

Abstract
Immunotherapies hold immense potential for achieving durable potency and long-term survival opportunities in cancer therapy. As vital biological mediators, peptides with high tissue penetration and superior selectivity offer significant promise for enhancing cancer immunotherapies (CITs). However, physicochemical peptide features such as conformation and stability pose challenges to their on-target efficacy. This review provides a comprehensive overview of recent advancements in therapeutic peptides targeting key steps of the cancer-immunity cycle (CIC), including tumor antigen presentation, immune cell regulation, and immune checkpoint signaling. Particular attention is given to the opportunities and challenges associated with these peptides in boosting CIC within the context of clinical progress. Furthermore, possible future developments in this field are also discussed to provide insights into emerging CITs with robust efficacy and safety profiles.
Key words: Cancer-immunity cycle, Oncolytic peptide, Peptide neoantigen vaccine, Peptide mimotope, Immune checkpoint blockade, Cancer immunotherapy, Synthetic long peptide vaccine, Peptide-major histocompatibility complex
Graphical abstract
This review focuses on the recent advances in the roles, mechanisms, ongoing clinical trials, and challenges involving therapeutic peptides that target cancer–immunity cycle.
1. Introduction
Cancer immunotherapies (CITs) have revolutionized cancer treatments by providing potential for systemic therapies with high specificity, lasting efficacy, and long-term survival opportunities1. The current landscape of CITs is dominated by several modalities, including adoptive cell transfer, immune checkpoint inhibitors (ICIs), oncolytic viruses, cancer vaccines, and cytokine therapies. These approaches represent significant milestones in the field of anticancer treatment2. Despite these notable advances, most currently available CITs still have limitations, such as low objective response rates, inevitable side effects, time-consuming and technically challenging production processes, and high costs3. Therefore, novel agents that are efficient and safe could significantly improve cancer treatment efficacy and become an emerging alternative to current drugs.
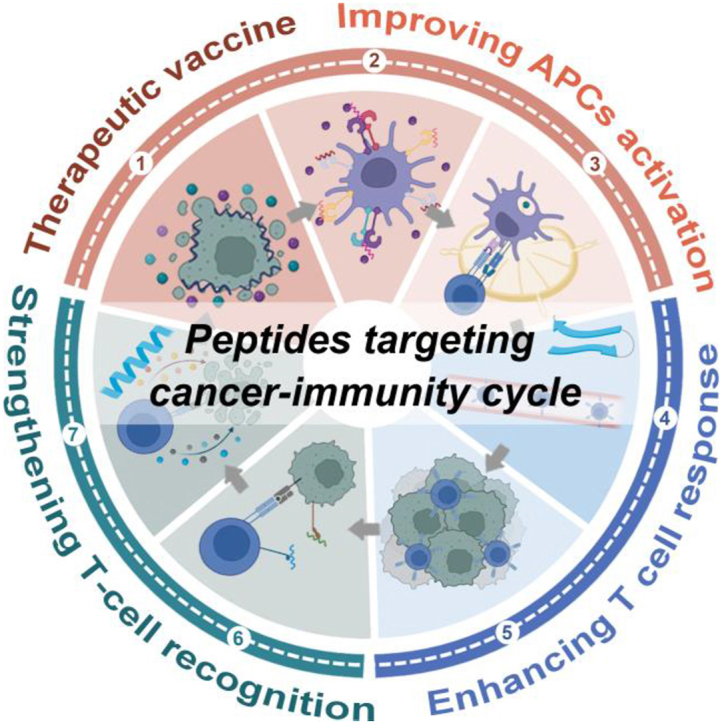
Peptides serve as natural ligands for numerous cell surface receptors and play critical physiological roles by orchestrating intracellular signal transduction pathways4. Thus, peptides, particularly endogenous ones, are frequently employed as viable and efficacious lead compounds in the pursuit of new drug discovery5. Currently, there are approximately 100 approved peptide drugs on the global market alongside hundreds of peptides undergoing clinical trials or preclinical studies6,7. It is estimated that the peptide pharmaceutical market will reach a value of around USD 48 billion by 2025, exhibiting a compound annual growth rate of 9.47. Compared to biomacromolecules such as recombinant proteins or antibodies commonly employed in CITs, peptides exhibit reduced immunogenicity, enhanced tissue penetration, and facile modification. As crucial biological mediators, peptides also demonstrate superior selectivity and specificity in inhibiting protein–protein interactions compared to small molecule drugs. Consequently, peptides possess immense potential for augmenting the efficacy of CITs by precisely activating and sustaining the so-called cancer-immunity cycle (CIC), which encompasses a series of functional sequential events describing how the immune system activates and eliminates cancer cells (Fig. 1)8, 9, 10. Additionally, low-molecular-weight peptides are generally easy to prepare, modify, store, and utilize at reduced costs, thereby facilitating their sustainable development. This review presents the roles of the peptides in CIC and various peptide-based strategies developed for achieving tumor-specific immunotherapy. Particular emphasis is placed on recent preclinical and clinical advancements related to these peptides that enhance CIC, as well as the opportunities and challenges in this field.
Figure 1.

Schematic symphony of peptides in cancer-immunity cycle (CIC). CIC can be divided into seven steps (numbered 1–7): 1) antigens release from dying tumor cells, 2) APCs uptake tumor antigens, 3) T-cell priming and activation in local lymph nodes, 4) trafficking of T cells to tumors via blood vessels, 5) infiltration of T cells into tumor tissues, 6) recognition of tumor cells by T cells, and 7) T-cell-mediated killing of tumor cells in the tumor microenvironment; then, dying tumor cells further contribute to a new round of CIC. Among them, peptides are mainly involved in four steps of the above cycle: (A) therapeutic vaccines, (B) improving APC activation, (C) enhancing T-cell response, and (D) strengthening T-cell recognition.
2. Peptides as immunogenic cell death (ICD) inducers
2.1. Effect of ICD in the CIC
The emission of five key signaling hallmarks characterizes ICD, which is triggered by tumor-associated antigens (TAAs) and danger-associated molecular pattern molecules (DAMPs) released from dying tumor cells. During ICD, secreted ATP acts as a “come-to-me signal” to recruit and activate immature dendritic cells (DCs); Annexin A1 guides the migration of immature DCs toward dying cells by a “find me” signal. Exposed CRT functions as an “eat-me” signal for antigen uptake by immature DCs associated with dead cells. HMGB1 release promotes DC maturation, while interferon I secretion stimulates immune cell activation through binding to surface receptors, thereby initiating an adaptive immune response11.
Although traditional chemotherapeutics, radiotherapy, and phototherapy have been shown to induce ICD, their effectiveness is limited by nonspecific cytotoxicity, scarce tumor antigens, inferior tumor penetration ability, or hypoxia12. In recent years, oncolytic viruses have generated tremendous excitement in the field of ICD-induced immunotherapy since talimogene laherparepvec was approved for advanced melanoma therapy by the FDA in 201513. However, oncolytic viruses inevitably face multiple concerns in terms of safety, storage, and administration14.
2.2. Oncolytic peptide-induced ICD
Oncolytic peptides, a class of emerging oncolytic alternatives derived from α-helical natural antimicrobial peptides, exhibit lower immunogenicity and production costs than oncolytic viruses. They possess distinct advantages based on the relatively homogeneous surface properties of tumor cells despite intratumoral heterogeneity15. Cationic charges and hydrophobic networks are two critical attributes that enable oncolytic peptides to selectively bind to anionic components (such as phosphatidylserine, O-glycosylated mucin, sialoganglioside, and heparin) expressed on tumor cell membranes through electrostatic adsorption. This binding interaction provides a foundation for the selective action of oncolytic peptides against tumor cells. The mechanisms underlying membrane-cavity formation can be categorized into three major models: barrel-stave, toroidal, and carpet models16. For instance, in the “barrel-stave” mechanism, peptides, such as melittin, form a bundle that inserts into the cell membrane once it reaches a certain concentration17. In the “toroidal” mechanism, peptides, such as maganins, induce the formation of various pores that cause thinning and curvature of the membrane18. In the “carpet” mechanism, peptides, including LL-37, cover the surface and disrupt the membrane structure like surfactants19. Oligomerization or structural rearrangement of tumor cell membranes can lead to rapid membrane lysis and accidental cell death, resulting in the release of abundant TAAs and DAMPs independent of intratumoral heterogeneity20,21. Subsequently, mature DCs present captured TAAs to naïve T cells, leading to their differentiation. In addition to disrupting the cell membrane, oncolytic peptides can also translocate into intracellular compartments, including the mitochondrial outer membrane, endoplasmic reticulum (ER), and Golgi apparatus, thereby triggering accidental cell death. For instance, LL-37 activated the intrinsic apoptotic pathway by inducing the release of apoptosis-inducing factor from the mitochondrial to the cytosol, resulting in mitochondrial outer-membrane permeabilization22. The peptide PFR derived from lactotransferrin targeted the ER and induced ER stress, elevation of cytoplasmic calcium levels, and the production of mitochondrial reactive oxygen species that ultimately lead to tumor cell death23. LTX-401 is an oncolytic peptidomimetic derivative that selectively targets the Golgi apparatus causing prominent cytoplasmic vacuolization and ICD24.
LTX-315, which is derived from the host defense peptide bovine lactoferricin, has been extensively studied and is considered a representative oncolytic peptide due to its ability to induce complete tumor regression and systemic immune responses in various carcinomas25. This effect was achieved through selective rapid membrane disruption and subsequent stimulation of ICD25. In addition, recent findings have identified ATP11B as a novel potential target of LTX-315 for inhibiting programmed cell death 1 ligand 1 (PD-L1) expression in pancreatic tumor cells25. Clinical evidence also demonstrated that LTX-315 can remodel the tumor microenvironment (TME) and increase intralesional CD8+ T cells in 86% of evaluated patients (12 out of 14), resulting in reduced a volume of injected tumors in 29% of patients26. Although intratumoral injection of LTX-315 caused vascular disorders and hypersensitivity/anaphylaxis in patients with advanced solid tumors, all treatment-related adverse events and toxicities were manageable and reversible26.
Additionally, ongoing clinical trials of several other oncolytic peptides for advanced solid tumors have shown promising results (Table 1). The preference of oncolytic peptides for targeting tumor cells minimizes nonspecific toxicity to normal tissues and holds the potential for overcoming drug resistance. However, the limited applicability of intratumoral administration restricts the use of oncolytic peptides for inaccessible or metastatic cancers.
Table 1.
