

Abstract
The global crisis of antimicrobial resistance (AMR) necessitates the development of broad-spectrum antibacterial drugs effective against multi-drug resistant (MDR) pathogens. BWC0977, a Novel Bacterial Topoisomerase Inhibitor (NBTI) selectively inhibits bacterial DNA replication via inhibition of DNA gyrase and topoisomerase IV. BWC0977 exhibited a minimum inhibitory concentration (MIC90) of 0.03–2 µg/mL against a global panel of MDR Gram-negative bacteria including Enterobacterales and non-fermenters, Gram-positive bacteria, anaerobes and biothreat pathogens. BWC0977 retains activity against isolates resistant to fluoroquinolones (FQs), carbapenems and colistin and demonstrates efficacy against multiple pathogens in two rodent species with significantly higher drug levels in the epithelial lining fluid of infected lungs. In healthy volunteers, single-ascending doses of BWC0977 administered intravenously (https://clinicaltrials.gov/study/NCT05088421) was found to be safe, well tolerated (primary endpoint) and achieved dose-proportional exposures (secondary endpoint) consistent with modelled data from preclinical studies. Here, we show that BWC0977 has the potential to treat a range of critical-care infections including MDR bacterial pneumonias.
Subject terms: Drug discovery and development, Bacterial infection
In this work, the authors probe the efficacy of BWC0977, a bacterial topoisomerase inhibitor, in pre-clinical animal models, also demonstrating that BWC0977 is safe and well tolerated in healthy human volunteers, in a phase 1 trial.
Introduction
The World Health Organization (WHO) has categorically identified AMR as one of the top ten global public health threats impacting humanity. Despite concerted global efforts, MDR infections caused 4.95 million deaths in 2019, with a disproportionate burden on the low- and middle-income countries (LMICs)1. Unless AMR is tackled aggressively, an estimated 10 million lives will be lost annually by 20502. Many of these pathogens pose a serious threat to patients in hospitals and critical-care settings by causing life-threatening conditions like complicated intra-abdominal infections (cIAI), bloodstream infections and pneumonia3–5. Since the introduction of fluoroquinolones (FQs) in the 1980s, there has not been a broad-spectrum class of drugs that are effective against multiple pathogenic bacteria. Currently, there are very limited options to treat infections caused by MDR pathogens6,7.
NBTIs are dual-acting agents that inhibit the bacterial DNA gyrase and topoisomerase IV8–11. Currently, there are two NBTIs – Gepotidacin and Zoliflodacin undergoing clinical trials12,13. The NBTIs work via a unique mechanism of inhibition by occupying target binding sites that are distinct from the FQs, and therefore lack cross-resistance to FQs14. Gepotidacin is an oral, first-in-class antibacterial (triazaacenaphthylene) being developed by GlaxoSmithKline for treating uncomplicated urinary tract infections (uUTI) and gonorrhea15,16. Gepotidacin has completed three, Phase-2 clinical trials for treating acute bacterial skin and skin structure infections (ABSSSI)17, uncomplicated urogenital gonorrhea (uUG) caused by Neisseria gonorrhoeae18, and uUTI19. The second NBTI, Zoliflodacin, a spiropyrimidinetrione developed by Entasis Therapeutics, has completed a Phase-3 trial as an oral treatment for uUG20. In this study, zoliflodacin demonstrated statistical non-inferiority of microbiological cure at the urogenital site versus treatment with intramuscular injection of ceftriaxone and oral azithromycin21.
Here, we show the discovery of BWC0977, a clinical-stage NBTI that demonstrates broad-spectrum activity against the major WHO published list of “global priority” pathogens22. These include carbapenem-resistant Enterobacteriaceae, carbapenem-resistant Pseudomonas aeruginosa, carbapenem-resistant Acinetobacter baumannii and methicillin-resistant Staphylococcus aureus22.
Results
The initial hit that led to the discovery of BWC0977 was identified from a whole cell phenotypic screen against E. coli ATCC25922, E. coli ΔacrB JW0451-2 and E. coli ΔtolC JW5503-1. The chemical library consisted of approximately 3000 compounds selected for structural diversity from commercial databases such as eMolecule, REAL database from Enamine, Aurora Fine Chemicals catalogue and custom synthesized fragments. The diverse set of compounds was procured from various libraries, including Enamine Diversity Library, Aurora Building Blocks 1, Lifechemical Screening and Fragment Library, Analyticon Focused Library, and Ottava Chemical Fragment and Diversity Library. Structural analyses based on literature search revealed that the early hit had similarity to known NBTIs8. A schematic of the medicinal chemistry optimization and structure-activity relationship (SAR) of the lead series is shown in Fig. 1.
Fig. 1. Structure-Activity Relationship (SAR) leading to the selection of BWC0977 as the clinical candidate.

The synthesis of the screening hit, compound 1 and the progressive synthesis of different compounds based on medicinal chemistry inputs towards improving the compound properties. The latter included enhanced potency, broad-spectrum antibacterial activity, optimal physico-chemical properties while mitigating tox liability leading to the selection of the clinical candidate, compound 9 or BWC0977. Abbreviations: Eco, Escherichia coli; Pae, Pseudomonas aeruginosa; Aba, Acinetobacter baumannii; Kpn, Klebsiella pneumoniae.
The screening hit, compound 1 exhibited a minimum inhibitory concentration (MIC) of 80–160 µg/ml against key Gram-negative and Gram-positive pathogens with micromolar E. coli gyrase and topoIV IC50s (half maximal inhibitory concentration of 13.2–27 µM). By employing rational drug design using the published gyrase crystal structure23, compounds 2 & 3 were found to display >100-fold improvement in E. coli gyrase supercoiling and topoIV decatenation IC50 (106–110 nM for gyrase, 398–360 nM for topoIV) over compound 1 with concomitant improvement in broad-spectrum MIC ( >40-fold). The oxindole left-hand side (LHS) ring system present in compounds 2 & 3 was designed to interact with the double stranded DNA (dsDNA) portion of gyrase complex to bring in broad-spectrum MIC. Shifting the secondary amine center along the linear linker carbon chain led to increased basicity and improved MIC (compound 2 vs. compound 3). Reducing the basicity of the linker amine length with 4-atom linker between the LHS and oxazolidinone ring system resulted in compound 4 exhibiting potent broad-spectrum MIC and iso-potent inhibition of gyrase and topoIV (36−57 nM). The expansion of oxindole ring of compound 4 into the quinolone ring system resulted in compound 5, with a 2-4-fold improved MIC, while maintaining potent enzyme inhibition (Fig. 1, Supplementary Table 1).
However, early leads from the oxazolidinyl alkyl amines series had significant activity against the cardiac Kv11.1, ion channel (referred as the human Ether-à-go-go-Related Gene channel, “hERG”) with compound 2-5 exhibiting hERG IC50 of 9–25 µM. Further medicinal chemistry efforts were focused on reducing the hERG inhibition, while simultaneously retaining or improving broad-spectrum MIC. To mitigate hERG inhibition, an additional nitrogen was introduced into the right-hand side (RHS) ring, such as a pyrimido-oxazinone ring to modulate the polarity and reduce pKa of the oxazolidinone amine. This breakthrough strategy reduced the hERG inhibition, albeit with a loss of broad-spectrum MIC (compound 5 vs compound 6). Compound 6 exhibited >10-fold increase in hERG IC50 (204 µM) with increased MIC against Gram-negative pathogens (Supplementary Table 1). The reduced hERG inhibition of compound 6 could be attributed to a combination of reduced basicity (pKa), lipophilicity (logD) and polar nature of the RHS ring, which is likely to disturb the binding of compound 6 to the hERG channel. Shifting the nitrogen in RHS ring system to pyrazino-oxazinone with an additional nitrogen in LHS ring system resulted in compound 7. Compound 7 with a quinoxalinone LHS ring system showed similar antibacterial spectrum as compound 5 with 8-fold increase in MIC against A. baumannii (0.25 µg/ml) and 4-fold increase in hERG IC50 (compound 5 vs 7). To design a compound with the right balance of broad-spectrum MIC and hERG inhibition, we removed the nitrogen on LHS ring in compound 7 with R and S configuration on the oxazolidinone ring, resulting in compounds 8 and 9. Both compounds exhibited similar broad-spectrum MIC, but with 2–3 fold difference in hERG inhibition, indicating that chirality at C-5 carbon of oxazolidinone ring does not influence the Gram-negative MIC activity, but impacts the hERG inhibition. Compounds with S-configuration (compound 9) showed 2.2-fold increase in hERG IC50 versus R-configuration (IC50 60 µM for compound 8 and 131 µM for compound 9) (Supplementary Table 1). Compound 9 showed convergence of reduced hERG inhibition and potent broad-spectrum activity. Additionally, the gyrase and topoIV potency for optimized compounds (compounds 4-9) demonstrated >1500-fold selectivity over human topoII enzyme. Encouraged by an improvement in broad-spectrum MIC, reduced hERG inhibition liability with desirable physicochemical properties, compound 9, re-designated as BWC0977 was nominated as the clinical candidate. The current study provides evidence for the microbiological spectrum, in vivo efficacy and pharmacokinetic-pharmacodynamic (PK-PD) driver in rodent models of infection, safety, and the well-tolerated dose proportional PK of BWC0977 in healthy human volunteers.
Antibacterial spectrum against MDR pathogens
BWC0977 was tested as per the Clinical Laboratory Standards Institute (CLSI) protocol24 against a panel of ATCC strains and was found to inhibit Gram-positive bacteria (S. aureus, E. faecalis) with median MICs of 0.01 µg/mL and 0.06 µg/mL, respectively and Gram-negative bacteria (E. coli, K. pneumoniae, P. aeruginosa, A. baumannii, E. cloacae and P. mirabilis) with median MIC of 0.03, 0.03, 0.25, 0.06, 0.06, and 0.12 µg/mL, respectively. In a study against a global panel of MDR pathogens (total of 2945 clinical isolates) collected in 2019, BWC0977 demonstrated potent activity with MIC90 (MIC value at which ≥90% of strains within a test population are inhibited i.e., the 90th percentile) against A. baumannii (1 μg/mL), P. aeruginosa (1 μg/mL), E. coli (0.5 μg/mL), K. pneumoniae (2 μg/mL), E. cloacae (2 μg/mL), Citrobacter species (1 μg/mL), Proteus species (0.5 μg/mL), M. morganii (1 μg/mL), and S. marcescens (1 μg/mL). The comparator drugs (ciprofloxacin, meropenem, meropenem-vaborbactam, cefiderocol and tobramycin) had considerably higher MIC90 values. Majority of the Enterobacterales tested (Table 1) & Gram-positives (Supplementary Table 2) were found to be resistant to currently prescribed drugs.
Table 1.
Determination of antibacterial potency [MIC90 (µg/ml)] of BWC0977 and other known antibiotics performed at the International Health Management Associates (IHMA) against a global collection of Gram-negative drug-resistant clinical isolates
| A. baumannii | P. aeruginosa | E. coli | K. pneumoniae | E. cloacae | Proteus spp. | Citrobacter spp. | S. maltophilia | M. morganii | S. marcescens | N. gonorrhoeae | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| N | 298 | 300 | 300 | 300 | 152 | 152 | 151 | 150 | 42 | 45 | 90 |
| BWC0977 | 1 | 1 | 0.5 | 2 | 2 | 0.5 | 1 | 0.25 | 1 | 1 | ≤ 0.016 |
| Ciprofloxacin | > 32 | 16 | ND | > 32 | ND | ND | ND | 16 | ND | ND | > 1 |
| Levofloxacin | > 32 | 32 | 16 | > 32 | 64 | 8 | 2 | 8 | 16 | 1 | ND |
| aCefiderocol | 4 | 2 | 2 | 2 | 4 | 0.5 | 2 | 2 | 1 | 1 | ND |
| Cefepime | > 32 | > 32 | > 32 | > 32 | 64 | 16 | > 32 | > 32 | > 32 | > 32 | ND |
| Cefpodoxime | ND | ND | > 32 | > 32 | 64 | > 32 | > 32 | ND | > 32 | > 32 | ND |
| Ceftriaxone | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | 0.06 |
| Ceftazidime | > 32 | >Z 32 | 64 | > 64 | 128 | 2 | > 64 | > 32 | 16 | 8 | ND |
| Ceftazidime-Avibactam | ND | > 16 | 0.25 | 1 | 2 | 0.06 | 0.5 | ND | 0.125 | 1 | ND |
| Aztreonam | ND | > 16 | > 64 | > 64 | 128 | 0.5 | 64 | ND | 16 | 32 | ND |
| Aztreonam/Avibactam | ND | > 16 | 0.125 | 0.25 | 0.5 | ≤ 0.016 | 0.25 | ND | 0.06 | 0.25 | ND |
| Piperacillin/Tazobactam | > 128 | 128 | ND | ND | 0.06 | ND | ND | > 128 | ND | ND | ND |
| Meropenem | > 64 | 32 | ≤ 0.06 | 32 | 8 | 0.125 | ≤ 0.06 | > 64 | 0.125 | 0.125 | ND |
| Meropenem-vaborbactam | > 32 | 32 | ≤ 0.03 | 16 | 0.25 | 0.125 | ≤ 0.03 | > 32 | 0.06 | 0.125 | ND |
| Colistin | 0.5 | 1 | 0.25 | 0.5 | 2 | > 32 | 0.5 | > 32 | 64 | > 32 | ND |
| Amikacin | >64 | 64 | 8 | 32 | 4 | 4 | 4 | > 64 | 8 | 8 | ND |
| Gentamicin | > 32 | > 32 | > 32 | > 32 | 1 | 16 | 8 | > 32 | 64 | 16 | ND |
| Doxycycline | 64 | 32 | 32 | 32 | 16 | > 32 | 16 | 4 | 64 | 16 | ND |
| Eravacycline | ND | ND | 0.25 | 1 | ND | 2 | 0.5 | ND | 1 | 2 | ND |
| Tobramycin | > 32 | > 32 | 16 | > 32 | 32 | 8 | 8 | > 32 | 16 | 32 | ND |
| Plazomicin | ND | ND | 1 | 0.5 | 0.5 | 4 | 0.5 | ND | 8 | 2 | ND |
This study was part of a CARB-X screening programme to evaluate the activity of BWC0977 against a global panel of MDR bacterial pathogens (total of 2945 isolates) collected in 2019. In the case of N. gonorrhoea, the collection comprised of isolates identified from 1997 to 2021. BWC0977 showed excellent potency and broad-spectrum activity against these isolates (Gram-negative bacteria: Table 1, Gram-positive bacteria - Supplement section - Table 2). aCefiderocol MICs against same isolates were generated in a different study.
Across a global panel of sequential clinical isolates (Supplementary Table 3), BWC0977 demonstrated potent activity against 7187 Gram-negative pathogens with MIC90 as follows: 0.5 µg/mL against E. coli, 1 µg/mL against P. aeruginosa, 2 µg/mL against K. pneumoniae, 0.5 µg/mL against A. baumannii, 1 µg/mL against E. cloacae (Supplementary Tables 4) and 1404 Gram-positive pathogens, 0.03 µg/mL against S. aureus including MRSA and 0.06 µg/mL Enterococcal species (Supplementary Table 5). Moreover, BWC0977 demonstrated excellent activity against the priority biothreat pathogens: Bacillus anthracis, Yersinia pestis, Burkholderia mallei, Burkholderia pseudomallei, and Francisella tularensis, several MDR pathogens associated with cystic fibrosis (Burkholderia cepacia), and key anaerobic pathogens (Supplementary Tables 4, 5).
