

Abstract
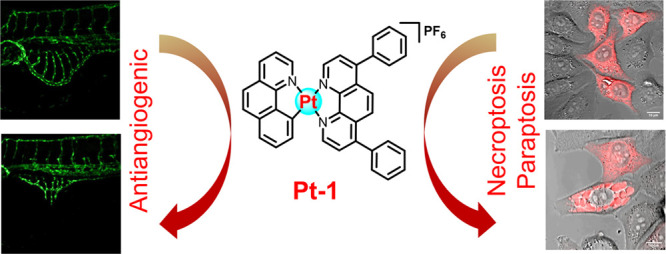
Cancer is a multifaceted disease involving various pathological processes, including uncontrolled proliferation, development of resistance, angiogenesis, metastasis, etc. Therefore, chemotherapeutic agents capable of simultaneously inhibiting proliferation, circumventing chemoresistance, and inhibiting angiogenesis can address multiple aspects of cancer progression. We recently identified a highly promising kinetically inert platinum antitumor agent, namely, Pt-1, that can circumvent cisplatin resistance and showed negligible nephrotoxicity. In this study, we explored the antiangiogenic potential and elucidated the detailed mechanism of cell death through which it exerts its antitumor activity. Pt-1 strongly inhibited angiogenesis in a zebrafish in vivo model at its therapeutically relevant nontoxic dose. Further, Pt-1 exerted antitumor activity through necroptosis- and paraptosis-mediated cell death. Taken together, the combination of antitumor activity with antiangiogenic property in Pt-1 makes it a highly promising antitumor candidate.
Keywords: Antiangiogenic, Necroptosis, Paraptosis, Chemotherapeutic, Platinum
Cancers continue to pose a substantial global health challenges with approximately 20 million new cases and 10 million deaths reported in 2020.1 Platinum (Pt) drugs, such as cisplatin, carboplatin, and oxaliplatin, have revolutionized cancer treatment, significantly improving treatment outcomes across various cancer types.2−4 Notably, following the incorporation of cisplatin into the treatment protocol, cure rates of testicular cancer have surpassed 95%, evidencing the remarkable success of Pt-complexes.2 The generalized mechanism of action of functional Pt drugs involves the formation of irreversible covalent adducts with DNA, RNA, or proteins, disrupting essential cellular processes and ultimately induction of apoptosis. Despite spectacular clinical success, Pt-drugs face several challenges, including innate or acquired resistance5 and the occurrence of systemic toxicities.2,3 Further, none of the clinically approved Pt drugs present antiangiogenic properties. Overcoming these obstacles is crucial for enhancing the effectiveness, widening the applicability, and improving the safety profile of Pt-based chemotherapy.
Considering the strong correlation between kinetic lability and deficiencies of Pt-drugs, we very recently reported a kinetically inert Pt-complex, Pt-1 (Figure 1), with potent antitumor activity.6Pt-1 showed nanomolar IC50 values (0.07–0.92 μM) against various cancer cells and overcame cisplatin resistance in lung, prostate, and ovarian cancer cells. The presence of the strong σ donating N^N and C^N chelating ligands impart kinetic inertness to Pt-1, rendering it unreactive toward forming covalent adducts with biomolecules. In sharp contrast to cisplatin, which is deactivated by biothiols such as glutathione (GSH), Pt-1 demonstrated resilience against GSH deactivation, thus overcoming resistance caused by GSH upregulation. In the cisplatin-sensitive A549 lung xenograft model, Pt-1 showed efficacy similar to that of cisplatin but without any nephrotoxicity. Intriguingly, in the cisplatin-resistant SKOV3 orthotopic xenograft model, Pt-1 exerted 2.5 times greater efficacy than did cisplatin. Preliminary mechanistic studies indicated that Pt-1 accumulates in the nucleus and mitochondria, intercalates with DNA, and depletes the mitochondrial membrane potential, leading to the elevation of oxidative stress. In a few recent reports, cytotoxicity and aggregation behaviors of structurally similar Pt complexes were investigated but without in-depth mechanistic study or evaluation of antiangiogenic potential.7−10 Although the antitumor activity of Pt-1 is highly encouraging,6 its antiangiogenic potential and the mechanism of cell death remained elusive.
Figure 1.

Structures of the kinetically labile Pt drug cisplatin and the kinetically inert candidate drug Pt-1.
