

Abstract
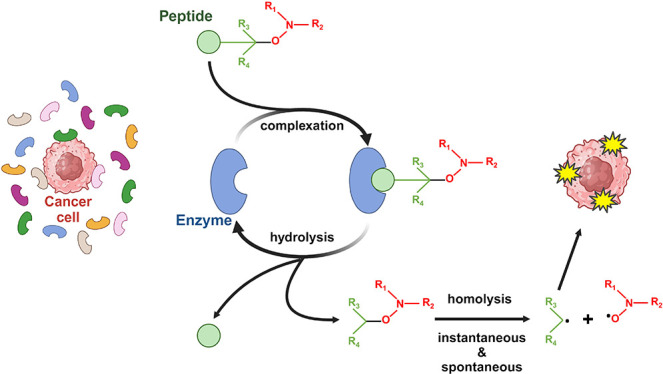
In search of better specificity and lower chances of resistance, protease-activatable alkoxyamine prodrugs to fight cancer have been proposed. These molecules are made of a peptide linked to an alkoxyamine. Proteolysis of the peptide converts the stable prodrug at 37 °C to a metastable alkoxyamine that spontaneously homolyzes into two free radicals: a stable nitroxide and a very reactive alkyl radical. The alkyl radical induces apoptosis in the surrounding cells by inducing random chemical alterations. Here, we show that varying the peptide moiety from succinyl-Ala-Ala-Pro-Val- to PyroGlu-Gly-Arg- or PyroGlu-Gly-Lys- is effective in switching the activating enzyme from elastase to urokinase. Furthermore, these prodrugs induce the death of HT-1080 cells, a cell line that secretes several active proteases in culture. This cytotoxic activity can be suppressed by protease inhibitors and does not affect cell lines devoid of active urokinase. We thus provide examples of alkoxyamine prodrugs that are efficiently activated by the limited intrinsic protease activity and that succeed in the destruction of cancer cell lines and cancer cells from tumor explants.
Introduction
Cancer chemotherapies need new modalities to avoid side effects and resistance. Prodrugs have long been identified as a way to deliver anticancer drugs that would minimize side effects.1,2 Ideally, a prodrug is a nontoxic molecule that is activated in situ only in the presence of a chosen activator. The most obvious activators are enzymes, since they usually have a specificity of action and a specificity of location. Furthermore, substrate turnover provides a continuous action as long as the prodrug is present. For tumors, targeting the overexpression of some glycosidases has been carried out3−5 to design drugs that are less toxic before but more toxic after activation by the targeted enzymes. Proteases have emerged as the most promising activators. Due to their potentially deleterious effects, proteolytic activities are tightly controlled in healthy tissues by means of inactive proenzymes that need to be activated by another protease, compartmentalization as in the lysosomes, and the presence of high concentrations of inhibitors. A total of 151 inhibitors correspond to about 500 proteases present in the genome.6 Thus, in healthy tissues, the lifetime of a protease is usually very short. For example, only by the control of the inhibitors, it is estimated that the half-lives of neutrophil elastase are about 5 ms in the upper respiratory tract and 50 ms in the lower respiratory tract.7 By contrast, persistent activities in the tumor microenvironment are well documented in tumors of many origins.1,8,9 Matrix metalloproteinase activities and overexpression involving MMP-2, MMP-9, MMP-7, and MT1-MMP (MMP-14) have been abundantly associated with cancer. Overexpression of cathepsin B and other cathepsins is associated with many tumors.10 Serine proteases are also involved. Urokinase activity is involved in a cascade of protease activation present in the tumor microenvironment.11 Active prostate-specific antigen is expressed at a high level in prostate cancer bone metastasis.12 Another serine protease, fibroblast activation protein, is active and overexpressed in many types of tumors,13 while neutrophil elastase is associated with tumor settlement and growth.14−16
Recently, protease-activatable alkoxyamines were proposed. Alkoxyamines are metastable molecules that spontaneously homolyze into two free radicals, a stable nitroxide and a highly reactive alkyl radical (Scheme 1).17,18 By acting on R1, R2, or R3, the molecules can be tuned to undergo rapid homolysis at 37 °C. Conveniently, by adding a peptide at a key position, the metastable alkoxyamines become stable with half-lives reaching weeks or months in physiological conditions. Alkoxyamines were proposed as antitumor treatment because the released alkyl radical is cytotoxic. Thus, in a proof-of-concept study, a construction was designed and tested, which targets neutrophil elastase activity19 (Scheme 2).
Scheme 1. General Scheme for Protease Activation of Alkoxyamine Prodrugs.

Scheme 2. The Succinyl-(Ala)2-Pro-Val Peptide Was Linked to an Anilide-Based Alkoxyamine to Gain Stability.
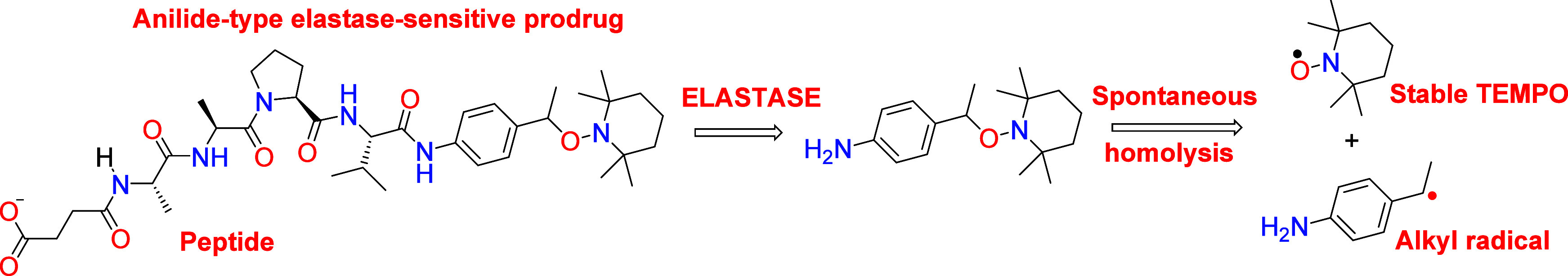
The prodrug was then activated by neutrophil elastase.
