

Abstract
CRISPR base editors can introduce point mutations into DNA precisely, and cytosine base editors (CBEs) catalyze C to T transitions. While CBEs have been thoroughly explored in cell culture and organisms such as mice, little is known about DNA base editing in insects. In this study, we evaluated germline editing rates of three different CBEs expressed under actin (ubiquitous) or nanos (germline) promoters utilizing Drosophila melanogaster. The original Rattus norvegicus-derived cytosine deaminase APOBEC1 (rAPO-1) displayed high base editing rates (~99%) with undetectable indel formation. Additionally, we show that base editors can be used for generating male sterility and female lethality. Overall, this study highlights the importance of promoter choice and sex-specific transmission for efficient base editing in flies while providing new insights for future genetic biocontrol designs in insects.
Subject terms: CRISPR-Cas9 genome editing, Genetic engineering, Genetics
Testing three types of cytosine base editors in fruit flies demonstrates the potential usage of these base editors in genetic biocontrol strategies, with the rAPO-1 achieving high editing efficiency (~99%) with no detectable INDEL mutations.
Introduction
Many of the recent advances in the field of biotechnology are due to progress in genome-modifying enzymes such as the CRISPR/Cas9 system1. Many CRISPR adaptations have been developed for different purposes, including CRISPR base editors that can substitute one nucleotide for another at a key location to introduce a point mutation2,3.
One of the primary motivations for developing base editors was to be able to introduce a specific nucleotide change without creating double-stranded breaks (DSBs)4. Before base editors, the homology-directed repair (HDR) mechanism was exploited to make precise changes to a DNA sequence, where a donor DNA template encoding the desired nucleotide substitution was supplied in conjunction with CRISPR/Cas9. In this method, the Cas9 creates the DSB, which can be repaired by HDR with the donor DNA functioning as the repair template. However, while HDR can install the desired mutation, it typically occurs at very low efficiency, with other repair pathways, such as non-homologous end-joining (NHEJ) generating insertions or deletions at the cut site5,6.
Instead, CRISPR-based editors can modify a single nucleotide without the requirement for any exogenous DNA or DNA double-strand breaks. This is achieved through fusing the modified nickase Cas9, which introduces single-strand breaks, to a base editor domain that deaminates the DNA and induces a point mutation using the endogenous DNA repair machinery7. Adenosine base editors (ABEs) can create A-to-G substitutions while C-to-T edits can be produced by cytosine base editors (CBEs)2,3. When using CBEs, the molecular mechanism of this approach starts with the formation of an R-loop, which exposes cytosines to the cytidine deaminase to be converted to uracil via enzymatic hydrolysis; this uracil is then recognized by eukaryotic and prokaryotic DNA polymerases as a thymidine, resulting in G:C to A:T transitions2,3. The deamination of cytidine happens at a specific interval within the R-loop, termed the editing window, meaning that other cytosines within the target window can be edited – forming bystander edits. However, base editing windows can be adjusted with certain modifications such as linker length between the base editor domain and the nickase Cas98 to meet the experimental necessities.
While this technology has been applied from agriculture to human therapy2,9,10, these approaches have not been explored extensively in insects and these species could represent an excellent option for developing genetic biocontrol or targeted genetic intervention strategies11. For example, insect species could be protected from pesticides or climate change by spreading a pesticide resistance12 or heat tolerance allele13. Base editors may be uniquely suited to spread these mutations as part of an Allele Sail14. An Allele Sail uses a genome editor to create DNA edits, which permit the development of viable and fertile offspring. This creates a unique dynamic where there is Mendelian inheritance of the editor, while the frequency of the edits increases at a super-Mendelian rate. Using agent-based modeling it has been shown that edits can reach very frequencies with a single, low-frequency release. Also, we could spread beneficial single nucleotide polymorphisms (SNPs) that can make mosquitoes resistant to Plasmodium to reduce malaria’s disease impact15,16. One way to propagate mutations into a population would be the use of the proposed Y-linked editors to disrupt the fitness of female progenies for population suppression17. Another option could be linking base editors to CRISPR-gene drives and harnessing the intrinsic super-Mendelian inheritance rates of these self-replicating elements18–20 to spread SNPs of interest.
While preparing this manuscript, two studies have demonstrated transgenic expression of CBEs and ABEs in the model organism D. melanogaster targeting the germline21,22. Thakkar and colleagues targeted the white gene using first-generation CBEs and ABEs previously optimized in human cell culture2,3, and reported an average germline editing rate of 70%21. In the second study, Doll and colleagues evaluated an evolved version of the Rattus norvegicus APOBEC1 (CBEevoAPOBEC1), and an evolved version of the Petromyzon marinus cytidine deaminase 1 (CDA1) protein (CBEevoCDA1)23. Here, they showed germline transmission rates of ~99% efficiency when using CBEevoCDA1 while the CBEevoAPOBEC1 showed lower base editing rates (70–95%)22. Interestingly, both studies required the elevated temperature of 28–29 °C to achieve higher editing rates in specific situations, which is not ideal, as the optimal survival and preferred temperature in Drosophila is 25 °C24,25. Therefore, we sought to identify different base editor domains displaying high editing rates (90–100%) at 25 °C and using two different promoters.
Specifically, we examined the efficiency of three different CBEs at 25 °C: (i) the original Rattus norvegicus-derived cytosine deaminase APOBEC1 (rAPO-1)2, (ii) an optimized version of the original APOBEC1 (ancBE4)26, and (iii) the AID*Δ variant of the human AID cytidine deaminase(AID*Δ)27. Importantly, all our base editors displayed complete editing efficiency (>99%) when using the ubiquitous actin promoter at 25 °C. Also, the AID*Δ displayed a wider editing window compared to the rAPO-1 and the ancBE4, which can be useful for applications looking to introduce multiple mutations at a target site. Additionally, we examined the influence of maternal or paternal transmission of the CBEs and found that our extremely efficient CBEs were precise, though indel formation increased when the CBEs were transmitted maternally. Lastly, our base editor system was able to produce male sterility and female lethality by targeting β-Tubulin (β-Tub) and sex-lethal (sxl) genes, respectively. Indeed, our studies expand the CBE repertoire and bring new information about the fine workings of base editing in insects.