Angiogenesis, the process of creating new blood vessels, plays a pivotal role in the progression of solid tumor growth and metastasis.11,12 As tumors grow, they require a steady blood supply to provide oxygen and nutrients and remove metabolic waste products. Angiogenesis enables tumors to establish this vascular network by stimulating the growth of new blood vessels from existing ones. Moreover, angiogenesis facilitates the spread of cancer cells to distant sites by providing routes for their dissemination through the bloodstream. Therefore, chemotherapeutic agents with antiangiogenic properties hold significant promise for cancer treatment. In addition to annihilating fast-growing cancer cells, these agents can directly inhibit the formation of new blood vessels within tumors, thereby disrupting the intricate network of vessels that sustain tumor growth and metastasis.13 This dual mechanism of antitumor action can lead to enhanced efficacy in suppressing tumor progression and improving patient outcomes. Of note, many FDA-approved antiangiogenic medications are often used in clinics to treat solid tumors alone or in conjunction with other chemotherapeutics.14
The majority of chemotherapeutics including clinical Pt drugs act by triggering apoptosis.2 However, cancer cells often develop mechanisms to evade apoptosis, thereby reducing the effectiveness of apoptotic drugs.5 As such, apoptosis resistance poses a significant challenge in cancer treatment. Thus, development of chemotherapeutics capable of inducing nonapoptotic cell death such as paraptosis and necroptosis holds significant potential for effective treatment of cancer patients.
Herein, we report the potent antiangiogenic property of Pt-1, in addition to characterizing its cell death mechanism. Utilizing a zebrafish model, we demonstrated that the complex exhibited potent in vivo antiangiogenic activity at doses relevant to its therapeutic application without inducing toxicity to the animals. Further, necroptosis and paraptosis were identified as the two major cell death mechanisms triggered by Pt-1. The ability to trigger nonapoptotic cell death empowers Pt-1 with the capability to overcome resistance to apoptosis.
Both in vitro and in vivo methods can be employed to assess the antiangiogenic potential of antitumor (candidate) drugs.15 Typically, in vitro cell-based assays are relatively easy and cost-effective but focus on particular stages of angiogenesis and serve for initial assessments, whereas in vivo models better replicate the dynamic microenvironment of living tissues, offering more relevant insights. We chose zebrafish to assess in vivo antiangiogenic properties of Pt-1. The zebrafish embryo is a preferred vertebrate model to investigate pharmacological efficacy and the toxicity of small molecules due to its high fecundity, in vitro fertilization, optical transparency, relative ease of maintenance, and notably high genetic and physiological similarity to humans.16,17 Before testing the antiangiogenic potential, we assessed the toxicity of Pt-1, if any, on zebrafish embryos (Wild Type AB). Viability and the change in morphology during the development of the zebrafish embryos between 26 to 72 hpf (hpf; hour post fertilization) incubated in different concentrations (1/3/5/8 μM) of Pt-1 were analyzed (Figure 2a). Survival analysis did not show any lethality of the embryo treated with ≤8 μM Pt-1. Animals treated with varied Pt-1 concentrations (≤8 μM) showed 100% viability in all the experiments (n = 4) (Figure 2b). Most importantly, morphologically, ≤8 μM Pt-1 treated embryos remain indistinguishable from their DMSO-treated vehicle control (Figures 2c and S1). Taken together, our analysis showed that Pt-1 is a nontoxic candidate drug at therapeutically relevant low μM concentration.
Figure 2.

Pt-1 is nontoxic to zebrafish embryos. (a) Schematic representation of experimental procedure. (b) Histogram showing animal viability at 72 hpf (n = 4, 20 embryos per test condition). (c) Bright-field images of the individual embryos at 72 hpf, treated with either vehicle control or Pt-1 (1/3/5/8 μM). Scale bar 200 μm. See Figure S1 for the extended image.
We next explored whether Pt-1 has antiangiogenic properties. The effect of Pt-1 on angiogenesis was examined by investigating the growth of subintestinal venous plexus (SIVP) formation in zebrafish embryos in the presence of different concentrations of Pt-1. The SIVP is a group of primarily venous angiogenic vessels that deliver yolk-derived nutrients to growing embryo. Later on, SIVP assists in supplying blood to the digestive system in both larval and adult animals.18 As a readily accessible vascular bed, the analysis of growing SIVP has been utilized to screen pro- or antiangiogenic small molecules.19 The development of SIVP starts from PCV (posterior cardinal vein) at around 28 hpf and continues growing ventrally.20 To evaluate the effect of Pt-1 on the SIVP formation, animals at 26 hpf, i.e., the period preceding the commencement of SIVP formation, were treated with vehicle or Pt-1 (1/3/5/8 μM, Figure 3a). Analysis of the total area covered by the SIVP and the number of compartments developed in the SIVP at 72 hpf showed that Pt-1 has a dose-dependent antiangiogenic effect on SIVP development (Figure 3b–d), indicating Pt-1 possesses strong antiangiogenic activity. Overall, our in vivo study showed that Pt-1 is nontoxic to zebrafish at its therapeutically relevant low micromolar concentration and can function as an antiangiogenic agent.
Figure 3.

Pt-1 showed a dose-dependent antiangiogenic effect on SIVP development in zebrafish embryos. (a) Schematic representation of the experimental procedure. (b) Maximum intensity projections of the confocal optical sections of SIVP of Tg(etv2:EGFP) embryos at 72 hpf that expressed EGFP in the endothelial cells. Dotted lines surround the area enclosed by the SIVP, while arrowheads point to specific SIVP compartments. Scale bar 50 μm. (c) A dot plot showing the area covered by the SIVP in each animal (n = 20). (d) Quantification of the number of compartments in each SIVP (n = 20). Data are represented in the mean ± s.e.m. format, and the corresponding P values denote *p ≤ 0.05, ***p ≤ 0.001, ****p ≤ 0.0001.
