

Abstract
Somatic mutations are the cause of cancer and have been implicated in other, noncancerous diseases and aging. While clonally expanded mutations can be studied by deep sequencing of bulk DNA, very few somatic mutations expand clonally, and most are unique to each cell. We describe a detailed protocol for single-cell whole-genome sequencing to discover and analyze somatic mutations in tissues and organs. The protocol comprises single-cell multiple displacement amplification (SCMDA), which ensures efficiency and high fidelity in amplification, and the SCcaller software tool to call single-nucleotide variations and small insertions and deletions from the sequencing data by filtering out amplification artifacts. With SCMDA and SCcaller at its core, this protocol describes a complete procedure for the comprehensive analysis of somatic mutations in a single cell, covering (1) single-cell or nucleus isolation, (2) single-cell or nucleus whole-genome amplification, (3) library preparation and sequencing, and (4) computational analyses, including alignment, variant calling, and mutation burden estimation. Methods are also provided for mutation annotation, hotspot discovery and signature analysis. The protocol takes 12–15 h from single-cell isolation to library preparation and 3–7 d of data processing. Compared with other single-cell amplification methods or single-molecular sequencing, it provides high genomic coverage, high accuracy in single-nucleotide variation and small insertions and deletion calling from the same single-cell genome, and fewer processing steps. SCMDA and SCcaller require basic experience in molecular biology and bioinformatics. The protocol can be utilized for studying mutagenesis and genome mosaicism in normal and diseased human and animal tissues under various conditions.
Introduction
Somatic mutations have since long been implicated as a cause of both cancer and aging1-3. By the 1970s, a concept of repeated cycles of somatic mutation and selection as the cause of cancer was generally accepted and experimentally confirmed4. However, a causal relationship of somatic mutations with aging proved more difficult to ascertain. Indeed, tumors are clones, and advances in sequencing technology soon allowed the characterization of their unique mutational landscapes5,6. By contrast, somatic mutations in normal tissues during aging could only be analyzed when clonally expanded, as in clonal hematopoiesis7,8. Postzygotic clonal expansion of somatic mutations has now been demonstrated as a cause of many Mendelian diseases9,10. However, most somatic mutations in postmitotic tissues are unique for each cell and do not clonally amplify. They cannot be easily detected because sequencing bulk DNA at 30× depth cannot discover mutations at extremely low allelic fractions.
To increase detection sensitivity, ultra-high-depth sequencing of bulk DNA, e.g., 300×, has been utilized11. This allows detecting mutations of typically >1% allelic fraction, i.e., ~0.5% of cells in a diploid cell population. However, in the context of 3 × 1013 cells of a human or over 1011 cells of an organ12,13, 0.5% of cells is still a huge clone. Even by limiting the amount of input bulk DNA to a few hundreds or thousands of cells, e.g., using laser-capture microdissection, a very limited number of clonally expanded mutations have been detected from normal tissues14.
An ideal approach to discover and quantify mutations in normal somatic cells is single-cell or clone sequencing. Stem cells, as well as a few other cell types, can be clonally expanded in vitro. The derived single-cell clones inherit all somatic mutations of the original single stem cells. In these clones, the somatic mutations of their original cells present in the same allelic fractions as germline mutations, and can be discovered by sequencing the clones at 30× (ref. 15). Indeed, somatic mutations in tumors can be detected because they are clonally expanded. However, only few cell types clonally expand in vivo or can be clonally expanded in vitro. The gold standard in this respect would be to sequence genomes of single cells. However, sequencing a single cell requires whole-genome amplification (WGA), which is error prone16. To address this challenge, we developed single-cell multiple displacement amplification (SCMDA) to avoid the most dominant type of amplification artifacts caused by heat-induced cytosine deamination, and a software tool, SCcaller, to filter out remaining artifacts17. Together, this allows to identify mutations accurately from single-cell sequencing data.
Using these approaches in a range of cell types, including lymphocytes, hepatocytes and bronchial epithelial cells, we conclusively demonstrated that somatic mutations accumulate in human cells in vivo. We have shown that the number of mutations varies from several hundred at birth to several thousand per cell at old age18-20. In addition, we applied the methods to analyze the effect of genetic (BRCA1 and BRCA2 variants) or environmental (tobacco smoking) factors on mutation burden in humans, and the effects of species-specific maximum life span on somatic mutation rate20-23. Here, we provide a complete, up-to-date protocol for the comprehensive analysis of somatic mutations in a single cell, which consists of four main stages: (1) single-cell or nucleus isolation, (2) single-cell or nucleus WGA, (3) library preparation and sequencing, and (4) data analyses, including sequence data alignment, variant calling and estimating mutation burden (Fig. 1). In addition, for the data analyses, we provide our highly integrated SCcaller-pipeline24. This data analysis pipeline allows users to process the data through 17 substeps with only two command lines, rather than going through each substep one by one. The discovered mutations can be annotated and utilized to discover mutation hotspots and mutation signatures, as described in the ‘Anticipated results’ section.
Fig. 1 ∣. A schematic overview of the single-cell whole-genome sequencing protocol.
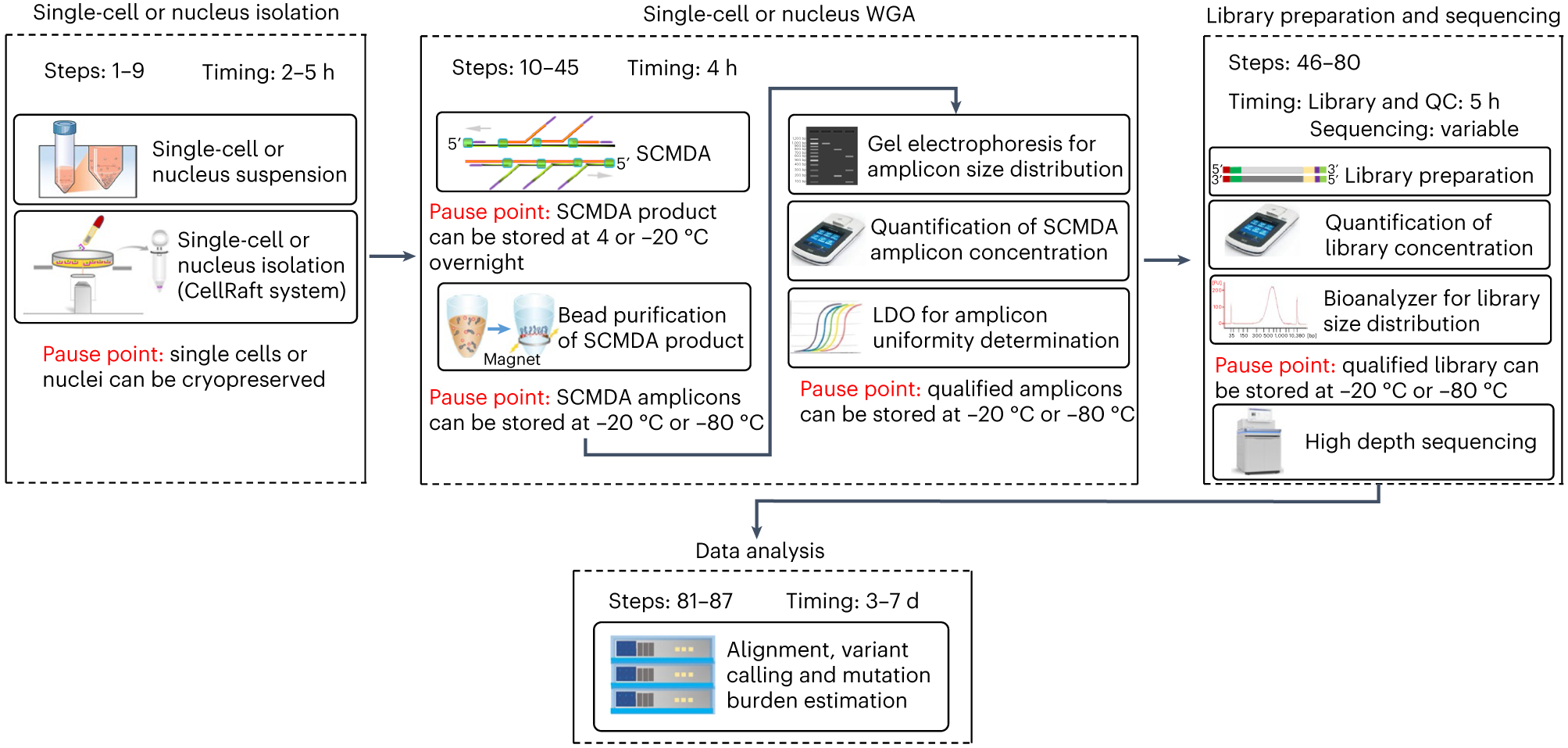
The entire protocol includes four stages: single-cell or nucleus isolation (Steps 1–9), single-cell or nucleus WGA (Steps 10–45), library preparation (Steps 46–80), and sequencing and data analysis (Steps 81–87). Major steps, timing and pause points of each stage are presented in the figure.
Development of the protocol
WGA is the critical step in single-cell whole-genome sequencing. As the human genome is huge, i.e., ~3.2 × 109 base pairs, amplifying it from a single cell (6 pg of DNA) to a sequenceable amount requires both high accuracy and efficiency. One amplification strategy is multiple displacement amplification (MDA)25. However, in conventional MDA-based single-cell amplification, as we have shown, most observed somatic single-nucleotide variations (SNVs) are artificial GC > AT transitions17. These artifacts are due to heat-induced cytosine deamination, for example, when cell lysis is done at high temperature. Although simply performing cell lysis with alkaline on ice can address this problem, its efficiency is extremely low: only a few percent of single cells can be successfully amplified. This is probably caused by the possible renaturation of single-stranded DNA after lysis and before MDA. In SCMDA, to increase amplification efficiency and accuracy, we add exo-resistant random primers (hexamers) before cell lysis and DNA denaturation, which is performed on ice. As the primers are very small, they are the first to hybridize with the single-strand DNA templets, when the cell lysis and denaturation are terminated (i.e., neutralized). This prevents renaturation of the original DNA templates from the single cell and promotes amplification efficiency to ~50–80% successful rate, depending on sample quality (Fig. 2).
Fig. 2 ∣. A schematic illustration of the principle and protocol of SCMDA.
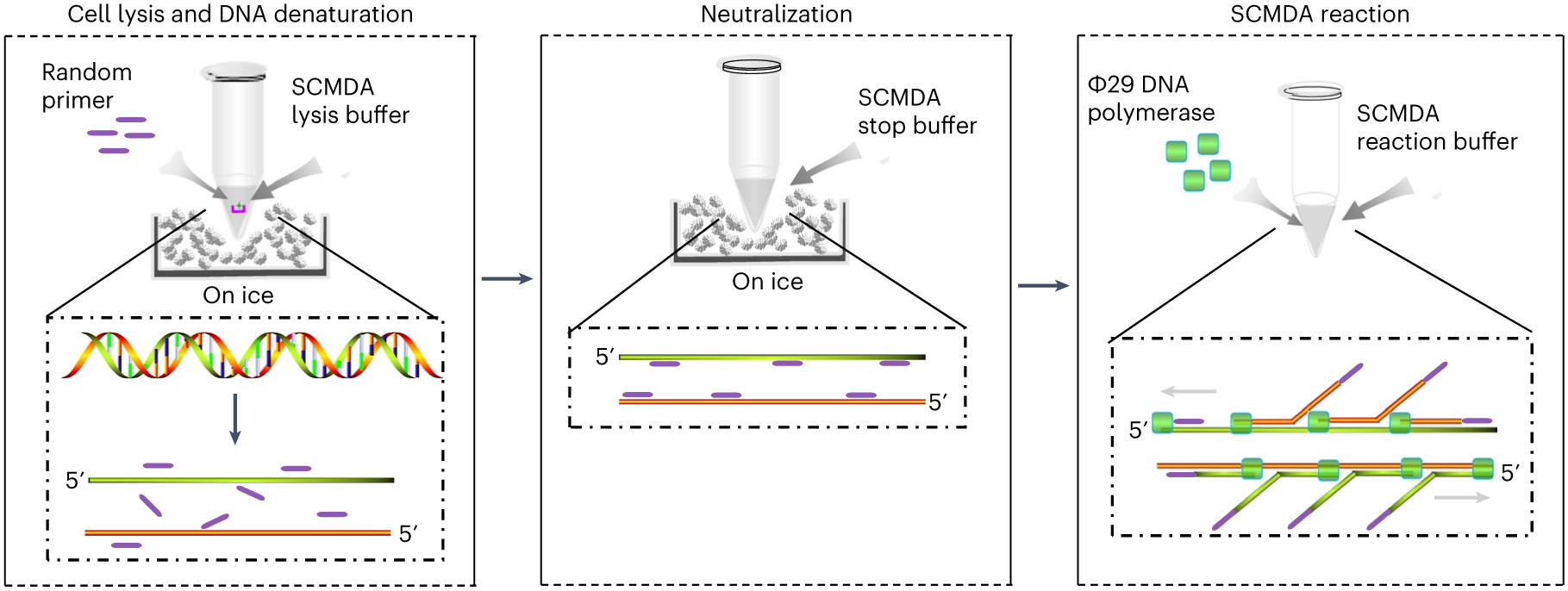
SCMDA includes three major stages: cell lysis and DNA denaturation (left), neutralization (middle) and SCMDA reaction (right). Cell lysis and DNA denaturation: a single cell (represented by a green star) on a cellraft (represented by a pink u-shape symbol) is collected into a PCR tube and placed on ice. Random primers (represented by a purple line) and SCMDA lysis buffer are added into the tube to start cell lysis and DNA denaturation on ice. Neutralization: SCMDA stop buffer is added into the PCR tube to neutralize the reaction of cell lysis and DNA denaturation. Random primers bind to single-strand DNA and prevent renaturation of the two original DNA strands. SCMDA reaction: Φ29 DNA polymerase (represented by a green square) and SCMDA reaction buffer are added into the PCR tube for SCMDA.
To further remove remaining artifacts, e.g., errors by the amplification enzyme, we developed a variant calling tool, SCcaller, for analyzing single-cell whole-genome sequencing data17. SCcaller models allelic amplification bias across the genome using known germline variants to adjust expected allelic fractions in a likelihood ratio test that distinguishes true mutations from artifacts and filters out the latter. SCcaller was originally developed for calling SNVs and validated by demonstrating that substantial overlap of somatic mutations was observed between nonamplified single-cell clones and amplified single cells from the same clones. In our latest update (version 2.0.0), SCcaller can now be used for calling insertions and deletions (INDELs) from the same single-cell sequencing data. The principle of using known germline variants for assisting somatic mutation calling has been applied later in linked-read analysis to accurately identify somatic SNVs using read-level phasing25,26.