The prodrug was activatable by neutrophil elastase and rapidly released the TEMPO nitroxide and an alkyl radical. The alkyl radical was able to rapidly trigger apoptosis in the U87 glioblastoma cell line in the presence of added neutrophil elastase while being nontoxic in the absence of the added enzyme. In principle, the targeting enzyme can be chosen by replacing the stabilizing peptide. It is also possible to graft a sugar to target glycosidases. This promising approach nevertheless raised a new question: are the proteolytic activities found in the tumor microenvironment sufficient to activate such prodrugs? While these activities are present in any type of cancer in which they were searched, quantitative data about these activities cannot be found in the literature because they are technically challenging. Thus, the test of cancer cell lines in culture seems a necessary option to assess the efficiency of alkoxyamine prodrugs. A further difficulty is that most cancer cell lines do not produce active proteases in the culture. For example, U87 cells produce inactive MMPs that are activated only in the conditions of zymography (autoactivation) or by adding trypsin (enzymatic activation) or APMA (chemical activation). This is due to the absence of a probable cascade of protease activation that is present in the cellular diversity of a tumor.11 However, some cell lines secrete active proteases.
In this paper, we designed three peptide-linked alkoxyamines addressing very different protease activities and we tested the effect on several tumor cell lines in culture or on primary cultures from tumor explants. Their efficiency and specificity are discussed.
Results
Prodrug Synthesis
All molecules are based on an anilide-TEMPO scaffold, because previous studies showed the complete stability of the prodrugs and the quick homolysis of the activated form. Stabilizing peptides were varied according to the targeted protease. Synthesis of Suc-Ala-Ala-Pro-Val-anilide-TEMPO was described earlier.19
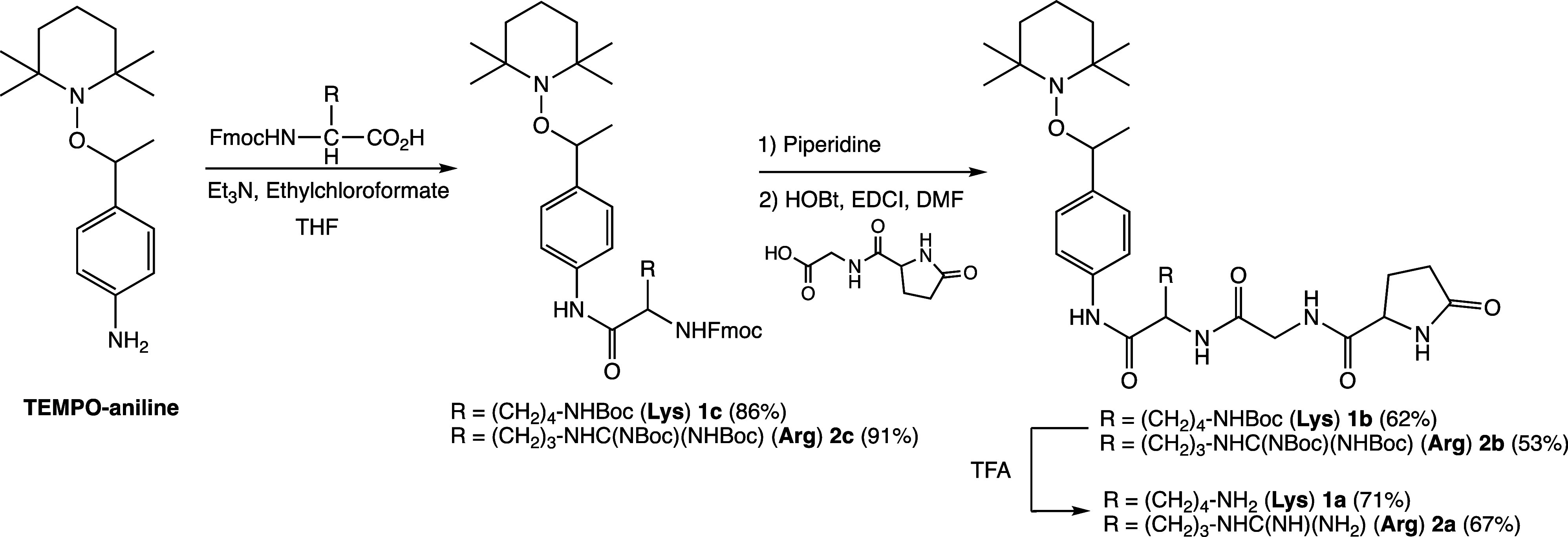
Alkoxyamines 1a and 2a are prepared through a 3-step procedure displayed in Scheme 3. Hence, the starting material TEMPO-aniline was prepared as already reported.20 The peptide was attached via a 2-step procedure. That is, the lysine for 1a or the arginine for 2a is first attached, and then the protected pGlu-Gly dipeptide is attached. This 2-step-procedure is more efficient than to attach the tripeptide.
Scheme 3. Synthesis of Two Prodrugs pGlu-Gly-Lys-anilide-TEMPO (1a) and pGlu-Gly-Arg-anilide-TEMPO (2a).
Values of homolysis rate constant kd show low values, affording activation energies Ea of 124.8 kJ/mol for 1a and 126.5 kJ/mol for 2a in water (see the Supporting Information for more details). Consequently, in the absence of enzymes and except specific acidic conditions for hydrolysis, all molecules prepared and investigated in this article are stable at 40 °C during the experimental time of 48 h at least.
Prodrugs pGlu-Gly-Lys-anilide-TEMPO (1a) and pGlu-Gly-Arg-anilide-TEMPO (2a) Targeting Urokinase
The urokinase-type plasminogen activator (uPA) is overexpressed and active in many types of cancer. With its receptor (uPAR), it participates in a cascade of protease activation also involving plasmin and matrix metalloproteinases.11,20 At the P1 site, it accommodates preferentially the amino acid Lys or Arg.21 We synthesized two prodrugs for uPA, one with the peptide Pyro-Glu-Gly-Lys- and one with the peptide Pyro-Glu-Gly-Arg-. Figure 1A,B shows the release of TEMPO with time in the presence or absence of uPA from these two substrates. The prodrug with arginine in the P1 position is fairly stable in the absence of uPA, while the prodrug with lysine is completely stable. In the presence of uPA, both prodrugs are activated. Figure 2 shows the Michaelis–Menten curves for the activation of each prodrug. For pGlu-Gly-Arg-anilide-TEMPO 2a, Km was 2 mM and kcat was 7.5 × 10–2 s–1, yielding a kcat/Km value of 30.6 M–1 s–1. For pGlu-Gly-Lys-anilide-TEMPO 1a, Km was over 14 mM, but kcat was apparently higher than that for pGlu-Gly-Arg-anilide-TEMPO but not measurable in our experimental conditions. However, since Km was high, we were in the condition [prodrug] ≪ Km; thus, direct measurement of kcat/Km was possible using the simplified Michaelis equation vinitial = kcat/Km × [E][S0]. This yielded a similar value of 32.5 M–1 s–1. Thus, at working concentrations, both substrates were valid.
