

Abstract
Oral squamous cell carcinoma (OSCC) is one of the common malignant tumors in the head and neck, characterized by high malignancy, rapid growth and metastasis, high invasive ability, and high mortality. In recent years, surgery combined with chemotherapy or radiotherapy remains the preferred clinical treatment for OSCC, despite considerable advances in diagnostic and therapeutic techniques. Hence, new targeted therapy is urgently needed. Histone modification affects the function of massive cells through histone acetyltransferase and histone deacetylase. Accompanied by the progress of some diseases, especially tumors, these proteins often show abnormal functions, and by reversing these abnormalities with drugs or gene therapy, the cancer phenotype can even be restored to normal. As a result, they are potential drug targets. This article reviewed the role of the histone dynamic process of acetylation modifications and their associated active modifying enzymes in the pathogenesis and progress of OSCC. Moreover, we explored the value of histone acetylation modification as a potential therapeutic target and the new progress of related drugs in clinical treatment.
1. Introduction
Oral cancer is one of the most common malignancies occurring in the maxillofacial region, with a mortality rate close to 50% and an overall 5-year survival rate of about 60%. 90% of them are squamous cell carcinoma. Oral squamous cell carcinoma (OSCC) is the sixth most common malignant tumor in the world. It occurs mostly in adults aged 40‒60 years in China, with more males than females. Alcohol, tobacco, and HPV infection are the most important risk factors for OSCC [1]. In recent years, the conventional therapies for OSCC have evolved into various modalities, including surgery, chemotherapy, radiotherapy, and combination therapy based on the diagnostic stage [2], but none of them has brought significant outcomes to the prognosis of OSCC. This is mainly due to the special anatomical structure of the oral cavity that provides great ease for metastasis. Most of the administered patients with progression to the middle and late stage have already missed the best treatment opportunities, with serious prognostic implications. Hence, the early diagnosis, treatment, and inhibition of metastasis are critical to control this disease. Moreover, it has great clinical significance to investigate the molecular biological mechanisms of occurrence, development, and metastasis of the disease and to discover new therapeutic targets for the intervention in those critical molecules and processes to achieve early diagnosis and treatment and suppress metastasis.
The progression of OSCC is mainly caused by the genetic or epigenetic alterations in proto-oncogenes, antioncogenes, and dysregulation of molecular networks. Histone modification is one of the most critical epigenetic regulations for gene expression by posttranslational modification of the histones [3, 4]. Posttranslational modifications of proteins are essential for organisms, while the metabolism and metabolites can control the activity of growth-related signaling pathways to regulate cell growth through covalent modifications of proteins [5]. The histone acetyltransferases (HATs) and the histone deacetylases (HDACs) are responsible for controlling the histone acetylation level and chromatin accessibility [3, 6]. Acetylation of the ɛ-amino group on the histone lysine residues plays a key role in cell growth activity and gene transcription. Epigenetic and posttranslational modifications have emerged as new targets for anticancer therapy. This review focused on the relationship between the acetylation/deacetylation of histones and the OSCC and the potential therapeutic targets. Figure 1 shows the pattern of acetylation function and lists the reported sites in OSCC.
Figure 1.
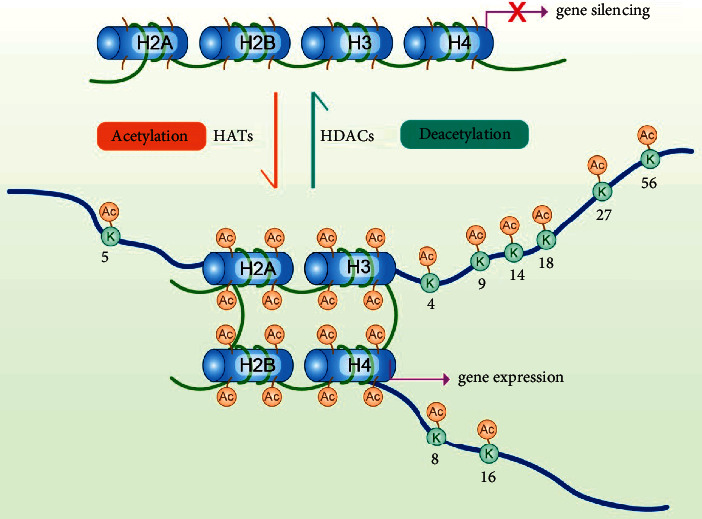
The dynamic process of reversible acetylation. Commonly functioning acetylation sites in oral squamous carcinoma.
2. Histone Modification
The concept that epigenetic abnormality is a possible sign of cancer has now been verified over the past decades. Several studies have suggested that the multimutations and the copy number alterations in the epigenetic modifiers (including acetyltransferase, deacetylase, methyltransferase, demethylase, and kinase) might jointly promote the progression of OSCC [7]. The nucleosome made up of approximately 147 bp of DNA is the basic unit of chromatin. It envelops a 2-copy histone octamer containing 4 kinds of histones (i.e., H2A, H2B, H3, and H4). Chromatin-modifying enzymes dynamically perform the posttranslational modifications of histones and DNA through tightly regulated mechanism [4, 8]. Posttranslational modification of histone is also an important mechanism regulating the structure and function of chromatin, which acts as a critical role in the emergence and progression of cancer through regulating the genetic transcription, chromatin remodeling, and nuclear structure [9]. Various posttranslational modifications exist in the eukaryotic animal cells, which usually include phosphorylation, ubiquitination, glycosylation, methylation, and acetylation. The histone tail projection of the nucleosome octamer experiences several posttranslational modifications including the added chemical groups like acetyl, phosphate, and methyl. Less common modifications include ubiquitination, sulfonation, and ribosylation, occurring on the residues of lysine, arginine, and serine on histones [10].
2.1. Acetylation
Various posttranslational modifications can regulate the function of histones, including the reversible acetylation of lysine terminal 1 group on histones. Acetylation is a universal protein modification that modulates a variety of cellular events including the cell cycle, cell metabolism, gene transcription, signal transduction, and RNA splicing [11]. Histone acetylation can disrupt or remove the nucleosomes from the transcription region and loosen the chromatin to facilitate the entry of proteins to the DNA to be replicated or transcribed. Studies of proteomics stated that acetylation can occur in every corner of the cell on thousands of proteins [12]. Histone acetylation is a widely studied posttranslational modification that can be reversed through the transfer of acetyl groups to lysine residues by HATs and its further removal by HDACs.
Histone acetyltransferase can attach the acetyl groups to the lysine residues in both histones and nonhistones, which can be divided into 2 groups according to their intracellular location and substrate specificity. The first group, named HAT, is located only in the nucleosomes and that has the effect on chromatin remodeling by modifying the chromatin-related histones. The other group, named B HAT, is located both in nucleosomes and cytoplasm and that can acetylate free soluble histones [13]. The common HATs are mainly composed of three families: the GCN5/PCAF family, the CBP/p300 subfamily, and the MYST family (MOZ, MOF, TIP60, and HBO1) [14] (see Table 1). The largest subfamily of histone acetyltransferase (MYST family) contains a high conserved MYST domain, and this structure consists of a zinc finger and an acetyl-CoA binding motif. The histone acetyltransferase has some extra structures including the plant homologous structural domain-linked zinc fingers (MORF and MOZ) and chromophores (TIP60 and MOF) [15], which are parts of the protein-complexes involved in the pro- and anticancer activities [16]. Another group of N-acetyltransferase (GNAT) family related to GCN5 is represented by the p300/CREB binding protein (CBP), binding factor (PCAF), HAT1, and GCN5, which contains bromo polysaccharides and adds an acetylated group of lysine to histone H2B, H3, and H4 [17, 18]. The CBP/p300 subfamily contains many small domains that interact with many other proteins containing disordered domains, including p53 and NF-κB [19].
