

Abstract
The interaction of coenzyme with thermostable homotetrameric NAD(H)-dependent alcohol dehydrogenase from the thermoacidophilic sulphur-dependent crenarchaeon Sulfolobus solfataricus (SsADH) and its N249Y (Asn-249→Tyr) mutant was studied using the high fluorescence sensitivity of its tryptophan residues Trp-95 and Trp-117 to the binding of coenzyme moieties. Fluorescence quenching studies performed at 25 °C show that SsADH exhibits linearity in the NAD(H) binding [the Hill coefficient (h)∼1) at pH 9.8 and at moderate ionic strength, in addition to positive co-operativity (h=2.0–2.4) at pH 7.8 and 6.8, and at pH 9.8 in the presence of salt. Furthermore, NADH binding is positively co-operative below 20 °C (h∼3) and negatively co-operative at 40–50 °C (h∼0.7), as determined at moderate ionic strength and pH 9.8. Steady-state kinetic measurements show that SsADH displays standard Michaelis–Menten kinetics between 35 and 45 °C, but exhibits positive and negative co-operativity for NADH oxidation below (h=3.3 at 20 °C) and above (h=0.7 at 70–80 °C) this range of temperatures respectively. However, N249Y SsADH displays non-co-operative behaviour in coenzyme binding under the same experimental conditions used for the wild-type enzyme. In loop 270–275 of the coenzyme domain and segments at the interface of dimer A–B, analyses of the wild-type and mutant SsADH structures identified the structural elements involved in the intersubunit communication and suggested a possible structural basis for co-operativity. This is the first report of co-operativity in a tetrameric ADH and of temperature-induced co-operativity in a thermophilic enzyme.
Keywords: archaeal alcohol dehydrogenase, coenzyme binding, co-operativity, fluorescence quenching, Hill coefficient, inter-subunit communication
Abbreviations: 3D, three-dimensional; ADH, alcohol dehydrogenase; SsADH, Sulfolobus solfataricus ADH; apo SsADH, coenzyme-free SsADH; ETX, 2-ethoxyethanol; h, Hill coefficient; holo SsADH, SsADH–NAD(H) complex; mSsADH, N249Y mutant of SsADH
INTRODUCTION
Studies of thermophilic enzymes and their homologous counterparts from mesophilic sources have yielded a wealth of information on the structural basis of protein function and stability under extreme conditions (reviewed in [1,2]), and have also highlighted the close relationship between conformational flexibility and enzyme function [3,4], as well as between structural fluctuations and thermal stability [5]. However, little is known about the function of thermoenzymes at temperatures far below their optimal temperature level due to the drop in catalytic efficiency, which limits kinetic investigations. Hydrogen tunnelling studies across a sizeable temperature range (65 °C to 5 °C) have nonetheless been successfully performed using ADH (alcohol dehydrogenase) from Bacillus stearothermophilus [6]. It is well known that temperature exerts a considerable influence on the balanced interplay of structural flexibility and rigidity, affecting ligand-binding ability and catalytic rate. The rigidification of an oligomeric thermophilic enzyme at low temperature, resulting in restrictions of the conformational fluctuations necessary for catalytic function [3], may also affect the subtle communication occurring between the subunits. It is expected that this effect is more pronounced in enzymes whose subunit operates through the relative motions of domains. Dehydrogenases present these characteristics, displaying a predominantly hinged domain motion for their function [7].
ADH from Sulfolobus solfataricus (SsADH), an aerobic crenarchaeon that grows optimally at 80 °C and pH≈3 and maintains its cytoplasmic pH at approx. 6.5, is a thermophilic NAD+-dependent homotetrameric zinc 150-kDa enzyme endowed with remarkable thermostability and broad substrate specificity. Cloning and over-expression in Escherichia coli of the SsADH gene, as well as the kinetic and thermostability properties of the enzyme, have been reviewed previously [8]. The 3D (three-dimensional) structure of the free enzyme and that of the enzyme–coenzyme–2-ethoxyethanol complex have been recently determined at 1.85 and 2.3 Å resolution respectively [9,10]. One interesting feature is that the domain closure occurring on coenzyme binding is more similar by extension (i.e. an 11 ° rotation) to that observed for the classical horse liver ADH (10 ° rotation) than for ADHs from bacterial counterparts (2.5 ° rotation) [10]. The 3D structure of the apo form of the mutant N249Y (Asn-249 →Tyr, mSsADH) has also been solved [11]. The substitution located in the coenzyme-binding domain decreases the affinity for both coenzyme and substrate, resulting in greater catalytic activity with respect to that of the parent protein [11]. The wild-type SsADH has a high affinity for NAD+ and NADH, as shown in fluorescence binding studies, and the Kd values are 0.10 and 0.05 μM respectively. Although the coenzyme binding is near to linearity under the experimental conditions used, i.e. pH 9.8, 25 °C [8], preliminary binding studies performed at pH 7.8 and 25 °C yielded a Hill coefficient (h) value of near 2, suggesting that the NAD(H) binding to SsADH is of a somewhat more complex nature [12].
In the present paper, the high fluorescence sensitivity of the two tryptophan residues per subunit, Trp-95 and Trp-117, to the binding of coenzyme moieties is used for coenzyme-binding studies at different pH, ionic strength and temperature. Although our results suggest that NAD+ and NADH binding to SsADH displays positive and negative co-operativity below 25 °C and above 35 °C respectively, no co-operative behaviour occurs with mSsADH. Steady-state kinetics performed in the temperature range of 20–80 °C showed that the wild-type enzyme exhibits kinetic co-operativity for coenzyme and progressively changes from positive to negative as the temperature is increased, whereas the mutant enzyme exhibits hyperbolic kinetics. An analysis of the SsADH and mSsADH structures identifies the structural segments involved in intersubunit communication and suggests a possible co-operativity mechanism. This change in the binding mechanism may be of interest regarding the conditions in vivo. To our knowledge, this is the first report of co-operativity in a tetrameric ADH and of a temperature-induced co-operativity in a thermophilic enzyme.
EXPERIMENTAL
Chemicals
NAD+ and NADH were purchased from Applichem (Darmstadt, Germany). Other chemicals were A grade substances from Sigma (St. Louis, MO, U.S.A.) or Applichem. NADH solutions were prepared daily in 20 mM Tris/HCl, pH 8.2, using nitrogen-purged water, whereas stock solutions of NAD+ were prepared in water, adjusted to approx. pH 6.5 with dilute NaOH and stored frozen at −20 °C. The purity of the coenzyme solutions was checked by determining the absorption quotients as indicated by the manufacturer. All solutions were made up with MilliQ water.
Recombinant protein purification
The wild-type and mutant N249Y SsADH were expressed and purified from E. coli, as described previously [8]. The complete removal of endogenous NAD(H) traces tightly bound to the wild-type SsADH was obtained by exhaustive dialysis, as described previously [8].
Protein concentration determination
Protein concentration was determined with a Bio-Rad protein assay kit using BSA as a standard and introducing a correction factor deduced from quantitative amino acid analysis. The correction factor used is in agreement with that calculated by spectrophotometric [8] and spectrofluorimetric titration of the active site of SsADH with NAD+ in the presence of excess pyrazole. In the present study, titration of apo SsADH (coenzyme-free SsADH) (typically 0.1–0.3 μM tetramer in the presence of 50 mM glycine/NaOH, pH 9.8, and 5 mM pyrazole) was performed, monitoring the quenching of the fluorescence (λexcitation=280 nm) that occurs following the addition of NAD+. The active-site concentration determined by the intersection of the titration plot gave an almost identical result to that deduced from the colorimetric method. Throughout the text ‘μM enzyme’ refers to the tetramer concentration.
