

Abstract
Human coronavirus (HCoV) 229E is a group 1 coronavirus and is specific to humans. So far, no animal model is available to study the pathogenesis of infection by HCoV-229E. We show here that the expression of aminopeptidase N (APN, also termed CD13), the receptor for HCoV-229E, is required but not sufficient to confer susceptibility in vivo. HCoV-229E infection was facilitated by crossing APN transgenic mice into signal transducers and activators of transcription (Stat) 1 null mice and by adaptation of HCoV-229E to grow in primary APN transgenic, Stat1 null fibroblasts. Double transgenic mice allow the study of human coronavirus group 1 infections in an animal model, in particular, viral tropism, replication, recombination, and spread in an immunocompromised situation. Furthermore, these mice provide an important tool for the evaluation of biosafety and efficacy of coronavirus-based vectors.
Keywords: human aminopeptidase N, CD13, genetically engineered humanized mouse
Coronaviruses are enveloped, positive-sense RNA viruses that infect mammalian and avian species. The Coronaviridae, together with the Arteriviridae and Ronaviridae families, compose the Nidovirales order (1). Human coronaviruses (HCoVs) are classified into groups 1 and 2, differentiated in sequence and antigenicity (2). Two HCoV species (HCoV-229E and -OC43) that have been known about for a long time cause mild respiratory infections (3, 4), whereas the recently discovered HCoV-NL63 strain (5–7) causes more severe respiratory infections, and the severe and acute respiratory syndrome (SARS) coronavirus (8, 9) causes severe respiratory and enteric infections with high mortality.
Coronavirus spike (S) glycoprotein is recognized by the cell surface receptor and is a dominant factor for tissue and species specificity (10, 11). Group 1 coronavirus uses aminopeptidase N (APN) as the receptor (12, 13). HCoV-229E, a member of this group, binds to human APN (hAPN) in rafts and enters the cell through caveolae (14). APN, also identified as the leukocyte surface differentiation antigen CD13 (15), is a membrane-bound exopeptidase of 150 kDa that is constitutively expressed as dimers on the surface of a wide variety of cells (16, 17).
Recently, several infectious cDNA clones of porcine, murine, and human coronaviruses have been isolated (18–21), facilitating reverse genetics of coronavirus to study the mechanism of replication and the development of vectors for vaccine development and gene therapy (22). Although testing of coronavirus vector efficacy in domestic and laboratory animal models is amenable, animal models to test coronavirus-derived human vectors have only been described for SARS coronavirus (23, 24) and HCoV-OC43 (25, 26).
This paper reports the generation of transgenic mice susceptible to HCoV-229E. Primary embryonic fibroblasts (PEFs) expressing transgenic hAPN were susceptible to HCoV-229E in vitro; in contrast, the transgenic mice were not susceptible to in vivo infection by the same virus. Two families of IFNs (IFN type I or αβ and type II or γ) are involved in antiviral host responses (27). A common property of both IFN types is the phosphorylation and activation of signal transducers and activators of transcription (Stat) 1 (28). Stat1-deficient mice are highly susceptible to microbial and viral infections because of impaired IFN responses (29, 30). To overcome the resistance of hAPN transgenic (hAPN+/+) mice to HCoV-229E infection, the generation of immunocompromised hAPN+/+ mice was taken as a strategy. Transgenic mice expressing hAPN deficient in the Stat1 gene were generated. PEFs from both transgenic hAPN+/+ or double transgenic hAPN+/+Stat1-/- mice were highly susceptible to HCoV-229E infection in vitro. Nevertheless, only hAPN transgenic mice deficient in the Stat1 gene permitted the in vivo replication of HCoV-229E adapted to grow in cells from double transgenic mice hAPN+/+Stat1-/-.
Materials and Methods
Construction of hAPN Transgene. hAPN cDNA (GenBank accession no. X13276), kindly provided by J. Olsen (University of Copenhagen, Copenhagen) (31), was cloned under control of 11-kb 5′ APN regulatory sequences (GenBank accession nos. AF176122, AF176123, and AF176124). The 3′ regulatory sequences were provided by a 1.7-kb fragment of the human β-globin gene (GenBank accession no. UO1317, nt 62553–64242) (Fig. 1A). The 5′ regulatory sequences, the APN cDNA, and all cloning junctions were sequenced with the Big Dye Terminator DNA sequencing kit (PerkinElmer) on an Applied Biosystems PRISM 377 automated DNA sequencer.
Fig. 1.

