

Abstract
The response of proteins to sequences of femtosecond infrared pulses provides a multidimensional view into their equilibrium distribution of structures and snapshot pictures of fast-triggered dynamical events. Analyzing these experiments requires advanced computational tools for assigning regions in the resulting multi-dimensional correlation plots to specific secondary-structure elements and their couplings. A differential sensitivity analysis technique based on a perturbation of the local (real space) Hamiltonian is developed to achieve that goal. Application to the amide I region of a small globular protein reveals regions associated with the α-helix, β-sheet, and their coupling. Comparison of signals generated in different directions shows that the double-quantum-coherence signal has a higher sensitivity to the couplings compared with the single-quantum-coherence (photon echo) technique.
Keywords: protein structure determination, two-dimensional infrared
Probing protein structure and folding pathways is one of the central problems of modern structural biology. Elaborate NMR pulse sequences are routinely used in the study of milli-second dynamical events in complex biomolecules with high spatial resolution (1). Optical response functions are particularly sensitive to structural features such as bond strengths, backbone angles, dipole moments, and polarizabilities, as well as the environmental factors that influence the structure such as hydrogen bond strengths and electric fields. Femtosecond optical techniques, thus, offer a good complimentary tool for monitoring fast protein motions and unraveling the underlying couplings between functional groups (2) in real time.
Two-dimensional techniques introduced into NMR in the 1970s revolutionized its resolution and utility (1). Similarly, significant progress has been made over the past decade in developing 2D infrared (2DIR) spectroscopy, based on the application of sequences of femtosecond pulses, into a powerful tool for probing protein structure and providing snapshots of fast-triggered protein folding events (3–6).
Spreading the information in two dimensions greatly enhances the resolution. Furthermore, 2DIR carries additional information regarding the fluctuations of two-exciton-state frequencies that is not available from linear (1D) spectra and provides a direct look at correlations among various segments of the protein. Cross peaks of 2DIR signals of equilibrated structures provide new structural constraints analogous to the nuclear Overhauser effect in 2D NMR. Structural information is extracted from multidimensional NMR data by using extensive simulations (7), through “direct inversion” based on constraint fits guided by simulations or structural optimizations. Their infrared analogues are less resolved, and anharmonic vibrational Hamiltonians are much more complex than spin Hamiltonians, making simulations an even more essential part of the analysis apparatus. Thanks to the ultrafast time scale, the simulation of 2DIR signals with atomic-level details only requires nanosecond trajectories, allowing the microscopic interpretation of spectra and providing a test for the simulated structural trajectories. Most 2DIR simulations carried out so far focused on small peptides with <10 residues (8–10), which do not have a unique stable structure. 2DIR signatures of ideal infinite size secondary structures were investigated recently (12, 13). Globular proteins are made of several secondary-structure segments, and relating their coherent vibrational response to the ideal motifs constitutes an open computational challenge.
In this article, we demonstrate how sensitivity analysis (14), in conjunction with a local basis Hamiltonian, may be used for assigning various regions in the spectra of a globular protein to specific secondary structures, and show how overlapping regions may be better separated in higher dimensions. This technique may further be applied for selecting the proper pulse sequence for specific structure application. We consider two three-pulse coherent four-wave mixing techniques, whereby three incoming pulses with propagation wave vectors k1, k2, and k3 interact with the protein to generate a coherent signal in the directions kI = –k1 + k2 + k3 and kIII = k1 + k2 – k3. The pulse sequence and time delays (t1, t2, and t3) are shown in Fig. 1 Top Right Upper.
Fig. 1.

The molecular structure of TB6 and pulse sequence configuration. (Top Left) The molecular structure of TB6. The Purple sections are the helices, the yellow sections are the β-strands, and the green and white sections are randomly coiling structures. (Top Right Upper) Pulse configuration for heterodyne four-wave mixing techniques. k1, k2, and k3 are the input pulses, and ks is the signal generated, which is in the same direction as the detection pulse k4.(Top Right Lower) The pulse sequence for coherent 2D experiments. ti are the time intervals between the pulses, and τi is the peak time of pulse i. (Middle) Red, linear absorption; black, AS(α); green, AS(β); blue, AS(γ); purple dash, the difference of the additive and the actual spectrum. (Bottom) Motif content of the vibrational eigenstates of TB6. Red, α; green, β; blue, random coil. The plot is vs. mode number; the (nonuniform) frequency scale is given at the top.