Results
Generating transgenic D. melanogaster for expression of CBEs and guide RNA (gRNAs)
To evaluate base editing in flies, we made a set of genetic constructs that expressed three different CBEs under two distinct promoters for a total of 6 conditions. These constructs carry the following features: First, a Cas9D10A nickase with the nuclear localization signal (NLS) derived from simian virus 40 (SV40) T antigen26,28. This modified nuclease was fused to either of the three different CBE domains: (i) the original Rattus norvegicus-derived cytosine deaminase (rAPO-1)2, (ii) its ancestrally reconstructed version ancBE4max base editor (ancBE4)26, or (iii) the AID*Δ variant of the human AID cytidine deaminase (AID*Δ)26. These base editors were expressed by either the ubiquitous promoter actin (act) or the well-known germline nanos (nos) promoter22,29,30 (Fig. 1A). We employed the virus-derived p10 terminator for the act-expressed CBEs due to its demonstrated great protein expression31 (Fig. 1A). We used the endogenous nos 3′ untranslated region (UTR) for the remaining constructs containing the nos promoter. Lastly, all base editor constructs were site-specifically integrated onto the 2ND chromosome using phiC31-mediated integration32 while using the mini-white selectable marker to identify positive transformants33 (Fig. 1A). As additional notes, all CBEs were fused to the N-terminal end of the nickase, which is key for efficient editing2. Contrary to most base editing designs, we excluded the uracil DNA glycosylase inhibitor (UGI) in our constructs as flies tolerate high levels of uracil in DNA34 compared to humans10.
Fig. 1. Diagrams of the cytosine base editor and ebony-gRNA constructs.

A Two promoters were used, a nos promoter for germline-specific expression and an actin promoter for ubiquitous base editor expression. Nos-5′ nos promoter, nos-3 nos terminator, p10-3′ p10 terminator, actin-5′ actin promoter, mini-w mini-white selectable marker, PhiC31 AttB recognition site for PhiC31-mediated integration. AmpR ampicillin resistance gene, ori bacterial origin of replication. B Diagram of the ebony-gRNA construct. dU6-1 and dU6-3 are both Drosophila U6 promoters for ubiquitous gRNA expression. C A schematic of the gRNAs used to target an ebony exon. The gRNA sequence is highlighted in gray, PAM (red) and the target bases (orange) for each gRNA are also highlighted.
Then, we also generated one fly line containing two different gRNAs designed to introduce premature stop codons into the endogenous ebony gene (Fig. 1B, C) when combined with the CBE transgenic flies. Both gRNAs were driven by validated U6 promoters19,29,35 driving ubiquitous expression of the separate gRNAs. The recessive ebony gene controls the amount of dopamine in cuticle formation, with disruption leading to a darker body color compared to wild type36. Also, the dual gRNA construct was also integrated at the same 2nd chromosome locus in a separate strain (Fig. 1B).
Actin and nanos CBEs display high germline editing rates
To assess the editing efficiency of our six different base editor scenarios, we conducted a base editing efficiency assay based on the ebony (dark color) phenotype. Flies homozygous for each CBE transgene were crossed to flies homozygous for the transgene expressing the gRNAs targeting ebony for stop codon generation at two different target sites. The resulting F1 transheterozygote animals were then crossed to a fly strain homozygous for a null mutation to ebony (Fig. 2A)36. At the F2 level, if base editors are able to introduce a stop codon by using any of the two gRNAs, those modifications will be represented by ebony mutant individuals displaying dark body color. If no base editing occurs, flies will present a wild-type body color due to heterozygosity (Fig. 2A).
Fig. 2. Phenotyping assay to evaluate germline editing rates.

A Schematic of the base editing assay for germline transmission. F0 males or F0 females homozygous for either the base editor or gRNAs were crossed together. F1 males or female transheterozygotes, which contained both transgenes (base editor and gRNAs) were crossed to a homozygous ebony mutant strain. The base editing efficiency in F1 individuals was obtained by scoring for the ebony phenotype in the F2 progeny. The green triangles in the F1 individuals indicate the chromosome where base editing could occur, e* CBE-mediated edit, e+ wild-type ebony, e– ebony mutation from mutant strain, Chr. chromosome, ebony-gRNAs construct expressing two gRNAs for introducing a premature stop codon into ebony. B–E Germline base editing rates of base-edited alleles as a percentage of F2 flies exhibiting the ebony phenotype (assessed from a total of at least 10 independent crosses, with the % base editing efficiency and total number of flies scored (N)). The effect of different CBE transmission modes was assessed by examining the percentage of ebony offspring through the four transmission pathways.
The overall phenotyping data indicated close to complete base editing efficiency for all actin-expressed CBEs, though the nos promoter seemed to be less efficient (Fig. 2B–E).
While the nos promoter is germline-specific, the act promoter has also shown high expression in both male and female germline cells37. In fact, the act promoter has produced higher editing rates in various CRISPR-based technologies, such as gene drives29, compared to nos, suggesting its overall higher performance. Yet, prior genome editing studies have shown sex differences in editing efficiency due to maternal deposition or zygotic gRNA expression38. Also, differences in DNA repair during gametogenesis between sexes39–41 could influence base editing outcomes. To reflect this greater importance, the transmission pathways are referred to in terms of what sex is transmitting the CBE: ‘F0 male/F1 male’ refers to the male transmitting the CBE in both the F0 and F1 generation. Then, we explored the role of sex differences by conducting crosses with both maternal and paternal F0 base editor parents, along with both maternal and paternal F1 transheterozygote parents (Fig. 2A).
When evaluating F1 male transmission of a CBE, we detected lower editing for F0 male/F1 male transmission (Fig. 2B) compared to F0 female/F1 male transmission (Fig. 2C) when using nos promoter; yet, base editing with the act promoter was not affected by gender transmission differences. This was most pronounced for nos-rAPO-1 where editing increased by 15.8%, with a slightly lower increase seen for nos-ancBE4 (11.1%). The nos-AID*∆ reached high editing rates with 99.1%, an increase of 7.5% (Fig. 2B, C). These observations might be due to F0 maternal deposition producing a higher quantity of the CBE compared to what is observed in the F0 male/F1 male condition (Fig. 2B, C).
With respect to the effect of CBE transmission in females, we found a relevant influence in F1 female transmission (Fig. 2D, E). We saw that the lowest performing base editor, the nos-rAPO-1 was 73.6% with F0 male/F1 male transmission, compared to 96.0% efficient with F0 female/F1 female transmission. This may arise through maternal deposition of a CBE and gRNA allowing unedited alleles to be edited in the F2 generation, essentially extending the window of possible editing as well as producing CBEs at a sensitive developmental time that potentially favors CBE-mediated gene inactivation. The female germline may also favor the generation of inactivated alleles, perhaps due to sex differences in DNA repair.
Lastly, we provided images of all F1 and F2 individuals within all conditions tested. Interestingly, flies at the F1 stage that expressed base editors from the actin promoter displayed an ebony phenotype while those expressing from the nos promoter displayed a wild-type phenotype. This indicates the higher expression of the actin promoter in somatic tissues compared to nos, which is more germline-specific42. Whereas, all F2 individuals displayed ebony phenotype for both promoters, as expected (Supplementary File - Supplementary Fig. 1).
Overall, our results suggest that female over male transmission of the transgenes is preferred to obtain higher base editing efficiencies. Also, we show that act-CBE’s are less influenced by differences in sex transmission of the transgenes as they show almost 100% efficiency in all conditions (Fig. 2B–E). Most likely, act-CBE’s displayed higher editing efficiencies due to their ubiquitous expression that should allow for a wider time window for base editing.