Clinical trials of representative peptides participated in CIC.
| Sort | Agent | Conditions | Phase | Status | Combination | NCT numberb |
|---|---|---|---|---|---|---|
| Oncolytic peptide | CyPep-1 | Advanced solid tumor malignancy | I/II | Recruiting | Use alone or combined with pembrolizumab | NCT04260529 |
| Advanced head and neck squamous cell, carcinoma, advanced breast cancer, and advanced melanoma | I/II | Recruiting | Use alone or combined with pembrolizumab | NCT05383170 | ||
| EP-100 | Ovarian cancer | II | Completed | Use alone or combined with paclitaxel | NCT01485848 | |
| Advanced solid tumors | I | Completed | Use alone | NCT00949559 | ||
| LL-37 | Melanoma | Completed | Use alone | NCT02225366 | ||
| LTX-315 | Transdermal accessible tumors | I | Completed | Use alone | NCT01058616 | |
| Melanoma, breast cancer, head and neck cancer, lymphoma, and triple-negative breast cancer | I | Completed | Use alone or combined with ipilimumab or pembrolizumab | NCT01986426 | ||
| Carcinoma | I | Completed | Combine with a cancer vaccine (GV1001) | NCT01223209 | ||
| Advanced solid tumor | II | Recruiting | Combine with pembrolizumab | NCT04796194 | ||
| Cancer of the skin and basal cell | II | Recruiting | Use alone | NCT05188729 | ||
| Soft tissue sarcoma | II | Active, not recruiting | Combined with TILs | NCT03725605 | ||
| Synthetic long peptide vaccines | NeoVax | Melanoma | I | Completed | Combined only with poly-ICLC | NCT01970358 |
| Glioblastoma | I | Recruiting | Combined with radiation therapy, Pembrolizumab and Temozolomide | NCT02287428 | ||
| Follicular lymphoma | I | Recruiting | Combined with Rituximab and Pembrolizumab | NCT03361852 | ||
| Lymphocytic leukemia | I | Recruiting | Combined with Cyclophosphamide and Pembrolizumab | NCT03219450 | ||
| Melanoma | I | Recruiting | Combined with Nivolumab Ipilimumab and Montanide | NCT03929029 | ||
| Ovarian cancer | I | Recruiting | Combined with Nivolumab | NCT04024878 | ||
| Kidney cancer | I | Recruiting | Combined with Ipilimumab | NCT02950766 | ||
| Melanoma and metastatic melanoma | I | Recruiting | Combined with CDX-301 and Nivolumab | NCT04930783 | ||
| GAPVAC | Glioblastoma | I | Completed | Combined with Poly-ICLC and GM-CSF | NCT02149225 | |
| Melanoma | I | Completed | Combined with poly-ICLC | NCT01970358 | ||
| Immunomodulator peptide | Pam3Cys-GDPKHPKSF (XS15) | Glioblastoma Multiforme of brain | I | Recruiting | Use alone | NCT04842513 |
| Point mutation antigenic peptide (In the past ten years) | MART-126–35(27L)a gp100209–217(210M)a | Melanoma (Skin) | I | Completed | Combined with BMS-936558 and Montanide ISA 51 VG | NCT01176461 |
| gp100209–217(210M)a | Recurrent melanoma | II | Completed | Combined with HPV 16 E7:12-20 | NCT00003895 | |
| Recurrent melanoma | III | Completed | Combined with Aldesleuk and Montanide ISA 51 VG | NCT00019682 | ||
| Melanoma | II | Completed | Combined with Leuprolide and MAGE-3 Peptide | NCT00254397 | ||
| NY-ESO-1157–165(165V)a | Melanoma (Skin) | I | Active, not recruiting | Combined with Nivolumab, gp100:280-288 (288V), Montanide ISA 51 vegetable grade (VG), and Ipilimumab | NCT01176474 | |
| Target immune checkpoint peptide | IO103 | Multiple myeloma | I | Completed | Combined with Montanide | NCT03042793 |
| Metastatic melanoma | I/II | Recruiting | PD-L1(IO103)/IDO(IO102) peptide vaccine combined with Nivolumab | NCT03047928 | ||
| Chronic lymphocytic leukemia | II | Completed | PD-L1(IO103), PD-L2(IO120) peptides with Montanide ISA51 | NCT03939234 | ||
| Oropharynx/larynx/hypopharynx/oral cavity squamous cell carcinoma | II | Recruiting | PD-L1(IO103)/IDO(IO102) peptide vaccine | NCT04445064 | ||
| Basal cell carcinoma | II | Completed | Use alone | NCT03714529 | ||
| Lung cancer non-small cell, head and neck squamous cell, carcinoma, urothelial carcinoma bladder | II | Recruiting | PD-L1(IO103)/IDO(IO102) peptide vaccine combined with pembrolizumab | NCT05077709 | ||
| Melanoma, squamous cell carcinoma of the head and neck | II | Not yet recruiting | PD-L1(IO103)/IDO(IO102) peptide vaccine combined with pembrolizumab | NCT05280314 | ||
| Metastatic melanoma, unresectable melanoma | III | Recruiting | PD-L1(IO103)/IDO(IO102) peptide vaccine combined with pembrolizumab | NCT05155254 |
Sequence: MART-126–35 (27L): ELAGIGILTV, gp100209–217 (210M): IMDQVPFSV, NY-ESO-1157–165 (165V): SLLMWITQV.
Clinical trials registered with the US National Library of Medicine at clinicaltrials.gov.
3. Peptide-based neoantigen vaccines
In the past decade, active immunotherapy utilizing de novo mutated tumor neoantigens has emerged as a personalized therapeutic strategy for cancer patients, which is distinct from passive immunotherapy induced by oncolytic peptides27.
3.1. Tumor specific antigens/neoantigens
The selection of antigens plays a crucial role in determining the efficacy of therapeutic vaccines. Early therapeutic vaccines primarily composed of TAAs have shown immune tolerance due to their expression in normal tissues, leading to unsatisfactory results in clinical trials28,29. In contrast, tumor-specific antigens/neoantigens, derived from nonsynonymous mutations, gene alterations, and genetic information are exclusively expressed in cancerous tissues30,31. Stimulation of antigen-specific T-cell responses by neoantigens prevents “off-target” damage to nonmalignant tissues and central tolerance of T cells toward self-epitopes. Consequently, neoantigens hold promise as cancer vaccines capable of eliciting a more specific and durable T-cell immune response32,33.
3.2. Synthetic long peptide (SLP) vaccines
Peptide vaccines, nucleic acid vaccines, and neoantigen-pulsed DC vaccines represent the three platforms of personalized neoantigen vaccines34,35. Among these platforms, neoantigen-derived peptides offer distinct advantages in terms of their explicit sequence, ease of preparation, and stable chemical properties compared to other vaccine forms. Peptide vaccines are typically designed as SLPs containing multiple sequences (25–35 amino acids)36,37, which can be more efficiently endocytosed and processed by antigen-presenting cells (APCs) within lysosomes. Subsequently, cleaved short peptides (9–11 residues) and long peptides (14–16 residues) are loaded onto major histocompatibility complex (MHC)-I and MHC-II molecules of APCs, respectively. These complexes then present the antigens to CD8+ or CD4+ T cells in a double-signal manner, thereby eliciting a potent T-cell response38. However, MHC-I-restricted short peptide-based vaccines alone may not be sufficient to induce robust CD8+ T-cell responses due to the relatively weak efficacy of MHC-I peptides39.
Based on whole-exome DNA sequencing of matched tumor and normal cells, NeoVax (Table 1), a personalized SLP vaccine targeting up to 20 individualized tumor neoantigens, was synthesized by predicting mutated peptides with high-affinity for binding to autologous human leukocyte antigen (HLA) molecules. In a phase I clinical study, four of six patients with high-mutation-rate melanoma showed no disease recurrence at the two-year follow-up after NeoVax vaccination. Additionally, two other patients with recurrent melanoma achieved complete tumor regression following treatment with a programmed cell death 1 (PD-1) antibody40,41. Compared to previous TAA vaccines, NeoVax demonstrated enhanced tumor specificity and immunogenicity32. Furthermore, when NeoVax was administered as an immunotherapy for glioblastoma, an immunologically “cold” tumor type characterized by insufficient mutation burden (Table 1), neoantigen-specific T-cell responses were induced within intracranial glioblastoma tumors42. Unfortunately, all glioblastoma patients experienced subsequent tumor recurrence and ultimately succumbed to progressive disease, indicating considerable challenges for the CITs of glioblastoma and the limitations of NeoVax immunotherapy43.
4. Immunomodulatory adjuvants based on peptides
4.1. The role of DCs in the CIC
Successful CITs entail activation of innate immune responses, leading to DC maturation, tumor antigen presentation, and subsequent activation of a cascade of adaptive immunity, ultimately leading to activation of antigen-specific T cells44. As professional APCs, DCs possess the ability to process endogenous proteins via the proteasome and present antigens to cytotoxic T cells in an MHC class I-restricted manner, while exogenous proteins within late endosomes undergo processing through phagocytosis and endocytosis, delivering antigens to helper T cells in an MHC class II-restricted way45, 46, 47. However, immature DCs can induce T-cell conversion into anergic and regulatory/inhibitory T cells when exposed to antigens48. In contrast, only mature DCs with costimulatory signals facilitate the differentiation of specific effector T cells, highlighting the significance of DC maturation in immune therapy49. Thus, the magnitude of the T-cell immune response primarily relies on the use of vaccine adjuvants; however, due to the potential risks associated with induced systemic inflammation, only a limited number of FDA-approved adjuvants have been used in humans since 193950. In terms of the underlying mechanism, in addition to self-derived DAMPs, vaccine adjuvants can also mimic pathogen-associated molecular patterns (PAMPs) and transmit danger signals to DCs through pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), C-type lectin receptors, and NOD-like receptors51. Consequently, PRR agonists have been extensively employed as potent vaccine adjuvants for inducing functional maturation of DCs49.
4.2. Peptide-mediated DC maturation
TLRs are the most extensively studied family of PRRs that specifically recognize PAMPs in microbial species52. Among the TLR family members, TLR2 has garnered significant attention as a potential therapeutic target for immune adjuvants due to its ability to sensitively detect diverse PAMPs derived from various pathogens on both mouse and human conventional DCs (cDCs)53. By forming heterodimers with either TLR1 or TLR6, TLR2 expands the repertoire of ligands against pathogens by recognizing lipopeptides or lipoproteins produced by microorganisms54 (Figure 2, Figure 3). Consequently, lipopeptide agonists, represented by MALP-2, Pam2CSK4, and Pam3CSK4, were consecutively reported and applied to enhance the efficacy of cancer vaccination and tumor rejection55. However, two major intrinsic challenges significantly hinder the utilization of these lipopeptides under physiological conditions: poor esterase stability and susceptibility to oxidation56,57. Increasing attention has been given to the development of chemically and metabolically stable TLR2 agonists as immune adjuvants. Wu group56 reported a newly designed Pam3CSK4 derivative SUP3 by replacing two ester groups with carbamates, substituting serine with glycine, and eliminating the fourth lysine residue (Fig. 2). SUP3 maintained its specificity and affinity for TLR2 while demonstrating remarkable improvements in stability and eliciting a more robust antitumor response in mice without inducing excessive inflammation compared to Pam3CSK456. Moreover, a novel Pam3Cys derivative called XS15 (Table 1) was evaluated as an effective adjuvant in a healthy human volunteer. The combination of XS15 and multi-peptide vaccination in a water-in-oil emulsion elicited long-lasting antigen-specific T-cell responses in circulation for more than one year following a single vaccination58. These results highlight the potential of XS15 as an adjuvant for cancer vaccination, leading to the ongoing phase I clinical trial recruiting patients with newly diagnosed HLA-A2-positive MGMT-methylated glioblastoma (NCT04842513). Of note, XS15 has also been employed in COVID-19 vaccine development to induce SARS-CoV-2 T-cell immunity59.