Table 1.
The major HAT families.
| Family | Dominant members | Characteristics |
|---|---|---|
| GANAT | HAT1 | Contains bromo polysaccharides and conducts the acetylation of lysine on histone H2B, H3, and H4 |
| GCN5 | ||
| PCAF | ||
|
| ||
| MYST | TIP60 | Contains a high conserved MYST domain, which consists of a zinc finger and an acetyl coenzyme, a binding motif |
| MOF | ||
| MOZ | ||
| MORF | ||
| HBO1 | ||
|
| ||
| P300/CPB | P300/CPB | Contains many small domains that interact with many other proteins containing disordered transactivation domains |
HATs catalyze the transfer of acetyl groups to the target lysine (Lys/K) residues on the histone tail, thereby neutralizing the positive lysine charge, loosening condensed chromatin, promoting activation of gene transcription, and exposing binding sites for “binding motif proteins” that recognize the modifications on the histone [20]. Apart from the histone charge modification, histone acetylation can regulate the intracellular pH. Interestingly, many tumor cells show low histone acetylation levels and the acidic internal environment. Besides, poor prognosis in cancer patients has also been associated with low intracellular pH [21]. Different HATs can simultaneously act as tumor suppressors and oncogenes, which mean the balance of acetylation is critical to the stability of cells [22]. For example, as a kind of HAT, Tip60 can regulate ataxia-telangiectasia mutated (ATM) and DNA damage response pathways to participate in tumorigenesis, and it can also activate the transcription of p53 and Myc. ATM is a key regulator of DNA double strand break repair. Upregulated Tip60 can activate ATM through acetylation to promote DNA damage repair and inhibit tumor cell growth [23]. The reduction of Tip60 expression leads to genomic instability and apoptotic signaling cascade impairment, promoting the onset of malignant transformation of tumors [24] (see Tables1 and 2).
Table 2.
The major HDAC families.
| Family | Dominant members | Characteristics |
|---|---|---|
| I | HDAC1, 2, 3, 8 | Widely expressed in human cell lines and nuclear tissues |
| II | IIa: HDAC4, 5, 7, 9 | Tissue specific expression, shuttling between nucleus and cytoplasm |
| IIb: HDAC6, 10 | ||
| III | Sirt1, 2, 3, 4, 5, 6, 7 | NAD+ dependent, with a very unique catalytic mechanism for deacetylation |
| IV | HDAC11 | Deacetylate different histone sites, resulting in reduced substrate specificity |
2.2. Deacetylation
Contrary to the HATs, the HDACs remove acetyl groups from the high acetylated histones and repress the gene transcription. There are 4 HDACs in mammals, including the Zn2+-dependent class I (i.e., Rpd3-like enzyme) consisting of HDAC1, HDAC2, HDAC3, and HDAC8; the class II (i.e., Hda1-like enzyme) consisting of a subclass of IIa (HDAC4, HDAC5, HDAC7, and HDAC9 belong to class IIa) and IIb (HDAC6 and HDAC10 belong to class IIb); the class III (i.e., Sir-like enzyme) consisting of 7 SIRTs referred as the NAD-dependent deacetylase; the class IV of only HDAC11 with homologous sequences of class I and II [24](see Table 2).
HDAC regulates tumorigenesis through mechanisms like activating oncogene signaling pathways and downregulating tumor suppressor genes. HDACs have low substrate specialty with each kind of them deacetylating multiple sites on histone. Although mutation of HDACs is rare, overexpression of HDACs is still common in cancer [10, 25]. HDACs interfere with the transcription of oncogenes and antioncogenes by removing the acetyl groups from histones and reversing the acetylation of chromatins. Furthermore, HDACs can catalyze the deacetylation of many nonhistone proteins, which control a series of biological processes including the development and progression of cancer [26]. These processes are involved in the cell cycle, apoptosis, DNA damage response, metastasis, angiogenesis, autophagy, and other honeycomb shaping process [27].
2.3. Histone Acetylation and Oral Cancer
Accumulating scientific evidence have presented that epigenetic alterations, including chromatin remodeling, noncoding RNAs, DNA methylation, and histone covalent modifications, are generally involved in oral carcinogenesis and treatment tolerance. Epigenetic modifications contribute to the formation of cell plasticity and cancer stem cells (CSCs) during tumor progression [3]. Especially the dysregulation of histone acetylation leads to the disorder of the activity of different genes, resulting in malignant transformation-related events. Due to the reversibility and low-abundance of acetylation, it is a challenge to identify a large number of acetylation sites. Few literature have indicated changes in histone acetylation on specific lysine residues in head and neck squamous cell carcinoma (HNSCC) [28]. The reported main acetylation sites are listed in Table 3.
Table 3.
Histone acetylation sites and expression in OSCC.
| Sites | Expression | Acetylase | Function | References |
|---|---|---|---|---|
| H2AK5 | Hyperacetylation | Tip60 | Hyperacetylation is associated with cancer promotion | [29] |
| H3K4 | Hypoacetylation | P300/Tip60 | Low acetylation level is positively correlated with malignancy | [28] |
| H3K9 | Hypoacetylation | P300 | CSC accumulation leads to hypoacetylation and promotes tumor; its absence is a hallmark of chemoresistance | [30, 31] |
| Hyperacetylation | High acetylation expression detected in oral cancer | [29] | ||
| H3K14 | Hyperacetylation | MOZ/MORF | P300-mediated hyperacetylation promotes tumor growth | [29] |
| H3K18 | Hyperacetylation | P300 | High acetylation levels are associated with poor prognosis | [28] |
| H3K27 | Hyperacetylation | GCN5 | Hyperacetylation enhances PD-L1 expression to promote tumor infiltration, metastasis, and recurrence | [32] |
| Hyperacetylation | Hyperacetylation activates PLAC2 to promote tumor proliferation | [33] | ||
| H3K56 | Hyperacetylation | P300/CPB | Hyperacetylation is associated with cancer promotion | [29] |
| H4K8 | Hypoacetylation | P300/CPB | Hypoacetylation is associated with cancer promotion | [29] |
| H4K16 | Hypoacetylation | hMOF/Tip60 | Hypoacetylation is associated with cancer promotion | [29] |
| Hyperacetylation | hMOF promotes acetylation to enhance oral cancer cell growth | [33] |
Most theories confirmed that in HNSCC, low level histone acetylation makes the nucleus smaller, which reduces the DNA damage repair proteins flowing into the nucleus and enhances the resistance to intercalation agents [34], with the most studied being H3K9ac. Some studies have demonstrated that the deletion of histone H3K9ac indicates the occurrence of chemoresistance and is associated with the NF-κB signaling and the accumulation of CSC [35]. The decrease of H3K9ac is related to the activation of epithelial-mesenchymal transition (EMT) and the increased cell proliferation during the process of oral carcinogenesis, indicating the H3K9ac is involved in the progression of HNSCC and is coexpressed with the mesenchymal vimentin prior to the invasion of HNSCC [30]. In oral cancer, the low level of H3K9 acetylation indicates a bad prognosis. The expression level of H3K9Ac is expressed at lower levels in OSCC than in oral leukoplakia, an oral precancerous lesion. In survival analysis, low expression of H3K9Ac was associated with a poorer prognosis in OSCC [31]. Interestingly, in contrast, some studies have found the hyperacetylation of histone H3 (mainly H3K14 and H3K9) in samples from patients with OSCC as well as the hyperacetylation of H2AK5 and H3K56 and hypoacetylation of H4K8 and H4K16 [29]. Several reports have reported the acetylation of special lysine residues in oral cancer. For example, both low and high levels of H3K4ac have been reported to be correlated with the progression and the poor prognosis of OSCC. The hypoacetylation of H3K4 is associated with the advanced OSCC tumor staging, primary tumor (T), regional lymph nodes (N), and perineural infiltration (PNI), while the H3K18ac is positively correlated with tumor stage [28]. Upregulation of H3K27ac was found to be associated with the activation of the placenta-specific protein 2 (PLAC2) gene in OSCC. PLAC2 promoted the progression of OSCC by modulating the Wnt/β-catenin signaling pathway. Compared with normal oral epithelial keratinocytes, PLAC2 is more abundant in oral squamous cell carcinoma cells (CAL-27 and SCC-9) [33]. Moreover, the non-long-strand coding RNA lncRNA MX1-215 can directly interact with the H3K27 acetylase GCN5, interrupting the combination of GCN5 and H3K27, interfering the transcription of PD-L1 and LGALS9 mediated by the acetylation of H3K27, and decreasing the acetylation level and expression level of PD-L1 and galectin-9 on the tumor cells [32]. It is also found that the hMOF (males absent on the first) can participate in the activation of transcription through acetylation of H4K16 and regulate the growth of cancer cells by the multicomb histone enhancer of zeste homolog 2 in OSCC. The upregulation of hMOF indicates poorer overall survival and disease-free survival [36].