Figure 1.

Kinetics of TEMPO release in the presence or absence of uPA for substrates pGlu-Gly-Arg-anilide-TEMPO (A) and pGlu-Gly-Lys-anilide-TEMPO (B) at 37 °C. TEMPO release is the marker of alkoxyamine activation, which is either spontaneous or catalytic.
Figure 2.

Michaelis–Menten plots for the substrates pGlu-Gly-Arg-anilide-TEMPO (A) and pGlu-Gly-Lys-anilide-TEMPO (B) in the presence of 10 μM uPA and 2 μM hemin to scavenge the freed alkyl radicals and shift the equilibrium between the alkoxyamine and the released alkyl radicals and TEMPO molecules in favor of homolysis.
To test the effect of these prodrugs on cancer cells, we chose the HT-1080 cell line because it secretes active uPA in culture,22 while most cell lines secrete or express inactive proenzymes.
In Figure 3, both prodrugs display a dose-dependent effect, suggesting that enough uPA is present in the medium. The prodrug pGlu-Gly-Lys-anilide-TEMPO was also applied on the U87 glioblastoma cell line but was inactive unless uPA was added in the medium, which further stresses that a specific enzyme activity is mandatory (Figure 4). Interestingly, the half effect is similar to the one observed with the amount of enzyme secreted by HT-1080, suggesting that the intrinsically secreted enzyme concentration is not limiting.
Figure 3.

Viability of HT-1080 cells vs pGlu-Gly-Arg-anilide-TEMPO and pGlu-Gly-Lys-anilide-TEMPO concentrations. All viability tests were done following the same sequence: as described in Materials and Methods, cells were first phased for 24 h by FCS starvation, then the alkoxyamine prodrug was added, and FCS was added 1 hour later. Viability readings were done 48 h after alkoxyamine addition by adding WST-1 and reading at 20 min incubation. Readings were done in duplicate, and data are shown as the mean ± SD.
Figure 4.

Viability of U87 cells versus pGlu-Gly-Lys-anilide-TEMPO concentration in the presence or absence of uPA (1 μM).
Then, the human adenocarcinoma cell line HT-29 was challenged with both prodrugs. HT-1080 and HT-29 cells both synthesize uPA, but in the case of HT-29, the enzyme is not secreted or secreted only as the inactive proenzyme.23Figure 5 clearly shows that none of the uPA-activatable prodrugs have an effect on HT-29 cells.
Figure 5.

Viability of HT-29 cells versus pGlu-Gly-Arg-anilide-TEMPO and pGlu-Gly-Lys-anilide-TEMPO concentrations, showing that a medium deprived of uPA does not activate the prodrug.
Since tumors are not only made of cancer cell clones but also contain fibroblasts and abundant infiltrated immune cells, we tested the two prodrugs on HT-1080 tumor explants. Figure 6 shows that both prodrugs have an activity, but cell death is not completely achievable as it is with the pure cell lines. As for the pure cell line, pGlu-Gly-Arg-anilide-TEMPO seems to achieve a more complete effect. This may be related to the better Km of this prodrug as an uPA substrate.
Figure 6.

Effect of the concentration of pGlu-Gly-Arg-anilide-TEMPO and pGlu-Gly-Lys-anilide-TEMPO on cells from the HT-1080 tumor explant.
Thus, these prodrugs activatable by uPA are both active on the HT-1080 uPA-secreting cell line but not on nonsecreting cell lines, thus achieving a specific effect. The presence of physiological concentrations of uPA is sufficient to activate the prodrugs. However, the concentration required for the half effect is twice the one required with succinyl-Ala-Ala-Pro-Val-anilide-TEMPO in the presence of added neutrophil elastase as published earlier.19
Prodrug Suc-Ala-Ala-Pro-Val-anilide-TEMPO Targeting Neutrophil Elastase
This substrate was initially intended to produce a proof of concept by inducing apoptosis in U87 glioblastoma cells in the presence of neutrophil elastase but not in its absence.19 However, the setup was not physiological since the enzyme was added. It showed, however, that the prodrug itself was not cytotoxic and that it could be enzymatically activated. Here, we tested this prodrug on HT-1080 cells. Although there is no report on the presence of active neutrophil elastase in these cell cultures, the proven presence of active uPA is likely to activate other enzymes.
Figure 7 shows that in the absence of any added enzyme, HT-1080 cells are killed in a dose-dependent manner as opposed to U87 cells19 with a half effect of around 200 μM, which is thus similar to the 150 μM half effect observed with the addition of 0.68 μM neutrophil elastase on U87 cells. Eglin C, α1-antitrypsin, E64, aprotinin, and batimastat were unable to rescue the cells (not shown). Interestingly, ilomastat, another potent metalloproteinase inhibitor, saved 100% of the cells even in the presence of the highest concentrations of the prodrug. This suggests that not only neutrophil elastase is not involved but also most probably no other serine protease or cysteine protease. However, the clear inhibition of the cytotoxicity in the presence of ilomastat suggests the action of a metalloproteinase.
Figure 7.

Dose-dependent viability of HT-1080 cells versus Suc-Ala-Ala-Pro-Val-anilide-TEMPO in the presence or absence of the metalloproteinase inhibitor ilomastat.
These results are confirmed with cells from HT-1080 tumor explants (see Figure 8). As opposed to the prodrugs activatable by uPA, 100% of the cells from the tumors are killed. Ilomastat again saves 100% of these cells.
Figure 8.

Viability of cells from HT-1080 tumor explants versus Suc-Ala-Ala-Pro-Val-anilide-TEMPO: Protection by the ilomastat is preserved for all cells. Eglin C does not protect.