Apart from the error prevention steps in SCMDA and SCcaller discussed above, we have also implemented and updated other essential steps in single-cell whole-genome sequencing. For single-cell isolation, we used the CellRaft system. We initially applied this only to adherent cell types, such as fibroblasts17,21,22, but later also to other cell types and nuclei (described in detail in the ‘Experimental design’ and ‘Procedure’). For preparing sequencing libraries, we initially used a PCR-free library preparation method17,21, and then showed that PCR-based library preparation can also be applied18,22. For sequencing platforms, we and others have demonstrated the feasibility of using Illumina Hiseq, Illumina Novaseq and BGI systems17,18,22,27. For sequence alignment, we applied and kept up to date the Genome Analysis Toolkit (GATK) best practice pipeline for analyzing human samples18 and implemented a modified version for comparison across different species22, sometimes without a fully synthesized reference genome, or without a germline variant database, e.g., the Single Nucleotide Polymorphism database (dbSNP) for humans. For downstream analysis, we calculate the mutation burden per single cell by adjusting variant calling accuracy and sequencing coverage of the single cells and implemented hotspot and signature analyses. The above improvements were included after the initial development of SCMDA and SCcaller18-23,28.
Applications of the method
The protocol was developed for the quantitative analyses of low-abundant, somatic mutations in normal, nonclonal single cells, such as total mutation burden, genome distribution of the mutations and the mutational signature. This can inform the sources of somatic mutations, their interactions within a single genome and their possible functional impact on human aging and disease, including but not limited to cancer. Other uses of the protocol in the field of mutagenesis include: (i) genetic toxicology for assessing mutagenicity of new and existing compounds, e.g., drugs, and (ii) assessing genome sequence integrity of specific cell lines, e.g., stem cell lines for therapeutic purposes. In addition, the protocol can also be adapted for analyzing somatic and/or germline mutations of biospecimens for which only very little material is available, e.g., early stage embryos, or in forensic science.
Comparison with other methods
As mentioned above, ultra-deep sequencing of bulk DNA, e.g., of laser-capture microdissection-collected microbiopsies, is the conventional approach for studying somatic mutations in normal, nontumor tissues. However, because the error rates of base calling from a single sequencing read after alignment are substantially higher than the actual frequency of mutation per base pair, it typically requires multiple sequencing reads to confidently call a true mutation and not a sequencing or alignment error. In bulk sequencing, different reads of the same genomic locus come from different cells; therefore, only clonally expanded mutations can be detected in this way. Since clonally expanded mutations are only a tiny fraction of all de novo somatic mutations in normal, nontumor tissues subject to growth selection, they are not representative for the overall somatic mutation burden in cells.
Increasing the accuracy of bulk sequencing is one way to address the problem above. If the number of sequencing errors per base pair (bp) in bulk sequencing is reduced and substantially smaller than the number of mutations per bp, mutations do not have to be clonally expanded to be detected. ‘Single-molecule sequencing’, e.g., Duplex sequencing29, NanoSeq30 and SMM-seq31, has been developed on the basis of the idea above. These methods uniquely tag complementary strands of DNA fragments, or keep them together (SMM-seq), before sequencing. After sequencing, the complementary strands of each fragment are recombined computationally, with mutations only considered as true mutations when found on both strands opposite each other. This strategy has become a cost-efficient way to quantify the mutation burden for bulk DNA.
The disadvantage of these single-molecule approaches is the lack of information about mutations in the same genome. Single-cell whole-genome sequencing is unique and critical for mutation analysis of normal tissues, because it offers the possibility of discovering most, if not all, mutations in the same single-cell genome. This allows the quantification of the mutation burden per cell, the discovery of mutational hotspots, the establishment of cell lineages32,33 and the analysis of interactions between mutations within a cell. In addition, single-cell sequencing is critically important in cases when only few cells are available, e.g., in studying oocytes or early embryonic cells. We provide a comparison of the three strategies, i.e., ultra-deep sequencing of bulk DNA, single-molecule sequencing and single-cell whole-genome sequencing (Table 1).
Table 1 ∣.
Somatic mutations and their detection methods
| Somatic mutation | Burden in a normal somatic cell |
Variant allele fraction (VAF) in a tissue |
Potential effect | Deep bulk sequencing |
Single-molecule sequencing (e.g., Duplex sequencing) |
Single-cell whole-genome sequencing (e.g., SCMDA) |
|---|---|---|---|---|---|---|
| Clonally expanded in development | Very rare | High | Mendelian diseases | Discovers mutations with a VAF >1% | (a) Discovers mutations of all VAFs; (b) is the most cost efficient | (a) Discovers mutations of all VAFs; (b) allows lineage construction; (c) determines mutation burden per cell |
| Clonally expanded during cell turnover with age | Rare | Low | Cancer and possibly other diseases | |||
| Not/barely clonally expanded | Hundreds to thousands | Extremely low | Functional decline in aging and age-related diseases | Not possible |
Single-cell whole-genome sequencing requires amplification. In the past few years, several WGA methods have been developed. They include: (i) PCR-based methods, e.g., degenerate oligonucleotide-primed PCR34; (ii) MDA-based methods35; (iii) hybrid methods, e.g., multiple annealing and looping based amplification cycles (MALBAC)36; and (iv) transposon-based methods, e.g., linear amplification via transposon insertion (LIANTI)37.These have been reviewed previously38. In brief, MDA offers higher genome coverage, lower allele dropout and lower false positive rates in SNV calling, while degenerate oligonucleotide-primed PCR and MALBAC have less bias in read distribution across the genome and subsequently higher accuracy in discovering copy number variations (CNVs) and aneuploidies. However, as discussed above, the error rate of conventional MDA for single-cell sequencing is still too high for reliable identification of somatic SNVs because it has never been designed for accurately calling variants. It requires high temperature for DNA denaturation39 and subsequently introduces substantial amount of cytosine deamination. SCMDA avoids the high-temperature DNA denaturation step and further suppresses its false positive rate17. More recently, transposon-based amplification methods have been developed, for example, LIANTI37 and Multiplexed End-Tagging Amplification of Complementary Strands40, which also can correctly estimate number of somatic SNVs and CNVs per cell. Compared with the transposon-based amplification methods, SCMDA still shows higher genome coverage. In addition, the procedure of SCMDA takes 4 h, including all quality control (QC) measurements, and has been applied to many studies by us and others17-23,27,41-43.
Limitations
SCMDA has some limitations. First, while the method can be applied to cell populations, including viably frozen cells or cells isolated from fresh tissue, as well as nuclei extracted from flash-frozen tissue, it may not be applicable to cells or nuclei extracted from formalin-fixed paraffin-embedded tissues. It remains to be tested whether tissue fixation protocols are compatible with SCMDA. Second, as discussed above, SCMDA is not an ideal approach for detecting CNVs and aneuploidies, compared with other single-cell WGA methods, e.g., MALBAC36 and LIANTI37. This is due to the unequal sequencing depth inherent to MDA, which also means that mutations from regions of low sequencing depth may not always be identified sensitively.
Limitations specific for SCcaller are its dependence on heterozygous single-nucleotide polymorphism (SNP) information for identifying locus-specific amplification bias and, therefore, eliminating potential artifacts in calling true variants. While this is generally not an issue when studying humans, it may become an issue when, for example, cells from inbred mice or rats are being used, for which SCcaller utilizes a default bias value typically observed in amplified human cells. Nevertheless, even in inbred animals SNPs are present, albeit at low frequency, and we have successfully used SCMDA for studying somatic mutation frequency in mice. When studying embryos, if heterozygous SNP information is not available, e.g., due to the lack of cells for bulk sequencing, one could pool sequencing data of multiple single embryo cells as surrogate for bulk total genomic DNA, but this requires modification of the data analysis pipeline, which is not covered in this protocol. SCcaller also assumes that the genome of a single cell is diploid, and it remains to be optimized and tested for cells of other ploidy levels (e.g., haploid, and tetraploid cells). Finally, our current pipeline does not detect genome structural variations, including large insertions, large deletions, inversions, translocations and retrotranspositions. This is because of an abundance of chimeric artifacts occurring during the MDA reaction. This may be resolved in future by applying long-read sequencing of single-cell amplicons and/or developing new computational software tools.
Experimental design
Overview
Here, we describe a complete protocol for using single-cell or single-nucleus whole-genome sequencing in discovering and analyzing somatic mutations in human and animal tissues. It includes four consecutive stages (Fig. 1): (i) single-cell or nucleus isolation (Steps 1–9 and Box 1); (ii) single-cell or nucleus WGA (Steps 10–45); (iii) library preparation and sequencing (Steps 46–80); and (iv) computational analyses, including sequence data alignment, variant calling and estimating mutation burden (Steps 81–87). To distinguish somatic and germline mutations, whole-genome sequencing of bulk DNA of the same subjects is also required (bulk DNA extraction is described in Box 2). Below, we describe the design of these stages in details.
BOX 1. Single-cell or nucleus isolation using FACS.
● TIMING 0.5–1 h for 96 cells or nuclei
FACS is an alternative method to the CellRaft system for single-cell or nucleus isolation. Below, we briefly describe its procedure.
Label 0.1 mL or 0.2 mL eight-strip PCR tubes (with cap strips) or 96-well PCR plate in a PCR workstation. All the PCR tubes or plates should be presterile and certified free of DNA, DNase and pyrogens.
Add 2.5 μL PBS into each tube or well with multichannel pipettes.
Quick spin the tubes or plates to make sure 2.5 μL PBS is at the bottom of each tube of well.
Keep the tubes with caps or plates with covers on ice until required.
-
Isolate single cells using FACS. All settings for cell isolation depends on FACS equipment and your experiment.
▲ CRITICAL STEP Please optimize the position between the collection tube or plate and nozzle to ensure that the target cell is deposited in the middle of the tube.
After sorting, quickly spin the tubes or plates to collect the isolated cells at the bottom.
-
Put the tubes or plates on dry ice immediately.
■ PAUSE POINT Transfer the tubes or plates containing single cells to −80 °C freezer and keep them at −80 °C until use (no longer than 1 year).
Proceed to Step 10 of the procedure for single-cell or nucleus WGA.
BOX 2. Bulk DNA extraction.
● TIMING 15 min
Multiple kits are available for bulk genome extraction. Here we describe our protocol according to the manufacturer’s instruction of Quick-DNA MiniPrep kit from ZYMO Research.
-
Suspend the target bulk cells (e.g., fibroblast, 0.5 × 106) in 100 μL of PBS.
▲ CRITICAL STEP The input cell number or the volume and type of suspension buffer varies depending on target cell or tissue types. For more details about cell number for different cell types, please see the instructions of the kit (https://files.zymoresearch.com/protocols/_d3024_d3025_quick-dna_miniprep_kit.pdf)
Add four volumes of genomic lysis buffer to every volume of cell solution, e.g., add 400 μL of genomic lysis buffer to 100 μL cell solution. Vortex for 5 s and keep the mixture at room temperature for 8 min.
Transfer the mixture to a Zymo-Spin IICR column in a collection tube. Centrifuge at 12,000g for 1 min, transfer the Zymo-Spin IICR column to a new collection tube and discard the flowthrough with the old collection tube.
Add 200 μL of DNA prewash buffer into the Zymo-Spin IICR column and centrifuge at 12,000g for 1 min.
Add 500 μL of g-DNA wash buffer to the Zymo-Spin IICR column. Centrifuge at 12,000g for 1 min.
Discard the flowthrough from the collection tube, put the Zymo-Spin IICR column back on the collection tube and centrifuge at 12,000g for 1 min to remove extra liquid from the Zymo-Spin IICR column.
-
Transfer the Zymo-Spin IICR column to a new 1.5 mL LoBind tube. Add 50 μL nuclease-free water in the center of the Zymo-Spin IICR Column. Incubate 5 min at room temperature and centrifuge at 12,000g for 1 min.
▲ CRITICAL STEP To improve DNA recovery, warm the aliquot of nuclease-free water or DNA elution buffer to 60–70 °C using a Thermomixer.
-
Quantify the eluted DNA as for amplicons, with Qubit (Steps 29–34) and run the gel electrophoresis to check DNA quality (Steps 35–39).
■ PAUSE POINT The eluted DNA can be safely stored at −20 °C or −80 °C for future use.
Single-cell or nucleus isolation (Steps 1–9)
The protocol starts with a single-cell or nucleus suspension. Methods of choice for creating a single-cell or nucleus suspension depend on the tissue type and its status; please refer to its corresponding protocols in the literature. To capture single cells or nuclei from these suspensions, we use a CellRaft system (or the CellRaft Air system, which is an automatic version of the CellRaft system and does not affect the protocol; Cell Microsystems). The key technology in the CellRaft systems is the CellRaft (CytoSort) array, which is composed of tens of thousands of magnetic micro ‘rafts’. Each raft can be released from the bulk array and transferred to a PCR tube or plate using a magnetic wand.
For adherent cells, e.g., human fibroblasts, we prepare a 4 mL suspension of ~5,000 cells and let the cells attach to the surface of a CytoSort array (200 × 200 μm, single reservoir) before capture. Rafts containing a single cell are identified under a microscope and transferred to a PCR tube with 2.5 μL phosphate-buffered saline (PBS). Unless immediately used, these are then frozen on dry ice and stored at −80 °C. For nonadherent cells, e.g., human lymphocytes or nuclei, we coat a CytoSort array with gelatin, poly-l-lysine or equivalents, before plating the cells or nuclei onto the array. This step helps the cells or nuclei adhere to the rafts. The capture rate of a CytoSort array is ~60%, i.e., up to 3,000 cells can be isolated from an input of 5,000 cells, according to the user manual from Cell Microsystem (https://cellmicrosystems.com/wp-content/uploads/2022/09/PD-015-rev04-CellRaft-Array-User-Manual.pdf).
Besides the CellRaft system, other methods can be applied to isolate single cells. We previously used hand-held capillaries44,45 and fluorescence-activated cell sorting (FACS)19,45. The advantage of the CellRaft is that users can better observe and transfer cells without substantial damage. Its limitation is that it takes more hands-on time than FACS and can only work with a more limited range of fluorescence markers, even with the upgraded CellRaft Air System. For cells exceeding the size of a raft (two options: 100 × 100 μm, or 200 × 200 μm), we recommend extracting nuclei from the cells for isolation.