Fluorescence binding studies
The fluorescence spectra of SsADH were obtained with a JASCO FP-777 spectrofluorometer, using a quartz cuvette containing 500 μl of enzyme solution thermostated at 25 °C. Both the enzyme solution and buffers were filtered through a 0.45 μm filter (Millipore) and all spectra were corrected for buffer fluorescence. NAD+ and NADH binding to SsADH and mSsADH were studied by monitoring the intrinsic fluorescence decrease in the protein occurring after coenzyme addition, or by monitoring the increase in emission of NADH excited at 340 nm due to the formation of the enzyme–coenzyme complex. In all cases, 2-μl aliquots of 10–30 μM NAD+ or NADH for SsADH or 3 mM NADH for mSsADH were added to 500 μl of the enzyme solution (0.1 μM tetramer) until the lowest change in fluorescence intensity (ΔF) was obtained. The enzyme was pre-incubated in each mixture for 20 min before titration.
The treatment of binding data to determine the dissociation constant, Kd, was performed using GraFit software (version 3.09b, Erithacus Software Ltd., Staines, U.K.) as follows. ΔFmax (maximal fluorescence change with complete saturation) was evaluated from the linear part of the double-reciprocal plot 1/ΔF versus 1/L, where L is the concentration of coenzyme added; the molar fraction (α) was obtained at each coenzyme concentration from the ratio ΔF/ΔFmax. The quantities αE0 (where E0 is the initial enzyme normality) and L-αE0 give the concentration of bound (Lb) and free (Lf) coenzymes in solution respectively. This formula is based on the assumption that fluorescence change is directly proportional to the amount of enzyme–coenzyme complex, and that the enzyme binds four ligand molecules per molecule. The Scatchard equation was used for the binding data analysis:
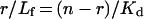 |
(1) |
where r is the number of moles of ligand bond per mole of enzyme at a given concentration of free ligand Lf, n is the number of binding sites per mole of enzyme at infinite Lf, and Kd is the dissociation constant of the enzyme–ligand complex [14]. When there is no interaction between multiple binding sites, i.e. when the sites are identical and independent, the r/Lf versus r plot is linear. Where plots of r/Lf versus r were non-linear, thus indicating co-operative interaction between the protein subunits, they were linearized through the use of an appropriate exponent for Lf [15]. The exponent was determined from a Hill plot of the binding data:
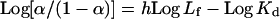 |
(2) |
where h and Kd are determined from the slope and intercept of the curve, respectively. h is the linearizing exponent in the equation:
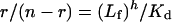 |
(3) |
which is basically a variant of eqn 1. Although each of the two plotting methods, Scatchard and Hill, contains the same intrinsic information concerning the binding equilibrium, the Hill plot was preferred for plotting data showing positive co-operativity [14]. Adair constants were determined by fitting the coenzyme binding data to the Adair equation for a molecule with four binding sites using GraFit software (eqn 4):
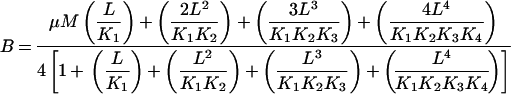 |
(4) |
where B is the amount bound, L is the concentration of free ligand, μM is the tetramer concentration, and Ki are the stepwise Adair equation constants expressed as dissociation constants. Intrinsic site dissociation constants Ki′ were calculated from the Adair constants using the following statistical relationships ([16]; eqns 5–8):
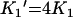 |
(5) |
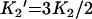 |
(6) |
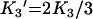 |
(7) |
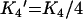 |
(8) |
The A280 value of the final titration mixtures was less than 0.1 for SsADH, so that the inner filter effect was neglected. In the case of mSsADH, A280>1 and the fluorescence data were corrected for the inner-filter effect according to the Brand and Witholt procedure [17]. All titrations were performed in triplicate. The variation coefficients for the Kd and h estimates were less than 5% and 5–10% for SsADH and mSsADH respectively.
Kinetics
SsADH activity assays at temperatures above 40 °C were performed spectrophotometrically as described by Raia et al. [8] using a Cary 1E spectrophotometer. A fluorimetric assay was used to detect poor SsADH activity at temperatures below 40 °C using the JASCO FP-777 spectrofluorometer equipped with an external thermo-cryostated water bath. The change in NADH fluorescence was recorded as a function of time, with λexcitation and λemission fixed at 340 and 463 nm respectively. In both methods, the reaction began when the coenzyme solution (5–20 μl) was added to a quartz or UV-grade methacrylate cuvette pre-incubated for 3 min at the determined temperature and containing 5 to 30 μg of enzyme and 0.25 mM benzaldehyde in 50 mM glycine/NaOH, pH 9.8. The kinetic constants of mSsADH for NAD+ were spectrophotometrically determined between 30 °C and 65 °C at the fixed concentrations of 20 mM cyclohexanol and various concentrations of coenzyme in 50 mM glycine/NaOH, pH 9.8. Correction of the ΔA340 value for high temperature NAD+ degradation was introduced as reported previously [8]. Kinetic parameter measurements were carried out in duplicate or triplicate and data were fitted either to the Michaelis–Menten equation or to the Hill equation using GraFit software. The variation coefficients were approx. 5% for Vmax, 15% for Km and 5–16% for h estimates. The thermodynamic parameters governing the enzyme reaction and the interaction between coenzyme and enzyme were determined using the van't Hoff analysis [18]. The values of ΔH‡, ΔG‡ and ΔS‡ refer to 1 mol per binding site.
CD and fluorescence studies
CD data was collected by means of a JASCO J-710 spectropolarimeter equipped with computer-controlled temperature cuvette holders and kept under a constant N2 flux. Far-UV CD spectra in the 190–250 nm region were recorded with 1.0-mm path-length cells containing 0.5 μM tetramer in 50 mM sodium phosphate, pH 6.8, or 17 mM sodium pyrophosphate, pH 9.8, in the absence and presence of 0.1 M NaCl. The holo SsADH form [SsADH–NAD(H) complex] was prepared by adding 20 μM NADH to the apo SsADH solution. The enzyme solutions were filtered through a 0.45 μm filter before the measurements were taken. The spectra were recorded in the temperature mode every 5 °C over the range of 15 °C–70 °C with a scan rate of 1.0 °C/min, and the final spectra, which represented the average of at least three tracings, were corrected for the buffer. Ellipticity, [θ], expressed in terms of mean residue ellipticity (deg·cm2·dmol−1) at 222 nm, was calculated from the equation [θ]=θ/10×C×l, where θ is ellipticity measured in mdeg, C represents the mean residue molar concentration (mol/l) and l represents the optical path-length in cm. The effect of temperature on SsADH and mSsADH fluorescence was investigated up to an instrumental limit of 65 °C as reported previously [19], using 0.15 μM enzyme concentration in 50 mM glycine/NaOH, pH 9.8.
Structural analysis
Structural analysis was carried out on the basis of the atom co-ordinates of the apo (PDB entry 1JVB, [9]) and holo (PDB entry 1R37, [10]) forms of SsADH and mSsADH (PDB entry 1NTO, [11]) using the Swiss-Pdb Viewer, version 3.7 program [20]. The crystallographic structure of mSsADH in the holo form has not been solved to date, given its very low affinity for coenzymes. The solvent exposure of residues was evaluated using the DSSP program [21]. This method, which determines the residue accessibility value to a solvent in Å2, establishes whether a residue is either exposed or buried if its accessibility is greater or less than 16% compared with the accessibility of a free residue.
