

Abstract
The 35-residue subdomain of the villin headpiece (HP35) is a small ultrafast folding protein that is being intensely studied by experiments, theory, and simulations. We have solved the x-ray structures of HP35 and its fastest folding mutant [K24 norleucine (nL)] to atomic resolution and compared their experimentally measured folding kinetics by using laser temperature jump. The structures, which are in different space groups, are almost identical to each other but differ significantly from previously solved NMR structures. Hence, the differences between the x-ray and NMR structures are probably not caused by lattice contacts or crystal/solution differences, but reflect the higher accuracy of the x-ray structures. The x-ray structures reveal important details of packing of the hydrophobic core and some additional features, such as cross-helical H bonds. Comparison of the x-ray structures indicates that the nL substitution produces only local perturbations. Consequently, the finding that the small stabilization by the mutation is completely reflected in an increased folding rate suggests that this region of the protein is as structured in the transition state as in the folded structure. It is therefore a target for engineering to increase the folding rate of the subdomain from ≈0.5 μs–1 for the nL mutant to the estimated theoretical speed limit of ≈3 μs–1.
Keywords: kinetics, temperature jump, NMR
Recent developments in both experimental and computational methods have opened exciting new possibilities for combining experiments with all-atom molecular dynamics (MD) simulations of protein folding. The dramatic improvement in time resolution in protein folding kinetic studies brought about by the introduction of nanosecond optical triggering methods (1–4), together with the use of distributed computing, now make it possible to directly compare MD simulations with experiments on ultrafast folding proteins (5). Such simulations are important because, if accurate, they contain a complete atomic description of the folding mechanism. The computational cost of folding simulations demands small size and ultrafast folding. HP35 is a subdomain of the headpiece of actin-binding protein villin and is the smallest naturally occurring polypeptide that folds autonomously without any cofactor or disulfide bond (6). Unlike other “miniproteins” (7), HP35 is highly thermostable (6) and, as revealed by the NMR structure, globular with a well packed hydrophobic core (8). These properties resemble much larger single-domain helical proteins and have made HP36 (HP35 plus an N-terminal Met) the focus of several all-atom folding simulations (9–17), which have thus far been based on the NMR structure.
Because of the delicate balance of stabilization forces involved in protein folding an accurate structure for the folded state is essential for MD simulations of folding and protein redesign calculations. We have previously shown that HP35 is one of the fastest folding proteins, with a folding rate (≈0.2 μs–1) approaching its theoretical limit (3 μs–1) (18). Mutations that further accelerate HP35 folding would represent a significant advantage for atomic-level MD simulations and might eliminate the free energy barrier separating the folded and unfolded states (19). For such “downhill” folders (20), all of the intermediate structures along the folding pathways would be populated and could, in principle, be studied by spectroscopic methods (21). So far, we have determined folding thermodynamics and kinetics for seven mutants (ref. 22 and J.K., W.A.E., and J.H., unpublished work), including mutants of the three core Phes. However, only the mutation K24 norleucine (nL) increased the folding rate. To facilitate MD simulations and gain insight into how to further increase the folding rate, we have used x-ray crystallography to obtain high-resolution structures of HP35 and its K24nL variant.
Materials and Methods
Peptide Preparation. HP35 (WT-LSDEDFKAVFGMTRSAFANLPLWKQQHLKKEKGLF) and its K24nL variant were synthesized by standard solid-phase chemistry on Applied Biosystems peptide synthesizer AB433A and purified by HPLC to >95% purity as judged by MS. This peptide is the reference molecule and differs from the true “wild type” by an N27H mutation. When protonated, H27 quenches the fluorescence of W23 as this part of the helix forms, thus providing a probe for folding (22).
Crystallization. Small aliquots of the dried powder for WT or K24nL mutants were weighed and resuspended at 100 mg/ml or 50 mg/ml in 10 mM Tris (pH 7.4). Crystals were obtained by using prefilled screens from Nextal Biotechnology (Montreal) and appeared from 2 μl of sitting drops (1 μl of protein plus 1 μl of precipitant) after 1–4 days at 20°C. WT crystals (space group P3121) grew from conditions containing sulfate or citrate. A derivative data set was collected from one crystal grown in condition Classics-87 (pH 6.4) and soaked for 1 min in the same condition plus 350 mM NaI and 20% glycerol. Native data sets were collected from a WT crystal grown in condition AMSO4-35 (pH 6.7) and from K24nL crystals (space group C2221) grown in conditions PEG-96 (pH 5.1) or PEG-70 (pH 7.0). Native WT or K24nL crystals were cryoprotected in their precipitants plus 20% sucrose or glycerol, respectively.