Generation and characterization of hAPN transgenic mice. (A) Structure of the hAPN expression cassette (not to scale). The gray boxes in the human β-globin fragment represent exons. The position of primers for detection of transgene integration and expression is indicated. MP, myeloid promoter; E, enhancer; EP, epithelial promoter. (B) Detection of hAPN protein in tissues of transgenic mice. hAPN protein was immunoprecipitated from tissue homogenates by using monoclonal anti-hAPN antibodies and detected by Western blotting using polyclonal anti-hAPN antibodies. (Upper) Lanes 1, 2, and 3, show liver, lung, and spleen, respectively. No recombinant protein was detected in tissues derived from nontransgenic animals (lanes 4–6). (Lower) To control the amount of protein load per lane, simultaneous immunoprecipitations of Stat1 from the tissue homogenates were performed. (C) Immunohistochemical localization of hAPN in the small intestine of transgenic mice. Tissue sections were incubated with polyclonal anti-mouse APN (1 and 2) and polyclonal anti-hAPN antibodies (3 and 4), visualized with avidin-biotin-peroxidase, and counterstained with hematoxylin. The recombinant protein was detected in the brush border of the absorptive cells of the small intestine from transgenic (3) but not from nontransgenic (4) animals. Endogenous APN protein was confirmed in the brush border of the absorptive cells of the small intestine from both genotypes (1 and 2). (Scale bars, 10 μm.)
Generation and Detection of Transgenic Mice. The gene construct was purified from vector sequences (pKOV920, Lexicon Genetics, The Woodlands, TX) by ultracentrifugation through a sodium chloride step gradient (32). Gain-of-function gene transfer was performed by microinjection of purified DNA into the pronuclei of ICR mouse zygotes (33). Transgenic mice were identified by PCR of genomic DNA with primers hAPNex19: 5′-TAT GGT GGT GGC TCG TTC TC-3′ (GenBank accession no. X13276, nt 2695–2714) and huβglobR: 5′-CAA GAA AGC GAG CTT AGT GAT AC-3′ (GenBank accession no. UO1317, nt 63623–63601) amplifying a transgene-specific 1.5-kb fragment.
Real-time quantitative PCR was used for determination of homozygosity in transgenic animals (34). For the transgene, the primers were huβglob-forward 5′-GCC CAT CAC TTT GGC AAA GA-3′ and huβglob-reverse 5′-GCC ACA CCA GCC ACC ACT-3′ (GenBank accession no. UO1317, nt 63512–63531 and nt 63583–63565) amplifying a product of 71 bp. The probe was 5′-FAM-CCA CCA GTG CAG GCT GCC TAT CAG A-TAMRA-3′ (GenBank accession no. UO1317, nt 63539–63563). The mouse tyrosinase gene was used as a reference with the primers muTyr1-forward 5′-CGA GCC TGT GCC TCC TCT AA-3′ and muTyr1-reverse 5′-CTC CCA TCA CCC ATC CAT G-3′ (GenBank accession no. D00439, nt 2608–2627 and nt 2675–2657), which amplified a 67-bp fragment. The probe was 5′-FAM-CTT GTT GGC AAA AGA ATG CTG CCC A-TAMRA-3′ (GenBank accession no. D00439, nt 2631–2654).
Immunoprecipitation and Western Blotting. Small-intestine samples were immediately rinsed in PBS containing leupetin, peptin, and aprotinin (5 μg/ml each) before homogenizing in lysis buffer (0.5% Triton X-100/50 mM Tris·Cl, pH 8.0/10% glycerol/0.1 mM EDTA/80 mM NaCl); other tissues were lysed immediately. Immunoprecipitation and Western blotting were performed as described in ref. 28 by using anti-hAPN antibodies (clone WM-15 from Serotec and clone WM-47 from Sigma-Aldrich for immunoprecipitation; polyclonal anti-human antibodies were provided by J. Olsen) and anti-mouse Stat1 antibodies (Santa Cruz Biotechnology). Immune complexes were generated with peroxidase-conjugated goat anti-rabbit IgG (Sigma-Aldrich) and visualized by using enhanced chemiluminescence reagent (Amersham Pharmacia Biotech).
Immunohistochemistry. Small-intestine and renal tissue samples were fixed in 4% buffered formalin and embedded in paraffin. Serial 3-μm sections were mounted on silan-coated slides. For immunohistochemistry, sections were deparaffinized, and endogenous peroxidases were blocked with 0.6% H2O2 in methanol for 15 min. Antigen retrieval was achieved by using microwave heating in citrate buffer (pH 6.0) for twice for 5 min, and slides were incubated with 1.5% horse serum for 30 min at room temperature. Primary antibodies, polyclonal anti-hAPN, and polyclonal anti-mouse APN (dilution 1:50, Santa Cruz Biotechnology) were applied overnight at 4°C. Subsequently, a biotinylated anti-goat secondary antibody was used, and the antigen–antibody complex was detected with an avidin-biotin-peroxidase kit (Vectastain ABC kit, Vector Laboratories). Antibody binding was detected with the chromogen diaminobenzidine (Sigma-Aldrich) and counterstained with Mayer's hematoxylin.
Cells and Viruses. Stable transfected mouse epithelial cells epH4 (35) were generated as described in ref. 28 and used for testing the transgene construct. Murine WT and transgenic PEFs (hAPN+/+ and hAPN+/+Stat1-/-) were obtained from day-11 embryos by digestion with trypsin and cultured in DMEM supplemented with 10% FCS. MRC-5 fibroblast cells (no. CCL-171, American Type Culture Collection) were kindly provided by R. Randall (University of Edinburgh, Edinburgh) and maintained in modified Eagle's medium supplemented with 10% FCS, nonessential amino acids, glutamine, and antibiotics. HCoV-229E (VR-740, American Type Culture Collection), kindly provided by K. Holmes (University of Colorado Health Science Center, Denver), was propagated and titrated at 32°C, unless specified otherwise, by using swine testis cells expressing hAPN (kindly provided by K. Holmes).