RESULTS
Fluorescence properties
The SsADH subunit contains two tryptophan residues, Trp-95 and Trp-117, located within the catalytic domain and thirteen tyrosine residues dispersed throughout the structure [9]. Steady-state fluorescence and structural studies recently published [19,22] have established the structural basis of dependence of the SsADH intrinsic fluorescence properties on the peculiarities of its fluorophores location. Following excitation at 280 nm, the apo SsADH fluorescence emission is dominated by tryptophan fluorescence with a relatively blue (λmax=320 nm) spectrum. A sophisticated theoretical analysis of the tryptophan and tyrosine position [22] determined that only four tyrosine residues located in the coenzyme-binding domain contribute directly to SsADH fluorescence, whereas the other nine residues have no significant effect due to non-radiative energy transfer to the tryptophan residues. One important characteristic is that the addition of saturating concentrations of NADH to the SsADH apo form results in 58% quenching of intrinsic fluorescence, with a negligible blue shift of λmax and a concurrent appearance of a second peak at about 422 nm due to the radiationless energy transfer between the enzyme and reduced nicotinamide ring. After excitation at 295 and 280 nm, NAD+ binding to the apo-enzyme also results in protein fluorescence quenching accompanied by a negligible blue shift, thus suggesting that there is no significant change in the apolarity of the environment surrounding the tryptophan residue(s) [19]. This is in agreement with the fact that the two fluorophores belong to a hydrophobic cluster, which includes not only Pro-94, Pro-115 and Phe-49, but also Trp-95 and Trp-117. Moreover, the accessibility values to the relative solvent of the Trp-95 side chain are approx. 1, 3.5 and 1%, whereas those of the Trp-117 side chain are approx. 7, 18 and 10% for the apo SsADH, holo SsADH and mSsADH respectively. The indole rings of Trp-95 and Trp-117 lie at distances of approx. 8 Å and 11 Å respectively, from the nicotinamide moiety of the coenzyme, whereas within the monomer they are fairly close to each other, i.e. 5.8 Å and 5.6 Å for the apo and holo form respectively [9,10]. However, unlike the two tryptophans, which lie so close in the monomer, the distances between Trp-95 and Trp-117 of monomer A and their counterparts in monomers B, C and D are between 30 Å and 50 Å. This indicates that the energy transfer process between tryptophan and NADH occurs more efficiently within the same subunit than among subunits. Lastly, Trp-95 and Trp-117 residues are included in a hydrophobic cluster and both contribute towards transferring the energy to the reduced coenzyme, although presumably to a varying extent. More importantly, the tryptophan fluorescence of the archaeal ADH is highly responsive to the subtle structural changes that occur in the protein molecule upon coenzyme binding.
Fluorescence binding studies
Binding of the oxidized and reduced form of the coenzyme was studied at different pH, ionic strength and temperature. Figures 1(A) and 1(B) shows the Scatchard and Hill plots of NADH binding to SsADH obtained at 25 °C in 50 mM glycine/NaOH, pH 9.8. The NADH binding is almost non-co-operative in this medium with the h value of 1.15±0.01 and Kd values of 0.12 and 0.20 μM calculated from Hill and Scatchard plots respectively. When fitting the binding data to the Adair equation (Figure 1B, inset, continuous line), the intrinsic dissociation constants K1′=0.30±0.076, K2′=0.11±0.04, K3′=0.27±0.09 and K4′=0.07±0.03 μM are provided. These can be derived in turn from the Adair constants. Figures 1(C) and 1(D) show the Scatchard and Hill plots for the NADH binding obtained in 50 mM Tris/HCl, pH 7.8. The Scatchard plot presents the typical form of a system exhibiting positive co-operativity with h=1.86±0.02 and Kd=0.077 and 0.072 μM calculated from the Hill and linearized Scatchard plots respectively. An analysis of the binding data with the Adair equation (Figure 1D, inset, continuous line) and the application of eqns 5–8 yields the intrinsic dissociation constants K1′=1.2±0.15, K2′=0.36±0.07, K3′=0.14±0.02 and K4′=0.05±0.01 μM. NADH fluorescence enhancement at 423 nm, which occurs after excitation at 340 nm (results not shown), yielded the same qualitative appearance and quantitative analysis of NADH binding obtained by monitoring intrinsic protein fluorescence quenching. This practically rules out the possibility that the curved Scatchard plot is the result of an artefact connected to enzyme purity or complications associated with fluorescence measurement. Table 1 lists the Kd, intrinsic site dissociation constants and h values determined for NAD+ and NADH at pH 6.8 to 9.8 using different buffers at 25 °C. The values of Kd from the Hill plot only are reported in order to facilitate a homogeneous evaluation of data. The positive co-operativity for NAD+ and NADH increases as pH decreases from 9.8 to 6.8. The affinity of the enzyme for NAD+ increases as pH increases, whereas the affinity for NADH does not change significantly. Similar behaviour for horse liver ADH has been attributed to pH-dependent variations in electrostatic attractions acting as a steering force behind the coenzyme-docking process [23]. Although the intrinsic site dissociation constants suggest a high degree of positive co-operativity in the binding to the second site between pH 6.8 and 8.8 for both NADH and NAD+, there is almost no co-operativity between the four subunits observed at pH 9.8 for both coenzyme forms. However, a slight positive co-operativity characterizes the NAD+ binding to the third and fourth site at pH 6.8. Different buffers provide similar results, indicating that the co-operativity phenomenon does not depend on the chemical species of the buffer. In subsequent investigations, glycine/NaOH was preferred to the Tris/HCl buffer due to the large dissociation constant shift in temperature of the latter and also because it is a substrate (hydrogen donor) of ADH.
Figure 1. Binding of coenzyme to SsADH at 25 °C and two different pH values.

(A) Scatchard plot for the interaction of NADH with SsADH in 50 mM glycine/NaOH, pH 9.8, at 25 °C. (B) Hill plot of the same data; the inset shows the solid line of the data fitted to the Adair equation for four binding sites. Enzyme concentration, 0.15 μM. (C) Scatchard plot for the interaction of NADH with SsADH in 50 mM Tris/HCl, pH 7.8, at 25 °C. The inset depicts the same data using the linearizing exponent h=1.9 determined by the Hill plot shown in (D). The inset in (D) shows the solid line of the data fit to the Adair equation for four binding sites. Enzyme concentration, 0.10 μM.
Table 1. Effect of pH on NADH and NAD+ binding to SsADH.
The values of the constants are given in μM±S.D.
| pH | h | Kd | K1′ | K2′ | K3′ | K4′ |
|---|---|---|---|---|---|---|
| NADH | ||||||
| 6.8* | 2.0 | 0.047 | 3.1±0.8 | 0.09±0.03 | 0.13±0.04 | 0.16±0.07 |
| 7.8† | 1.9 | 0.077 | 1.2±0.15 | 0.36±0.07 | 0.14±0.02 | 0.05±0.03 |
| 8.8† | 1.5 | 0.024 | 0.31±0.06 | 0.04±0.01 | 0.05±0.01 | 0.03±0.08 |
| 9.8‡ | 1.1 | 0.085 | 0.30±0.05 | 0.22±0.05 | 0.07±0.01 | 0.10±0.04 |
| NAD+ | ||||||
| 6.8* | 2.3 | 0.50 | 6.9±1.7 | 0.99±0.36 | 0.40±0.15 | 0.20±0.09 |
| 7.8† | 2.4 | 0.094 | 3.4±0.6 | 0.94±0.20 | 0.23±0.04 | 0.23±0.06 |
| 8.8† | 1.4 | 0.080 | 0.94±0.32 | 0.08±0.05 | 0.14±0.03 | 0.24±0.15 |
| 9.8‡ | 1.1 | 0.072 | 0.14±0.02 | 0.16±0.04 | 0.066±0.014 | 0.24±0.19 |
* 50 mM sodium phosphate.
† 50 mM Tris/HCl.
‡ 50 mM glycine/NaOH.