Both additive and a differential sensitivity analysis techniques were applied to reveal signatures of couplings among secondary motifs and assign different spectral regions to the structures. The former compares the actual spectra with additive spectra obtained by switching off the couplings among different secondary structure motifs. In the latter, we examine derivatives of the spectra with respect to various parameters of the local Hamiltonian. Our simulations show that 2DIR techniques are much more sensitive to the couplings and offer better resolution for the secondary structures and their interactions compared with the linear absorption. We further demonstrate that (t2, t3) correlation plots of kIII are more sensitive to the couplings compared with (t1, t3) correlations in kI.
In The Vibrational Exciton Hamiltonian and Simulation Protocol, we introduce the vibrational Hamiltonian and the simulation protocol. The additive sensitivity analysis is presented in Test of Secondary Structure Additivity of Coherent Spectra, and the differential analysis is developed in Differential Sensitivity Analysis. We conclude with a discussion of the results and future perspectives.
The Vibrational Exciton Hamiltonian and Simulation Protocol
Vibrational motions and spectra are commonly described by normal modes. These collective coordinates, which provide a convenient zero-order approximation for the vibrational eigenstates sand frequencies, are delocalized and do not directly connect with the structure; it is much easier to trace the structural origin of spectral features by using local vibrational coordinates (15).
Our simulations were carried out for the coherent third-order IR response of a 74-residue protein domain, TB6 (Fig. 1 Top Left) (16). TB6 is one of the 11 genetic mutations in TB domains from human fibrillin 1, which cause the Marfan syndrome. There are 10 residues in its α-helix section (α), 22 β-sheets (β), and 42 randomly coiled residues (γ). This protein was chosen because it is large enough to have well defined secondary structures, yet sufficiently small to make the simulations feasible (13).
We start with the vibrational exciton Hamiltonian (11, 12).
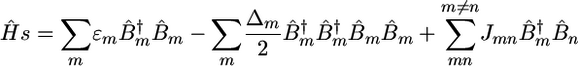 |
Here, 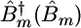 is the creation (annihilation) operator for the mth amide I mode, localized within the amide units (O
is the creation (annihilation) operator for the mth amide I mode, localized within the amide units (O C—N—H segments), with frequency εm and diagonal anharmonicity Δm. These operators satisfy the Bose commutation relations
C—N—H segments), with frequency εm and diagonal anharmonicity Δm. These operators satisfy the Bose commutation relations 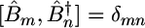 . Jmn is the harmonic intermode coupling.
. Jmn is the harmonic intermode coupling.
All parameters of the Hamiltonian fluctuate because of conformational changes of the backbone, and solvent and side-chain dynamics. The vibrational system is thus perturbed by a time-dependent electric field causing line broadening and frequency shifts. The present study focuses on the primary peak pattern. We thus calculated the averaged values of all frequency shifts and did not model the line broadening microscopically. A Lorenzian lineshape with a 20-cm–1 homogeneous linewidth was assumed for all transitions.