Figure 2.

Structures of the human TLR2/TLR6-Pam2CSK4 complex (PDB: 3A79), human TLR2/TLR1-Pam3CSK4 complex (PDB: 2z7x), and SUP3. TLR1, TLR2, and TLR6 are depicted in blue, grey, and green respectively. Additionally, the Pam2CSK4 and Pam3CSK4 are shown by sticks in yellow and pink. The hydrogen bonds are marked by red dotted lines and their interaction residues are labeled accordingly.
Figure 3.

Signaling pathways induced by Clec9a, Clec10a, TLR2/TLR1, and TLR2/TLR6. (A) Clec9a possesses a hemi-immunoreceptor tyrosine-based activation motif (HITAM) cytoplasmic tail in the signaling pathway that contains a highly conserved tyrosine. Upon phosphorylation of this tyrosine, it facilitates the recruitment and binding of spleen tyrosine kinase (SYK). Consequently, the phagocytosed dead cell fragments are transported to a recycling endosome compartment. (B) TLR2 specifically triggers the MyD88-dependent pathway, recruiting interleukin-1 receptor-associated kinases (IRAKs) to the TLR1/TLR2 and TLR2/TLR6 complexes. Subsequently, phosphorylated and activated IRAK binds to tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6), thereby activating the IκB kinase (IKK) complex for promoting NF-κB translocation into the cell nucleus. Additionally, activation of the mitogen-activated protein kinase (MAPK) pathway can also occur, facilitating AP-1 activation. Ultimately, these aforementioned signaling molecules induce inflammatory cytokines and chemokines in a concerted manner. (C) When combined with GalNAc ligands, human Clec10a can trigger extracellular regulated protein kinase (ERK)-dependent pathways, leading to the phosphorylation of ERK and subsequent activation of p90-RSK, cAMP, and response-element binding protein (CREB). This cascade ultimately enhances the secretion of IL-12 and TNF-α.
Among all subsets of DCs, cDCs have demonstrated superiority in antigen presentation and immune enhancement compared to other DC subsets60,61. Due to the remarkable impact of cDCs on antigen presentation and immunopotentiation61, C-type lectin-like receptor 9A (Clec9a) has emerged as a crucial target for delivering cancer antigens to DCs. Clec9a is highly expressed on cDCs in lymphoid organs, blood, and peripheral tissues such as tumors. It specifically recognizes and phagocytoses fragments from dead cells, leading to the subsequent routing of dead cell-associated antigens into the cross-priming pathway (Fig. 3)62,63. Therefore, Clec9a-based peptides offer potential tools for the direct delivery of cancer antigens without the need for adjuvants, thereby preventing the inflammatory side effects associated with adjuvants. CBP-12 was developed as a 12-mer peptide carrier with high affinity for the Clec9a (KD = 0.61 ± 0.09 μmol/L) via an in silico-aided method. CBP-12-conjugated peptide vaccines enhanced uptake and cross-presentation of antigens, leading to stimulation of specific CD8+ T-cell responses and inhibition of tumor growth, even under adjuvant-free conditions64. Ca2+-dependent lectin-type receptor family member 10A (Clec10a) is another essential pathogen-recognition receptor for DC maturation and is characterized by high recognition for those structures containing terminal N-acetyl galactosamine (GalNAc)65. Generally, glycans containing terminal GalNAc residues are referred to as Tn antigens (GalNAc-Ser/Thr) and are highly expressed in nearly all cancer cells (Fig. 3). These antigens can trigger a cascade of immune events, including DC maturation and T-cell activation66. Using a phage display library with GalNAc-specific lectin receptor analogs, researchers developed sv6D, a tetravalent compound, with a tri-lysine core based on a 6-mer peptide. At a concentration of 10 nmol/L, sv6D demonstrated a significant cellular response. Subcutaneous injection of Sv6D led to substantial expansion of mature DCs in the peritoneal cavity and prolonged survival time in mice bearing ovarian tumors without any observed toxicity67.
5. Peptide mimotopes for enhanced T-cell response
Peptides function as epitopes and elicit a robust adaptive response in the antigen processing and presentation pathway68. The processed antigens are displayed on the surface of target cells or APC as peptide–MHC complex, which interacts with and binds to T cells, initiating T-cell recognition. Subsequently, the antigen recognition signal is transduced into T cells through CD3. Upon receiving additional synergistic stimulation signals, T cells can be activated and differentiate into effector T cells and memory T cells. CD8+ cytotoxic T cells then exert their primary antitumor immune effect by recognizing antigenic peptides presented by MHC I molecules and eliminating cancer cells. Furthermore, accumulating evidence has demonstrated that CD4+ T cells can also eliminate autologous tumors through the MHC II pathway while impeding cancer cell cycle progression to play an antiproliferative role69,70. Simultaneously, CD4+ T cells can secrete cytokines such as tumor necrosis factor and IFN-γ to facilitate the activation, differentiation, and proliferation of cytotoxic T lymphocytes, thereby augmenting their killing capacity71, 72, 73. However, previous clinical trials using peptide antigens have encountered setbacks due to their limited efficacy and affinity74, as T-cell responses are typically influenced by the affinities between antigenic peptides and MHC molecules. Therefore, peptide mimotopes that function as epitope analogs have been developed to enhance the effectiveness of epitope-based vaccines and elicit a robust antigen-specific T-cell response73.
5.1. Improvement of peptide-MHC binding affinity
Based on the crystal structures of MHC molecules and epitope peptides, which elucidate their critical protein–protein interaction modes (Fig. 4), anchor residues of epitope peptides can be further modified using artificial or natural amino acids to generate mimotopes, thereby improving antigenicity and binding affinity73. The peptide binding grooves of both the MHC-I and MHC-II molecules are flanked by two α-helices and one beta-pleated sheet. Epitope peptides, in an extended conformation, interact with six pockets (from A to F) within the grooves. In MHC-I molecules, the last residue of the epitope peptide is buried within pocket F, whereas in MHC-II molecules, the epitope peptide extends beyond the F pocket. Through comprehensive analysis of MHC molecules, consensus peptide binding motifs have been identified for numerous alleles and utilized to predict high-affinity peptides through continuously improved algorithms. One such instance was the MUC1 epitope, an ideal tumor antigen for various tumor types, but MUC1 epitopes have not yet been approved for phase III clinical trials. A possible explanation lies in the low affinities of wild-type MUC1 epitopes for MHC molecules. Through computational prediction algorithms, three modified MUC1 peptides were generated by introducing point mutations at position 1 (P1) and position 2 (P2) residues of the MUC1 wild-type epitopes. These altered MUC1 mimotopes exhibited enhanced affinities for MHC-I HLA-A0201 and demonstrated stronger immunogenicity compared to their respective wild-type MUC1 epitopes, suggesting their potential utility as antitumor peptide vaccines75. However, modifications to the anchor residues of the typical melanoma-associated MHC-I-restricted epitopes gp100209–21776 (Table 1) and Melan-A/MART-126–3577 (Table 1) did not generate T cells with improved functionality or cross-reactivity compared to wild-type epitopes. This finding suggested that mutagenesis may sometimes be limited by a loss of peptide specificity, highlighting the importance of precise T-cell receptor (TCR)-pMHC interactions for generating a specific T-cell repertoire78,79.
Figure 4.

Binding of epitope peptides to the classic MHC I and MHC II molecules. The human MHC I class molecule (HLA-A∗0201, blue) and MHC II class molecule (HLR-DR1, purple) are bound to the peptide LLFGYPVYV (left, PDB ID:1HHK) and the peptide PKYVKQNTLKLAT (right, PDB ID:1FYT), respectively. Stick views of peptides are used to display their binding grooves with MHC I and MHC II molecules. The arrows indicate the critical positions for binding with MHC molecules (in pink) and binding to TCRs (in rose red), numbered P1–P9. The function of the unnumbered pockets depends on the specific MHC molecules. Figures were generated by PyMOL.
5.2. Improvement of TCR-pMHC binding affinity
The TCR-pMHC interaction plays a crucial role in antigen-specific adaptive immunity, thus another avenue for mimotope development is to increase the binding affinity between pMHC molecules and TCRs80. One extensively studied tumor antigen is NY-ESO-1, which is presented by HLA-A∗0201 in complex with the TCR. By analyzing the structure of TCR-like antibodies bound to the NY-ESO-1157–165 peptide (SLLMWITQC), noncritical residues at position 9 (P9) of NY-ESO-1157–165 were mutated, resulting in an analog called NY-ESO-1157–165V (SLLMWITQV) (Table 1). This analog exhibited a 20-fold improvement in binding affinity81 and a 100-fold increase in potency for activating both CD4+ and CD8+ T cells82. Wei et al.83 screened peptide libraries to identify residue substitutions in GP70423–431 (SPSYVYHQF), which were derived from an endogenous retrovirus known as MuLV and restricted by the MHC-I molecule. They discovered that alanine substitution at position 5 within GP70423–431 induced significant conformational changes during TCR engagement with the wide-type peptide, leading to enhanced stability of the pMHC–TCR complex. These findings elucidate the efficacy of modified GP70423–431 as a vaccine candidate83.
Collectively, these findings suggest that ideal peptide mimotopes should be efficiently presented by MHC molecules, and accurately recognized by TCRs. Although the mutation of weak epitope peptides to generate high-affinity and effective mimotopes remains challenging, methods for predicting and enhancing the binding of peptide mimotopes to MHC or TCR are currently under development80,84.
6. Peptide-mediated immune checkpoint blockade (ICB)
The amplitude and quality of the antitumor immune response rely on the balance between inhibitory signals and costimulatory signals. In CIC, dysregulation of effector immune cells caused by immune checkpoint activation leads to the evasion of cancer cells from immune surveillance85. To counteract this negative feedback, immunotherapeutic antibodies have been developed to block various immune checkpoints, including the well-studied cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), PD-1, and its ligand PD-L1. However, a drawback of these antibodies is the emergence of severe immune-related adverse events (irAEs) in patients86. In contrast to therapeutic antibodies, peptides offer promising alternatives with enhanced tumor penetration and reduced systemic retention and irAEs. Of note, numerous peptide-based ICIs have been extensively employed in nanomedicines and combination therapies for CITs (Fig. 5)87.
Figure 5.

Peptides targeting immune checkpoints. There are three pairs of immune checkpoints in the interaction between APCs and T cells: 1) PD-L1 or PD-L2/PD-1, 2) LAG-3/MHC, and 3) CD28/CTLA-4. Regarding the interaction between T cells and tumor cells, five pairs of immune checkpoints associated with peptides are shown. Among them, 4) VISTA/VSIG-3, 5) TIGIT/CD155, and 6) BTLA/HVEM are new immune checkpoints. In addition, 7) CD47/SIRPα represents a pair of immune checkpoints in the interaction between macrophages and tumor cells. The plus sign (pink) indicates a positive effect on the antitumor response, while the minus sign (gray) indicates an inhibitory effect. The peptides targeting these immune checkpoints are highlighted in yellow.
