under transmitted light condition. The projected area, which served as an indicator, was calculated using FIJI-ImageJ software.
Immunohistochemistry for Histological Samples.
Organoids grown in the Matrigel were harvested by disrupting the Matrigel with PBS and subsequently centrifuged. Organoids were then embedded in HistoGel (Thermo Scientific) and placed into tissue cassettes. After the HistoGel solidified at RT for about 5-8 min, the histological samples were then fixed with 10% formalin (VWR) for 1.5 h and then stored in 70% ethanol prior to downstream paraffin embedding. Paraffin embedding, tissue block preparation, the slicing of tissues into 5 μm thickness and immunohistochemistry staining were carried out in the Histology core facility of Swanson biotechnology center at the Koch Institute using standard protocols. Antibodies used and their dilutions are listed in SI Appendix, Table S3.
Whole-Mount Organoid Immunofluorescence Staining and EdU Incorporation, and Confocal MZicroscopy.
Organoids were grown in Matrigel, seeded in the center of a well in the 1.5 coverslip glass-bottom 12-well plate (Mattek). To pulse label cells with 5’-ethynyl-2’-deoxyuridine (EdU), the live organoids were incubated in culture medium containing 10 µM EdU for 2 h. Immediately following the incubation with EdU, cells were fixed and permeabilized using the procedure described above. Click-iT EdU Imaging Kit (Invitrogen) was used to stain for EdU prior to antibody incubations.
For immunofluorescent microscopy, exponentially growing organoids were then fixed with 4% paraformaldehyde in PBS for 20 min at room temperature. Then, cells were washed with PBS with 3% BSA once followed by permeabilization in PBS with 0.5% Triton for 30 min at RT. The organoids were incubated with primary antibody overnight at 4 °C, and with secondary fluor-conjugated antibody for 50 min to 1 h at room temperature in the dark. All antibodies (diluted in PBS with 3% BSA) and dilutions are listed in SI Appendix, Table S3. The organoids were stained with DAPI for 10 min in the dark before being mounted in the Prolong Gold Antifade Mountant medium (Thermo Fisher) for imaging.
The Nikon Spinning-Disk Confocal Microscope equipped with a Yokogawa spinning disk and Andor Clara camera, controlled by NIS-element software, was used to acquire the images of the organoids. The images were analyzed using FIJI-ImageJ software.
Comet Assay.
Single cells were isolated using the TrypLE method as described previously. The suspended cells were counted using a hemocytometer. The comet assays were conducted using the Trevigen Comet Assay Kit (R&D Systems) under alkaline comet assay conditions. DNA was then stained with SYBR Gold Nucleic Acid Gel Stain (Invitrogen) at room temperature for 30 min in the dark and washed twice with H2O (30 min each time on a rocking platform). Images were taken under the FITC channel with a Nikon Plan Apo 4X/0.2 objective, ORCA-ER camera, and NIS-Elements software. Exposure times (same for the same cell type) were determined using the autoexposure function of NIS-Element software. The percentage of DNA in comet tails was analyzed using the automated OpenComet tool (63) in FIJI (ImageJ). At least 100 comets from two biological replicates per genotype were analyzed.
RNA Isolation, Sequencing, and GSEA.
Organoids from the same Matrigel dome were harvested for each RNA extraction using the RNeasy kit (Qiagen). Library preparation for RNA sequencing was performed using the published high-throughput protocol (64) with experimental modification and standard sequencing and data analysis listed in Additional Methods in SI Appendix. RNA sequencing and data analysis were performed at the MIT BioMicro Center. GSEA/2.0.13 was then run projecting expression matrices to c2 cp MSigDB to identify enrichment patterns.
Plasmid Handling and Lentiviral and Adenoviral Transduction.
All plasmids were extracted using the QIAGEN Plasmid Maxi Prep kit. Lentiviral packaging plasmids, pMD2.G (RRID:Addgene 12259) and psPAX2 (RRID:Addgene_12260), were gifts from Didier Trono. pLX304 vectors were gifts from Alejandro Sweet-Cordero. pLX304-RAD21(mouse) was purchased from Gene Universal Inc. The Vectors expressing Cdt1-mCherry were gifts from Marianna Trakala (65, 66). Lentiviral vector preparations and packaging were done following previously published procedures (16). Organoid cells were trypsinized using TrypLE to obtain single cells, which were then plated on Matrigel. These cells were infected with concentrated lentivirus with least 107 infectious units per milliliter (IFU/mL) determined by using Lenti-X GoStix (Takara ClonTech). Specifically, 30,000 single cells were infected with 500µL lentiviral particles for 3 h at 37 °C in the tissue culture incubator. After removing the lentivirus, a second layer of Matrigel was added, and cells were cultured in normal growth medium for 24 h before commencing drug selection. Drug selection typically involved the addition of the drug, blasticidin (4 µm/mL) for 5 to 6 d until uninfected cells did not survive. Single organoid clones that survived the drug selection were selected for subsequent experiments.