Structure Determination. Data were collected at 95° K on a Rigaku (Tokyo) Raxis IV++ detector with CuKα radiation or at the Advanced Photon Source (Argonne National Laboratory, Argonne, IL) beamline Southeast Regional Collaborative Access Team 22-ID. The initial structure was solved with solve (23) and resolve (24) by using the anomalous signal from the soaked iodide. It was used as the molecular replacement search model in epmr (25) for subsequent structures. All structures were refined with cns (26) and shelxl (27), and all ≈1-Å structures were refined with individual anisotropic displacement parameters for all atoms and riding hydrogens for protein atoms (Table 1). Some residues were modeled with alternate conformations based on examination of 2 Fo – Fc and Fo – Fc maps. Solvent-accessible surface area (SASA) and RMS deviation (RMSD) were calculated with cns (26). The percent accessibility is defined as the SASA (1.4-Å probe) of a residue in the peptide fragment divided by the SASA of the same residue in isolation without its neighbors.
Table 1. X-ray data and refinement statistics.
| WT (P3121)
|
nL (C2221)
|
||||
|---|---|---|---|---|---|
| pH 6.4 | pH 6.4 | pH 6.7 | pH 5.1 | pH 7.0 | |
| Unit cell, a b c, Å | 32.0 | 32.0 | 57.0 | 19.7 40.2 | 75.4 |
| Wavelength, Å | 1.542 | 0.900 | 0.900 | 1.542 | 0.900 |
| Resolution Å, 30 | 1.35 | 1.00 | 1.07 | 1.55 | 0.95 |
| Last shell | 1.40 | 1.04 | 1.11 | 1.61 | 0.98 |
| Rsym, % | 4.5 | 10.0 | 4.3 | 6.6 | 6.4 |
| Last shell | 12.8 | 48.1 | 42.3 | 15.3 | 21.3 |
| I/δ | 45.5 | 14.4 | 50.3 | 38.7 | 20.5 |
| Last shell | 10.4 | 4.0 | 4.1 | 14.2 | 5.3 |
| Completeness, % | 93.2 | 99.7 | 99.9 | 93.5 | 90.1 |
| Last shell | 49.6 | 99.8 | 99.9 | 58.5 | 57.6 |
| Redundancy | 43.0 | 25.8 | 19.8 | 26.4 | 16.3 |
| Rano, %/FOM | 4.8/0.69 | ||||
| No. reflections | 18,859 | 15,534 | 4,299 | 17,315 | |
| Rwork, % | 18.24 | 15.47 | 17.69 | 14.53 | |
| Rfree, % | 20.52 | 16.15 | 21.29 | 17.06 | |
| No. atoms, protein/solvent | 319/91 | 374/51 | 300/51 | 302/39 | |
| B factors, Å2, protein/all | 17/18 | 16/18 | 14/15 | 13/15 | |
| RMS bonds, Å | 0.014 | 0.015 | 0.009 | 0.016 | |
| RMS angles, Å | 0.039 | 0.043 | 0.026 | 0.030 | |
| Luzzati error, Å | 0.08 | 0.07 | 0.12 | 0.05 | |
Rsym, Σh|I—〈I〉|/ΣhI, where I and 〈I〉 are the intensity and mean intensity of each reflection (h = hk1), respectively. Rano, Σh, —h∥FPH—〈FPH〉∥/Σh,—h(|FPH|). FOM, 〈P(α)eiα/P(α)〉, where α is the phase and P(α) is the phase probability. Rwork, Rfree, ΣhFo—Fc|/ΣhFo. Atoms with alternate conformations are counted individually.
Folding Thermodynamics and Kinetics. Equilibrium fluorescence and CD measurements were carried out at pH 4.8 on peptides at concentrations of 20 and 200 μM, respectively. The exact sample concentrations were determined by Trp absorbance at 280 nm (ε = 5,500 M–1·cm–1). Kinetic measurements were done by using laser temperature jump (T jump) apparatus as described (28). In brief, the 10-ns, 7- to 10-K T jump is generated by focusing the 10-mJ, 10-ns pulse at 1,540 nm (obtained by Raman shifting the 1,064-nm fundamental of NdYAG laser with 500 psi of CH4) to a 250-μm spot on the sample cell. The fluorescence is excited with a frequency-doubled Ar+ laser, whose 264-nm output is focused to a 20-μm spot inside the heated volume. The Trp emission is passed through a band-pass filter and focused on a photomultiplier. The preamplified signal is read by a digitizing oscilloscope interfaced to a computer. Kinetic traces for the nL mutant were measured every 5°C from 45°C to 80°C. The T-jump sizes were calibrated by using the known temperature dependence of the quantum yield of N-acetyl tryptophanamide (ΦNATA = 0.14 at 293 K). Typically an average of 2,048 shots was collected for each temperature at a repetition rate of 10/3 Hz.