Amplicon sequencing of the target sites confirms stop codon abundance and phenotypical analysis
To establish a correlation between our phenotype data and genotypes, we conducted amplicon sequencing (Amp-Seq) of both target sites for each condition tested. In brief, we pooled flies from two independent experiments for each condition assessed, and all samples contained both the wildtype (unedited) and ebony (potentially edited) F2 individuals, and the average number of flies per sample was 82 individuals (Supplementary File - Supplementary Table 4).
From our sequencing data, we first sought to establish how closely the percentage of reads with at least one stop codon matched the phenotype data. In our base editing assay, 50% of alleles are inherited from the homozygous null ebony mutant, which is not able to undergo F1 germline editing (Fig. 2A). Importantly, the mutation carried by the ebony null mutant employed for F1 crosses, falls outside of the region sequenced, allowing for equal representation in the F2 progeny sequencing. Therefore, we would expect the F1 germline editing of ebony to amount to a maximum of 50%. If we then assume that all phenotypically ebony F2 flies inherited a base editor-mediated stop codon, we can predict the stop codon percentage based on the phenotype data.
Indeed, the predicted stop codon percentage (in orange) aligns with the observed stop codon percentage for all samples that fall below the theoretical threshold of 50%, which is consistent with the CBE-mediated stop codon (in blue) creating the ebony phenotype (Fig. 3A–D). However, the stop codon abundance exceeds 50% only when maternal transmission of the actin-expressed CBEs at the F1 level occurs (Fig. 3C, D). These results suggest that maternal deposition of the base editor and gRNA transgenes allows for editing of the incoming F1 alleles from the null homozygous parent (Fig. 3E), even when both transgenes do not cohabit within the F2 progenies. Lastly, we examined the contribution of each gRNA separately to extend the stop codon analysis. Indeed, both gRNAs introduced stop codons at the target nucleotide. However, gRNA2 was more active for every base editor and condition tested, suggesting its major contribution to the observed phenotypes (Supplementary File - Supplementary Fig. 2).
Fig. 3. Stop codon molecular analysis of editing by CBEs.

A–D The percentage of reads with a stop codon compared to the predicted percentage of ebony flies from the Amp-Seq subset of the phenotyping assay. The stop codon percentage at either gRNA1/gRNA2 was determined from the Amp-Seq data (n = 2, the total number of flies used per cross is detailed in Supplementary Table 4, data is individual measurements (points)). The predicted ebony percentage was the percentage of flies from the Amp-Seq data subset that were phenotypically ebony in our experiments from Fig. 2. E Diagram of how maternal deposition of a CBE may increase the stop codon abundance above the theoretical editing threshold of 50%.
Drosophila base editors displayed high efficiency and product purity
The CBEs’ average capacity to introduce C-to-T transitions is known as product purity43. This precision consists of three common CBE by-products: indels, C-to-G, and C-to-A edits. We have evaluated the product purity of the base editors tested and the presence of such by-products in all conditions.
As mentioned, our gRNA transgene carries 2 different gRNAs targeting the ebony gene at two different genomic regions to introduce a stop codon and produce gene disruption. The first gRNA contains a C (named C1), which when converted to T, produces a stop codon. The second gRNA instead contains two Cs (C1 and C2) that are susceptible to being converted and generate a stop codon (Fig. 4A). Here, we focus on these three nucleotides for our purity and by-product analysis.
Fig. 4. Frequency of different types of base substitution at the target C bases.

A The gRNA sequences for gRNA1 and gRNA2 are detailed (black), with the target cytidines (orange) numbered and the PAM sequence in red (data presented as the mean frequency of reads with the specified substitution out of all reads harboring a non-C nucleotide at the target base; n = 2, the total number of flies used per cross is detailed in Supplementary Table 4). B–E The effect of different CBE transmission modes on base substitution frequency was assessed by examining the four transmission pathways.
In general, we observed a very high frequency (90–100%) of C-to-T edits for all base editors and modes of base editor transmission (Fig. 4B–E). Specifically, when evaluating F1 male transmission and gRNA1, we saw high product purity edits with low frequencies of C-to-G or C-to-A substitutions except for the F0 female/F1 male transmission where act-AID*∆ and nos-rAPO-1 showed ~5% of undesired C-to-G transitions (Fig. 4C). Interestingly, we observed a different trend when examining gRNA2-C1; in this case, we observed widespread C-to-G or C-to-A substitutions ranging from 5 to 10% in both F0 male/F1 male and F0 female/F1 male transmission scenarios (Fig. 4c). It is important to note that the susceptible C to be mutated in gRNA1 is located in position 5 (PAM distal), while gRNA2-C1 is placed in position 4 (Fig. 4A), and it has been shown in human cell culture that product purity is enhanced when the susceptible C is in PAM-distal position 543,44. In line with these observations, gRNA2-C2 displayed high product purity in both F1 male transmission scenarios except for act-AID*∆, and this gRNA2-C2 is also located in PAM-distal position 5, as occurs in gRNA1-C1 (Fig. 4A–C). Additionally, we noted wider editing for the AID*∆ domain, in line with previous studies45 (Supplementary File - Supplementary Fig. 3).
Similarly, high product purity was observed when evaluating F1 female transmission conditions using both promoters and the three base editors tested, though the gRNA2-C1 displayed higher rates of undesired by-products compared to gRNA1 and gRNA-C2 (Fig. 4D, E). These observations are in line with the F1 male transmission outcomes and suggest that the targeted position of C is critical to inducing cleaner base substitution.
Regarding indel rates, we observed a range of 0% to ~20% within the transmission conditions tested (Fig. 5A). Specifically, the AID*∆ base editor displayed higher indel rates (~10–20%) than the rAPO-1 (0–5%) and the ancBE4 (0–10%) when the transgenes were inherited from F1 females (Fig. 5A – blue and purple bars). However, we observed 0% indel with base editor domains when the base editor machinery was inherited from F1 males (Fig. 5A – red and green bars). Also, it is important to note that nanos promoter produced fewer indels than the actin promoter with the three different base editor domains tested (Fig. 5A). Overall, we identified the nos-rAPO-1 as the indel-free condition, independently of the base editing transmission mode (Fig. 5A). Therefore, our indel analysis indicates that nos is less prone to generate indels compared to the actin promoter. Also, more indels were generated when the base editing components were inherited from F1 females, suggesting differences in DNA repair between both genders. While indels created by CRISPR base editors have not been deeply explored in insects so far, previous CRISPR-based approaches such as gene drive seeking to spread engineered alleles also displayed higher rates of indels when the CRISPR elements were inherited through females29,46,47.
Fig. 5. Analysis of indels caused by cytidine base editing.