Single-cell or single-nucleus WGA (Steps 10–45)
The protocol uses SCMDA to perform single-cell or single-nucleus WGA. This starts with a single-cell or nucleus deposited in 2.5 μL PBS in a PCR tube, which undergoes lysis followed by the denaturation of its DNA on ice with alkaline (Steps 13–15). The genome is then amplified with Φ29 DNA polymerase. Amplification primers are added before cell lysis to promote amplification efficiency. After SCMDA, amplicon products are subject to three QC steps: quantifying the yield with Qubit (Steps 29–34), analysis of fragment size with gel imaging (Steps 35–39) and assessment of amplification uniformity using a locus dropout (LDO) test (Steps 40–45), which we perform using quantitative PCR on eight loci chosen from its corresponding species-specific reference genome17,21,22,44. Of note, a limitation of SCMDA- and MDA-based amplification methods in general is that they do not work with formalin-fixed paraffin-embedded specimens, probably because the crosslinking of the fixative inhibits amplification.
Library preparation and sequencing (Steps 46–80)
We recommend targeting a sequencing depth across the whole genome of ~30× for each single cell and the bulk DNA sample (Box 2). As most next-generation sequencing platforms now generate high-quality sequencing reads, the choice of sequencing platform is mostly driven by cost considerations. We recommend using the Illumina NovaSeq 6000 platform with a 150 bp pair-end option and requesting 90–100 GB of data (i.e., 300–333 million read pairs) per single human cell. Although not all sequencing reads can be perfectly aligned to the reference genome, this typically yields 25–30× of whole-genome coverage with high mapping quality. For other species, the requirement of sequencing data should be adjusted according to the size of their reference genomes. Besides Illumina platforms, others have successfully applied the BGISEQ-500 platform with 100 bp paired-end reads27. The choice of library preparation method is dependent on the choice of sequencing platform. We did not observe substantial differences between PCR-based and PCR-free libraries. Here, we describe the use of NEBNext Ultra II FS DNA library prep kit for sequencing with Illumina NovaSeq, but other library preparation methods can be used.
Data analysis using SCcaller-pipeline (Steps 81–87)
In this protocol, we provide a highly integrated data processing pipeline, SCcaller-pipeline, which put together the 17 substeps from raw fastq files to somatic mutations and mutation burden into two commands (Fig. 3).
Fig. 3 ∣. Flowcharts of the SCcaller-pipeline.
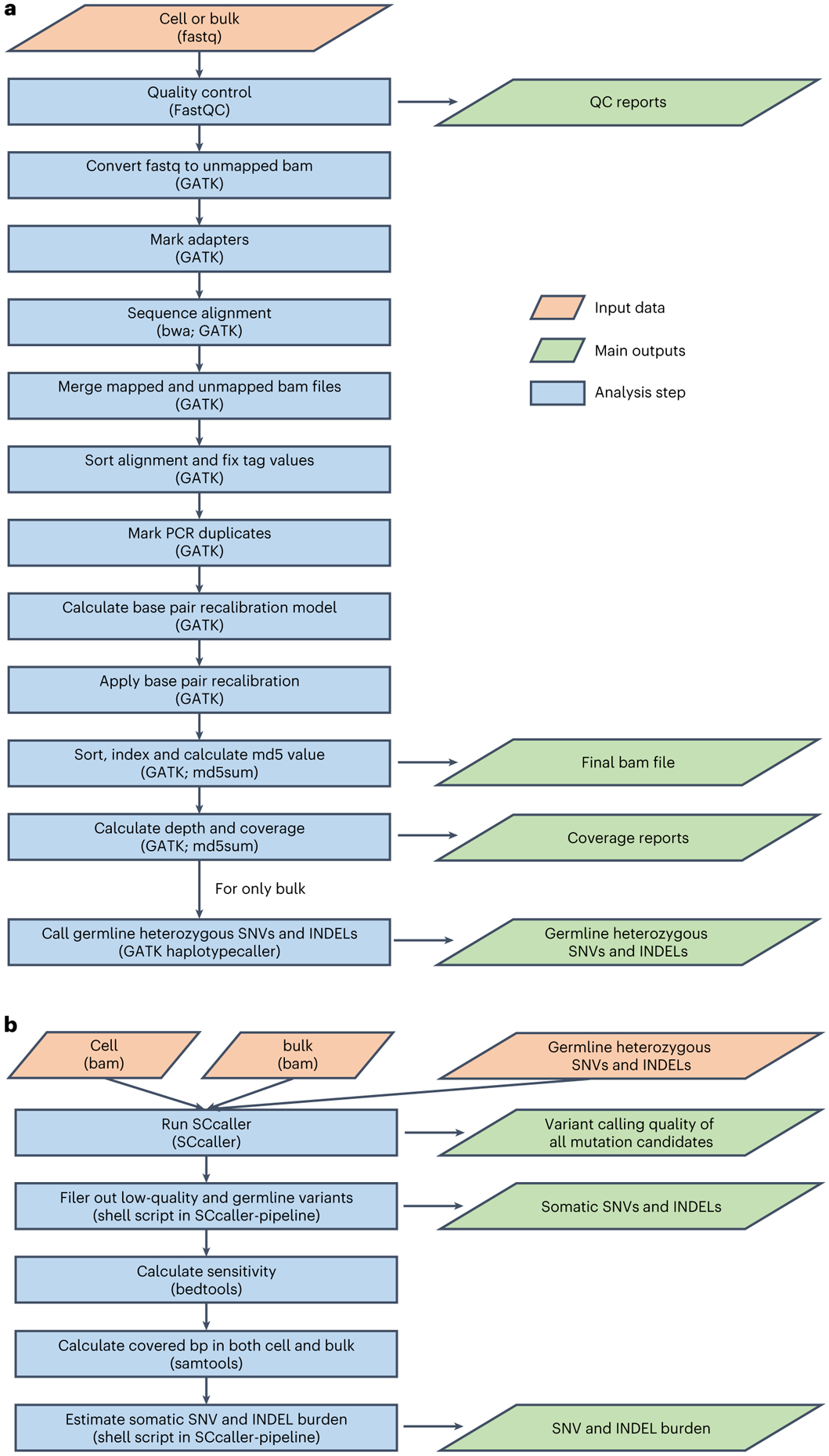
The pipeline includes two major stages (i.e., commands). a, QC, sequence alignment and germline variant calling. This command should be performed for single-cell sequencing data and bulk sequencing data separately. For single-cell sequencing data, it takes fastq files of a sample (either bulk or single cell) as input and outputs aligned bam file as well as reports of QC and genome coverage. For bulk sequencing data, in addition to the above, it outputs a list of germline heterozygous SNPs and INDELs. b, Somatic mutation calling and mutation burden estimation. This command takes bam files of a single cell and its corresponding bulk DNA, and the list of germline heterozygous SNPs and INDELs (all are outputs from a) as input, and outputs somatic mutations, their variant calling qualities, and estimated mutation burden (i.e., the number of mutations per cell after adjusting variant calling accuracy and genomic coverage).
QC, sequence alignment and germline variant calling (Steps 82–84).
These steps should be performed on single-cell and bulk sequencing data separately. For QC, we use FastQC on raw sequencing data (fastq files)46. For alignment to the reference genome, we use the GATK best practice ‘Data pre-processing for variant discovery’ workflow47. Of note, recalibrating base quality scores (RBQS) requires an input of one or more datasets of known germline variants, ideally reported in population-based studies, e.g., the 1000 Genomes Project for humans. No such dataset is available for most other species. To address this issue, as recommended by the GATK team, we suggest first skipping the RBQS stage, using the GATK HaplotypeCaller to call a draft set of germline variants from bulk DNA of all samples of the species and using the draft set to perform RBQS to generate the final aligned and recalibrated bam files of single cells and the bulk DNA (for more details, see ref. 22). Finally, germline SNVs and INDELs are called from the aligned and recalibrated bam file of the bulk DNA using HaplotypeCaller.
Variant calling and estimating mutation burden (Steps 85–87).
For calling somatic SNVs and INDELs from single cells, we use the SCcaller and call somatic mutations from genomic regions with at least 20× depth for both single cells and bulk17 (Fig. 4). Somatic variants of the single cells are called, with the following input files, which are output files from Steps 82–84: (i) bam file of the single cell, (ii) vcf file of its heterogenous germline variants and (iii) bam file of its corresponding bulk DNA. To estimate the mutation burden per single cell (termed ‘mutation frequency’ in our previous papers), we adjust the number of observed somatic mutations for variation in sequencing coverage and variant calling sensitivity. The latter is estimated by calling germline heterozygous SNVs and INDELs in single cells. As we and others shown previously, mutation burden increases with age15,18,48, and elevated mutation burden has also been associated with environmental factors (e.g., smoking)20 or genetic factors (e.g., BRCA1 or BRCA2 germline variants)23.
Fig. 4 ∣. Flowchart of the SCcaller algorithm.
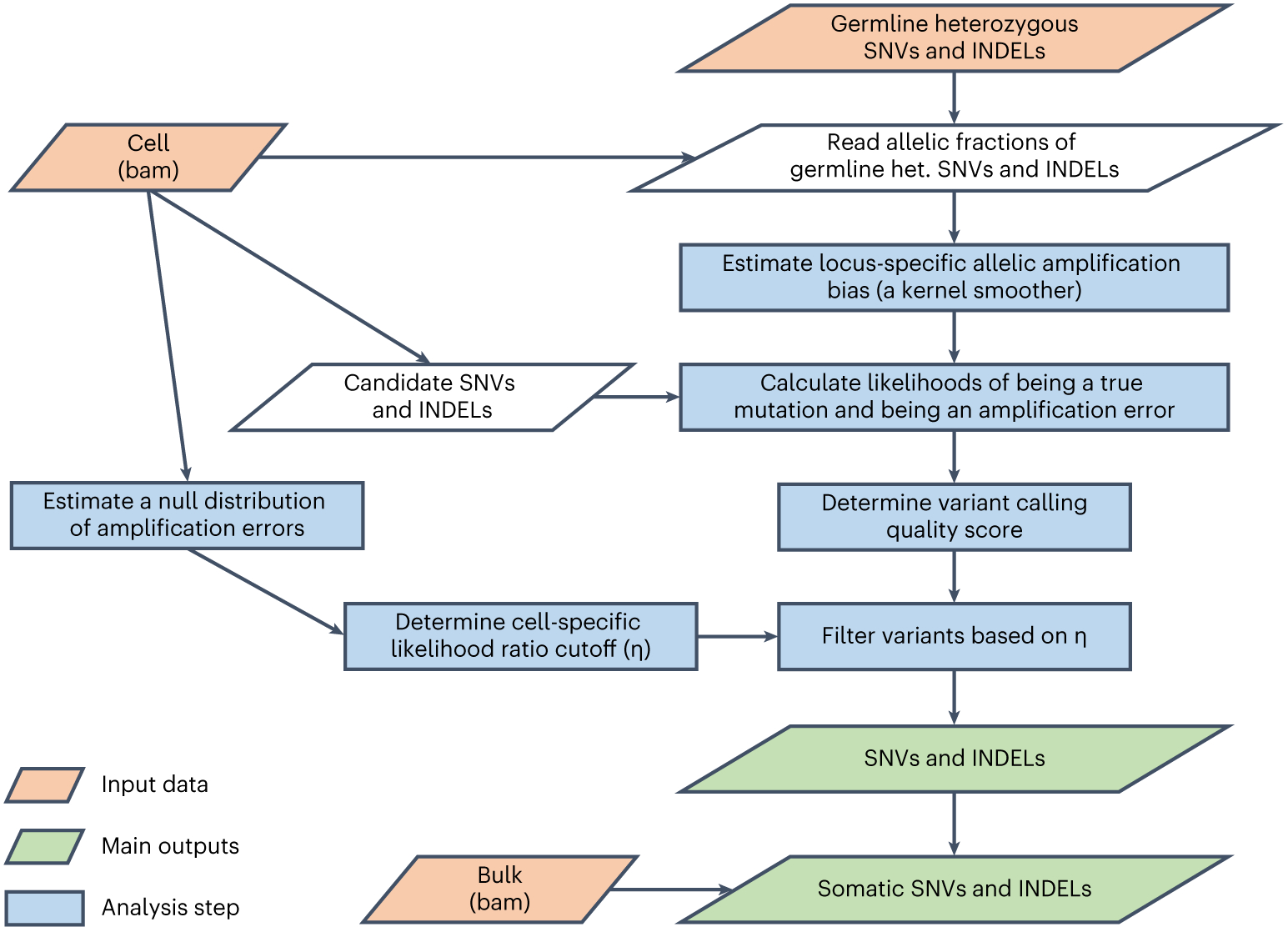
In brief, a heterozygous (het.) mutation should ideally present in 50% of sequencing reads of a diploid cell, but in real single-cell sequencing data, it often deviates from 50% substantially, because the two copies of chromosome are often not amplified to an equivalent amount. In single-cell sequencing data, SCcaller estimates such allelic amplification bias for every position in the genome from the allelic amplification bias observed for known germline heterozygous SNPs and INDELSs close to the position, and adjusts the bias in calling somatic variations: a true somatic mutation should be observed with a similar allelic fraction of sequencing reads as its neighboring germline heterozygous SNPs and INDELs (which may not be 50%). Please see ref. 17 for more descriptions.
Materials
Biological materials
-
Target cells or nuclei of interest in a single-cell or nucleus suspension. We have used this protocol on fibroblasts, lymphocytes, hepatocytes and epithelial cells of humans, and fibroblasts of several rodent species17-23.
▲ CAUTION Any experiments using human material must conform to relevant institutional and national regulations, and informed consent must be obtained. For the studies we present here, work was covered by Einstein-Montefiore Institutional Review Board, Lonza Walkersville Inc., Kerafast Inc., AllCells, and STEMCELL Technologies.
Reagents
Single-cell or nucleus isolation
PBS, pH 7.4, 1× (Gibco, cat. no. 10010-023)
FBS (Thermo Fisher Scientific, cat. no. 26140079)
Penicillin–streptomycin, 10,000 U/mL (Thermo Fisher Scientific, cat. no. 15140-122)
Examples for tissue culturing or cell isolation reagents depending on cell type (Table 2)
Table 2 ∣.