2.4. Histone Deacetylation and Oral Cancer
Changes in the expression of multiple HDACs have been reported to be closely related to the regulation of cell cycle-related genes, cell invasion, apoptosis, angiogenesis, differentiation, and migration in a variety of cancers [37, 38]. For example, several studies have shown that the HDAC2 is overexpressed in OSCC, leading to increased stability of HIF-1α and the increased invasion and migration of HNSCC. The high abundance of HDAC2 is associated with the T and N states in the late stage [39]. Sakuma et al. [40] also found that HDAC6 is overexpressed in late HNSCC, indicating the activity of HDAC6 may associate with the tumor invasiveness of oral cancer. Besides, some researchers also found that HDAC6 catalyzes the deacetylation of α-tubulin and increases cell mobility and tumor metastasis [41]. The HDAC7 is also overexpressed in head and neck tumor, whose accumulation can activate proto oncogene c-MYC and promote cell proliferation [42]. The accumulation of HDAC8 induces the proliferation of cancer cells by inhibiting the activation and autophagy of Caspase in OSCC. In contrast, silencing of HDAC8 significantly inhibited OSCC cell proliferation, invasion, and metastasis [42]. HDAC9 has been reported to have oncogenic effects in OSCC by targeting proapoptotic genes to promote tumor growth. The low expressed HDAC9 inhibited the proliferation of SCC116 cells, increased apoptosis, and induced the G0/G1 arrest. The overexpressed HDAC9 positively correlates with the OS and promotes OSCC by targeting the transcription factors of MEF2D and NR4A1/Nur77 (proapoptotic MEF2 targets) [43].
The SIRT family belongs to the NAD+-dependent histone deacetylase class III and is involved in the cell cycle, transcriptional regulation, and metabolism [44]. Contrary to the HDAC, the SIRT acts as a tumor suppressor in cancer, preventing the occurrence of DNA damage and oxidative stress [45]. The SIRT1, SIRT2, SIRT3, SIRT5, and SIRT7 are reduced in advanced HNSCC and have the potential to be used as prognostic markers [46]. While SIRT6 accumulates in peripheral blood of HNSCC patients [47], SIRT1 is the mammalian homolog of the chromatin silencing factor Sir2 in S. cerevisiae, which is expressed at elevated levels and catalytic activity in OSCC cells [48]. The latest study has presented that the SIRT7 inhibits the EMT in the metastasis of OSCC by promoting the deacetylation of SMAD4, therefore, reducing the proliferation and invasion of OSCC cells in vitro [49]. In OSCC, the posttranscriptional regulation of SIRT3 is induced by miR-31, which targets the SIRT3 to destroy the mitochondrial structure and increase the oxidative stress response in oral cancer. The downregulation of SIRT3 reduces the migration and invasion of cancer cells enhanced by miR-31 [50]. The reported main histone deacetylase are listed in Table 4.
Table 4.
Histone deacetylase and expression in OSCC.
| Name | Expression | Function | References |
|---|---|---|---|
| HDAC2 | High expression | Overexpressed in OSCC, resulting in protein HIF-1 α, resulting in increased invasion and migration of HNSCC | [39] |
| HDAC6 | High expression | Send α-tubulin deacetylation, thus promoting the process of tumor metastasis development | [40, 41] |
| HDAC7 | High expression | Activate c-myc and promote the proliferation of oral tumor cells | [42] |
| HDAC8 | High expression | Inhibition of caspase activation and autophagy in OSCC | [42] |
| HDAC9 | High expression | Promote tumor growth by targeting proapoptotic genes | [43] |
| SIRT1, 2, 3, 5, 7 | Low expression | Inhibit tumor and prevent DNA damage and oxidative stress | [45, 46] |
3. The Role of Histone Deacetylase Inhibitors in the Treatment of Oral Squamous Carcinoma
Owing to the reversibility of acetylation modifications, it is clear that the changes observed in the development of cancer have become attractive targets for therapy with the thriving research on the histone deacetylase inhibitor (HDACi)-related drugs. Although the HDACi has been applied to the treatment of hematologic malignancies successfully, its clinical efficacy as an independent drug in the treatment of solid tumors remains limited. The HDACi is able to achieve the best clinical efficacy only by combining with other treatments, including radiotherapy and chemotherapy, for synergistic or additive effects [51]. Current research data suggested that the HDACi has the best effect on HNSCC when administered together with other therapeutic drugs [52, 53].
The HDACi counteracts the abnormal acetylation state of proteins in cancer cells and reactivates the expression of tumor suppressors, thus inducing cell cycle arrest, apoptosis, differentiation, and inhibiting angiogenesis and metastasis [8]. Besides, the tumor cells are more sensitive to the apoptosis induced by HDACi than normal cells [54]. Based on their chemical structures, HDACi is classified into four classes, including hydroxamic acid, cyclic peptides, short-chain fatty acids, and benzamide. Most of them have already been developed as anticancer drugs with different specificity, efficiency, and pharmacokinetic and toxicological properties [8, 51–53, 55, 56]. Studies on the anticancer effects of HDAC in oral squamous cell carcinoma are presented in Table 5.
Table 5.