Detection of Viral Antigens by Immunofluorescence and RT-PCR. PEFs were infected (multiplicity of infection = 0.5), incubated for 20 h, and mixed with methanol, and the presence of HCoV-229E was identified with a S-specific monoclonal antibody (5–11H.6) kindly provided by P. Talbot (Institut National de la Recherche Scientifique–Institut Armand-Frappier, Quebec) (36) by using conventional procedures (37). Detection of virus replication by RT-PCR was studied after the expression of S protein mRNA by using primers virus-forward 5′-GTT GCT TTT TAG ACT TTG TGT CTA C-3′ and virus-reverse 5′-CAA TCA CCT CTC CCA GTA CC-3′ (nt 34–58 and nt 20883–20864, GenBank accession no. AF304460). The expression of β-actin mRNA was monitored as an internal control by using primers β-actin-forward 5′-CGG GAG ATC GTG CGG GAC AT-3′ and β-actin-reverse 5′-AGC ACC GTG TTG GCG TAG AG-3′ (nt 703–722 and nt 983–964, GenBank accession no. A4550069).
Infection of hAPN+/+ and hAPN+/+Stat1-/- Mice with HCoV-229E. Mice were initially infected with HCoV-229E grown in MRC-5 cells at 32°C. The in vivo infection did not result in productive virus infection. To overcome this limitation, two new stocks of the virus were produced by propagating the virus for four passages at either 32°C or 37°C on PEFs derived from hAPN+/+Stat1-/- cells. The “adapted” virus stocks were named HCoV-229E-32 and HCoV-229E-37, respectively. Mice were infected with a total of 5 × 107 plaque-forming units (pfu) in 1 ml by different routes (intragastric, 0.5 ml; oral, 0.1 ml; intranasal, 0.05 ml; i.p., 0.25 ml) or by the intranasal route with 2 × 108 pfu per mouse. Two mice were infected per dose and were killed at the indicated days postinfection. Tissues were collected to determine virus titer and for histopathology at various times after infection. All animal experiments were carried out in accordance with protocols approved by the Austrian and Spanish laws and European directives.
Results
Generation of hAPN Transgenic Mice. APN transcription is regulated by two different promoters (Fig. 1A). A distal promoter is located 8 kb upstream of the translation initiation site and controls the APN expression in myeloid and fibroblast cells (38). The proximal promoter is active in epithelial cells (39). To generate transgenic mice expressing the receptor for HCoV-229E mimicking its natural distribution, the 3.4-kb cDNA of hAPN was put under control of 11-kb genomic sequences containing all known APN-specific regulatory elements. Splice sites and polyadenylation signal for the cDNA were provided by a 1.7-kb human β-globin fragment (Fig. 1A). Correct splicing of the transgene and production of hAPN in murine cells was demonstrated by RT-PCR and Western blotting by using stably transfected epH4 cells (data not shown).
Microinjection of hAPN expression cassette DNA into mouse zygotes resulted in 11 transgenic founder animals, of which 10 transmitted the transgene to progeny (nos. 270, 275, 313, 315, 735, 746, 795, 861, 884, and 902). The presence of the hAPN gene in litters was monitored by PCR, and integration of intact transgene copies was confirmed by Southern blotting (data not shown). Mouse lines 270 and 861 expressed the highest levels of hAPN mRNA (data not shown) and were used in most of the assays. Hemi- and homozygous animals were healthy and showed no negative effects of transgene expression or insertional mutagenesis (40).
Although PEFs from the transgenic mice expressing hAPN were susceptible to infection by HCoV-229E, the mice providing the cells were not infected by the same virus (data not shown). To increase the replication of HCoV-229E, double transgenic mice were obtained by crossing hAPN homozygous males from lines 270 and 861 to immunocompromised Stat1-/- females. F2 progeny was screened for deficiency in both Stat1 alleles by an established PCR assay (30) and for homozygosity for hAPN (hAPN+/+; see above). Double transgenesis (hAPN+/+Stat1-/-) did not result in detectable side effects in the animals.