The fundamental frequency of each of the 74 local modes is given by εm(t) = ε – δεm(t), where ε = 1,707 cm–1 is the frequency of isolated NMA molecule in the gas phase (17, 18). The water molecules as well as all protein atoms other than the amide units are treated as a bath, which generates the fluctuating electrostatic field responsible for the frequency shift δεm(t). The instantaneous frequency shift was calculated for each mode by using the linear correlation formula with the instantaneous electric potential (19). The point charges of the peptide unit atoms (OC—N—H) were obtained from ref. 19, and those of the other protein atoms were taken from the CHARMM27 force field (20). The TIP3 model was used for the water charges and the simulation. An ensemble of δεm(t) was generated by using the simulated structural trajectory obtained from a molecular dynamics simulation starting with the NMR resolved structure from the Protein Data Bank (ID code 1APJ), which includes all of the atoms of one TB6 and 13,608 water molecules. The simulation was carried out by using the charmm package with the CHARMM27 force field (20). A 10-Å cutoff was used for nonbonded interactions. The long-range electrostatic interactions were calculated using the Ewald Sum method (21). The TB6 domain was imbedded in a 76-Å cube of TIP3 water. The cutoff distance for the Lennard–Jones forces was set to 12 Å. All water molecules overlapping with the protein were removed. An adopted basis Newton–Raphson energy minimization (5 × 104 steps) (20) was performed to release the internal tension. In this and all steps that follow, the protein backbone atom coordinates were held fixed at the NMR resolved structure. After energy minimization, the system was heated to 300 K in 20 ps, and a 5-ns equilibration in NPT (constant number, pressure, and temperature) ensemble (22) with 1-fs time steps was performed to get the correct density of the system. The extended system method was used to hold the temperature and pressure constant (22), and the final box length was 74.67 Å. This was followed by a 10-ns equilibration of the system in the NVE (constant number, volume, and energy) ensemble with a 2-fs time step [the SHAKE algorithm (23) was used in the NVE ensemble equilibration to constrain the bonds connected with hydrogen atoms]. A 1-ns NVE dynamics was run with 2-fs time steps. One hundred snapshots of the frequency shifts δεm(t) were recorded, and the average value of εm(t) for each amide I mode was then used as εm in the Hamiltonian Eq. 1.
The average values and variances of εm for the various modes of each secondary structure type are given in Table 1, and the entire distributions are presented in the supporting information, which is published on the PNAS web site. The α modes have a smaller red shift and a narrower distribution than β. The couplings of nearest covalently bonded modes were obtained from Tasumi's (17, 18) ab initio map, and all other couplings were calculated by using the electrostatic transition dipole coupling model. The distributions of the couplings Jmn between various motifs are presented in the supporting information. The average values and variances are given in Table 2. Note that there are many very weak couplings (<1 cm–1), which do not affect the spectra. Jαα is the strongest: Over 50% of the Jαα couplings are larger than 1 cm–1, as opposed to 33% for Jββ and 10% for Jγγ. The anharmonicity Δm = 16 cm–1 was assumed to be identical for all residues. The dipole coupling with the optical field E(t) is 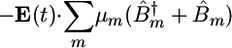 , where μm is the transition dipole moment of the m peptide unit (17, 18). All transition dipoles were taken to have the same magnitude.
, where μm is the transition dipole moment of the m peptide unit (17, 18). All transition dipoles were taken to have the same magnitude.
Table 1. The averages and variances of local mode frequencies.
| Frequency |
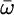 , cm-1 , cm-1
|
δ, cm-1 |
|---|---|---|
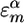 (t) (t) |
1,684.7 | 22.71 |
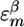 (t) (t) |
1,667.8 | 29.83 |
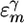 (t) (t) |
1,681.2 | 42.09 |
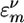 (t) is the frequency of the mth mode of ν type at time t.
(t) is the frequency of the mth mode of ν type at time t.
Table 2. The averages and variances of the absolute value of the couplings.
| Coupling | |J̄|, cm-1 | δ(|J|), cm-1 |
|---|---|---|
| Jαα | 3.13 | 3.88 |
| Jββ | 1.78 | 3.32 |
| Jγγ | 0.60 | 1.43 |
| Jαβ | 0.47 | 1.32 |
| Jαγ | 0.92 | 1.97 |
| Jβγ | 0.46 | 1.31 |
Following the procedure used in our previous study (11, 12), the Hamiltonian matrix was constructed by using a harmonic basis set that includes the ground state |0〉, the 74 one-exciton excited states 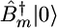 , and the 2,774 two-exciton excited states
, and the 2,774 two-exciton excited states 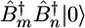 . Numerical diagonalization of the Hamiltonian yielded the one-exciton and two-exciton eigenstates. The motif content of each of the single-exciton vibrational states was calculated by summing the squares of its expansion coefficients for all of the modes for each structural type (α, β, and γ) in the local basis. These are displayed in Fig. 1 Bottom. The α region (red) is the most compact (1,653–1,689 cm–1). The β region (green) is broader (1,637–1,704 cm–1), and γ states are spread all over the band. Note that both α and β have some states outside the main regions, reflecting the nonideal, twisted structure of the various motifs.