Killing kinetics and resistance frequency studies
BWC0977 exhibited significant bactericidal activity in vitro against a panel of susceptible and MDR strains of E. coli, A. baumannii, K. pneumoniae, P. aeruginosa and E. cloacae when sequentially sampled following exposure at 0.5X to 16X MIC and the viable colony forming units (CFUs) enumerated at various time intervals. There was limited improvement in bacterial kill with increasing concentrations, but significant kill over time (Supplementary Figure 1). The in vitro resistance frequency of BWC0977 at 4X MIC was <1 × 10−9 against E. coli ATCC 25922 and P. aeruginosa ATCC 27853, 2.5 × 10−9 for A. baumannii ATCC 19606. Sequencing of the gyrA and parC gene of resistant colonies revealed no mutations in the key NBTI binding sites. The resistance frequency against MDR strains (sourced from ATCC) E. coli BAA-2471, P. aeruginosa BAA-2797, A. baumannii BAA-2885 was 4 × 10−9, < 1 × 10−9 and 2.5 × 10−8, respectively. None of the resistant clones from these studies were cross-resistant to other antibiotics including FQs. However, sequencing mutant colonies from E. coli BAA-2471 showed additional mutations: G235A in parC (D79N in DNA topoIV subunit A) and C1315A or A1322T in parE (P439T or K441I in DNA topoIV subunit B).
Efficacy in neutropenic mouse thigh infection model
A 26-hour neutropenic murine thigh infection model with P. aeruginosa NCTC 13921 (MIC 0.25 µg/ml) was employed for dose-ranging and fractionation25. Dose-ranging studies were repeated thrice to generate robust data sets for modelling and the effective dose (ED50) was estimated to be 58 mg/kg q8h. Mice were dosed with BWC0977 subcutaneously 2 h post-infection and bacterial density (CFU/g) versus 2 h baseline control was used as the endpoint. For fractionation, 160 mg/kg once every 24 h, 80 mg/kg every 12 h, 40 mg/kg every 6 h were administered and samples taken at 8, 14, 26 h post-infection. The effect observed was the same irrespective of the regimen, indicating that free area-under-the-curve over MIC (fAUC/MIC) as the efficacy driver (Fig. 2A). Several wild-type and MDR isolates of P. aeruginosa, A. baumannii, K. pneumoniae and E. coli with MICs ranging from 0.06−0.5 mg/L were tested in dose-ranging studies (Supplementary Table 6). In these studies, BWC0977 administered every 8 h demonstrated excellent dose-response, while achieving logarithmic bacterial killing (Fig. 2B).
Fig. 2. Dose-fractionation and determination of PK-PD drivers of efficacy for BWC0977, either administered subcutaneously in a neutropenic P. aeruginosa infected mice thigh model (A & B) or administered via intravenous infusion in a neutropenic P. aeruginosa infected rat lung infection model (C & D).

Dose-fractionation of BWC0977 performed in P. aeruginosa infected mice (per dose n = 4 /cage, 8 thigh samples, right & left of each). A BWC0977 was administered subcutaneously at 160 mg/kg total daily dose delivered as a single, two doses or 4 doses over a 24 h period and the CFU burden monitored at various time points. Polymyxin (25 mg/kg) was used as the comparator drug. Data plotted as mean ± SD. B Aggregate plot of BWC0977 efficacy following dose-ranging studies (per dose n = 4/cage, 8 thigh samples, right & left of each) in a neutropenic mice thigh model infected with multiple isolates of P. aeruginosa, A. baumannii, K. pneumoniae and E. coli with pre-existing multidrug resistance mechanisms (Supplementary Table 6). Data plotted as mean ± SD. C Multiple isolates of P. aeruginosa, A. baumannii, K. pneumoniae and E. coli with different resistance mechanisms (Supplementary Table 7) were used for dose-ranging studies and the ΔLog10CFU/gm lung from each animal (n = 2 per treatment group) is plotted as a function of fAUC24/MIC for ease of comparison across isolates. D The PK-PD index that best described the in vivo efficacy of BWC0977 against P. aeruginosa in the neutropenic rat lung infection model. Dose-fractionation was carried out with BWC0977 as per the details shown in Supplementary Table 8. Log10CFU/gm lung from each animal is plotted as a function of fAUC24/MIC, fCmax/MIC and %T > MIC. fAUC24/MIC was identified as the appropriate driver translating to BWC0977 efficacy.
Efficacy in neutropenic rat lung infection model
BWC0977 demonstrated excellent bactericidal dose-response against multiple MDR isolates (includes isolates resistant to ciprofloxacin, colistin and carbapenems) of P. aeruginosa, A. baumannii, K. pneumoniae and E. coli (Fig. 2C; Supplementary Table 7). Dose-fractionation study was designed in the rat inhalation model infected with P. aeruginosa ATCC27853. BWC0977 was administered as a 2-hour intravenous infusion, starting 2 h post-infection with total doses of 450, 400, 350, 300, 150, 75, 40, 20, 10 & 5 mg/kg, fractionated as q24h, q12h, or q8h over a 24-hour period. The PK-PD indices for the various doses administered are shown in Supplementary Table 8. The PK-PD index that best described the in vivo efficacy of BWC0977 against P. aeruginosa was fAUC/MIC (r2 = 0.90), followed by %T > MIC (r2 = 0.83), and fCmax/MIC (r2 = 0.74) (Fig. 2D). Similar PK-PD indices were obtained with A. baumannii ATCC19606 (Supplementary Figure 2A; fAUC/MIC r2 = 0.84, % t > MIC r2 = 0.81, fCmax/MIC r2 = 0.67); K. pneumoniae SKB067 (Supplementary Figure 2B; fAUC/MIC r2 = 0.91, fCmax/MIC r2 = 0.80, %T > MIC r2 = 0.67).
Absorption, Distribution, Metabolism & Excretion (ADME)
BWC0977 shows plasma protein binding in mice (86%), rats (93%), guinea pigs (84%), dogs (94%), cynomolgus monkeys (91%), and humans (94%). It neither partitions into red blood cells nor crosses the blood-brain barrier. BWC0977 primarily undergoes metabolism by the CYP3A4 enzyme; 80% in reaction phenotyping assay. Intrinsic clearance (CLint) values predicted a low clearance [<30% of liver blood flow (LBF)] in rat, dog, monkey, and human hepatocytes. The predicted human plasma CLp is 0.33 L/hr/kg and the predicted % human LBF (1.24 L/hr/kg) is 28%. BWC0977 has low potential to undergo drug-drug interaction (DDI) as it does not inhibit the CYP1A2, CYP2C9, CYP2C19, CYP2D, CYP3A4 or CYP3A5 enzymes up to 100 µM or induce the CYP1A2, CYP2B6, or CYP3A4 enzymes up to 100 µM. BWC0977 is a substrate of the MATE-2K and OAT-3 human transporters; kinetic rate constants Km and Vmax of MATE2-K transport in HEK293 cells were 469 µM and 111 pmol/mg/min, respectively. Similarly, the kinetic rate constants Km and Vmax of OAT3 transport in HEK293 were 121 µM and 76.1pmol/mg/min, respectively. BWC0977 is a P-glycoprotein (P-gp) substrate and exhibits low permeability and is neither a substrate nor an inhibitor of the bile salt export pump (BSEP) up to 50 µM.
Linear PK was observed in mice, rats and dogs following single and multiple-dose intravenous administration with good distribution across tissues and dose-proportionality in Cmax and exposure (Supplementary Table 9). No accumulation was observed in PK studies following multiple-dose administration, and this was further confirmed in the 14-day Good Laboratory Practice (GLP) studies in rat and dog. Similar elimination half-life (t1/2) of 5−6 h was observed across species. In rats, about 1.5% of the total dose administered was observed unchanged in the bile. In rats and dogs, the proportion of unchanged drug excreted in urine was 27% and 10%, respectively.
Epithelial lining fluid concentrations in mice and rats
BWC0977 was administered subcutaneously at 10, 40, 80 and 120 mg/kg, q24h in neutropenic CD-1 mice infected intramuscularly with P. aeruginosa NCTC 13921 in the thigh. Using the plasma protein binding value of 87%, the free plasma levels were calculated. The concentration of BWC0977 was significantly higher in the ELF compared to the free plasma levels (Fig. 3A, B; Supplementary Table 10).
Fig. 3. Determination of pharmacokinetics (PK) in the plasma and epithelial lining fluid (ELF) in neutropenic mice and rats following infection with Pseudomonas aeruginosa.

A In neutropenic CD-1 mice, (n = 3/cage, per dose for each time point) thigh infection model, plasma PK was determined following subcutaneous administration of 10, 40, 80 and 120 mg/kg of BWC0977 q24h following infection with Pseudomonas aeruginosa NCTC 13921 and plasma samples taken at 0, 0.5, 1, 2, 4, 6, 8, and 24 h post-dosing (n = 3/cage, per dose for each time point). Data plotted as mean ± SD. (B) The ELF was obtained by instilling 2 ml of sterile saline into the lungs of mice and removing saline from the lungs twice at 0, 0.5, 1, 2, 4, 6, 8 h post-dosing. Plasma protein binding was estimated to be ~87%. Data plotted as mean ± SD. C In the neutropenic rat (n = 3) lung infection model, 100 mg/kg of BWC0977 was administered following infection with P. aeruginosa ATCC27853. The ELF and plasma samples were taken at 0.5, 1 h (during infusion), and post-infusion at 1.25, 1.5, 2, 3, 5, 9, 25 h post-dosing (n = 3, per dose for each time point). The ELF was obtained by instilling 2 ml of sterile saline into the lungs and removing saline from the lungs. The free plasma levels were calculated from the total plasma levels using the plasma protein binding value of 94%. For all evaluations mean per dose/time point ± SD is plotted.
BWC0977 was administered intravenously at 100 mg/kg over 1-hour in neutropenic rats infected with P. aeruginosa ATCC27853 and the ELF and plasma samples were taken at various time points post-infusion26,27. The concentration of BWC0977 was significantly higher in the ELF compared to the free plasma levels (Fig. 3C; Supplementary Table 11).
Pre-clinical safety studies in rats and dogs
BWC0977 displayed a hERG IC50 of 131 µM in an automated patch-clamp and 49.7 µM in the manual patch-clamp assay. It did not inhibit other cardiac ion channels at concentrations up to 300 µM. In a dog cardiovascular study, after 1-hour infusion of 30 or 60 mg/kg, an increase of 4–11 msec in QTc was observed within 3−6 h with notable increase in heart rate at both dose levels. The no-observed-adverse-effect-level (NOAEL) in rats was 75 mg/kg/day and based on clinical observations demonstrated AUC0–24 of 52 µg/ml*hr with corresponding Cmax of 26 µg/ml. There were no microscopic findings in any organs attributable to systemic BWC0977 toxicity in rats up to the highest dose level (250 mg/kg/day) assessed, with corresponding AUC0–24 of 355 µg/ml*hr. In dogs, the NOAEL was determined as 40 mg/kg/day due to clinical observations. Microscopic changes consistent with hepatobiliary toxicity was observed at higher dose levels and the AUC0–24 achieved at 40 mg/kg/day in dogs was 90 µg/ml*hr, with corresponding Cmax of 25 µg/ml.
Safety & PK following administration of BWC0977 in healthy human volunteers
Human PK values were predicted based on the 2-compartment open model which was the best-fit model of mean PK data following intravenous bolus/infusion of different doses of BWC0977 in mice, rats, guinea pigs and dogs. Each of the PK parameters estimated was scaled as per body weight of the pre-clinical species using simple allometry and linear-regression analysis. Allometric coefficient and intercept were determined and used to estimate the human PK parameters as per simple allometric approach for humans (assumed weight of 70 kg). The predicted human PK parameters of CL, CLD2, V1 and V2 were 20.17 L/hr, 1.002 L/hr, 40.84 L, and 23.13 L, respectively.
In a single-ascending dose study (https://clinicaltrials.gov/study/NCT05088421) in healthy volunteers, BWC0977 was generally well tolerated following single IV infusion dose of 120, 240, 480, 720 or 1050 mg (Fig. 4A). The plasma Cmax and AUC increased proportionally with dose (Fig. 4B; Supplementary Table 12). The average apparent terminal half-life increased gradually with dose, from 3.3 h for 120 mg to 8.4 h for 1050 mg of BWC0977. The amount of drug excreted in urine during the 48 h interval was less than dose-proportional to increasing dose, with fraction of dose excreted unchanged in urine gradually decreasing from 35% for 120 mg to 20% for 1050 mg (Fig. 4C). Across the dose levels, mean clearance (CL) and VD at steady state (Vss) was 19.62–21.08 L/hr and 44.03–51.50 L/hr, respectively. There was good concordance between the predicted and observed PK parameters (Fig. 4D, E).
Fig. 4. Plasma and urine pharmacokinetics of BWC0977 following 2-hr intravenous administration in the single-ascending dose (SAD) study in healthy human volunteers. Modelling and prediction of human PK parameters based on experimental data from preclinical species.

The PK of BWC0977 administered as a single intravenous infusion at doses 120, 240, 480, 720 or 1050 mg in healthy volunteers (n = 6 per dose) over 120 min. A The time-concentration response in plasma observed in the ascending dose groups. Mean ± SD from 6 subjects plotted versus time. B Dose proportionality observed with respect to plasma Cmax and AUC. Mean ± SD from 6 subjects plotted versus dose (mgs). (C) The amount of drug excreted in urine in the 48 h interval following dose administration was less than dose proportional to increasing dose, with the fraction of dose excreted in urine gradually decreasing from 35% for 120 mg BWC0977 to 20% for 1050 mg BWC0977. Mean ± SD from 6 subjects plotted versus dose (mgs). A model built on simple allometric scaling was used to predict the human PK parameters: Cmax (D) & AUC (E) based on a 2-compartment open model. Mean ± SD from 6 subjects plotted for each observed parameter. This was the best-fit model of the mean PK data generated following intravenous bolus / infusion administration of different doses of BWC0977 into mice, rats, guinea pigs and dogs. There was good concordance between the predicted and observed parameters.
In the multiple-ascending dose study (https://clinicaltrials.gov/study/NCT05942820), two subjects completed 12/14 and 14/14 doses with the test substance (240 mg) as planned with no evidence of erythema or phlebitis but with minimal pain following drug administration. Bedside ultrasound at the end of the dosing revealed small non-occlusive thrombus proximal to the infusion site in both volunteers. There were no systemic adverse events. The blood profile for biochemical parameters for various organ functions including coagulation parameters were found to be normal. There was no evidence of a thrombus on the other arm where the volunteers were cannulated for PK sampling. Yet, due to the evidence of thrombus at the infusion site, we decided not to progress further until we get to the root cause of thrombus formation. It is important to rule out that BWC0977 per se did not have a clotting risk (the extensive preclinical GLP toxicology studies in rats and dogs did not reveal any evidence of such a risk). Therefore, we have stopped C002-2023-01 and are currently developing a new formulation.
Discussion
There has been a steady decline in the antibiotics pipeline due to lack of investment, broken market dynamics for new antibiotics and the exit of pharmaceutical companies from this space28. Globally, bulk of antibiotic discovery and development are happening in small biotech companies29. Given the scientific challenges in developing affordable, point-of-care diagnostics, there is a dire need for broad-spectrum antibacterials to save human lives and help clinicians to empirically treat critically-ill patients with suspected MDR infections30,31. The discovery of BWC0977 with broad-spectrum activity against MDR pathogens, desirable safety profile and key attributes supporting human studies is unique among the NBTIs32–34.