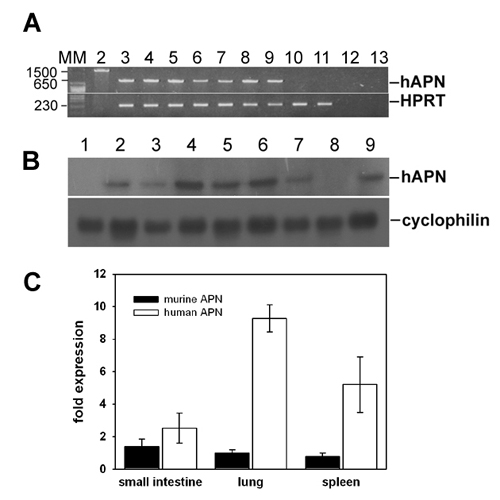
Detection of Transgenic hAPN mRNA and Protein. Expression of hAPN mRNA in mouse tissues was monitored by RT-PCR, Northern blotting, and real-time RT-PCR (see the supporting information, which is published on the PNAS web site). Sequencing of transgene-specific RT-PCR products revealed the correct expression of hAPN mRNA (data not shown). As reported for humans (17), hAPN mRNA in mice was found to be ubiquitously expressed with the expected length. Real-time RT-PCR was performed to compare the endogenous with the transgene APN level in different tissues. hAPN levels were similar to (small intestine and spleen) or slightly exceeded (lung) the endogenous APN level.
hAPN protein expression was measured in different tissues (liver, lung, spleen, kidney, small intestine, and heart) by Western blotting and immunoprecipitation. Tissues from all nine hAPN transgenic lines expressed a recombinant protein with the expected molecular mass for hAPN (150 kDa) (Fig. 1B). Crossing of hAPN+/+ transgenic mice to Stat1-/- mice did not alter the hAPN expression (data not shown).
hAPN expression was detected by immunohistochemistry in the small intestine (Fig. 1C3) and renal tissue (data not shown) from transgenic, but not from nontransgenic, animals (Fig. 1C4). In contrast, mouse APN-specific polyclonal antibody resulted in positive staining of the brush border membranes from both transgenic and nontransgenic mice (Fig. 1C 1 and 2), indicating that endogenous APN expression was not perturbed in transgenic mice.
Susceptibility to HCoV-229E in Cell Culture. The susceptibility of PEFs from hAPN+/+ transgenic lines 270 and 861 and of the corresponding double mutant lines hAPN+/+Stat1-/- 270 and 861 to infection by HCoV-229E grown in MRC-5 cells at 32°C was studied. HCoV-229E-infected cultures were maintained at 32°C, because it was found that this virus strain grows better at this temperature than at 37°C. Virus growth was first confirmed by immunof luorescence in PEFs from both hAPN+/+Stat1+/+ and hAPN+/+Stat1-/- mice (Fig. 2A). PEFs from WT (hAPN-/-Stat1+/+) and hAPN-/-Stat1-/- mice were not infected, whereas the positive control MRC-5 was immunostained. Furthermore, synthesis of HCoV-229E S gene mRNA was detected by RT-PCR in PEFs from hAPN+/+ mice for at least six passages but not in hAPN-/- PEFs (Fig. 2B).
Fig. 2.

Susceptibility of PEFs from transgenic mice to HCoV-229E infection. (A) Cells from the indicated transgenic mice lines or from control cells (MRC-5 and ICR WT mice) were infected with HCoV-229E grown in MRC-5 cells at 32°C. Cells were incubated at 32°C, and virus was detected by immunofluorescence using monoclonal antibody 5–11H.6 specific for HCoV-229E S protein. (B) (Upper) Growth of HCoV-229E was monitored by studying the expression of a HCoV-229E S gene mRNA. (Lower) The expression of β-actin mRNA was analyzed as a control. hAPN+/+ and hAPN-/-, transgenic mice homozygous for hAPN or not carrying this transgene; Stat1+/+ and Stat1-/-, WT mice or Stat1-deficient mice; MM, molecular markers in bp.
HCoV-229E growth kinetics was studied in PEFs derived from prototype transgenic mice embryos at 32°C and 37°C (Fig. 3A). HCoV-229E did not grow in APN-negative PEFs, as expected (Fig. 3A a, b, c, and d). HCoV-229E grew better at 32°C in PEFs of the two hAPN+/+Stat+/+ lines tested (Fig. 3Aa) than at 37°C (Fig. 3Ab). In contrast, temperature shift from 32°C to 37°C had a reduced effect on HCoV-229E growth in PEFs from double transgenic mice hAPN+/+Stat1-/- 270 (Fig. 3A c and d) or MRC-5 cells (Fig. 3A a and b), indicating that temperature sensitivity was cell type-dependent. Because the effect of temperature on virus growth was observed in Stat1+/+ cells but not in Stat1-/- cells, it is likely that the different sensitivity of the cell types to virus infection is related to Stat1 functions.
Fig. 3.

Growth kinetics of HCoV-229E in vitro. (A) PEFs from the indicated transgenic mice or MRC-5 cells were infected (multiplicity of infection = 1) with HCoV-229E grown in MRC-5 cells at 32°C. (B) PEFs from hAPN+/+Stat1+/+ (a and b) and hAPN+/+Stat1-/- (c and d) mice were infected (multiplicity of infection = 1) with HCoV-229E or HCoV-229E-37, respectively. In A and B, infected cells were incubated at 32°C (a and c) or 37°C (b and d) for the indicated times. Virus growth was evaluated by determining the plaque forming units in swine testis-hAPN cells.