Examples of media and coatings for single-cell isolation with CellRaft
| Cell type | Culture mediuma | Manufacturer, cat. no. |
Coating | Recommended concentration |
Manufacturer, cat. no. |
|---|---|---|---|---|---|
| Fibroblast | FGM | Lonza, CC4126 | n.a | n.a | n.a |
| Hepatocyte | Hepatocyte plating medium | Lonza, MP-100 | Collagen type I | 10 μg/mL in PBS | Sigma, C3867 |
| Lymphocyte | RPMI 1640 | Gibco, 11875093 | Gelatin type B | 2% solution in H2O | Sigma, G1393 |
| Nuclei (e.g., neuronal) | PBS + 5% FBS | See reagent list | Poly-l-lysine | 0.01% solution in H2O | Sigma, P4707 |
We list the basic medium here. Different supplements may be used depending on culture purposes or conditions.
SCMDA
-
HCl, 6 N (Thermo Fisher Scientific, cat. no. 24308)
▲ CAUTION HCl, 6 N can cause skin burns and eye damage. Handle using appropriate protective gloves, clothing and eye protection. When breaking open a glass ampule, protect fingers and consider using an ampule breaker.
-
KOH, 2 N (G-Biosciences, cat. no. R008)
▲ CAUTION KOH, 2 N can cause skin burns and eye damage. Handle using appropriate protective gloves, clothing, and eye protection.
-
DTT, 1 M (Thermo Fisher Scientific, cat. no. P2325)
▲ CAUTION DTT can cause skin and eye irritation. Handle using appropriate protective gloves, clothing and eye protection.
Ethylenediaminetetraacetic acid, pH 8.0, 0.5 M (Sigma, cat. no. 03690)
Tris–HCl, pH 7.5, 1 M (Fisher Scientific, cat. no. BP1757)
Nuclease-free water (Qiagen, cat. no. 129114)
Exo-resistant random primer (Thermo Fisher Scientific, cat. no. SO181)
Amplification reaction buffer (Qiagen, cat. no. 150035)
DNA polymerase Φ29 (Qiagen, cat. no. 150035)
DNA purification
AMPure beads reagent XP (Beckman Coulter Life Sciences, cat. no. A63881)
Ethanol 100% (DeconTM Labs, cat. no. 04-355-450)
Bulk DNA extraction
Quick-DNA MiniPrep kit (ZYMO Research, cat. no. D3024)
-
β-Mercaptoethanol (BME; Sigma, cat. no. M6250)
▲ CAUTION BME is toxic and may cause damage to organs if swallowed or inhaled, and is fatal in contact with skin. Wear protective gloves, clothing, and eye and face protection.
DNA quantification
Qubit dsDNA HS assay kit (Thermo Fisher Scientific, cat. no. Q32854)
Gel electrophoresis
Agarose (Sigma, cat. no. A6013)
TAE buffer, 50× (Thermo Fisher Scientific, cat. no. B49)
TrackIt 1 Kb Plus DNA ladder and loading buffer (Thermo Fisher Scientific, cat. no. 10488085)
GelRed Nucleic Acid Stain, 10,000× (Thomas Scientific, cat. no. C755G17)
LDO
Fast SYBR Green master mix (Thermo Fisher Scientific, cat. no. 4385612)
LDO primers (IDT, Table 3)
Table 3 ∣.
LDO primers for human sample
| Chr | Forward | Reverse |
|---|---|---|
| 1p | TTTGATGGAGAAATCCGAGG | CTGACTCGGAGAGCAGGAC |
| 1q | GGTAGGATGATTCTAGAATGCCA | GCCCAAATTGGCTTCTTTTT |
| 4p | AACTGAATGGCAGTGAAAACA | CCCTAGCCTGTCATTGCTG |
| 4q | TATAGCCCACCTGACCCAAG | CTGTCATCACTGTCTACTTCCTCTC |
| 10p | GTTCTGCTGCCTCTACACAGG | ATCCTTCTGTGAACTCTCAAATCC |
| 10q | CTTCCTGACCTGTTTGCAGT | CTTCAGTGCACAGAATGCAG |
| 12p | CCACACACTCTGGTTTTATAAAGC | TTTTTCTCCTGCATCCATGG |
| 12q | TCCTCATTGTTGGGGATGAT | TGGCCAAAAATAGAAGCCAT |
Library preparation
NEBNext Ultra II FS DNA Library Prep kit (NEB, cat. no. E7805)
Multiplex Oligos for Illumina, index primers set 1 (NEB, cat. no. E7335; more options available from NEB)
Bioanalyzer
Agilent DNA high-sensitivity kit (Agilent, cat. no. 5067-4626)
Equipment
Single-cell or nucleus isolation
CO2 tissue culture incubator (Thermo Fisher Scientific, cat. no. 13-998-125)
Class II, type A2 biosafety cabinets (Thermo Fisher Scientific, cat. no. 13-261-222)
Centrifuge 5910R (Eppendorf, cat. no. 2231000770)
Centrifuge 5425 (Eppendorf, cat. no. 5405000107)
MyFuge 12 tabletop centrifuge (Benmark Scientific, cat. no. C1012)
CellRaft System (Cell Microsystem, cat. no. CRK) or CellRaft Air System (Cell Microsystem, cat. no. AIR-1)
CellRaft (CytoSort) Array (Cell Microsystem, cat. no. CS-200S-5)
Inverted microscope (Olympus, cat. no. CKX53F3)
Upright microscope (VWR, cat. no. 10836-004)
Cellometer Auto T4 (Nexcelom, cat. no. Auto T4)
Disposable hemacytometer (Nexcelom, cat. no. CP2)
Vortex mixer (Fisher Scientific, cat. no. S38786)
Easypet 3 (Eppendorf, cat. no. 4430000018)
15 mL and 50 mL conical tubes (Thermo Fisher Scientific, cat. nos. 339651 and 339653)
5 mL, 10 mL, 25 mL and 50 mL serological pipettes (Midsci, cat. nos. PR-SERO-5, PR-SERO-10, PR-SERO-25 and PR-SERO-50)
2 μL, 10 μL, 20 μL, 200 μL and 1,000 μL single-channel pipettes (Rainin, cat. nos. 17014413, 17014409, 17014412, 17014411 and 17014407)
10 μL, 20 μL, 200 μL and 1,250 μL filter tips (Thomas Scientific, cat. nos. CX13210, CX13211, CX13212 and CX13214)
-
Eight-tube strips, 0.2 mL (VWR, cat. nos. 89126-686)
▲ CRITICAL Tubes for single-cell isolation should be DNase, RNase, DNA, RNA and pyrogen-free and presterile.
SCMDA
UV PCR workstation (Airclean System, cat. no. 36-101-8894)
CoolSafe cooling chamber for 0.2 mL PCR tubes (USA Scientific, cat. no. 9124-0100)
-
1.5 mL DNA LoBind tubes (Eppendorf, cat. no. 0030108418)
▲ CRITICAL Tubes are certified PCR clean purity grade: free of human DNA, DNase, RNase and PCR inhibitors.
SimpliAmp Thermal Cycler (Thermo Fisher Scientific, cat. no. A24811)
DNA purification
NEBNext magnetic separation rack (NEB, cat. no. S1515S)
Eight-channel pipette, 10–100 μL (optional, USA Scientific, cat. no. 7108-1100)
Reservoir, 10 mL, sterile (optional, USA Scientific, cat. no. 1930-2100)
Bulk DNA extraction
Thermomixer (Eppendorf, cat. no. 5384000020)
DNA quantification
Gel electrophoresis
Erlenmeyer flask, 250 mL (Thermo Fisher Scientific, cat. no. FB500250)
Countertop microwave (Sumsung, cat. no. 8578027)
Owl EasyCast B2 Mini Gel Electrophoresis System (Thermo Fisher Scientific, cat. no. B2)
Smart Doc Gel Imaging System F (Thomas Scientific, cat. no. 1177×50)
Milli-Q water purification system (Millipore, cat. no. ZMQP60001)
LDO
QuantStudio 3 real-time PCR system (Thermo Fisher Scientific, cat. no. A28566)
MicroAmp fast optical 96-well reaction plate with barcode, 0.1 mL (Thermo Fisher Scientific, cat. no. 4346906)
MicroAmp optical adhesive film (Thermo Fisher Scientific, cat. no. 4311971)
Eight-channel pipette, 0.5–10 μL (Eppendorf, cat. no. 3125000010)
Bioanalyzer
2100 Bioanalyzer instrument (Agilent, cat. no. G2939BA)
Data analysis: hardware
-
A workstation or computer cluster running a Linux system
▲ CRITICAL We recommend 8 central processing unit cores and 64 GB random-access memory for sequence alignment and variant calling. For disk space, 15 GB is required for the reference files, and another 200 GB is required per sample for sequence alignment of single cells or bulk DNA.
Data analysis: software
▲ CAUTION When updating software tools, cross-compare results to older versions using test data.
bwa v0.7.17 (https://bio-bwa.sourceforge.net/)
bedtools v2.30.0 (https://bedtools.readthedocs.io/en/latest/)
FastQC v0.11.9 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/)
GATK v4.3.0.0 (https://gatk.broadinstitute.org/hc/en-us)
Java v1.8 (https://www.java.com/en/)
python v2.7.18 (https://www.python.org/)
samtools v1.9 (http://www.htslib.org/)
SCcaller v2.0.0 (https://github.com/biosinodx/SCcaller)
Data analysis: pipeline
SCcaller-pipeline v1.0.0 (https://github.com/XiaoDongLab/SCcaller-pipeline)
Data analysis: reference files
▲ CRITICAL Here, we list only human references files (version GRCh38). For other species, look for reference files from the National Center for Biotechnology Information or ENSEMBL database.
▲ CRITICAL All the reference files can be downloaded from the cloud storage of the Broad Institute at: https://console.cloud.google.com/storage/browser/genomics-public-data/references/hg38/v0
Reference genome: Homo_sapiens_assembly38.fasta
dbSNP dataset: Homo_sapiens_assembly38.dbsnp138.vcf
- INDEL datasets:
- Homo_sapiens_assembly38.known_indels.vcf
- Mills_and_1000G_gold_standard.indels.hg38.vcf
Reagent setup
Coating reagent for CytoSort array
If the stock concentration of the coating reagent is the same as the recommended concentration in the working solution as shown in Table 2, please use the coating reagent directly. Otherwise, dilute the coating reagents in the corresponding liquid, as indicated in Table 2, to the recommended concentration. The diluted working solution should be prepared fresh and used immediately.
Lysis buffer from Quick-DNA MiniPrep kit
Add BME to the genomic lysis buffer to a final dilution of 0.5% (vol/vol), i.e., 250 μL per 50 mL. The BME genomic lysis buffer can be stored at room temperature (18–25 °C) until the expiration date of the BME or the genomic lysis buffer, whichever occurs first.
Fresh 80% (vol/vol) ethanol for bead purification
Dilute 100% (vol/vol) ethanol with nuclease-free water to make 80% (vol/vol) ethanol. Prepare fresh 80% (vol/vol) ethanol for each batch of experiment. The fresh 80% (vol/vol) ethanol should be prepared at room temperature and used immediately.
LDO primers
Reconstitute the oligonucleotides (Table 3) to 100 μM stock solutions in water and make 10 μM working solutions with water. The 100 μM stock solutions can be stored at −80 °C for long-term storage. The 10 μM working solutions can be stored at −20 °C and used in a year (recommended).
Agarose gel
Add 1 g agarose into 100 mL purified water from a Millipore system to a final 1% (wt/vol) gel solution. Microwave for 2–3 min until the agarose is completely dissolved. When the agarose gel solution cools to ~50 °C, add 10 μL of GelRed (10,000×) to a final 1× working solution. Pour the agarose gel into a gel tray with the well comb in place. The agarose gel with GelRed should be use immediately. However, any remaining unused agarose gel can be saved in a zip-sealing bag with 1× TAE buffer (~1 mL) and stored in the dark at 4 °C.
▲ CAUTION After dissolving in the microwaves, the agarose solution is hot. Be careful to take out the flask after microwave and avoid eruptive boiling when stir the flask.
▲ CRITICAL Pour agarose gels slowly to avoid bubbles.
1× TAE buffer
Dilute the 50× TAE buffer to 1× TAE buffer with purified water. 1× TAE buffer should be used immediately. However, unused 1× TAE buffer can be stored at room temperature for several weeks. If the buffer becomes cloudy, discontinue using it.
Gel–dye mix for bioanalyzer
Allow high-sensitivity DNA dye to concentrate and the high-sensitivity DNA gel matrix to warm to room temperature for 30 min to 1 h. Add 15 μL of high-sensitivity DNA dye concentrate to a high-sensitivity DNA gel matrix vial. Vortex the solution well and spin down. Transfer the mixture to a spin filter. Centrifuge at 2,250g for 10 min. Protect the solution from light. Store at 4 °C. Use the prepared gel–dye mix within 6 weeks.
▲ CRITICAL It is very important to balance the centrifuge when spinning gel–dye mix in the filter tube. Usually, we add 380 μL of water in a 1.5 mL Eppendorf tube as a balance.
SCMDA lysis buffer
Prepare the SCMDA lysis buffer as detailed in the table below.
| Reagent | Volume (mL) | Final concentration (mM) |
|---|---|---|
| KOH (2 M) | 2 | 400 |
| DTT (1 M) | 1 | 100 |
| Ethylenediaminetetraacetic acid (0.5 M) | 0.2 | 10 |
| Nuclease-free water | 6.8 | – |
| Total volume | 10 | – |
SCMDA lysis buffer can be aliquoted in small volumes, e.g., 100 μL, depending on usage, and stored at −20 °C for 3 months. Avoid multiple freeze–thaw cycles, which reduce its efficiency.
SCMDA stop buffer
Prepare SCMDA stop buffer as detailed in the table below.
| Reagent | Volume (mL) | Final concentration (mM) |
|---|---|---|
| HCl (2 M) | 2 | 400 |
| Tris–HCl (1 M) | 6 | 600 |
| Nuclease-free water | 2 | – |
| Total volume | 10 | – |
SCMDA stop buffer can be aliquoted in small volumes, e.g., 100 μL, depending on usage, and stored at −20 °C for 3 months. Avoid multiple freeze–thaw cycles, which reduce its efficiency.
Exo-resistant random primers
Aliquot the random primers in small volumes depending on usage and store it at −20 °C until the expiration date on the original tube of random primers. Avoid multiple freeze–thaw cycles, which reduce its efficiency.