Results
Comparison of X-Ray and NMR Structures. The x-ray structure of HP35 was solved and refined to atomic resolution. The WT structure consists of a three-helix cluster surrounding a compact central core of three Phes (Fig. 1) and differs significantly from the NMR structures of HP36 solved in isolation (Protein Data Bank ID code 1VII) (8) or within the larger headpiece (Protein Data Bank ID code 1QQV) (29). These structures were RMS-fitted by using their main-chain atoms (the most accurate part of each residue), and coordinate differences (RMSD) were calculated. The main-chain and all-atom RMSDs between WT (pH 6.7) and 1VII are 1.7 and 2.0 Å, respectively (Fig. 2a and Table 2). Similar comparison of the subdomain fragment (residues 42–75) of 1QQV with WT (pH 6.7) reveals corresponding RMSDs of 1.3 and 2.3 Å (Fig. 2c and Table 2). The main-chain RMSD between NMR and x-ray structures are at least three times larger than that among x-ray structures (0.4–0.5 Å). These NMR–x-ray differences are particularly striking when only helix α3 is superimposed, showing that α1 and α2 adopt a different orientation relative to α3 in the x-ray and NMR structures (Fig. 2b). A similar result is obtained when either of the other two helices is fitted (fits not shown). As might be expected, the differences in helix position and orientation result in significant differences in the packing of the core and in helix–helix interactions.
Fig. 1.

Structure of WT (pH 6.7). (a and b) Front (a) and back (b) views. Uncharged, acidic, and basic residues are shown in black, red, and blue, respectively. Lighter shades of the same color show alternate conformations. Dashed lines depict H bonds <3.5 Å. (c) Detail of the hydrophobic core. Van der Waals contacts <4 Å are depicted as dashed lines, with each Phe and its neighbors in the same color. For clarity, only side-chain or main-chain atoms of some residues and hydrogens of the core Phes are shown. (d) A representative region of the final 2 Fo – Fc map, contoured at 1.6 σ, showing density of D5–F6.
Fig. 2.

Comparison of x-ray and NMR structures. (a) 1VII (red) fitted to WT (pH 6.7) (green) by residues 1–34 with only selective side chains is shown. Dashed lines are H bonds as in Fig. 1. (b) The same structures are fitted by residues 23–33 to highlight their dramatic differences. (c) RMS fit as with a, but between WT (pH 6.7) (green) and 1QQV (red).
Table 2. RMSD values of nonhydrogen atoms in residues 1–34.
| WT (pH 6.7) | nL (pH 7.0) | 1VII (pH 3.7) | |
|---|---|---|---|
| WT (pH 6.4) | 0.42 (0.71) | ||
| nL (pH 7.0) | 0.51 (1.26) | ||
| nL (pH 5.1) | 0.45 (1.34) | 0.23 (0.51) | 1.61 (2.04) |
| 1VII (pH 3.7) | 1.67 (1.97) | ||
| 1QQV (pH 7.0) | 1.26 (2.34) | 1.99 (2.79) |
RMSD of residues 1-34 for main-chain (or all) atoms. Only selective values are given, as most comparisons between NMR (1VII and 1QQV) and x-ray (WT and nL) structures are redundant and provide no additional information.
Structural Basis of HP35 Stability. At this resolution it is possible to describe in atomic detail a number of structural features that contribute to the stabilization of the folded state. Although helices α1 (residues 2–11) and α2 (residues 13–20) are much shorter than α3 (residues 21–33), peptide fragments from which α3 has been removed still have considerable structure as judged by NMR and CD (30), suggesting these two helices together play a more important role in establishing the proper fold.
Three highly conserved Phes, F6, F10, and F17 (31, 32), form the bulk of the hydrophobic core. All are located within α1 and α2 and are presumably primarily responsible for the structural stability of this fragment (30). They are almost completely occluded from solvent by residues V9, G11, M12, L20, K24, L28, and K32 on one side (Fig. 1a) and residues R14, Q25, K29, and L34 on the opposite side (Fig. 1b). In the WT (pH 6.7), residue F17 is the most buried (2.1% solvent accessibility, Table 3), followed by F6 (2.5%) and F10 (7.6%). These results are consistent with previously published mutagenesis data that showed that F17L, F6L, or F10L mutants are significantly less thermally stable than WT, with melting points that are lowered by 27°C, 17°C, or 12°C, respectively (31). These Phes are positioned such that their aromatic rings are either perpendicular (F6 with F17) or rotated ≈45° (F10 with F6 and F17) to each other (Fig. 1c). As observed in a survey of high-resolution x-ray crystal structures (33), these orientations are energetically optimal for interactions between the edge C—H hydrogens of one ring and the π electron cloud of another (34, 35). The Phe–Phe interactions between α1 and α2 are critical for establishing the villin fold, as double mutations that disrupt interactions within a single helix (F6L/F10L) lower the stability of the folded peptide, but do not alter its tertiary structure as determined by gel filtration (31). On the other hand, mutations that disrupt interactions between helices (F6L/F17L or F10L/F17L) result in a disordered structure (31).