The same virus infecting PEFs from hAPN+/+ mice failed to infect hAPN+/+Stat1+/+, hAPN+/+Stat1-/-, hAPN-/-Stat1-/-, and WT mice in vivo. In an attempt to improve HCoV-229E growth in hAPN+/+Stat1-/- mice, the virus originally grown in MRC-5 cells at 32°C was passaged four times in PEFs from hAPN+/+Stat1-/- 270 mice either at 32°C or 37°C, giving rise to HCoV-229E-32 and HCoV-229E-37, respectively. The growth kinetics of the passaged viruses were analyzed at 32°C and 37°C in comparison to the original HCoV-229E. HCoV-229E-37 showed a slightly higher titer at 37°C in hAPN+/+Stat1+/+ PEFs (Fig. 3Bb) and faster growth kinetics in hAPN+/+Stat1-/- PEFs (Fig. 3Bd). Nevertheless, because adapted HCoV-229E-37 also grew slightly better at 32°C than the nonadapted virus in both hAPN+/+Stat1+/+ and hAPN+/+-Stat1-/- PEFs (Fig. 3B a and c), the higher growth cannot be attributed to a better fitness at 37°C. In fact, HCoV-229E-32 also grew in hAPN+/+Stat1-/- mice (data not shown), reinforcing the concept that adaptation to grow in cells from transgenic mice was important, but the temperature maintained during this adaptation was not.
The adaptation of HCoV-229E could be due to any mutation of the virus genome. Frequently, the ability to infect a host and virus tropism is related to a change in the S glycoprotein recognized by the cell surface receptor (11, 41, 42). Therefore, we decided to compare the sequence of the S genes from the adapted and nonadapted HCoV-229E. Two nucleotide differences leading to amino acid changes in position 278 (T > I) and 814 (N > A) were observed. Reverse genetic approaches are under way to determine whether these nucleotide changes are responsible for HCoV-229E adaptation.
Susceptibility to HCoV-229E in Vivo. HCoV-229E-37 was tested for in vivo virus growth in hAPN+/+Stat1+/+, hAPN+/+Stat1-/-, hAPN-/-Stat1-/-, and WT mice by simultaneous inoculation through different routes (oral, intranasal, intragastric, and i.p.) and through the nasal route only, because HCoV-229E is considered a respiratory pathogen. Mice were killed at 1–4, 8, and 16 days postinoculation. Lung, liver, spleen, and small intestine were harvested and used for analysis. HCoV-229E-37 was only isolated from tissues derived from double transgenic mice hAPN+/+Stat1-/- (Fig. 4A). No virus growth was detected in hAPN-/- mice, even if they were Stat1-/-, indicating that the hAPN+/+ genotype was essential.
Fig. 4.

Susceptibility of transgenic mice to HCoV-229E-37. Mice with the indicated genotype were infected by different routes (oral, intragastric, intranasal, and i.p.) with HCoV-229E-37. At the indicated times postinfection, lung, intestine, spleen, and liver tissues were collected, and virus titers were determined by using a plaque assay. (A) The results shown represent medium values of titers in the lungs of the different mouse strains. (B) Virus growth in different tissues of hAPN+/+Stat1-/- mice infected with HCoV-229E-37. Virus growth could not be detected in the tissues of nontransgenic animals. To simplify the figure, only nontransgenic lung is shown. (C) RT-PCR analysis of HCoV-229E-37 growth in different tissues from the indicated transgenic mice by studying the expression of HCoV-229E S gene mRNA. The position of the expected amplified DNA is indicated. ST-hAPN, swine testis cells transformed with the gene encoding hAPN; S+ and S-, Stat1+/+ and Stat1-/-mice, respectively; Stat1 (+) and (-), presence or absence of functional Stat1 alleles; hAPN (+) and (-), presence or absence of homozygous hAPN transgenes; MM, molecular markers in bp. (D) Hematoxylin/eosin-stained lung sections 8 days after HCoV-229E-37 infection. Global severe inflammatory reactions with massive neutrophilic infiltrations were prominent in the lung of hAPN+/+Stat1-/- mice (3 and 4), whereas no histopathological changes could be detected in the lung of nontransgenic mice (1 and 2). (Scale bars: 1 and 3, 100 μm; 2 and 4, 200 μm.)
Upon infection through different routes, the virus was found in large amounts in the lungs and in the gut (Fig. 4B). In addition, the virus was found in the spleen and, during the first day postinfection, in the liver. Because both organs are involved in virus clearance, it is not possible to rule out that detection of the virus was due to accumulation rather than replication.
Upon administration of the virus exclusively by the nasal route, virus growth was detected in the lung of double transgenic mice with titers of up to 6 × 103 plaque-forming units/g of tissue at day 3 postinfection (data not shown). No virus was found in the gut, liver, or spleen in nontransgenic or single transgenic animals.
Tissue extracts from infected mice were used to amplify potentially present virus on swine testis cells expressing hAPN, and virus presence was confirmed by RT-PCR only for hAPN+/+Stat1-/- mice (Fig. 4C). Furthermore, no virus was detected by RT-PCR in hAPN+/+Stat1-/- mice, even after this amplification step in tissue extracts from mice infected with the “nonadapted” virus (data not shown).