Antitumor effects of HDACis in OSCC.
| Class | Name | Target | Mechanism | References |
|---|---|---|---|---|
| Hydroxamic acids | SAHA | HDAC I, HDAC II, HDAC IV | Induces hyperacetylation of H2A and H3 and inhibits tumor activity | [57] |
| Upregulate E calcineurin and ErbB3, downregulate vimentin, EGFR and ErbB2, and enhance the antitumor effect of defibrotide | [58] | |||
| TSA | HDAC I, HDAC II | Induced apoptosis of tumor cells in combination with PS-341 | [59] | |
| Combine with ATRA to inhibit tumor growth | [60] | |||
| LBH589 | HDAC I, HDAC II, HDAC IV | Induce high acetylation of H3 and H4, inhibit tumor growth and induce apoptosis, and lead to G1 phase block | [61] | |
| Upregulate the expression of p21 and induce G2/M arrest and cancer cell apoptosis | [62] | |||
|
| ||||
| Cyclopeptides | Apicidin | HDAC1, HDAC3 | Competitive combination of HDAC and antitumor proliferation | [63] |
| Increase the level of LC3-II and increase the apoptosis and autophagy of tumor cells | [64] | |||
| FK228 | HDAC1, HDAC2 | Reduce Ki67 staining and inhibit tumor growth | [65] | |
| Induce high expression of hTERT and inhibit tumor growth | [66] | |||
| Trapoxin | HDAC8 | Inhibit tumor cell proliferation as a potential anticancer compound | [67] | |
|
| ||||
| Short-chain fatty acids | VPA | HDAC I, HDAC II | Promote H3, H4 acetylation and inhibit oncogene expression | [68] |
| Increase G1 arrest and apoptosis of tumor cells | [69] | |||
| PBA | HDAC I, HDAC II | Promote tumor cell apoptosis and inhibit EMT transformation | [70] | |
| Reduce TNF-α level and promote DNA repair | [71] | |||
| NaBu | HDAC I, HDAC IIa | Induce cell cycle arrest, related to the increased expression of kip1 | [72] | |
|
| ||||
| Benzamides | MS-275 | HDAC I | Make tumor cells stagnate in G0/G1 phase, enhance H3 and H4 acetylation, and promote apoptosis | [73] |
| Increase cisplatin cytotoxicity | [74] | |||
| Inhibit tumor migration and invasion by activating mir-107 and miR-138 | [75] | |||
| MGCD0103 | HDAC I, HDAC IV | Has better radiotherapy sensitization effect | [76] | |
3.1. The Class of Hydroxamic Acid
The hydroxamic acid class of HDACi has been extensively studied in recent years, which mainly consists of rings, aliphatic chains, and hydroxamic acids for the surface recognition region, linkage region, and metal binding region (zinc combining region), respectively. It mainly contains SAHA (suberoylanilide hydroxamic acid), trichostatin A (TSA), and panobinostat, belinostat.
SAHA is a nonselective HDACi and the first FDA-approved HDACi that can be used alone or in combination as clinical treatment since October 2006 [77]. A research report found that SAHA can enhance the acetylation of H2A and H3 and inhibit the anticancer activity in vitro and xenotransplantation models by inducing cell activity reduction, caspase dependent apoptosis, and tumor growth inhibition [57]. In addition, powerful antiproliferative and synergistic effects of SAHA and gefitinib were noted. The SAHA enhances the antitumor function of gefitinib by upregulating the E-cadherin and ErbB3, downregulating the vimentin, EGFR, and ErbB2, and the restoration of mesenchyme to an epithelial phenotype [58]. Studies also reflected that the SAHA can enhance the cellular chemosensitivity to cisplatin, thereby mediating apoptosis in oral squamous carcinoma cells [78].
It has been verified that TSA is a natural inhibitor of HDAC class I and II. The changes in chromatin acetylation of HNSCC unexpectedly triggered a decrease in CSC. The hyperacetylation of chromatin in HNSCC induced by TSA can interrupt the generation of CSC and destroy the ability of stem cells to maintain tumor globules. It is also demonstrated that the TSA can decrease the enzyme activity of ALDH (a recognized marker of CSCs) [30]. The SAHA and TSA also reduce the CSC markers of CD44 and ABCG2, the expression of genes related to cell stemness, and the EMT phenotype in the oral cancer [79]. The combined application of TSA and PS-341 (bortezomib), a proteasome inhibitor, was also studied in HNSCC. Although TSA alone did not induce apoptosis, it can activate cystathionine to significantly enhance the degree of apoptosis induced by PS-341 [59]. The combination therapy of TSA and all-trans retinoic acid (ATRA) synergistically inhibited the growth of tumor cells and strongly induced transcriptional and activation of target genes, thereby restoring the tumor sensitivity of HNSCC cell lines to retinoic acid [60]. HDACi reportedly appears to be valuable in the treatment of radiosensitized tumors in solid tumors, including HNSCC. TSA, SAHA, M344 (SAHA analogue), and desmethyl peptide (FR90228) regulate the cellular response to ionizing radiation and promote apoptosis and cell cycle arrest in HNSCC [80]. Recent treatments have shown that the TSA combined with 5-Aza-dC or RG108 can significantly reduce the vitality of HSC-2 cells and Ca922 cells. The TSA combined with DZNep also reduces the vitality of Ca922 cells, and the combination of TSA and the combination of TSA with other epigenetic inhibitors is also therapeutic for OSCC [81].
Panobinostat, an FDA-approved hydroxamic acid analogue (LBH589) for the treatment of refractory/recurrent multiple myeloma, induces hyperacetylation of histones H3 and H4 [82]. The LBH589 can inhibit cell growth and induce the G1 arrest and the apoptosis of OSCC by increasing the inhibition of transcription factor specificity protein 1 [61]. In vitro studies performed by flow cytometry on laryngopharyngeal (FaDu) and oral (CAL-27, SCC-15, UM-SCC-1, and UM-SCC-47) OSCC cell lines showed that treatment of these cells with LBH589 upregulated p21 and induced G2/M phase block and cell death [62].
3.2. The Class of Cyclic Peptide
The cyclic peptide class of HDACi can be divided into 2 categories according to the functional groups: one category contains (2S, 9S)-2-amino-9, 10-epoxy-8-oxodecanoic acid (Aoe), and epoxy ketone, while the other category does not contain the Aoe structure, which is mainly represented by apicidin, FK228, and trapoxin.
Apicidin is a cyclic tetrapeptide isolated from Fusarium, whose decanoic acid side chain, macrocyclic structure, and tryptophan side chain competitively bind HDAC. It has a significant antitumor function involving the antiproliferative activity and differentiation inducing activity toward the cancer cells [63]. Moreover, the OSCC cell lines treated with apicidin presented elevated levels of LC3-II, G2/M arrest, apoptosis, and autophagy as assessed by the MTT, DAPI staining, and flow cytometry [64].
Romidepsin (FK228) is a natural HDACi separated from Chromobacterium violaceum, which can induce the inhibition of HDAC with the characteristics of p21 (Waf1/Cip1) and reduce the staining of Ki67. Despite the limitations of single romidepsin treatment in the treatment of HNSCC, it still can effectively inhibit the tumor-related HDAC [65]. As an HDAC inhibitor, the FK228 induces the telomerase reverse transcriptase (hTERT) gene through a complex mechanism that involves the inhibition of histone deacetylation and other transcription factors except for the c-myc [66].