Table 3. Residue-specific interactions.
| % access*
|
Neighboring residues in van der Waals contacts
|
% Identity
|
||
|---|---|---|---|---|
| Residue | WT | K24nL | ||
| F6 | 2.5 | 1.9 | F10, R14, F17 | 100 |
| F10 | 7.6 | 3.8 | F6, V9, F17, K29, K32, L34 | 91 |
| F17 | 2.1 | 4.9 | F6, F10, M12, L20, Q25, L28, K29 | 87 |
| L20 | 16.9 | 17.5 | M12, F17, P21, K24, L28 | 74 |
| L28 | 17.3 | 5.9 | F10, F17, L20, K24, H27 | 70 |
| L34 | 17.7 | 22.4 | F10 | 96 |
| F35 | 66.7 | 53.3 | L1, F6, F10, K29, L34 | 100 |
| D3 | 38.7 | 37.9 | R14 | 61(D/E) |
| R14 | 28.7 | 31.1 | F6, L1, D3 | 70(R/K) |
| Q25 | 12.0 | 10.7 | F17, L22, A18, L20 | 57(39 K/R) |
| K24 | 23.7 | 17.1 | L20, P21, W23, H27, L28 | 87(9 R) |
| K29 | 16.7 | 13.1 | F10, F17, Q25, Q26, F35 | 91(9 R) |
Van der Waals interactions <4 Å, with those between the core Phes and residues residing in a different helix are given in italics. Interactions that are observed in the x-ray structures but not in the NMR structure 1VII are shown in bold. The sequence identity is based on a comparison of villin from 23 species (8).
Values are given for WT (pH 6.7) and K24nL (pH 7.0); values for the other two structures are similar.
The core is further stabilized by van der Waals contacts between these Phes and the side chains of 10 conserved residues (Table 3) (32). Some of these contacts are also observed in the NMR structure 1VII, but a significant number (given in bold letters in Table 3) are not, again illustrating the differences between the x-ray and NMR structures. These contacts involve nine interhelix and one intrahelix residue pairs. Some of these residues are closely packed against neighboring side chains and significantly buried. In particular, the conserved L20 packs against M12, F17, P21, K24, and L28, and L28 packs against F10, F17, L20, K24, and H27 (Fig. 1a). The survey of the villin and advillin family (32) shows that, with the exception of R14 and Q25, these nine amino acids can be replaced with equivalent ones as long as the contacting carbons are preserved. For example, the Leus at positions 20, 28, and 34 can also be a Thr, Ile, Met, Qln, Glu, or Phe (32).
The x-ray structures also reveal interhelical H bonds that may be important for stabilizing the subdomain. α1 and α2 are connected by H bonds between R14 of α2 and L1 and D3 of α1 (Fig. 1b and Table 4). The guanidinium group of R14 is perfectly oriented to donate H bonds to the peptide oxygen of L1 and the carboxyl oxygen of D3. Among all members of the villin family, this residue is basic in nature (32), and α1 and α2 could still be stabilized by a H bond or salt bridge when the Arg is replaced by a Lys. Similarly, residue 3 is acidic in nature, but its identity is less critical, as a nonacidic group here would not affect the remaining H bond between R14 and L1. In ≈30% of the cases, van der Waals interactions presumably replace H bonds when hydrophobic residues reside at positions 3 and 14. Likewise, α2–α3 interhelical H bonds are found in the x-ray structures between A18 and L20 of α2 and Q25 of α3 (Fig. 1b and Table 4). As with R14, the amino nitrogen of Q25 is optimally oriented to donate a H bond to the peptide oxygens of A18 and L20. This residue is either a Gln, Arg, or Lys among the villin family, and the H-bond pattern is presumably preserved with either a Lys or Arg (32).
Table 4. Interhelix H bonds in x-ray structures.
| First atom | Second atom | WT | nL | Second atom | WT | nL |
|---|---|---|---|---|---|---|
| R14_NH2 | L1_O | 2.7 | 2.6 | |||
| R14_NH1 | D3_OD1 | — | 3.1 | L1_O | — | 2.8 |
| R14_Nε | D3_OD1 | 2.7 | — | |||
| Q25_NH2 | A18_O | 2.9 | 3.6 | L20_O | 3.3 | 2.8 |
Distances for WT (pH 6.7) and nL (pH 7.0).