All hAPN+/+Stat1-/- mice that were challenged survived at least for 18 days after infection. Clinically, susceptible mice showed a mild loss of weight and slight temperature increase. Organs of hAPN+/+Stat1-/- mice challenged by means of different routes showed hemorrhagic areas in lung and small intestine, mainly between days 2 and 4 postinfection. Spleens from mice inoculated by different routes (but not mice intranasally inoculated) had a size 3- to 4-fold higher than mock-infected mice. Furthermore, in the lung and small intestine of these mice, coronavirus particles could be detected by using electron microscopy (data not shown). Histopathology of intranasally infected hAPN+/+Stat1-/- mice revealed changes consistent with a viral infection of the lung (Fig. 4D).
Overall, these data indicated that to infect mice with HCoV-229E, several requirements were needed: (i) expression of hAPN in mice, (ii) impairment of the immune system (Stat1-/-), and (iii) “adaptation” of HCoV-229E to grow in hAPN+/+Stat1-/- cells.
Discussion
HCoV-229E is specific to humans. To generate an animal model for HCoV-229E infection, several lines of hAPN transgenic mice were produced. hAPN was expressed ubiquitously in transgenic mice, with a pattern similar to that described in human, as shown by semiquantitative and quantitative hAPN mRNA detection and by protein analyses. Although PEFs from hAPN+/+Stat1+/+ and hAPN+/+Stat-/- mice were susceptible to infection by HCoV-229E, susceptibility to HCoV-229E was only possible in immunocompromised mice (hAPN+/+ Stat1-/-) by using a HCoV-229E adapted to grow in PEFs of hAPN+/+Stat1-/- in vitro.
PEFs from hAPN-expressing mice were susceptible to HCoV-229E, indicating functionality of the transgene receptor. In contrast, PEFs from WT or Stat1-/- mice were resistant to HCoV-229E infection, indicating that expression of hAPN was an essential determinant of HCoV-229E host range and that susceptibility of the transgenic PEFs to HCoV-229E was not solely due to the IFN unresponsiveness or to the adaptation of the virus to murine PEFs during virus passage within these cells. Our results confirmed previous studies that demonstrated that transfection of nonpermissive cells with hAPN expression vectors is sufficient to confer susceptibility to HCoV-229E in vitro (13, 43).
The adaptation of the virus to grow in hAPN+/+Stat1-/- mice did not imply adaptation to high (37°C) temperature because (i) the virus passed at both 32°C and 37°C infected the mice and (ii) both viruses grew up to similar extent at 37°C. In addition, virus adaptation was not conferred by the use of a murine receptor; susceptibility was still dependent on the presence of hAPN in mice.
A previous attempt to generate APN transgenic mice susceptible to group 1 coronavirus failed (44). Our successful development of a susceptible model was probably due to the use of comprehensive APN regulatory elements, the generation of double transgenic hAPN+/+Stat1-/- mice, and the use of a virus that was adapted to grow in PEFs from these mice. The molecular basis of HCoV-229E adaptation remains to be defined. It could be related to a better fitness of the viral S protein to interact with the hAPN receptor. Alternatively, it could be due to the lack of activation of antiviral genes in Stat1-/- mice.
Virus entry and spread in mammals is restricted by innate host defense mechanisms, including the IFNs (45). Clearance of transmissible gastroenteritis virus, another group 1 coronavirus, has been associated with IFN production (46). The requirement for perturbing the innate immunity by disruption of IFN signaling was also shown in a murine model susceptible to measles virus. As observed in our model, murine cells expressing huCD46 were permissive in vitro (47). However, mice providing these cells remained refractory to measles virus infection. Crossing huCD46 transgenic mice to IFNAR1-/- mice rendered susceptibility to measles virus in vivo.
Infectious cDNAs of different group 1 coronaviruses have been obtained. Because coronavirus tissue specificity and species specificity can be engineered, the same expression systems can be used to target expression to different organs and animals species, including humans (22). The humanized mice described here will allow studies of virus and viral vector tropism, replication, recombination, and spread in an immunocompromised situation.
Crossing hAPN+/+ mice with a collection of mice having immune deficiencies of a different nature (reviewed in ref. 48) will be helpful to determine the compartment of the immune system responsible for the resistance to HCoV-229E infection and also to determine the biosafety of coronavirus-based vectors.
Supplementary Material
Acknowledgments
We thank Sabine Müller (VBC Genomics, Vienna) for isolating the APN regulatory sequences, Magda Helmreich and Waltraud Tschulenk (University of Veterinary Medicine, Vienna) for expert technical assistance, and Birgit Strobl (University of Veterinary Medicine, Vienna) for helpful comments on the manuscript. This work was supported by European Community Grants QLK2-CT-2001-00874 (to M.M.) and QLK2–2001-01050 (to L.E.), the Austrian Federal Ministry for Education, Science, and Culture (GZ 200.074/1-VI/1a/2002), and the Spanish Commission of Science and Technology.