. Numerical diagonalization of the Hamiltonian yielded the one-exciton and two-exciton eigenstates. The motif content of each of the single-exciton vibrational states was calculated by summing the squares of its expansion coefficients for all of the modes for each structural type (α, β, and γ) in the local basis. These are displayed in Fig. 1 Bottom. The α region (red) is the most compact (1,653–1,689 cm–1). The β region (green) is broader (1,637–1,704 cm–1), and γ states are spread all over the band. Note that both α and β have some states outside the main regions, reflecting the nonideal, twisted structure of the various motifs.
Test of Secondary Structure Additivity of Coherent Spectra
The linear and the third-order response function were calculated by multiple summations over the eigenstates by using equations 2–5 of ref. 15. The rotating wave approximation was used to select the dominant resonant terms for the response functions of the kI = –k1 + k2 + k3 and the kIII = k1 + k2 – k3 techniques (15). The response tensors were rotationally averaged with respect to the optical field by using equation 13 of ref. 24 (also see appendix A in ref. 12, and ref. 25). All calculations were performed for parallel (zzzz) pulse polarizations. The linear absorption was calculated as the imaginary part of the Fourier transform of the linear response function. The kI signal was calculated assuming that pulses k2 and k3 are time coincident setting t2 = 0. The S(kI) signal was calculated as the absolute value of the double Fourier transform of the response function with respect to t1 and t3 and is displayed as the corresponding conjugate frequency variables ω1 and ω3 (equation 9 of ref. 12). the S(kIII) signal was calculated in a similar way for time coincident pulses k1 and k2 setting t1 = 0 (equation 10 of ref. 12). It is displayed as ω2 and ω3, which are the Fourier conjugate to t2 and t3.
The calculated linear absorption is displayed in Fig. 1 Middle (red solid line). To investigate its sensitivity to the couplings between secondary structures, we have calculated the additive spectrum for each secondary structure separately [AS(α), AS(β), and AS(γ)] by turning off all inter-secondary-structure couplings. The total additive spectrum, AS(α) + AS(β) + AS(γ), and its deviation from the actual spectrum (purple dash line) depicted in the figure illustrate that the linear absorption is pretty much additive and is hardly affected by intermode couplings among different motifs.
S(kI) and S(kIII) are shown in Fig. 2. 2DIR clearly improves the resolution compared with the linear absorption: The kI spectrum shows some new features, but the resolution is greatly enhanced for kIII, which reveals several well separated peaks. The coherences obtained in the two techniques can be understood by using the double-sided Feynman diagrams (11, 12) shown in Fig. 3 that represent the evolution of the vibrational density matrix in the various time intervals. Three Feynman diagrams i, ii, and iii contribute to kI. In all of these diagrams, the density matrix is in a single quantum coherence |0〉〈e| between the ground state and the singly excited state during t1. During t3 it is in the conjugate coherence |e〉〈0| for i and ii, and in a coherence between the one and two exciton manifolds |f〉〈e| for iii. During both time intervals, the density matrix is in a single quantum coherence, which induces the correlations along the ω1 and ω3 axis. kIII is described by two Feynman diagrams, iv and v, and shows two-quantum coherences between ground state and the two-exciton band |f〉〈0| during the t2 interval. During t3, we have a single quantum coherence |e′〉〈0| for iv and |f〉〈e′| for v. The (ω2, ω3) frequency correlation plots will then show double quantum coherences along ω2. The improved resolution of kIII stems from the absence of the diagonal peaks that dominate the kI signal and obscure the off-diagonal (cross) peaks, and from doubling the frequency bandwidth of two-quantum coherences. We have anticipated these advantages of kIII based on the analogy with corresponding NMR pulse sequences (26–28). The additive third-order spectra are also displayed in Fig. 2. To gain a better insight into the additivity, we had further calculated the relative difference spectra (RDS), defined as the difference between the actual and the additive spectrum divided by the intensity of the strongest peak in the actual spectrum. The RDS for kI and kIII are shown in Fig. 4 Top. Several peak-shift features, indicated by adjacent positive (red or yellow) and negative (blue) peak pairs, can be observed for kIII but not for kI. The relative magnitudes of the highest RDS peak (0.039 for the linear absorption, 0.19 for kI, and 0.65 for kIII) indicate that the linear spectrum is pretty much additive, whereas the nonlinear techniques kI and kIII are increasingly nonadditive and consequently much more sensitive to the couplings among secondary structures.