Although N249Y substitution causes a substantial decrease in affinity for NAD+ and NADH [24], NADH was preferred for binding studies because of its lower Kd value. The Hill coefficient values at 25 °C for the mutant enzyme were 0.96±0.04 and 1.1±0.06 in 50 mM glycine/NaOH, pH 9.8, and 50 mM sodium phosphate, pH 6.8, respectively. The corresponding Kd values were 460±52 μM and 3.9±1.3 mM respectively.
Table 2 shows the intrinsic site dissociation constant, Kd, and h values for the NAD+ and NADH binding in different ammonium sulphate and sodium chloride concentrations in 50 mM glycine/NaOH, pH 9.8, and at 25 °C. Although the affinity of SsADH for the coenzyme does not noticeably change by increasing the ionic strength, the degree of positive co-operativity increases to approx. 2. The intrinsic site dissociation constants suggest a high degree of positive co-operativity in binding to the second site for both NAD+ and NADH in the presence of high salt concentration, followed by no significant co-operativity between the remaining sites. However, the binding of the second NAD+ molecule in the presence of 20 and 100 mM ammonium sulphate makes it more difficult for the next molecule to bind, which in turn makes it easier for the fourth to bind, i.e. the intrinsic constants combine in such a way as to produce a mixture of positive and negative co-operativity. A similar mechanism characterizes NADH binding in the presence of 150 mM NaCl (Table 2), where the Hill plot appears slightly convex with a change in slope at 50% saturation (results not shown) and yielding two values of h, 1.4 and 1.2.
Table 2. Effect of salt on NADH and NAD+ binding to SsADH.
The values of the constants are given in μM±S.D. The compositions of the buffers are as follows: A, 20 mM glycine/NaOH, pH 9.8; B, 50 mM glycine/NaOH, pH 9.8; C, 50 mM Tris/HCl, pH 8.8, at 25 °C. AS, ammonium sulphate.
| Buffer | h | Kd | K1′ | K2′ | K3′ | K4′ |
|---|---|---|---|---|---|---|
| NADH | ||||||
| A | 0.9 | 0.087 | 0.07±0.01 | 0.073±0.007 | 0.058±0.004 | 0.194±0.015 |
| B | 1.1 | 0.12 | 0.30±0.076 | 0.11±0.04 | 0.27±0.09 | 0.07±0.03 |
| B+20 mM AS | 1.3 | 0.043 | 0.34±0.05 | 0.05±0.01 | 0.06±0.01 | 0.06±0.012 |
| B+100 mM AS | 1.8 | 0.076 | 1.73±0.20 | 0.168±0.025 | 0.145±0.017 | 0.138±0.025 |
| C+100 mM AS | 1.6 | 0.049 | 0.98±0.08 | 0.069±0.006 | 0.13±0.01 | 0.10±0.01 |
| B+25 mM NaCl | 1.1 | 0.082 | 0.015±0.001 | 0.17±0.03 | 0.031±0.004 | 0.16±0.06 |
| B+150 mM NaCl | 1.4 (1.2)* | 0.066 (0.092)* | 0.75±0.22 | 0.061±0.028 | 0.267±0.061 | 0.12±0.11 |
| B+1 M NaCl | 1.6 | 0.12 | 0.31±0.06 | 0.04±0.01 | 0.05±0.01 | 0.035±0.008 |
| NAD+ | ||||||
| B | 1.1 | 0.072 | 0.14±0.02 | 0.16±0.04 | 0.066±0.014 | 0.24±0.19 |
| B+20 mM AS | 1.4 | 0.14 | 0.83±0.17 | 0.138±0.043 | 0.39±0.12 | 0.06±0.02 |
| B+100 mM AS | 1.8 | 0.66 | 13.4±1.6 | 0.195±0.02 | 1.15±0.11 | 0.53±0.08 |
| B+30 mM NaCl | 1.3 | 0.20 | 0.76±0.09 | 0.27±0.06 | 0.34±0.05 | 0.30±0.13 |
| B+150 mM NaCl | 1.5 | 0.28 | 218±83 | 0.44±0.16 | 0.0020±0.0007 | 0.934±0.82 |
* The values not in parentheses refer to the binding curve below 50% saturation, and those in parentheses refer to the binding curve above 50% saturation.
The effect of salt on NADH binding to mSsADH was different to that observed for SsADH. In fact, at 25 °C and in 50 mM glycine/NaOH, pH 9.8, containing 100 mM ammonium sulphate, the Hill coefficient and the Kd value for the mutant enzyme were 1.2±0.2 and 280±32 μM respectively.
The ionic strength of the buffers (Table 1) barely doubles when pH decreases from pH 9.8 to 6.8, whereas that of 50 mM glycine/NaOH, pH 9.8 increases approx. 14-fold following NaCl addition from 0.025 up to 1M (Table 2), thus suggesting that the co-operativity degree is more affected by pH than by ionic strength.
The effect of temperature on coenzyme binding to SsADH was investigated between 17 °C and 50 °C in 50 mM glycine/NaOH, pH 9.8. Figure 2(A) (continuous line) shows a typical binding curve for NADH at 17 °C obtained by fitting the experimental data to the Adair equation. The binding curve exhibits sigmoidicity at a low NADH concentration, corresponding to the positive co-operativity shown by the convex Scatchard plot of the same data (Figure 2A, inset). Plotting the data of Figure 2(A) in a Hill plot gives h values of 2.9 and 1.4 below and above 50% saturation respectively (Figure 2B). The presence of two slopes in the Hill plot with a change at approx. 50% saturation was observed for NADH binding at higher temperatures up to about 25 °C. Above this temperature, SsADH displays a co-operativity degree which changes from slightly positive to negative until 50 °C. Figure 2(C) shows a typical binding curve for NADH at 39 °C obtained according to Adair data analysis, whereas the inset shows a concave Scatchard plot, which is indicative of negative co-operativity. Hill analysis yields a h value of 0.79±0.02 and a Kd value of 0.079 μM (Figure 2D).
Figure 2. Binding of coenzyme to SsADH at pH 9.8 and two different temperature values.

(A) Positive co-operative binding of NADH to SsADH at 17 °C, pH 9.8. The continuous line is the data fit to the Adair equation (eqn 4) for four binding sites. Eqns (5–8) yield the intrinsic site dissociation constants K1′=5.38±0.65, K2′=0.13±0.02, K3′=0.08±0.01 and K4′=0.10±0.01 μM. (A, inset) and (B) are the Scatchard and convex Hill plots of the same data respectively. The slopes labelled 1 and 2 correspond to h=2.97±0.28 and Kd=0.014 μM, and h=1.36±0.02 and Kd=0.11 μM respectively. Enzyme concentration, 0.13 μM. (C) Negative co-operative binding of NADH to SsADH at 39 °C, pH 9.8. The continuous line is the data fit to the Adair equation (eqn 4) for four binding sites. The intrinsic site dissociation constants derived by eqns (5–8) are K1′=0.004±0.003, K2′=0.04±0.02, K3′=0.06±0.02 and K4′=0.08±0.04 μM. (C, inset) and (D) are the Scatchard and Hill plots of the same data respectively. The latter yields h=0.79±0.02 and Kd=0.080 μM. Enzyme concentration, 0.15 μM.