The activity of succinyl-Ala-Ala-Pro-Val-anilide-TEMPO on HT-1080 cells was unexpected because the secretion of neutrophil elastase by these cells has never been reported. This was confirmed by the addition of eglin C and α1-antitrypsin, which do not protect the cells. Interestingly, ilomastat, a metalloproteinase inhibitor, was able to save the cells at all prodrug concentrations tested. The presence of active MMP-2, MMP-9, and MT1-MMP in HT-1080 cell cultures is well documented.24 However, a search in the MEROPS protease activity database and our own enzymatic and docking experiments (not shown) tend to exclude these proteases because the requirements at the P′ positions of the substrate are not met by our prodrug. We also experimentally verified that active MMP-2 does not hydrolyze the succinyl-Ala-Ala-Pro-Val peptide from the alkoxyamine using analytical HPLC even after overnight incubation at 37 °C (Supporting Information, Figure S26). On the other hand, members of another metalloproteinase family called “ADAM” are also inhibited by Ilomastat. Furthermore, these proteases can tolerate many structures at the P′ positions of their substrates as opposed to MMPs, which are more restrictive.25 Proteases of this family have been found active on the surface of HT-1080 cells26 and have been found upregulated in many cancer types27 and thus could be involved in the effect that we observed. The fact that ilomastat but not batimastat protects the cells is not sufficient to further identify the protease involved. A dedicated work will be required to identify this protease. Clearly, however, this enzyme activity may become a selective target for anticancer drugs.
Discussion
Recently, we developed prodrugs based on an alkoxyamine scaffold that could be activated by neutrophil elastase. These prodrugs were able to induce apoptosis of U87 cells in the presence of neutrophil elastase, while they appeared nontoxic in the absence of the enzyme. The stabilization of the prodrug by a peptide was meant to render the prodrug versatile, i.e., adapted to be activatable by a variety of proteases simply by changing the peptide sequence. This would be a step toward personalized medicine by adapting the prodrug to the enzyme activities present in each type of tumor. Hence, we had two goals in this paper: first, to verify that the prodrug could be adapted to a very different protease activity and, second, to verify that intrinsic protease activities of some tumor cells are sufficient to activate the prodrug at a rate that leads to cell death.
The first goal was achieved since peptides Pyro-Glu-Gly-Lys- and Pyro-Glu-Gly-Arg- made the prodrug activatable by urokinase. This means that the presence of the alkoxyamine moiety in the P′ positions does not interfere with the accommodation of the peptide inside the enzyme reactive site. Thus, it may be possible to target other serine proteases of interest for cancer such as PSA or FAP by adapting the peptide moiety. Our results also suggest that metalloproteases can be activators of our prodrugs. However, specifically targeting activation by MMPs will require modification of the alkoxyamine moiety since the main substrate selection pocket is in the P′ position for this enzyme class.
The second important result of this work is that cells secreting their own active proteases produce enough enzyme activity to activate the alkoxyamines. Here, we tested HT-1080 cells in a culture or from tumor explants. HT-1080 cells are known to secrete active urokinase, which in turn probably activates a series of other proteases that are secreted as proenzymes. The resulting activities were sufficient to activate the prodrugs based on the Pyro-Glu-Gly-Lys-, Pyro-Glu-Gly-Arg-, and succinyl-Ala-Ala-Pro-Val- peptides and to kill the cell line in culture. Remarkably, the cells from the corresponding tumor explants were also killed by the prodrugs, although they are a mixture of HT-1080 and tumor-infiltrating cells. These results suggest a way to kill not only the cancerous cells but also their necessary cellular environment.
The strategy employing protease-activated prodrugs is meant to be more specific than antimitotic chemotherapy. It is likely to be specific because proteolytic activity is tightly regulated in healthy tissues. However, proteolytic activities are sometimes redundant, meaning that a similar activity can take place in another organ that is not to be treated. Such a situation can be handled by a careful comparative study of the substrate specificities. If the same specificity in the P1 position is not unusual, it is rarely the case in the P2 and P3 positions. Thus, modifying the peptide would probably solve the unwanted activation of the prodrug.
At the moment, our alkoxyamines are efficient in the 150 μM range. The intrinsic enzyme activity seems to be sufficient for optimal processing of the prodrugs. Thus, the best lead to lower efficient concentration is probably to modify the alkyl radical moiety for more chemical aggressiveness. Better cell-penetrating properties of this alkyl radical could also generate more lethal alteration, provided that the initial prodrug does not penetrate the cells. Finally, targeting membrane-bound proteinases could enhance efficiency by generating the alkyl radical very close to the membrane, thus increasing the rate and the accumulation of deleterious chemical alterations.
Materials and Methods
Synthesis
Solvents and reactants for the preparation of alkoxyamines were used as received. Routine reaction monitoring was performed using silica gel 60 F254 TLC plates; spots were visualized upon exposure to UV light and a phosphomolybdic acid solution in EtOH, followed by heating. Purifications were performed on a Reveleris X2 Flash System BUCHI, Switzerland. Cartouches flash Reveleris and GraceResolv: silica 40 μm. 1H and 13C NMR spectra were recorded in CDCl3, CD3OD, and DMSO-d6 on a 300 or 400 MHz spectrometer. Chemical shifts (δ) in parts per million were reported using residual nondeuterated solvents as internal references for 1H and 13C NMR spectra. High-resolution mass spectra (HRMS) were obtained on a SYNAPT G2 HDMS (Waters) spectrometer equipped with a pneumatically assisted atmospheric pressure ionization source. Positive mode electrospray ionization was used on samples: electrospray voltage, 2800 V; opening voltage, 20 V; nebulizer gas pressure (nitrogen), 800 L/h. Low-resolution mass spectra were recorded on the ion trap AB SCIEX 3200 QTRAP equipped with an electrospray source. The parent ion [M + H]+ is quoted. The purity of final compounds was >95% as assessed by NMR spectra for all compounds and by HPLC for compounds 1a and 2a.
Coupling of Lysine or Arginine with TEMPO-aniline
To a colorless solution of protected amino acids, FmocNH-Lys(NHBoc)-OH (2.9 g, 8.87 mmol, 1.2 equiv) or FmocNH-Arg((NH)C(NBoc)(NHBoc))-OH (4.7 g, 7.96 mmol, 1.2 equiv) in THF (30 mL), ClCO2Et (1.22 mL, 9.41 mmol, 1.3 equiv), and Et3N (1.31 mL, 9.41 mmol, 1.3 equiv) were added at −15 °C. After stirring for 15 min at −15 °C, TEMPO-anilide (1 equiv) dissolved in THF (20 mL) was added at −15 °C to the colorless suspension by cannulation. The mixture was stirred overnight at room temperature and concentrated in vacuo. The residue was treated with 1 M aq. HCl (20 mL), extracted with EtOAc (250 mL), washed with sat. NaHCO3 (30 mL) and brine (20 mL), and dried over anhydrous MgSO4. The crude product was purified by flash chromatography to obtain 1c (86%) or 2c (91%).