0.1× TE buffer
Dilute the 1× TE buffer to 0.1× TE buffer with nuclease-free water. The 1× TE buffer is provided from the NEBNext Ultra II FS DNA library prep kit. The 0.1× TE buffer can be store at −20 °C until the expiration date of the 1× TE buffer.
Equipment setup
Data analysis: pipeline setup
This pipeline requires corresponding versions of software tools and reference genome files that can be downloaded directly. Load all required software tools in the Linux environment before processing the pipeline file. The current pipeline files are provided to utilize the Sun Grid Engine (SGE) job scheduler of a computer cluster. To use under other job schedulers, e.g., Portable Batch System or slurm, modify the pipeline files for their specific options.
Procedure
Single-cell or nucleus isolation
● TIMING 2–5 h for 96 cells or nuclei
▲ CRITICAL Here, we demonstrate the usage of the CellRaft system to capture single cells for depositing in tubes. These steps should also be followed if using the CellRaft Air system. In Box 1 we describe the use of FACS as an alternative isolation method. Other methods can be applied as mentioned above.
Following the manufacturer’s instruction, wet a CytoSort array: add 3 mL PBS or tissue-culture media, which may vary depending on cell types, to the array for 3 min at room temperature.
Aspirate PBS or media.
Repeat Steps 1–2 a further two times.
- Apply coatings to the CytoSort array.
- For nonadherent cells or nuclei, apply cell culture coatings to the array (Table 2) and incubate at 37 °C for 1 h.
-
For adherent cells, follow the same procedure as used for seeding cells in a cell culture dish.▲ CRITICAL STEP The array has been treated for tissue culture by the manufacturer and can be used for adherent cells directly without coatings.
-
Prepare 5,000 cells in 4 mL PBS with 5% FBS (vol/vol) or tissue-culture media, which may vary depending on cell types, mix thoroughly and transfer the solution into the CytoSort array.
▲ CRITICAL STEP This is for a 200 μm single reservoir array of 10,000 microwells. Adjust volume and cell number if using a different CytoSort array.
-
Incubate the array to allow cells to settle and adhere. The duration of this step can vary from 10 min to several hours depending on cell type and culture conditions. For example, fibroblasts and plateable hepatocytes can settle and adhere on the array at 37 °C in 3 h. For nonadherent cells or nuclei, spin the array at 150g for 30 s twice and check with a microscope to confirm that the cells or nuclei have attached to the array.
◆ TROUBLESHOOTING
-
To prepare tubes for single cell or nucleus isolation, add 2.5 μL of PBS to each 0.2 mL PCR tube.
▲ CRITICAL STEP The PCR tubes must be presterile and certified free of DNA, DNase and pyrogens. Make sure that the 2.5 μL PBS is at the bottom of each collection tube.
Aspirate the old PBS or media added in the Step 5. To remove dead and floating cells, floating nuclei and cell-free DNA before picking single cells, add 1 ml of fresh and warm PBS with 5% (vol/vol) FBS or tissue-culture media to wash the array and then aspirate the liquid. Repeat the washing process two additional times. After that, add 4 mL fresh and warm PBS with 5% (vol/vol) FBS or tissue-culture media into the array.
-
Following the manufacturer’s instructions for the CellRaft system (or CellRaft Air system), deposit single cells into the PCR tubes containing 2.5 μL PBS and check the presence of a single raft in the PCR tubes with an upright microscope. Once confirmed, put the PCR tubes on dry ice immediately.
▲ CAUTION Dry ice is much colder than regular ice, and can burn the skin similar to frostbite. You should wear insulated gloves when handling it.
■ PAUSE POINT Single cells in the PCR tubes can be stored at −80 °C until use (no longer than 1 year).
Single-cell or nucleus WGA
● TIMING 2 h
-
10.
Take out the aliquoted SCMDA lysis buffer and stop buffer (see ‘Reagent setup’) from the −20 °C freezer and keep them on ice.
-
11.
Thaw exo-resistant random primers on ice.
-
12.
Take out the PCR tubes containing single cells from the −80 °C freezer, perform a quick spin and place them on ice. In addition, add 1 ng of bulk DNA (sBox 2) diluted in 2.5 μL PBS into a PCR tube as a positive control and add 2.5 μL PBS into another PCR tube as a negative control.
-
13.
After the random primer is completely thawed, quickly vortex, spin down and add 1 μL into each single cell sample.
▲ CRITICAL STEP Do not pipette up and down the mixture of the primer and single cell sample.
-
14.
Add 3 μL of completed thawed and well mixed SCMDA lysis buffer along the wall of PCR tube of each single cell sample, flick after adding the buffer for all the samples in the batch, perform a quick spin and leave the tubes on ice for 10 min for cell lysis and DNA denaturation.
-
15.
Add 3 μL of completed thawed and well mixed SCMDA stop buffer to neutralize the reaction of cell lysis and DNA denaturation, flick, quick spin, and keep it on ice.
▲ CRITICAL STEP To ensure that cell lysis is stopped right after 10 min, add SCMDA stop Buffer directly into the reaction mixture. If processing multiple samples simultaneously, use a multichannel pipette for this step.
-
16.Prepare the SCMDA amplification mix for each cell on ice as follows.
Reagent Volume (μL) Amplification reaction buffer 30 DNA polymerase Φ29 2 -
17.
Add 32 μL of the amplification mixture into each reaction, flick and spin down.
-
18.Run the SCMDA reaction according to the following cycling conditions with the heated lid set to 70 °C:
Cycle no. Temperature (°C) Time (min) 1 30 90 2 65 3 3 4 ∞
Bead purification
● TIMING 30 min
-
19.
Place AMPure beads at room temperature for at least 30 min before use. Vortex AMPure beads to fully resuspend.
-
20.
Add 75.6 μL of resuspended AMPure beads (1.8× of sample volume) into 42 μL of SCMDA product in the PCR tube. Mix well by pipetting up and down at least ten times.
-
21.
Incubate samples on the bench at room temperature for at least 5 min.
-
22.
Place the PCR tube on an appropriate magnetic stand to allow the beads to settle to the magnet and separate from the supernatant.
-
23.
After 5 min or when the solution is clear, carefully remove and discard the clear supernatant.
▲ CRITICAL STEP Be careful not to disturb the beads when remove the supernatant. Substantial bead loss will result in reduced yield.
◆ TROUBLESHOOTING
-
24.
Keep the PCR tube on the magnetic stand, add 200 μL of 80% (vol/vol) freshly prepared ethanol, and incubate at room temperature for 30 s. Carefully remove and discard the ethanol supernatant.
▲ CRITICAL STEP To avoid moving the beads, add the 80% (vol/vol) ethanol along the tube wall of the opposite side to the beads. Be careful not to discard the beads.
-
25.
Repeat Step 24. Then, remove the remaining liquid with a p10 or p20 pipette.
▲ CRITICAL STEP Make sure that all visible liquid is removed after the second wash.
-
26.
Air dry the beads while the tube is on the magnetic stand with its cap open.
▲ CRITICAL STEP Be careful to not over-dry the beads, which may cause lower recovery of product.
-
27.
Remove the PCR tube from the magnetic stand. Add 32 μL of nuclease-free water to elute SCMDA product from the beads. Mix thoroughly by pipetting ten times to resuspend the beads. Incubate at room temperature for 5 min.
-
28.
Place the tube on the magnetic stand. After 5 min, or when the solution is clear, transfer 30 μL of supernatant to a new 1.5 mL DNA LoBind tubes. This is the final purified SCMDA product (called amplicons).
■ PAUSE POINT Amplicons can be safely stored at −20 °C until further processing (no longer than 1 year). For long term storage (longer than a year), store the amplicons at −80 °C.
DNA quantification
● TIMING 20 min
-
29.
Dilute 1 μL of SCMDA amplicon with 19 μL of nuclease-free water for 1:20 dilution and mix thoroughly.
-
30.
Prepare the Qubit working solution by diluting the Qubit dsDNA HS reagent 1:200 in Qubit dsDNA HS buffer, e.g., add 1 μL of concentrated HS reagent into 199 μL of HS buffer.
▲ CRITICAL STEP Place the HS reagent at room temperature to thaw completely before use and protect it from light.
-
31.
Add 199 μL of Qubit working solution and 1 μL of diluted SCMDA amplicon to each individual assay tube.
▲ CRITICAL STEP Mix the solution thoroughly by vortexing and be careful not to create bubbles.
-
32.
Add 190 μL of Qubit working solution to each of the tubes used for the standards. Add 10 μL of each of the standard samples provided in the Qubit dsDNA HS assay kit (Standard #1 and #2) into the tubes respectively.
▲ CRITICAL STEP Mix the solution thoroughly by vortexing and be careful not to create bubbles.
-
33.
Incubate all the tubes at room temperature for 2 min. Protect samples from the light.
-
34.
Read the concentrations of the standards and the samples following on-screen instructions from the Qubit 4 Fluorometer.
◆ TROUBLESHOOTING
Agarose gel electrophoresis
● TIMING 45 min
-
35.
Add 0.5 μL of loading buffer to 4.5 μL of the diluted SCMDA amplicons (from Step 37). If the loading buffer is in not 10×, adjust volumes accordingly.
-
36.
Place the agarose gel into the electrophoresis unit and add 1× TAE buffer until the gel is covered.
-
37.
Carefully load the DNA ladder and samples into wells of the gel.
-
38.
Run the gel at 100 V for 40 min.
-
39.
Visualize and image gel picture.
◆ TROUBLESHOOTING
LDO test
● TIMING 1 h
▲ CRITICAL LDO and agarose gel electrophoresis can be performed simultaneously.
-
40.
Dilute the SCMDA amplicon (from Step 28 or 29) and unamplified genomic DNA (as positive control, from Box 2) to 1 ng/μL in 20 μL per sample. Prepare 20 μL nuclease-free water as a negative control.
-
41.
Prepare a 96-well plate and the sufficient PCR mixture for each well (see table below). In our assay, we test eight loci on four different chromosomes (Table 3).
▲ CRITICAL STEP In the LDO reaction, primers are a mixture of forward primer and reverse primer for each target locus. The final concentration of each primer should be 1 μM.Reagent Volume per sample (μL) Fast SYBR Green master mix 5 Locus-specific primers (1 μM) 1 Nuclease-free water 2 SCMDA sample (1 ng/μL) or genomic DNA (1 ng/μL) or H2O 2 Total volume 10 -
42.
Seal the plate and spin down using 280 g for 1 min.
-
43.Run the LDO qPCR according to the following cycling conditions with fast mode.
Cycle no. Temperature (°C) Time (s) 1 95 20 2–41 (40 cycles) 95 3 60 30 -
44.Calculate the relative uniformity values (RUVs) for the eight loci of each amplicon. RUVs are calculated as,
where denotes the value of the locus in cell , and denotes the value of the same locus in the unamplified bulk DNA. -
45.
Select amplicons based on the calculated RUVs. If its RUVs of six of the eight loci are between 0.25 and 4, the amplicon is considered of acceptable quality, and is selected for library construction.
◆ TROUBLESHOOTING
Library preparation
● TIMING 4h
▲ CRITICAL Multiple sequencing library preparation methods can be chosen even for the same sequencing platform. We chose the NEBNext Ultra II FS DNA library prep kit as an example for the Illumina NovaSeq Platform. This library prep kit was designed for 100–500 ng purified genomic DNA with a starting volume of 26 μL. We have modified several steps for SCMDA amplicons and bulk genomic DNA (for bulk DNA extraction, see Box 2) as described below.
-
46.
Prepare 400 ng of SCMDA amplicon per cell, which is eluted in Step 28 and has passed the LDO test (Step 45), in a final volume of 13 μL 1× TE buffer provided in the library prep kit in a 0.2 mL PCR tube. Prepare bulk genomic DNA in the same way.
-
47.For each library preparation, add the NEBNext Ultra II FS reaction buffer for fragmentation/end prep and enzyme mixture into a same 0.2 mL PCR tube containing diluted SCMDA amplicon or genomic DNA as follows. All the mixture in a 0.2 mL PCR tube is called FS reaction mixture.
Reagent Volume (μL) SCMDA sample or genomic DNA 13 (~400 ng) NEBNext Ultra II FS reaction buffer 3.5 NEBNext Ultra II FS enzyme mix 1 Total volume 17.5 ▲ CRITICAL STEP Ensure that the Ultra II FS reaction buffer is completely thawed. Vortex Ultra II FS enzyme mix before use.
■ PAUSE POINT If needed, samples can be stored at −20 °C; however, this may result in ~20% loss in yield (see instruction manual from NEB).
-
48.Vortex the tube with the FS reaction mixture for 5 s, quickly spin down, keep in a thermocycler, and run the following program with the heated lid set to 75 °C.
Cycle no. Temperature (°C) Time (min) 1 37 12 2 65 30 3 4 ∞ ▲ CRITICAL STEP Incubation time may vary depending on the targeted fragment sizes. Please refer to the instruction of NEBNext Ultra II FS DNA library prep kit (https://www.neb.com/protocols/2017/10/25/protocol-for-fs-dna-library-prep-kit-e7805-e6177-with-inputs-greater-than-or-equal-to-100-ng). The targeted fragment size is determined based on sequencing read length.
-
49.Take out the PCR tube from the thermocycler and place it on ice and add the following components into the FS reaction mixture, pipetting the entire volume up and down to mix thoroughly.
Reagent Volume (μL) FS reaction mixture 17.5 NEBNext Ultra II ligation master mix 15 NEBNext ligation enhancer 0.5 NEBNext adaptor for Illuminaa 1.25 Total volume 34.25 aThe adaptor is provided in a separate kit (NEB, Multiplex Oligos for Illumina). -
50.
Spin down quickly and incubate at 20 °C for 15 min in a thermocycler with its heated lid off.
-
51.
Take out the PCR tube from the thermocycle, keep it on ice and add 1.5 μL of USER enzyme, which is contained in the same kit as the adaptor (e.g., NEB, Multiplex Oligos for Illumina).
-
52.
Mix well and incubate at 37 °C for 15 min in a thermocycler with its heated lid set to 50 °C.
■ PAUSE POINT Samples can be stored overnight at −20 °C.
-
53.
Keep the PCR tube at room temperature and add 14.3 μL of 0.1× TE buffer to bring the volume of the reaction up to 50 μL.
-
54.
Vortex AMPure XP beads to resuspend and add 13 μL (~0.25× sample volume) of the resuspended beads to the above solution. Mix well by pipetting.