Author contributions: L.E. and M.M. designed research; C.L., C.M.S., S.M., T.K., and P.P. performed research; C.L., C.M.S., and T.K. contributed new reagents/analytic tools; C.L., M.E., and I.W. analyzed data; and C.L., L.E., and M.M. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: HCoV, human coronavirus; APN, aminopeptidase N; hAPN, human APN; S, coronavirus spike; PEF, primary embryonic fibroblast; Stat, signal transducers and activators of transcription.
See Commentary on page 8073.
References
- 1.Enjuanes, L., Spaan, W., Snijder, E. & Cavanagh, D. (2000) in Virus Taxonomy: Classification and Nomenclature of Viruses, eds. van Regenmortel, M. H. V., Fauquet, C. M., Bishop, D. H. L., Carsten, E. B., Estes, M. K., Lemon, S. M., Mayo, M. A., McGeoch, D. J., Pringle, C. R. & Wickner, R. B. (Academic, New York), pp. 827-834.
- 2.Enjuanes, L., Brian, D., Cavanagh, D., Holmes, K., Lai, M. M. C., Laude, H., Masters, P., Rottier, P., Siddell, S. G., Spaan, W. J. M., et al. (2000) in Virus Taxonomy: Classification and Nomenclature of Viruses, eds. van Regenmortel, M. H. V., Fauquet, C. M., Bishop, D. H. L., Carsten, E. B., Estes, M. K., Lemon, S. M., Mayo, M. A., McGeoch, D. J., Pringle, C. R. & Wickner, R. B. (Academic, New York), pp. 835-849.
- 3.Denison, M. R. (1999) in Viral Infections of the Respiratory Tract, eds. Dolin, R. & Wringht, P. F. (Dekker, New York), Vol. 127, pp. 253-280. [Google Scholar]
- 4.McIntosh, K. (1996) in Fields Virology, eds. Fields, B. N., Knipe, D. M. & Howley, P. M. (Lippincott, Philadelphia), 3rd Ed., pp. 1095-1103.
- 5.van der Hoek, L., Pyrc, K., Jebbink, M. F., Vermeulen-Oost, W., Berkhout, R. J., Wolthers, K. C., Wertheim-van Dillen, P. M., Kaandorp, J., Spaargaren, J. & Berkhout, B. (2004) Nat. Med. 10, 368-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woo, P. C., Lau, S. K., Chu, C. M., Chan, K. H., Tsoi, H. W., Huang, Y., Wong, B. H., Poon, R. W., Cai, J. J., Luk, W. K., et al. (2005) J. Virol. 79, 884-895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Esper, F., Weibel, C., Ferguson, D., Landry, M. L. & Kahn, J. S. (2005) J. Infect. Dis. 191, 492-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peiris, J. S., Lai, S. T., Poon, L. L., Guan, Y., Yam, L. Y., Lim, W., Nicholls, J., Yee, W. K., Yan, W. W., Cheung, M. T., et al. (2003) Lancet 361, 1319-1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rota, P. A., Oberste, M. S., Monroe, S. S., Nix, W. A., Campagnoli, R., Icenogle, J. P., Penaranda, S., Bankamp, B., Maher, K., Chen, M. H., et al. (2003) Science 300, 1394-1399. [DOI] [PubMed] [Google Scholar]
- 10.Kuo, L., Godeke, G. J., Raamsman, M. J., Masters, P. S. & Rottier, P. J. (2000) J. Virol. 74, 1393-1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanchez, C. M., Izeta, A., Sanchez-Morgado, J. M., Alonso, S., Sola, I., Balasch, M., Plana-Duran, J. & Enjuanes, L. (1999) J. Virol. 73, 7607-7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delmas, B., Gelfi, J., L'Haridon, R., Vogel, L. K., Sjostrom, H., Noren, O. & Laude, H. (1992) Nature 357, 417-420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeager, C. L., Ashmun, R. A., Williams, R. K., Cardellichio, C. B., Shapiro, L. H., Look, A. T. & Holmes, K. V. (1992) Nature 357, 420-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nomura, R., Kiyota, A., Suzaki, E., Kataoka, K., Ohe, Y., Miyamoto, K., Senda, T. & Fujimoto, T. (2004) J. Virol. 78, 8701-8708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Look, A. T., Ashmun, R. A., Shapiro, L. H. & Peiper, S. C. (1989) J. Clin. Invest. 83, 1299-1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olsen, J., Kokholm, K., Noren, O. & Sjostrom, H. (1997) Adv. Exp. Med. Biol. 421, 47-57. [DOI] [PubMed] [Google Scholar]
- 17.Riemann, D., Kehlen, A. & Langner, J. (1999) Immunol. Today 20, 83-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Almazan, F., Gonzalez, J. M., Penzes, Z., Izeta, A., Calvo, E., Plana-Duran, J. & Enjuanes, L. (2000) Proc. Natl. Acad. Sci. USA 97, 5516-5521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Casais, R., Thiel, V., Siddell, S. G., Cavanagh, D. & Britton, P. (2001) J. Virol. 75, 12359-12369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thiel, V., Herold, J., Schelle, B. & Siddell, S. G. (2001) J. Gen. Virol. 82, 1273-1281. [DOI] [PubMed] [Google Scholar]
- 21.Yount, B., Curtis, K. M. & Baric, R. S. (2000) J. Virol. 74, 10600-10611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Enjuanes, L., Almazan, F. & Ortego, J. (2003) in Gene Transfer and Expression in Mammalian Cells, ed. Makrides, S. C. (Elsevier Science, Amsterdam), pp. 151-168.