Our preliminary mechanistic studies revealed Pt-1 accumulates in the nucleus and mitochondria, intercalates with DNA, and elevates the level of ROS in HeLa cells.6 Apoptosis, necrosis (or necroptosis), autophagy, and paraptosis are the common forms of cell death triggered by antitumor metal complexes that target mitochondria and induce oxidative stress. To characterize the mechanism of cell death induced by Pt-1, HeLa cells were treated with increasing concentrations of Pt-1, doubly stained with annexin-V-APC (AV, an apoptosis marker) and propidium iodide (PI, a necrosis marker), and analyzed with quantitative flow cytometry.21 Worthy of note, as all cellular assays were performed using ≤24 h incubation time, concentrations of Pt-1 used were 0.5 μM (0.4 × IC50/24h), 1 μM (0.8 × IC50/24h), or 2 μM (1.6 × IC50/24h) based on the IC50/24h value (1.2 μM) of Pt-1 against HeLa cells in a 24 h incubation assay. As shown in Figure 4a (Figure S2 for dot plots), no significant increase in AV+ apoptotic population was observed in Pt-1 treated cells irrespective of its dose (0% in untreated vs 5–8% in treated), suggestive of nonapoptotic cell death. Importantly, Pt-1 caused an increase in the population of PI+/AV– necrotic cells (0% in untreated vs 16–18% in treated cells). Absence of apoptosis was further confirmed by Western blot analysis (Figure 4b). Cisplatin treatment resulted in clear upregulation of apoptotic signaling proteins such as cleaved PARP, p53, and cleaved caspase-3 in HeLa cells. In sharp contrast, no significant change in the expression level of any apoptotic proteins was observed in cells treated with Pt-1.
Figure 4.

(a) Flow cytometry analysis of HeLa cells left untreated or treated with increasing concentrations of Pt-1 (0.5–2 μM, (0.4–1.6) × IC50/24h) for 24 h followed by stained with annexin-V and propidium iodide (PI). (b) Western blot analysis of apoptosis marker proteins in HeLa cells treated with 1 μM (0.8 × IC50/24h) compound Pt-1 or 10 μM cisplatin (0.7 × IC50/24h).19 Uncropped blots are presented in Figure S5.
Then, to test whether Pt-1 elicits programmed necrosis or necroptosis, we determined the level of receptor interacting protein (RIP) kinases RIP1 and RIP3 in HeLa cells treated with Pt-1. RIP1 and/or RIP3 are the two key players involved in the necroptosis signaling pathway.22,23 Western blot analysis revealed, similar to the positive control shikonin,24Pt-1 also increased the level of both RIP1 and RIP3 in HeLa cells (Figure 5), confirming Pt-1 evoked necroptosis not necrosis. Along with mitochondrial damage, oxidative stress, and ER-stress, hyperactivation of poly(ADP–ribose) polymerase (PARP) was suggested to be an important upstream event in necroptosis. Severe damage to DNA activates PARP, an important nuclear enzyme that participates in DNA repair, which can trigger upregulation of RIP1.25,26 As shown in Figure 4b, Pt-1 treatment caused significant upregulation of PARP, indicating DNA damage caused by Pt-1 either directly or through ROS generation or by both. This data is in line with the fact that Pt-1 has the ability to bind to DNA as well as induce ROS generation in HeLa cells.6 Nevertheless, these data revealed upregulation PARP is one of the upstream events in the RIP1-mediated necroptosis induced by Pt-1.
Figure 5.

Western blot analysis of necroptosis marker proteins in HeLa cells. Shikonin was employed as a positive control. Uncropped blots are presented in Figure S6.
Careful analysis of the flow cytometry data (Figure 4a) indicated that PI+ necroptotic population did not increase with increasing dose of Pt-1 from 0.5 μM to 1 or 2 μM, suggestive of other modes of cell death perhaps operating in parallel. In fact, during imaging experiments, we noticed treatment with Pt-1 caused progressive cytoplasmic vacuolization in HeLa cells (Figure 6a). Vacuole formation is an important morphological feature associated with autophagic and/or paraptotic cell death.27,28 Autophagy is a programed cell death regulated via conversion of LC3-I to LC3-II protein.23 Our Western blot analysis confirmed that the ratio of LC3B–II to LC3BI remained unaltered when cells are treated with Pt-1, excluding the possibility of autophagic cell death (Figure 6b).
Figure 6.

(a) Fluorescence microscopy images of untreated or Pt-1 treated HeLa cells. Nucleus was stained with Hoechst 33258 (λex = 377 nm, λem = 447 nm). Cytosolic vacuoles is indicated by red arrows. (b) Western blot analysis of autophagy marker proteins in HeLa cells treated with Pt-1. Uncropped blots are presented in Figure S6.