▲ CRITICAL STEP The volume of AMPure XP beads provided here is for a library of 350 bp insert size. For libraries with different insert size, the appropriate volumes of beads are different. See NEBNext Ultra II FS DNA library prep kit for a recommendation.
-
55.
Incubate at room temperature for 5 min.
-
56.
Place the PCR tube on a magnetic stand to separate beads from the supernatant.
-
57.
Wait for 5 min or until the solution is clear, and then transfer the supernatant (~63 μL) containing DNA to a new tube without disturbing beads. Discard the beads.
▲ CRITICAL STEP Be careful to keep the supernatant and discard the beads, which is the opposite operation to the previous bead purification for SCMDA product.
-
58.
Add 5 μL resuspended beads to the supernatant and mix well.
-
59.
Incubate at room temperature for 5 min.
-
60.
Place the tube on the magnetic stand to separate the beads from the supernatant.
-
61.
Remove and discard the supernatant after 5 min or when the solution is clear.
-
62.
Repeat Steps 24–26.
-
63.
Remove the PCR tube from the magnetic stand. Add 8.5 μL of 0.1× TE buffer to elute the beads. Mix thoroughly by pipetting ten times to resuspend the beads. Incubate at room temperature for 5 min.
-
64.
Place the tube on the magnetic stand. When the solution is clear, transfer 7.5 μL of the supernatant to a new PCR tube.
■ PAUSE POINT Samples can be safely stored at −20 °C.
-
65.Prepare the PCR enrichment mix by combining the following components:
Reagent Volume (μL) Adaptor-ligated DNA fragments 7.5 NEBNext Ultra II Q5 master mix 12.5 Index Primer/i7 Primer 2.5 Universal PCR Primer/i5 Primer 2.5 Total volume 25 ▲ CRITICAL STEP Here, we use single index primers. The i5 primer is a universal PCR primer without an index; the i7 primer provides an index to distinguish samples. Label each sample with different i7 index and record it carefully. To use with dual index primers, refer to the NEB kit.
-
66.Place the tube with PCR enrichment mix on a thermocycler and run the following PCR program with the heated lid set to 105 °C.
Cycle step Temperature (°C) Time (s) Cycles Initial denaturation 98 30 1 Denaturation 98 10 5 Annealing/extension 65 75 Final extension 65 5 min 1 Hold 4 ∞ 1 ▲ CRITICAL STEP Perform the minimum number of PCR cycles as required for a sufficient yield to minimize PCR duplication. Preparing a library using the NEB kit requires a minimum of three PCR cycles to add the complete adaptor sequences. We typically add another two cycles for library enrichment. Refer to the NEB kit for the recommendation for cycle numbers with a different amount of input DNA.
-
67.
Add 16.3 μL (~0.65× of sample volume) resuspended beads to the PCR reaction and mix well.
-
68.
Repeat Steps 21–26.
-
69.
Remove the tube from the magnetic stand. Elute the DNA library from beads by adding 15 μL of 0.1× TE buffer.
-
70.
Mix well by pipetting. Incubate at room temperature for 5 min.
-
71.
Place the tube on the magnetic stand. After 5 min or when the solution is clear, transfer 14 μL of the supernatant to a new 1.5 mL DNA LoBind tube. This is the final library.
■ PAUSE POINT Samples can be safely stored at −20 °C.
Library quantification
● TIMING 20 min
-
72.
Repeat Steps 29–34.
◆ TROUBLESHOOTING
Assess library quality and size distribution on a Bioanalyzer
● TIMING 1 h
-
73.
Dilute libraries to the range of 1 ng/μL to 5 ng/μL in 2 μL.
-
74.
Load 1 μL of the diluted library on a DNA high-sensitivity chip following the instruction of Bioanalyzer 2100 for DNA sample.
-
75.
Record the library size.
◆ TROUBLESHOOTING
Next-generation sequencing
● TIMING variable
-
76.
Choose and decide the sequencer mode and the requirement of your data amount. We sequence eight to ten of SCMDA or bulk libraries in one lane of Novaseq 6000 with Novogene. Each lane outputs 800–900 Gb of data.
-
77.
Prepare the library for sequencing: dilute library to ~5 ng/μL in 10 μL of 0.1× TE buffer using a 1.5 mL DNA LoBind tube.
▲ CRITICAL STEP The amount of library prepared for sequencing varies depending on the data amount required. Check the minimum requirement with the sequencing facility.
-
78.
Keep the diluted library at −20 °C before shipping. Ship the frozen library with dry ice.
▲ CAUTION Dry ice is much colder than regular ice, and can burn the skin similarly to frostbite. You should wear insulated gloves when handling it.
-
79.
Provide sample information including library type, index sequences, insert size, library elution buffer, data amount requested, etc. to the sequencing facility.
-
80.
Follow the instruction from the sequencing facility for the remaining steps. For example, confirmation for the QC report of library or sequencing data.
Data analysis
▲ CRITICAL Refer to our latest update of SCcaller-pipeline on GitHub24 for the most recent version of the data analysis pipeline. The timing indicated below may vary substantially depending on performance of the workstation or computer cluster.
-
81.
Download raw sequencing data (in fastq format) from the sequencing facility.
QC, sequence alignment and germline variant calling
● TIMING 2–5 d
-
82.
Download pipeline file ‘sccaller_pipeline_1.sh’ to the working directory and enter the directory.
-
83.
Deposit fastq files under folder ./fastq/ as $sample_id_1.fq.gz $sample_id_2.fq.gz for the pair-end reads for a specific sample.
-
84.Submit this job to a computer cluster as the following (shown for SGE):
- For single cell:
qsub sccaller_pipeline_1.sh $sample_id cell
- For bulk DNA:
qsub sccaller_pipeline_1.sh $sample_id bulk
▲ CRITICAL STEP For species without a well-studied list of germline variants, e.g., the 1000 Genomes for humans, refer to ref. 22 for sequence alignment, which was also discussed in the ‘Experimental design’ section.
Table 4 ∣.
Substeps in data analysis pipeline
| Step | Software (main option) | Note |
|---|---|---|
| 82.1 | FastQC | QC of raw sequencing read |
| 82.2 | GATK FastqToSam | Convert fastq files to unmapped bam file |
| 82.3 | GATK MarkIlluminaAdapters | Mark sequencing adapters in unmapped bam file |
| 82.4 | GATK SamToFastq; bwa; samtools | Sequence alignment |
| 82.5 | GATK MergeBamAlignment | Merge mapped bam file with unmapped bam file |
| 82.6 | GATK SortSam; GATK SetNmMdAndUqTags | Sort BAM file by coordinate order and fix tag values for NM and UQ |
| 82.7 | GATK MarkDuplicates | Mark duplicate reads to avoid counting non-independent observations |
| 82.8 | GATK BaseRecalibrator | Generate base quality score recalibration model |
| 82.9 | GATK ApplyBQSR | Apply base quality score recalibration model |
| 82.10 | GATK SortSam; GATK BuildBamIndex; md5sum | Sort by coordinate, index and calculate md5 |
| 82.11 | samtools | Calculate depth and coverage |
| 82.12 | GATK HaplotypeCaller | Call germline heterozygous SNVs and INDELs (only for bulk DNA) |
| 85.1 | SCcaller | Run SCcaller on all candidate mutations |
| 85.2 | n.a | Filter out low-quality and germline calls and keep only somatic mutations |
| 85.3 | bedtools | Calculate sensitivity |
| 85.4 | samtools | Calculate coverage of both bulk and single cell |
| 85.5 | n.a | Estimate SNV and INDEL burdens per single cell |
Table 5 ∣.
Result files after sequence alignment
| Directory | File(s) | Note and usage |
|---|---|---|
| ./${sample_id} | ${sample_id}_1 (or 2) _fastqc.html (or .zip) | FastQC reports |
| ./${sample_id} | ${sample_id}.markilluminaadapters_metrics.txt | Markadaptor report |
| ./${sample_id} | ${sample_id}.bwa.stderr.log | Alignment report |
| ./${sample_id} | ${sample_id}.hg38.duplicate_metrics | Markduplicate report |
| ./${sample_id} | ${sample_id}.hg38.recal_data.csv | RBQS report |
| ./${sample_id} | ${sample_id}.hg38.gatk4.bam | Alignment bam file; input of the next step |
| ./${sample_id} | ${sample_id}.hg38.gatk4.bai | bam index |
| ./${sample_id} | ${sample_id}.hg38.gatk4.bam.md5 | bam md5 |
| ./coverage | summary_coverage.csv | Summary of sequencing coverage |
| ./ht | ${sample_id}.gvcf.gz | Germline variants (vcf; only for bulk DNA) |
| ./ht | ${sample_id}.gvcf.gz.tbi | Index of germline variant vcf file (only for bulk DNA) |
| ./ht | ${sample_id}.hg38.hsnp.biallelic.dbsnp.vcf | Heterozygous germline SNVs reported in dbSNP (vcf; only for bulk DNA); input of the next step |
| ./ht | ${sample_id}.hg38.hindel.biallelic.dbsnp.vcf | Heterozygous germline INDELs reported in dbSNP (vcf; only for bulk DNA); input of the next step |
| ./sensitivity | ${sample_id}.hsnp.biallelic.dbsnp.20x.sorted.bed | Heterozygous germline SNVs reported in dbSNP (bed; only for bulk DNA); input of the next step |
| ./sensitivity | ${sample_id}.hindel.biallelic.dbsnp.20x.sorted.bed | Heterozygous germline INDELs reported in dbSNP (bed; only for bulk DNA); input of the next step |
Variant calling
● TIMING 1–2 d
-
85.
Download pipeline file ‘sccaller_pipeline_2.sh’ to the working directory and enter the directory.
-
86.
Download SCcaller file ‘sccaller_v2.0.0.py’ to the working directory and enter the directory.
-
87.Submit this job to a computer cluster as the following (shown for SGE):
qsub sccaller_pipeline_2.sh $bulk_id $cell_id
$cell_id is the $sample_id of the single cell; and $bulk_id is the $sample_id of its corresponding bulk DNA. The above pipeline integrates the 5 substeps (from 85.1 to 85.5) as shown in Fig. 3b and Table 4. This creates the following result files as presented in Table 6.▲ CRITICAL STEP For cells or bulk DNA with substantially lower sequencing depth than 30×, mutation burden may not be estimated accurately. This is because SCcaller by its default setting requires at least four variant-supporting reads and 20 total reads from a locus to call a variant (to avoid sequencing errors, and to distinguish homozygous and heterozygous variants), and a sequencing depth of 30× typically allows ~50% the genome covered by 20× or more in the data (Supplementary Table 1). In Supplementary Tables 1-5, we demonstrate the genomic coverage, sensitivity and false positive per base pair, which were estimated from our previous 30× single-cell sequencing data18.
Table 6 ∣.
Result files after variant calling
| Directory | File(s) | Note and usage |
|---|---|---|
| ./sccaller | ${cell_id}.sccaller.vcf | SCcaller results for all candidate variant positions |
| ./sccaller | ${cell_id}.somatic.snv.vcf | SCcaller results for somatic SNVs |
| ./sccaller | ${cell_id}.somatic.indel.vcf | SCcaller results for somatic INDELs |
| ./sccaller | sc_${cell_id}.sccaller_01to-1.log | SCcaller log file |
| ./sensitivity | sensitivity.txt | Estimated variant calling sensitivity based on germline heterozygous SNVs and INDELs |
| ./coverage | summary_coverage_both.csv | No. base pairs covered with at least 20× in both single cell and its bulk |
| ./mutationburden | mutation_burden.csv | A summary of coverage, sensitivity, and SNV and INDEL burdens |
Troubleshooting
Troubleshooting advice can be found in Table 7.
Table 7 ∣.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 6 | For nonadherent cells or nuclei, after centrifugation, there are still many floating cells or nuclei, not attached to the array | The array was not coated well, e.g., coating concentration or temperature is not correct or timing is not enough | Check if the coating concentration and incubator temperature are correct. Incubate the array with cells or nuclei at 37 °C for another 1–3 h and spin down again |
| 23 | Bead solution is not clear even after 5 min | Magnets are not strong enough | Change to a new or stronger magnetic stand |
| The initial sample volume or total volume is too high | Divide the initial samples into two tubes for purification | ||
| Bead ratio is too high | Keep the tube on the stand and pipette bead solution to speed up separation between bead and supernatant | ||
| 34 | SCMDA product yield is low | Reagent efficiency is low | If the yield of positive control is also low, prepare new lysis and stop buffer and use a new kit and random primers |
| The quality of input cell or nucleus is low, e.g., dead cell, or damaged cell or nuclei | If positive control is normal, repeat SCMDA or re-isolate single cells or nuclei | ||
| 39 | SCMDA product has smaller size | Cell or nucleus may be damaged or genome is degraded | Avoid harmful conditions when isolating single cells or nuclei (Cellraft system is recommended) |
| Bead ratio is not correct | Double check bead ratio and its efficiency | ||
| 45 | RUV is not between 0.25 and 0.4 | Quantification or dilution is not correct | Check the absolute Ct value of each locus in the same cell or positive control. If all the values are low (<18) or high (>35), it indicates that quantification or dilution has problem. Repeat quantification or dilution |
| The quality of input cell or nucleus is low: genome was degraded, which causes amplification bias | Check the absolute Ct value. If positive control is normal but RUVs of two or more loci of the cell are not in the range—indicating amplification nonuniformity, repeat SCMDA or repeat from single-cell isolation | ||
| 72 | Library yield is low | The initial input of SCMDA product is low | Make sure that the concentration is quantified using Qubit (other quantification methods may get a different concentration of the same sample) |
| Bead purification recovery is low | Mix beads and samples well when purification, and be careful not to over-dry | ||
| Number of cycles for PCR enrichment is low | Increase by one or two PCR cycles on the last PCR enrichment step | ||
| 75 | Library insert size is not expected | SCMDA product quality is low | Remake a library using a different SCMDA product |
| Fragmentation time is not optimized | Test different fragmentation times | ||
| Bead ratio is not correct | Optimize bead ratio | ||
| All above | Perform size selection with beads or gel if still using this library |
Timing
Steps 1–9, Single-cell or nucleus isolation, 2–5 h
Steps 10–18, SCMDA, 2 h
Steps 19–28, Purification of SCMDA amplicons, 30 min
Steps 29–34, Quantification of purified SCMDA amplicons, 20 min
Steps 35–39, Agarose gel electrophoresis, 45 min
Steps 40–45, LDO, 1 h (LDO and agarose gel electrophoresis can be performed simultaneously)
Steps 46–71, Library preparation, 4 h
Step 72, Quantification of library, 20 min
Steps 73–75, Running bioanalyzer, 1 h
Steps 76–80, Sequencing, timing varies depending on data amount, sequencer or vendor
Steps 81–87, Data analysis, 3–7 d
Anticipated results
Single-cell or nucleus isolation
In Fig. 5, we provide examples of single human fibroblast, lymphocyte, hepatocyte and neuronal nuclei on a single ‘raft’ of a CytoSort array under a microscope.