Adenoviral particles containing a Cre recombinase (AAV-Cre) were obtained from The University of Iowa Carver College of Medicine Viral Vector core. AAV-Cre viruses are at a concentration over 107 IFU/mL. Cre gene is under the control of a CMV promoter. For adenoviral transduction, 4,000 cells were transferred to an Eppendorf tube and pelleted, and the medium was carefully removed. Next, 0.5 µL of viral particles was added to the cell pellet. The cells were gently resuspended and incubated at room temperature for 10 min in the cell culture hood. Cells were resuspended in 40 µL Matrigel and organoids were cultured using the standard protocol described above.
Western Blot.
Organoids embedded in Matrigel were gently washed twice with ice-cold PBS without disrupting the Matrigel domes. The Matrigel domes were broken by pipetting up and down with PBS. Organoids were pelleted by centrifuging at 400 g for 3 min. Organoid pellets were then lysed in RIPA buffer (Thermo Fisher Scientific) supplemented with protease inhibitor cocktails and phosphatase inhibitor (PhosStop, Roche) according to the manufacturer’s protocol. Typically, 4 domes of organoids were lysed in 200μL RIPA buffer. Protein extracts were quantified by Bradford (Bio-Rad) and equal amounts subjected to SDS-PAGE (4% to 12%, Bio-Rad or Invitrogen). Proteins were transferred to a nitrocellulose membrane using iBlot 2 Dry Blotting System (Thermo Fisher Scientific). Blots were blocked using OneBlock Western blocking buffer (Genesee Scientific) and incubated with primary antibodies at 4 °C overnight in the same blocking buffer. Horseradish peroxidase (HRP)-conjugated or fluorophore-conjugated antibodies were used as secondary antibodies in the bocking buffer at room temperature for 1 h and detected by ChemiDoc MP Imaging System (Bio-Rad) respectively. Signal quantification was performed using FIJI-ImageJ gel analysis software. Detailed information on antibodies and their dilutions are listed in SI Appendix, Table S3.
Karyotyping.
DNA from 10,000 cells was extracted by using the QIAmp DNA Mini Kit (Qiagen). Karyotyping sequencing of the DNA and data analysis were provided at the MIT BioMicro Center (Additional Methods in SI Appendix).
Apoptosis Profiling Assay.
The assay was performed on the organoids at passage three after the induction of T-ERG. Organoids were grown for six days after seeding in Matrigel before protein extraction for the assay. Three independent replicates were pooled for the assay. Organoids from each replicate were harvested from four independent Matrigel domes. Cells were counted prior to the assay. The apoptosis profiling assay was conducted using the Proteome Profiler Mouse Apoptosis Array (R&D Systems). Signal intensity of each apoptotic marker was normalized to both internal assay control and the number of cells harvested.
Disclaimer.
The contents of this publication are the sole responsibility of the author(s) and do not necessarily reflect the views, opinions, or policies of Uniformed Services University of the Health Sciences, The Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc., the Department of Defense, the Departments of the Army, Navy, or Air Force. Mention of trade names, commercial products, or organizations does not imply endorsement by the U.S. Government.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank the Koch Institute’s Robert A. Swanson (1969) Biotechnology Center for technical support, specifically the Integrated Genomics and Bioinformatics (composed of the Genomics Facility and the Barbara K. Ostrom (1978) Bioinformatics Facility), Histology (The Hope Babette Tang (1983) Histology Facility), Microscopy, High Throughput Sciences and Flow Cytometry core facilities. We thank Jacqueline Lees’ lab members and scientists from Center for Prostate Disease Research for providing general research suggestions. We thank Erica Burds and Christopher Jamieson for funding management for this project. We thank the participants and staff of the HPFS and the PHS for their valuable contributions. In particular, we would like to recognize the contributions of Siobhan Saint-Surin, Betsy Frost-Hawes, Ann Fisher, Ruifeng Li, Maggie Bristol, and Bailey Vaselkiv. This project was supported by the Bridge Project, a partnership between the Koch Institute for Integrative Cancer Research at MIT and the Dana-Farber/Harvard Cancer Center (PIs: A.A. and L.A.M.). X.A.S. was supported by the Virginia and D.K. Ludwig Fund for Cancer Research, a Jane Coffin Childs Memorial Fellowship, and the Center for Prostate Disease Research core funding (HU0001-23-2-0008). K.H.S. and L.A.M. were supported by a PEER Award by the Zhu Family Center for Global Cancer Prevention. K.H.S., K.L.P., T.L.L., W.A., and L.A.M. are Prostate Cancer Foundation Young Investigators. The Health Professionals Follow-up Study is supported by the National Cancer Institute (NCI U01 CA167552). This work was supported in part by grants P01 CA228696, 5P30 CA014051, P30 CA006516, and P30 CA008748 from the NCI. A.A. was supported as an investigator of the Howard Hughes Medical Institute, from the NIH via R01 HD085866 and by the Paul F. Glenn Center for Biology of Aging Research at MIT. M.G.V.H. acknowledges support from the MIT Center for Precision Cancer Medicine, the Ludwig Center at MIT and the NIH (R35 CA242379).