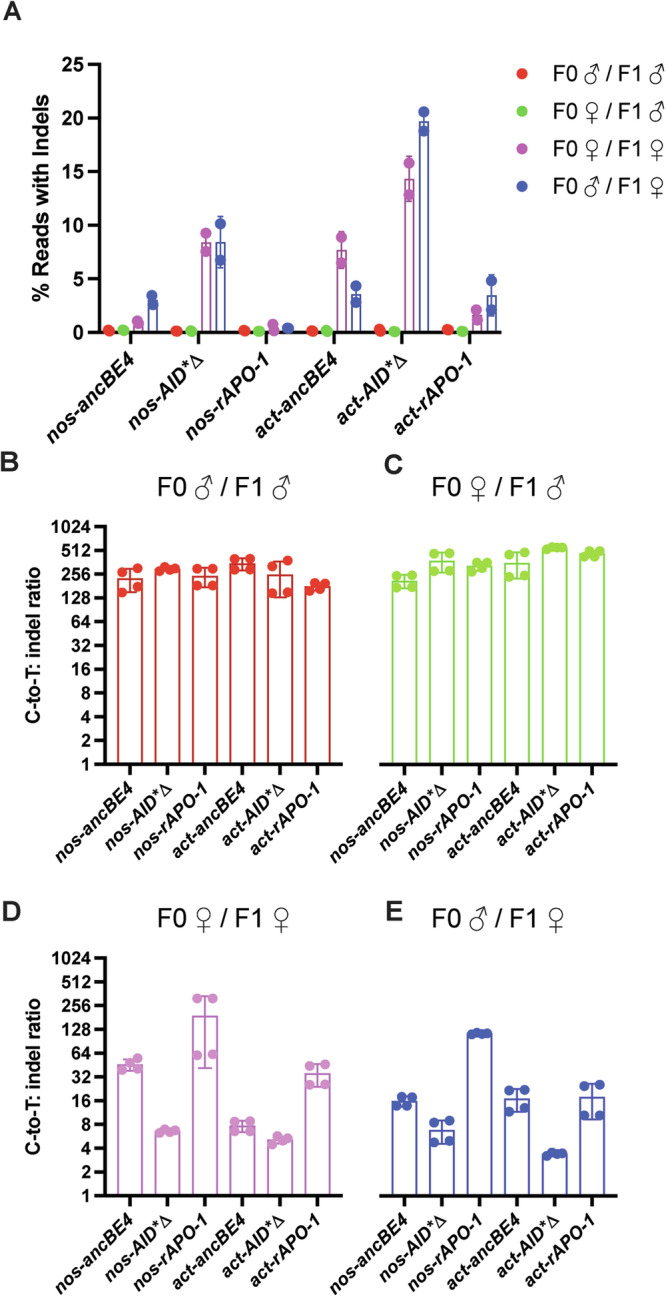
A The rate of indel formation seen in Amp-Seq reads for all CBEs and transmission pathways (n = 2, the total number of flies used per cross is detailed in Supplementary Table 4, data is mean ± s.d.). B–E The C-to-T:indel ratio at every cytidine base that had at least 5% base conversion across all CBEs. The ratio was calculated from all the Amp-Seq reads for that sample (n = 4, data is mean ± s.d.).
To further assess the performance of our base editor designs, we calculated the C-to-T:indel ratio for cytidine bases that had at least 5% base conversion across all CBEs. Here, a higher ratio is preferred as an indication of overall base editor efficiency. There were only two cytidines that fit this criterion, the two target cytidines of gRNA2. In line with our previous results and previous work22, the best performance was detected when the base editing components were inherited through the male at the F1 level; here, ratios of 128–512 were observed (Fig. 5B–E). Additionally, our analysis employing females at the F1 level displayed dramatically lower indel ratios, except for nos-rAPO-1, which maintained a high C-to-T: indel ratio (Fig. 5D, E). These results confirm that either differences in DNA repair in the female germline or maternal deposition are producing greater indel formation.
CRISPR base editors can produce male sterility and female lethality
While previous work focused on targeting pigmentation and essential genes22, we wondered whether base editors could be used to generate different phenotypes such as male sterility and female lethality. As we mentioned in the introduction, CRISPR base editors could be employed for genetic biocontrol approaches such as precise genetic sterile insect technique (pgSIT)48,49, gene drives20, or sex-linked genome editors17 for population control.
To test this idea, we generated two additional gRNA lines containing two gRNAs each targeting β-Tubulin (β-Tub) or sex-lethal (sxl) loci since their disruption causes mutant male flies to be sterile and selective lethality of females48,49, respectively. Specifically, these gRNAs were designed to introduce stop codons, as we did before in our ebony experiments. Then, we decided to use our actin-rAPO as a base editing source since this line was highly efficient and generated a very low frequency of indels.
First, we crossed females carrying the β-Tub-gRNAs to males expressing the rAPO domain under the act promoter (Fig. 6A). Then, the F1 males carrying both transgenes were single-pair crossed to wild-type females to evaluate male sterility in the F2 progeny (Fig. 6A). If males carrying both transgenes were sterile, we should not observe any individuals developing to adults in the F2 generation. Indeed, we observed sterility in 7 out of 8 experiments as we did not observe adults in those 7 tubes (Fig. 6A). To molecularly confirm stop codon generation in β-Tub mutant individuals, we pooled all males into one tube and performed amplicon sequencing. We analyzed nucleotide distribution across the gRNA and displayed the percentage of thymidines observed within the total sequencing reads (Fig. 6B). We observed that both gRNAs targeting β-Tub were able to introduce stop codons with base editing frequencies ranging from 40% to 94% (Fig. 6B), which correlates with the observed sterility phenotypes.
Fig. 6. CRISPR base editors induce male sterility and female lethality.

A Females carrying gRNAs targeting β-Tub gene for stop codon generation were crossed to males carrying the base editing machinery. Male individuals hatching from these crosses were single-pair crossed to wild-type females to evaluate male sterility. B C-to-T transitions to generate stop codons at both target sites into the β-Tub gene were analyzed via deep sequencing. The percentage of mutations is depicted. C Females expressing gRNAs targeting the sxl gene were crossed to the base editor line to evaluate female lethality in the F1 generation. D C-to-T transitions to generate stop codons at both target sites into the sxl gene from males carrying the base editing domain (APOBEC) and the sxl-gRNA transgenes. The percentage of mutations is depicted. (Ref: reference sequence; β-Tub and sxl indicate the mutated sequences). C-to-T mutations are highlighted in red. PAM sequences are labeled in blue. F0, F1, and F2 indicate the individual's generation.