Then, we investigated if the cytosolic vacuoles are related to paraptosis, a newly discovered cell death mechanism.29 Worthy of note, various metal-based chemotherapeutics which cause mitochondrial dysfunction and generate ROS were shown to induce ER stress-mediated paraptosis.21,30 Extensive cytosolic vacuolization, ER-stress, ER and/or mitochondria dilation, and Ca2+ overload mitochondria are important hallmarks of paraptosis.28
We have recently shown that Pt-1 causes mitochondrial damage and induces oxidative stress through production of superoxide radicals.6 Given the strong interconnection between intracellular redox-imbalance and endoplasmic reticulum (ER) where a variety of intercellular redox reactions are housed, we investigated the effect of Pt-1 on ER.31,32 Along with protein folding/trafficking, ER also plays key role in regulating Ca2+ homeostasis, lipid biosynthesis, and redox signaling in cells.30 Western blot analysis revealed, similar to known ER-stress inducer celastrol,28 treatment with Pt-1 led to accumulation of unfolded proteins (ubiquitinated for proteasomal degradation) in HeLa cells (Figure 7a). Accumulation of unfolded proteins is known to trigger ER-stress through unfolded protein response (UPR) primarily mediated by three ER transmembrane proteins, such as IRE1α, ATF-6, and PERK.30 As shown in Figure 7b, treatment with Pt-1 caused ER-stress induction through all three arms as evidenced from upregulation of IRE1α, ATF-6, and p-elf2α (a downstream protein in the PERK pathway).
Figure 7.

Western blot analysis of unfolded ubiquitinated proteins (a) and ER-stress marker proteins (b) in HeLa cells treated with Pt-1 (1 μM, 0.8 × IC50/24h) or celastrol (positive control). Uncropped blots are presented in Figures S7 and S8.
Next, we examined the effect of Pt-1 on the morphology of cells and organelles by using transmission electron microscopy (TEM) and confocal microscopy. As shown in Figure 8, treatment with Pt-1 resulted in extensive cytoplasmic vacuolization and severely dilated mitochondria and ER. However, the nucleus remained intact. This is in good agreement with paraptotic morphological features.28,33
Figure 8.

Representative TEM images of untreated and Pt-1 treated HeLa cells. The bottom panel shows a magnified view of selected square boxes.
Further, confocal imaging was performed to investigate whether vacuoles are of mitochondrial and/or ER origin. As cyclometalated Pt(II) complexes can be luminescent, we first recorded the absorption and emission spectra of Pt-1 (see Supporting Information for spectra). Our data suggested that the potential interference of Pt-1 with imaging in the green/red channel can be ruled out (details provided in the Supporting Information). HeLa cells with transiently GFP-labeled mitochondria or RFP-labeled ER were treated with Pt-1 following a recently reported protocol from our group.21 Morphology was analyzed using confocal microscopy, and results are presented in Figure 9a. In sharp contrast to mitochondria of untreated cells that appeared filamentous and elongated, treatment with 4 resulted in dilation of mitochondria as evidenced by the appearance of puncta like green fluorescence accumulated in the perinuclear region of cells (Figure 9a). However, no green fluorescence was observed inside the vacuoles, confirming that the vacuoles are not of mitochondrial origin. Similarly, while ER morphology in the untreated ER-RFP cells appeared reticular, the ER in Pt-1 treated cells was found to be severely dilated, and importantly the red fluorescence is precisely localized inside the cytoplasmic vacuoles (Figure 9a). This confirmed that the vacuoles are of ER origin and filled with dilated ER.
Figure 9.

(a) Comparison of morphology of untreated, Pt-1 (2 μM, 8 h) treated HeLa cells with GFP labeled mitochondria (Mito-GFP cells, λex = 488 nm, λem = 500–576 nm) and RFP labeled ER (ER-RFP cells, λex = 543 nm, λem = 584–645 nm). Cytoplasmic vacuoles are indicated by white arrows, Scale bar = 10 μm. (b) Confocal microscopy of HeLa cells left untreated or treated with Pt-1 (2 μM, 8 h) or positive control celastrol (2 μM, 8 h) and then stained with mitochondrial Ca2+ probe Rhod-2AM (λex = 543 nm, λem = 555–601 nm). (c) Quantification of mitochondrial Ca2+ in HeLa cells left untreated or treated with Pt-1 (2 μM, 8 h) or celastrol (2 μM, 8 h). Data are presented as average ± SD of CTCF (n ≥ 7). The asterisk denotes difference that is statistically significant (*** p < 0.001).
Further, since paraptosis requires protein synthesis, we investigated the effect of cycloheximide (a protein synthesis inhibitor) on vacuolization. HeLa cells were treated with Pt-1 either in the absence or presence of cycloheximide. Morphological analysis confirmed that cycloheximide effectively inhibited cytosolic vacuolization caused by compound Pt-1 (Figure S3).