Trapoxin, a fungal product, was found to induce morphological reversion from transformed to normal in sis-transformed NIH3T3 fibroblasts [83]. Low concentration of trapoxin irreversibly inhibited the deacetylation of acetylated histone molecules through the covalent binding of epoxides and histone deacetylase. The process caused accumulation of highly acetylated core histone in a variety of mammalian cell lines [84]. Other findings have found that the product of binding trapoxin to histone deacetylase HDAC8 helps inhibit tumor cell proliferation as a potential anticancer compound [67].
3.3. The Class of Short-Chain Fatty Acid
The short-chain fatty acid class of HDACi has a relatively simple structure including a carboxyl group that binds the metal ions. It mainly contains sodium butyrate, valproic acid, and phenylbutyric acid.
Valproic acid (VPA), which is often widely used as a broad-spectrum antiepileptic drug and mood stabilizer in clinical practice, has been reported to be a class I and II HDAC inhibitor that promotes hyperacetylation of the N-terminal chains of histones H3 and H4 and nonhistones, thereby altering chromatin structure and preventing restricted expression of oncogenes. Moreover, several researchers have reported that the enhanced function of HDAC can stabilize the DNMT, while the VPA degrades the DNMT1 by multiple biochemical mechanisms including acetylation [68]. Because of its HDAC inhibitory activity, VPA has been a safe treatment for epilepsy for many years, and therefore, VPA is considered a good candidate for antitumor therapy in patients with metastatic or recurrent HNCs. In a clinical study, Gan et al. [52] have found that VPA inhibits the growth of HNSCC cell lines as a monotherapy or in combination with other anticancer drugs at physiological doses. The VPA therapy can increase the G1 arrest, apoptosis, and the expression of small ubiquitin-related modifiers. VPA treatment resulted in reduced tumor volume and increased apoptosis in xenograft models [69].
As an ammonia scavenger in the treatment of urea cycle diseases, phenylbutyric acid (PBA) leads to cell apoptosis, differentiation, and cell cycle arrest. Radiation-exposed patients receiving PBA have relatively low levels of mucosal oxidative stress and TNF-α, accompanied by mild oral mucositis, which promotes DNA repair and survival [71]. Sodium phenylbutyrate has a proapoptotic effect on tumor cells, inhibiting transforming growth factor-β-related epithelial-mesenchymal transition, with a decrease in the mesenchymal marker N-calmodulin and an increase in the epithelial marker E-calmodulin [70]. The PBA-derived HDACs (S)-HDAC42 present a higher activity of antiproliferation than SAHA, which can induce cell apoptosis by elevating the p21and p27, reducing the levels of CDK6 in the G1 phase, the cyclin D1 and the phosphorylation of Akt. Accordingly, it showed high potency in inhibiting the growth of OSCC in a Ca922 xenograft nude mouse model [85].
The short-chain fatty acid derivative sodium butyrate (NaBu) is another class I and IIa HDAC inhibitor belonging to aliphatic fatty acids. It is presented by the flow cytometry and immunocytochemical analysis that the NaBu significantly inhibits the proliferation of Tca8113 in a time and dose-dependent manner, and the NaBu therapy can inhibit the in vitro growth of OSCC cell lines and induce the cell cycle arrest, which may be related to the increased expression of p27 [72].
3.4. The Class of Benzamide
The benzamide class of HDACi has not been extensively studied as the first three HDAC inhibitors, which mainly contain the MS-275 (entinostat) and MGCD0103 (mocetinostat).
Entinostat is a bioavailable class I HDACi with a long half-life. In vitro and in vivo studies suggested that entinostat alone or in combination may be a promising agent for the treatment of OSCC due to its antiproliferative and proapoptotic effects. Administration of entinostat could reduce the proliferation of OSCC cells, leading to the G0/G1 arrest and massive apoptosis of tumor cells. Meanwhile, an increase in reactive oxygen species and a significant reduction in CSCs was observed as well. Entinostat also caused an increase in acetylated histone H3 or H4 and changes in the expression of cell cycle-related proteins (e.g., p21) [73]. The synergistic effect of HDACi and cisplatin enhanced the induction of cytotoxicity and apoptosis compared with using cisplatin alone, while the MS-275 also played the same role in the cytotoxicity of cisplatin during the treatment of OSCC. [74]. Combination treatment also activated the miR-138 and miR-107, leading to interrupting the following migration and invasion of tumor cells [75].
Preprocessing of the OSCC cell lines with the MGCD0103 and 5-aza20-deoxycytidine prior to radiotherapy resulted in better radiosensitization compared to the preprocessing by 5-Aza-dC alone [76].
4. Summary and Outlook
Histones have many common epigenetic modifications, and acetylation is one of the most common, which regulates DNA transcription and interferes with gene expression. The deacetylation is opposite of acetylation. The balance of those two reactions is necessary to ensure the integrity of chromatin. Therefore, histone acetylation in cancer cells can play a double role in cancer progression, which may be involved in suppressing the silencing of oncogenes and enhancing the expression of oncogenes. Evidence accumulated from a large amount of experimental data suggests that overexpression of HDAC may be closely associated with the development and progression of OSCC, and HDAC can be considered as a potential anticancer agent for OSCC. Further study on histone acetylation modification may be helpful to discover new therapies for oral cancer.
Acknowledgments
This work was supported by the Beijing Natural Science Foundation (7222177) and National Natural Science Foundation of China (81602534).
Contributor Information
Yong Wang, Email: fgwangyong@163.com.
Jun Wen, Email: drwenjun@sina.com.
Data Availability
The data used to support the findings of this study are available from the corresponding authors upon request.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Authors' Contributions
Ying Lu collected the related studies and drafted the manuscript. Jinjin Yang participated in the design of the review. Junwen Zhu and Shuyuan Luo helped to publish the review. Yao Shu, Xuan Zou, and Qiao Ruan prepared the tables and the figure. Jun Wen and Yong Wang reviewed and revised the first manuscript. All authors read and approved the final manuscript. Ying Lu and Jinjin Yang contributed equally to this work.