6.1. Design strategies of peptide ICIs
Peptide ICIs are specifically designed to target critical protein–protein interaction interfaces known as “hot spots” or epitopes, with the aim of blocking immune checkpoints. These essential motifs can be categorized into three types: (a) short-chain sequences comprising consecutive peptide epitopes; (b) secondary structural epitopes, such as a side of the face from the α-helix that can bind to the hydrophobic groove in complementary residues; (c) tertiary structural epitopes targeting both sides of the protein–protein interaction interface88. Based on these structural features, the rational design principles for peptide ICIs can be summarized as follows. (a) If the binding motif between receptors and ligands is known, specific fragments from the interaction interface can be utilized to obtain the desired peptide. (b) In cases where only one of these proteins is known, a “Peptide Walking” strategy could be employed to develop a self-inhibitory peptide. (c) When the motif is unknown, ICI peptides can be generated through computational de novo sequencing methods or screening phage-display libraries for potential targets8.
6.2. Peptides targeting classic immune checkpoints
The PD-1/PD-L1 signaling pathway plays an essential role in maintaining immune homeostasis. Engagement of PD-L1/PD-L2 and PD-1 triggers phosphorylation of the ITIM and ITSM motifs on PD-1. Subsequently, SH1 and SH2 containing tyrosine phosphatase 1/2 (SHP-1, SHP-2) are recruited by PD-1 to inhibit the downstream PI3K/AKT pathway. This leads to the suppression of T-cell proliferation, survival, as well as protein synthesis and IL-2 production. In addition, the interaction between PD-1 and PD-L1 also enhances PTEN activity to further restrain the PI3K pathway (Fig. 5). Tumor cells often exploit the negative feedback mechanism mediated by PD-1/PD-L1 to evade immune surveillance and achieve immune escape. On the other hand, IFN-γ secreted by tumor-infiltrating lymphocytes could induce the upregulation of PD-L1 expression on tumor cells through the IFNGR-1/2- JAK1/2-STAT1/3-IRF-1 axis. Thus, blockade of either PD-1 or PD-L1 could augment CIT efficacy while prolonging the duration of the immune response89, 90, 91. To mitigate irAEs, the peptide NP-12 (Table 2) was developed by utilizing critical binding epitopes as the first PD-1-targeting peptide, demonstrating remarkable antitumor activity in vivo92. CLP002 (Table 2), a 12-residue anti-PD-L1 peptide inhibitor, was developed through phage display biopanning and exhibited superior tumor penetration compared to that of the PD-L1 antibody93. To enhance the proteolytic resistance of the peptide, DPPA-1 (Table 2), the first d-enantiomeric peptide antagonist, was designed by combining mirror-image phage display and chemical protein synthesis techniques94 and subsequently was optimized to yield OPBP-1 (Table 2) with improved antitumor efficacy via point mutation and molecular dynamics simulation approaches95. Notably, due to the potent antitumor activities and enzymatic resistance of OPBP-1 in vivo, N,N,N-trimethyl chitosan was used to fabricate hydrogel for the oral delivery of OPBP-1. The OPBP-1-based hydrogel exhibited high bioavailability and significant antitumor efficacy with negligible toxicity, opening a new prospect for the oral administration of ICIs in cancer immunotherapy95.
Table 2.
Targets, names, and sequences of representative peptides targeting immune checkpoints.
| Target | Peptide name | Sequence | Ref. |
|---|---|---|---|
| PD-1 | NP-12 | H-SNTSESFK(H-SNTSESF)FRVTQLAPKAQIKE-NH2 | 92 |
| PD-L1 | CLP002 | WHRSYYTWNLNT | 93 |
| DPPA-1 | H-nyskptdrqyhf-NH2a | 94 | |
| OPBP-1 | gqsehhmrvysfa | 95 | |
| CTLA-4 | ERY2-4 | Ac-C1AWGQAILEGELAWLEGGGGGAGQLADLKRQLAWWKQAC1-NH2b | 99 |
| TIGIT | DTBP-3 | H-ggytfhwhrlnp-NH2a | 106 |
| BTLA | HVEM(14–39) | Ac-ESC1PKC2SPGYRVKEAC1GELTGTVC2EP-NH2b | 108 |
| HVEM | gD (1–36)(K10C–T29C) | Ac-KYALVDASLC1MADPNRFRGKDLPVLDQLC1DPPGVRR-NH2b | 110 |
| VISTA | AP1049 | H-SSAC1DWIKRSC1H-NH2b | 115 |
| LAG-3 | C25 | H-C1VPMTYRAC1b-OH | 118 |
| CD47 | Pep-20 | AWSATWSNYWRH | 122 |
Lowercase letters indicate d-amino acids.
Cn: two cysteines with the same number form a corresponding disulfide bond.
CD28 binds to its ligand CD80 (B7-1)/CD86 (B7-2) to amplify TCR signaling and subsequently activate T cells. CTLA-4, derived from naïve T cells, inhibits AKT phosphorylation by activating PP2A and competitively binds to CD80/CD86, resulting in the suppression of T-cell function96. Although anti-CTLA-4 antibodies have been proven to increase the survival of patients with melanoma, head and neck squamous cell cancer, renal cell carcinoma, and non-small cell lung cancer97, antibody-dependent cellular toxicity-mediated natural killer (NK) cells may also eliminate CTLA-4 expressing regulatory T cells (Tregs) and cytotoxic T lymphocytes98. Under these circumstances, ERY2-4 (Table 2) was identified through yeast-displayed libraries with a KD value of 196.8 ± 2.3 nmol/L for CTLA-4; it effectively disrupted the interaction between CTLA-4 and B7 without the limitations of antibody-dependent cellular toxicity or cross-reactivity with CD2899.
6.3. Peptides targeting new immune checkpoints
Due to insufficient antigens and extraordinarily high/low PD-L1 expression, the majority of tumors are unresponsive to PD-1/PD-L1 blockade therapy, with a response rate below 30%100. The limited therapeutic efficacy and emergence of acquired drug resistance have prompted a gradual shift in research focus toward alternative ICB agents101,102, such as T-cell immunoglobulin and the ITIM domain (TIGIT), B and T cell lymphocyte attenuator (BTLA), V-domain Ig suppressor of T cell activation (VISTA), and lymphocyte activation gene-3 (LAG-3) (Fig. 5).
TIGIT, which is expressed on Tregs, activated T cells, and NK cells, competes with CD226 for binding to their ligand PVR (CD-155 or Nectin-2) to transmit inhibitory signals103, 104, 105. The identification of DPPA-1 facilitated the discovery of DTBP-3 (Table 2), the first peptide capable of blocking TIGIT, offering a potential candidate for overcoming anti-PD-1 resistance in vivo106.
BTLA, expressed on human CD8+ T lymphocytes, interacts with herpes virus entry mediator (HVEM) to form an immunosuppressing TME, in which dysfunctional T cells continuously express BTLA107,108. Based on the BTLA/HVEM complex, the disulfide-linked peptides HVEM (14–39)109 and gD (1–36) (K10C–T29C)110 were designed as inhibitors of BTLA/HVEM interactions with a KD = 250 nmol/L and demonstrated the ability to effectively block the formation of the BTLA/HVEM complex in 293T cells (Table 2).
VISTA functions as a stimulatory ligand for APCs, promoting immune activation, while also acting as a negative ligand for T cells, suppressing their proliferation, activation, and cytokine production111. It is expressed mainly on myeloid cells and T lymphocytes, particularly on naïve Foxp3+ Tregs and CD4+ T cells112. Studies have indicated that VISTA can potentially interact with inhibitory proteins such as VSIG3 but is also regarded as a self-signaling receptor113,114. The peptide AP1049 (Table 2) was designed to target the critical regions called “hot spots” of VISTA and exhibited superior efficacy in driving T-cell proliferation compared to anti-PD-L1 antibodies and anti-VISTA antibodies115.
LAG-3, also known as CD223, is a transmembrane protein primarily expressed in T cells, NK cells, and Tregs116. It predominantly attenuates the effector function of CIT, while enhancing the inhibitory function of Tregs117. As a promising complement to the LAG-3 antibody, the cyclic peptide C25 (Table 2) was developed through phase display to specifically bind with high affinity to LAG-3 and elicit potent antitumor effects by CD8+ T cells in mice bearing CT26, B16, and B16-OVA cells. Simultaneously, the infiltration of Tregs into the tumor significantly decreased118.
In addition to DCs and T cells, macrophages are also involved in CIC by facilitating the phagocytosis of tumor cells through the blockade of cluster of differentiation 47 (CD47). This mechanism enhances the cross-presentation of tumor antigens and promotes antigen-specific CD8+ T-cell responses (Fig. 5)119. CD47, which acts as a “don't eat me” signal, is often overexpressed in tumor cells to evade immune surveillance120. However, CD47 antibodies used in clinical trials have shown potential side effects such as anemia121. Thus, a CD47-targeting peptide with a low molecular weight, Pep-20 (Table 2), was screened for specific binding to the human CD47-IgV domain and disrupting the interaction between CD47 and SIRPα by effectively interfering with tyrosine phosphorylation of SIRPα. Importantly, blocking CD47 with Pep-20 initiated an antitumor T-cell immune response without significant blood toxicity compared to blocking CD47 with an anti-CD47 antibody122.
7. Peptide vaccines targeting immunosuppressive cells
Treating cancer patients with immune-modulatory vaccines targeting tumor immune escape mechanisms is a novel, generalizable strategy. For example, PD-L1- and IDO-specific T cells are easily and rapidly activated to kill PD-L1+ and/or IDO+ tumor/immunosuppressive cells, yet they exhibit strict regulation without inducing autoimmune reactions123. IO103, a PD-L1-derived peptide epitope with 19 amino acids, was developed to effectively stimulate the cytotoxicity of PD-L1-specific T cells against both PD-L1-positive cancer cells and immune-suppressive cells124. In clinical trials, IO103 demonstrated significant immune modulatory effects in multiple myeloma124 and basal cell carcinoma125, leading to complete regression in certain cancer patients with low-level and reversible adverse reactions. Moreover, the combination of the PD-L1/IDO (IO103/IO102) peptide vaccine with an anti-PD-1 antibody and Montanide adjuvant achieved an unprecedentedly high objective response rate of 80%, including 43% complete responses, along with a remarkable median progression-free survival of 26 months in metastatic melanoma patients123 (Table 1).