During the hit-to-lead program, the weak enzyme inhibition of compound 1 against E. coli gyrase and topoIV was attributed to the lack of contact with double-stranded DNA (dsDNA), which is critical for NBTIs to exert their antibacterial activity35. A review of the literature and structural similarity analyses showed that the screening hit (compound 1) is very similar to NBTI that have been published in literature34,35. By deciphering the structure-activity relationship of the NBTI class from literature, the initial medicinal chemistry design strategy focused on the design of a LHS ring system capable of interacting with the DNA base pairs. Our design strategy to introduce the oxindole/quinolone ring to enable optimal interaction with dsDNA part of gyrase complex in compounds 2-5 resulted in significant improvements in enzymatic potency and broad-spectrum MIC activity. This further reinforced the critical need for a NBTI to contact dsDNA and interact with the gyrase and topoIV. Surprisingly, the improved potency seen with compounds 2-5 came at a cost of hERG liability, which necessitated further medicinal chemistry optimization. Shifting the basic nitrogen along the 4-atom linker chain by connecting both the LHS and oxazolidinone ring system with calculated pKa of 7.27–8.51 was found to be compatible for potency. Efforts to mitigate hERG inhibition while improving the broad-spectrum MIC was accomplished by lowering of lipophilicity (logD) with reduced basicity using nitrogen insertion in the RHS ring. The creation of pyrimido-oxazinone RHS ring, helped in mitigating the hERG inhibition in compound 6 versus compound 5. Encouraged by this strategy, the lipophilicity was modulated with a nitrogen shift on RHS ring and chirality variation at C-5 of the oxazolidinone ring with compound 8 and 9. Changing the pyrimido-oxazinone RHS into a pyrazino-oxazinone ring in compound 8 and 9 with moderate increase in lipophilicity provided the breakthrough convergence of improved antibacterial MIC with hERG attenuation. The convergence is likely due to the increased bacterial permeability and effective disruption of pyrazino-oxazinone ring on the hERG channel. The stereo-selectivity at C-5 carbon of oxazolidinone ring had minimal impact on potency, indicating that chirality does not influence the binding of BWC0977. The SAR modification leading to the discovery of BWC0977 is presented (Supplementary Figure 3).
We employed molecular docking to probe the binding mode of BWC0977 with E. coli gyrase and topoIV (Fig. 5A, B). The modelling predicts a critical hydrogen-bond interaction with RHS ring amide-NH on the pyrazino-oxazinone ring and D82 carboxylate at GyrA dimer interface and Van der Waals contact between -CH2 portion of oxazinone ring in BWC0977 and sidechain of M120. Such interactions have been reported for other NBTIs (PDB ID:5BS336 and 6QTK37). However, the oxazolidinone ring carbonyl of BWC0977 also forms a unique hydrogen-bond interaction with sidechain -NH of R121 (PDB ID:7FVT32). On the other hand, LHS portion of BWC0977 adopts its role as DNA stacker, a signature feature observed across NBTIs. The unique spatial arrangement of BWC0977’s key interaction residues, situated at least 4-atoms away from the ciprofloxacin ligand atoms, imparts a unique mode of binding and allows BWC0977 to circumvent cross-resistance to FQs. To validate the molecular docking-predicted binding mode of BWC0977, two independent replicas of MD simulations (100 ns) were performed with E. coli gyrase and TopoIV. The MD simulation results revealed no significant difference compared to the initial binding mode, demonstrating the stability of the binding mode over the 100 nanosecond simulation period (Supplementary Figure 4 & Supplementary Figure 5).
Fig. 5. Molecular docking to identify unique residues of (A) Gyrase and (B) Topoisomerase IV involved in key interactions with BWC0977.

The crystal bound conformations of ciprofloxacin mapped to EcGyrase (A), and EcTopoIV (B). The residues which form key ion-mediated interactions are shown as sticks and labelled. Mg2+ ions are shown as black spheres and ion-mediated interactions are highlighted with broken lines. The BWC0977 binding orientation determined by molecular docking is also shown as sticks to illustrate the variable binding pockets. All key interaction residues of BWC0977 are minimum 4 atoms away from the ciprofloxacin ligand atoms. Also, the DNA bases where BWC0977 left-hand side (LHS) ring stack are different from ciprofloxacin interacting DNA base pairs. Therefore, none of the BWC0977 interacting amino acid residues are directly involved in ciprofloxacin binding, implicating an unlikely possibility of development of cross-resistance.
Furthermore, BWC0977 exhibits excellent broad-spectrum activity across a global panel of MDR pathogens with varying resistance mechanisms including resistance to FQs38. FQ resistance arises due to specific mutations on target gene (gyrA), target protection (qnr), target modifications (aac6) or due to efflux (qep)39. In vitro studies with BWC0977, clearly indicate that these changes do not affect its activity and BWC0977 retains its inhibition of target enzyme and antibacterial activity even under pre-existing mutations, while FQs lose their inhibition and MICs (Fig. 6). Given its dual target inhibition, BWC0977 displays a low rate of spontaneous resistance frequency across diverse Gram-negative pathogens (<10−9).
Fig. 6. Non-overlapping MIC (µg/ml) between ciprofloxacin (•) and BWC0977 (Δ) across clinical isolates.

Scatter plot of individual MICs of 35–40 fluoroquinolone-resistant clinical isolates of E. coli, A. baumannii and P. aeruginosa, and 98 fluoroquinolone-resistant clinical isolates of K. pneumoniae. The MIC values of BWC0977 and ciprofloxacin are non-overlapping, indicating the lack of cross-resistance between ciprofloxacin and BWC0977, a finding that will support the use of BWC0977 for treating infections caused by fluoroquinolone-resistant bacterial pathogens.
We used mice (thigh) and rat (lung) models of infection to establish the PK-PD drivers of efficacy. These studies showed significant reduction with all the dosing groups in bacterial load compared to the control animals. The fractionated regimens suggested the PK-PD driver to be fAUC/MIC. BWC0977 showed a higher volume of distribution (VD), ~20-fold higher exposures in ELF compared to free plasma in neutropenic P. aeruginosa infected mice and rat lungs. The high VD and ELF exposures drive the profound bactericidal effect seen in the rodent lung infection models and this has the potential to translate into clinical benefit in treating various bacterial pneumonias.
Detailed metabolic studies showed CYP3A4 to be the major metabolizing enzyme and hepatic metabolism of BWC0977 to be a minor contributor for its elimination. The dose-limiting toxicities observed in rat and dog studies were clinical observations primarily consisting of decreased activity, fast or laboured respiration, likely attributable to Cmax. These observations are unlikely to be centrally mediated, since BWC0977 does not cross the blood-brain-barrier. The observations are most likely attributable to peripheral inhibition of acetylcholine esterase (AChE), which was one among the four (AChE, MAO-B, Adrenergic α1 A, 5-HT1A) hits from the Cerep-88 secondary pharmacology screen. The inhibition of AChE by BWC0977 in vitro at 30 μM (14.05 μg/mL) was rapidly reversed upon a 100-fold dilution to 0.3 μM (0.14 μg/mL), in the presence of excess AChE. There were no treatment-related trends in any of the clinical biochemical tests, vital signs, physical examinations, and electrocardiogram (ECG) assessments between BWC0977 and placebo. As cardiovascular changes can be readily monitored by ECG, the transient nature of changes in parameters, the ability to modulate the Cmax with duration of infusion, and by fractionating the daily dose, these effects were not considered to pose significant risk to humans.
Given the global spread of MDR pathogens implicated in healthcare-associated infections (HAI), BWC0977 offers an attractive choice as a broad-spectrum antibiotic to physicians treating critically-ill patients40. The in vitro spectrum of activity of BWC0977 and results from the other preclinical studies presented here, are supportive of BWC0977 advancing into clinical trials to treat cUTI, HABP, VABP and cIAI.
Cystic fibrosis (CF) patients experience frequent bacterial infections due to a range of pathogens that evolve into extremely drug-resistant clones over time41–43. The spectrum of activity against pathogens from CF patients (B. cepacia, MRSA and P. aeruginosa), coupled with its high pulmonary drug levels also support trials in patients with CF.
Additionally, BWC0977 has demonstrated potent MIC against priority biothreat pathogens and efficacy in murine infection models including Burkholderia pseudomallei (data not shown). HABP and VABP are the greatest threats to hospitalized patients, posing severe treatment challenges to physicians. These patients are mostly in intensive-care units, where MDR pathogens can readily spread and cause life-threatening infections44. Under such situations, BWC0977 could become the treatment of choice due to its higher pulmonary drug levels and significant killing of MDR pathogens in rodent models of pneumonia. BWC0977 demonstrated significant bactericidal efficacy against multiple drug-susceptible and drug-resistant strains in the rodent thigh and lung infection models.
Based on the promising safety and tolerability data from the first-in-human SAD study, BWC0977 was further investigated in a multiple-ascending dose (MAD) study (https://clinicaltrials.gov/study/NCT05942820). In the MAD study, we encountered infusion site reactions (ISRs). The MAD study was terminated, and current efforts are ongoing to develop a new formulation that will minimise the risk of ISRs. The successful completion of MAD study with a new formulation will guide the clinical development of BWC0977 in human patients to treat a broad variety of MDR infections. A limitation of this study is the probability of target attainment (PTA) which needs to be established from the MAD studies to determine the frequency of drug dosing required to achieve efficacy in a patient population harbouring various target pathogens.
In early human studies, BWC0977 appears to be well tolerated at exposures required to achieve efficacy. Based on its potent antimicrobial activity against a diverse set of MDR pathogens, well-characterized and monitorable preclinical safety risks, BWC0977 appears to be differentiated among other NBTIs as well as newer drugs either in the market or in clinical development45.
Methods
The research presented in this manuscript complies with all relevant ethical regulations. This includes ethical approval for all the animal studies employed in this work, and the informed consent and approvals associated with the Phase I clinical trial presented in this work.
Ethics approval for preclinical animal studies
Dose finding studies and efficacy of BWC0977 in the neutropenic murine thigh infection model, pharmacokinetics of BWC0977 in neutropenic mouse thigh model infected with Pseudomonas aeruginosa NCTC 13921, and epithelial lining fluid concentration determination in neutropenic mouse thigh model infected with Pseudomonas aeruginosa NCTC 13921 were conducted in the laboratory of Professor William Hope by the Antimicrobial Pharmacodynamics and Therapeutics Group (Department of Molecular and Clinical Pharmacology), University of Liverpool, United Kingdom. These experiments were conducted under UK Home Office project License PAC022930 which was renewed in 2022 as PP3585942. These licenses and all experiments conducted under them are approved by the University of Liverpool Animal Welfare Ethics Review Board.
Pharmacokinetics of BWC0977 in plasma and pulmonary epithelial lining fluid in a neutropenic lung infection model in rats, dose fractionation and dose response studies of BWC0977 in rat lung infection model, and determination of PK-PD index for BWC0977 in the rat lung infection model were conducted at TheraIndx Lifesciences Private Limited, Bangalore, after obtaining permission from the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), New Delhi, India and approval from the Institutional Animal Ethics Committee (IAEC). Approval numbers: IAEC/08/2018/078, IAEC/13/2020/159, IAEC/22/2022/254, IAEC/22/2022/255, and IAEC/24/2023/269.
The dog pharmacokinetic study, rat and dog safety and toxicokinetic studies were performed in accordance with the agreed Protocol and with Covance Laboratories Limited standard operating procedures. Aspects of the studies performed at Covance, Harrogate were conducted in compliance with the United Kingdom Good Laboratory Practice (GLP) Monitoring Authority, Medicines and Healthcare Products Regulatory Agency (MHRA) Good Laboratory Practice Regulations 1999, Statutory Instrument 1999 No. 3106, as amended by the Good Laboratory Practice (GLP) - (Codification Amendments Etc.), Regulations 2004, Statutory Instrument 2004 No. 994 and the Organisation for Economic Co-operation and Development (OECD) Principles of Good Laboratory Practice ENV/MC/CHEM (98) 17 (revised 1997, issued January 1998).
The pharmacokinetic studies in rats & mice, and the bile duct cannulation (BDC) study in rats were performed at Syngene International, India, were carried out on naïve animals as per the provisions of the Institutional Animal Ethics Committee (IAEC) approvals, viz., SYNGENE/IAEC/800/01-2017, SYNGENE/IAEC/937/05-2018, SYNGENE/IAEC/1005/11-2018, and SYNGENE/IAEC/1147/02-2020.
Guinea pig pharmacokinetic study in male Guinea pigs (BWR-001 / PKMGP-001) was conducted under the approved protocol PK-PRO-715-06 by Suven Life Sciences Institutional Animal Ethics Committee Meeting dated 18-May-2019 and facility is registered for research and breeder; registration number as 769/PO/RcBi/SL/03/CPCSEA; dated 14-Jan-2016. The studies were approved by the Animal Welfare Ethical Review Board (A WERB) from the Suven Life Sciences Limited, Pashamylaram, India.
Ethics Approval for First in human studies
The two first in human trials were conducted in CMAX, Adelaide, Australia.
C001-2020-01 (https://clinicaltrials.gov/study/NCT05088421): A randomized, double-blind, placebo-controlled, Phase 1 study of the safety, tolerability and pharmacokinetics of single and multiple ascending doses of BWC0977 in healthy adult volunteers.
C002-2023-01 (https://clinicaltrials.gov/study/NCT05942820): A randomized, double-blind, placebo-controlled, Phase 1 study of the safety, tolerability, and pharmacokinetics of single and multiple ascending doses of BWC0977 in healthy adult volunteers.
These human studies were initiated following approval by the Bellberry Human Research Ethics Committee (HREC) of the trial design, protocol and the key criteria specified for the study. The Bellberry Human Research Ethics committee (HREC) has scientifically and ethically reviewed this study. This Bellberry HREC is constituted and operates in accordance with the National Health and Medical Research Council’s National Statement on Ethical Conduct in Human Research (2007, incorporating all updates). Bellberry Human Research Ethics Committee does not disclose personal details of its reviewing members. Please note that the Principal Investigator and Co-Investigators were not members of the Bellberry Human Research Ethics Committee that reviewed this study.
Determination of the Minimum Inhibitory Concentration (MIC)
MIC was determined based on the Clinical Laboratory Standards Institute (CLSI) guidelines24 in cation-adjusted Mueller Hinton broth (MHB) using broth microdilution methodology in 96-well microtitre plates (ThermoFisher Scientific, Catalogue No. 130188). Briefly, the inoculum used for all the experiments was derived from a single seed lot, maintained as a glycerol (20%) stock at −80 oC. To revive the cultures, it was first sub-cultured on LB plates for isolated colonies and a single colony inoculated into LB broth, grown for 16–18 h at 37 oC and appropriately diluted in cation-adjusted Mueller Hinton broth (3–7 × 105 CFU/ml). BWC0977 stocks (4 mg/ml) and serial dilutions were prepared in dimethylsulfoxide (DMSO). To 147 μl of bacterial culture, 3 μL of compound from each of the dilutions was added to the wells of 96-well microtitre plates. A 10-concentration range was set up with a start concentration of 4 μg/ml, and two fold serial dilutions resulting in final concentrations of 2 μg/ml, 1μg/ml, 500 ng, 250, 125, 62.5, 31.25, 15.6 and 7.8 ng/ml. For reference inhibitors, the specific solvents used are listed in Supplementary Table 13. Media control, culture control and appropriate reference drug controls were included in each assay. The plates were incubated at 37 oC for 16–18 h. Growth was monitored by checking the absorbance at 600 nm (A600). MIC is the concentration that resulted in ≥80% growth inhibition. MIC90 (µg/ml) values of BWC0977 against multiple bacterial isolates were determined using a total of 8591 isolates comprising 7187 Gram-negative and 1404 Gram-positive organisms. This global collection comprised of geographically diverse set of clinical isolates and was tested in different laboratories [St. John’s Medical College & Hospital and Narayana Health, Bangalore India, JMI Laboratories, International Health Management Associates (IHMA), Colorado State University, University of Michigan, Seattle Children Hospital, University of Alabama, Birmingham, University of Texas Health Science Center at San Antonio / National Institute of Allergy and Infectious Diseases (NIAID), Walter Reed Army Institute of Research (WRAIR), Maryland, USA and United States Military Research Institute of Infectious Diseases (USAMRIID)]. In addition, a CARB-X sponsored MIC90 study of both Gram-positive and Gram-negative bacteria (total of 2,945 isolates) was conducted at IHMA.