Author contributions
X.A.S., K.H.S., M.G.V.H., A.A., and L.A.M. designed research; X.A.S., K.H.S., D.R.S., Z.L., K.L.P., T.L.L., W.A., E.G.D., and L.A.M. performed research; X.A.S., K.H.S., D.M., P.A.S., K.L.P., T.L.L., W.A., E.G.D., K.L., T.J., S.H., M.L., and L.A.M. analyzed data; X.A.S. led collaborations; X.A.S., K.H.S., M.G.V.H., A.A., and L.A.M. conceptualized and initiated this project; D.R.S. developed the organoid cultures; M.B. interpreted experimental data; and X.A.S. and K.H.S. wrote the paper.
Competing interests
M.G.V.H. discloses that he is a scientific advisor for Agios Pharmaceuticals, Auron Therapeutics, iTeos Therapeutics, Droia Ventures, Lime Therapeutics, Pretzel Therapeutics, and Sage Therapeutics. L.A.M. reports research funding from Janssen and Astra Zeneca; serves on the scientific advisory board and holds equity interest in Convergent Therapeutics; and was a consultant to Bayer Pharmaceuticals. W.A. discloses honoraria from Roche, Medscape, Aptitude Health, Clinical Education Alliance, OncLive/MJH Life Sciences, touchIME, Pfizer, theMedNet; serves as a consulting or advisory role in Clovis Oncology, Janssen, Overcoming Resistance In Cancer (ORIC) Pharmaceuticals, Daiichi Sankyo, AstraZeneca/MedImmune, Pfizer, Laekna Therapeutics, MOMA Therapeutics, Endeavor BioMedicines.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
Experimental data are available upon request to the lead author. The organoid RNA and DNA sequencing data from this study are available on Gene Expression Omnibus (accession number GSE265776) (67). Prostate cancer data from the HPFS and PHS are available through an HPFS project proposal as described at https://www.hsph.harvard.edu/hpfs/for-collaborators; transcriptome data are available on Gene Expression Omnibus (accession number GSE62872) (68).
Supporting Information
References
- 1.Beroukhim R., et al. , The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weaver B. A., Cleveland D. W., Does aneuploidy cause cancer? Curr. Opin. Cell Biol. 18, 658–667 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Taylor A. M., et al. , Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell 33, 676–689.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stopsack K. H., et al. , Aneuploidy drives lethal progression in prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 116, 11390–11395 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.El Gammal A. T., et al. , Chromosome 8p deletions and 8q gains are associated with tumor progression and poor prognosis in prostate cancer. Clin. Cancer Res. 16, 56–64 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Stopsack K. H., et al. , Differences in prostate cancer genomes by self-reported race: Contributions of genetic ancestry, modifiable cancer risk factors, and clinical factors. Clin. Cancer Res. 28, 318–326 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alshalalfa M., et al. , Chromosome 8q arm overexpression is associated with worse prostate cancer prognosis. Urol. Oncol. 41, 106.e17–106.e23 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matejcic M., et al. , Germline variation at 8q24 and prostate cancer risk in men of European ancestry. Nat. Commun. 9, 4616 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ben-David U., Amon A., Context is everything: Aneuploidy in cancer. Nat. Rev. Genet. 21, 44–62 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Li R., Zhu J., Effects of aneuploidy on cell behaviour and function. Nat. Rev. Mol. Cell Biol. 23, 250–265 (2022). [DOI] [PubMed] [Google Scholar]
- 11.Davoli T., et al. , Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 155, 948–962 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng H., Zhang N., Pati D., Cohesin subunit RAD21: From biology to disease. Gene 758, 144966 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Easton D. F., Eeles R. A., Genome-wide association studies in cancer. Hum. Mol. Genet. 17, R109–115 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Grisanzio C., Freedman M. L., Chromosome 8q24-associated cancers and MYC. Genes Cancer 1, 555–559 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim J. H., et al. , Integrative analysis of genomic aberrations associated with prostate cancer progression. Cancer Res. 67, 8229–8239 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Su X. A., et al. , RAD21 is a driver of chromosome 8 gain in Ewing sarcoma to mitigate replication stress. Genes Dev. 35, 556–572 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tomlins S. A., et al. , Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 310, 644–648 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Clark J. P., Cooper C. S., ETS gene fusions in prostate cancer. Nat. Rev. Urol. 6, 429–439 (2009). [DOI] [PubMed] [Google Scholar]