8. Current challenges and future opportunities for peptides targeting CIC
Although much progress has been achieved, current peptides targeting CIC in clinical trials have limitations such as conformational flexibility and susceptibility to proteolysis, which results in a restricted application of local administration. To address these challenges, various modification methods for peptide stabilization, including stapling chemistry126, d-amino acid substitution106, PEGylation127, and self-assembly128, can be employed. We developed oncolytic peptides derived from wasp venom (StAno) with significantly enhanced structural stability and antitumor activity through a side-chain-retention stapling modification. Strikingly, the leading StAno exhibited superior oncolytic potency compared to that of LTX-315, resulting in a 50% long-term survival rate in mice bearing B16F10 tumors129. To improve the circulation half-life and tumor targeting of oncolytic peptides, we further conjugated a peptide chimera by coupling a D-type PD-L1 targeting peptide with a PEG-functionalized oncolytic peptide via a matrix metalloproteinase-2-sensitive linker130. This all-in-one polymer chimera offers a promising application for tumor targeting and optimizing the pharmacokinetic properties of both oncolytic peptides and peptide ICIs. In addition to improving the bioavailability of therapeutic vaccines, such as oncolytic peptides, this innovative design effectively connected two critical steps (tumor antigen release and recognition of cancer cells by T cells) in CIC, thereby overcoming the immune evasion mechanisms that develop in the TME. Given the fact that numerous vaccine-based peptides have undergone extensive preclinical investigations but ultimately proven ineffective during clinical trials, this design may provide a paradigm for enhancing the efficacy of current therapeutic vaccines. Moreover, despite advancements in preclinical and clinical studies involving immune agonists, these agents have demonstrated modest therapeutic efficacy due to issues related to size constraints, epitope orientation binding affinity low stability, and undesired profile when administered in vivo131. Due to the same backbone and noncovalent interactions between residues, peptides possess the ability to self-assemble into nanomaterials that exhibit exceptional resistance against proteases while demonstrating polyvalent efficacy toward specific targets132. Wang's group133 reported the development of a polyvalent peptide CD40 nanoagonist (PVA-CD40) with a preorganized self-assembly structure, which effectively activates DCs for an amplified and durable antitumor immune response. When combined with an anti-PD-1 antibody, PVA-CD40 demonstrated significant inhibition of tumor growth in 4T1 mice, resulting in an optimal survival rate of up to 37%. In contrast, none of the mice survived when treated with clinically relevant combination therapy involving CD40 antibody and anti-PD-1 treatment. This proof-of-concept study highlights the potential clinical translation of self-assembled peptides in CITs. Furthermore, rapid advancements in artificial intelligence and machine learning algorithms also facilitate the discovery and optimization of immunoregulatory peptides134. We anticipate that these novel approaches can further enhance peptide functionality and therapeutic efficacy by specifically targeting CIC.
9. Conclusions
Since CIT was listed as a breakthrough in 2013 by the journal Science and subsequent acknowledgment through the 2018 Nobel Prize in Physiology or Medicine, it has garnered significant attention from both academic and industry scientists who have made notable advancements in immunomodulators, cell therapies, cancer vaccines, oncolytic viruses, and CD3-targeted bispecific antibodies135. Building upon these milestones, CITs hold immense potential as promising cancer treatment modalities and have demonstrated success in clinical applications136. Notably, CIC is a crucial aspect of CITs that drives the development of diverse strategies to regulate stimulatory and inhibitory factors within tumors. Here, we focused on elucidating how peptides exert their prominent roles in regulating CIC as an emerging form of cancer immunotherapy. Despite the inherent limitations of peptides, including poor stability, extensive chemical and pharmaceutical investigations have been undertaken to propose promising strategies for overcoming these challenges. Furthermore, the implementation of novel therapeutic approaches serves as a viable strategy to enhance the effectiveness of peptides. For instance, targeted protein degradation technology has been widely utilized to degrade pathogenic proteins across different tumor types while exhibiting favorable attributes in clinical settings137, 138, 139, 140. Peptide-based protein degraders targeting immune checkpoints (e.g., PD-L1, CD155, and CD47) may serve as future directions for better CITs while overcoming drug resistance caused by inhibitors.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 82322073, 82173846, and 82304533), CAMS Innovation Fund for Medical Sciences (CIFMS) (No. 2023-I2M-3-009, China), Oriental Scholars of Shanghai Universities (TP2022081, China), China Postdoctoral Science Foundation (No. 2021M702215), Jiangxi Province Thousand Talents Program (jxsq2023102168, China), Young Talent Lifting Project of Young Talent Lifting Project of China Association of Chinese Medicine [No. CACM-(2021-QNRC2-A08)], Shanghai Rising-Star Program (No. 22QA1409100, China), Shanghai Sailing Program (No. 22YF1445000, China), 2021 Shanghai Science and Technology Innovation Action Plan (No. 21S11902800, China), Three-Year Action Plan for Three-year Action Plan for Shanghai TCM Development and Inheritance Program [No. ZY(2021-2023)-0208 and ZY(2021-2023)-0401, China], High level Key Discipline of National Administration of Traditional Chinese Medicine (No. zyyzdxk-2023071, China), Innovation Team and Talents Cultivation Program of High level Key Discipline of National Administration of Traditional Chinese Medicine (No. ZYYCXTD-D-202004, China), Key project at central government level: The ability establishment of sustainable use for valuable Chinese medicine resources (No. 2060302, China) and Innovation team of high-level local universities in Shanghai: Strategic Innovation Team of TCM Chemical Biology (China). All figures were created with BioRender.com.
Author contributions
Xiaokun Zhang: Conceptualization, Writing – original draft. Ye Wu: Conceptualization, Writing – original draft. Jiayi Lin: Resources. Shengxin Lu: Resources. Xinchen Lu: Resources. Aoyu Cheng: Resources. Hongzhuan Chen: Writing – review & editing, Funding acquisition. Weidong Zhang: Writing – review & editing, Funding acquisition. Xin Luan: Funding acquisition, Writing – review & editing.
Conflicts of interest
The authors declare no conflicts of interest.
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Contributor Information
Hongzhuan Chen, Email: hongzhuan_chen@hotmail.com.
Weidong Zhang, Email: wdzhangy@hotmail.com.
Xin Luan, Email: luanxin@shutcm.edu.cn.
References
- 1.Kirchhammer N., Trefny M.P., Auf der Maur P., Läubli H., Zippelius A. Combination cancer immunotherapies: emerging treatment strategies adapted to the tumor microenvironment. Sci Transl Med. 2022;14 doi: 10.1126/scitranslmed.abo3605. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Y., Zhang Z. The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol. 2020;17:807–821. doi: 10.1038/s41423-020-0488-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hegde P.S., Chen D.S. Top 10 challenges in cancer immunotherapy. Immunity. 2020;52:17–35. doi: 10.1016/j.immuni.2019.12.011. [DOI] [PubMed] [Google Scholar]
- 4.Mukherjee A.G., Wanjari U.R., Gopalakrishnan A.V., Bradu P., Biswas A., Ganesan R., et al. Evolving strategies and application of proteins and peptide therapeutics in cancer treatment. Biomed Pharmacother. 2023;163 doi: 10.1016/j.biopha.2023.114832. [DOI] [PubMed] [Google Scholar]
- 5.Hanna C.C., Hermant Y.O., Harris P.W.R., Brimble M.A. Discovery, synthesis, and optimization of peptide-based antibiotics. Acc Chem Res. 2021;54:1878–1890. doi: 10.1021/acs.accounts.0c00841. [DOI] [PubMed] [Google Scholar]
- 6.Muttenthaler M., King G.F., Adams D.J., Alewood P.F. Trends in peptide drug discovery. Nat Rev Drug Discov. 2021;20:309–325. doi: 10.1038/s41573-020-00135-8. [DOI] [PubMed] [Google Scholar]
- 7.Timur S.S., Gursoy R.N. The role of peptide-based therapeutics in oncotherapy. J Drug Target. 2021;29:1048–1062. doi: 10.1080/1061186X.2021.1906884. [DOI] [PubMed] [Google Scholar]
- 8.Li Q., Shi Z., Zhang F., Zeng W., Zhu D., Mei L. Symphony of nanomaterials and immunotherapy based on the cancer-immunity cycle. Acta Pharm Sin B. 2022;12:107–134. doi: 10.1016/j.apsb.2021.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang L., Huang Y., Lindstrom A.R., Lin T.Y., Lam K.S., Li Y. Peptide-based materials for cancer immunotherapy. Theranostics. 2019;9:7807–7825. doi: 10.7150/thno.37194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang L., Wang N., Zhang W., Cheng X., Yan Z., Shao G., et al. Therapeutic peptides: current applications and future directions. Signal Transduct Targeted Ther. 2022;7:48. doi: 10.1038/s41392-022-00904-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang T., Huang X., Zhang G., Hong Z., Bai X., Liang T. Advantages of targeting the tumor immune microenvironment over blocking immune checkpoint in cancer immunotherapy. Signal Transduct Targeted Ther. 2021;6:72. doi: 10.1038/s41392-020-00449-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Z., Lai X., Fu S., Ren L., Cai H., Zhang H., et al. Immunogenic cell death activates the tumor immune microenvironment to boost the immunotherapy efficiency. Adv Sci. 2022;9 doi: 10.1002/advs.202201734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ledford H. Cancer-fighting viruses win approval. Nature. 2015;526:622–623. doi: 10.1038/526622a. [DOI] [PubMed] [Google Scholar]