These H bonds are consistently observed in structures that were crystallized in different space groups. However, they are only partially observed in the two NMR structures (Fig. 2). In 1VII, the terminal NH2 and carbonyl atoms need to be swapped before Q25 can donate H bonds to the peptide oxygens of A18 and L20. Also, the amino group of R14 donates a H bond to the peptide oxygen of L1 but its Nε group is both in the wrong orientation and too far away to donate a H bond to the carboxyl oxygen of D3. In 1QQV, the amino group of Q25 donates a H bond to the peptide oxygen of F17 instead. Its guanidinium group of R14 is too far away to donate a H bond to peptide oxygen of L1 and not properly oriented to donate any H bond to the carboxyl oxygens of D3.
Folding Kinetics and Structure of nL Mutant. As pointed out by McKnight et al. (8), K24 is one of the most buried side chains of HP35. In an attempt to stabilize, and possibly speed up folding, we replaced this Lys with nL. A two-state fit of the thermal unfolding curve (Fig. 3a), using an empirical baseline, shows that the removal of the charged amino group produces a slight stabilization of the structure by ≈0.5 kcal/mol. The T-jump kinetics are well described by a biexponential function, with a nearly temperature-independent 70-ns fast phase very similar to that observed for the WT (Fig. 3b) (22) and a slower phase that corresponds to the overall unfolding/refolding transition (22). No slower kinetic phases exist, as evidenced by the excellent agreement between the equilibrium Trp quantum yield and the quantum yield after the complete relaxation of the microsecond kinetic phase (Fig. 3 a Inset and b) (36). As found previously for WT (22), the two-state analysis for K24nL of the slower exponential kinetic phase yields Arrhenius temperature dependence for the folding and unfolding rates (Fig. 3c). The folding time for K24nL is 1.7 ± 0.3 μs at 300 K, a 2.5-fold acceleration compared with WT.
Fig. 3.

Thermodynamic and kinetic measurements. (a) Equilibrium thermal unfolding of WT (empty symbols) and nL (filled symbols) were monitored by the molar ellipticity at 222 nm and Trp fluorescence quantum yield (Φ, Inset). Black continuous lines are fits to the data using a two-state model {Keq = [U]/[F] = exp(–ΔH/R(1/T – 1/Tm)} (22). (Inset) Red squares show agreement of Φ values at equilibrium and after complete relaxation, indicating no slower kinetic phases. For nL, ΔH = –25 kcal/mol, Tm = 350 K, ΔS = 70 cal/(K mol), and ΔGfolding,300K = –3.6 kcal/mol (for WT, ΔGfolding,300K = –3.1 kcal/mol). (b) Φ for nL after a 10-ns T jump from 333 to 343 K (dashed blue line is equilibrium value at 343 K). The observed kinetics are biphasic, with the detail of the fast phase (Inset). (c) Two-state analysis of the slower relaxation phase (krel, black circles), which corresponds to overall unfolding/refolding kinetics (22). For nL, folding (blue circles): 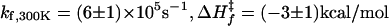 ; unfolding (red circles):
; unfolding (red circles): 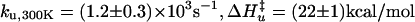 . (Inset) The same analysis for WT (22).
. (Inset) The same analysis for WT (22).
To gain structural insight into the origin of the increased folding rate, we also solved the x-ray structure of K24nL to 1-Å resolution. The mutant crystallizes in a different space group, presumably because the deletion of the amino group removes the salt bridge in the WT crystal between K24 and D5 of symmetry-related molecules. The aliphatic side chains of two symmetry-related K24 residues sandwich the aromatic ring of W23, which adopts two equally occupied conformations (Fig. 4a), one where its symmetry-related rings overlap each other on a 2-fold axis, and another where its ring interacts with the symmetry-related ring of H27. Nevertheless, the high resolution permits a detailed comparison between the WT and K24nL structures (Fig. 4 b–d and Table 2). The main-chain atoms differ by only 0.5 Å, and all atoms differ by 1.3 Å (excluding F35); the larger RMSD for all atoms is in large part caused by the side chains adopting different conformations because of crystal packing.
Fig. 4.

Packing of WT (pH 6.7) and structure of K24nL (pH 7.0). (a) A view down the crystallographic 2-fold axis in WT, with the Trp and Lys side chains in CPK. K24–D5 and W23–H27 interactions between symmetry-related molecules are depicted by dashed lines. (b) Front view of K24nL as in Fig. 1a. Note that the side chain of nL24 is compact here but extended in Fig. 1a. (c) Overlap of WT (green) and nL (red), but showing only the Phes, K29, and sulfate anions. (d) Same overlap as in c, but rendered as in Fig. 1b for all atoms.