- 19.The Cancer Genome Atlas Research Network, The molecular taxonomy of primary prostate cancer. Cell 163, 1011–1025 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stopsack K. H., et al. , Oncogenic genomic alterations, clinical phenotypes, and outcomes in metastatic castration-sensitive prostate cancer. Clin. Cancer Res. 26, 3230–3238 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salles D. C., et al. , Assessment of MYC/PTEN status by gene-protein assay in grade group 2 prostate biopsies. J. Mol. Diagn. 23, 1030–1041 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pettersson A., et al. , Myc overexpression at the protein and mRNA level and cancer outcomes among men treated with radical prostatectomy for prostate cancer. Cancer Epidemiol. Biomarkers Prev. 27, 201–207 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stark J. R., et al. , Gleason score and lethal prostate cancer: Does 3 + 4 = 4 + 3? J. Clin. Oncol. 27, 3459–3464 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahearn T. U., et al. , A prospective investigation of PTEN Loss and ERG expression in lethal prostate cancer. J. Natl. Cancer Inst. 108, djv346 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stopsack K. H., et al. , p53 immunohistochemistry to identify very high-risk primary prostate cancer: A prospective cohort study with three decades of follow-up. Eur. Urol. Oncol. 6, 110–112 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guedes L. B., et al. , Analytic, preanalytic, and clinical validation of p53 IHC for detection of TP53 missense mutation in prostate cancer. Clin Cancer Res. 23, 4693–4703 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baena E., et al. , ETV1 directs androgen metabolism and confers aggressive prostate cancer in targeted mice and patients. Genes Dev. 27, 683–698 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Linn D. E., Penney K. L., Bronson R. T., Mucci L. A., Li Z., Deletion of interstitial genes between TMPRSS2 and ERG promotes prostate cancer progression. Cancer Res. 76, 1869–1881 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klezovitch O., et al. , A causal role for ERG in neoplastic transformation of prostate epithelium. Proc. Natl. Acad. Sci. U.S.A. 105, 2105–2110 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tomlins S. A., et al. , Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia 10, 177–188 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bose R., et al. , ERF mutations reveal a balance of ETS factors controlling prostate oncogenesis. Nature 546, 671–675 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deneen B., Denny C. T., Loss of p16 pathways stabilizes EWS/FLI1 expression and complements EWS/FLI1 mediated transformation. Oncogene 20, 6731–6741 (2001). [DOI] [PubMed] [Google Scholar]
- 33.Lessnick S. L., Dacwag C. S., Golub T. R., The Ewing’s sarcoma oncoprotein EWS/FLI induces a p53-dependent growth arrest in primary human fibroblasts. Cancer Cell 1, 393–401 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Subramanian A., et al. , Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mootha V. K., et al. , PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Kotsantis P., Petermann E., Boulton S. J., Mechanisms of oncogene-induced replication stress: Jigsaw falling into place. Cancer Discov 8, 537–555 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karthaus W. R., et al. , Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell 159, 163–175 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Franke T. F., Hornik C. P., Segev L., Shostak G. A., Sugimoto C., PI3K/Akt and apoptosis: Size matters. Oncogene 22, 8983–8998 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Vidotto T., et al. , Association of self-identified race and genetic ancestry with the immunogenomic landscape of primary prostate cancer. JCI Insight 8, e162409 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lunardi A., et al. , Suppression of CHK1 by ETS family members promotes DNA damage response bypass and tumorigenesis. Cancer Discov. 5, 550–563 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arnould C., et al. , Loop extrusion as a mechanism for formation of DNA damage repair foci. Nature 590, 660–665 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meisenberg C., et al. , Repression of transcription at DNA breaks requires cohesin throughout interphase and prevents genome instability. Mol. Cell 73, 212–223 e217 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Piazza A., et al. , Cohesin regulates homology search during recombinational DNA repair. Nat. Cell Biol. 23, 1176–1186 (2021). [DOI] [PubMed] [Google Scholar]