Second, we crossed females carrying the sxl-gRNAs to males expressing the rAPO domain under the act promoter; this genetic cross was repeated 4 times independently, and the average percentage observed for each category within these experiments is depicted (Fig. 6C). Here, we should not observe females carrying both transgenes in the F1 generation since sxl disruption causes female lethality. Importantly, we observed a skewed ratio of females in all our experiments. There are four possible genotypes in the F1 generation of Fig. 6C, so that if there is no female lethality, we would expect each of the four genotypes to contribute 25% to the population. Yet, in the F1 population, we observed only 7% of females carrying both the sxl and act-rAPO (Fig. 6C), indicating that our system is capable of inducing female lethality. Most importantly, we crossed these female escapees to wild-type males and confirmed these individuals were not fertile. Then, we decided to sequence males containing both transgenes (APOBEC and sxl-gRNAs) since these individuals should carry the C-to-T mutation to stop codon introduction while being viable and suitable for molecular characterization, as the targeted exon is only critical for female development. For this experiment, we pooled eight males from one of our experiments to perform deep sequencing. Here, we observed 85% base editing rates at one target site, while the second gRNA seemed to be almost inactive; we only observed a 1% editing rate in this case (Fig. 6D). Therefore, these results suggest that the observed female lethality should be mainly imposed by the activity of the first gRNA. Also, we sequenced the surviving female escapees to evaluate if gene editing occurred in these individuals. In this case, we pooled seven female escapees from one of our experiments and performed deep sequencing of these infertile females. Here, our sequencing analysis confirmed that our system is able to mutate the sxl gene of these females at one of the target sites with base editing rates of 75%. Yet, our second gRNA showed low activity (Supplementary File - Supplementary Fig. 4), as observed when sequencing the males. Indeed, our active gRNA was enough to induce female lethality or sterility within the individual escapees. Importantly, we quantified the product purity when targeting these genes and confirmed that C-to-T substitutions are the most prevalent event with no indels generation (Supplementary File - Supplementary Fig. 5), as observed when targeting the ebony gene. Overall, these experiments expand the applicability of base editors and demonstrate that these systems could be utilized for genetic biocontrol purposes.
Discussion
In this study, we have characterized an expression system for a variety of CBEs showing ~99% base editing rates when targeting the ebony locus. Importantly, the extremely high efficiency was maintained across all four possible modes of CBE transmission at 25 °C.
Given the similarities between our work and previous studies21,22, we consider it appropriate to discuss in more detail the results from both works since they describe relevant information for future base editing approaches seeking to manipulate insect DNA. It is important to note that we tested three base editing domains that are different from Doll and colleagues. First, we tested the original Rattus norvegicus-derived cytosine deaminase APOBEC1 (rAPO-1), which represents the first CRISPR base editor described2, while the previous work evaluated an evolved version of the Rattus norvegicus APOBEC1 (CBEevoAPOBEC1)22. While our rAPO-1 was highly efficient at 25 °C, the CBEevoAPOBEC1 displayed temperature sensitivity and the highest rates were achieved at 29 °C. These differences could be attributed to the fact that CBEevoAPOBEC1 was created by phage-assisted continuous evolution (PACE)50, which requires temperatures of 37 °C, and could cause the original rAPO-1 to become more sensitive to temperature. We also tested the ancBE4 domain, which is a newer version of the rAPO-1 obtained by codon usage optimization using mammalian cell culture26. We did not observe major differences between our rAPO-1 and ancBE4 in terms of editing efficiency, though the rAPO-1 generated fewer indels compared to the ancBE4. It is assumed that synonymous mutations in mammals are generally considered free from natural selection, imposing little to no impact on fitness51. However, the usage of synonymous codons in animals such as Drosophila or worms is under selective pressure, significantly affecting the fitness of these organisms52,53. This could be the reason why, while ancBE4 was preferred over rAPO-1 in previous work using mammalian cells26, our results suggest that rAPO-1 is a better choice for base editing in flies.
Doll and colleagues22 tested a second domain, which is an evolved version of the Petromyzon marinus cytidine deaminase 1 (CDA1) protein (CBEevoCDA1)45,54; here, the CBEevoCDA1 displayed temperature-tolerance showing high base editing rates (90–99%) at 25 °C and 29 °C. In line with previous cell culture studies45, this domain displayed a wider base editing window similar to what we observed using the AID*∆ domain. It is important to note that the CDA1 from lampreys is an ortholog of the AID vertebrate domain55, and could be the reason for their similarities in terms of base editing dynamics. Therefore, both domains are suitable for efficient base editing in flies at 25 °C.
Takkar and colleagues employed two CBE domains: (i) the same APOBEC1 (rAPO-1) tested in this work, and (ii) the CBEevoCDA1 tested by Doll and colleagues22. However, their study reported lower base editing rates (~70%) compared to ours and the studies by Doll and colleagues. These differences could be due to the nuclease used: while we and Doll and colleagues employed the regular nickase Cas9 tested in previous human cell culture studies2,3, Takkar and colleagues tested a modified version, xCas956, which could be less active, as previously shown in Drosophila57.
In this work, we also carefully explored differences in editing rates when the CBE was transmitted maternally compared to paternally at the F0 and F1 generations. In general, we observed ~99% editing rates when the CBEs were inherited through females, instead, lower editing rates were observed when the base editing machinery was passed through males. These results are in line with other CRISPR-based methods such as gene drives were higher super-Mendelian rates are observed in females29,58.
In terms of base editing precision and formation of undesired by-products, it is important to highlight that our work analyzed germline outcomes based on F2 individuals from our genetic crosses; Doll et al. also employed F1 flies for this analysis22, which is indicative of somatic modifications. Also, Takker and colleagues analyzed indels from F2 individuals but using Sanger sequencing21, which is a different method compared to our deep sequencing analysis. Overall, we show very high precision when CBEs were transmitted through F1 males, with suitable C-to-T:indel ratios exceeding 150 for all CBEs. Indeed, our work and previous studies analyzing somatic outcomes in flies22 display improved C-to-T:indel ratios compared to other organisms, where base editing with CBEevoAPOBEC1 presented C-to-T:indel ratios between 5 and 2545,59,60. However, we observed C-to-T:indel ratios at undesired rates with F1 female CBEs transmission, except for the nos-rAPO-1 CBE. The higher indel formation rates for maternal CBE transmission compared to paternal transmission can be explained by maternal deposition of the CBE into the developing embryo, where indels could be predominantly repaired by non-homologous end joining (NHEJ)61. Intriguingly, other strategies such as prime editing reported no sex differences for non-precise edits between F1 male or F1 female transmission62,63, though multiple parameters such as targeted gene, distinct gRNAs or promoters could influence these results. Importantly, these findings underscore the importance of examining sex differences in genome editing when using CBEs in insects.
Indeed, our rAPO-1 was the most valuable domain as it displayed high editing rates, product purity, and no indels. While rAPO-1 can be suitable for base editing requiring narrow windows, the previous CBEevoCDA1 and the AID*∆ domain tested in this work could be better options when wider base editing windows are needed. It is important to note though that we have tested a limited number of gRNAs in this work. While the ebony gRNAs showed high germline base editing efficiencies under almost any condition when analyzing F2 individuals, gRNAs targeting the β-Tub and sxl genes displayed lower efficiencies. It is important to highlight that in this case, we are analyzing somatic base editing rates as we analyze F1 individuals. Overall, these results indicate that the selection of gRNAs is crucial for effective base editing.