During the course of paraptosis, dilated ER releases Ca2+, which is influxed in mitochondria, leading to mitochondrial Ca2+ overload.28 Therefore, we quantified the mitochondrial Ca2+ level of untreated and Pt-1 treated HeLa cells using a mitochondrial Ca2+ probe Rhod-2AM. As shown in Figure 9b,c, similar to the known paraptosis inducer celastrol, treatment with Pt-1 dramatically increased mitochondrial Ca2+ level. Taken together, along with necroptosis, the above evidence unambiguously confirmed paraptosis as another mode of cell death induced by Pt-1.
Further, to obtain preliminary insight into the mechanism of cell death in vivo, Western blot analysis was performed with the A549 tumors excised from the mice used recently in our in vivo efficacy study.6 Consistent with our results obtained from in vitro cellular assays, RIP1 and RIP3 as well as ER-stress marker proteins p-elf2α and IRE1α proteins were dramatically upregulated in tumors of mice that received Pt-1 treatment as compared to tumors of vehicle-treated mice, suggesting tumor growth inhibition in vivo by Pt-1 was related to necroptosis and/or paraptosis mediated cell death (Figure S4).
Intrigued by the nonapoptotic cell death induction by Pt-1, we investigated its suitability for treatment of apoptosis resistance tumors using an in vitro model. Apoptosis resistance arises from defective apoptotic signaling of tumor cells, which makes the tumor nonresponsive to commonly used apoptotic drugs including Pt drugs.34−37 It is important to mention here that Pt complexes with a nonapoptotic mechanism of action are relatively rare.21,38 Since the mechanism of cell death induced by Pt-1 is independent of apoptotic signaling mediated by pro-apoptotic proteins such as p53, Bax, Bak, caspase, etc., Pt-1 was expected to circumvent apoptosis resistance. To verify this, we used a well-established apoptosis resistant model comprising of wild type (Bax-Bak-WT) and Bax-Bak double knock out (Bax-Bak-DKO) murine embryonic fibroblasts (MEFs).19,21,37 As presented in Figure 10, Pt-1 showed comparable potency in both wild-type Bax-Bak-WT (IC50 = 0.42 ± 0.05 μM) and apoptosis-resistant Bax-Bak-DKO (0.36 ± 0.01 μM) cells. In sharp contrast, the potency of apoptotic drugs cisplatin and etoposide reduced (2–3)-fold in apoptosis-resistant Bax-Bak-DKO cells (IC50 for cisplatin and etoposide, 0.92 ± 0.02 μM and 2.15 ± 0.05 μM, respectively) as compared to the wild-type Bax-Bak-WT cells (IC50 for cisplatin and etoposide, 0.41 ± 0.03 μM and 0.76 ± 0.03 μM, respectively).21 This data highlight the potential of Pt-1 for treatment of apoptotic-resistant tumors.
Figure 10.
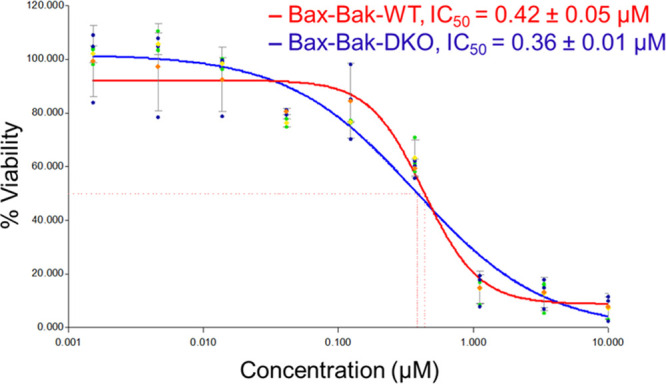
IC50 values and dose–response plots of Pt-1 in Bax-Bak-WT and Bax-Bak-DKO MEFs (48 h for MTT assay).
In summary, this study identified a potent in vivo antiangiogenic property of in vivo active antitumor agent Pt-1 at its therapeutically relevant nontoxic dose. The antiangiogenic property of a molecule can help to inhibit the growth of tumors and the spread of cancer. Further, Pt-1 exerts its antitumor activity through necroptosis and paraptosis mediated cell death in vitro as well as in vivo. These important findings lay a solid background for further development of kinetically inert Pt antitumor agents as dual chemotheraputic and antiangiogenic agents for highly effective anticancer therapy.
Acknowledgments
The authors gratefully acknowledge the financial support under the project no. RTI4003 from the Department of Atomic Energy (DAE) and Tata Institute of Fundamental Research (TIFR), India. Zebrafish work was supported by the DBT, New Delhi (BT/PR26241/GET/119/244/2017).
Glossary
Abbreviations
- IC50
half inhibitory concentration
- hpf
hours postfertilization
- SIVP
subintestinal venous plexus
- MEF
murine embryonic fibroblast
- MTT
methylthiazolyldiphenyl-tetrazolium bromide
- PI
Propidium iodide
- TEM
Transmission elctron microscopy.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.4c00207.