- 14.Raja J., Ludwig J.M., Gettinger S.N., Schalper K.A., Kim H.S. Oncolytic virus immunotherapy: future prospects for oncology. J Immunother Cancer. 2018;6:140. doi: 10.1186/s40425-018-0458-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luan X., Wu Y., Shen Y.W., Zhang H., Zhou Y.D., Chen H.Z., et al. Cytotoxic and antitumor peptides as novel chemotherapeutics. Nat Prod Rep. 2021;38:7–17. doi: 10.1039/d0np00019a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gan B.H., Gaynord J., Rowe S.M., Deingruber T., Spring D.R. The multifaceted nature of antimicrobial peptides: current synthetic chemistry approaches and future directions. Chem Soc Rev. 2021;50:7820–7880. doi: 10.1039/d0cs00729c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mulder K.C., Lima L.A., Miranda V.J., Dias S.C., Franco O.L. Current scenario of peptide-based drugs: the key roles of cationic antitumor and antiviral peptides. Front Microbiol. 2013;4:321. doi: 10.3389/fmicb.2013.00321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kabelka I., Vácha R. Advances in molecular understanding of α-helical membrane-active peptides. Acc Chem Res. 2021;54:2196–2204. doi: 10.1021/acs.accounts.1c00047. [DOI] [PubMed] [Google Scholar]
- 19.Majewska M., Zamlynny V., Pieta I.S., Nowakowski R., Pieta P. Interaction of LL-37 human cathelicidin peptide with a model microbial-like lipid membrane. Bioelectrochemistry. 2021;141 doi: 10.1016/j.bioelechem.2021.107842. [DOI] [PubMed] [Google Scholar]
- 20.Vitale I., Yamazaki T., Wennerberg E., Sveinbjornsson B., Rekdal O., Demaria S., et al. Targeting cancer heterogeneity with immune responses driven by oncolytic peptides. Trends Cancer. 2021;7:557–572. doi: 10.1016/j.trecan.2020.12.012. [DOI] [PubMed] [Google Scholar]
- 21.Galluzzi L., Vitale I., Aaronson S.A., Abrams J.M., Adam D., Agostinis P., et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25:486–541. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bankell E., Liu X., Lundqvist M., Svensson D., Swärd K., Sparr E., et al. The antimicrobial peptide LL-37 triggers release of apoptosis-inducing factor and shows direct effects on mitochondria. Biochem Biophys Rep. 2022;29 doi: 10.1016/j.bbrep.2021.101192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lv Y., Shao G., Zhang Q., Wang X., Meng Y., Wang L., et al. The antimicrobial peptide PFR induces necroptosis mediated by ER stress and elevated cytoplasmic calcium and mitochondrial ROS levels: cooperation with Ara-C to act against acute myeloid leukemia. Signal Transduct Targeted Ther. 2019;4:38. doi: 10.1038/s41392-019-0073-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou H., Sauvat A., Gomes-da-Silva L.C., Durand S., Forveille S., Iribarren K., et al. The oncolytic compound LTX-401 targets the Golgi apparatus. Cell Death Differ. 2016;23:2031–2041. doi: 10.1038/cdd.2016.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang T., Huang X., Zhang G., Lu M., Hong Z., Wang M., et al. Oncolytic peptide LTX-315 induces anti-pancreatic cancer immunity by targeting the ATP11B–PD-L1 axis. J Immunother Cancer. 2022;10 doi: 10.1136/jitc-2021-004129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spicer J., Marabelle A., Baurain J.F., Jebsen N.L., Jossang D.E., Awada A., et al. Safety, antitumor activity, and T-cell responses in a dose-ranging phase I trial of the oncolytic peptide LTX-315 in patients with solid tumors. Clin Cancer Res. 2021;27:2755–2763. doi: 10.1158/1078-0432.CCR-20-3435. [DOI] [PubMed] [Google Scholar]
- 27.Liu J., Fu M., Wang M., Wan D., Wei Y., Wei X. Cancer vaccines as promising immuno-therapeutics: platforms and current progress. J Hematol Oncol. 2022;15:28. doi: 10.1186/s13045-022-01247-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Janelle V., Rulleau C., Del Testa S., Carli C., Delisle J.S. T-cell immunotherapies targeting histocompatibility and tumor antigens in hematological malignancies. Front Immunol. 2020;11:276. doi: 10.3389/fimmu.2020.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shae D., Baljon J.J., Wehbe M., Becker K.W., Sheehy T.L., Wilson J.T. At the bench: engineering the next generation of cancer vaccines. J Leukoc Biol. 2020;108:1435–1453. doi: 10.1002/JLB.5BT0119-016R. [DOI] [PubMed] [Google Scholar]
- 30.Vormehr M., Tureci O., Sahin U. Harnessing tumor mutations for truly individualized cancer vaccines. Annu Rev Med. 2019;70:395–407. doi: 10.1146/annurev-med-042617-101816. [DOI] [PubMed] [Google Scholar]
- 31.Jou J., Harrington K.J., Zocca M.B., Ehrnrooth E., Cohen E.E.W. The changing landscape of therapeutic cancer vaccines-novel platforms and neoantigen identification. Clin Cancer Res. 2021;27:689–703. doi: 10.1158/1078-0432.CCR-20-0245. [DOI] [PubMed] [Google Scholar]
- 32.Blass E., Ott P.A. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat Rev Clin Oncol. 2021;18:215–229. doi: 10.1038/s41571-020-00460-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xie N., Shen G., Gao W., Huang Z., Huang C., Fu L. Neoantigens: promising targets for cancer therapy. Signal Transduct Targeted Ther. 2023;8:9. doi: 10.1038/s41392-022-01270-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fritah H., Rovelli R., Chiang C.L., Kandalaft L.E. The current clinical landscape of personalized cancer vaccines. Cancer Treat Rev. 2022;106 doi: 10.1016/j.ctrv.2022.102383. [DOI] [PubMed] [Google Scholar]
- 35.Saxena M., van der Burg S.H., Melief C.J.M., Bhardwaj N. Therapeutic cancer vaccines. Nat Rev Cancer. 2021;21:360–378. doi: 10.1038/s41568-021-00346-0. [DOI] [PubMed] [Google Scholar]
- 36.Supabphol S., Li L., Goedegebuure S.P., Gillanders W.E. Neoantigen vaccine platforms in clinical development: understanding the future of personalized immunotherapy. Expet Opin Invest Drugs. 2021;30:529–541. doi: 10.1080/13543784.2021.1896702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sobhani N., Scaggiante B., Morris R., Chai D., Catalano M., Tardiel-Cyril D.R., et al. Therapeutic cancer vaccines: from biological mechanisms and engineering to ongoing clinical trials. Cancer Treat Rev. 2022;109 doi: 10.1016/j.ctrv.2022.102429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao X., Pan X., Wang Y., Zhang Y. Targeting neoantigens for cancer immunotherapy. Biomark Res. 2021;9:61. doi: 10.1186/s40364-021-00315-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bai S., Jiang H., Song Y., Zhu Y., Qin M., He C., et al. Aluminum nanoparticles deliver a dual-epitope peptide for enhanced anti-tumor immunotherapy. J Control Release. 2022;344:134–146. doi: 10.1016/j.jconrel.2022.02.027. [DOI] [PubMed] [Google Scholar]
- 40.Ott P.A., Hu Z., Keskin D.B., Shukla S.A., Sun J., Bozym D.J., et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217–221. doi: 10.1038/nature22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu Z., Leet D.E., Allesoe R.L., Oliveira G., Li S., Luoma A.M., et al. Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nat Med. 2021;27:515–525. doi: 10.1038/s41591-020-01206-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keskin D.B., Anandappa A.J., Sun J., Tirosh I., Mathewson N.D., Li S., et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2019;565:234–239. doi: 10.1038/s41586-018-0792-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peng M., Mo Y., Wang Y., Wu P., Zhang Y., Xiong F., et al. Neoantigen vaccine: an emerging tumor immunotherapy. Mol Cancer. 2019;18:128. doi: 10.1186/s12943-019-1055-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y., Su L., Morin M.D., Jones B.T., Mifune Y., Shi H., et al. Adjuvant effect of the novel TLR1/TLR2 agonist Diprovocim synergizes with anti-PD-L1 to eliminate melanoma in mice. Proc Natl Acad Sci U S A. 2018;115:e8698–e8706. doi: 10.1073/pnas.1809232115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van Kaer L., Parekh V.V., Postoak J.L., Wu L. Role of autophagy in MHC class I-restricted antigen presentation. Mol Immunol. 2019;113:2–5. doi: 10.1016/j.molimm.2017.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Amon L., Lehmann C.H.K., Baranska A., Schoen J., Heger L., Dudziak D. Transcriptional control of dendritic cell development and functions. Int Rev Cell Mol Biol. 2019;349:55–151. doi: 10.1016/bs.ircmb.2019.10.001. [DOI] [PubMed] [Google Scholar]
- 47.Lee M.Y., Jeon J.W., Sievers C., Allen C.T. Antigen processing and presentation in cancer immunotherapy. J Immunother Cancer. 2020;8 doi: 10.1136/jitc-2020-001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lutz M.B. Induction of CD4+ regulatory and polarized effector/helper T cells by dendritic cells. Immune Netw. 2016;16:13–25. doi: 10.4110/in.2016.16.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu Y., Shi Y., You J. Strategy and clinical application of up-regulating cross presentation by DCs in anti-tumor therapy. J Control Release. 2022;341:184–205. doi: 10.1016/j.jconrel.2021.11.011. [DOI] [PubMed] [Google Scholar]
- 50.Zhang X., Yang B., Ni Q., Chen X. Materials engineering strategies for cancer vaccine adjuvant development. Chem Soc Rev. 2023;52:2886–2910. doi: 10.1039/d2cs00647b. [DOI] [PubMed] [Google Scholar]
- 51.Borriello F., Poli V., Shrock E., Spreafico R., Liu X., Pishesha N., et al. An adjuvant strategy enabled by modulation of the physical properties of microbial ligands expands antigen immunogenicity. Cell. 2022;185 doi: 10.1016/j.cell.2022.01.009. 614-29.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaur A., Baldwin J., Brar D., Salunke D.B., Petrovsky N. Toll-like receptor (TLR) agonists as a driving force behind next-generation vaccine adjuvants and cancer therapeutics. Curr Opin Chem Biol. 2022;70 doi: 10.1016/j.cbpa.2022.102172. [DOI] [PubMed] [Google Scholar]
- 53.Simpson M.E., Petri W.A., Jr. TLR2 as a therapeutic target in bacterial infection. Trends Mol Med. 2020;26:715–717. doi: 10.1016/j.molmed.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mansouri A., Yousef M.S., Kowsar R., Usui N., Akthar I., Miyamoto A. Sperm activate TLR2/TLR1 heterodimerization to induce a weak proinflammatory response in the bovine uterus. Front Immunol. 2023;14 doi: 10.3389/fimmu.2023.1158090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakao M., Sugaya M., Fujita H., Miyagaki T., Morimura S., Shibata S., et al. TLR2 deficiency exacerbates imiquimod-induced psoriasis-like skin inflammation through decrease in regulatory T cells and impaired IL-10 production. Int J Mol Sci. 2020;21:8560. doi: 10.3390/ijms21228560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guo X., Wu N., Shang Y., Liu X., Wu T., Zhou Y., et al. The novel toll-like receptor 2 agonist SUP3 enhances antigen presentation and T cell activation by dendritic cells. Front Immunol. 2017;8:158. doi: 10.3389/fimmu.2017.00158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kaur A., Kaushik D., Piplani S., Mehta S.K., Petrovsky N., Salunke D.B. TLR2 agonistic small molecules: detailed structure–activity relationship, applications, and future prospects. J Med Chem. 2021;64:233–278. doi: 10.1021/acs.jmedchem.0c01627. [DOI] [PubMed] [Google Scholar]