Fig. 5 ∣. Examples of single cells or nuclei on CytoArray under a microscope.

a, A fibroblast. b, A lymphocyte. c, A hepatocyte. d, A neuronal nucleus. Scale bar, 100 μm.
Single-cell or nucleus WGA
As described in the ‘Procedure’ section, we perform QC on the amplicons, i.e., the product of the single-cell or nucleus WGA. DNA concentration of the amplicons can vary depending on several factors including their species, cell quality, lysis efficiency and amplification efficiency. Although only very little, e.g., 400 ng for the NEB kit, is enough for library preparation, good-quality amplicons of a human diploid cell are typically more than 10 μg, and amplicons less than 5 μg is not recommended for library preparation and sequencing. Fragment size of the amplicon is typically enriched over 10 kb (Fig. 6). For the eight-locus LDO test, we expect good-quality amplicons to have a minimum of six of the eight loci of RUVs between 0.25 and 4.
Fig. 6 ∣. Examples of gel electrophoresis result of SCMDA amplicons.
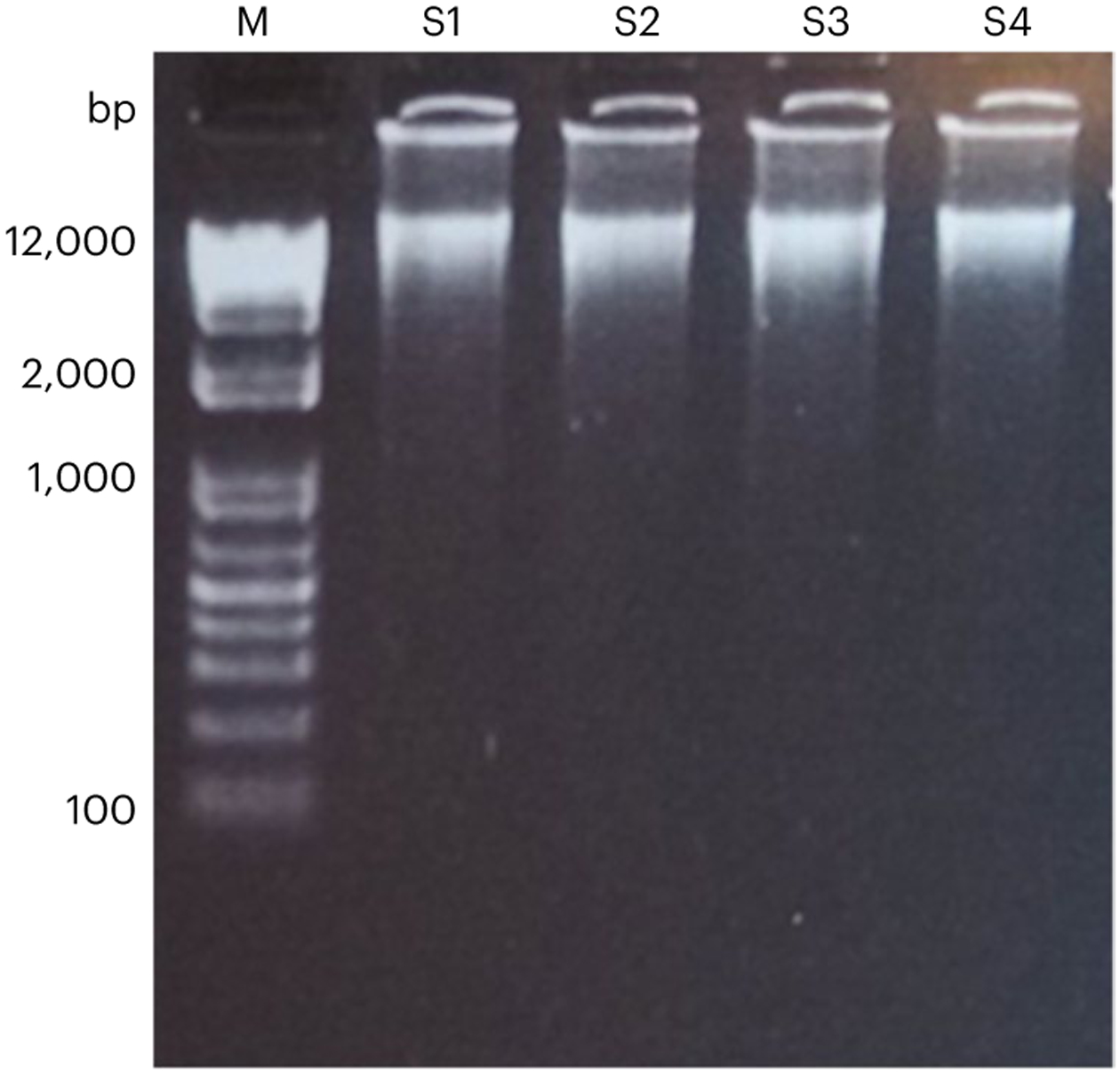
M: DNA ladder. S1–S4: four examples of SCMDA amplicons.
Library preparation and sequencing
For library preparation using the NEBNext Ultra II FS DNA library prep kit, we expect a good-quality library to have a yield of 300–600 ng and a peak fragment size of 500–600 bp, as measured by a bioanalyzer (Fig. 7).
Fig. 7 ∣. An example of library size distribution of SCMDA amplicons.
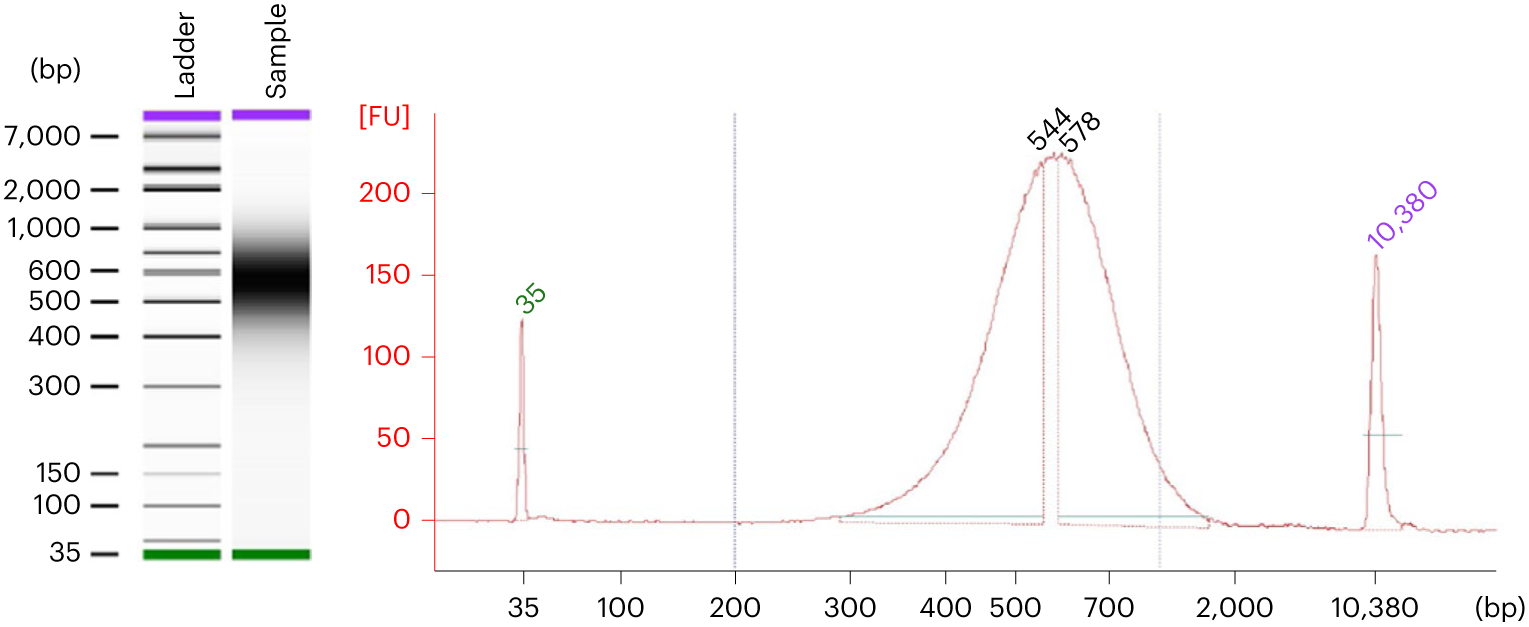
The size distribution is checked using Bioanalyzer 2100 and High-Sensitivity DNA Kit. A densitometry plot (a gel-like image) of the library size distribution (left) and electropherograms of the library size distribution (right) of the same sample as shown on the left. FU, fluorescence units. The peaks at 35 and 10,380 bp are the lower and upper size markers.
After sequencing with an Illumina NovaSeq 6000 platform, we expect ~300 million 150 bp pair-end reads per single cell or bulk DNA sample. Good-quality sequencing data typically have (i) over 80% bases of quality scores ≥30, (ii) an approximately even distribution of base pair quality score and GC content among different positions in the reads, and (iii) no substantial adapter contaminations, as measured by FastQC.
Sequencing alignment, variant calling and estimating mutation burden
Our data analysis pipeline generates a list of result files as presented in Tables 5 and 6 for alignment and variant calling separately. After sequence alignment, the fractions of the single-cell genome and the bulk genome covered with a sequencing depth of at least 20× are reported in the ‘summary_coverage.csv’ file (Table 8). The fraction of genome covered by at least 20× in both a single cell and its corresponding bulk DNA is reported in the ‘summary_coverage_both.csv’ file (Table 9). For cells of substantially less sequencing coverage (e.g., 30% genome covered with 20× depth), first check whether enough sequencing data was obtained for both single cells and their bulk DNA. If so, this suggests severe allelic dropouts, which are rare for amplicons of sufficient yield that passed the LDO test. For most bulk DNA, typically >1 million heterozygous germline SNVs and >100,000 heterozygous germline INDELs are observed from its sequencing data and reported by the dbSNP database before. Somatic SNVs and INDELs of a cell are deposited in the ‘$cell_id.somatic.snv.vcf’ and ‘$cell_id.somatic. indel.vcf’ files, respectively. For estimating mutation burden, the ‘mutation_burden.csv’ file reports a complete summary of the result (Table 10).
Table 8 ∣.
An example of the ‘summary_coverage.csv’ file
| Sample ID | No. total bases in aligned dataa |
No. bases in the reference genome covered by a min. depth of | ||||
|---|---|---|---|---|---|---|
| 20× | 15× | 10× | 5× | 1× | ||
| LZBC01B1b | 91,148,877,023 | 1,790,601,190 | 2,116,261,277 | 2,430,865,011 | 2,686,213,358 | 2,820,884,367 |
| LZBC01B2b | 87,353,451,553 | 1,645,030,218 | 1,970,657,709 | 2,310,831,238 | 2,621,406,394 | 2,807,940,655 |
| LZBC01B3b | 91,506,866,980 | 1,739,792,848 | 2,063,011,469 | 2,385,735,441 | 2,661,458,027 | 2,815,446,172 |
| LZBC01B4b | 83,416,723,257 | 1,550,224,661 | 1,885,944,822 | 2,249,341,364 | 2,592,982,296 | 2,803,939,071 |
| LZBC01BBc | 69,274,174,573 | 2,208,462,812 | 2,613,799,770 | 2,762,866,918 | 2,821,643,386 | 2,844,741,054 |
Dividing this value by the number of bases in the reference genome is the average sequencing depth across the genome.
Examples of scWGS data.
An example of bulk WGS data.
Table 9 ∣.
An example of the ‘summary_coverage_both.csv’ file
| Cell ID | No. bases in the reference genome covered by at least 20× in both the single cell and its corresponding bulk |
|---|---|
| LZBC01B1 | 1,079,630,695 |
| LZBC01B2 | 992,580,939 |
| LZBC01B3 | 1,048,853,241 |
| LZBC01B4 | 926,174,542 |
Table 10 ∣.
An example of the ‘mutation_burden.csv’ file
| Bulk ID | Cell ID | Size of reference genome (bp) |
No. bases in the reference genome covered by at least 20× in both the single cell and its corresponding bulk |
SNV sensitivity |
INDEL sensitivity |
No. observed SNVs |
No. observed INDELs |
SNV burden |
INDEL burden |
|---|---|---|---|---|---|---|---|---|---|
| LZBC01BB | LZBC01B1 | 3,217,346,917 | 1,079,630,695 | 0.84 | 0.81 | 462 | 17 | 1647.5 | 62.5 |
| LZBC01BB | LZBC01B2 | 3,217,346,917 | 992,580,939 | 0.82 | 0.80 | 445 | 13 | 1764.4 | 52.4 |
| LZBC01BB | LZBC01B3 | 3,217,346,917 | 1,048,853,241 | 0.83 | 0.81 | 523 | 15 | 1939.7 | 57.1 |
| LZBC01BB | LZBC01B4 | 3,217,346,917 | 926,174,542 | 0.82 | 0.80 | 419 | 14 | 1778.2 | 60.5 |
Measurement of amplification bias
Compared with conventional bulk DNA sequencing, the unique challenge to our protocol is the potential bias in genome amplification. We implemented the LDO test as a QC step. As an alternative to LDO, one may consider applying low depth sequencing (e.g., 1× depth) to the single-cell libraries, and use the PaSD-qc software for QC and selection of libraries of good quality49.
From the data analysis, we recommend using two sets of measurement. First, the ‘summary_coverage.csv’ file provides sequencing coverage of the single cells, and typically, one would expect ~50% of the genome covered with 20× or more (examples are presented in Table 8 and Supplementary Table 1). Second, the ‘$cell_id.sccaller.vcf’ file provides η values observed for each chromosome or contig of the single-cell data. The η values estimated by SCcaller indicate bias in genome amplification. A smaller fraction of genome covered by 20× and a smaller η value indicate higher bias, and subsequently a higher chance of misclassifying amplification errors as mutations. In Fig. 8, we provide η values of all cells in three major datasets including human lymphocytes18, hepatocytes19 and lung epithelium cells20 for users to compare with their own data. As shown, a η value around 0.2 is expected. If substantially small fraction of genome is covered and the average η value across all chromosomes is substantially small, we suggest to carefully confirm whether wet-laboratory experiment (especially single-cell isolation and amplification) was performed correctly according to the protocol, and repeat the experiment if necessary.