Detailed experimental procedures related to biological assays, additional data and figures related to flowcytometry, zebrafish toxicity, Absorption and emission spectra of Pt-1, uncropped blots (PDF)
Author Contributions
M.P. designed and supervised the project. M.M., S.C., and T.R.P. performed synthesis, characterization, and cellular experiments and interpreted the data. C.P. designed and supervised the zebrafish study. S.D. conducted experiments on zebrafish and analyzed the data. S.C. and P.R. performed TEM imaging and analyzed the data. All authors contributed to preparing and editing the manuscript and approved the submission.
The authors declare no competing financial interest.
Special Issue
Published as part of ACS Medicinal Chemistry Lettersspecial issue “Celebrating the 25th Anniversary of the Chemical Research Society of India”.
Supplementary Material
References
- Sung H.; Ferlay J.; Siegel R. L.; Laversanne M.; Soerjomataram I.; Jemal A.; Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- Johnstone T. C.; Suntharalingam K.; Lippard S. J. The next generation of platinum drugs: Targeted Pt(II) agents, nanoparticle delivery, and Pt(IV) prodrugs. Chem. Rev. 2016, 116, 3436–3486. 10.1021/acs.chemrev.5b00597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheff D. M.; Hall M. D. A Drug of Such Damned Nature.1 Challenges and Opportunities in Translational Platinum Drug Research. J. Med. Chem. 2017, 60, 4517–4532. 10.1021/acs.jmedchem.6b01351. [DOI] [PubMed] [Google Scholar]
- Rottenberg S.; Disler C.; Perego P. The rediscovery of platinum-based cancer therapy. Nat. Rev. Cancer 2021, 21, 37–50. 10.1038/s41568-020-00308-y. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Kang Y.; Chen L.; Wang H.; Liu J.; Zeng S.; Yu L. The drug-resistance mechanisms of five platinum-based antitumor agents. Front. Pharmacol. 2020, 11, 343. 10.3389/fphar.2020.00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda T. R.; M M.; Vaidya S. P.; Chhatar S.; Sinha S.; Mehrotra M.; Chakraborty S.; Gadre S.; Duari P.; Ray P.; Patra M. The Power of Kinetic Inertness in Improving Platinum Anticancer Therapy by Circumventing Resistance and Ameliorating Nephrotoxicity. Angew. Chem., Int. Ed. 2023, 62, e202303958 10.1002/anie.202303958. [DOI] [PubMed] [Google Scholar]
- Wang W.; Wang P.; Liao X.; Yang B.; Gao C.; Yang J. A Series of Planar Phosphorescent Cyclometalated Platinum(II) Complexes as New Anticancer Theranostic Agents That Induce Oncosis. J. Med. Chem. 2023, 66, 13103–13115. 10.1021/acs.jmedchem.3c01126. [DOI] [PubMed] [Google Scholar]
- Wang W.; Wang P.; Shen F.; Gao C.; Yang J. Turn-on Near-Infrared Phosphorescent Recognition of Anion Based on Self-Assembly of Cyclometalated Platinum Complexes That Induce Oncosis and Monitor Living Cells. ACS Nano 2024, 18, 5656–5671. 10.1021/acsnano.3c11366. [DOI] [PubMed] [Google Scholar]
- McGhie B. S.; Sakoff J.; Gilbert J.; Gordon C. P.; Aldrich-Wright J. R. Novel Planar Pt(II) Cyclometallated Cytotoxic Complexes with G-Quadruplex Stabilisation and Luminescent Properties. Int. J. Mol. Sci. 2022, 23, 10469. 10.3390/ijms231810469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGhie B. S.; Sakoff J.; Gilbert J.; Gordon C. P.; Aldrich-Wright J. R. Synthesis and Characterisation of Fluorescent Novel Pt(II) Cyclometallated Complexes with Anticancer Activity. Int. J. Mol. Sci. 2023, 24, 8049. 10.3390/ijms24098049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro M. M. Antiangiogenic Cancer Drug Using the Zebrafish Model. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1846–1853. 10.1161/ATVBAHA.114.303221. [DOI] [PubMed] [Google Scholar]
- Makrilia N.; Lappa T.; Xyla V.; Nikolaidis I.; Syrigos K. The role of angiogenesis in solid tumours: an overview. Eur. J. Int. Med. 2009, 20, 663–671. 10.1016/j.ejim.2009.07.009. [DOI] [PubMed] [Google Scholar]