Fig. 2.

Two-dimensional coherent vibrational spectra of TB6. (Left) Sensitivity analysis for kI. In the left column is shown the total simulated spectrum S(kI) and its derivatives with respect to the various secondary structures, DS(α), DS(β), and DS(γ). In the right column is shown the additive spectrum obtained by neglecting inter secondary structure couplings AS(kI), and the contributions of each motifs to the spectrum [AS(α), AS(β), and AS(γ)]. (Right) Sensitivity analysis for kIII.In the left column is shown the total simulated spectrum S(kIII) and its derivatives with respect to the various secondary structures, DS(α), DS(β), and DS(γ). In the right column is shown the additive spectrum obtained by neglecting inter secondary structure couplings, AS(kIII), and the contributions of each motifs to the spectrum [AS(α), AS(β), and AS(γ)].
Fig. 3.

Double-sided Feynman diagrams for the two-pulse sequences simulated in this work. By varying the time delays and signal direction, we can selectively probe desired vibrational coherences in the system. i–iii are the Feynman diagrams for the kI technique, where pulses k2 and k3 are coincident and we vary the intervals t1and t3. The 2D signals are given by the absolute value of the double Fourier transform with the conjugate frequencies ω1 and ω3, respectively. iv and v are the Feynman diagrams for kIII, where pulses k1 and k2 are time coincident, we vary the intervals t2 and t3, and ω2 and ω3 are the conjugate frequencies. The excitonic level scheme shown in vi has the ground state, |0〉, single-exciton manifold, |e〉, and two-exciton states, |f〉.
Fig. 4.

Characteristic regions in the 2DlR for various secondary structures in TB6. (Top) RDS of kI (Left) and kIII (Right). (Middle) The main regions of 2DIR spectra for α (black) and β (red) structures in kI (Left) and kIII (Right). (Bottom) Differential sensitivity spectra with respect to Jαβ for kI (Left) and kIII (Right).
Differential Sensitivity Analysis
Dissecting 2D spectra into the various motif regions is essential for their effective use for probing folding dynamics. Examination of the additive spectra AS(ν) (Fig. 2 Right) is the simplest way to accomplish that goal. However, this may be too crude when interstructure couplings are strong and the spectrum is highly nonadditive. In this case, the additive spectrum does not provide a good zero-order reference because the vibrational eigenstates are delocalized across the various motifs and are not well represented by the localized eigenstates used in the additive spectra. A more gentle way to characterize the regions in the spectra and assign them to different secondary structures is provided by the differential sensitivity analysis (DS): We make a small shift ην to the energies 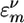 of all modes belonging to the νth motif (ην should be much smaller than all Jmn). The difference of the perturbed and the unperturbed spectrum reflects its sensitivity to this perturbation, and its spectral region can then be assigned to the structure of type ν. Obviously, DS(ν) of each motif depends on the couplings with other motifs. This is an inevitable consequence of the nonadditivity of the spectra when various segments interact strongly. DS(ν) may, thus, not be viewed as the “pure” spectrum of motif ν but rather as the spectral feature which is strongly affected by that motif.