Within this main-chain RMSD limit and with two exceptions, the WT and K24nL structures can be considered identical, considering that each pair of structures in the same space group (WT at pH 6.4 and 6.7 and K24nL at pH 5.1 and 7.0) differ from each other by similar RMSD values (Table 2). The first significant structural difference is the conformation of the aromatic ring of F35, which has previously been shown to be both flexible (8) and neutral in affecting stability or folding rate (22). When the electropositive pocket that is created by R14 and K29 is exposed (the ring of F35 is flipped out in WT), small negatively charged anions are observed (Fig. 1b). However, in K24nL, this ring is flipped inward, bumping K29 upward and relaxing the K29–F17 contact (Fig. 4c). Although F17 is perturbed ≈2.3 Å, this perturbation causes minimal changes in the overall structure, as the ring of F17 simply swivels around the Cα—Cβ bond (the main-chain RMSD for F17 is only 0.5 Å) and minor adjustments are made to the local packing.
The other significant structural change is the region of mutated residue 24. Removal of the charged amino group allows the aliphatic carbons to become more buried in the hydrophobic environment (Figs. 1a, 4b, and 5a). Burial alters its interaction with neighboring side chains, in particular with L28, which results in a decrease in their SASA (Table 3 and Fig. 5b). There are also changes of comparable magnitude in SASA of surface Lys and Arg residues, but these are probably caused by changes in conformation of solvent-exposed atoms that result from differences in lattice contacts in the two space groups (Fig. 5c).
Fig. 5.

Local perturbation by nL substitution. (a) A close-up view of residue 24 in WT (pH 6.7, blue) and K24nL (pH 7.0, yellow), showing the local perturbation of L28 by the K24nL mutation. (b) Plot of the difference in SASA for each residue between the two structures. Red and green bars correspond to residues that are involved or not involved in crystal contacts, respectively. (c) Differences in crystal contacts between the two structures are depicted in this molecular surface rendering, with residues involved in crystal contacts in red (observed in only one of two structures) or blue (observed in both structures) and residues not involved in any crystal contact in green.
Discussion
We have solved the x-ray structures of HP35 (WT and K24nL) to atomic resolution. Folding thermodynamics and kinetics for K24nL were experimentally studied and compared with our previously determined values for WT (22). The K24nL mutation was found to slightly stabilize the folded structure and accelerate the folding rate 2.5-fold.
These accurate atomic-resolution x-ray structures are important for the current effort to simulate folding by all-atom MD methods (9–17), which up to now have used the NMR structure 1VII (8). Comparison of the x-ray structures with 1VII show significant differences. 1VII differs from WT at position 27 (N27H) and contains an N-terminal Met. However, thermal unfolding studies show nearly identical between the thermodynamic parameters of WT and 1VII (22), suggesting that the differences between the x-ray and NMR structures do not arise from the two amino acid differences (Table 2 and Fig. 2). Furthermore, the x-ray structures of WT versus K24nL show only two local changes, one at the K24nL substitution and another at the C terminus that is caused by a change in anion binding and conformation of F35. The K24nL structure is almost identical to WT despite being in a different space group (Fig. 4 c and d). The main-chain RMSD between WT and K24nL is comparable to that between WT at different pHs. This finding suggests that the differences between x-ray and NMR structures do not represent intrinsic differences in structure between crystal and solution environments, but are most likely caused by the less-accurate NMR structure. For example, all residues in the x-ray structures adopt φ–ψ angles in the “most-favored” region of the Ramachandran plot, whereas only 67% and 87% adopt most-favored angles in the NMR structures 1VII and 1QQV, respectively. The K24nL structures determined at pH 7.0 and 5.1 (Table 2) are virtually identical so the pH at which the structures were determined probably does not account for the structural differences between WT (pH 6.7) and 1VII (pH 3.7). Furthermore, comparable differences are observed between x-ray and NMR structures determined at pH values that are close to each other (1VII with K24nL (pH 5.1) and 1QQV (pH 7.0) with WT (pH 6.7) (Table 2).
Interhelical H bonds may play an important role in defining the HP35 fold. In particular, the helix α3 seems to be interacting only weakly with the hydrophobic core (none of the three core Phes is in α3). As mentioned above, experimental evidence suggests that the isolated α3 is completely disordered, whereas a subdomain lacking α3 still retains residual structure. In modeling of folding of small helical proteins using a diffusion-collision model, Islam et al. (37) found that α3 of HP35 does not fold until intrinsic helical propensities for its residues are increased to unrealistic values. The nature of stabilization of α3 is therefore not obvious, and the observed interhelical H bonds may be crucial, especially because the folding free energy of HP35 at 300 K is only 3.1 kcal/mol.