Figure 3(A) shows the effect of temperature on NADH binding investigated at two pH values for SsADH and mSsADH. The Hill coefficient for the wild-type enzyme at pH 9.8 decreases from 2.9 to 0.6 as temperature increases from 17 °C to 48 °C, whereas at pH 7.8 the Hill coefficient decreases from 2.8 to 1.1 as temperature increases from 20 °C to 40 °C. It is worth noting that the decrease in pH of one unit increases the degree of positive cooperativity along the temperature range. However, no co-operativity seems to occur for NADH binding to the mutant enzyme in the 18–48 °C temperature range, although a slight positive co-operativity is displayed at high temperatures. Figure 3(B) shows the effect of temperature in the 17–40 °C range, pH 9.8, on each intrinsic dissociation constant characterizing the NADH binding to each of the four SsADH sites. Although the binding of the first coenzyme molecule proves difficult at temperatures below 25 °C, it is easier for the next molecule to bind, after which binding to the remaining sites occurs without any apparent co-operativity between the subunits. At temperatures around 25 °C, the four constants do not significantly differ from each other, suggesting no co-operativity between the subunits in coenzyme binding. An Adair analysis of NADH binding at pH 7.8 yields similar results with K1′>K2′≈K3′≈K4′ (results not shown). Figure 3(B) shows the dependence of the dissociation constant on temperature calculated for SsADH and mSsADH from data giving the Hill coefficient values shown in Figure 3(A). The affinity of NADH for SsADH decreases approx. 10-fold as temperature increases from 17 ° to around 25 °C, after which it does not change significantly, hence ΔH‡≈0 kJ/mol, i.e. the binding is predominantly entropy-driven. The ΔG‡ and ΔS‡ values calculated for binding at 25 °C are −39.0 kJ/mol and 0.131 kJ/mol×K respectively; the former value is consistent with a very tight coenzyme–SsADH complex, whereas the latter reflects a major conformational change associated with the release of solvent molecules following the burial of a partially hydrophobic surface. Although a similar entropic contribution to the free energy was reported for NADH binding to horse liver ADH (ΔH‡=0.0 kJ/mol, ΔG‡=−36.8 kJ/mol, ΔS‡=0.123 kJ/mol×K; [25]), the normal situation for dehydrogenases is one in which the binding of NAD+ and NADH has large negative enthalpy and entropy terms typical of enthalpy–entropy compensation [25]. However, the binding of NADH to mSsADH at 25 °C is both enthalpically (ΔH‡∼32.0 kJ/mol) and entropically (ΔS‡=0.171 kJ/mol×K) unfavourable, with a ΔG‡ value of −19.0 kJ/mol which is consistent with a very weak coenzyme–SsADH complex. Control assays established that SsADH and mSsADH remained fully active under all the conditions adopted during fluorescence titration, thus indicating the remarkable robustness of the two enzymes [24].
Figure 3. Dependence of NADH binding to SsADH and mSsADH on temperature.

(A) h values were determined for SsADH in 50 mM glycine/NaOH, pH 9.8, (●) and 50 mM Tris/HCl, pH 7.8, (○), and for mSsADH (▲) in 50 mM glycine/NaOH, pH 9.8, at the indicated temperatures by fluorescence quenching as described in the Experimental section. pH values of the buffers were controlled at each temperature. (B) The intrinsic site dissociation constants describing the binding to four binding sites for SsADH. (●) K1′, (○) K2′, (▼) K3′ and (△) K4′. The inset in (B) shows the Kd for SsADH (●) and mSsADH (▲) reported against temperature. The results are expressed as the means±S.D. and are smaller than the symbol when absent in the Figure.
Kinetics
The kinetics displayed by SsADH for NADH oxidation is clearly sigmoidal at 20 °C and progressively changes to non-co-operative as the temperature is raised to 40 °C, after which it becomes almost negatively co-operative at higher temperatures (results not shown). Conversely, mSsADH displays hyperbolic kinetics for NAD+ reduction within the temperature range investigated (results not shown). Figure 4 shows the Hill coefficient values calculated from kinetic data as a function of temperature. Steady-state kinetic data broadly parallel the binding data (Figure 3A), indicating that true homotropic binding co-operativity (rather than a random kinetic mechanism [26]) is responsible for the co-operative kinetic behaviour observed. The SsADH affinity for NADH and its catalytic efficiency remain almost constant around 40 °C, after which both change (Figure 4, inset), with the latter increasing considerably, whereas the coenzyme binding becomes progressively less strong.
Figure 4. Dependence of kinetic co-operativity of SsADH and mSsADH on temperature.

Hill coefficient values for SsADH (●) were calculated from the steady-state kinetic parameters determined with a saturating concentration of benzaldehyde and variable NADH concentrations in 50 mM glycine/NaOH, pH 9.8, using fluorescence and spectrophotometric methods at temperatures below and above 40 °C respectively. The Hill coefficient values for mSsADH (○) were calculated from the steady-state kinetic parameters determined with a saturating concentration of cyclohexanol and variable NAD+ concentrations in 50 mM glycine/NaOH, pH 9.8, as described under the Experimental section. Inset, the dependence of Km (○) and kcat (△) of SsADH for NADH on temperature. The results are expressed as the means±S.D. and are smaller than the symbol when absent in the Figure.
Secondary and tertiary structure spectroscopic studies
CD spectra were recorded in the amide region to investigate whether the secondary structure of SsADH and mSsADH was affected by temperature, pH and salt, using the conditions yielding no co-operativity (pH 9.8, 25 °C) and positive co-operativity (pH 6.8, 25 °C, or pH 9.8 plus 0.1 M NaCl, 25 °C). Figure 5 summarizes the changes in CD ellipticity at 222 nm for both apo and holo forms of SsADH, as well as mSsADH at increasing temperatures. The molar ellipticity of apoSsADH and mSsADH increases by 20% following a temperature increase from 15 °C to 70 °C, whereas the molar ellipticity of holo SsADH increases by only 10%, thus reflecting a degree of structural tightening induced by the coenzyme binding. Figure 5 (inset) shows that the helical content of the three proteins at 25 °C is not significantly affected by pH or by the presence of salt.
Figure 5. Effect of temperature on the secondary structure of SsADH and mSsADH.

The dichroic activity at 222 nm for SsADH in the absence (●) and presence of NADH (○), and for N249Y mutant (△), was measured at various temperatures in 17 mM sodium pyrophosphate, pH 9.8. Inset, the dichroic activity of the three ADH forms as measured at 25 °C in 50 mM sodium phosphate, pH 6.8 (open bars), and 17 mM sodium pyrophosphate, pH 9.8, in the absence (dotted bars), and presence (hatched bars) of 0.1 M NaCl. Experimental tracings were accumulated and corrected for blanks by computer calculations, as described in the Experimental section. Enzymes and NADH concentrations were 0.5 and 20 μM respectively. The results are expressed as the means±S.D. for three independent experiments.
As the temperature rose from 10 °C to 65 °C, the fluorescence intensity of both SsADH and mSsADH decreased linearly by approx. 50% following excitation at 280 nm, and approx. 40% following excitation at 297 nm, whereas the position of the emission maximum shifted by 1.5±0.5 nm toward a longer wavelength for both the enzymes (results not shown). The slight red shift reflects the rigidity and hydrophobic character of the aromatic cluster where Trp-95 and Trp-117 are located. This would seem to indicate that a gradual thermal expansion of the molecule over the temperature range could permit only a very limited access of the solvent molecule to the tryptophan residue.
These results suggest that no apparent change in the secondary and tertiary structure of the SsADH molecule accompanies the change in the co-operativity degree.