References
- 1.Hashibe M., Brennan P., Chuang S. C., et al. Interaction between tobacco and alcohol use and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Cancer Epidemiology Biomarkers & Prevention . 2009;18(2):541–550. doi: 10.1158/1055-9965.epi-08-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Algazi A. P., Grandis J. R. Head and neck cancer in 2016: a watershed year for improvements in treatment? Nature Reviews Clinical Oncology . 2017;14(2):76–78. doi: 10.1038/nrclinonc.2016.196. [DOI] [PubMed] [Google Scholar]
- 3.Castilho R. M., Squarize C. H., Almeida L. O. Epigenetic modifications and head and neck cancer: implications for tumor progression and resistance to therapy. International Journal of Molecular Sciences . 2017;18(7):p. 1506. doi: 10.3390/ijms18071506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Audia J. E., Campbell R. M. Histone modifications and cancer. Cold Spring Harbor Perspectives in Biology . 2016;8(4) doi: 10.1101/cshperspect.a019521.a019521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Figlia G., Willnow P., Teleman A. A. Metabolites regulate cell signaling and growth via covalent modification of proteins. Developmental Cell . 2020;54(2):156–170. doi: 10.1016/j.devcel.2020.06.036. [DOI] [PubMed] [Google Scholar]
- 6.West A. C., Johnstone R. W. New and emerging HDAC inhibitors for cancer treatment. Journal of Clinical Investigation . 2014;124(1):30–39. doi: 10.1172/jci69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin D., Abba M. C., Molinolo A. A., et al. The head and neck cancer cell oncogenome: a platform for the development of precision molecular therapies. Oncotarget . 2014;5(19):8906–8923. doi: 10.18632/oncotarget.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dawson M. A., Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell . 2012;150(1):12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 9.Rothbart S. B., Strahl B. D. Interpreting the language of histone and DNA modifications. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms . 2014;1839(8):627–643. doi: 10.1016/j.bbagrm.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chi P., Allis C. D., Wang G. G. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nature Reviews Cancer . 2010;10(7):457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Q., Zhang Y., Yang C., et al. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science . 2010;327(5968):1004–1007. doi: 10.1126/science.1179687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao S., Xu W., Jiang W., et al. Regulation of cellular metabolism by protein lysine acetylation. Science . 2010;327(5968):1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Friedmann D. R., Marmorstein R. Structure and mechanism of non-histone protein acetyltransferase enzymes. FEBS Journal . 2013;280(22):5570–5581. doi: 10.1111/febs.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Narita T., Weinert B. T., Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nature Reviews Molecular Cell Biology . 2019;20(3):156–174. doi: 10.1038/s41580-018-0081-3. [DOI] [PubMed] [Google Scholar]
- 15.Su J., Wang F., Cai Y., Jin J. The functional analysis of histone acetyltransferase MOF in tumorigenesis. International Journal of Molecular Sciences . 2016;17(1):p. 99. doi: 10.3390/ijms17010099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Avvakumov N., Côté J. The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene . 2007;26(37):5395–5407. doi: 10.1038/sj.onc.1210608. [DOI] [PubMed] [Google Scholar]
- 17.Salah Ud-Din A. I., Tikhomirova A., Roujeinikova A. Structure and functional diversity of GCN5-relatedN-acetyltransferases (GNAT) International Journal of Molecular Sciences . 2016;17(7):p. 1018. doi: 10.3390/ijms17071018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ngo L., Brown T., Zheng Y. G. Bisubstrate inhibitors to target histone acetyltransferase 1. Chemical Biology & Drug Design . 2019;93(5):865–873. doi: 10.1111/cbdd.13476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dyson H. J., Wright P. E. Role of intrinsic protein disorder in the function and interactions of the transcriptional coactivators CREB-binding protein (CBP) and p300. Journal of Biological Chemistry . 2016;291(13):6714–6722. doi: 10.1074/jbc.r115.692020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heintzman N. D., Stuart R. K., Hon G., et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nature Genetics . 2007;39(3):311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 21.Stricker S. H., Köferle A., Beck S. From profiles to function in epigenomics. Nature Reviews Genetics . 2017;18(1):51–66. doi: 10.1038/nrg.2016.138. [DOI] [PubMed] [Google Scholar]
- 22.Jacobson R. H., Ladurner A. G., King D. S., Tjian R. Structure and function of a human TAFII250 double bromodomain module. Science . 2000;288(5470):1422–1425. doi: 10.1126/science.288.5470.1422. [DOI] [PubMed] [Google Scholar]
- 23.Zhang K., Wu Q., Liu W., et al. FAM135B sustains the reservoir of Tip60-ATM assembly to promote DNA damage response. Clinical and Translational Medicine . 2022;12(8):p. e945. doi: 10.1002/ctm2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sykes S. M., Mellert H. S., Holbert M. A., et al. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Molecular Cell . 2006;24(6):841–851. doi: 10.1016/j.molcel.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bannister A. J., Kouzarides T. Regulation of chromatin by histone modifications. Cell Research . 2011;21(3):381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y., Seto E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med . 2016;6(10) doi: 10.1101/cshperspect.a026831.a026831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reichert N., Choukrallah M. A., Matthias P. Multiple roles of class I HDACs in proliferation, differentiation, and development. Cellular and Molecular Life Sciences . 2012;69(13):2173–2187. doi: 10.1007/s00018-012-0921-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Y. W., Kao S. Y., Wang H. J., Yang M. H. Histone modification patterns correlate with patient outcome in oral squamous cell carcinoma. Cancer . 2013;119(24):4259–4267. doi: 10.1002/cncr.28356. [DOI] [PubMed] [Google Scholar]
- 29.Arif M., Vedamurthy B. M., Choudhari R., et al. Nitric oxide-mediated histone hyperacetylation in oral cancer: target for a water-soluble HAT inhibitor, CTK7A. Chemistry & Biology . 2010;17(8):903–913. doi: 10.1016/j.chembiol.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 30.Giudice F. S., Pinto D. S., Nör J. E., Squarize C. H., Castilho R. M. Inhibition of histone deacetylase impacts cancer stem cells and induces epithelial-mesenchyme transition of head and neck cancer. PLoS One . 2013;8(3) doi: 10.1371/journal.pone.0058672.e58672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Webber L. P., Wagner V. P., Curra M., et al. Hypoacetylation of acetyl-histone H3 (H3K9ac) as marker of poor prognosis in oral cancer. Histopathology . 2017;71(2):278–286. doi: 10.1111/his.13218. [DOI] [PubMed] [Google Scholar]