- 44.Strom L., Lindroos H. B., Shirahige K., Sjogren C., Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol. Cell 16, 1003–1015 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Potts P. R., Porteus M. H., Yu H., Human SMC5/6 complex promotes sister chromatid homologous recombination by recruiting the SMC1/3 cohesin complex to double-strand breaks. EMBO J. 25, 3377–3388 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Unal E., et al. , DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol. Cell 16, 991–1002 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Tittel-Elmer M., et al. , Cohesin association to replication sites depends on rad50 and promotes fork restart. Mol. Cell 48, 98–108 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deng P., et al. , RAD21 amplification epigenetically suppresses interferon signaling to promote immune evasion in ovarian cancer. J. Clin. Invest. 132, e159628 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sfanos K. S., et al. , Phenotypic analysis of prostate-infiltrating lymphocytes reveals TH17 and Treg skewing. Clin. Cancer Res. 14, 3254–3261 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stopsack K. H., et al. , Tumor protein expression of the DNA repair gene BRCA1 and lethal prostate cancer. Carcinogenesis 41, 904–908 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dominguez-Sola D., Gautier J., MYC and the control of DNA replication. Cold Spring Harb. Perspect. Med. 4, a014423 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rohban S., Cerutti A., Morelli M. J., d’Adda di Fagagna F., Campaner S., The cohesin complex prevents Myc-induced replication stress. Cell Death Dis. 8, e2956 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Toyoshima M., et al. , Functional genomics identifies therapeutic targets for MYC-driven cancer. Proc. Natl. Acad. Sci. U.S.A. 109, 9545–9550 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sinnott J. A., et al. , Prognostic utility of a new mRNA expression signature of gleason score. Clin. Cancer Res. 23, 81–87 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pettersson A., et al. , The TMPRSS2:ERG rearrangement, ERG expression, and prostate cancer outcomes: A cohort study and meta-analysis. Cancer Epidemiol. Biomarkers Prev. 21, 1497–1509 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Flavin R., et al. , SPINK1 protein expression and prostate cancer progression. Clin. Cancer Res. 20, 4904–4911 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stopsack K. H., et al. , Extent, impact, and mitigation of batch effects in tumor biomarker studies using tissue microarrays. Elife 10, e71265 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cheng D. T., et al. , Memorial sloan kettering-integrated mutation profiling of actionable cancer targets MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J. Mol. Diagn. 17, 251–264 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Keegan N. M., et al. , Clinical annotations for prostate cancer research: Defining data elements, creating a reproducible analytical pipeline, and assessing data quality. Prostate 82, 1107–1116 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chakravarty D., et al. , Oncokb: A precision oncology knowledge base. JCO Precis. Oncol. 2017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Spurr L. F., et al. , Quantification of aneuploidy in targeted sequencing data using ASCETS. Bioinformatics 37, 2461–2463 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Drost J., et al. , Organoid culture systems for prostate epithelial and cancer tissue. Nat. Protoc 11, 347–358 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gyori B. M., Venkatachalam G., Thiagarajan P. S., Hsu D., Clement M. V., OpenComet: An automated tool for comet assay image analysis. Redox Biol. 2, 457–465 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mildrum S., et al. , High-throughput minitaturized RNA-seq library preparation. J. Biomol. Tech. 31, 151–156 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sakaue-Sawano A., et al. , Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132, 487–498 (2008). [DOI] [PubMed] [Google Scholar]
- 66.Abe T., et al. , Visualization of cell cycle in mouse embryos with Fucci2 reporter directed by Rosa26 promoter. Development 140, 237–246 (2013). [DOI] [PubMed] [Google Scholar]
- 67.Su X. A., et al. , Data from “RAD21 promotes oncogenesis and lethal progression of prostate cancer.” Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE265776. Deposited 24 April 2024. [DOI] [PMC free article] [PubMed]
- 68.Penney K., et al. , Data from “Association of Prostate Cancer Risk Variants with Gene Expression in Normal and Tumor Tissue”. Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE62872. Deposited 30 October 2014. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
Experimental data are available upon request to the lead author. The organoid RNA and DNA sequencing data from this study are available on Gene Expression Omnibus (accession number GSE265776) (67). Prostate cancer data from the HPFS and PHS are available through an HPFS project proposal as described at https://www.hsph.harvard.edu/hpfs/for-collaborators; transcriptome data are available on Gene Expression Omnibus (accession number GSE62872) (68).