Lastly, we explored the efficiency of base editors when targeting sex-determination genes employed in population suppression stratetiges48. We observed almost full penetrance when targeting the sxl gene. Though some females containing both transgenes were observed, though these individuals were sterile and therefore could not reproduce. All these observations were confirmed via deep sequencing since stop codons were detected at one target site with editing rates of 75–85%. Regarding the β-Tub locus, we observed maximum base editing rates of 94% after sequencing males containing both the rAPO-1 and β-Tub-gRNAs transgenes. Phenotypically, 7 out of 8 independent males displayed sterility as they were not able to produce viable progeny when crossed to wild-type females. To reach full penetrance and bypass the presence of fertile males, we could design gRNA-transgenes editing more than two target sites. In principle, our base editor system could be easily converted into a “base editor pgSIT” system, where generated sterile males could be used for mass release. Current Cas9-based pgSIT strategies introduce a variety of DNA breaks that produce sterile males containing distinct mutations48, which could potentially impose different fitness cost effects within the male population. Instead, using base editors one could generate a more predictable and homogeneous genetic makeup within the sterile male population while avoiding potential fitness cost concerns.
In summary, we characterized three new and highly efficient CBEs for their use in flies. These base editors displayed high rates of C-to-T transitions with low or undetectable indel rates. Also, we have identified sex differences in editing efficiency and in precision through varying the paternal or maternal transmission of the CBE to show the importance of accounting for transmission of the base editor components. Lastly, we show the possibility of adapting base editors to generate male sterility and female lethality, opening new avenues for next-generation pgSIT systems. Indeed, future studies will seek to translate these technologies to mosquitoes for propagating beneficial or deleterious alleles as part of genetic biocontrol strategies for vector control. Overall, our work expands the base editing toolbox for genome editing while bringing new insights into the biology of base editing in insects.
Material and methods
Genetic construct assembly
NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs) was used to assemble all constructs. All plasmids are listed in Supplementary File - Supplementary Table 1 and all primers are listed in Supplementary File - Supplementary Table 2. After assembly, plasmids were transformed into One Shot™ Stbl3™ Chemically Competent E. coli (Invitrogen). The desired transformants were confirmed by restriction analysis and Sanger sequencing. The final DNA sequence information for the constructs is available on NCBI; accession numbers are provided for each construct in the Supplementary Information File (Supplementary Table 1).
Insect rearing and generation of transgenic fly strains
Ebony mutant flies were from the Bloomington Stock Center (BDSC: 1658). Enzyme expression plasmids were purified using the ZymoPURE II Plasmid Midiprep Kit (Zymo Research #D4200) and sent to BestGene Inc. (Chino Hills, CA) for ϕC31-mediated integration. All strains were maintained on a cornmeal diet based on the Bloomington standard Nutri-Fly formulation (catalog number 66-113; Genesee Scientific). Flies were reared in a controlled environment room at 25.0 °C, 75% humidity, and a 12 h light/dark cycle with a 30 min transition period. Biosafety approval was granted by the Institutional Biosafety Committee of Macquarie University (#5049).
Base editing efficiency assay
To assess the editing efficiency of the six different versions of base editors we conducted a base editing efficiency assay (Fig. 2). Flies homozygous for a CBE transgene were crossed to flies homozygous for the pCFD4-2xebony transgene. The resulting F1 transheterozygote was then crossed to a fly strain homozygous for a null mutation to ebony (BDSC: #1658). We then scored the F2 progeny, where offspring inheriting two inactivated copies of the ebony gene would be dark-bodied. Every stage of the experiment was conducted at 25 °C. For evaluating the ability of our base editor system to generate male sterility and female lethality, we crossed the actin-rAPO-1 to two different lines expressing either gRNAs targeting β-Tub or sxl loci, as described in Fig. 6.
Stop codon abundance data computational analysis
For each aligned read sequence, we identified stop codons that were in the same location as the gRNAs. This was done by translating each read sequence and identifying the position of every stop codon in each read. To account for gaps in the alignment between each read and the reference sequence, the position of every stop codon in each read was adjusted. Finally, we compared the position of every stop codon in each read to the position of the gRNAs and identified those that were in the same location as the gRNA.
Deep sequencing
Genotyping of the base editing region was conducted with Amp-Seq. Pooled DNA was extracted with a Quick-DNA Tissue/Insect Miniprep Kit (Zymogen). Then, the target region was amplified by PCR (Q5 Hot Start High-Fidelity DNA Polymerase, New England Biolabs) with primers with partial Illumina® adapter sequences. The PCR amplicon was purified through a gel digest. The concentrations of the purified products were normalized following measurement with a Qubit 4 Fluorometer (Invitrogen). The samples were submitted to Azenta (Amplicon-EZ) for sequencing. Then, sequencing reads were demultiplexed, trimmed, and fastq files were used as input for CRISPR editing analysis using CRISPResso264. A minimum of 100,000 total reads aligned in all our experiments using CRISPResso2. Primers employed for deep sequencing analysis can be found in Supplementary File - Supplementary Table 2.
Statistics and reproducibility
Flies were fed on standard cornmeal. All experimental crosses were maintained at 25 °C. Flies were anesthetized with CO2 when scoring phenotypes and preparing crosses. For all experimental crosses, virgin females were collected as pupae and crossed after hatching. F0 crosses were made in pools of 3–5 males crossed to 3–5 females. F1 females were single-pair crossed. After all the flies emerged, F2 flies were scored for sex, body, and induced-sex-determination phenotypes using a Leica M165 F2 stereomicroscope. The experiments were randomized. Experimental crosses where contamination impeded proper fly development were excluded from the analyses. Crosses were performed in shatter-proof polypropylene vials (Genesee Scientific Cat. #32-120). All graphs were generated using GraphPad Prism 9 and Adobe Illustrator. Statistical analyses were performed using GraphPad Prism 9.
Supplementary information
Acknowledgements
We thank Sara Sanz Juste and Alena Bishop for their scientific feedback.
Author contributions
V.L.D.A. and M.M. conceived the project. V.L.D.A. and M.C. designed the plasmids. M.C. obtained all Drosophila transgenic lines tested in this work. M.C., C.N., H.N., and S.B. performed the fly experiments. A.T. carried out the computational analysis. V.L.D.A. and M.C. performed the figure visualizations. V.L.D.A. and M.C. wrote the manuscript, which was edited by all the authors.
Peer review
Peer review information
Communications Biology thanks Ji-Long Liu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Jun Wei Pek and Mengtan Xing. A peer review file is available.
Data availability
The plasmids generated in this study have been deposited in the GenBank database (https://www.ncbi.nlm.nih.gov/genbank/) with the following accession numbers: PP576060, PP576061, PP576062, PP576063, PP576064, PP576065, PP576066, PP576067, PP576068. A custom script was used to analyze the stop codon abundance and can be found here: (https://github.com/aidantay/Fly_CRISPResso_analysis). Furthermore, all Supplementary Information covering the raw phenotypical scoring data collected in the base editing experiments targeting ebony is provided as a table (Supplementary Table 4) within the Supplementary Information file. For other resources such as transgenic flies, they are available upon request after completing the material transfer agreement (MTA).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Maciej Maselko, Email: maciej.maselko@mq.edu.au.