Determination of the spontaneous resistance frequency of BWC0977
The spontaneous resistance frequency of BWC0977 was determined and the resistant colonies of Gram-negative bacteria were characterized using microbiology and molecular techniques. Towards this aim, mid-logarithmic phase cultures of Gram-negative bacteria (109 CFU/mL) were plated on Luria-Bertani agar plates containing BWC0977 equivalent to 2x, 4x, 8x and 16x MIC. Plates were incubated for 24–36 h at 37 oC, and the resistance frequency determined by counting the number of colony forming units (CFUs) on agar plates46. BWC0977 MICs for the colonies isolated from drug containing plates were determined and the gyrA and parC gene sequenced to confirm any target site mutations. The resistance frequency of ciprofloxacin was determined in parallel using the same methodology.
Killing kinetics assay
Killing kinetics was determined by enumerating the number of bacterial survivors at various time points following compound exposure. Approximately 107 CFU/ml cells (A600 of 0.1) were treated with various concentrations (2-fold serial dilutions, 9 concentrations, with start of either 256 to 1 μg/ml, 64 to 0.25 μg/ml, 32 to 0.125 μg/ml, 16–0.06 μg/ml, 8 to 0.03 μg/ml, 4 to 0.015 μg/ml, or 2 to 0.008 μg/ml μg/ml) of compounds in a 96-well microtitre plate47. At various time intervals (0,1,3,6,9,12,24 h), 30 μl/150 μl of treated cultures was serially diluted and plated for bacterial survivors on LB agar plates. The plates were incubated at 37 oC for 16–18 h, and the bacterial colonies enumerated. Multiple wells of each concentration with total volume of 150μl (column-wise) was prepared in the same plate. Sampling was done for the same concentration every time from a different well for the various time points. The extent of bactericidality was monitored against a panel of susceptible and MDR strains of E. coli, A. baumannii, K. pneumoniae, P. aeruginosa and E. cloacae. The viable colony forming units (CFUs) were enumerated, expressed as Δlog10CFU for each drug treatment per timepoint in comparison to start log10CFU and the plots generated using GraphPad Prism.
Dose finding studies and efficacy of BWC0977 in the neutropenic murine thigh infection model
These studies were conducted in the laboratory of Professor William Hope by the Antimicrobial Pharmacodynamics and Therapeutics Group (Department of Molecular and Clinical Pharmacology), University of Liverpool, United Kingdom. For the dose finding study, BWC0977 was administered by subcutaneous (SC) injection at 40 mg/kg every 6 h, 80 mg/kg every 12 h or 160 mg/kg every 24 h using a 26 h neutropenic murine dual thigh infection model infected with P. aeruginosa NCTC 13921. Polymyxin B (SC, 25 mg/kg q6h) served as a comparator for this study. Mice were infected intramuscularly with P. aeruginosa NCTC 13921 two hours prior to treatment with BWC0977. Inoculum was adjusted to ~ 1 × 107 CFU /ml and BWC0977 (in 10% ascorbic acid, pH adjusted to 4.6 with sodium bicarbonate) was administered subcutaneously at fractionated dose levels, 40 mg/kg q6h x 4, 80 mg/kg q12 x 2 or 160 mg/kg q24h x1. Animals were euthanized at 2 h post-infection (pre-treatment controls) or serially at 8, 14 and 26 h post-infection (untreated controls and all BWC0977-treated groups); polymyxin B treated group was euthanized at 26 hours post-infection only.
To investigate the efficacy of BWC0977 in the 26 hour neutropenic murine dual thigh infection model following infection with different Gram-negative bacteria, multiple doses (Supplementary Table 14) were administered every 8 h (q8h). Mice (male CD-1, n = 3 per group) were infected intramuscularly with a Gram-negative bacterial inoculum. To make the inoculum, bacteria was revived from −80 °C by streaking onto Mueller Hinton agar and incubating for 16–24 h. Single colonies were picked from the agar plate, emulsified into 30 ml Mueller Hinton broth, and placed on shaking incubator overnight. The overnight culture was centrifuged at 4000 x g for 5 min to produce a bacterial pellet. The supernatant was removed, and the pellet resuspended in sterile PBS and adjusted to the correct OD450 (determined from bacterial growth curve studies). Further dilutions were made in PBS to reach the target CFU/mL (as determined by inoculum finding studies). Two hours post-infection, treatment with BWC0977 was administered subcutaneously (SC) at doses (Supplementary Table 14) q8h and comparator control. Animals were euthanized at 2-hour post infection (pre-treatment controls), or 26 h post-infection (vehicle control and all treated groups) except for animals that reached their clinical endpoints as follows: one animal in the vehicle control group at 20.5 h post-infection, and one animal at 10 mg/kg at 23 h post-infection. Data was collected from all animals at the time of euthanasia. All animals were monitored, and health scored throughout, increasing in frequency, as needed, when clinical observations started to develop e.g. piloerection, hunched posture etc. All laboratory animal experiments were conducted under UK Home Office project License PAC022930 which was renewed in 2022 as PP3585942. These licenses and all experiments conducted under them were approved by the University of Liverpool Animal Welfare Ethics Review Board.
Plasma protein binding assay
Protein binding was measured using the equilibrium dialysis method48. Compound was added to 10% plasma giving a concentration of 10 µM and dialysed with isotonic buffer for 18 h at 37 oC. The plasma and buffer solutions were analysed using generic LC-UV-MS and the first apparent binding constant for the compound derived. The binding constant was then used to determine the % free fraction in 100% plasma.
The percentage of plasma bound/unbound fraction preparation was calculated as follows:
Wherein,
Tc = Total plasma concentration was determined by the calculated concentration on the plasma side of the chamber
Fc = Total plasma concentration was determined by the calculated concentration on the buffer side of the chamber
To = Total compound concentration determined before analysis
Inhibition of supercoiling activity of E. coli gyrase
The E. coli gyrase supercoiling activity assays were performed using reagents obtained from Inspiralis Limited, Norfolk. UK). Test compounds were pre-incubated with 1 nM enzyme at 24 oC for 10 min. Reaction was initiated by the addition of 60 ng relaxed pBR322 DNA, and the incubation was continued at 37 oC for 40 min. Reactions were terminated using a mixture of Proteinase K (3 μL 2% SDS + 0.8 μL 20 mg/mL Proteinase K), followed by 30 min incubation at 37 °C. Samples were mixed with 4 μL STEB [40 % (w/v) sucrose, 100 mM Tris-HCl (pH 8), 10 mM EDTA, 0.5 mg/mL bromophenol blue], and resolved by gel electrophoresis (2.5 V/cm for 3 h) on 0.8 % agarose gel in 1X TAE buffer. Gels were stained with 0.8 µg/mL ethidium bromide (EtBr) for 10 min and the DNA bands were imaged using a gel documentation system. The band intensities were quantified using the Quantity One basic software. The compound IC50 values were determined using non-linear regression, four parameter curve-fit using GraphPad Prism software.
Inhibition of decatenation activity of E. coli topoisomerase IV
The E. coli topoisomerase IV decatenation reactions were performed using 2.5 nM enzyme and 60 ng kinetoplast DNA (kDNA) obtained from Inspiralis. The assay protocol followed was like the above-described supercoiling assays.
Homology Modelling of E. coli Gyrase and Topo IV Complexes
The sequences of E. coli gyrA, gyrB, parC and parE were extracted from UniprotKB. The NBTI class of oxabicyclooctane derivative bound SaGyrase crystal structure (PDB ID: 5BS3) was utilized to build EcGyrase complex. In the case of EcTopoIV, A. baumannii TOPO IV crystal structure (PDB ID: 2XKK) served as a template. SWISS-MODEL49 web server was utilized to build the homology models of independent chains and superimposition protocols of PyMOL software were employed to construct the tetrameric complexes. The coordinates of dsDNA molecules extracted from template crystal structures was used to generate the complete complexes.
Input Compound Structure Preparation
BWC0977 was considered for molecular docking, the 2-D structure was sketched using MarvinSketch program and exported in a.SDF file format. The ligand molecules were prepared in LigPrep50 module of the Schrodinger suite. The structure was expected to be at or near a local energy minimum, but not necessarily either a global minimum or the optimal conformation for binding to the target. The prepared ligands by LigPrep (output) were considered for multi-conformation generation by Macromodel conformation search module i.e., Mixed-torsional/Low-mode sampling (MTLM). This model combines a Monte Carlo method of exploring torsional space that efficiently locates the widely separated minima on the potential energy surface with a low-mode conformational search method along the energetically “soft” degrees of freedom. The OPLS_2005 force field was used, and energy minimization was 500 steps of the TNCG method. The energy window for saving structures was set to 21 kJ/mol. The redundancy threshold was 0.5 Å RMSD, and any redundant conformers were removed. The maximum number of conformers to be generated was set to 25 and default parameters were used for the remaining options.
Glide Molecular Docking
The Glide molecular docking51 of Schrodinger suite was utilized to predict the binding mode of BWC0977 with homology modelled EcGyrase and EcTopoIV protein 3-D structures. Bound co-crystal NBTI ligand (PDB ID: 5BS3) was superposed into the modelled complexes of EcGyrase and EcTopoIV to specify the NBTI binding pocket. The docking grid was restricted to NBTI pocket based on the extracted NBTI bound ligand. Before starting the Glide grid generation for molecular docking, homology model structures were processed using the protein preparation wizard. The Glide-SP (standard precision) algorithm utilizes pre-computed grids generated using receptor sites defined by centroids of the crystallographic ligands. The docking protocol starts with the systematic conformational expansion of the ligand, followed by placement at the receptor site. Minimization of ligand in the field of receptor was carried out using the OPLS-AA force field with the default distance-dependent dielectric parameters. The lowest energy poses are then subjected to a Monte Carlo procedure that samples nearby torsional minima. Different compounds can then be ranked using GlideScore, a modified version of the ChemScore function that includes terms for steric clashes and buried polar groups. Default Van der Waal’s scaling was used (1.0 for the receptor and 0.8 for the ligand). Advanced settings were edited to increase the pose sampling. A total of 10,000 poses (default 5000) per ligand were set for the initial phase of docking and the poses per ligand per energy minimization raised to 1000 from 400. The output from the conformation generation method (25 conformers) was considered as the input for molecular docking. A total of 10 poses per ligand were saved as output for post-docking analyses.
Molecular Dynamics
Initial binding orientations of BWC0977 generated by molecular docking approach with modelled structures of EcGyr and EcTopoIV were considered further for Molecular Dynamics simulations. Three completely independent replicas were launched for each system. The individual pieces of the system i.e., protein, water molecule (TIP3P), Mg2+ and Na+Cl− ions at a concentration of 0.15 mM were built and assembled. Proteins and ions were described by the ff99SB52 and the parameter for BWC0977 was obtained with the GAFF253. The energy minimization, equilibration and MD protocol was carried out with the PMEMD program of AMBER18 MD package54. After initial energy minimization, successive steps of NVT and NPT (300 K, 1 bar) MD were performed, with progressive removal of position restraints applied to the protein atoms. Then, simulations were run for 100 ns (NPT ensemble, 300 K, 1 bar) and the first 50 nanoseconds were considered as equilibration and discarded; the last 50 nanoseconds of each replica were retained for analysis. A time step of 2 femtoseconds was used in the production phase and PME (Particle Mesh Ewald)55 was employed for the treatment of long-range electrostatic interactions, with the application of a switch function between 1.0 and 1.2 nm. Temperature was kept at 300 K by the Nose-Hoover scheme56,57 using a time constant for coupling of 1 picosecond. Pressure was maintained at 1 bar by a semi-isotropic Parrinello-Rahman barostat58, with coupling time constant of 5 picoseconds and compressibility of 4.5 × 10−5 bar−1. All bonds to hydrogen atoms were constrained by the LINCS algorithm59. CPPTRAJ module of AmberTools19 program was used for trajectory analysis. Before analysis, ions, and water molecules beyond 5 Å of the ligand position were removed.
Pharmacokinetics of BWC0977 in neutropenic mouse thigh model infected with Pseudomonas aeruginosa NCTC 13921
The objective of this study was to describe the concentrations of BWC0977 in mouse plasma in a neutropenic murine, P. aeruginosa strain NCTC 13921 infected thigh model. Male CD-1 mice were infected with P. aeruginosa NCTC 13921 in both thighs and BWC0977 administered via a single subcutaneous (SC) injection at 10, 40, 80, or 120 mg/kg. Terminal blood samples (3/time-point) were collected for concentration analysis of BWC0977 at 0.5, 1, 2, 4, 6, 8, and 24 h post-dosing. Using the plasma protein binding value of 87%, the free plasma levels were calculated. BWC0977 concentration analysis in mouse plasma was conducted using LC/MS/MS, with a lower limit of quantitation (LLOQ) of 0.25 μg/mL for plasma. Descriptive parameters were calculated (mean and median concentrations) of which dose-proportionality and other descriptive characteristics were determined.
Epithelial lining fluid concentrations in neutropenic mouse thigh model infected with Pseudomonas aeruginosa NCTC 13921
BWC0977 was administered subcutaneously at 10, 40, 80 and 120 mg/kg, q24h in neutropenic CD-1 mice infected intramuscularly with P. aeruginosa NCTC 13921 in the thigh. The ELF was obtained by instilling sterile saline into the lungs and removing saline from the lungs at 0, 0.5, 1, 2, 4, 6, 8 h post-dosing.
Pharmacokinetics of BWC0977 in plasma and pulmonary epithelial lining fluid in a neutropenic lung infection model in rats
100 mg/kg of BWC0977 was administered intravenously for 60 min once to neutropenic rats (male, Wistar) infected with P. aeruginosa ATCC27853 (MIC − 0.25 µg/ml) and plasma samples were taken at 0.5 and 1 hour (during infusion) and post-infusion at 1.25, 1.5, 2.0, 3.0, 5.0, 9.0 and 25 h. Similarly, the epithelial lining fluid (ELF) was obtained by instilling 2 ml of sterile saline into the lungs and removing saline from the lungs. PK data analysis was done using the WinNonlin® software.