1c
1H NMR (400 MHz, MeOD) δ 7.79 (d, J = 7.5 Hz, 2H), 7.67 (t, J = 7.9 Hz, 2H), 7.52 (d, J = 8.2 Hz, 2H), 7.38 (t, J = 7.5 Hz, 2H), 7.33–7.14 (m, 4H), 4.77 (q, J = 6.6 Hz, 1H), 4.40 (d, J = 6.8 Hz, 2H), 4.33–4.12 (m, 2H), 3.19–2.89 (m, 2H), 1.98–1.65 (m, 2H), 1.64–1.38 (m, 8H), 1.46 (d, J = 6.7 Hz, 3H), 1.42 (s, 9H), 1.35–1.27 (m, 5H), 1.24–1.12 (m, 3H), 1.09–0.99 (m, 3H), 0.66 (s, 3H). 13C NMR (101 MHz, MeOD) δ 173.2 (C), 158.5 (2 × C), 145.3 (C), 145.2 (C), 143.0 (C), 142.6 (2 × C), 138.3 (C), 128.7 (2 × CH aro), 128.2 (4 × CH aro), 126.2 (2 × CH aro), 121.2 (2 × CH aro), 120.9 (2 × CH aro), 84.1 (CH), 79.9 (C), 67.9 (CH2), 60.9 (2 × C), 57.1 (CH), 48.5 (CH), 41.4 (2 × CH2), 41.0 (CH2), 34.8 (2 × CH3), 33.1 (CH2), 30.6 (CH2), 28.8 (3 × CH3), 24.2 (CH2), 23.7 (CH3), 20.8 (2 × CH3), 18.1 (CH2). HRMS m/z (ESI) calcd for C43H59N4O6+ [M + H]+, 727.4429; found, 727.4428.
2c
1H NMR (400 MHz, CDCl3) δ 7.65 (d, J = 7.5 Hz, 2H), 7.51 (dd, J = 10.2, 7.3 Hz, 2H), 7.35 (d, J = 7.3 Hz, 2H), 7.28 (t, J = 7.5 Hz, 2H), 7.23–7.06 (m, 4H), 4.64 (q, J = 6.6 Hz, 1H), 4.51–4.19 (m, 3H), 4.12 (t, J = 7.0 Hz, 2H), 3.59–3.16 (m, 2H), 2.20–1.52 (m, 4H), 1.53–1.52 (m, 6H), 1.40 (s, 9H), 1.35 (d, J = 6.7 Hz, 3H), 1.32 (s, 9H), 1.19 (s, 3H), 1.06 (s, 3H), 0.99–0.89 (m, 3H), 0.57 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 170.0 (C), 163.0 (C), 156.4 (C), 156.4 (C), 153 (m, (C)), 143.6 (C), 143.5 (C), 142.2 (C), 141.1 (2 × C), 135.9 (C), 127.5 (2 × CH aro), 126.9 (4 × CH aro), 125.0 (CH aro), 124.9 (CH aro), 120.3 (CH aro), 120.2 (CH aro), 119.8 (2 × CH aro), 83.2 (C), 82.6 (CH), 79.4 (C), 66.8 (CH2), 59.5 (2 × C), 54.8 (CH), 47.0 (CH), 40.2 (2 × CH2), 39.6 (CH2), 34.2 (2 × CH3), 29.0 (CH2), 28.1 (3 × CH3), 27.9 (3 × CH3), 25.6 (CH2), 23.4 (CH3), 20.1 (2 × CH3), 17.0 (CH2). HRMS m/z (ESI) calcd for C48H67N6O8+ [M + H]+, 855.5015; found, 855.5013.
Deprotection of the Fmoc Group and Peptide Coupling with pGlu-Gly-OH
Alkoxyamine 1c or 2c was dissolved in piperidine (2 mL per g of product) and stirred for 2 h at room temperature. The excess of piperidine was removed in vacuo, and a short chromatographic column was carried out to remove Fmoc residue with 100% DCM. The deprotected product was obtained with a mixture of 10% methanol and 90% DCM quantitatively. Then, deprotected alkoxyamines (1 equiv) were solubilized in DMF (2 mL per 500 mg of alkoxyamine), a dipeptide (pGlu-Gly-OH) was added (1.1 equiv), and then HOBt (1.1 equiv) was added. After 15 min at room temperature, EDCI was added (1.1 equiv), and the reaction was stirred overnight at room temperature and concentrated in vacuo. The residue was treated with 1 M aq. HCl (20 mL), extracted three times with DCM (50 mL), washed with sat. NaHCO3 (50 mL) and brine (50 mL), and dried over anhydrous MgSO4. The crude product was purified by flash chromatography to obtain 1b (62%) or 2b (53%).
1b
1H NMR (400 MHz, MeOD) δ 7.92–7.60 (m, 2H), 7.42 (d, J = 8.7 Hz, 2H), 5.42 (q, J = 6.5 Hz, 4H), 4.47 (dd, J = 8.8, 5.2 Hz, 1H), 4.25 (dd, J = 8.8, 4.5 Hz, 1H), 3.96 (s, 2H), 3.06 (t, J = 6.7 Hz, 2H), 2.58–2.37 (m, 2H), 2.38–2.25 (m, 1H), 2.28–2.06 (m, 1H), 2.08–1.74 (m, 8H), 1.72 (d, J = 6.6 Hz, 3H), 1.66 (s, 3H), 1.58–1.38 (m, 4H), 1.52 (s, 3H), 1.44 (s, 9H), 1.33 (s, 3H), 0.95 (s, 3H). 13C NMR (101 MHz, MeOD) δ 181.5 (C), 175.7 (C), 172.7 (C), 171.6 (C), 158.6 (C), 140.1 (C), 138.1 (C), 128.5 (2 × CH aro), 121.5 (2 × CH aro), 87.9 (CH), 79.9 (C), 71.1 (C), 70.5 (C), 58.1 (CH), 55.5 (CH), 43.6 (CH2), 41.1 (CH2), 38.8 (2 × CH2), 32.8 (CH2), 30.6 (CH2), 30.5 (CH2), 30.3 (2 × CH3), 28.8 (3 × CH3), 26.3 (CH2), 24.2 (CH2), 23.5 (CH3), 20.8 (CH3), 20.7 (CH3), 16.6 (CH2). HRMS m/z (ESI) calcd for C35H57N6O7+ [M + H]+, 673.4283; found, 673.4285.