Structure analysis
The SsADH 3D tetramer is a dimer of dimers, denoted as A–B and C–D. Each subunit is in contact with the three others, creating a tight packing arrangement [9]. An analysis of the apo structure reveals that there are 22, 3 and 5 hydrophobic residues and 13, 5 and 2 polar residues at A–B, A–C and A–D interfaces respectively, whose side chains form van der Waals contacts and salt bridges, as well as a number of hydrogen bonds. In order to formulate a hypothesis for a co-operative intersubunit communication mechanism, we considered the possibility that some of the above residues were also involved in the catalysis mechanism of the enzyme. Inspection of the apo and holo structures highlights several residues endowed with these characteristics located at the A–B interface and apparently none at the A–C and A–D interfaces. Figures 6 and 7 summarize the results of this analysis. Most of the residues listed in Figure 6 are distributed along the segments 46–62 and 269–296 shown in Figure 7(B). More importantly, Phe-49, Leu-272, Leu-295 and Val-296 of subunit A, and Leu-286 of subunit B, form the substrate-binding hydrophobic pocket together with other residues of subunit A, Ser-40, Trp-95, Ile-120 and Trp-117 [10]. Furthermore, Val-270, Gly-271, Leu-272, Phe-273, Ser-294 and Val-296 of subunit A, as well as Thr-285 of subunit B, make a number of hydrogen bonds and van der Waals contacts with the nicotinamide-ribose moiety of the coenzyme [10]. SsADH undergoes a substantial conformational change on coenzyme binding, characterized by a large rotation (11 °) of the catalytic domain relative to the coenzyme domain [10], and by a structural rearrangement of loops 46–62 and 270–275 at the domain interface. Indeed, the movement of the two loops are correlated due to the van der Waals interaction between Gly-50 and Gly-274, and between Phe-49 and Leu-272. Furthermore, the Phe-273 side chain swings about 8 Å following the apo–holo transition (Figure 7C), thus allowing loop 46–62 to rearrange and extensively interact through a complementary hydrophobic surface with the helix αF region of residues 280–286 of the nearby subunit B. This interface is lined by residues Pro-282B, Leu-283B, Thr-285B and Leu-286B [10]. The two β-strands, βS and βF at the A–B interface (Figure 7B) run anti-parallel, and the gap between them is bridged by specific side chain–side chain contacts that provide most of the interactions between A and B subunits. In particular, Glu-288 (Figures 7A and 7B) forms salt-links with His-110, Arg-161 and Lys-165 (Figure 6) of the second subunit, contributing significantly to the hyperthermostability of the archaeal enzyme [9]. The conformational rearrangement in the apo–holo transition leaves the position of the two β-strands, βS and βF, and helix αF essentially unchanged, with the exception of the segment 270–275.
Figure 6. Representation of the intersubunit interactions in the dimer A–B of apo SsADH.

Due to the crystallographic 222 symmetry of the molecule, a symmetrical interaction (jA–iB) exists for each interaction (iA–jB) between residue i of subunit A and residue j of subunit B. The contacts are listed only once. Residues whose hydrophobic side chains interact within 4.5 Å are shown in italics. Residues involved in electrostatic interactions are shown in bold [9].
Figure 7. Structural analysis of SsADH and mSsADH.

(A) Partial view of the SsADH holo structure looking at the A–B dimer of the homotetramer, illustrating the helix αF and β-strands, βS and βF of subunit A (red) and B (blue) at dimer interface (centre of Figure). The side chains of the residues involved in the interface interactions are coloured in pink (subunit A) and in yellow (subunit B). The coenzyme molecule and the Trp-95 and Trp-117 side chains are coloured in cyan and green respectively. The orange spheres represent the catalytic and structural zinc atoms. (B) α-Carbon traces of the 46–62 and 269–296 segments from both apo (blue) and holo (red) form of the subunit A, and apo (yellow) and holo (green) form of the subunit B of SsADH, are shown superimposed. Residues 276–279 and 289–294 belong to strand βS and βF respectively, connected by helix αF 280–288. The coenzyme NAD(H) and 2-ethoxyethanol (ETX) molecules are shown in grey. The red and green sphere represents the catalytic zinc in the holo form. (C) Comparison of the regions 46–62, 248–250 and 270–275 of the mutant N249Y SsADH (red) and the apo (blue) and holo (yellow) forms of wild-type SsADH structure. Residues 270–273 interact directly with the NADH molecule (cyan) [10]. In the SsADH apo form, the Phe-49 side chain occupies the same position as the ETX molecule (pink) in the ternary complex structure. The swing of the Phe-273 side chain occurring upon apo–holo transition is shown, as well as its position in the mSsADH apo structure. The movement of the Phe-273 side chain in SsADH allows loop 46–62 to rearrange and extensively interact with the helix αF region of residues 280–286 of the nearby subunit B [10]. Segment 46–62 is one of the most flexible parts of SsADH structure. It was found to be very disordered in the mutant N249Y apo structure due to the conformational changes induced by the mutation [10,11]. Important contacts between this loop and the segment 280–286 of the near subunit B are presumably disturbed or lost in the mutant structure, precluding proper intersubunit communication. This Figure was prepared using VMD [38] and Persistence of Vision Raytracer (http://www.povray.org/) (A, C), MolScript [39] and Raster3D [40] (B).
A detailed comparative analysis was reported previously [11] on both SsADH and mSsADH structures in order to explain the effect of N249Y substitution on active site. The substitution N249Y in the coenzyme domain generates a structural rearrangement of residues 247–250, accompanied by an adjustment in the conformation of segment 270–275 (between βE and βS) and of segment 280–283, the N-terminal part of helix αF ([11], Figure 7C). The main consequence of these changes is the loss of important interactions that contribute towards locking the adenine and nicotinamide moieties into the interdomain cleft, as well as a large increase in the disorder of loop 46–62 [10,11].
DISCUSSION
Structural analysis and experimental studies indicate that the binding of coenzyme to SsADH results in major conformational change and significant quenching of the protein fluorescence. The quenching induced by NADH binding can be attributed to a radiation-less energy transfer that is determined mainly by the distance and reciprocal orientation between the donor and acceptor [17]. However, the quenching induced by NAD+ binding may be correlated to the large conformational change occurring in the protein, since the structural rearrangements can bring quencher groups, such as amide or carboxyl, closer to the indole ring and/or render the distance and reciprocal orientation between the acceptor and donor less favourable to a non-radiative energy transfer.
The binding of four molecules of coenzyme to SsADH at pH values close to 10, in relatively low ionic strength and temperatures of around 30 °C, exhibits a Hill coefficient near to unity, indicating that the four binding sites are independent, i.e. there are no co-operative interactions between subunits. At lower temperatures and pH values and in relatively high ionic strength, the enzyme clearly exhibits positively co-operative behaviour. Although kinetic data accordingly indicate that co-operativity in SsADH is positive and negative at lower and higher temperatures respectively, the mutant N249Y SsADH displays non-co-operative behaviour in coenzyme binding by fluorescence quenching, as well as by kinetic studies under the same experimental conditions used for the wild-type enzyme.
Structural analyses suggest a plausible mechanism for the intersubunit communication of the co-operative event. Loop 270–275 plays a major role, since it is involved in the coenzyme binding, as well as in interactions with loop 46–62 in the same subunit and with the residues 280–285 of the helix αF of the adjacent subunit. The latter interact in turn with the residues of loop 46–62 of the first subunit. Therefore, it seems likely that helix αF senses the movements associated with the coenzyme binding to the first subunit, and that these subtle motions can propagate via interface contacts to segments 270–275 and 294–296 of the second subunit (Figure 7B). It is not unreasonable to hypothesize that the absence of co-operativity in mSsADH is mainly due to the loss of important interactions being mediated by residues 270–275 in the wild-type enzyme. Yet it is worth noting is that a mutation to only one residue involved in coenzyme binding is sufficient to produce an enzyme with improved catalytic properties and slightly greater thermal resistance [24] whose binding sites prove independent in coenzyme binding.