- 58.Rammensee H.G., Wiesmuller K.H., Chandran P.A., Zelba H., Rusch E., Gouttefangeas C., et al. A new synthetic toll-like receptor 1/2 ligand is an efficient adjuvant for peptide vaccination in a human volunteer. J Immunother Cancer. 2019;7:307. doi: 10.1186/s40425-019-0796-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heitmann J.S., Bilich T., Tandler C., Nelde A., Maringer Y., Marconato M., et al. A COVID-19 peptide vaccine for the induction of SARS-CoV-2 T cell immunity. Nature. 2022;601:617–622. doi: 10.1038/s41586-021-04232-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eisenbarth S.C. Dendritic cell subsets in T cell programming: location dictates function. Nat Rev Immunol. 2019;19:89–103. doi: 10.1038/s41577-018-0088-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Balan S., Bhardwaj N. Cross-presentation of tumor antigens is ruled by synaptic transfer of vesicles among dendritic cell subsets. Cancer Cell. 2020;37:751–753. doi: 10.1016/j.ccell.2020.05.013. [DOI] [PubMed] [Google Scholar]
- 62.Cueto F.J., Del Fresno C., Sancho D. DNGR-1, a dendritic cell-specific sensor of tissue damage that dually modulates immunity and inflammation. Front Immunol. 2019;10:3146. doi: 10.3389/fimmu.2019.03146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Drouin M., Saenz J., Chiffoleau E. C-type lectin-like receptors: head or tail in cell death immunity. Front Immunol. 2020;11:251. doi: 10.3389/fimmu.2020.00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gou S., Wang S., Liu W., Chen G., Zhang D., Du J., et al. Adjuvant-free peptide vaccine targeting Clec9a on dendritic cells can induce robust antitumor immune response through Syk/IL-21 axis. Theranostics. 2021;11:7308–7321. doi: 10.7150/thno.56406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hoober J.K. ASGR1 and its enigmatic relative, CLEC10A. Int J Mol Sci. 2020;21:4818. doi: 10.3390/ijms21144818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zaal A., Li R.J.E., Lübbers J., Bruijns S.C.M., Kalay H., van Kooyk Y., et al. Activation of the C-type lectin MGL by terminal GalNAc ligands reduces the glycolytic activity of human dendritic cells. Front Immunol. 2020;11:305. doi: 10.3389/fimmu.2020.00305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Eggink L.L., Roby K.F., Cote R., Kenneth Hoober J. An innovative immunotherapeutic strategy for ovarian cancer: CLEC10A and glycomimetic peptides. J Immunother Cancer. 2018;6:28. doi: 10.1186/s40425-018-0339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mpakali A., Stratikos E. The role of antigen processing and presentation in cancer and the efficacy of immune checkpoint inhibitor immunotherapy. Cancers. 2021;13:134. doi: 10.3390/cancers13010134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oh D.Y., Kwek S.S., Raju S.S., Li T., McCarthy E., Chow E., et al. Intratumoral CD4+ T cells mediate anti-tumor cytotoxicity in human bladder cancer. Cell. 2020;181 doi: 10.1016/j.cell.2020.05.017. 1612-25.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Seung E., Xing Z., Wu L., Rao E., Cortez-Retamozo V., Ospina B., et al. A trispecific antibody targeting HER2 and T cells inhibits breast cancer growth via CD4 cells. Nature. 2022;603:328–334. doi: 10.1038/s41586-022-04439-0. [DOI] [PubMed] [Google Scholar]
- 71.Leko V., Rosenberg S.A. Identifying and targeting human tumor antigens for T cell-based immunotherapy of solid tumors. Cancer Cell. 2020;38:454–472. doi: 10.1016/j.ccell.2020.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sim M.J.W., Sun P.D. T cell recognition of tumor neoantigens and insights into T cell immunotherapy. Front Immunol. 2022;13 doi: 10.3389/fimmu.2022.833017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Slansky J.E., Nakayama M. Peptide mimotopes alter T cell function in cancer and autoimmunity. Semin Immunol. 2020;47 doi: 10.1016/j.smim.2020.101395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhu S.Y., Yu K.D. Breast cancer vaccines: disappointing or promising? Front Immunol. 2022;13 doi: 10.3389/fimmu.2022.828386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yu H., Ye C., Li J., Pan C., Lin W., Chen H., et al. An altered HLA-A0201-restricted MUC1 epitope that could induce more efficient anti-tumor effects against gastric cancer. Exp Cell Res. 2020;390 doi: 10.1016/j.yexcr.2020.111953. [DOI] [PubMed] [Google Scholar]
- 76.Ayres C.M., Baker B.M. Peptide-dependent tuning of major histocompatibility complex motional properties and the consequences for cellular immunity. Curr Opin Immunol. 2022;76 doi: 10.1016/j.coi.2022.102184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Baumgaertner P., Schmidt J., Costa-Nunes C.M., Bordry N., Guillaume P., Luescher I., et al. CD8 T cell function and cross-reactivity explored by stepwise increased peptide-HLA versus TCR affinity. Front Immunol. 2022;13 doi: 10.3389/fimmu.2022.973986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brennick C.A., George M.M., Srivastava P.K., Karandikar S.H. Prediction of cancer neoepitopes needs new rules. Semin Immunol. 2020;47 doi: 10.1016/j.smim.2020.101387. [DOI] [PubMed] [Google Scholar]
- 79.Smith A.R., Alonso J.A., Ayres C.M., Singh N.K., Hellman L.M., Baker B.M. Structurally silent peptide anchor modifications allosterically modulate T cell recognition in a receptor-dependent manner. Proc Natl Acad Sci U S A. 2021;118 doi: 10.1073/pnas.2018125118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Joglekar A.V., Li G. T cell antigen discovery. Nat Methods. 2021;18:873–880. doi: 10.1038/s41592-020-0867-z. [DOI] [PubMed] [Google Scholar]
- 81.Stewart-Jones G., Wadle A., Hombach A., Shenderov E., Held G., Fischer E., et al. Rational development of high-affinity T-cell receptor-like antibodies. Proc Natl Acad Sci U S A. 2009;106:5784–5788. doi: 10.1073/pnas.0901425106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shenderov E., Kandasamy M., Gileadi U., Chen J., Shepherd D., Gibbs J., et al. Generation and characterization of HLA-A2 transgenic mice expressing the human TCR 1G4 specific for the HLA-A2 restricted NY-ESO-1157–165 tumor-specific peptide. J Immunother Cancer. 2021;9 doi: 10.1136/jitc-2021-002544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wei P., Jordan K.R., Buhrman J.D., Lei J., Deng H., Marrack P., et al. Structures suggest an approach for converting weak self-peptide tumor antigens into superagonists for CD8 T cells in cancer. Proc Natl Acad Sci U S A. 2021;118 doi: 10.1073/pnas.2100588118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Singh S.P., Mishra B.N. Major histocompatibility complex linked databases and prediction tools for designing vaccines. Hum Immunol. 2016;77:295–306. doi: 10.1016/j.humimm.2015.11.012. [DOI] [PubMed] [Google Scholar]
- 85.Lucibello G., Mograbi B., Milano G., Hofman P., Brest P. PD-L1 regulation revisited: impact on immunotherapeutic strategies. Trends Mol Med. 2021;27:868–881. doi: 10.1016/j.molmed.2021.06.005. [DOI] [PubMed] [Google Scholar]
- 86.Martins F., Sofiya L., Sykiotis G.P., Lamine F., Maillard M., Fraga M., et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol. 2019;16:563–580. doi: 10.1038/s41571-019-0218-0. [DOI] [PubMed] [Google Scholar]
- 87.Lee Y., Song S., Yang S., Kim J., Moon Y., Shim N., et al. Photo-induced crosslinked and anti-PD-L1 peptide incorporated liposomes to promote PD-L1 multivalent binding for effective immune checkpoint blockade therapy. Acta Pharm Sin B. 2024;14:1428–1440. doi: 10.1016/j.apsb.2023.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cheng S.S., Yang G.J., Wang W., Leung C.H., Ma D.L. The design and development of covalent protein–protein interaction inhibitors for cancer treatment. J Hematol Oncol. 2020;13:26. doi: 10.1186/s13045-020-00850-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen X., Pan X., Zhang W., Guo H., Cheng S., He Q., et al. Epigenetic strategies synergize with PD-L1/PD-1 targeted cancer immunotherapies to enhance antitumor responses. Acta Pharm Sin B. 2020;10:723–733. doi: 10.1016/j.apsb.2019.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wu Q., Jiang L., Li S.C., He Q.J., Yang B., Cao J. Small molecule inhibitors targeting the PD-1/PD-L1 signaling pathway. Acta Pharmacol Sin. 2021;42:1–9. doi: 10.1038/s41401-020-0366-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lenouvel D., González-Moles M.Á., Talbaoui A., Ramos-García P., González-Ruiz L., Ruiz-Ávila, et al. An update of knowledge on PD-L1 in head and neck cancers: physiologic, prognostic and therapeutic perspectives. Oral Dis. 2020;26:511–526. doi: 10.1111/odi.13088. [DOI] [PubMed] [Google Scholar]
- 92.Sasikumar P.G., Ramachandra R.K., Adurthi S., Dhudashiya A.A., Vadlamani S., Vemula K., et al. A rationally designed peptide antagonist of the PD-1 signaling pathway as an immunomodulatory agent for cancer therapy. Mol Cancer Therapeut. 2019;18:1081–1091. doi: 10.1158/1535-7163.MCT-18-0737. [DOI] [PubMed] [Google Scholar]
- 93.Liu H., Zhao Z., Zhang L., Li Y., Jain A., Barve A., et al. Discovery of low-molecular weight anti-PD-L1 peptides for cancer immunotherapy. J Immunother Cancer. 2019;7:270. doi: 10.1186/s40425-019-0705-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chang H.N., Liu B.Y., Qi Y.K., Zhou Y., Chen Y.P., Pan K.M., et al. Blocking of the PD-1/PD-L1 interaction by a D-peptide antagonist for cancer immunotherapy. Angew Chem Int Ed Engl. 2015;54:11760–11764. doi: 10.1002/anie.201506225. [DOI] [PubMed] [Google Scholar]
- 95.Li W., Zhu X., Zhou X., Wang X., Zhai W., Li B., et al. An orally available PD-1/PD-L1 blocking peptide OPBP-1-loaded trimethyl chitosan hydrogel for cancer immunotherapy. J Control Release. 2021;334:376–388. doi: 10.1016/j.jconrel.2021.04.036. [DOI] [PubMed] [Google Scholar]
- 96.Nakane S., Imamura K., Hisanaga R., Ishihara K., Saito A. Systemic administration of cytotoxic T lymphocyte-associated antigen 4 (CTLA-4)-Ig abrogates alveolar bone resorption in induced periodontitis through inhibition of osteoclast differentiation and activation: an experimental investigation. J Periodontal Res. 2021;56:972–981. doi: 10.1111/jre.12909. [DOI] [PubMed] [Google Scholar]
- 97.Atkins M.B., Clark J.I., Quinn D.I. Immune checkpoint inhibitors in advanced renal cell carcinoma: experience to date and future directions. Ann Oncol. 2017;28:1484–1494. doi: 10.1093/annonc/mdx151. [DOI] [PubMed] [Google Scholar]