of all modes belonging to the νth motif (ην should be much smaller than all Jmn). The difference of the perturbed and the unperturbed spectrum reflects its sensitivity to this perturbation, and its spectral region can then be assigned to the structure of type ν. Obviously, DS(ν) of each motif depends on the couplings with other motifs. This is an inevitable consequence of the nonadditivity of the spectra when various segments interact strongly. DS(ν) may, thus, not be viewed as the “pure” spectrum of motif ν but rather as the spectral feature which is strongly affected by that motif.
The lower six panels of kI in Fig. 2 show that for kI, the DS and AS peak positions are very close. This is because the couplings between different amide I modes are not very strong. However, the DS spectra have additional peaks. Also, because the AS represent the spectra of the isolated structures and the DS include their couplings with other parts of the protein, we observe peak shifts. Consequently, the two overlapping peaks in AS for α are well separated in DS. Similar trends are observed in the kIII spectra shown in Fig. 2. We have operationally defined the frequency regime bounded by the 35% contour of the highest peak of DS(α) in Fig. 2 as the α regime. This is shown by the black contour in the middle panels of Fig. 4. The β regime defined similarly is given by the red contour. These two regions are clearly better separated in kIII than in kI.
This differential sensitivity analysis can also be used to identify the regions corresponding to inter-secondary-structure couplings: By perturbing the off-diagonal elements in the local Hamiltonian, it should be possible to probe directly the effect of couplings on the spectra. This should identify the spectral domains that are sensitive to real-space interactions, and to molecular structure. Fig. 4 Bottom shows that the DS of the kIII technique with respect to Jαβ is clearly more localized than kI. Because of the large variance of the couplings, we applied a different perturbation protocol, whereby all Jαβ were multiplied by a factor 1.001. A comparison of Fig. 4 Middle with Fig. 4 Bottom shows that for both techniques, the strong peaks of DS with respect to Jαβ appear around the region where the α and β features overlap. The peak intensity is determined by the ratio of the coupling and the energy difference of the two local modes. When the coupling is much weaker than the energy difference (the weak coupling limit), the changes of the eigenstate energies are also small and the difference spectrum is weak. The difference spectrum signal will increase as this ratio increases. In the strong coupling limit, the eigenstates will have similar contributions from both local modes and should lie within the overlapping region of α and β.
Discussion
We have demonstrated how sensitivity analysis of simulations carried out by using a Hamiltonian expanded in local vibrational coordinates may be used to dissect 2DIR spectra of globular proteins and identify the regions that contain signatures of different secondary structures or specific couplings. This tool, which is widely used for inverting data to obtain optimal model parameters (14), could help in the design of new pulse sequences. In TB6, the α and β regions of kI strongly overlap, and the cross peaks are congested and buried under strong and broad diagonal peaks. The kIII technique shows a higher sensitivity to intersecondary-structure couplings and a better resolution. The effects of random coil (γ) are strong because it is the dominant structure content. However, the average Jαα and Jββ are much stronger than Jγγ, and we expect α and β features to dominate the 2DIR spectra for other proteins where they are more abundant. The γ feature may be filtered out, for example, by isotope labeling of all of the random coil units of a protein with known structure.
In our simulations, kIII shows better separation of peaks compared with kI. The advantages of kIII stem from the absence of coherences between ground and one exciton states along the ω2 axis and the expanded frequency range, which improves the frequency resolution (26–28). kIII has strong “diagonal” peaks created by four interactions with the same transition dipole. These peaks, which could overlap with the cross peaks, carry direct information about anharmonicity (peak shift), which is an additional ingredient in resolving the structure. This ingredient is hidden in the amplitude of the diagonal peak in kI. One advantage of kI (also known as the photon echo) is its capacity to eliminate certain types of inhomogeneous broadening (static frequency fluctuations), thus generating narrower lineshapes than kIII. This was recently demonstrated for ruthenium dicarbonyl complex (29).