Determination of the structural differences between WT and K24nL are important for engineering HP35 to fold faster. All of the increase in the folding equilibrium constant is reflected in the increased folding rate (φ ≡ Δlnkf/ΔlnKf ≈ 1.3 ± 0.3). The simplest structural interpretation, introduced by Fersht and coworkers (38, 39), is that the residue at this position is making the same interactions in the transition-state structures as in the native state (assuming the mutation does not affect the denatured state). This interpretation requires that the mutation only perturbs the structure locally (40). The x-ray structures of WT and K24nL support the assumption of a local perturbation by the mutation, which allows an increase in burial of the aliphatic side chain of nL compared to Lys. This decrease in SASA would be expected to be a stabilizing factor, consistent with our experimental finding. Loss of the charge is not expected to have any global effect on the structure, because previous thermodynamic studies showed minimal change in stability upon changing the ionization state of H27 (22). Stabilizing effects, attributed to the increased hydrophobicity and increased folding rate upon removal of partially buried changed groups, have also been observed for another ultrafast folding helical protein, the albumin binding domain 1prb7–53 (41, 42).
Kubelka et al. (18) have used both empirical and theoretical arguments to suggest that a protein's folding “speed limit” (43) is approximately (N/100 μs)–1, where N is the number of residues, (0.4 μs)–1 for HP35. This estimate suggests that it is possible to accelerate the folding of HP35 by almost an order of magnitude compared with the K24nL mutant. The finding of a high φ value for this residue, with support for the interpretation from the x-ray structures, is a valuable clue that suggests this region of the protein should be a target for future engineering aimed at further accelerating its folding.
The neighboring residue, K29, is a potential target that may further increase the folding rate. Like K24, this residue is completely surrounded by hydrophobic residues (Table 4 and Figs. 1b and 4c) and removal of its charged amino group, as in a K29nL substitution, is expected to have similar structural and thermodynamic consequences. That is, the aliphatic side chain of nL is expected to interact more closely with its neighbors, leading to a decrease in SASA.
Acknowledgments
We thank Ming Yau for synthesis and purification of the villin peptides and Jessica Bell for help with synchrotron data collection. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract W-31-109-Eng-38.
Abbreviations: nL, norleucine; SASA, solvent-accessible surface area; MD, molecular dynamics; T jump, temperature jump; RMSD, RMS deviation.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 1WY3, 1WY4, 1YRF, and 1YRI).
References
- 1.Jones, C. M., Henry, E. R., Hu, Y., Chan, C. K., Luck, S. D., Bhuyan, A., Roder, H., Hofrichter, J. & Eaton, W. A. (1993) Proc. Natl. Acad. Sci. USA 90, 11860–11864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Callender, R. H., Dyer, R. B., Gilmanshin, R. & Woodruff, W. H. (1998) Annu. Rev. Phys. Chem. 49, 173–202. [DOI] [PubMed] [Google Scholar]
- 3.Gruebele, M. (1999) Annu. Rev. Phys. Chem. 50, 485–516. [DOI] [PubMed] [Google Scholar]
- 4.Eaton, W. A., Munoz, V., Hagen, S. J., Jas, G. S., Lapidus, L. J., Henry, E. R. & Hofrichter, J. (2000) Annu. Rev. Biophys. Biomol. Struct. 29, 327–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Snow, C. D., Nguyen, H., Pande, V. S. & Gruebele, M. (2002) Nature 420, 102–106. [DOI] [PubMed] [Google Scholar]
- 6.McKnight, C. J., Doering, D. S., Matsudaira, P. T. & Kim, P. S. (1996) J. Mol. Biol. 260, 126–134. [DOI] [PubMed] [Google Scholar]
- 7.Imperiali, B. & Ottesen, J. J. (1999) J. Pept. Res. 54, 177–184. [DOI] [PubMed] [Google Scholar]
- 8.McKnight, C. J., Matsudaira, P. T. & Kim, P. S. (1997) Nat. Struct. Biol. 4, 180–184. [DOI] [PubMed] [Google Scholar]