Víctor López Del Amo, Email: victor.lopezdelamo@uth.tmc.edu.
Supplementary information
The online version contains supplementary material available at 10.1038/s42003-024-06848-5.
References
- 1.Barrangou, R., Sontheimer, E. J. & Marraffini, L. A. CRISPR: Biology and Applications (John Wiley & Sons, 2022).
- 2.Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. & Liu, D. R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature533, 420–424 (2016). 10.1038/nature17946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gaudelli, N. M. et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature551, 464–471 (2017). 10.1038/nature24644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Porto, E. M. & Komor, A. C. In the business of base editors: evolution from bench to bedside. PLoS Biol.21, e3002071 (2023). 10.1371/journal.pbio.3002071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uddin, F., Rudin, C. M. & Sen, T. CRISPR gene therapy: applications, limitations, and implications for the future. Front. Oncol.10, 1387 (2020). 10.3389/fonc.2020.01387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gratz, S. J. et al. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics194, 1029–1035 (2013). 10.1534/genetics.113.152710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evanoff, M. & Komor, A. C. Base editors: modular tools for the introduction of point mutations in living cells. Emerg. Top. Life Sci.3, 483–491 (2019). 10.1042/ETLS20190088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan, J., Zhang, F., Karcher, D. & Bock, R. Author Correction: engineering of high-precision base editors for site-specific single nucleotide replacement. Nat. Commun.10, 2019 (2019). 10.1038/s41467-019-10069-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mishra, R., Joshi, R. K. & Zhao, K. Base editing in crops: current advances, limitations and future implications. Plant Biotechnol. J.364, 292 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rees, H. A. & Liu, D. R. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet.19, 770–788 (2018). 10.1038/s41576-018-0059-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kosch, T. A. et al. Genetic approaches for increasing fitness in endangered species. Trends Ecol. Evol.37, 332–345 (2022). 10.1016/j.tree.2021.12.003 [DOI] [PubMed] [Google Scholar]
- 12.Chang, X. et al. Genomic variant analyses in pyrethroid resistant and susceptible malaria vector. G310, 2185–2193 (2020). 10.1534/g3.120.401279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rolandi, C., Lighton, J. R. B., de la Vega, G. J., Schilman, P. E. & Mensch, J. Genetic variation for tolerance to high temperatures in a population of Drosophila melanogaster. Ecol. Evol.8, 10374–10383 (2018). 10.1002/ece3.4409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson, M.L., Hay, B.A. & Maselko, M. Altering traits and fates of wild populations with Mendelian DNA sequence modifying Allele Sails. Nat Commun15, 6665 (2024) [DOI] [PMC free article] [PubMed]
- 15.Harris, C. et al. Polymorphisms in Anopheles gambiae immune genes associated with natural resistance to Plasmodium falciparum. PLoS Pathog.6, e1001112 (2010). 10.1371/journal.ppat.1001112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horton, A. A. et al. Identification of three single nucleotide polymorphisms in Anopheles gambiae immune signaling genes that are associated with natural Plasmodium falciparum infection. Malar. J.9, 160 (2010). 10.1186/1475-2875-9-160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burt, A. & Deredec, A. Self-limiting population genetic control with sex-linked genome editors. Proc. Biol. Sci. 285, 20180776 (2018). [DOI] [PMC free article] [PubMed]
- 18.Gantz, V. M. & Bier, E. Genome editing. The mutagenic chain reaction: a method for converting heterozygous to homozygous mutations. Science348, 442–444 (2015). 10.1126/science.aaa5945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.López Del Amo, V., Juste, S. S. & Gantz, V. M. A nickase Cas9 gene-drive system promotes super-Mendelian inheritance in Drosophila. Cell Rep.39, 110843 (2022). 10.1016/j.celrep.2022.110843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bier, E. Gene drives gaining speed. Nat. Rev. Genet.23, 5–22 (2022). 10.1038/s41576-021-00386-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thakkar, N., Hejzlarova, A., Brabec, V. & Dolezel, D. Germline editing of using CRISPR-Cas9-based cytosine and adenine base editors. CRISPR J.6, 557–569 (2023). 10.1089/crispr.2023.0026 [DOI] [PubMed] [Google Scholar]
- 22.Doll, R. M., Boutros, M. & Port, F. A temperature-tolerant CRISPR base editor mediates highly efficient and precise gene editing in Drosophila. Sci. Adv.9, eadj1568 (2023). 10.1126/sciadv.adj1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thuronyi, B. W. et al. Continuous evolution of base editors with expanded target compatibility and improved activity. Nat. Biotechnol.37, 1070–1079 (2019). 10.1038/s41587-019-0193-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sayeed, O. & Benzer, S. Behavioral genetics of thermosensation and hygrosensation in Drosophila. Proc. Natl Acad. Sci. USA93, 6079–6084 (1996). 10.1073/pnas.93.12.6079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamada, F. N. et al. An internal thermal sensor controlling temperature preference in Drosophila. Nature454, 217–220 (2008). 10.1038/nature07001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koblan, L. W. et al. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol.36, 843–846 (2018). 10.1038/nbt.4172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hess, G. T. et al. Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat. Methods13, 1036–1042 (2016). 10.1038/nmeth.4038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shui, S., Wang, S. & Liu, J. Systematic investigation of the effects of multiple SV40 nuclear localization signal fusion on the genome editing activity of purified SpCas9. Bioengineering9, 83 (2022) [DOI] [PMC free article] [PubMed]
- 29.López Del Amo, V. et al. A transcomplementing gene drive provides a flexible platform for laboratory investigation and potential field deployment. Nat. Commun.11, 352 (2020). 10.1038/s41467-019-13977-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ali, I. et al. Cis-regulatory elements affecting the Nanos gene promoter in the germline stem cells. J. Biotechnol.145, 323–329 (2010). 10.1016/j.jbiotec.2009.12.011 [DOI] [PubMed] [Google Scholar]
- 31.Pfeiffer, B. D., Truman, J. W. & Rubin, G. M. Using translational enhancers to increase transgene expression in Drosophila. Proc. Natl Acad. Sci. USA109, 6626–6631 (2012). 10.1073/pnas.1204520109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Venken, K. J. T., He, Y., Hoskins, R. A. & Bellen, H. J. P. acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science314, 1747–1751 (2006). 10.1126/science.1134426 [DOI] [PubMed] [Google Scholar]