Hydrogen bonds and electrostatic interactions are formed exothermically and are therefore stronger at low temperatures. In contrast, hydrophobic bonds are formed endothermically and are weaker at low temperatures. It has been argued that salt bridges destabilize or only mildly stabilize the folded state of a protein at room temperature as a result of the increase in the electrostatic desolvation penalty incurred when forming a salt bridge [27]. Presumably, the changes in ionic strength, temperature and pH result in intersubunit communication perturbations in SsADH through the cleavage or weakening and/or formation or strengthening of one or more interactions involved in the co-operative effects. Because of this, the binding of the first coenzyme molecule induces a structural change in the adjacent subunit, which can improve the binding of the second molecule or render it difficult. The propagation of perturbations occurs through local structural changes that are so subtle that they cannot be detected by CD and fluorescence measurements.
Analyses using the Adair equation of the NADH binding at low temperatures (Figure 2) yields the relationship K1′>K2′≈K3′≈K4′. This suggests that strong positive co-operativity is stimulated by the binding of the first coenzyme molecule and allows the subsequent molecules to bind more easily. Taken together, these results seem to suggest that SsADH behaviour may be explained by the general ligand-induced model [28,29] in which the subunit interaction constants combine to produce positive or negative co-operativity depending on the experimental conditions. The very high affinity between enzyme and coenzyme at low temperatures is due to the decrease in flexibility of the coenzyme-binding site concurrent with the tight packing increase achieved by the tetramer structure. Presumably, this tightening allows the cooperative interactions to propagate in such a way as to favour coenzyme binding, not only to the neighbouring subunit in the same dimer, but also to the subunit of the adjacent dimer, hence the Hill coefficient is near 3. In the SsADH tetramer, A–C and B–D pairs interact through catalytic domain regions, with intermolecular contacts contributed mostly by the residues of the structural zinc lobe 95–117. The opposite monomers, A–D and B–C, also interact through coenzyme domain residues, as well as through the α4 helix and the loop that follows in the catalytic domain [9]. However, no residue of those participating in the A–C and A–D interactions is involved in either the coenzyme or the substrate binding, thus suggesting that any co-operative communications to a third subunit (C or D) take place by means of a more complex pattern of interacting residues.
Many studies on a variety of protein structures have shown that in addition to other environmental conditions (hydration state, pH or ion concentration), temperature considerably affects the balanced interplay of structural flexibility and rigidity ([30] and references therein). A sufficient level of internal conformational flexibility is required for proper functioning and sufficient rigidity is also required to maintain the specific fold of the macromolecule. The simultaneous increase in both Km and kcat values (Figure 4) occurring above 40 °C is thus consistent with the requirement of a somewhat flexible protein architecture in order to bind and properly accommodate the substrate at the active site to perform the catalytic event and to release the product [31]. Although major function impairment occurs at temperatures below 40 °C, where SsADH nevertheless achieves positive co-operativity, slight negative co-operativity occurs at higher temperatures where the enzyme demonstrates its greater catalytic efficiency.
To summarize, our studies provide evidence for temperature-induced co-operativity which has not been described to date for any thermophilic enzyme. Within the class of dehydrogenases, tetrameric yeast ADH does not display any co-operativity in coenzyme binding in the 5–40 °C temperature range [32] and neither does dimeric horse liver ADH, which binds the coenzyme linearly at 25 °C [23,33,34]. Interestingly, the 3D structure of dimeric equine ADH shows that none of the residues involved in the coenzyme binding participate in forming the interactions at dimer interface. However, tetrameric phosphorylating glyceraldehyde-3-phosphate dehydrogenase from yeast binds NAD+ with positive co-operativity, whereas the enzymes from E. coli and B. stearothermophilus exhibit negative co-operativity [35]. S. solfataricus nicotinamide-mononucleotide adenylyltransferase, a hexameric thermophilic enzyme involved in the NAD+ biosynthetic pathway, exhibits non-linear kinetic behaviour at 70 °C, with apparently negative co-operativity with respect to ATP (h=0.29) and NMN (h=0.65) [36].
A positively co-operative enzyme at low substrate concentrations is always more sensitive to changes in flux than a Michaelis–Menten or negatively co-operative system. This feature may prove significant for enzymes involved in energy-generating pathways, which usually operate at fluxes far below full capacity. Therefore rapid fluctuations of substrates or co-factors will interfere only minimally with the all-important task of energy generation [37]. Although a protein must be considered in its physiological context in order to allow realistic conclusions to be made concerning external perturbation and adaptive patterns, it is thought that the binding mechanism devised by the archaeal ADH may be the response to environmental fluctuations, pH, ionic strength and temperature, and/or to subtle changes in the intracellular milieu. Thus a temperature of 15–20 °C, at which SsADH displays positive co-operativity, represents a downshift of over 60 °C from the optimal growth temperature of Sulfolobus cells, which is around 80 °C. This temperature shift accounts for a rapid cessation of cellular activity as reported by Hjort and Bernander [41], who also observed that cultures of Sulfolobus species held at room temperature or in ice-cold water resumed growth and cell cycle progression upon return to 79 °C. Temperature shifts of this magnitude are seldom experienced by Sulfolobus species in their natural habitats and in this context it is tempting to speculate that co-operativity is a device with which the internal flux of the cell responds to such shifts and controls a metabolic process.
Acknowledgments
We thank Professor Edward P. Whitehead for his helpful discussion and Dr L. Esposito for help with structural analyses.
References
- 1.Jaenicke R., Böhm G. The stability of proteins in extreme environments. Curr. Opin. Struct. Biol. 1998;8:738–748. doi: 10.1016/s0959-440x(98)80094-8. [DOI] [PubMed] [Google Scholar]
- 2.Danson M. J., Hough D. W. Structure, function and stability of enzymes from the Archaea. Trends Microbiol. 1998;6:307–314. doi: 10.1016/s0966-842x(98)01316-x. [DOI] [PubMed] [Google Scholar]
- 3.Závodszky P., Kardos J., Svingor Á., Petsko G. A. Adjustment of conformational flexibility is a key event in the thermal adaptation of proteins. Proc. Natl. Acad. Sci. U.S.A. 1998;95:7406–7411. doi: 10.1073/pnas.95.13.7406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kohen A., Klinman J. P. Protein flexibility correlates with degree of hydrogen tunnelling in thermophilic and mesophilic alcohol dehydrogenases. J. Am. Chem. Soc. 2000;122:10738–10739. [Google Scholar]
- 5.Fitter J., Heberle J. Structural equilibrium fluctuations in mesophilic and thermophilic α-amylase. Biophys. J. 2000;79:1629–1636. doi: 10.1016/S0006-3495(00)76413-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kohen A., Cannio R., Bartolucci S., Klinman J. P. Enzyme dynamics and hydrogen tunnelling in a thermophilic alcohol dehydrogenase. Nature (London) 1999;299:496–499. doi: 10.1038/20981. [DOI] [PubMed] [Google Scholar]
- 7.Gerstein M., Lek A. M., Chothia C. Structural mechanisms for domain movements in proteins. Biochemistry. 1994;33:6739–6749. doi: 10.1021/bi00188a001. [DOI] [PubMed] [Google Scholar]
- 8.Raia C. A., Giordano A., Rossi M. Alcohol dehydrogenase from Sulfolobus solfataricus. Methods Enzymol. 2001;331:176–195. doi: 10.1016/s0076-6879(01)31056-x. [DOI] [PubMed] [Google Scholar]
- 9.Esposito L., Sica F., Raia C. A., Giordano A., Rossi M., Mazzarella L., Zagari A. Crystal structure of the alcohol dehydrogenase from the hyperthermophilic Archaeon Sulfolobus solfataricus at 1.85 Å resolution. J. Mol. Biol. 2002;318:463–477. doi: 10.1016/S0022-2836(02)00088-8. [DOI] [PubMed] [Google Scholar]
- 10.Esposito L., Bruno I., Sica F., Raia C. A., Giordano A., Rossi M., Mazzarella L., Zagari A. Crystal structure of a ternary complex of the alcohol dehydrogenase from Sulfolobus solfataricus. Biochemistry. 2003;42:14397–14407. doi: 10.1021/bi035271b. [DOI] [PubMed] [Google Scholar]
- 11.Esposito L., Bruno I., Sica F., Raia C. A., Giordano A., Rossi M., Mazzarella L., Zagari A. Structural study of a single-point mutant of Sulfolobus solfataricus alcohol dehydrogenase with enhanced activity. FEBS Lett. 2003;539:14–18. doi: 10.1016/s0014-5793(03)00173-x. [DOI] [PubMed] [Google Scholar]
- 12.Giordano G., Rossi M., Raia C. A. Proc. 4th Int. Congress on Extremophiles. Italy: Naples; 2002. Evidence for co-operativity in S, solfataricus alcohol dehydrogenase. 22–26 September 2002, Abstract P191. [Google Scholar]
- 13. Reference deleted.