- 32.Ma H., Chang H., Yang W., Lu Y., Hu J., Jin S. A novel IFNα-induced long noncoding RNA negatively regulates immunosuppression by interrupting H3K27 acetylation in head and neck squamous cell carcinoma. Molecular Cancer . 2020;19(1):p. 4. doi: 10.1186/s12943-019-1123-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen F., Qi S., Zhang X., Wu J., Yang X., Wang R. lncRNA PLAC2 activated by H3K27 acetylation promotes cell proliferation and invasion via the activation of Wnt/β catenin pathway in oral squamous cell carcinoma. International Journal of Oncology . 2019;54(4):1183–1194. doi: 10.3892/ijo.2019.4707. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Almeida L. O., Abrahao A. C., Rosselli-Murai L. K., et al. NFκB mediates cisplatin resistance through histone modifications in head and neck squamous cell carcinoma (HNSCC) FEBS Open Bio . 2013;4(1):96–104. doi: 10.1016/j.fob.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guimarães D. M., Almeida L. O., Martins M. D., et al. Sensitizing mucoepidermoid carcinomas to chemotherapy by targeted disruption of cancer stem cells. Oncotarget . 2016;7(27):42447–42460. doi: 10.18632/oncotarget.9884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Q., Sun H., Shu Y., Zou X., Zhao Y., Ge C. hMOF (human males absent on the first), an oncogenic protein of human oral tongue squamous cell carcinoma, targeting EZH2 (enhancer of zeste homolog 2) Cell Proliferation . 2015;48(4):436–442. doi: 10.1111/cpr.12177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ropero S., Esteller M. The role of histone deacetylases (HDACs) in human cancer. Molecular Oncology . 2007;1(1):19–25. doi: 10.1016/j.molonc.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Witt O., Deubzer H. E., Milde T., Oehme I. HDAC family: what are the cancer relevant targets? Cancer Letters . 2009;277(1):8–21. doi: 10.1016/j.canlet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 39.Chang H. H., Chiang C. P., Hung H. C., Lin C. Y., Deng Y. T., Kuo M. Y. P. Histone deacetylase 2 expression predicts poorer prognosis in oral cancer patients. Oral Oncology . 2009;45(7):610–614. doi: 10.1016/j.oraloncology.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 40.Sakuma T., Uzawa K., Onda T., et al. Aberrant expression of histone deacetylase 6 in oral squamous cell carcinoma. International Journal of Oncology . 2006;29(1):117–124. doi: 10.3892/ijo.29.1.117. [DOI] [PubMed] [Google Scholar]
- 41.Gasche J. A., Goel A. Epigenetic mechanisms in oral carcinogenesis. Future Oncology . 2012;8(11):1407–1425. doi: 10.2217/fon.12.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ahn M. Y., Yoon J. H. Histone deacetylase 7 silencing induces apoptosis and autophagy in salivary mucoepidermoid carcinoma cells. Journal of Oral Pathology & Medicine . 2017;46(4):276–283. doi: 10.1111/jop.12560. [DOI] [PubMed] [Google Scholar]
- 43.Rastogi B., Raut S. K., Panda N. K., Rattan V., Radotra B. D., Khullar M. Overexpression of HDAC9 promotes oral squamous cell carcinoma growth, regulates cell cycle progression, and inhibits apoptosis. Molecular and Cellular Biochemistry . 2016;415(1-2):183–196. doi: 10.1007/s11010-016-2690-5. [DOI] [PubMed] [Google Scholar]
- 44.Haigis M. C., Guarente L. P. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes & Development . 2006;20(21):2913–2921. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- 45.Noguchi A., Li X., Kubota A., et al. SIRT1 expression is associated with good prognosis for head and neck squamous cell carcinoma patients. Oral Surgery, Oral Medicine, Oral Pathology and Oral Radiology . 2013;115(3):385–392. doi: 10.1016/j.oooo.2012.12.013. [DOI] [PubMed] [Google Scholar]
- 46.Lai C. C., Lin P. M., Lin S. F., et al. Altered expression of SIRT gene family in head and neck squamous cell carcinoma. Tumor Biology . 2013;34(3):1847–1854. doi: 10.1007/s13277-013-0726-y. [DOI] [PubMed] [Google Scholar]
- 47.Lv J. F., Hu L., Zhuo W., Zhang C. M., Zhou H. H., Fan L. Epigenetic alternations and cancer chemotherapy response. Cancer Chemotherapy and Pharmacology . 2016;77(4):673–684. doi: 10.1007/s00280-015-2951-0. [DOI] [PubMed] [Google Scholar]
- 48.Xiong P., Li Y. X., Tang Y. T., Chen H. G. Proteomic analyses of Sirt1-mediated cisplatin resistance in OSCC cell line. The Protein Journal . 2011;30(7):499–508. doi: 10.1007/s10930-011-9354-9. [DOI] [PubMed] [Google Scholar]
- 49.Li W., Zhu D., Qin S. SIRT7 suppresses the epithelial-to-mesenchymal transition in oral squamous cell carcinoma metastasis by promoting SMAD4 deacetylation. Journal of Experimental & Clinical Cancer Research . 2018;37(1):p. 148. doi: 10.1186/s13046-018-0819-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kao Y. Y., Chou C. H., Yeh L. Y., et al. MicroRNA miR-31 targets SIRT3 to disrupt mitochondrial activity and increase oxidative stress in oral carcinoma. Cancer Letters . 2019;456:40–48. doi: 10.1016/j.canlet.2019.04.028. [DOI] [PubMed] [Google Scholar]
- 51.Martins M. D., Castilho R. M. Histones: controlling tumor signaling circuitry. Journal of Carcinogenesis & Mutagenesis . 2013;1(5):1–12. doi: 10.4172/2157-2518.S5-001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gan C. P., Hamid S., Hor S. Y., et al. Valproic acid: growth inhibition of head and neck cancer by induction of terminal differentiation and senescence. Head & Neck . 2012;34(3):344–353. doi: 10.1002/hed.21734. [DOI] [PubMed] [Google Scholar]
- 53.Gillenwater A. M., Zhong M., Lotan R. Histone deacetylase inhibitor suberoylanilide hydroxamic acid induces apoptosis through both mitochondrial and Fas (Cd95) signaling in head and neck squamous carcinoma cells. Molecular Cancer Therapeutics . 2007;6(11):2967–2975. doi: 10.1158/1535-7163.mct-04-0344. [DOI] [PubMed] [Google Scholar]
- 54.Ungerstedt J. S., Sowa Y., Xu W. S., et al. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proceedings of the National Academy of Sciences of the United States of America . 2005;102(3):673–678. doi: 10.1073/pnas.0408732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lindsay C., Seikaly H., Biron V. L. Epigenetics of oropharyngeal squamous cell carcinoma: opportunities for novel chemotherapeutic targets. Journal of Otolaryngology - Head & Neck Surgery . 2017;46(1):p. 9. doi: 10.1186/s40463-017-0185-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ceccacci E., Minucci S. Inhibition of histone deacetylases in cancer therapy: lessons from leukaemia. British Journal of Cancer . 2016;114(6):605–611. doi: 10.1038/bjc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kumar B., Yadav A., Lang J. C., Teknos T. N., Kumar P. Suberoylanilide hydroxamic acid (SAHA) reverses chemoresistance in head and neck cancer cells by targeting cancer stem cells via the downregulation of nanog. Genes Cancer . 2015;6(3-4):169–181. doi: 10.18632/genesandcancer.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bruzzese F., Leone A., Rocco M., et al. HDAC inhibitor vorinostat enhances the antitumor effect of gefitinib in squamous cell carcinoma of head and neck by modulating ErbB receptor expression and reverting EMT. Journal of Cellular Physiology . 2011;226(9):2378–2390. doi: 10.1002/jcp.22574. [DOI] [PubMed] [Google Scholar]
- 59.Kim J., Guan J., Chang I., Chen X., Han D., Wang C. Y. PS-341 and histone deacetylase inhibitor synergistically induce apoptosis in head and neck squamous cell carcinoma cells. Molecular Cancer Therapeutics . 2010;9(7):1977–1984. doi: 10.1158/1535-7163.mct-10-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Whang Y. M., Choi E. J., Seo J. H., Kim J. S., Yoo Y. D., Kim Y. H. Hyperacetylation enhances the growth-inhibitory effect of all-trans retinoic acid by the restoration of retinoic acid receptor beta expression in head and neck squamous carcinoma (HNSCC) cells. Cancer Chemotherapy and Pharmacology . 2005;56(5):543–555. doi: 10.1007/s00280-004-0970-3. [DOI] [PubMed] [Google Scholar]