Fig. 8 ∣. Violin plots of the η values of 262 single cells in three studies.
Average η value of autosomes per cell was calculated for each single cell in the three studies18-20. Box plot elements are defined as follows: center line indicates median, box limits indicate upper and lower quartiles, whiskers indicate 1.5× interquartile range and points indicate the outliers of the box plots.
Mutation annotation
From the observed somatic SNVs and INDELs, we annotate these variants to understand if they occur in functionally important genomic regions or loci. This not only provides direct evidence whether mutations have occurred in specific single cells, but also demonstrates if mutations enrich in some genes, genetic pathways or genetic elements (e.g., enhancers) of interest in the population of cells. We recommend performing the annotation in two steps. First, apply ANNOVAR (Annotate Variation) or its webserver wANNOVA50,51. In brief, ANNOVAR provides three types of annotations: gene-based, region-based and filter-based annotations. Its protocol was published previously with Nature Protocols52 and is well documented on its website21. Second, considering that only a fraction of the genome is functionally active in a specific somatic cell type, we recommend annotating whether the mutations are located in functionally active regions of the corresponding cell type, using public repositories, e.g., the ENCODE project53,54, or bulk RNA sequencing and ATAC sequencing of cells of the same cell population, as we have shown in a previous study18.
Mutation hotspot analysis
It has first been shown that mutations may cluster at specific genomic loci in tumors55,56. As we have shown previously, single-cell whole-genome sequencing identifies mutation hotspots in normal, human B lymphocytes18. These hypermutation events are known as mutation hotspots—‘shower’ or ‘kataegis’—and are likely to be caused by specific mechanisms. To identify mutation hotspots, we suggest using the ‘ClusteredMutations’ package in R57,58. Of note, its default parameters including (i) the distance between two mutations and (ii) the minimum number of mutations per hotspot, were optimized for analyzing bulk sequencing of tumor samples and are over-stringent for analyzing normal somatic cells. One can pool observed mutations from all cells of the same population or experimental group and choose a less stringent criteria for the analysis. For identified mutation hotspots, one can annotate their genomic loci by overlapping them with gene annotations using the University of California Santa Cruz genome browser59,60.
Mutational signature analysis
Specific mutagens are known to preferably cause specific base pair alterations. For example, tobacco smoking increases the rate of C > A transition, as shown in lung cancers61. By bringing also the two flanking bases of a mutation, e.g., ACA > AAA, into the analysis, mathematical decomposition of these different types of mutations can reveal common and unique mutational patterns across samples, called mutational signatures62. These mutational signatures are well-known indicators of mechanisms that cause the mutations in both tumors and normal cells5,18,20,63. Multiple methods have previously been developed to discover mutational signatures, and we have implemented several of these methods in our webserver of the SomaMutDB database64. For more in depth analysis we recommend using the SigProfiler5, which has the mutational signatures documented at its website, well-supported by its authors on GitHub65-68.
Supplementary Material
Key points.
Protocol for single-cell whole-genome sequencing to discover and analyze somatic mutations in tissues and organs, using single-cell multiple displacement amplification alongside the SCcaller software.
Compared with bulk sequencing approaches, single-cell whole-genome sequencing allows discovery of most, if not all, mutations in the same single-cell genome. This enables quantification of the mutation burden per cell, discovery of mutational hotspots and establishment of cell lineages.
Acknowledgements
L.Z. is supported by the American Federation for Aging Research (the Sagol Network GerOmic Award for Junior Faculty). J.V. is supported by the US National Institutes of Health awards (P01 AG017242, P01 AG047200, P30 AG038072, U01 ES029519, U01 HL145560 and U19 AG056278). X.D. is supported by the NIH awards (R00 AG056656, P01 HL160476, U54 AG076041 and U54 AG079754).
Related links
Key references using this protocol
- Dong X. et al. Nat. Methods 14, 491–493 (2017): 10.1038/nmeth.4227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. et al. Proc. Natl Acad. Sci. USA 116, 9014–9019 (2019): 10.1073/pnas.1902510116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. et al. Sci. Adv 7, eabj3284 (2021): 10.1126/sciadv.abj3284 [DOI] [PMC free article] [PubMed] [Google Scholar]
Footnotes
Competing interests
L.Z., M.L., A.Y.M., J.V. and X.D. are co-founders and shareholders of SingulOmics Corp. C.M. declares no conflict of interest.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41596-023-00914-8.
Data availability
Raw sequencing data for SCMDA-amplified single cells can be obtained from our previous publications: human fibroblasts17, human B lymphocytes18, human hepatocytes19, human lung epithelium cells20, human mammary epithelial cells23 and fibroblasts of multiple rodent species21,22.
Code availability
The source code for the data analysis pipeline and its documentations is freely available on GitHub via ref. 24 and at https://github.com/XiaoDongLab/SCcaller-pipeline.
References
- 1.Failla G. The aging process and cancerogenesis. Ann. N. Y. Acad. Sci 71, 1124–1140 (1958). [DOI] [PubMed] [Google Scholar]
- 2.Szilard L. On the nature of the aging process. Proc. Natl Acad. Sci. USA 45, 30–45 (1959). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vijg J & Dong X Pathogenic mechanisms of somatic mutation and genome mosaicism in aging. Cell 182, 12–23 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nowell PC The clonal evolution of tumor cell populations. Science 194, 23–28 (1976). [DOI] [PubMed] [Google Scholar]
- 5.Alexandrov LB et al. The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 578, 82–93 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaiswal S. et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med 371, 2488–2498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kessler MD et al. Common and rare variant associations with clonal haematopoiesis phenotypes. Nature 612, 301–309 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Erickson RP Somatic gene mutation and human disease other than cancer. Mutat. Res 543, 125–136 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Erickson RP Somatic gene mutation and human disease other than cancer: an update. Mutat. Res 705, 96–106 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Bae T. et al. Analysis of somatic mutations in 131 human brains reveals aging-associated hypermutability. Science 377, 511–517 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bianconi E. et al. An estimation of the number of cells in the human body. Ann. Hum. Biol 40, 463–471 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Sender R, Fuchs S & Milo R Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 14, e1002533 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brunner SF et al. Somatic mutations and clonal dynamics in healthy and cirrhotic human liver. Nature 574, 538–542 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blokzijl F. et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 538, 260–264 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martincorena I & Campbell PJ Somatic mutation in cancer and normal cells. Science 349, 1483–1489 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Dong X. et al. Accurate identification of single-nucleotide variants in whole-genome-amplified single cells. Nat. Methods 14, 491–493 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang L. et al. Single-cell whole-genome sequencing reveals the functional landscape of somatic mutations in B lymphocytes across the human lifespan. Proc. Natl Acad. Sci. USA 116, 9014–9019 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brazhnik K. et al. Single-cell analysis reveals different age-related somatic mutation profiles between stem and differentiated cells in human liver. Sci. Adv 6, eaax2659 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang Z. et al. Single-cell analysis of somatic mutations in human bronchial epithelial cells in relation to aging and smoking. Nat. Genet 54, 492–498 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Milholland B. et al. Differences between germline and somatic mutation rates in humans and mice. Nat. Commun 8, 15183 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang L. et al. Maintenance of genome sequence integrity in long- and short-lived rodent species. Sci. Adv 7, eabj3284 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun S. et al. Single-cell analysis of somatic mutation burden in mammary epithelial cells of pathogenic BRCA1/2 mutation carriers. J. Clin. Invest 132 10.1172/jci148113 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang L. et al. Single-cell whole-genome sequencing for discovering somatic mutations. GitHub 10.5281/zenodo.7826180 (2023). [DOI] [Google Scholar]
- 25.Luquette LJ, Bohrson CL, Sherman MA & Park PJ Identification of somatic mutations in single cell DNA-seq using a spatial model of allelic imbalance. Nat. Commun 10, 3908 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bohrson CL et al. Linked-read analysis identifies mutations in single-cell DNA-sequencing data. Nat. Genet 51, 749–754 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Min S. et al. Absence of coding somatic single nucleotide variants within well-known candidate genes in late-onset sporadic Alzheimer’s disease based on the analysis of multi-omics data. Neurobiol. Aging 108, 207–209 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang L. et al. Analysis of somatic mutations in senescent cells using single-cell whole-genome sequencing. AgingBio 1, 1–6, 10.59368/agingbio.20230005 (2023). [DOI] [Google Scholar]
- 29.Schmitt MW et al. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl Acad. Sci. USA 109, 14508–14513 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abascal F. et al. Somatic mutation landscapes at single-molecule resolution. Nature 593, 405–410 (2021). [DOI] [PubMed] [Google Scholar]
- 31.Maslov AY et al. Single-molecule, quantitative detection of low-abundance somatic mutations by high-throughput sequencing. Sci. Adv 8, eabm3259 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zafar H, Navin N, Chen K & Nakhleh L SiCloneFit: Bayesian inference of population structure, genotype, and phylogeny of tumor clones from single-cell genome sequencing data. Genome Res. 29, 1847–1859 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang F. et al. MEDALT: single-cell copy number lineage tracing enabling gene discovery. Genome Biol. 22, 70 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Telenius H. et al. Degenerate oligonucleotide-primed PCR: general amplification of target DNA by a single degenerate primer. Genomics 13, 718–725 (1992). [DOI] [PubMed] [Google Scholar]
- 35.Dean FB, Nelson JR, Giesler TL & Lasken RS Rapid amplification of plasmid and phage DNA using Phi 29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res. 11, 1095–1099 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zong C, Lu S, Chapman AR & Xie XS Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 338, 1622–1626 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen C. et al. Single-cell whole-genome analyses by linear amplification via transposon insertion (LIANTI). Science 356, 189–194 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang L, Ma F, Chapman A, Lu S & Xie XS Single-cell whole-genome amplification and sequencing: methodology and applications. Annu. Rev. Genomics Hum. Genet 16, 79–102 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Spits C. et al. Whole-genome multiple displacement amplification from single cells. Nat. Protoc 1, 1965–1970 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Xing D, Tan L, Chang CH, Li H & Xie XS Accurate SNV detection in single cells by transposon-based whole-genome amplification of complementary strands. Proc. Natl Acad. Sci. USA 10.1073/pnas.2013106118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sarangi V. et al. SCELLECTOR: ranking amplification bias in single cells using shallow sequencing. BMC Bioinforma. 21, 521 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Motyer A et al. The mutational landscape of single neurons and oligodendrocytes reveals evidence of inflammation-associated DNA damage in multiple sclerosis. Preprint at bioRxiv 10.1101/2022.04.30.490132 (2022). [DOI] [Google Scholar]
- 43.Bloom JC & Schimenti JC Sexually dimorphic DNA damage responses and mutation avoidance in the mouse germline. Genes Dev. 34, 1637–1649 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gundry M, Li W, Maqbool SB & Vijg J Direct, genome-wide assessment of DNA mutations in single cells. Nucleic Acids Res. 40, 2032–2040 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gravina S, Dong X, Yu B & Vijg J Single-cell genome-wide bisulfite sequencing uncovers extensive heterogeneity in the mouse liver methylome. Genome Biol. 17, 150 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ailshire JA, Beltran-Sanchez H & Crimmins EM Becoming centenarians: disease and functioning trajectories of older US Adults as they survive to 100. J. Gerontol. Ser. A, Biol. Sci. Med. Sci 70, 193–201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van der Auwera GA & O'Connor BD Genomics in the Cloud: Using Docker, GATK, and WDL in Terra 1st edn (O'Reilly Media, 2020). [Google Scholar]
- 48.Lodato MA et al. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 359, 555–559 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sherman MA et al. PaSD-qc: quality control for single cell whole-genome sequencing data using power spectral density estimation. Nucleic Acids Res. 46, e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang K, Li M & Hakonarson H ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang X & Wang K wANNOVAR: annotating genetic variants for personal genomes via the web. J. Med. Genet 49, 433–436 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang H & Wang K Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc 10, 1556–1566 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Consortium EP An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mejia-Ramirez E & Florian MC Understanding intrinsic hematopoietic stem cell aging. Haematologica 105, 22–37 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang J et al. Evidence for mutation showers. Proc. Natl Acad. Sci. USA 104, 8403–8408 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nik-Zainal S et al. Mutational processes molding the genomes of 21 breast cancers. Cell 149, 979–993 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lora D. Location and visualization of clustered somatic mutations. v1.0.1 . CRAN-R Project https://cran.r-project.org/web/packages/ClusteredMutations/ClusteredMutations.pdf (2016). [Google Scholar]
- 58.Bhagat TD et al. miR-21 mediates hematopoietic suppression in MDS by activating TGF-beta signaling. Blood 121, 2875–2881 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kent WJ et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee BT et al. The UCSC Genome Browser database: 2022 update. Nucleic Acids Res. 50, D1115–d1122 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alexandrov LB et al. Mutational signatures associated with tobacco smoking in human cancer. Science 354, 618–622 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ & Stratton MR Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 3, 246–259 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alexandrov LB et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sun S, Wang Y, Maslov AY, Dong X & Vijg J SomaMutDB: a database of somatic mutations in normal human tissues. Nucleic Acids Res. 50, D1100–D1108 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Denkinger MD, Leins H, Schirmbeck R, Florian MC & Geiger H HSC aging and senescent immune remodeling. Trends Immunol. 36, 815–824 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Osorio FG et al. Somatic mutations reveal lineage relationships and age-related mutagenesis in human hematopoiesis. Cell Rep. 25, 2308–2316.e2304 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mitchell E. et al. Clonal dynamics of haematopoiesis across the human lifespan. Nature 606, 343–350 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rodrigues-Moreira S. et al. Low-dose irradiation promotes persistent oxidative stress and decreases self-renewal in hematopoietic stem cells. Cell Rep. 20, 3199–3211 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing data for SCMDA-amplified single cells can be obtained from our previous publications: human fibroblasts17, human B lymphocytes18, human hepatocytes19, human lung epithelium cells20, human mammary epithelial cells23 and fibroblasts of multiple rodent species21,22.
The source code for the data analysis pipeline and its documentations is freely available on GitHub via ref. 24 and at https://github.com/XiaoDongLab/SCcaller-pipeline.