Dose fractionation and dose response studies of BWC0977 in rats
For efficacy studies, prior to the start of the infection process, all animals were divided into groups of 2 or 3 animals each. Four days and one day before the date of infection, for inducing neutropenia, each rat was dosed intra-peritoneally with cyclophosphamide equivalent to 150 mg/kg and100 mg/kg respectively. Rats were then placed into an induction chamber and anaesthesia induced by exposing the animals to 3–5% isoflurane in an oxygen flow set at approximately 1 liter per minute. On the day of the infection, a 16−18 h Casein Soybean Digest (CSD) broth culture, was centrifuged and the cells resuspended in sterile normal saline to obtain ~ 109 CFU/ml. Infection was initiated by instilling 35 μl containing ~107 CFU/rat of the inoculum into each nostril of the anesthetized animal with the following strains: A. baumannii ATCC19606, A. baumannii SAC002 (a recent clinical isolate from Bangalore, India; ciprofloxacin & meropenem-resistant), E. coli ATCC BAA-2469 (ciprofloxacin & meropenem-resistant), E. coli ATCC BAA-2471 (ciprofloxacin & meropenem-resistant), E. coli SEC-015 (a recent clinical isolate from Bangalore, India; ciprofloxacin & meropenem-resistant), K. pneumoniae SKB067 (a recent clinical isolate from Bangalore, India; colistin, ciprofloxacin & meropenem-resistant), K. pneumoniae ATCC 13883, K. pneumoniae KPNIH1 (ciprofloxacin & meropenem-resistant), K. pneumoniae MKP103 (ciprofloxacin-resistant), P. aeruginosa ATCC 27853, P. aeruginosa SPA041 (a recent clinical isolate from Bangalore, India; ciprofloxacin-resistant). Four hours post-infection, animals were treated at a constant rate infusion with doses of either BWC0977 {4% L-Ascorbic acid + 1.8 % lactic acid + 2.5% niacinamide in WFI, pH 4.5 with sodium bicarbonate, 3 mg/kg, 10 mg/kg, 30 mg/kg, 100 mg/kg} by constant rate intravenous infusion over 2 h or meropenem (in saline,15 mg/kg bid, intravenous bolus dose) or ciprofloxacin (in MilliQ water, pH 4. 5 adjusted with HCl, 30 mg/kg bid, intravenous bolus dose). Rats were anaesthetized using ketamine 70 mg/kg IP + xylazine 20 mg/kg IP at a dose volume of 10 ml/kg, at the rate of 1 ml/hr, except untreated controls. Animals were euthanized at 4 h post-infection (pre-treatment controls), or 28 h post-infection (vehicle control and all treated groups). The euthanized animals were dipped into 70% ethanol for surface decontamination, lung muscles aseptically excised, weighed, placed into 2 ml broth, and homogenized. Serial ten-fold dilutions of the lung homogenates were prepared in sterile broth, plated onto agar plates and CFUs enumerated following 16−18 h of incubation at 37 °C. The individual log10CFU/g lung values were plotted against C/MIC, AUC/MIC and %T > MIC (24 h) and the relationship was assessed with the appropriate pharmacodynamic model using the GraphPad Prism software. The neutropenic rats were monitored throughout the study for general clinical signs.
Determination of PK-PD index for BWC0977 in the rat lung infection model
Dose-fractionation studies were carried out to identify the PK-PD index that correlates best with the efficacy of BWC0977 in the rat lung infection model using different Gram-negative pathogens. Neutropenic rats were infected intranasally with ~7 × 107 CFU/animal of P. aeruginosa ATCC27853. Four hours post-infection, animals were treated with varying total doses of 450, 400, 350, 300, 150, 75, 40, 2 0,1 0 & 5 mg/kg fractionated as q24h, q12h, or q8h over a 24 h period and administered as a 2 h intravenous infusion, over a period of 24 h. Meropenem (total dose of 30 mg/kg), administered as 15 mg/kg, i.v. bolus or twice daily served as a positive control to validate the study. The mean log10CFU/g lung was estimated in each group. The individual log10CFU/g lung values were plotted against C/MIC, AUC/MIC and %T > MIC (24 h) and the relationship was assessed using the appropriate pharmacodynamic model using the GraphPad Prism software.
Reaction phenotyping
To determine the major human CYP P450 isozymes (1A2, 2C9, 2C19, 2D6 and 3A4) responsible for metabolism of the test compound, using the method was reported earlier60. Working stock solutions (50 µM) of test compound (4 µL) was spiked to 356 µL of CYP isozymes to obtain a final test compound concentration of 0.5 µM and pre-incubated for 10 min at 37 oC. After pre-incubation, 45 µL of the pre-incubation mixture was precipitated with 200 µL of ice-cold acetonitrile containing internal standard (0 min sample) and 5 µL of 10 mM NADPH was added to the mixture. To the remaining mixture, 35 µL master stock solution of NADPH (10 mM) was added to the remaining pre-incubation mixture and incubated on a shaking water bath for 60 min at 37 oC. At each time point (0, 3, 6, 9, 12, 15, 20, 30, 45, and 60 min), 50 µL of incubation mixture was precipitated with 200 µL of ice-cold acetonitrile containing the internal standard. Samples were vortexed for 10 min and centrifuged at 1800 x g for 10 min. After centrifugation, 100 µL of supernatant was diluted with 100 µL of water and analysed by LC-MS/MS analysis.
Similarly, for without co-factor incubations, instead of 10 mM NADPH, 100 mM potassium phosphate buffer was added to the samples and incubated for 60 min at 37 oC. The 0 minute samples (50 µL) were precipitated immediately with 200 µL of acetonitrile containing the internal standard. After 30 and 60 min of incubation time, 50 µL of sample was precipitated with 200 µL of acetonitrile containing the internal standard. After precipitation, the samples were treated like the co-factor samples and analysed using LC-MS/MS.
In vitro - in vivo extrapolation (IVIVE) of clearance in humans and prediction of human pharmacokinetic parameters
The well-stirred model61 was used for predicting human CL using human hepatocyte Clint and free fraction (fu) in human plasma. Liver blood flow rates, liver weights, hepatocellularity and in vitro - in vivo correlation / extrapolation (IVIVC / E) templates were routinely employed.
Briefly, cryo-preserved hepatocytes were thawed and transferred into pre-warmed (maintained at 37 oC) buffer medium, and hepatocytes mixed by gently inverting the tube three times. The cell suspension was centrifuged at 50 x g at room temperature for 5 min. The supernatant was discarded, and cell pellet loosened by gently swirling the centrifuge tube and the hepatocytes resuspended in 2 mL of pre-warmed buffer. The total cell count was determined, and the number of viable cells enumerated by trypan blue dye exclusion method. The acceptable cell viability at the beginning of the assay was ~85%. The hepatocyte suspension was diluted with the buffer to attain a final concentration of 1 million cells/mL (1 × 106 cells/mL). The stock (10 mM) solution of test compounds and positive controls were prepared in DMSO. Subsequently, sub-stock (1 mM) solutions were prepared by diluting 10 µL of 10 mM stock solution with 90 µL of DMSO. The final working stock (1 µM) solution was prepared by diluting 2 µL of sub-stock solution with 1998 µL of incubation media. For the assay, working stock solution (1 µM) was spiked into hepatocyte incubation mixture to obtain a final concentration of 0.5 µM. The final organic content in the assay was <0.1%.
The stability assay was conducted in duplicate (n = 2). Manually, 200 µL of diluted hepatocyte suspension (1 × 106 cells/mL) was added to each well of a 48-well plate. 200 µL of test compound (1 µM) prepared was added in the incubation medium to each of the wells containing hepatocytes. Final concentration of hepatocytes and test compound in the assay were 0.5 × 106 cells/mL (0.5 million cells/mL) and 0.5 µM, respectively. The 48-well plate was placed in an incubator maintained at 37 oC, 5% CO2 atmosphere and 95% relative humidity. The hepatocyte mixture was incubated for 120 min with constant shaking at 250 rpm. At each time-point (0, 5, 10, 15, 30, 60, 90, and 120 min), 50 µL aliquot of hepatocyte mixture was added into 96-deep well plate and precipitated with 200 µL of acetonitrile containing an internal standard. The samples were mixed well in a vortex mixer and centrifuged for 10 min at 2000 x g. After centrifugation, the supernatant (100 µL) was separated and transferred to a fresh 96-well plate and diluted with 100 µL of water. The samples were analyzed using LCMS/MS method.
The metabolic stability was expressed as the percentage of parent remaining and calculated from the peak area ratio of NCE remaining after incubation (tx) compared to the time zero (t0) incubation. The percentage of parent test compound remaining at each time point was calculated by comparing the peak area ratio of test compound after incubation (tx) with peak area of time zero (t0) incubation. Similarly, half-life (t1⁄2) was calculated using the following equation:
The intrinsic clearance (Clint) was calculated using the following equations:
Scaling factors to represent hypocellularity million cells/gm liver and liver/kg body weight.
P-gp, BCRP and BSEP inhibition and substrate determination
Membrane vesicles were diluted in incubation medium and added to a 96-well incubation plate. To evaluate the test article as an inhibitor - solvent control, test article or inhibitors in DMSO were added to the membrane vesicles (1% v/v of the final reaction volume) and were pre-incubated for 15 min at 37 ± 2 °C. After pre-incubation, the incubation was initiated by the addition of probe substrate and MgATP (4 mM) or MgAMP (4 mM) in incubation medium and incubated for the designated time.
To evaluate the test article as a substrate - solvent control, or inhibitors in DMSO were added to the membrane vesicles (1% v/v of the final reaction volume) and were preincubated for 15 min at 37 ± 2 oC. After pre-incubation, the incubation was initiated by the addition of the probe substrate or the test article and MgATP (4 mM) or MgAMP (4 mM) in incubation medium and incubated for the designated time. At the end of incubation period, an aliquot was collected for recovery measurement. The final protein concentration was 50 μg / incubation. The incubation reaction was terminated by the addition of chilled washing mix and samples filtered using a filter plate. The filtered samples were washed five times with chilled washing mix and dried at room temperature for approximately two hours. Experimental conditions are summarized in Supplementary Tables 15, 16. For samples incubated with radiolabeled substrate, the substrate was extracted from the filtered vesicles with scintillation fluid and analyzed using a MicroBeta2 liquid scintillation counter. For samples incubated with unlabeled substrate, the substrate was extracted from the filtered vesicles with 50:50 v/v methanol:water containing internal standard and analyzed by LC MS/MS.
OATP, OAT, OCT and MATE substrate and Km / Vmax determination
Cells were plated onto standard 24-well tissue culture plates in cell culture medium 1 to 3 days prior to the experiment. OATP1B1, OATP1B3, MATE1, MATE2-K and control cells were incubated with butyric acid (10 mM) for 24 h prior to the experiment to induce transporter gene expression. Incubation of HEK293 cells were carried out in HBSS buffer containing sodium bicarbonate (4 mM) and HEPES (9 mM), pH 7.4 (OATP, OAT and OCT) or pH 8.5 (MATE).
Prior to the experiment, cell culture plates (transporter-expressing and control cells) were removed from the incubator, the cell culture medium was removed, and incubation medium was added to the plate to rinse the cell culture medium from the cells. To evaluate BWC0977 as a substrate, incubation medium was replaced with incubation medium containing the inhibitor or solvent control, and the plates were preincubated for 15 or 30 min. After pre-incubation, incubation medium was replaced with incubation medium containing the inhibitor or solvent control and BWC0977 or positive control substrate. Samples were incubated at 37 ± 2 °C in triplicate for the designated time. After incubation, incubation medium was removed with an aliquot collected to measure recovery, and cells were rinsed once with chilled PBS containing 0.2% w/v BSA and twice with chilled PBS. For samples incubated with radiolabeled substrates, the PBS was removed, and sodium hydroxide (0.1 M) was added with pipette mixing to extract the compound from the cells. An aliquot of the medium was added to a 96-well plate, diluted with scintillation fluid, and analyzed on a MicroBeta2 scintillation counter. The concentration of protein in incubations was determined with a BCA Protein Assay Kit. Experimental conditions are summarized in Supplementary Table 17.
For samples incubated with unlabeled substrates, the PBS was removed, and 50:50 v/v methanol:water containing an internal standard was added with pipette mixing to extract the compound from the cells for analysis using a LC-MS/MS. When the accumulation of unlabeled substrate was measured, the protein concentration in representative wells was measured as protein concentration that cannot be determined after extraction with organic solvent.
Samples were analyzed by multiple reaction monitoring LC-MS/MS methods developed at the testing facility. Analysis was performed with an appropriate SCIEX or Waters mass spectrometer equipped with a Shimadzu Nexera, Shimadzu Prominence or Waters Acquity LC system interfaced by electrospray ionization.
Pharmacokinetic studies in mice and rats
Intravenous (i.v) formulations for pharmacokinetic (PK) studies were formulated in different vehicles and excipients to achieve the desirable solubility for intravenous route of administration. An appropriate amount of the test compound was weighed and dissolved in the required volume of vehicle, followed by vortexing for a few seconds to dissolve the compound. Then the solution (or) suspension was sonicated at room temperature for 5 min to obtain a visually clear solution and or homogenous suspension. All the formulations were prepared freshly at room temperature before dosing. All these formulations were observed to be stable at room temperature for more than 24 h. The formulation details of each of the compound tested is shown in Supplementary Table 18.
The rodent PK studies were carried out in male Sprague-Dawley (SD) rats (8–12 weeks of age, weighing 280 ± 20 gm at the time of dosing) and CD1 mice (8−12 weeks of age, weighing 30–35 gm body weight at the time of dosing) to estimate the plasma clearance, VD and terminal half-life, area under curve (AUC) and peak plasma concentration (Cmax) and time of peak plasma concentration (Tmax) following intravenous routes of administration.
Rats were anaesthetized by using isoflurane. The jugular and femoral veins of rat were cannulated, and the study was performed 48 h post-cannulation.
At each time point, about 100 µL of rat blood was collected from the jugular vein into a labelled microfuge tube containing 200 mM K2EDTA solution (20 µL per mL of blood) and equivalent volume of heparinized saline was replaced following sample collection. Similarly, 25 µL of mice blood was collected from the saphenous vein into a labelled microfuge tube containing 200 mM K2EDTA solution. Serial blood sampling method was used for blood collection. Blood samples were collected at pre-dose, 0.25 h, 0.5, 1, 2, 4, 6, 8 and 24 h post-dosing. The blood samples were processed to obtain the plasma samples within 30 min of the scheduled sampling time. All plasma samples were stored at −70 °C until bioanalysis was performed.
Bile duct cannulation (BDC) study in rats
The rat BDC study was carried out in male Sprague-Dawley (SD) rats (8–12 weeks of age, weighing 280 ± 20 gm at the time of dosing) to estimate the plasma clearance, VD and terminal half-life, AUC and Cmax and Tmax following an intravenous route of administration. Rats were anaesthetized using isoflurane anaesthesia. The bile duct, jugular and femoral veins of rat were cannulated, and the study was performed 48 h post-cannulation. At each time point, about 100 µL of rat blood was collected from the jugular vein into a labelled microfuge tube containing 200 mM K2EDTA solution (20 µL per mL of blood) and equivalent volume of heparinized saline was replaced following sample collection. Bile, urine and faeces were collected at 0–2, 2–4, 4–8, 8–12 and 12–24 h post-dose administration. The blood samples were processed to obtain the plasma samples within 30 min of scheduled sampling time and other matrices (bile, urine and faeces) as well. All samples were stored at −70 °C until the bioanalysis was performed.
Guinea pig pharmacokinetic study
The formulation details of the compound tested is shown in Supplementary Table 18.The study was performed in fasted, male Dunkin Hartley guinea pigs. A total of 6 animals were used and were catheterized to the jugular vein for infusion and the femoral artery for blood collection. Test item formulations were prepared on the day of treatment. Blood samples were collected through catheterized femoral artery at pre-dose (0 min) and 0.25, 0.5, 1 (at the end of infusion), 1.083, 3, 5, 7, 9 and 24 h post-dose. At each time point, 0.3 mL of blood was withdrawn and transferred into pre-labelled 0.5 mL micro centrifuge tubes containing 10 μL of 1000 IU/mL heparin sodium as anticoagulant and mixed gently to facilitate mixing of anticoagulant with the blood. Blood samples were kept on ice bath until centrifugation. The collected blood samples were centrifuged at 2000 x g for 10 min at 4 °C. Plasma was separated and transferred into pre-labelled tubes and stored at −20 °C until analysis was performed.