In the present study, because of the high computational cost, we have only included homogeneous broadening by adding a 20-cm–1 linewidth to all transitions. The lineshapes of cross peaks resulting from overtones and combination bands are directly related to fluctuations of energy gaps between one- and two-exciton states, which may have arbitrary correlations with the fluctuations of fundamental frequencies. The cross peaks in kI and peaks in kIII can have contributions from a slow (inhomogeneous), fast (homogeneous), or intermediate type of fluctuations. Below, we briefly discuss the possible algorithms for incorporating line broadening microscopically. A high-level, second-order cumulant expansion of Gaussian fluctuations (CGF) approach based on the Brownian oscillator model (12) can calculate the line broadening functions directly from molecular dynamics trajectories. This approach does not require repeated diagonalization of the Hamiltonian because Gaussian fluctuations can be introduced analytically in the time domain. Because of the large number of required linebroadening functions, this approach has so far been applied only to short peptides and NMA (8–10). Slow fluctuations may be accounted for by performing an inhomogeneous averaging over many configurations. The bottleneck will be the repeated Hamiltonian diagonalizations and carrying out the multiple summations over eigenstates.
Full-scale inhomogeneous averaging over 100 configurations in TB6 will require 1,000 machine hours on an AMD Opteron 244. The present sensitivity analysis will require repeating this calculation 40 times. The nonlinear exciton equations (11, 12) provide the most promising method for large-scale inhomogeneous averaging. They use a single-exciton basis and view the nonlinear response in terms of scattering between excitons (quasi-particles) rather than transitions among eigenstates. The expensive computation of multiple exciton states is totally avoided. The bottleneck in this calculation is the calculation of a tetradic frequency-dependent exciton scattering matrix. Our recent calculations show that useful approximations that exploit the short-range nature of exciton couplings may reduce computational cost and will scale very favorably with protein size.
Sensitivity analysis can further be applied to connect chirally sensitive techniques such as vibrational IR circular dichroism and Raman optical activity (30) to structure, and to analyze hydrogen bonding networks. Once the various regions and cross peaks have been assigned, it should be possible to use pulse shaping learning algorithms (11, 31, 32) to selectively magnify or suppress the contributions of desired motifs, to study them separately. This could be particularly useful for probing protein folding pathways.
Supplementary Material
Acknowledgments
We thank Dr. Thomas la Cour Jansen for many helpful discussions. This work was supported by National Institutes of Health Grant 1R01 GM59230-03 and National Science Foundation Grant CHE-0132571.
Author contributions: W.Z., D.A., and S.M. designed research; W.Z. and D.A. performed research; W.Z. and D.A. analyzed data; and W.Z. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: 2DIR, 2D infrared.
References
- 1.Wuthrich, K. (1986) NMR of Proteins and Nucleic Acids (Wiley Interscience, New York).