2b
1H NMR (400 MHz, MeOD) δ 1H NMR (400 MHz, MeOD) δ 7.43 (d, J = 8.5 Hz, 2H), 7.15 (d, J = 8.6 Hz, 2H), 4.66 (q, J = 6.6 Hz, 1H), 4.41 (dd, J = 8.4, 5.3 Hz, 1H), 4.13 (dd, J = 8.5, 4.7 Hz, 1H), 3.86 (d, J = 8.0 Hz, 2H), 3.53–3.26 (m, 2H), 2.89–2.26 (m, 1H), 2.26–2.09 (m, 1H), 2.10–1.97 (m, 1H), 1.89–1.79 (m, 1H), 1.73–1.45 (m, 6H), 1.41 (s, 9H), 1.39–1.31 (m, 21H), 1.19 (s, 3H), 1.14–1.01 (m, 3H), 0.92 (s, 3H), 0.52 (s, 3H). 13C NMR (101 MHz, MeOD) δ 181.5 (C), 175.6 (C), 172.2 (C), 171.4 (C), 164.5 (C), 157.7 (C), 154.1 (C), 143.1 (C), 138.2 (C), 128.2 (2 × CH aro), 121.2 (2 × CH aro), 84.4 (C), 84.1 (CH), 80.4 (C), 60.9 (2 × C), 58.2 (CH), 55.1 (CH), 43.4 (CH2), 41.4 (2 × CH2), 41.2 (CH2), 34.8 (2 × CH3), 30.5 (CH2), 30.4 (CH2), 28.6 (3 × CH3), 28.2 (3 × CH3), 26.7 (CH2), 26.6 (CH2), 23.7 (CH3), 20.8 (2 × CH3), 18.2 (CH2). HRMS m/z (ESI) calcd for C40H65N8O9+ [M + H]+, 801.4869; found, 801.4868.
Boc Deprotection
Boc-protected alkoxyamine 1b or 2b (300 mg, 1 equiv) was dissolved in DCM (4 mL), and then TFA was added (10 equiv). After 4 h of agitation at room temperature, TFA was evaporated in vacuo by coevaporation for three times with toluene. The crude product was purified by flash chromatography (8:2 DCM/MeOH) to obtain 1a (71%) or 2a (67%).
1a
1H NMR (400 MHz, MeOD) δ 7.64 (d, J = 8.5 Hz, 2H), 7.36 (d, J = 8.6 Hz, 2H), 5.20 (q, J = 6.6 Hz, 1H), 4.53 (dd, J = 9.0, 5.0 Hz, 1H), 4.33–4.20 (m, 1H), 4.02–3.93 (m, 2H), 2.96 (t, J = 7.6 Hz, 2H), 2.56–2.37 (m, 2H), 2.37–2.27 (m, 1H), 2.22–2.09 (m, 1H), 2.02–1.91 (m, 1H), 1.89–1.63 (m, 8H), 1.60 (d, J = 6.6 Hz, 3H), 1.56–1.42 (m, 6H), 1.40–1.32 (m, 3H), 1.24–1.14 (m, 3H), 0.82 (s, 3H). 13C NMR (101 MHz, MeOD) δ 180.1 (C), 174.4 (C), 170.9 (C), 170.2 (C), 138.9 (C), 137.9 (C), 127.0 (2 × CH aro), 120.1 (2 × CH aro), 84.8 (CH), 65.3 (2 × C), 56.7 (CH), 53.8 (CH), 42.3 (CH2), 39.2 (CH2), 38.4 (2 × CH2), 31.2 (CH2), 30.7 (2 × CH3), 29.1 (CH2), 26.7 (CH2), 25.2 (CH2), 22.4 (CH2), 22.3 (CH3), 19.4 (2 × CH3), 15.9 (CH2). HRMS m/z (ESI) calcd for C30H49N6O5+ [M + H]+, 573.3759; found, 573.3760.
2a
1H NMR (400 MHz, MeOD) δ 7.75–7.67 (m, 2H), 7.49–7.37 (m, 2H), 5.46 (q, J = 6.5 Hz, 1H), 4.59–4.45 (m, 1H), 4.27 (dd, J = 8.8, 4.5 Hz, 1H), 3.95 (d, J = 2.2 Hz, 2H), 3.25 (tt, J = 7.0, 4.1 Hz, 2H), 2.54–2.39 (m, 2H), 2.36–2.24 (m, 1H), 2.22–2.10 (m, 1H), 2.09–1.69 (m, 10H), 1.70–1.56 (m, 5H), 1.73 (d, J = 6.5 Hz, 3H), 1.53 (s, 3H), 1.34 (s, 3H), 0.94 (s, 3H). 13C NMR (101 MHz, MeOD) δ 181.6 (C), 175.9 (C), 172.1 (C), 171.7 (C), 158.7 (C), 140.1 (C), 137.9 (C), 128.5 (2 × CH aro), 121.6 (2 × CH aro), 88.0 (CH), 71.6 (C), 70.9 (C), 58.0 (CH), 54.9 (CH), 43.7 (CH2), 42.0 (CH2), 38.6 (CH2), 38.5 (CH2), 30.5 (CH2), 30.3 (CH2), 29.9 (2 × CH3), 26.6 (CH2), 26.3 (CH2), 23.5 (CH3), 20.7 (CH3), 20.6 (CH3), 16.5 (CH2). HRMS m/z (ESI) calcd for C30H49N8O5+ [M + H]+, 601.3820; found, 601.3818.
Cell Culture
The HT-1080 human fibrosarcoma-derived epithelial cell line, HT-29 human adenocarcinoma, and the U87 human glioblastoma cell line were used in this work. These cell lines were purchased from ATCC (LGC standards, Molsheim, France). Cells were cultured in DMEM supplemented with 10% fetal calf serum and 1% penicillin–streptomycin in a humidified atmosphere with 5% CO2 at 37 °C. All the culture products were from Gibco Corporation.
Tumor Implantation and Tumor Growth
The protocol was described by Guilhon et al.28 This protocol yields cell populations containing various proportions of tumor-infiltrating cells.
Briefly, the HT-1080 cell line at around 60% of confluence was collected at a cell density of 5 × 104 cells/μL. Fifty microliters of the cell suspension was injected subcutaneously in the thigh of female Swiss Nude mice. After 2 weeks, the subcutaneous tumor reached a volume of around 1 mL. The animals were sacrificed, and the tumor was excised for cell culture.