Non-rodent (dog) pharmacokinetic study
The formulation details of the compound tested is shown in Supplementary Table 18. The dog PK study was carried out in male Beagle dogs of minimum 10 kg body weight to estimate the plasma clearance, VD and terminal half-life, area under curve (AUC) and peak plasma concentration (Cmax), time of peak plasma concentration (Tmax) following an intravenous infusion route of administration. Briefly, compound was administered to a group of 3 non-naïve male dogs. The first set of 3 animals received an intravenous infusion and following a washout period of 4 days, received a higher dose administration. Blood samples (1 mL) was collected from the jugular vein by venepuncture into tubes containing K2EDTA anticoagulant at the following sampling times: Pre-dose, 0.083 (5 min), 0.25, 0.5, 1, 2, 4, 8 and 24 h post-dosing. Immediately following collection, blood samples were inverted to ensure mixing with anticoagulant and placed on wet ice. As soon as practically possible, samples were centrifuged (2000 x g, 10 min, at 4 °C) and the resultant plasma decanted into appropriately labelled polypropylene tubes in 96-well plate format and stored in a freezer set to maintain a temperature of ≤ −65 °C, until analysis was performed.
Bioanalysis of plasma samples
Plasma samples (mice/rat/guinea pig/dog) were analysed using a fit-for purpose LC-MS/MS method. For guinea pig sample analysis (range was 40.1−20100 ng/mL) for mice, rats, and dogs (range was 1–3000 ng/mL).
Pharmacokinetic data analysis
Pharmacokinetic parameters were estimated using Phoenix® WinNonlin® version 6.4 or higher (Certara USA, Inc., Princeton, New Jersey). A non-compartmental approach consistent with the intravenous infusion route of administration was used for parameter estimation. The individual plasma concentration-time data was used for pharmacokinetic calculations. In addition to parameter estimates for individual animals, descriptive statistics (e.g., mean, standard deviation, coefficient of variation, median, min, max) have been reported, as appropriate.
hERG screening data
BWC0977 was tested for inhibition of the human ether-a-go-go related gene (hERG) K+ channel using QPatch HTX automated electrophysiology62. BWC0977 was solubilised to 100 mM in DMSO before dilution in HBPS to 300 μM. A 6-point concentration-response curve was generated using 3.16-fold serial dilutions from the top test concentration. Electrophysiological recordings were made from a Chinese hamster ovary cell line stably expressing the full-length hERG potassium channel. Single cell ionic currents were measured in whole-cell patch clamp configuration at room temperature (21−23 oC) using the QPatch HTX platform (Sophion). Intracellular solution contained (in mM): 120 KF, 20 KCl, 10 EGTA,10 HEPES and was buffered to pH7.3. The extracellular solution (HEPES-buffered physiological saline, HBPS) contained (in mM): 145 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 10 % glucose buffered to pH7.4. Cells were clamped at a holding potential of −80mV. Cells were stepped to +20 mV for 2 seconds then −40 mV for 3 seconds before returning to the holding potential. This sweep was repeated 10 times at 10 second intervals. hERG currents were measured from the tail step and referenced to the holding current. Compounds were then incubated for 2 min prior to a second measurement of ion channel current using an identical pulse train. The IC50 values were obtained from a 4-parameter logistic fit of the concentration-response data. Reference compound (cisapride) values were consistent with those reported in the literature.
Prediction of human PK parameters based on simple allometry of pre-clinical PK parameters estimated from PK modelling of combined species mixed effect model approach
The human PK profile was predicted based on four species data using Non-Linear Mixed Effect (NLME) model63. Specifically, total PK data from mice (0.023 kg), rat (0.282 kg), guinea pig (0.43 kg) and dog (10.5 kg) studies were extracted, and the combined species data were fit together to different 1, 2 and 3 compartment IV bolus/infusion PK models, respectively, following different weighting scheme. The best fit PK model among the fitting was a two-compartment IV bolus/ infusion PK model with a 1/Y2 predicted weighting scheme. Individual species data was fit to a two-compartment intravenous model and PK parameters such as V1, CL, V2, CLD2 were estimated. For all four species, individual PK parameters were individually scaled as per simple allometry and respective allometric exponents and coefficients were determined.
Combined species data was fit using NLME allometric model using Phoenix NLME (ver 6.4). Human PK parameters such as V1, CL, V2 and CLD2 were estimated along with corresponding exponents and allometric coefficients.
| 1 |
| 2 |
| 3 |
| 4 |
Following the 2-compartment model fitting and using predicted human PK parameters human profile (plasma concentrations vs time) the PK profile following different 80mg–1050mg single dose via IV infusion administration for 120 min was predicted.
First-in-human study of BWC0977 administered intravenously for 2 h as single ascending doses (SAD)
A Phase 1, randomized, double-blind, placebo-controlled, single dose escalation study to evaluate the safety, tolerability and PK of BWC0977 administered through IV infusion to healthy adult subjects has been completed in CMAX, Adelaide, Australia. The first in human trial was initiated following approval by the Human Research Ethics Committee (HREC) of the trial design, protocol and the key criteria specified for the study. Within each cohort of 8 subjects, efforts were made to randomize approximately equal numbers of males and females to either active or placebo (including both genders).
In single ascending dose (SAD) escalation study, 40 subjects in five dose cohorts of 8 subjects each (6 active, 2 placebo) were randomized to receive single IV infusion doses of BWC0977 or placebo infused over 120 ( ± 10 min) (Supplementary Table 19). Five dose levels of BWC0977 were assessed according to an ascending single-dose regimen (120 mg, 240 mg, 480 mg, 720 mg, 1050 mg). The starting dose is based on safety results from non-clinical studies.
All details pertaining to formulation, screening of subjects, written informed consent, admission and duration of hospitalization, evaluation parameters, inclusion and exclusion criteria, study endpoints, PK monitoring, statistical analysis and safety aspects of the trial are available at ClinicalTrials.gov ID: NCT05088421.
Chemistry: General chemical methods
All commercial reagents and solvents were used without further purification. Analytical thin-layer chromatography (TLC) was performed on SiO2 plates on alumina. Visualization was accomplished by UV irradiation at 254 and 220 nm. Purity of all the final derivatives for biological testing was confirmed to be >95%.
Evaporations were carried out by rotary evaporation in vacuo and work-up procedures were carried out after removal of residual solids by filtration; temperatures are quoted as °C; operations were carried out at room temperature, that is typically in the range 18−26 °C and without the exclusion of air unless otherwise stated, or unless the skilled person would otherwise work under an inert atmosphere; column chromatography (by the flash procedure) was used to purify compounds and was performed on Merck Kieselgel silica (Art. 9385) unless otherwise stated.
In general, the course of reactions was followed by TLC, HPLC, or LC/MS and reaction times are given for illustration only; yields are given for illustration only and are not necessarily the maximum attainable. The structure of the end products was generally confirmed by NMR and mass spectral techniques. Proton magnetic resonance spectra were generally determined in DMSO d6 unless otherwise stated, using a Bruker DRX 300 spectrometer or a Bruker DRX-400 spectrometer, operating at a field strength of 300 MHz or 400 MHz, respectively. In cases where the NMR spectrum is complex, only diagnostic signals are reported. Chemical shifts are reported in parts per million (ppm) downfield from tetramethylsilane as an external standard (δ scale) and peak multiplicities are shown, thus: s - singlet; d - doublet; dd - doublet of doublets; dt - doublet of triplets; dm - doublet of multiplets; t - triplet; m - multiplet; br - broad. Fast atom bombardment (FAB) mass spectral data were generally obtained using a Platform spectrometer (upplied by Micromass) run in electrospray and, where appropriate, either positive ion data or negative ion data were collected using Agilent 1100 series LC/MS equipped with Sedex 75ELSD. The lowest mass major ion is reported for molecules where isotope splitting results in multiple mass spectral peaks (for example when chlorine is present). Reverse-phase HPLC was carried out using YMC Pack ODS AQ (100×20 mmID, S 5 Å particle size, 12 nm pore size) on Agilent instruments; each intermediate was purified to the standard required for the subsequent stage and was characterized in sufficient detail to confirm that the assigned structure was correct; purity was assessed by HPLC, TLC, or NMR and identity was determined by infrared spectroscopy (IR), mass spectroscopy or NMR spectroscopy as appropriate. HRMS data was acquired using an Agilent 6520, Quadrupole-time of flight tandem mass spectrometer (Q-Tof MS/MS) coupled with an Agilent 1200 series HPLC system.
Synthesis of compound 1-9
Compound 1 (CAS: 2156619-18-8; 6-[5-(3-Aminopropyl)−2-oxo-3-oxazolidinyl]−2H-pyrido[3,2-b]−1,4-oxazin-3(4H)-one)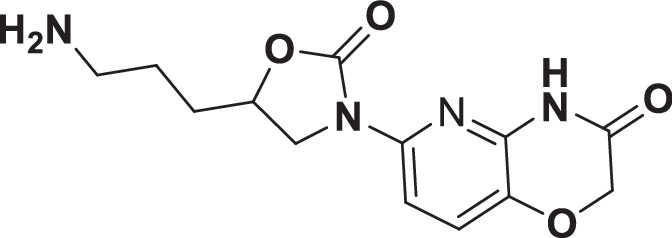
Compound 1 (CAS: 2156619-18-8) was synthesized as reported earlier in the patent WO2017199265A164. 1H NMR (400 MHz, DMSO-d6): δ 7.71 (bs, 1H), δ 7.59 (d, J = 8.6 Hz, 1H), 7.44 (d, J = 8.6 Hz, 1H), 4.68-4.75 (m, 1H), 4.61 (s, 2H), 4.22 (dd, J = 8.4, 10 Hz, 1H), 3.70 (dd, J = 6.6 Hz, 10 Hz, 1H), 2.85 (t, J = 8.0 Hz, 2H), 1.73-1.81 (m, 2H), 1.61-1.71 (m, 2H); LCMS calculated for C13H16N4O4, 292.30 Observed = 293.2; HRMS calculated for C13H16N4O4, 292.30, Observed = 293.1298;
Compound 2 (CAS: 2156618-99-2; 6-[5-[3-[[(6-Fluoro-2,3-dihydro-1-methyl-2-oxo-1H-indol-7-yl) methyl] amino]propyl]−2-oxo-3-oxazolidinyl]−2H-pyrido[3,2-b]−1,4-oxazin-3(4H)-one)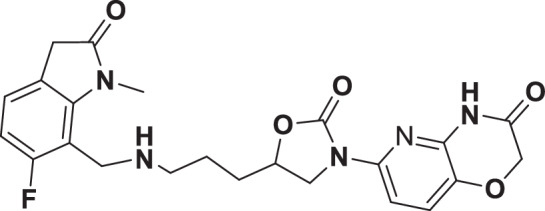
Compound 2 (CAS: 2156618-99-2) was synthesized as reported earlier in the patent WO2017199265A1. 1H NMR (400 MHz, MeOD): δ 8.44 (bs, 1H), 7.70 (d, J = 8.6 Hz, 1H), 7.36 (d, J = 8.6 Hz, 1H), 7.30–7.34 (m, 1H), 6.87–6.92 (m, 1H), 4.74-4.76 (m, 1H), 4.63 (s, 2H), 4.39 (s, 2H), 4.31–4.36 (m, 1H), 3.84–3.88 (m, 1H), 3.67–3.70 (m, 2H), 3.54–3.58 9 m, 5H), 3.08–3.10 (m, 2H), 1.84–1.90 (m, 4H); LCMS calculated for C23H24FN5O5, 469.47 Observed = 470.0; HRMS calculated for C23H24FN5O5, 469.47 Observed = 470.3601; HPLC = 87.33% (Zorbax Eclipse plus C18 RRHD(50×2.1) mm,1.8μ; Mobile phase A:0.1%TFA in WaterB:Acetonitrile);
Compound 3 (CAS: 2156619-07-5; 6-[(5S)−5-[2-[[2-(6-fluoro-2,3-dihydro-1-methyl-2-oxo-1H-indol-7-yl)ethyl]amino]ethyl]−2-oxo-3-oxazolidinyl]−2H-pyrido[3,2-b]−1,4-oxazin-3(4H)-one)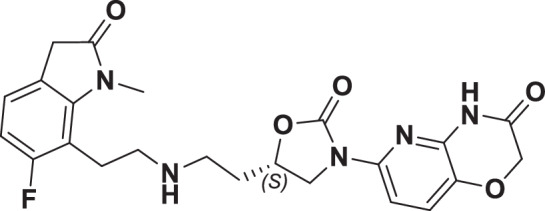
Compound 3 (CAS: 2156619-07-5) was synthesized as reported earlier in the patent WO2017199265A1. 1H NMR (400 MHz, DMSO-D6): δ 11.23 (s, 1H), 7.59 (d, 1H, J = 8.4 Hz), 7.44 (d, 1H, J = 8.8 Hz), 7.18–7.15 (m, 1H), 6.86–6.82 (m, 1H), 4.79–4.76 (m, 1H), 4.62 (s, 2H), 4.26–4.22 (m, 1H), 3.79–3.75 (m, 1H), 3.53 (s, 2H), 3.12–3.10 (m, 2H), 2.96 (s, br, 4H), 2.03 (s, br, 2H). LCMS Calc. for Calc. for C23H24FN5O5, 469.47; Obs 470.0; [M+ + H]. HRMS Calc. for C23H24FN5O5, 469.47; Obs 470.1190; HPLC Purity = 95.07% (HPLC Column: Atlantis dC18 (250*4.6) mm 5 µm, Mobile Phase A: 0.1% TFA in water, Mobile Phase B: Acetonitrile.).
Compound 4: (S)−6-(5-(((2-(6-fluoro-1-methyl-2-oxoindolin-7-yl) ethyl) amino) methyl)−2-oxooxazolidin-3-yl)−2H-pyrido[3,2-b] [1,4] oxazin-3(4H)-one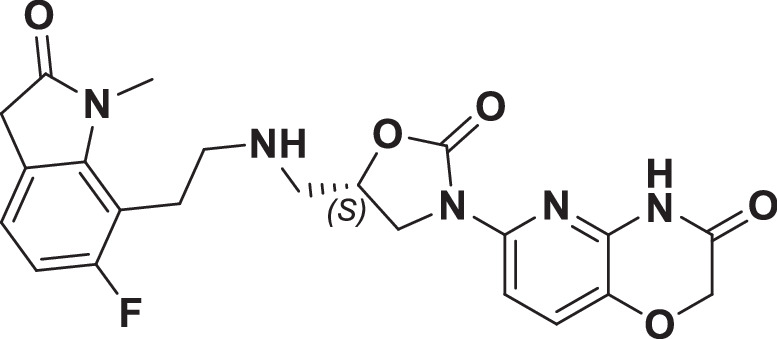
Compound 4 was synthesized using the procedure for compound 3 as reported earlier in the patent WO2017199265 A1 and WO2018225097 A1.
1H NMR (400 MHz, DMSO-D6): δ 11.20 (s, 1H), 7.60 (d, 1H, J = 8.8 Hz), 7.44 (d, 1H, J = 8.8 Hz), 7.12–7.09 (m, 1H), 6.82–6.77 (m, 1H), 4.77 (s, br, 1H), 4.62 (s, 2H), 4.15–4.11 (m, 1H), 3.87–3.83 (m, 1H), 3.49 (s, 2H), 3.38 (m, 3H), 3.03–3.01(m, 2H), 2.99–2.96 (m, 2H), 2.84–2.82 (m, 2H). LCMS Calc. for Calc. for C22H22FN5O5, 455.45; Obs :456.2; [M + + H]; HRMS Calc. for Calc. for C22H22FN5O5, 455.45; Obs :456.1166; HPLC Purity = 98.13% (HPLC Column: Atlantis dC18 (250*4.6) mm 5 µm, Mobile Phase A: 0.1% TFA in water, Mobile Phase B: Acetonitrile, RT = 8.90 min.