- 2.Kulbelka, J., Hofrichter, J. & Eaton, W. A. (2004) Curr. Opin. Struct. Biol. 14, 76–88. [DOI] [PubMed] [Google Scholar]
- 3.Mukamel, S. (2000) Annu. Rev. Phys. Chem. 51, 691–729. [DOI] [PubMed] [Google Scholar]
- 4.Bredenbeck, J., Helbing, J., Behrendt, R., Renner, C., Moroder, L., Wachtveitl, J. & Hamm, P. (2003) J. Phys. Chem. B 107, 8654–8660. [Google Scholar]
- 5.Asplund, M. C., Zanni, M. T. & Hochstrasser, R. M. (2000) Proc. Natl. Acad. Sci. USA 97, 8219–8224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Golonzka, O., Khalil, M., Demirdoven, N. & Tokmakoff, A. (2001) Phys. Rev. Lett. 86, 2154–2157. [DOI] [PubMed] [Google Scholar]
- 7.Lindorff-Larsen, K., Best, R. B., DePristo, M. A., Dobson, C. M. & Vendruscolo, M. (2005) Nature 433, 128–132. [DOI] [PubMed] [Google Scholar]
- 8.Kwac, K. & Cho, M. H. (2003) J. Chem. Phys. 119, 2247–2255. [Google Scholar]
- 9.Woutersen, S., Pfister, R., Hamm, P., Mu, Y. G., Kosov, D. S. & Stock, G. (2002) J. Chem. Phys. 117, 6833–6840. [Google Scholar]
- 10.Jansen, T. l. C., Zhuang, W. & Mukamel, S. (2004) J. Chem. Phys. 121, 10577–10598. [DOI] [PubMed] [Google Scholar]
- 11.Mukamel, S. & Abramavicius, D. (2004) Chem. Rev. 104, 2073–2098. [DOI] [PubMed] [Google Scholar]
- 12.Abramavicius, D., Zhuang, W. & Mukamel, S. (2004) J. Phys. Chem. B 108, 18034–18045. [Google Scholar]
- 13.Demirdoven, N., Cheatum, C. M., Chung, H. S., Khalil, M., Knoester, J. & Tokmakoff, A. (2004) J. Am. Chem. Soc. 126, 7981–7990. [DOI] [PubMed] [Google Scholar]
- 14.Tomovic, R. & Vukobratovic, M. (1972) General Sensitivity Theory (Elsevier, New York).
- 15.Dreyer, J., Moran, A. M. & Mukamel, S. (2003) J. Phys. Chem. B 107, 5967–5985. [Google Scholar]
- 16.Yuan, X. M., Downing, A. K., Knott, V. & Handford, P. A. (1997) EMBO J. 16, 6659–6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Torii, H. & Tasumi, M. (1998) J. Raman Spectrosc. 29, 81–86. [Google Scholar]
- 18.Torii, H. & Tasumi, M. (1992) J. Chem. Phys. 96, 3379–3387. [Google Scholar]
- 19.Cha, S. Y., Ham, S. H. & Cho, M. H. (2002) J. Chem. Phys. 117, 740–750. [Google Scholar]
- 20.Brooks, B. R., Bruccoleri, R. E., Olafson, B. D., States, D. J., Swaminathan, S. & Karplus, M. (1983) J. Comput. Chem. 4, 187–217. [Google Scholar]
- 21.Essmann, U., Perera, L., Berkowitz, M. L., Darden, T., Lee, H. & Pedersen, L. G. (1995) J. Chem. Phys. 103, 8577–8593. [Google Scholar]
- 22.Hoover, W. G. (1985) Phys. Rev. A 31, 1695–1697. [DOI] [PubMed] [Google Scholar]
- 23.Vangunsteren, W. F. & Berendsen, H. J. C. (1977) Mol. Phys. 34, 1311–1327. [Google Scholar]
- 24.Hochstrasser, R. M. (2001) Chem. Phys. 266, 273–284. [Google Scholar]
- 25.Andrews, D. L. & Thirunamachandran, T. (1977) J. Chem. Phys. 67, 5026–5033. [Google Scholar]
- 26.Leegwater, J. A. & Mukamel, S. (1994) J. Chem. Phys. 101, 7388–7398. [Google Scholar]
- 27.Scheurer, C. & Mukamel, S. (2001) J. Chem. Phys. 115, 4989–5004. [Google Scholar]
- 28.Mukamel, S. & Tortschanoff, A. (2002) Chem. Phys. Lett. 357, 327–335. [Google Scholar]
- 29.Fulmer, E. C., Mukherjee, P., Krummel, A. T. & Zanni, M. T. (2004) J. Chem. Phys. 120, 8067–8078. [DOI] [PubMed] [Google Scholar]
- 30.Barron, L. D. (2004) Molecular Light Scattering and Optical Activity (Cambridge Univ. Press, Cambridge, U.K.), 2nd Ed.
- 31.Abramavicius, D. & Mukamel, S. (2004) J. Chem. Phys. 120, 8373–8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levis, R. J. & Rabitz, H. A. (2002) J. Phys. Chem. A 106, 6427–6444. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.