All experiments on animals have been approved by the local (life licensing committee of Bordeaux France CE50) and the French national ethic committee under file number APAFIS 36693. Animals were kept in our animal facility with agreement number B33063930.
Tumor Dissociation
Excised subcutaneous tumors were kept at 4 °C in RPMI (Gibco). One millimeter-thick slices were cut, and discarding extra-tumoral conjunctive tissue, the slices were divided again in small pieces. Then, they were incubated for 20 min at 37 °C in a solution of DMEM with trypsin (2.5%, Gibco) and DNase I (0.1%, Roche Diagnostics GmbH). Next, the solution was discarded, and the pieces were triturated in a DNase solution (DMEM, DNase I (0.1%), and Hepes (0.3M) (Sigma-Aldrich)). After a resting step of 15 min at room temperature and 10 min at 4 °C, the unhomogenized fragments were discarded and the cell suspension was centrifuged at 180g for 5 min. The pellet was suspended in DMEM and 10% FCS, and the cells were seeded in multiwell plates (CELLSTAR, Greiner Bio-One, Germany) at a density of 25,000 cells per well.
Cell Treatment
After 48 h of culture and before the prodrug treatment, the medium was exchanged to unsupplemented medium during 24 h. The goal is to decrease the cell growth, stop the cell cycle, and have the cells in the same phase. Then, the inhibitors and prodrugs were applied in serum-free culture media. After 1 h of incubation, FCS (10% final) was added and the cultures were kept for 48 h. The inhibitor and prodrug concentrations appear in the legend of the figures.
Viability Test
The cell proliferation reagent WST-1 (Sigma) was used according to the manufacturer’s instructions. The reagents are tetrazolium salts that are cleaved to soluble formazan by the mitochondrial dehydrogenases of viable cells. Consequently, the absorbance of the generated product is proportional to the living cells. Briefly, the solution was added to the culture medium at a 1/10th dilution and incubated at 37 °C. The absorbances were recorded with a Multiskan SkyHigh at 450 and 600 nm as reference wavelengths. After checking the time of incubation, the 450 nm absorbance was linear for more than 30 min. The one at 600 nm was constant during the time of measure. All readings were done between 20 and 30 min.
Enzyme Kinetics
Enzymatic Activation of the Substrate pGlu-Gly-Arg/Lys-anilide-TEMPO
The urokinase-type plasminogen activator (Merck) was dissolved and aliquoted following the provider’s instructions. Alkoxyamines (1 mM) were reacted with 2 μM uPA in 50 mM Hepes (pH = 7.5), 5 mM NaCl, and 2 μM hemin (Santa Cruz Biotechnology) to scavenge the freed alkyl radicals. Since homolysis results in the release of a stable TEMPO free radical, the reaction was recorded over time with the EPR spectrometer EMX Nano (Bruker) in a volume of 50 μL. Acquisition parameters: B0 = 3423 G; sweep time, 10 s; sweep width, 80 G; attenuation, 6 dB; delay between scan, 30 s; amplitude modulation, 4 G; gain, 50 dB.
Michaelis Constants
In a volume of 50 μL, 10 μM uPA, 1 mM hemin, and several concentrations of the alkoxyamine prodrug from 0 to 7.5 mM were mixed. Kinetics were monitored at 37 °C by EPR spectrometry with the following parameters: B0 = 3428 G; sweep width, 50 G; sweep time, 4.15 s; attenuation, 20 dB; delay between scan, 5 ms; modulation amplitude, 1 G; gain, 40 dB. Initial velocities were measured and plotted versus concentration for the extraction of Km, kcat, and kcat/Km.
Acknowledgments
This project has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement no. 863099.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.4c05592.
1H, 13C, DEPT 135, and HRMS of 1c, 2c, 1b, 2b, 1a, and 2a; HPLC traces of pGlu-Gly-Lys-anilide-TEMPO 1a and pGlu-Gly-Arg-anilide-TEMPO 2a; kinetic analysis of alkoxyamine bond homolysis; proof for the inactivity of MMP-2 on suc-AAPV-anilide-TEMPO (PDF)
Author Contributions
∥ M.F. and S.S. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Choi K. Y.; Swierczewska M.; Lee S.; Chen X. Protease-activated drug development. Theranostics 2012, 2, 156–178. 10.7150/thno.4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Huo F.; Zhang Y.; Cheng F.; Yin C. Enzyme-activated prodrugs and their release mechanisms for the treatment of cancer. Journal of Materials Chemistry B 2022, 10, 5504–5519. 10.1039/D2TB00922F. [DOI] [PubMed] [Google Scholar]
- Renoux B.; Fangous L.; Hotten C.; Peraudeau E.; Eddhif B.; Poinot P.; Clarhaut J.; Papot S. A beta-glucuronidase-responsive albumin-binding prodrug programmed for the double release of monomethyl auristatin E. MedChemComm 2018, 9, 2068–2071. 10.1039/C8MD00466H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legigan T.; Clarhaut J.; Tranoy-Opalinski I.; Monvoisin A.; Renoux B.; Thomas M.; Le Pape A.; Lerondel S.; Papot S. The first generation of beta-galactosidase-responsive prodrugs designed for the selective treatment of solid tumors in prodrug monotherapy. Angew. Chem. 2012, 51, 11606–11610. 10.1002/anie.201204935. [DOI] [PubMed] [Google Scholar]
- Martin H.; Lazaro L. R.; Gunnlaugsson T.; Scanlan E. M. Glycosidase activated prodrugs for targeted cancer therapy. Chemical Society reviews 2022, 51, 9694–9716. 10.1039/D2CS00379A. [DOI] [PubMed] [Google Scholar]
- Wang L.; Main K.; Wang H.; Julien O.; Dufour A. Biochemical Tools for Tracking Proteolysis. Journal of proteome research 2021, 20, 5264–5279. 10.1021/acs.jproteome.1c00289. [DOI] [PubMed] [Google Scholar]
- Gauthier F.; Fryksmark U.; Ohlsson K.; Bieth J. G. Kinetics of the inhibition of leukocyte elastase by the bronchial inhibitor. Biochimica et biophysica acta 1982, 700, 178–183. 10.1016/0167-4838(82)90095-4. [DOI] [PubMed] [Google Scholar]