Compound 5 (CAS: 2254566-42-0; 6-[(5S)−5-[[[2-(7-Fluoro-1,2-dihydro-1-methyl-2-oxo-8-quinolinyl)ethyl]amino]methyl]−2-oxo-3-oxazolidinyl]−2H-pyrido[3,2-b]−1,4-oxazin-3(4H)-one)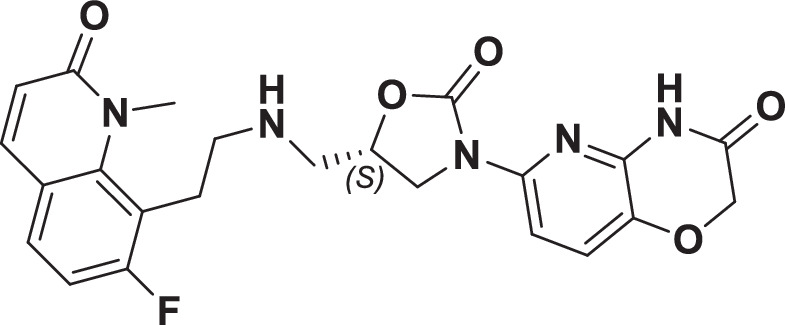
Compound 5 (CAS: 2254566-42-0) was synthesized as reported earlier in patent WO2018225097 A165.
1H-NMR (400 MHz, DMSO-d6): δ 11.19 (brs,1H), 7.84 (d, J = 9.16 Hz, 1H), 7.64–7.58 (m, 2H), 7.42 (d, J = 8.76 Hz, 1H), 7.13 (t, J = 9.12 Hz, 1H), 6.53 (d, J = 9.28 Hz, 1H), 4.70 (s, 1H), 4.61 (s, 2H), 4.11 (t, J = 9.40 Hz, 1H), 3.86–3.82 (m, 1H), 3.71 (s, 3H), 3.13–2.84 (m, 2H), 2.80–2.67 (m, 4H). HRMS Calc. for C23H22FN5O5, 467.46; Obs.: 468.1665. LCMS Calc. for C23H22FN5O5, 467.46; Obs.: 468.3 [M+ + H] +.
Compound 6 (CAS: 2254566-55-5); 2-[(5S)−5-[[[2-(7-Fluoro-1,2-dihydro-1-methyl-2-oxo-8-quinolinyl)ethyl]amino]methyl]−2-oxo-3-oxazolidinyl]−6H-pyrimido[5,4-b][1,4]oxazin-7(8H)-one)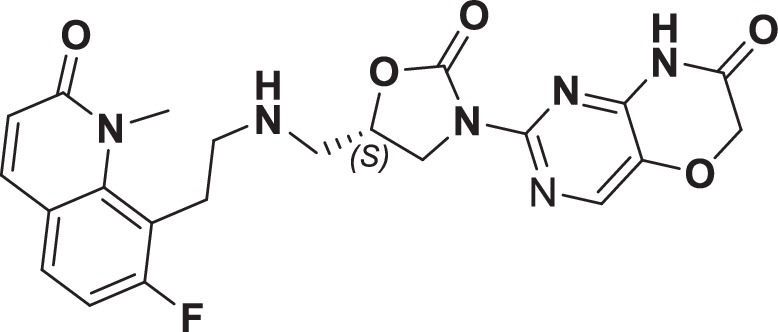
Compound 6 (CAS: 2254566-55-5) was synthesized as reported earlier in WO2018225097 A1 patent. 1H-NMR (400 MHz, DMSO-d6): δ 8.22 (s, 1H), 8.16 (s, 1H), 7.85 (d, J = 9.20 Hz, 1H), 7.64 (t, J = 6.80 Hz, 1H), 7.15 (t, J = 8.80 Hz, 1H), 6.55 (d, J = 9.20 Hz, 1H), 4.72 (s, 3H), 4.13 (t, J = 9.20 Hz, 1H), 3.89–3.85 (m, 1H), 3.73 (s, 3H), 3.17–3.15 (m, 2H), 2.92-2.84 (m, 4H).LC_MS: Calc. for C22H21FN6O5, 468.45; Obs. 469.1 [M + H] +.
Compound 7 (CAS: 2254566-91-9; 6-[(5S)−5-[[[2-(6-Fluoro-3,4-dihydro-4-methyl-3-oxo-5-quinoxalinyl)ethyl]amino]methyl]−2-oxo-3-oxazolidinyl]−2H-pyrazino[2,3-b]−1,4-oxazin-3(4H)-one)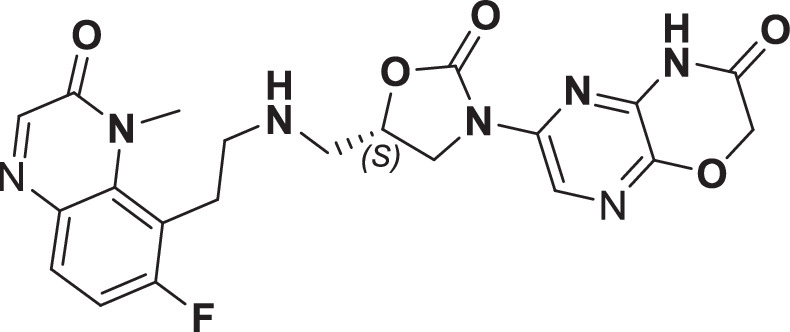
Compound 7 (CAS: 2254566-91-9) was synthesized as reported earlier in WO2018225097 A1. 1H-NMR (400 MHz, DMSO-d6): δ 11.61 (s, 1H), 8.38 (s, 1H), 8.16 (s, 2H), 7.72 (t, J = 6.64 Hz, 1H), 7.25 (t, J = 9.28 Hz, 1H), 4.87 (s, 2H), 4.77 (brs, 1H), 4.07 (t, J = 8.92 Hz, 1H), 4.05–3.75 (m, 4H), 3.17–3.10 (m, 2H), 2.91–2.85 (m, 4H). LCMS Calc. for C21H20FN7O5, 469.43; Obs.: 468.0 [M+-H]. HRMS Calc. for C21H20FN7O5, 469.43; Obs.: 470.0989. HPLC Purity = 93.26% (HPLC Column: Atlantis dC18 (250*4.6) mm 5 µm, Mobile Phase A: 0.1% AcOH in water, Mobile Phase B: Acetonitrile.
Compound 8 (CAS: 2254566-53-3; 6-[(5 R)−5-[[[2-(7-Fluoro-1,2-dihydro-1-methyl-2-oxo-8-quinolinyl)ethyl]amino]methyl]−2-oxo-3-oxazolidinyl]−2H-pyrazino[2,3-b]−1,4-oxazin-3(4H)-one)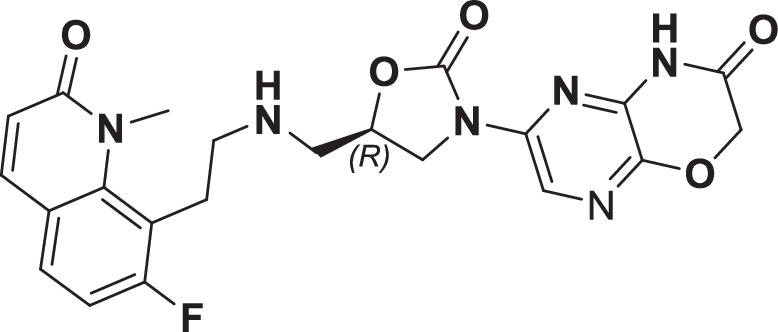
Compound 8 (CAS: 2254566-53-3) was synthesized as reported earlier in WO2018225097 A1. 1H NMR (400 MHz, DMSO-d6): δ 11.62 (s, 1H), 9.16 (brs, 1H), 8.40–8.38 (m,1H), 7.89–7.86 (m, 1H), 7.73–7.69 (m, 1H), 7.22–7.17 (m, 1H), 6.59–6.55 (m, 1H), 5.01 (brs, 1H), 4.88–4.87 (m, 2H), 4.23–4.19 (m, 1H), 3.83–3.79 (m, 1H), 3.73–3.72 (m, 3H),3.40–3.34 (m, 4H), 3.14 brs, 2H). LCMS Calc. for C22H21FN6O5, 468.45; Obs.: 467.1 [M+-H]. HRMS Calc. for C21H20FN7O5, 468.45; Obs.: 469.0989. HPLC Purity = 96.38% (HPLC Column: Atlantis dC18 (250*4.6) mm 5 µm, Mobile Phase A: 0.1% TFA in water, Mobile Phase B: Acetonitrile).
Compound 9 (CAS: 2254567-00-3; 6-[(5S)−5-[[[2-(7-Fluoro-1,2-dihydro-1-methyl-2-oxo-8-quinolinyl)ethyl]amino]methyl]−2-oxo-3-oxazolidinyl]−2H-pyrazino[2,3-b]−1,4-oxazin-3(4H)-one)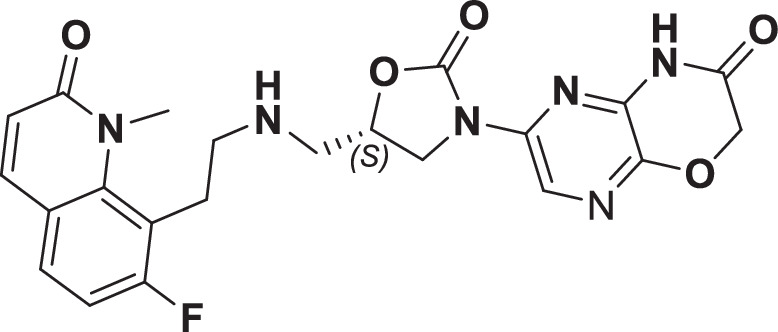
Compound 9 (CAS: 2254567-00-3) was synthesized as reported earlier in WO2018225097 A1. 1H NMR (400 MHz, DMSO- d6): δ 11.61 (bs, 1H), 8.38–8.37 (d, 1H, J = 3.2 Hz), 8.14 (s, 1H), 7.84–7.82 (d, 1H, J = 9.2 Hz), 7.64–7.60 (dd,1H, J1 = 8.8, J2 = 6.4 Hz), 7.15–7.10 (t, 1H, J = 8.8 Hz), 6.54–6.52 (d, 1H, J = 9.2 Hz), 4.85 (s, 2H), 4.79–4.76 (m, 1H), 4.11-4.06 (m, 1H), 3.83–3.79 (m, 1H), 3.71 (s, 3H), 3.16–3.13 (m, 2H), 2.95–2.91 (m, 2H), 2.89–2.82 (m, 2H). LCMS Calc. for C22H21FN6O5, 468.45; Obs.: 469.1; [M+ + H]. HRMS Calc. for C22H21FN6O5, 468.45; Obs.: 469.1077; HPLC Purity = 97.52%, Column: Atlantis dC18 (250 × 4.6) mm, 5μm, Mobile Phase A: 0.1% TFA in water, Mobile Phase B: Acetonitrile.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Source data
Acknowledgements
We thank Drs. Maria Uria Nickelson & Su Chiang, CARB-X, Boston University School of Law, Boston, USA for their valuable guidance throughout the entire period of work; Drs. Jason Cummings & Richard Slayden, Colorado State University, Fort Collins, USA for MIC determination on key biothreat pathogens; Drs. Adam Johnson, Adam Stevenson and Ana Jimenez-Valverde, University of Liverpool for assisting the mice thigh infection model studies; Drs. Maria McElmeel, Nathan Wiederhold & Thomas Patterson, The University of Texas Health Science Center at Houston, Texas, USA for MIC determination on key Gram-negative pathogens; Dr. Rafael Hernandez, Cystic Fibrosis Isolate Core, Seattle Children’s Research Institute of Seattle Children’s Hospital, Seattle, USA for MIC testing on key pathogens associated with cystic fibrosis; Dr. Ken Waites, University of Alabama at Birmingham, Birmingham, USA for MIC testing on mycoplasma strains; Dr. John Lipuma, Burkholderia cepacia Research Laboratory and Repository (BcRLR), University of Michigan Medical School, USA for MIC testing on Burkolderia species; Drs. James M. Meinig & Teresa Krackauer, United States Army Medical Research Institute of Infectious Diseases (USAMRIID), Fort Detrick, Maryland, USA for MIC testing on key biothreat pathogens; Drs. Malik Raynor, Supaksorn Chattagul, LTC Charlotte Lanteri, Walter Reed Army Institute of Research (WRAIR), USA for MIC testing on war-fighter pathogens; Dr. Erin Zeituni, National Institute of Allergy and Infectious Diseases (NIAID), Rockville, USA for facilitating the MIC, in vitro DMPK and safety testing at the pre-clinical services of NIAID; Swathi Purighalla at Narayana Health for help with procuring clinical isolates. Akshaya Ravishankar, Anirudh Shanbhag, Prakruthi Amar, Purnendu Bhowmik, Purnima Singh, Rajani Ravishankar, Riya Narjari, Daljeet Kaur, Anubha Nath and Savitha Prabhumurthy, Bugworks Research India Pvt. Ltd. Bangalore, India for assisting in assays. Research reported in this manuscript is in part supported by CARB-X. CARB-X’s funding for this project is sponsored by the Cooperative Agreement Number IDSEP160030 from ASPR/BARDA and by awards from Wellcome, Germany’s Federal Ministry of Education and Research, and the UK Global Antimicrobial Resistance Innovation Fund (GAMRIF) funded by the UK Government Department of Health and Social Care (DHSC). The content is solely the responsibility of the authors and does not necessarily represent the official views of CARB-X or any of its funders. Research reported in this manuscript is in part supported by BIRAC, Dept. of Biotechnology, Govt. of India via BIPP grant BT/BIPP0803/30/14.
Author contributions
Shahul Hameed led the medicinal chemistry and designed the chemical leads, including the clinical candidate BWC0977. Harish Kotakonda led the DMPK studies and is clinical project manager for the first-in-human study. Sreevalli Sharma, Maitrayee Sharma, Teby Thomas, and Vasanthi Ramachandran designed and executed the microbiology experiments: MIC, killing kinetics and cytotoxicity assays. Dhanashree Pillai, Savitha Raveendran, and Vasan Sambandamurthy designed and executed the resistance frequency, mutant characterization studies. Radha Nandishaiah, Nainesh Katagihallimath, and Santanu Datta designed and executed the biochemistry experiments pertaining to Gyrase and Topoisomerase IV enzymes. Ranga Rao led the synthetic chemistry efforts. Claire Sadler, Ian Slater, and Michael Morton designed and led the discovery toxicology studies. Abhijeeth Chandrasekaran is the clinical study monitor for the first-in-human study. Ed Griffen assisted in the medicinal chemistry aspects. Sambasiva Reddy and Venugopal Jonnalagadda assisted in the execution of the DMPK assays and the formulation development for the first-in-human studies. Nagakumar Bharatham and Suryanarayanan Venkatesan executed the molecular docking studies. Ramesh Jayaraman & Mahesh Nanjundappa designed and executed the rat ELF studies and PK-PD efficacy studies in the neutropenic rat lung infection model. Sreenath Rajagopal developed macro tools for the data analysis. Harikrishna Tumma led the GMP manufacturing of BWC0977 for the first-in-human studies. Amy Watters, Holly K. Becker, Jill M. Lindley, Robert Flamm, Michael Huband, Dan Sahm, Meredith Hackel, Tarun Mathur tested BWC0977 and its close analogs against a large panel of drug susceptible and resistant clinical isolates to determine MIC90. Ruwanthi Kolamunnage-Dona, Jennifer Unsworth, Laura Mcentee, Nikki Farrington and Shampa Das designed and executed the mice ELF studies and the PK-PD efficacy studies in neutropenic thigh infection model in mice. Dhanasekaran Manickam, Chandrashekara Narayana, Sivakandan Jayachandiran, Hrushikesava Reddy, and Sathya Shanker synthesized all the chemical leads including BWC0977. Vijay Richard and Savitha Nagaraj led the MIC determination of BWC0977 and its analogs against recent clinical isolates from India. John Tomayko led the design of the clinical studies including the drafting of the study protocols. Robert Clay led the regulatory aspects including submission to the Australian agencies for the first-in-human studies. V. Balasubramanian is the Principal Investigator who program managed the discovery of BWC0977.