- Bielenberg D. R.; Zetter B. R. The contribution of angiogenesis to the process of metastasis. Cancer J. 2015, 21, 267–273. 10.1097/PPO.0000000000000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Abd A. M.; Alamoudi A. J.; Abdel-Naim A. B.; Neamatallah T. A.; Ashour O. M. Anti-angiogenic agents for the treatment of solid tumors: Potential pathways, therapy and current strategies - A review. J. Adv. Res. 2017, 8, 591–605. 10.1016/j.jare.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stryker Z. I.; Rajabi M.; Davis P. J.; Mousa S. A. Evaluation of Angiogenesis Assays. Biomedicines 2019, 7, 37. 10.3390/biomedicines7020037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe K.; Clark M. D.; Torroja C. F.; Torrance J.; Berthelot C.; Muffato M.; Collins J. E.; Humphray S.; McLaren K.; Matthews L.; McLaren S.; Sealy I.; Caccamo M.; Churcher C.; Scott C.; Barrett J. C.; Koch R.; Rauch G.-J.; White S.; Chow W.; Kilian B.; Quintais L. T.; Guerra-Assunção J. A.; Zhou Y.; Gu Y.; Yen J.; Vogel J.-H.; Eyre T.; Redmond S.; Banerjee R.; Chi J.; Fu B.; Langley E.; Maguire S. F.; Laird G. K.; Lloyd D.; Kenyon E.; Donaldson S.; Sehra H.; Almeida-King J.; Loveland J.; Trevanion S.; Jones M.; Quail M.; Willey D.; Hunt A.; Burton J.; Sims S.; McLay K.; Plumb B.; Davis J.; Clee C.; Oliver K.; Clark R.; Riddle C.; Elliott D.; Threadgold G.; Harden G.; Ware D.; Begum S.; Mortimore B.; Kerry G.; Heath P.; Phillimore B.; Tracey A.; Corby N.; Dunn M.; Johnson C.; Wood J.; Clark S.; Pelan S.; Griffiths G.; Smith M.; Glithero R.; Howden P.; Barker N.; Lloyd C.; Stevens C.; Harley J.; Holt K.; Panagiotidis G.; Lovell J.; Beasley H.; Henderson C.; Gordon D.; Auger K.; Wright D.; Collins J.; Raisen C.; Dyer L.; Leung K.; Robertson L.; Ambridge K.; Leongamornlert D.; McGuire S.; Gilderthorp R.; Griffiths C.; Manthravadi D.; Nichol S.; Barker G.; Whitehead S.; Kay M.; Brown J.; Murnane C.; Gray E.; Humphries M.; Sycamore N.; Barker D.; Saunders D.; Wallis J.; Babbage A.; Hammond S.; Mashreghi-Mohammadi M.; Barr L.; Martin S.; Wray P.; Ellington A.; Matthews N.; Ellwood M.; Woodmansey R.; Clark G.; Cooper J. D.; Tromans A.; Grafham D.; Skuce C.; Pandian R.; Andrews R.; Harrison E.; Kimberley A.; Garnett J.; Fosker N.; Hall R.; Garner P.; Kelly D.; Bird C.; Palmer S.; Gehring I.; Berger A.; Dooley C. M.; Ersan-Ürün Z.; Eser C.; Geiger H.; Geisler M.; Karotki L.; Kirn A.; Konantz J.; Konantz M.; Oberländer M.; Rudolph-Geiger S.; Teucke M.; Lanz C.; Raddatz G.; Osoegawa K.; Zhu B.; Rapp A.; Widaa S.; Langford C.; Yang F.; Schuster S. C.; Carter N. P.; Harrow J.; Ning Z.; Herrero J.; Searle S. M. J.; Enright A.; Geisler R.; Plasterk R. H. A.; Lee C.; Westerfield M.; de Jong P. J.; Zon L. I.; Postlethwait J. H.; Nüsslein-Volhard C.; Hubbard T. J. P.; Crollius H. R.; Rogers J.; Stemple D. L. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. 10.1038/nature12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailone R. L.; Fukushima H. C. S.; Ventura Fernandes B. H.; De Aguiar L. K.; Corrêa T.; Janke H.; Grejo Setti P.; Roça R. D. O.; Borra R. C. Zebrafish as an alternative animal model in human and animal vaccination research. Lab. Anim. Res. 2020, 36, 13. 10.1186/s42826-020-00042-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isogai S.; Horiguchi M.; Weinstein B. M. The vascular anatomy of the developing zebrafish: an atlas of embryonic and early larval development. Dev. Biol. 2001, 230, 278–301. 10.1006/dbio.2000.9995. [DOI] [PubMed] [Google Scholar]
- M M.; Gadre S.; Chhatar S.; Chakraborty G.; Ahmed N.; Patra C.; Patra M. Potent Ruthenium–Ferrocene Bimetallic Antitumor Antiangiogenic Agent That Circumvents Platinum Resistance: From Synthesis and Mechanistic Studies to In Vivo Evaluation in Zebrafish. J. Med. Chem. 2022, 65, 16353–16371. 10.1021/acs.jmedchem.2c01174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goi M.; Childs S. J. Patterning mechanisms of the sub-intestinal venous plexus in zebrafish. Dev. Biol. 2016, 409, 114–128. 10.1016/j.ydbio.2015.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadre S.; Manikandan M.; Duari P.; Chhatar S.; Sharma A.; Khatri S.; Kode J.; Barkume M.; Kasinathan N. K.; Nagare M.; Patkar M.; Ingle A.; Kumar M.; Kolthur-Seetharam U.; Patra M. A rationally designed bimetallic platinum (II)-ferrocene antitumor agent induces non-apoptotic cell death and exerts in vivo efficacy. Chem.-Eur. J. 2022, 28, e202201259 10.1002/chem.202201259. [DOI] [PubMed] [Google Scholar]