- 33.Klemenz, R., Weber, U. & Gehring, W. J. The white gene as a marker in a new P-element vector for gene transfer in Drosophila. Nucleic Acids Res.15, 3947–3959 (1987). 10.1093/nar/15.10.3947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muha, V. et al. Uracil-containing DNA in Drosophila: stability, stage-specific accumulation, and developmental involvement. PLoS Genet.8, e1002738 (2012). 10.1371/journal.pgen.1002738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Port, F., Chen, H.-M., Lee, T. & Bullock, S. L. Optimized CRISPR/Cas tools for efficient germline and somatic genome engineering in Drosophila melanogaster. Proc. Natl Acad. Sci. USA111, E2967–E2976 (2014). 10.1073/pnas.1405500111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pool, J. E. & Aquadro, C. F. The genetic basis of adaptive pigmentation variation in Drosophila melanogaster. Mol. Ecol.16, 2844–2851 (2007). 10.1111/j.1365-294X.2007.03324.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li, H. et al. Fly Cell Atlas: a single-nucleus transcriptomic atlas of the adult fruit fly. Science375, eabk2432 (2022). 10.1126/science.abk2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin, C.-C. & Potter, C. J. Non-mendelian dominant maternal effects caused by CRISPR/Cas9 transgenic components in Drosophila melanogaster. G36, 3685–3691 (2016). 10.1534/g3.116.034884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rockenbach, M. K., Fraga, L. R., Kowalski, T. W. & Sanseverino, M. T. V. Expression profiles of meiotic genes in male vs. female gonads and gametes: insights into fertility issues. Front. Genet.14, 1125097 (2023). 10.3389/fgene.2023.1125097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morelli, M. A. & Cohen, P. E. Not all germ cells are created equal: aspects of sexual dimorphism in mammalian meiosis. Reproduction130, 761–781 (2005). 10.1530/rep.1.00865 [DOI] [PubMed] [Google Scholar]
- 41.García-Rodríguez, A., Gosálvez, J., Agarwal, A., Roy, R. & Johnston, S. DNA damage and repair in human reproductive cells. Int. J. Mol. Sci. 20, 31 (2018). [DOI] [PMC free article] [PubMed]
- 42.Forrest, K. M., Clark, I. E., Jain, R. A. & Gavis, E. R. Temporal complexity within a translational control element in the nanos mRNA. Development131, 5849–5857 (2004). 10.1242/dev.01460 [DOI] [PubMed] [Google Scholar]
- 43.Komor, A. C. et al. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci. Adv.3, eaao4774 (2017). 10.1126/sciadv.aao4774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zong, Y. et al. Efficient C-to-T base editing in plants using a fusion of nCas9 and human APOBEC3A. Nat. Biotechnol. 10.1038/nbt.4261 (2018). [DOI] [PubMed]
- 45.Thuronyi, B. W. et al. Publisher correction: continuous evolution of base editors with expanded target compatibility and improved activity. Nat. Biotechnol.37, 1091 (2019). 10.1038/s41587-019-0253-5 [DOI] [PubMed] [Google Scholar]
- 46.Gantz, V. M. et al. Highly efficient Cas9-mediated gene drive for population modification of the malaria vector mosquito Anopheles stephensi. Proc. Natl Acad. Sci. USA112, E6736–E6743 (2015). 10.1073/pnas.1521077112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hammond, A. M. et al. The creation and selection of mutations resistant to a gene drive over multiple generations in the malaria mosquito. PLoS Genet.13, e1007039 (2017). 10.1371/journal.pgen.1007039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kandul, N. P. et al. Transforming insect population control with precision guided sterile males with demonstration in flies. Nat. Commun.10, 84 (2019). 10.1038/s41467-018-07964-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li, M. et al. Suppressing mosquito populations with precision guided sterile males. Nat. Commun.12, 5374 (2021). 10.1038/s41467-021-25421-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Esvelt, K. M., Carlson, J. C. & Liu, D. R. A system for the continuous directed evolution of biomolecules. Nature472, 499–503 (2011). 10.1038/nature09929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chamary, J. V., Parmley, J. L. & Hurst, L. D. Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat. Rev. Genet.7, 98–108 (2006). 10.1038/nrg1770 [DOI] [PubMed] [Google Scholar]
- 52.Duret, L. Evolution of synonymous codon usage in metazoans. Curr. Opin. Genet. Dev.12, 640–649 (2002). 10.1016/S0959-437X(02)00353-2 [DOI] [PubMed] [Google Scholar]
- 53.Shields, D. C., Sharp, P. M., Higgins, D. G. & Wright, F. Silent’ sites in Drosophila genes are not neutral: evidence of selection among synonymous codons. Mol. Biol. Evol.5, 704–716 (1988). [DOI] [PubMed] [Google Scholar]
- 54.Nishida, K. et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science353, aaf8729 (2016). [DOI] [PubMed]
- 55.Holland, S. J. et al. Expansions, diversification, and interindividual copy number variations of AID/APOBEC family cytidine deaminase genes in lampreys. Proc. Natl Acad. Sci. USA115, E3211–E3220 (2018). 10.1073/pnas.1720871115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hu, J. H. et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature556, 57–63 (2018). 10.1038/nature26155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ni, X.-Y., Zhou, Z.-D., Huang, J. & Qiao, X. Targeted gene disruption by CRISPR/xCas9 system in Drosophila melanogaster. Arch. Insect Biochem. Physiol.104, e21662 (2020). 10.1002/arch.21662 [DOI] [PubMed] [Google Scholar]
- 58.Terradas, G. et al. Inherently confinable split-drive systems in Drosophila. Nat. Commun.12, 1480 (2021). 10.1038/s41467-021-21771-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yeh, W.-H. et al. In vivo base editing restores sensory transduction and transiently improves auditory function in a mouse model of recessive deafness. Sci. Transl. Med. 12, eaay9101 (2020). [DOI] [PMC free article] [PubMed]
- 60.Cornean, A. et al. Precise in vivo functional analysis of DNA variants with base editing using ACEofBASEs target prediction. eLife11, e72124 (2022). [DOI] [PMC free article] [PubMed]
- 61.Hammond, A. et al. Regulating the expression of gene drives is key to increasing their invasive potential and the mitigation of resistance. PLoS Genet.17, e1009321 (2021). 10.1371/journal.pgen.1009321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bosch, J. A. & Perrimon, N. Prime editing for precise genome engineering in Drosophila. Methods Mol. Biol.2540, 113–134 (2022). 10.1007/978-1-0716-2541-5_5 [DOI] [PubMed] [Google Scholar]
- 63.Bosch, J. A., Birchak, G. & Perrimon, N. Precise genome engineering in using prime editing. Proc. Natl Acad. Sci. USA118, e2021996118 (2021). [DOI] [PMC free article] [PubMed]
- 64.Clement, K. et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol.37, 224–226 (2019). 10.1038/s41587-019-0032-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The plasmids generated in this study have been deposited in the GenBank database (https://www.ncbi.nlm.nih.gov/genbank/) with the following accession numbers: PP576060, PP576061, PP576062, PP576063, PP576064, PP576065, PP576066, PP576067, PP576068. A custom script was used to analyze the stop codon abundance and can be found here: (https://github.com/aidantay/Fly_CRISPResso_analysis). Furthermore, all Supplementary Information covering the raw phenotypical scoring data collected in the base editing experiments targeting ebony is provided as a table (Supplementary Table 4) within the Supplementary Information file. For other resources such as transgenic flies, they are available upon request after completing the material transfer agreement (MTA).