Peer review
Peer review information
Nature Communications thanks Courtney Safir, Hang Nguyen, Khondaker Miraz Rahman, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Data availability
The raw data supporting killing kinetics, pharmacokinetics (pre-clinical and clinical), efficacy in animal models are provided as an excel file, “Source data.xlsx”. Source data are provided with this paper.
Competing interests
The authors Shahul Hameed P, Harish Kotakonda, Sreevalli Sharma, Radha Nandishaiah, Nainesh Katagihallimath, Ranga Rao, Abhijeeth Chandrasekaran, Ed Griffen, Dhanashree Pillai, Sambasiva Reddy, Nagakumar Bharatham, Suryanarayanan Venkatesan, Venugopal Jonnalagadda, Maitrayee Sharma Savitha Raveendran, Sreenath Rajagopal, Harikrishna Tumma, Santanu Datta, Vasan Sambandamurthy, Vasanthi Ramachandran, Robert Clay, John Tomayko, and V. Balasubramanian declare their competing interests as either current or former employees or consultants of Bugworks Research India Pvt. Ltd. Further, authors Shahul Hameed P., Nagakumar Bharatham, Nainesh Katagihallimath, Sreevalli Sharma, & Radha Nandishaiah are inventors on the patent WO2017199265A1, which covers compounds 1 – 4 disclosed in the manuscript; and authors Shahul Hameed P., Nagakumar Bharatham, Nainesh Katagihallimath, Sreevalli Sharma, Radha Nandishaiah, Vasanthi Ramachandran and V. Balasubramanian are inventors on the patent WO2018225097A1, which covers compounds 5– 9 disclosed in the manuscript. The authors Claire Sadler, Ian Slater, Michael Morton, Amy Watters, Holly Becker, Jill Lindley, Robert Flamm, Michael Huband, Dan Sahm, Meredith Hackel, Tarun Mathur, Ruwanthi Kolamunnage-Dona, Jennifer Unsworth, Laura Mcentee, Nikki Farrington, Dhanasekaran Manickam, Chandrashekara Narayana, Sivakandan Jayachandiran, Hrushikesava Reddy, Sathya Shanker, Ramesh Jayaraman, Mahesh Nanjundappa, Vijay Richard, Teby Thomas, Savitha Nagaraj, and Shampa Das declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-52557-2.
References
- 1.Murray, C. J. et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet399, 629–655 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jim O’Neill. Nat. Rev. Drug Discov. 15, 526 (2016). [DOI] [PubMed]
- 3.Tacconelli, E. et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis.18, 318–327 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Morris, S. & Cerceo, E. Trends, Epidemiology, and Management of Multi-Drug Resistant Gram-Negative Bacterial Infections in the Hospitalized Setting. Antibiot. (Basel). 9, 196 (2020). [DOI] [PMC free article] [PubMed]
- 5.Centers for Disease Control and Prevention (U.S.). Antibiotic Resistance Threats in the United States, 2019. https://stacks.cdc.gov/view/cdc/8253210.15620/cdc:82532 (2019).
- 6.Blaskovich, M. A. T. Antibiotics Special Issue: Challenges and Opportunities in Antibiotic Discovery and Development. ACS Infect. Dis.6, 1286–1288 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Paterson, D. L. Antibacterial agents active against Gram Negative Bacilli in phase I, II, or III clinical trials. Expert Opin. Investig. Drugs33, 371–387 (2024). [DOI] [PubMed] [Google Scholar]
- 8.Wiener, J. J. M. et al. Tetrahydroindazole inhibitors of bacterial type II topoisomerases. Part 2: SAR development and potency against multidrug-resistant strains. Bioorg. Med. Chem. Lett.17, 2718–2722 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Black, M. T. et al. Mechanism of action of the antibiotic NXL101, a novel nonfluoroquinolone inhibitor of bacterial type II topoisomerases. Antimicrob. Agents Chemother.52, 3339–3349 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bax, B. D. et al. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature466, 935–940 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Dougherty, T. J. et al. NBTI 5463 is a novel bacterial type II topoisomerase inhibitor with activity against gram-negative bacteria and in vivo efficacy. Antimicrob. Agents Chemother.58, 2657–2664 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biedenbach, D. J. et al. In Vitro Activity of Gepotidacin, a Novel Triazaacenaphthylene Bacterial Topoisomerase Inhibitor, against a Broad Spectrum of Bacterial Pathogens. Antimicrob. Agents Chemother.60, 1918–1923 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacobsson, S. et al. High in vitro activity of the novel spiropyrimidinetrione AZD0914, a DNA gyrase inhibitor, against multidrug-resistant Neisseria gonorrhoeae isolates suggests a new effective option for oral treatment of gonorrhea. Antimicrob. Agents Chemother.58, 5585–5588 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kokot, M. et al. Exploring Alternative Pathways to Target Bacterial Type II Topoisomerases Using NBTI Antibacterials: Beyond Halogen-Bonding Interactions. Antibiot. Basel Switz.12, 930 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scangarella-Oman, N. E. et al. Dose Selection for Phase III Clinical Evaluation of Gepotidacin (GSK2140944) in the Treatment of Uncomplicated Urinary Tract Infections. Antimicrob. Agents Chemother.66, e0149221 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scangarella-Oman, N. E. et al. Dose selection for a phase III study evaluating gepotidacin (GSK2140944) in the treatment of uncomplicated urogenital gonorrhoea. Sex. Transm. Infect.99, 64–69 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Riordan, W. et al. Efficacy, Safety, and Tolerability of Gepotidacin (GSK2140944) in the Treatment of Patients with Suspected or Confirmed Gram-Positive Acute Bacterial Skin and Skin Structure Infections. Antimicrob. Agents Chemother.61, e02095–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perry, C. R. et al. Efficacy and Safety of Gepotidacin as Treatment of Uncomplicated Urogenital Gonorrhea (EAGLE-1): Design of a Randomized, Comparator-Controlled, Phase 3 Study. Infect. Dis. Ther.12, 2307–2320 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wagenlehner, F. et al. Oral gepotidacin versus nitrofurantoin in patients with uncomplicated urinary tract infection (EAGLE-2 and EAGLE-3): two randomised, controlled, double-blind, double-dummy, phase 3, non-inferiority trials. Lancet403, 741–755 (2024). [DOI] [PubMed] [Google Scholar]
- 20.Taylor, S. N. et al. Single-Dose Zoliflodacin (ETX0914) for Treatment of Urogenital Gonorrhea. N. Engl. J. Med.379, 1835–1845 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Callaway, E. Groundbreaking’: first treatment targeting ‘super-gonorrhoea’ passes trial. Nature623, 236–236 (2023). [DOI] [PubMed] [Google Scholar]
- 22.Sati, H. et al. WHO Bacterial Priority Pathogens List. (2024).
- 23.Vanden Broeck, A., Lotz, C., Ortiz, J. & Lamour, V. Cryo-EM structure of the complete E. coli DNA gyrase nucleoprotein complex. Nat. Commun.10, 4935–4935 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weinstein, M. P. & Patel, J. B. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically: M07-A11. (Committee for Clinical Laboratory Standards, Wayne, PA, 2018).
- 25.McEntee, L. et al. Pharmacodynamics of Tebipenem: New Options for Oral Treatment of Multidrug-Resistant Gram-Negative Infections. Antimicrob. Agents Chemother.63, e00603–e00619 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang, D. B., Morrissey, I., Murphy, T., Hawser, S. & Wilcox, M. H. Efficacy evaluation of iclaprim in a neutropenic rat lung infection model with methicillin-resistant Staphylococcus aureus entrapped in alginate microspheres. Eur. J. Clin. Microbiol. Infect. Dis.37, 673–678 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Deguchi, Y., Sun, J., Tauchi, Y., Sakai, S. & Morimoto, K. Distribution Characteristics of Grepafloxacin, a Fluoroquinolone Antibiotic, in Lung Epithelial Lining Fluid and Alveolar Macrophage. Drug Metab. Pharmacokinet.18, 319–326 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Årdal, C. et al. Antibiotic development — economic, regulatory and societal challenges. Nat. Rev. Microbiol.18, 267–274 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Årdal, C. et al. Insights into early stage of antibiotic development in small- and medium-sized enterprises: a survey of targets, costs, and durations. J. Pharm. Policy Pract.11, 8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dheman, N. et al. An Analysis of Antibacterial Drug Development Trends in the United States, 1980–2019. Clin. Infect. Dis.73, e4444–e4450 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Balasubramanian, R., Van Boeckel, T. P., Carmeli, Y., Cosgrove, S. & Laxminarayan, R. Global incidence in hospital-associated infections resistant to antibiotics: An analysis of point prevalence surveys from 99 countries. PLoS Med20, e1004178–e1004178 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singh, S. B. et al. Tricyclic 1,5-naphthyridinone oxabicyclooctane-linked novel bacterial topoisomerase inhibitors as broad-spectrum antibacterial agents-SAR of left-hand-side moiety (Part-2). Bioorg. Med. Chem. Lett.25, 1831–1835 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Desai, J., S, S., Kumar, S. & Sharma, R. Novel Bacterial Topoisomerase inhibitors (NBTIs) – A comprehensive review. Eur. J. Med. Chem. Rep.3, 100017 (2021). [Google Scholar]
- 34.Kolarič, A., Anderluh, M. & Minovski, N. Two Decades of Successful SAR-Grounded Stories of the Novel Bacterial Topoisomerase Inhibitors (NBTIs). J. Med. Chem.63, 5664–5674 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kokot, M., Anderluh, M., Hrast, M. & Minovski, N. The Structural Features of Novel Bacterial Topoisomerase Inhibitors That Define Their Activity on Topoisomerase IV. J. Med. Chem.65, 6431–6440 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gibson, E. G., Bax, B., Chan, P. F. & Osheroff, N. Mechanistic and Structural Basis for the Actions of the Antibacterial Gepotidacin against Staphylococcus aureus Gyrase. ACS Infect. Dis.5, 570–581 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cumming, J. G. et al. Discovery of a Series of Indane-Containing NBTIs with Activity against Multidrug-Resistant Gram-Negative Pathogens. ACS Med. Chem. Lett.14, 993–998 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jacoby, G. A. Mechanisms of Resistance to Quinolones. Clin. Infect. Dis.41, S120–S126 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Redgrave, L. S., Sutton, S. B., Webber, M. A. & Piddock, L. J. V. Fluoroquinolone resistance: mechanisms, impact on bacteria, and role in evolutionary success. Trends Microbiol22, 438–445 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Ferrer, R. et al. Empiric Antibiotic Treatment Reduces Mortality in Severe Sepsis and Septic Shock From the First Hour. Crit. Care Med.42, 1749–1755 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Filkins, L. M. & O’Toole, G. A. Cystic Fibrosis Lung Infections: Polymicrobial, Complex, and Hard to Treat. PLoS Pathog.11, e1005258 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oliver, A., Cantón, R., Campo, P., Baquero, F. & Blázquez, J. High Frequency of Hypermutable Pseudomonas aeruginosa in Cystic Fibrosis Lung Infection. Science288, 1251–1253 (2000). [DOI] [PubMed] [Google Scholar]
- 43.Scoffone, V. C. et al. Burkholderia cenocepacia Infections in Cystic Fibrosis Patients: Drug Resistance and Therapeutic Approaches. Front. Microbiol.8, 1592–1592 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Modi, A. R. & Kovacs, C. S. Hospital-acquired and ventilator-associated pneumonia: Diagnosis, management, and prevention. Cleve. Clin. J. Med.87, 633–639 (2020). [DOI] [PubMed] [Google Scholar]
- 45.Butler, M. S. et al. Analysis of the Clinical Pipeline of Treatments for Drug-Resistant Bacterial Infections: Despite Progress, More Action Is Needed. Antimicrob. Agents Chemother.66, e0199121 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Penwell, W. F. et al. Molecular mechanisms of sulbactam antibacterial activity and resistance determinants in Acinetobacter baumannii. Antimicrob. Agents Chemother.59, 1680–1689 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hameed, P. S. et al. Nitrothiophene carboxamides, a novel narrow spectrum antibacterial series: Mechanism of action and Efficacy. Sci. Rep.8, 7263–7263 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kariv, I., Cao, H. & Oldenburg, K. R. Development of a high throughput equilibrium dialysis method. J. Pharm. Sci.90, 580–587 (2001). [DOI] [PubMed] [Google Scholar]
- 49.Waterhouse, A. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res46, W296–W303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen, I. J. & Foloppe, N. Drug-like Bioactive Structures and Conformational Coverage with the LigPrep/ConfGen Suite: Comparison to Programs MOE and Catalyst. J. Chem. Inf. Model.50, 822–839 (2010). [DOI] [PubMed] [Google Scholar]
- 51.Friesner, R. A. et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem.47, 1739–1749 (2004). [DOI] [PubMed] [Google Scholar]
- 52.Lindorff-Larsen, K. et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins78, 1950–1958 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A. & Case, D. A. Development and testing of a general amber force field. J. Comput. Chem.25, 1157–1174 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Case, D. A. et al. Amber 2018. Unpublished 10.13140/RG.2.2.31525.68321 (2018).
- 55.Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys.98, 10089–10092 (1993). [Google Scholar]
- 56.Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys.81, 511–519 (1984). [Google Scholar]
- 57.Hoover, W. G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A31, 1695–1697 (1985). [DOI] [PubMed] [Google Scholar]
- 58.Parrinello, M. & Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys.52, 7182–7190 (1981). [Google Scholar]
- 59.Hess, B., Bekker, H., Berendsen, H. J. C. & Fraaije, J. G. E. M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem.18, 1463–1472 (1997). [Google Scholar]
- 60.Crespi, C. L. & Penman, B. W. Use of cDNA-expressed human cytochrome P450 enzymes to study potential drug-drug interactions. Adv. Pharmacol. San. Diego Calif.43, 171–188 (1997). [DOI] [PubMed] [Google Scholar]
- 61.Smith, D. A., Waterbeemd, H. van de & Walker, D. K. Pharmacokinetics and Metabolism in Drug Design. (Wiley-VCH, Weinheim, New York, 2001).
- 62.Elkins, R. C. et al. Variability in high-throughput ion-channel screening data and consequences for cardiac safety assessment. J. Pharmacol. Toxicol. Methods68, 112–122 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cosson, V. F., Fuseau, E., Efthymiopoulos, C. & Bye, A. Mixed effect modeling of sumatriptan pharmacokinetics during drug development. I: Interspecies allometric scaling. J. Pharmacokinet. Biopharm.25, 149–167 (1997). [DOI] [PubMed] [Google Scholar]
- 64.Shahul Hameed P. M., Bharatham, N., Katagihallimath, N., Sharma, S. & Nandishaiah, R. Heterocyclic compounds useful as anti-bacterial agents and method for production. WO2017199265A1. (2017).
- 65.Shahul Hameed P. M. et al. Heterocyclic compounds useful as anti-bacterial agents and method for production thereof. WO2018225097A1. (2018).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data supporting killing kinetics, pharmacokinetics (pre-clinical and clinical), efficacy in animal models are provided as an excel file, “Source data.xlsx”. Source data are provided with this paper.