- 14.Dahlquist F. W. The meaning of Scatchard and Hill plots. Methods Enzymol. 1978;48:270–299. doi: 10.1016/s0076-6879(78)48015-2. [DOI] [PubMed] [Google Scholar]
- 15.Kuehn G. D., Barnes L. D., Atkinson D. E. Yeast diphosphopyridine nucleotide specific isocitrate dehydrogenase. Purification and some properties. Biochemistry. 1971;10:3945–3951. doi: 10.1021/bi00797a023. [DOI] [PubMed] [Google Scholar]
- 16.Palmer T. Understanding Enzymes. 4th edn. London: Prentice Hall/Ellis Horwood; 1995. The binding of ligand to proteins; pp. 107–127. [Google Scholar]
- 17.Brand L., Witholt B. Fluorescence measurements. Methods Enzymol. 1967;11:776–856. [Google Scholar]
- 18.Segel I. S. Enzyme Kinetics. New York: John Wiley and Sons; 1975. Effects of pH and temperature; pp. 926–941. [Google Scholar]
- 19.Giordano A., Raia C. A. Steady-state fluorescence properties of S. solfataricus alcohol dehydrogenase and its selenomethionyl derivative. J. Fluorescence. 2003;13:17–24. [Google Scholar]
- 20.Guex N., Peitsch M. C. SWISS-MODEL and the Swiss-PDB viewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 21.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2673. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 22.Giordano A., Russo C., Raia C. A., Kuznetsova I. M., Stepanenko O. V., Turoverov K. K. Highly UV-absorbing complex in selenomethionine-substituted alcohol dehydrogenase from Sulfolobus solfataricus. J. Proteome Res. 2004;3:613–620. doi: 10.1021/pr034132d. [DOI] [PubMed] [Google Scholar]
- 23.Adolph H. W., Kiefer M., Cedergren-Zeppezauer E. Electrostatic effects in the kinetics of coenzyme binding to isozymes of alcohol dehydrogenase from horse liver. Biochemistry. 1997;36:8743–8754. doi: 10.1021/bi970398k. [DOI] [PubMed] [Google Scholar]
- 24.Giordano A., Cannio R., La Cara F., Bartolucci S., Rossi M., Raia C. A. Asn249→Tyr substitution at the coenzyme binding domain activates Sulfolobus solfataricus alcohol dehydrogenase and increases its thermal stability. Biochemistry. 1999;38:3043–3054. doi: 10.1021/bi982326e. [DOI] [PubMed] [Google Scholar]
- 25.Subramanian S., Ross P. D. Thermodynamics of binding of oxidized and reduced nicotinamide adenine dinucleotides, adenosine-5′-diphosphoribose, and 5′-iodosalicylate to dehydrogenases. Biochemistry. 1978;17:2193–2197. doi: 10.1021/bi00604a027. [DOI] [PubMed] [Google Scholar]
- 26.Hammes G. G. Enzyme Catalysis and Regulation. London: Academic Press; 1982. Regulation of enzyme catalysis; pp. 152–186. [Google Scholar]
- 27.Elcock A. H. The stability of salt bridges at high temperatures: implications for hyperthermophilic proteins. J. Mol. Biol. 1998;284:489–502. doi: 10.1006/jmbi.1998.2159. [DOI] [PubMed] [Google Scholar]
- 28.Cook R. A., Koshland D. E. Positive and negative co-operativity in yeast glyceraldehyde 3-phosphate dehydrogenase. Biochemistry. 1970;9:3337–3342. doi: 10.1021/bi00819a007. [DOI] [PubMed] [Google Scholar]
- 29.Koshland D. E., Hamadani K. Proteomics and models for enzyme cooperativity. J. Biol. Chem. 2002;277:46841–46844. doi: 10.1074/jbc.R200014200. [DOI] [PubMed] [Google Scholar]
- 30.Fontana A., De Filippis V., Polverino de Laureto P., Scaramella E., Zambonin M. Rigidity of thermophilic enzymes. In: Ballestreros A., Plou F. J., Iborra J. L., Halling P. J., editors. Stability and Stabilization in Biocatalysis. Vol. 5. Amsterdam: Elsevier Sciences; 1998. pp. 277–294. [Google Scholar]
- 31.Jaenicke R. Protein stability and molecular adaptation to extreme conditions. Eur. J. Biochem. 1991;202:715–728. doi: 10.1111/j.1432-1033.1991.tb16426.x. [DOI] [PubMed] [Google Scholar]
- 32.Karlovic D., Amiguet P., Bonner F. J., Luisi P. L. Spectroscopic investigation of binary and ternary coenzyme complexes of yeast alcohol dehydrogenase. Eur. J. Biochem. 1976;66:277–284. doi: 10.1111/j.1432-1033.1976.tb10517.x. [DOI] [PubMed] [Google Scholar]
- 33.Iweibo I., Weiner H. Role of zinc in horse liver alcohol dehydrogenase. Coenzyme and substrate binding. Biochemistry. 1972;11:1003–1018. doi: 10.1021/bi00756a009. [DOI] [PubMed] [Google Scholar]
- 34.Anderson D. C., Dahlquist F. W. Properties of bound trifluoroethanol complexes with horse liver alcohol dehydrogenase. Biochemistry. 1982;21:3569–3578. doi: 10.1021/bi00258a007. [DOI] [PubMed] [Google Scholar]
- 35.Roitel O., Sergienko E., Branlant G. Dimers generated from tetrameric phosphorylating glyceraldehyde-3-phosphate dehydrogenase from Bacillus stearothermophilus are inactive but exhibit cooperativity in NAD binding. Biochemistry. 1999;38:16084–16091. doi: 10.1021/bi9912802. [DOI] [PubMed] [Google Scholar]
- 36.Raffaelli N., Lorenzi T., Emanuelli M., Amici A., Ruggieri S., Magni G. Nicotinamide-mononucleotide adenylyltransferase from Sulfolobus solfataricus. Methods Enzymol. 2001;331:281–292. doi: 10.1016/s0076-6879(01)31065-0. [DOI] [PubMed] [Google Scholar]
- 37.Ghosh R. On the physiological significance of positive and negative co-operativity. J. Theor. Biol. 1981;93:395–401. doi: 10.1016/0022-5193(81)90111-9. [DOI] [PubMed] [Google Scholar]
- 38.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 39.Kraulis P. J. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 1991;24:946–950. [Google Scholar]
- 40.Merritt E. A., Bacon D. J. Raster3D: photorealistic molecular graphics. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 41.Hjort K., Bernander R. Changes in cell size and DNA content in Sulfolobus cultures during dilution and temperature shift experiments. J. Bacteriol. 1999;181:5669–5675. doi: 10.1128/jb.181.18.5669-5675.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]