- 9.Duan, Y., Wang, L. & Kollman, P. A. (1998) Proc. Natl. Acad. Sci. USA 95, 9897–9902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duan, Y. & Kollman, P. A. (1998) Science 282, 740–744. [DOI] [PubMed] [Google Scholar]
- 11.Shen, M. Y. & Freed, K. F. (2002) Proteins 49, 439–445. [DOI] [PubMed] [Google Scholar]
- 12.Fernandez, A., Shen, M. Y., Colubri, A., Sosnick, T. R., Berry, R. S. & Freed, K. F. (2003) Biochemistry 42, 664–671. [DOI] [PubMed] [Google Scholar]
- 13.Sullivan, D. C. & Kuntz, I. D. (2001) Proteins 42, 495–511.11170204 [Google Scholar]
- 14.Zagrovic, B., Snow, C. D., Shirts, M. R. & Pande, V. S. (2002) J. Mol. Biol. 323, 927–937. [DOI] [PubMed] [Google Scholar]
- 15.Kinnear, B. S., Jarrold, M. F. & Hansmann, U. H. (2004) J. Mol. Graphics Model 22, 397–403. [DOI] [PubMed] [Google Scholar]
- 16.Mukherjee, A. & Bagchi, B. (2004) J. Chem. Phys. 120, 1602–1612. [DOI] [PubMed] [Google Scholar]
- 17.Ripoll, D. R., Vila, J. A. & Scheraga, H. A. (2004) J. Mol. Biol. 339, 915–925. [DOI] [PubMed] [Google Scholar]
- 18.Kubelka, J., Hofrichter, J. & Eaton, W. A. (2004) Curr. Opin. Struct. Biol. 14, 76–88. [DOI] [PubMed] [Google Scholar]
- 19.Bryngelson, J. D., Onuchic, J. N., Socci, N. D. & Wolynes, P. G. (1995) Proteins 21, 167–195. [DOI] [PubMed] [Google Scholar]
- 20.Garcia-Mira, M. M., Sadqi, M., Fischer, N., Sanchez-Ruiz, J. M. & Munoz, V. (2002) Science 298, 2191–2195. [DOI] [PubMed] [Google Scholar]
- 21.Eaton, W. A. (1999) Proc. Natl. Acad. Sci. USA 96, 5897–5899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kubelka, J., Eaton, W. A. & Hofrichter, J. (2003) J. Mol. Biol. 329, 625–630. [DOI] [PubMed] [Google Scholar]
- 23.Terwilliger, T. C. & Berendzen, J. (1999) Acta Crystallogr. D 55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Terwilliger, T. C. (2000) Acta Crystallogr. D 56, 965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kissinger, C. R., Gehlhaar, D. K. & Fogel, D. B. (1999) Acta Crystallogr. D 55, 484–491. [DOI] [PubMed] [Google Scholar]
- 26.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905–921. [DOI] [PubMed] [Google Scholar]
- 27.Sheldrick, G. M. & Schneider, T. R. (1997) Methods Enzymol. 277, 319–343. [PubMed] [Google Scholar]
- 28.Thompson, P. A., Eaton, W. A. & Hofrichter, J. (1997) Biochemistry 36, 9200–9210. [DOI] [PubMed] [Google Scholar]
- 29.Vardar, D., Buckley, D. A., Frank, B. S. & McKnight, C. J. (1999) J. Mol. Biol. 294, 1299–1310. [DOI] [PubMed] [Google Scholar]
- 30.Tang, Y., Rigotti, D. J., Fairman, R. & Raleigh, D. P. (2004) Biochemistry 43, 3264–3272. [DOI] [PubMed] [Google Scholar]
- 31.Frank, B. S., Vardar, D., Buckley, D. A. & McKnight, C. J. (2002) Protein Sci. 11, 680–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vermeulen, W., Vanhaesebrouck, P., Van Troys, M., Verschueren, M., Fant, F., Goethals, M., Ampe, C., Martins, J. C. & Borremans, F. A. (2004) Protein Sci. 13, 1276–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burley, S. K. & Petsko, G. A. (1985) Science 229, 23–28. [DOI] [PubMed] [Google Scholar]
- 34.Burley, S. K. & Petsko, G. A. (1986) FEBS Lett. 203, 139–143. [DOI] [PubMed] [Google Scholar]
- 35.Hunter, C. A., Singh, J. & Thornton, J. M. (1991) J. Mol. Biol. 218, 837–846. [DOI] [PubMed] [Google Scholar]
- 36.Buscaglia, M., Lapidus, L. J., Eaton, W. A. & Hofrichter, J. (2005) J. Mol. Biol. 347, 657–664. [DOI] [PubMed] [Google Scholar]
- 37.Islam, S. A., Karplus, M. & Weaver, D. L. (2002) J. Mol. Biol. 318, 199–215. [DOI] [PubMed] [Google Scholar]
- 38.Fersht, A. R., Matouschek, A. & Serrano, L. (1992) J. Mol. Biol. 224, 771–782. [DOI] [PubMed] [Google Scholar]
- 39.Fersht, A. (1999) Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding (Freeman, New York).
- 40.Munoz, V. & Eaton, W. A. (1999) Proc. Natl. Acad. Sci. USA 96, 11311–11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu, Y. J., Fu, X. R., Wang, T., Tamura, A., Takada, S., Savan, J. G. & Gai, F. (2004) Chem. Phys. 307, 99–109. [Google Scholar]
- 42.Wang, T., Zhu, Y. J. & Gai, F. (2004) J. Phys. Chem. B 108, 3694–3697. [Google Scholar]
- 43.Hagen, S. J., Hofrichter, J., Szabo, A. & Eaton, W. A. (1996) Proc. Natl. Acad. Sci. USA 93, 11615–11617. [DOI] [PMC free article] [PubMed] [Google Scholar]