- 23.Martina, B. E., Haagmans, B. L., Kuiken, T., Fouchier, R. A., Rimmelzwaan, G. F., Van Amerongen, G., Peiris, J. S., Lim, W. & Osterhaus, A. D. (2003) Nature 425, 915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roberts, A., Vogel, L., Guarner, J., Hayes, N., Murphy, B., Zaki, S. & Subbarao, K. (2005) J. Virol. 79, 503-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pearson, J. & Mims, C. A. (1985) J. Virol. 53, 1016-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacomy, H. & Talbot, P. J. (2003) Virology 315, 20-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stark, G. R., Kerr, I. M., Williams, B. R., Silverman, R. H. & Schreiber, R. D. (1998) Annu. Rev. Biochem. 67, 227-264. [DOI] [PubMed] [Google Scholar]
- 28.Muller, M., Laxton, C., Briscoe, J., Schindler, C., Improta, T., Darnell, J. E., Jr., Stark, G. R. & Kerr, I. M. (1993) EMBO J. 12, 4221-4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Durbin, J. E., Hackenmiller, R., Simon, M. C. & Levy, D. E. (1996) Cell 84, 443-450. [DOI] [PubMed] [Google Scholar]
- 30.Meraz, M. A., White, J. M., Sheehan, K. C., Bach, E. A., Rodig, S. J., Dighe, A. S., Kaplan, D. H., Riley, J. K., Greenlund, A. C., Campbell, D., et al. (1996) Cell 84, 431-442. [DOI] [PubMed] [Google Scholar]
- 31.Olsen, J., Cowell, G. M., Konigshofer, E., Danielsen, E. M., Moller, J., Laustsen, L., Hansen, O. C., Welinder, K. G., Engberg, J., Hunziker, W., et al. (1988) FEBS Lett. 238, 307-314. [DOI] [PubMed] [Google Scholar]
- 32.Fink, P. S. (1991) BioTechniques 10, 446, 448, 450. [PubMed] [Google Scholar]
- 33.Brem, G., Brenig, B., Goodman, H. M., Selden, R. C., Graf, F., Kruff, B., Springmann, K., Hondele, J., Meyer, J., Winnacker, E.-L., et al. (1985) Zuchthygiene 20, 251-252. [Google Scholar]
- 34.Tesson, L., Heslan, J. M., Menoret, S. & Anegon, I. (2002) Transgenic Res. 11, 43-48. [DOI] [PubMed] [Google Scholar]
- 35.Fialka, I., Schwarz, H., Reichmann, E., Oft, M., Busslinger, M. & Beug, H. (1996) J. Cell Biol. 132, 1115-1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Talbot, P. J., Paquette, J. S., Ciurli, C., Antel, J. P. & Ouellet, F. (1996) Ann. Neurol. 39, 233-240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sola, I., Alonso, S., Zuniga, S., Balasch, M., Plana-Duran, J. & Enjuanes, L. (2003) J. Virol. 77, 4357-4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shapiro, L. H., Ashmun, R. A., Roberts, W. M. & Look, A. T. (1991) J. Biol. Chem. 266, 11999-12007. [PubMed] [Google Scholar]
- 39.Olsen, J., Laustsen, L., Karnstrom, U., Sjostrom, H. & Noren, O. (1991) J. Biol. Chem. 266, 18089-18096. [PubMed] [Google Scholar]
- 40.Reichart, U., Kappler, R., Scherthan, H., Wolf, E., Muller, M., Brem, G. & Aigner, B. (2000) Biochem. Biophys. Res. Commun. 269, 496-501. [DOI] [PubMed] [Google Scholar]
- 41.Leparc-Goffart, I., Hingley, S. T., Chua, M. M., Phillips, J., Lavi, E. & Weiss, S. R. (1998) J. Virol. 72, 9628-9636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bonavia, A., Zelus, B. D., Wentworth, D. E., Talbot, P. J. & Holmes, K. V. (2003) J. Virol. 77, 2530-2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tresnan, D. B., Levis, R. & Holmes, K. V. (1996) J. Virol. 70, 8669-8674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benbacer, L., Stinackre, M. G., Laude, H. & Delmas, B. (1998) Adv. Exp. Med. Biol. 440, 53-59. [DOI] [PubMed] [Google Scholar]
- 45.Hertzog, P. J., O'Neill, L. A. & Hamilton, J. A. (2003) Trends Immunol. 24, 534-539. [DOI] [PubMed] [Google Scholar]
- 46.Charley, B. & Laude, H. (1988) J. Virol. 62, 8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Manchester, M. & Rall, G. F. (2001) Trends Microbiol. 9, 19-23. [DOI] [PubMed] [Google Scholar]
- 48.Casanova, J. L. & Abel, L. (2004) Nat. Rev. Immunol. 4, 55-66. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}