- 98.Ha D., Tanaka A., Kibayashi T., Tanemura A., Sugiyama D., Wing J.B., et al. Differential control of human Treg and effector T cells in tumor immunity by Fc-engineered anti-CTLA-4 antibody. Proc Natl Acad Sci U S A. 2019;116:609–618. doi: 10.1073/pnas.1812186116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ramanayake Mudiyanselage T.M.R., Michigami M., Ye Z., Uyeda A., Inoue N., Sugiura K., et al. An immune-stimulatory helix–loop–helix peptide: selective inhibition of CTLA-4–B7 interaction. ACS Chem Biol. 2020;15:360–368. doi: 10.1021/acschembio.9b00743. [DOI] [PubMed] [Google Scholar]
- 100.Jiang Y., Zhao X., Fu J., Wang H. Progress and challenges in precise treatment of tumors with PD-1/PD-L1 blockade. Front Immunol. 2020;11:339. doi: 10.3389/fimmu.2020.00339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Marin-Acevedo J.A., Dholaria B., Soyano A.E., Knutson K.L., Chumsri S., Lou Y. Next generation of immune checkpoint therapy in cancer: new developments and challenges. J Hematol Oncol. 2018;11:39. doi: 10.1186/s13045-018-0582-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Marin-Acevedo J.A., Kimbrough E.O., Lou Y. Next generation of immune checkpoint inhibitors and beyond. J Hematol Oncol. 2021;14:45. doi: 10.1186/s13045-021-01056-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Deng C., Li W., Fei Y., Wang L., Chen Y., Zeng X., et al. Imbalance of the CD226/TIGIT immune checkpoint is involved in the pathogenesis of primary biliary cholangitis. Front Immunol. 2020;11:1619. doi: 10.3389/fimmu.2020.01619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lin Y.X., Hung M.C., Hsu J.L., Hsu J.M. The N-linked glycosylations of TIGIT Asn32 and Asn101 facilitate PVR/TIGIT interaction. Biochem Biophys Res Commun. 2021;562:9–14. doi: 10.1016/j.bbrc.2021.05.034. [DOI] [PubMed] [Google Scholar]
- 105.Dougall W.C., Kurtulus S., Smyth M.J., Anderson A.C. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev. 2017;276:112–120. doi: 10.1111/imr.12518. [DOI] [PubMed] [Google Scholar]
- 106.Zhou X., Zuo C., Li W., Shi W., Zhou X., Wang H., et al. A novel D-peptide identified by mirror-image phage display blocks TIGIT/PVR for cancer immunotherapy. Angew Chem Int Ed Engl. 2020;59:15114–15118. doi: 10.1002/anie.202002783. [DOI] [PubMed] [Google Scholar]
- 107.Yu X., Zheng Y., Mao R., Su Z., Zhang J. BTLA/HVEM signaling: milestones in research and role in chronic hepatitis B virus infection. Front Immunol. 2019;10:617. doi: 10.3389/fimmu.2019.00617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yu M., Zhao H., Miao Y., Luo S.Z., Xue S. Virtual evolution of HVEM segment for checkpoint inhibitor discovery. Int J Mol Sci. 2021;22:6638. doi: 10.3390/ijms22126638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Spodzieja M., Kuncewicz K., Sieradzan A., Karczynska A., Iwaszkiewicz J., Cesson V., et al. Disulfide-linked peptides for blocking BTLA/HVEM binding. Int J Mol Sci. 2020;21:636. doi: 10.3390/ijms21020636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kuncewicz K., Battin C., Sieradzan A., Karczynska A., Orlikowska M., Wardowska A., et al. Fragments of gD protein as inhibitors of BTLA/HVEM complex formation—design, synthesis, and cellular studies. Int J Mol Sci. 2020;21:8876. doi: 10.3390/ijms21228876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Xu W., Hiếu T., Malarkannan S., Wang L. The structure, expression, and multifaceted role of immune-checkpoint protein VISTA as a critical regulator of anti-tumor immunity, autoimmunity, and inflammation. Cell Mol Immunol. 2018;15:438–446. doi: 10.1038/cmi.2017.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.ElTanbouly M.A., Croteau W., Noelle R.J., Lines J.L. VISTA: a novel immunotherapy target for normalizing innate and adaptive immunity. Semin Immunol. 2019;42 doi: 10.1016/j.smim.2019.101308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang J., Wu G., Manick B., Hernandez V., Renelt M., Erickson C., et al. VSIG-3 as a ligand of VISTA inhibits human T-cell function. Immunology. 2019;156:74–85. doi: 10.1111/imm.13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xie X., Chen C., Chen W., Jiang J., Wang L., Li T., et al. Structural basis of VSIG3: the ligand for VISTA. Front Immunol. 2021;12 doi: 10.3389/fimmu.2021.625808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Noelle R.J., Wang L., Broughton T., Sempere L.F., Lines J.L., Noelle Randolph J., inventor. King's College London, assignee . 2016 Nov 17. VISTA antagonist and methods of use. United States Patent US 2015231215A1. [Google Scholar]
- 116.Lecocq Q., Keyaerts M., Devoogdt N., Breckpot K. The next-generation immune checkpoint LAG-3 and its therapeutic potential in oncology: third time's a charm. Int J Mol Sci. 2020;22:75. doi: 10.3390/ijms22010075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Anderson A.C., Joller N., Kuchroo V.K. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. 2016;44:989–1004. doi: 10.1016/j.immuni.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhai W., Zhou X., Wang H., Li W., Chen G., Sui X., et al. A novel cyclic peptide targeting LAG-3 for cancer immunotherapy by activating antigen-specific CD8+ T cell responses. Acta Pharm Sin B. 2020;10:1047–1060. doi: 10.1016/j.apsb.2020.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Feng M., Jiang W., Kim B.Y.S., Zhang C.C., Fu Y.X., Weissman I.L. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer. 2019;19:568–586. doi: 10.1038/s41568-019-0183-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Veillette A., Chen J. SIRPα–CD47 immune checkpoint blockade in anticancer therapy. Trends Immunol. 2018;39:173–184. doi: 10.1016/j.it.2017.12.005. [DOI] [PubMed] [Google Scholar]
- 121.Andrechak J.C., Dooling L.J., Discher D.E. The macrophage checkpoint CD47: SIRPα for recognition of ‘self’ cells: from clinical trials of blocking antibodies to mechanobiological fundamentals. Philos Trans R Soc Lond B Biol Sci. 2019;374 doi: 10.1098/rstb.2018.0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang H., Sun Y., Zhou X., Chen C., Jiao L., Li W., et al. CD47/SIRPα blocking peptide identification and synergistic effect with irradiation for cancer immunotherapy. J Immunother Cancer. 2020;8 doi: 10.1136/jitc-2020-000905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kjeldsen J.W., Lorentzen C.L., Martinenaite E., Ellebaek E., Donia M., Holmstroem R.B., et al. A phase 1/2 trial of an immune-modulatory vaccine against IDO/PD-L1 in combination with nivolumab in metastatic melanoma. Nat Med. 2021;27:2212–2223. doi: 10.1038/s41591-021-01544-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jorgensen N.G., Klausen U., Grauslund J.H., Helleberg C., Aagaard T.G., Do T.H., et al. Peptide vaccination against PD-L1 with IO103 a novel immune modulatory vaccine in multiple myeloma: a phase I first-in-human trial. Front Immunol. 2020;11 doi: 10.3389/fimmu.2020.595035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jørgensen N.G., Kaae J., Grauslund J.H., Met Ö., Nielsen S.L., Pedersen A.W., et al. Vaccination against PD-L1 with IO103 a novel immune modulatory vaccine in basal cell carcinoma: a phase IIa study. Cancers. 2021;13:911. doi: 10.3390/cancers13040911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Luong H.X., Bui H.T.P., Tung T.T. Application of the all-hydrocarbon stapling technique in the design of membrane-active peptides. J Med Chem. 2022;65:3026–3045. doi: 10.1021/acs.jmedchem.1c01744. [DOI] [PubMed] [Google Scholar]
- 127.Andrianov A.K. Noncovalent PEGylation of protein and peptide therapeutics. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2023;15 doi: 10.1002/wnan.1897. [DOI] [PubMed] [Google Scholar]
- 128.Zou P., Chen W.T., Sun T., Gao Y., Li L.L., Wang H. Recent advances: peptides and self-assembled peptide-nanosystems for antimicrobial therapy and diagnosis. Biomater Sci. 2020;8:4975–4996. doi: 10.1039/d0bm00789g. [DOI] [PubMed] [Google Scholar]
- 129.Wu Y., Lu D., Jiang Y.X., Jin J.M., Liu S.H., Chen L.L., et al. Stapled wasp venom-derived oncolytic peptides with side chains induce rapid membrane lysis and prolonged immune responses in melanoma. J Med Chem. 2021;64:5802–5815. doi: 10.1021/acs.jmedchem.0c02237. [DOI] [PubMed] [Google Scholar]
- 130.Lu L., Zhang H., Zhou Y.D., Lin J.Y., Gao W.D., Yang T., et al. Polymer chimera of stapled oncolytic peptide coupled with anti-PD-L1 peptide boosts immunotherapy of colorectal cancer. Theranostics. 2022;12:3456–3473. doi: 10.7150/thno.71129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Liu D., Guo P., McCarthy C., Wang B., Tao Y., Auguste D. Peptide density targets and impedes triple negative breast cancer metastasis. Nat Commun. 2018;9:2612. doi: 10.1038/s41467-018-05035-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yang J., An H.W., Wang H. Self-assembled peptide drug delivery systems. ACS Appl Bio Mater. 2021;4:24–46. doi: 10.1021/acsabm.0c00707. [DOI] [PubMed] [Google Scholar]
- 133.Mamuti M., Wang Y., Zhao Y.D., Wang J.Q., Wang J., Fan Y.L., et al. A polyvalent peptide CD40 nanoagonist for targeted modulation of dendritic cells and amplified cancer immunotherapy. Adv Mater. 2022;34 doi: 10.1002/adma.202109432. [DOI] [PubMed] [Google Scholar]
- 134.Basith S., Manavalan B., Hwan Shin T., Lee G. Machine intelligence in peptide therapeutics: a next-generation tool for rapid disease screening. Med Res Rev. 2020;40:1276–1314. doi: 10.1002/med.21658. [DOI] [PubMed] [Google Scholar]
- 135.Xin Yu J., Hubbard-Lucey V.M., Tang J. Immuno-oncology drug development goes global. Nat Rev Drug Discov. 2019;18:899–900. doi: 10.1038/d41573-019-00167-9. [DOI] [PubMed] [Google Scholar]
- 136.Kraehenbuehl L., Weng C.H., Eghbali S., Wolchok J.D., Merghoub T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat Rev Clin Oncol. 2022;19:37–50. doi: 10.1038/s41571-021-00552-7. [DOI] [PubMed] [Google Scholar]
- 137.Chirnomas D., Hornberger K.R., Crews C.M. Protein degraders enter the clinic—a new approach to cancer therapy. Nat Rev Clin Oncol. 2023;20:265–278. doi: 10.1038/s41571-023-00736-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jin J.M., Wu Y., Chen J.J., Shen Y.W., Zhang L.J., Zhang H., et al. The peptide PROTAC modality: a novel strategy for targeted protein ubiquitination. Theranostics. 2020;10:10141–10153. doi: 10.7150/thno.46985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lin J.Y., Jin J.M., Shen Y.W., Zhang L.J., Gong G., Bian H.T., et al. Emerging protein degradation strategies: expanding the scope to extracellular and membrane proteins. Theranostics. 2021;11:8337–8349. doi: 10.7150/thno.62686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lin J.Y., Liu H.J., Wu Y., Jin J.M., Zhou Y.D., Zhang H., et al. Targeted protein degradation technology and nanomedicine: powerful allies against cancer. Small. 2023;19 doi: 10.1002/smll.202207778. [DOI] [PubMed] [Google Scholar]