- 61.Jeon Y. J., Ko S. M., Cho J. H., Chae J. I., Shim J. H. The HDAC inhibitor, panobinostat, induces apoptosis by suppressing the expresssion of specificity protein 1 in oral squamous cell carcinoma. International Journal of Molecular Medicine . 2013;32(4):860–866. doi: 10.3892/ijmm.2013.1451. [DOI] [PubMed] [Google Scholar]
- 62.Prystowsky M. B., Adomako A., Smith R. V., et al. The histone deacetylase inhibitor LBH589 inhibits expression of mitotic genes causing G2/M arrest and cell death in head and neck squamous cell carcinoma cell lines. The Journal of Pathology . 2009;218(4):467–477. doi: 10.1002/path.2554. [DOI] [PubMed] [Google Scholar]
- 63.Han J. W., Ahn S. H., Park S. H., et al. Apicidin, a histone deacetylase inhibitor, inhibits proliferation of tumor cells via induction of p21WAF1/Cip1 and gelsolin. Cancer Research . 2000;60(21):6068–6074. [PubMed] [Google Scholar]
- 64.Ahn M. Y., Ahn S. G., Yoon J. H. Apicidin, a histone deaceylase inhibitor, induces both apoptosis and autophagy in human oral squamous carcinoma cells. Oral Oncology . 2011;47(11):1032–1038. doi: 10.1016/j.oraloncology.2011.07.027. [DOI] [PubMed] [Google Scholar]
- 65.Haigentz M., Jr, Kim M., Sarta C., et al. Phase II trial of the histone deacetylase inhibitor romidepsin in patients with recurrent/metastatic head and neck cancer. Oral Oncology . 2012;48(12):1281–1288. doi: 10.1016/j.oraloncology.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Murakami J., Asaumi J., Kawai N., et al. Effects of histone deacetylase inhibitor FR901228 on the expression level of telomerase reverse transcriptase in oral cancer. Cancer Chemotherapy and Pharmacology . 2005;56(1):22–28. doi: 10.1007/s00280-004-0976-x. [DOI] [PubMed] [Google Scholar]
- 67.Porter N. J., Christianson D. W. Binding of the microbial cyclic tetrapeptide trapoxin A to the class I histone deacetylase HDAC8. ACS Chemical Biology . 2017;12(9):2281–2286. doi: 10.1021/acschembio.7b00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brodie S. A., Li G., El-Kommos A., et al. Class I HDACs are mediators of smoke carcinogen-induced stabilization of DNMT1 and serve as promising targets for chemoprevention of lung cancer. Cancer Prevention Research . 2014;7(3):351–361. doi: 10.1158/1940-6207.capr-13-0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sang Z., Sun Y., Ruan H., Cheng Y., Ding X., Yu Y. Anticancer effects of valproic acid on oral squamous cell carcinoma via SUMOylation in vivo and in vitro. Experimental and Therapeutic Medicine . 2016;12(6):3979–3987. doi: 10.3892/etm.2016.3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Qian K., Sun L., Zhou G., et al. Sodium phenylbutyrate inhibits tumor growth and the epithelial-mesenchymal transition of oral squamous cell carcinoma in vitro and in vivo. Cancer Biotherapy and Radiopharmaceuticals . 2018;33(4):139–145. doi: 10.1089/cbr.2017.2418. [DOI] [PubMed] [Google Scholar]
- 71.Chung Y. L., Lee M. Y., Pui N. N. Epigenetic therapy using the histone deacetylase inhibitor for increasing therapeutic gain in oral cancer: prevention of radiation-induced oral mucositis and inhibition of chemical-induced oral carcinogenesis. Carcinogenesis . 2009;30(8):1387–1397. doi: 10.1093/carcin/bgp079. [DOI] [PubMed] [Google Scholar]
- 72.Gong L., Wang W. M., Ji Y., Wang Y., Li D. W. Zhonghua Kou Qiang Yi Xue Za Zhi . 2010;45(10):619–622. [PubMed] [Google Scholar]
- 73.Marques A. E. M., Nascimento Filho C. H. V., Marinho Bezerra T. M., Guerra E. N. S., Castilho R. M., Squarize C. H. Entinostat is a novel therapeutic agent to treat oral squamous cell carcinoma. Journal of Oral Pathology & Medicine . 2020;49(8):771–779. doi: 10.1111/jop.13039. [DOI] [PubMed] [Google Scholar]
- 74.Rikiishi H., Shinohara F., Sato T., Sato Y., Suzuki M., Echigo S. Chemosensitization of oral squamous cell carcinoma cells to cisplatin by histone deacetylase inhibitor suberoylanilide hydroxamic acid. International Journal of Oncology . 2007;30(5):1181–1188. doi: 10.3892/ijo.30.5.1181. [DOI] [PubMed] [Google Scholar]
- 75.Datta J., Islam M., Dutta S., Roy S., Pan Q., Teknos T. N. Suberoylanilide hydroxamic acid inhibits growth of head and neck cancer cell lines by reactivation of tumor suppressor microRNAs. Oral Oncology . 2016;56:32–39. doi: 10.1016/j.oraloncology.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.De Schutter H., Kimpe M., Isebaert S., Nuyts S. A systematic assessment of radiation dose enhancement by 5-Aza-2’-deoxycytidine and histone deacetylase inhibitors in head-and-neck squamous cell carcinoma. International Journal of Radiation Oncology, Biology, Physics . 2009;73(3):904–912. doi: 10.1016/j.ijrobp.2008.10.032. [DOI] [PubMed] [Google Scholar]
- 77.Rangwala S., Zhang C., Duvic M. HDAC inhibitors for the treatment of cutaneous T-cell lymphomas. Future Medicinal Chemistry . 2012;4(4):471–486. doi: 10.4155/fmc.12.6. [DOI] [PubMed] [Google Scholar]
- 78.Suzuki M., Endo M., Shinohara F., Echigo S., Rikiishi H. Enhancement of cisplatin cytotoxicity by SAHA involves endoplasmic reticulum stress-mediated apoptosis in oral squamous cell carcinoma cells. Cancer Chemotherapy and Pharmacology . 2009;64(6):1115–1122. doi: 10.1007/s00280-009-0969-x. [DOI] [PubMed] [Google Scholar]
- 79.Chikamatsu K., Ishii H., Murata T., et al. Alteration of cancer stem cell-like phenotype by histone deacetylase inhibitors in squamous cell carcinoma of the head and neck. Cancer Science . 2013;104(11):1468–1475. doi: 10.1111/cas.12271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang Y., Jung M., Dritschilo A., Jung M. Enhancement of radiation sensitivity of human squamous carcinoma cells by histone deacetylase inhibitors. Radiation Research . 2004;161(6):667–674. doi: 10.1667/rr3192. [DOI] [PubMed] [Google Scholar]
- 81.Ushio R., Hiroi M., Matsumoto A., Mori K., Yamamoto N., Ohmori Y. Enhanced cytotoxic effects in human oral squamous cell carcinoma cells treated with combined methyltransferase inhibitors and histone deacetylase inhibitors. Biomedicines . 2022;10(4):p. 763. doi: 10.3390/biomedicines10040763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xu M., Yin L., Cai Y., et al. Epigenetic regulation of integrin β6 transcription induced by TGF-β1 in human oral squamous cell carcinoma cells. Journal of Cellular Biochemistry . 2018;119(5):4193–4204. doi: 10.1002/jcb.26642. [DOI] [PubMed] [Google Scholar]
- 83.Yoshida H., Sugita K. A novel tetracyclic peptide, trapoxin, induces phenotypic change from transformed to normal in sis-oncogene-transformed NIH3T3 cells. Japanese Journal of Cancer Research . 1992;83(4):324–328. doi: 10.1111/j.1349-7006.1992.tb00109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kijima M., Yoshida M., Sugita K., Horinouchi S., Beppu T. Trapoxin an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase. Journal of Biological Chemistry . 1993;268(30):22429–22435. doi: 10.1016/s0021-9258(18)41547-5. [DOI] [PubMed] [Google Scholar]
- 85.Baek S. H. When signaling kinases meet histones and histone modifiers in the nucleus. Molecular Cell . 2011;42(3):274–284. doi: 10.1016/j.molcel.2011.03.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding authors upon request.