- Conrad M.; Angeli J. P. F.; Vandenabeele P.; Stockwell B. R. Regulated necrosis: disease relevance and therapeutic opportunities. Nat. Rev. Drug Discovery 2016, 15, 348–366. 10.1038/nrd.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D.; Kang R.; Berghe T. V.; Vandenabeele P.; Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. 10.1038/s41422-019-0164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.; Luo Y.; Zhao J.; Yang F.; Zhao H.; Fan W.; Ge P. Shikonin kills glioma cells through necroptosis mediated by RIP-1. PLoS One 2013, 8, e66326 10.1371/journal.pone.0066326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosna J.; Voigt S.; Mathieu S.; Lange A.; Thon L.; Davarnia P.; Herdegen T.; Linkermann A.; Rittger A.; Chan F. K.; Kabelitz D.; Schütze S.; Adam D. TNF-induced necroptosis and PARP-1-mediated necrosis represent distinct routes to programmed necrotic cell death. Cell. Mol. Life Sci. 2014, 71, 331–348. 10.1007/s00018-013-1381-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aredia F.; Scovassi A. I. Poly(ADP-ribose): A signaling molecule in different paradigms of cell death. Biochem. Pharmacol. 2014, 92, 157–163. 10.1016/j.bcp.2014.06.021. [DOI] [PubMed] [Google Scholar]
- Tan C.-P.; Lu Y.-Y.; Ji L.-N.; Mao Z.-W. Metallomics insights into the programmed cell death induced by metal-based anticancer compounds. Metallomics 2014, 6, 978–995. 10.1039/c3mt00225j. [DOI] [PubMed] [Google Scholar]
- Yoon M. J.; Lee A R.; Jeong S. A.; Kim Y.-S.; Kim J. Y.; Kwon Y.-J.; Choi K. S. Release of Ca2+ from the endoplasmic reticulum and its subsequent influx into mitochondria trigger celastrol-induced paraptosis in cancer cells. Oncotarget 2014, 5, 6816. 10.18632/oncotarget.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperandio S.; de Belle I.; Bredesen D. E. An alternative, nonapoptotic form of programmed cell death. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 14376–14381. 10.1073/pnas.97.26.14376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King A. P.; Wilson J. J. Endoplasmic reticulum stress: an arising target for metal-based anticancer agents. Chem. Soc. Rev. 2020, 49, 8113–8136. 10.1039/D0CS00259C. [DOI] [PubMed] [Google Scholar]
- Yoboue E. D.; Sitia R.; Simmen T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 2018, 9, 331. 10.1038/s41419-017-0033-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanders R. J. A.; Waterham H. R.; Ferdinandusse S.. Metabolic Interplay between Peroxisomes and Other Subcellular Organelles Including Mitochondria and the Endoplasmic Reticulum. Front. Cell Dev. Biol. 2016, 3. 10.3389/fcell.2015.00083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng K.; Zheng Y.; Xia W.; Mao Z.-W. Organometallic anti-tumor agents: targeting from biomolecules to dynamic bioprocesses. Chem. Soc. Rev. 2023, 52, 2790–2832. 10.1039/D2CS00757F. [DOI] [PubMed] [Google Scholar]
- Brasacchio D.; Busuttil R. A.; Noori T.; Johnstone R. W.; Boussioutas A.; Trapani J. A. Down-regulation of a pro-apoptotic pathway regulated by PCAF/ADA3 in early stage gastric cancer. Cell Death & Disease 2018, 9, 442. 10.1038/s41419-018-0470-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erler J. T.; Cawthorne C. J.; Williams K. J.; Koritzinsky M.; Wouters B. G.; Wilson C.; Miller C.; Demonacos C.; Stratford I. J.; Dive C. Hypoxia-mediated down-regulation of Bid and Bax in tumors occurs via hypoxia-inducible factor 1-dependent and -independent mechanisms and contributes to drug resistance. Mol. cell. biol. 2004, 24, 2875–2889. 10.1128/MCB.24.7.2875-2889.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olejniczak S. H.; Clements J. L.; Bangia N.; Hernandez-Ilizaliturri F. J.; Czuczman M. S. Down-regulation of Bax and Bak in immuno-chemotherapy resistant non-hodgkin’s lymphoma cells. Blood 2006, 108, 4758–4758. 10.1182/blood.V108.11.4758.4758. [DOI] [Google Scholar]
- Law B. Y. K.; Mok S. W. F.; Chan W. K.; Xu S. W.; Wu A. G.; Yao X. J.; Wang J. R.; Liu L.; Wong V. K. W. Hernandezine, a novel AMPK activator induces autophagic cell death in drug-resistant cancers. Oncotarget 2016, 7, 8090–8104. 10.18632/oncotarget.6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong D. Y. Q.; Lim J. H.; Ang W. H. Induction of targeted necrosis with HER2-targeted platinum(iv) anticancer prodrugs. Chem. Sci. 2015, 6, 3051–3056. 10.1039/C5SC00015G. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.