- Huang H.; Zhang H.; Onuma A. E.; Tsung A.. Neutrophil Elastase and Neutrophil Extracellular Traps in the Tumor Microenvironment, In: Birbrair A. (Ed.) Tumor Microenvironment: State of the Science, Springer International Publishing: Cham, 2020, pp. 13-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niland S.; Riscanevo A. X.; Eble J. A. Matrix Metalloproteinases Shape the Tumor Microenvironment in Cancer Progression. Int. J. Mol. Sci. 2021, 23, 146. 10.3390/ijms23010146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulkower K. I.; Butler C. C.; Linebaugh B. E.; Klaus J. L.; Keppler D.; Giranda V. L.; Sloane B. F. Fluorescent microplate assay for cancer cell-associated cathepsin B. Eur. J. Biochem. 2000, 267, 4165–4170. 10.1046/j.1432-1327.2000.01458.x. [DOI] [PubMed] [Google Scholar]
- Mekkawy A. H.; Pourgholami M. H.; Morris D. L. Involvement of urokinase-type plasminogen activator system in cancer: an overview. Medicinal research reviews 2014, 34, 918–956. 10.1002/med.21308. [DOI] [PubMed] [Google Scholar]
- LeBeau A. M.; Kostova M.; Craik C. S.; Denmeade S. R. Prostate-specific antigen: an overlooked candidate for the targeted treatment and selective imaging of prostate cancer. Biol. Chem. 2010, 391, 333–343. 10.1515/bc.2010.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubal M.; Vymolova B.; Matrasova I.; Vymola P.; Veprkova J.; Syrucek M.; Tomas R.; Vanickova Z.; Krepela E.; Konecna D.; Busek P.; Sedo A. Fibroblast activation protein as a potential theranostic target in brain metastases of diverse solid tumours. Pathology 2023, 55, 806–817. 10.1016/j.pathol.2023.05.003. [DOI] [PubMed] [Google Scholar]
- Nozawa F.; Hirota M.; Okabe A.; Shibata M.; Iwamura T.; Haga Y.; Ogawa M. Elastase activity enhances the adhesion of neutrophil and cancer cells to vascular endothelial cells. The Journal of surgical research 2000, 94, 153–158. 10.1006/jsre.2000.6002. [DOI] [PubMed] [Google Scholar]
- Deryugina E.; Carre A.; Ardi V.; Muramatsu T.; Schmidt J.; Pham C.; Quigley J. P. Neutrophil Elastase Facilitates Tumor Cell Intravasation and Early Metastatic Events, iScience 2020, 23, 101799 10.1016/j.isci.2020.101799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z.; Yang P. Role of imbalance between neutrophil elastase and alpha 1-antitrypsin in cancer development and progression, The Lancet. Oncology 2004, 5, 182–190. 10.1016/S1470-2045(04)01414-7. [DOI] [PubMed] [Google Scholar]
- Audran G.; Marque S. R. A.; Mellet P. Smart Alkoxyamines: A New Tool for Smart Applications. Accounts of chemical research 2020, 53, 2828–2840. 10.1021/acs.accounts.0c00457. [DOI] [PubMed] [Google Scholar]
- Audran G.; Bagryanskaya E. G.; Bikanga R.; Coote M. L.; Guselnikova O.; Hammill C. L.; Marque S. R. A.; Mellet P.; Postnikov P. S. Dynamic Covalent Bond: Modes of Activation of the C—ON Bond in Alkoxyamines. Prog. Polym. Sci. 2023, 144, 101726 10.1016/j.progpolymsci.2023.101726. [DOI] [Google Scholar]
- Seren S.; Joly J. P.; Voisin P.; Bouchaud V.; Audran G.; Marque S. R. A.; Mellet P. Neutrophil Elastase-Activatable Prodrugs Based on an Alkoxyamine Platform to Deliver Alkyl Radicals Cytotoxic to Tumor Cells. Journal of medicinal chemistry 2022, 65, 9253–9266. 10.1021/acs.jmedchem.2c00455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon B. J.; Kwaan H. C. Components of the Plasminogen-Plasmin System as Biologic Markers for Cancer. Advances in experimental medicine and biology 2015, 867, 145–156. 10.1007/978-94-017-7215-0_10. [DOI] [PubMed] [Google Scholar]
- Rijken D. C.; Groeneveld E. Substrate specificity of tissue-type and urokinase-type plasminogen activators. Biochemical and biophysical research communications 1991, 174, 432–438. 10.1016/0006-291X(91)91434-E. [DOI] [PubMed] [Google Scholar]
- Bansal V.; Roychoudhury P. K. Production and purification of urokinase: a comprehensive review. Protein expression and purification 2006, 45, 1–14. 10.1016/j.pep.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Hsiao J. K.; Law B.; Weissleder R.; Tung C. H. In-vivo imaging of tumor associated urokinase-type plasminogen activator activity. J. Biomed. Opt. 2006, 11, 034013 10.1117/1.2204029. [DOI] [PubMed] [Google Scholar]
- Yamahana H.; Terashima M.; Takatsuka R.; Asada C.; Suzuki T.; Uto Y.; Takino T. TGF-beta1 facilitates MT1-MMP-mediated proMMP-9 activation and invasion in oral squamous cell carcinoma cells. Biochemistry and biophysics reports 2021, 27, 101072 10.1016/j.bbrep.2021.101072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S.Chapter 248 - ADAM Metalloproteinases, In: Rawlings N. D.; Salvesen G. (Eds.) Handbook of Proteolytic Enzymes (Third Edition), Academic Press, 2013, pp 1086-1094. [Google Scholar]
- Toth M.; Sohail A.; Mobashery S.; Fridman R. MT1-MMP shedding involves an ADAM and is independent of its localization in lipid rafts. Biochemical and biophysical research communications 2006, 350, 377–384. 10.1016/j.bbrc.2006.09.052. [DOI] [PubMed] [Google Scholar]
- Murphy G. The ADAMs: signalling scissors in the tumour microenvironment. Nat. Rev. Cancer 2008, 8, 932–941. 10.1038/nrc2459. [DOI] [PubMed] [Google Scholar]
- Guilhon E.; Voisin P.; de Zwart J. A.; Quesson B.; Salomir R.; Maurange C.; Bouchaud V.; Smirnov P.; de Verneuil H.; Vekris A.; Canioni P.; Moonen C. T. Spatial and temporal control of transgene expression in vivo using a heat-sensitive promoter and MRI-guided focused ultrasound. The journal of gene medicine 2003, 5, 333–342. 10.1002/jgm.345. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.