

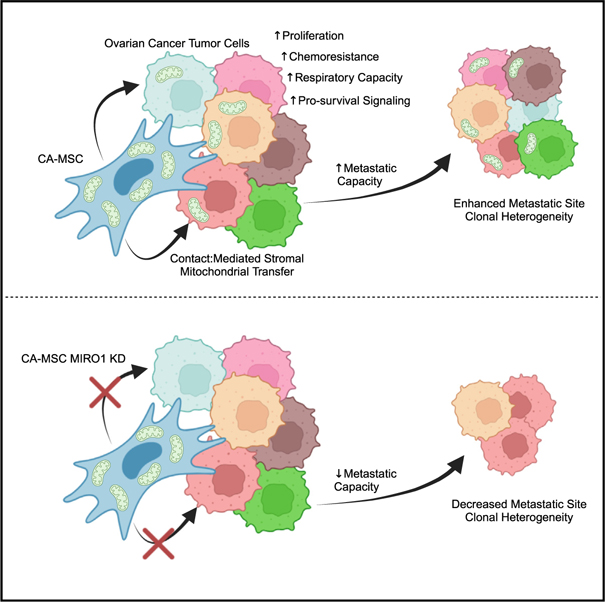
SUMMARY
Ovarian cancer is characterized by early metastatic spread. This study demonstrates that carcinoma-associated mesenchymal stromal cells (CA-MSCs) enhance metastasis by increasing tumor cell heterogeneity through mitochondrial donation. CA-MSC mitochondrial donation preferentially occurs in ovarian cancer cells with low levels of mitochondria (“mito poor”). CA-MSC mitochondrial donation rescues the phenotype of mito poor cells, restoring their proliferative capacity, resistance to chemotherapy, and cellular respiration. Receipt of CA-MSC-derived mitochondria induces tumor cell transcriptional changes leading to the secretion of ANGPTL3, which enhances the proliferation of tumor cells without CA-MSC mitochondria, thus amplifying the impact of mitochondrial transfer. Donated CA-MSC mitochondrial DNA persisted in recipient tumor cells for at least 14 days. CA-MSC mitochondrial donation occurs in vivo, enhancing tumor cell heterogeneity and decreasing mouse survival. Collectively, this work identifies CA-MSC mitochondrial transfer as a critical mediator of ovarian cancer cell survival, heterogeneity, and metastasis and presents a unique therapeutic target in ovarian cancer.
In brief
Frisbie et al. demonstrate that stromal-to-tumor cell mitochondrial transfer drives ovarian cancer metastasis and clonal heterogeneity. Carcinoma-associated mesenchymal stem cells donate mitochondria to vulnerable tumor cells, enhancing tumor cell proliferation, chemoresistance, and respiratory capacity and triggering ANGPTL3 secretion to further promote tumor growth. These findings offer a potential therapeutic target for ovarian cancer.
Graphical Abstract
INTRODUCTION
Ovarian cancer is the deadliest gynecologic malignancy in the Western world, notorious for its difficult detection, rapid metastatic spread, and development of chemotherapy resistance.1 A major mediator of metastasis and chemotherapy resistance is genetic heterogeneity of cancer cells within the tumor.2–4 Enhanced tumor cell (TC) clonal heterogeneity is an independent prognostic factor for decreased survival in ovarian cancer patients.5–7 The mechanisms that enhance TC clonal heterogeneity in ovarian cancer are unclear, although increasing evidence suggests that the tumor microenvironment plays a critical role in shaping TC behavior at both primary and metastatic sites.
Our previous work identified carcinoma-associated mesenchymal stromal cells (CA-MSCs) as an essential component of the ovarian tumor microenvironment supporting ovarian cancer growth, survival, chemotherapy resistance, and metastasis.8–11 Ovarian cancer CA-MSCs are multipotent stromal cells that originate from epigenetic reprogramming of naive resident tissue MSCs, inducing a phenotypic change that conveys pro-tumorigenic properties.8,11 A critical consequence of this pro-tumorigenic education is the high affinity of CA-MSCs to bind to adjacent ovarian cancer cells.11 CA-MSCs directly interact with ovarian cancer cells, forming heterocellular complexes that cometastasize to distant organs.11 The physical interaction between CA-MSCs and ovarian cancer cells appears to be a critical mediator of metastasis, although the precise mechanism remains undefined.
MSCs interact with adjacent cells through various mechanisms, including donating their mitochondria in times of stress. Horizontal mitochondrial transfer from MSCs to distressed cells is demonstrated in numerous disease models and contributes to stromal-mediated tissue revitalization in response to injury.12–16 Likewise, mitochondrial transfer can also contribute to malignancy in leukemia, breast, ovary, colon, and lung cancers, among others.16–20 Ovarian cancer cells disseminating into the peritoneal cavity must overcome the selective pressure of the peritoneal/ascites microenvironment to successfully metastasize; this selective pressure functions as a major bottleneck influencing cellular heterogeneity by ensuring that only the hardiest TCs survive.21–23 We thus hypothesized that CA-MSCs donate mitochondria to ovarian cancer cells to overcome these stresses and enable metastasis, in turn, driving increased heterogeneity at metastatic sites.
Here, we demonstrate that CA-MSCs enhance the proliferation, therapy resistance, spread, and distant organ colonization of ovarian TCs both in vitro and in vivo by donating mitochondria to metabolically vulnerable TCs, which in turn drives clonal heterogeneity. We further explore the downstream impact of donated mitochondria in recipient TCs, identifying mitochondrial-to-nuclear communication leading to pro-growth paracrine signaling via ANGPTL3 secretion, suggesting horizontal mitochondrial transfer has a broader effect outside of the recipient cell. This work presents mitochondrial transfer as an important mechanism that drives clonal heterogeneity during ovarian cancer progression.
RESULTS
CA-MSCs increase ovarian cancer cell heterogeneity
Previous work demonstrated that CA-MSCs enhance the proliferation, chemotherapy resistance, and metastasis of ovarian TCs in vitro and in vivo.9–11,24 To elucidate whether CA-MSCs also increase TC heterogeneity, a genomic DNA barcoding system was incorporated into the high-grade serous ovarian cancer (HGSC) cell line OVCAR3, such that each TC contained one unique genetic barcode, enabling identification of individual TC clones via DNA sequencing.
CA-MSCs were isolated from HGSC patient samples without prior treatment and validated as previously described.11,25 Using a tail-vein injection model, TCs alone or TCs + CA-MSCs were injected into NSG mice. CA-MSCs were injected in a 1:10 ratio with TCs based on our prior work demonstrating equivalent CA-MSC:TC ratios in human ascites complexes.11 A tail-vein model was used to bypass confounding effects of differential primary tumor growth, thus enabling unadulterated assessment of clones that have entered the circulation.26 After 21 days, when the first mouse met endpoint criteria, all mice were euthanized and sites of TC colonization were quantified at necropsy. Liver, lung, abdomen, ascites, and blood were isolated and used for DNA sequencing to identify TC clones. While both TC alone and TC + CA-MSC groups demonstrated 100% TC colonization of the lung, the TC + CA-MSC group demonstrated enhanced TC colonization/growth in the liver (100% vs. 57%), abdomen (100% vs. 0%), and malignant ascites (63% vs. 0%) vs. the TC-only group (Figure 1A). TC + CA-MSC co-injection versus TC alone resulted in significantly higher clonal heterogeneity within tumor-involved organ sites, including the liver, lung, and blood (Figures 1B–1D). While the TC-only group did not metastasize to the abdomen or yield malignant ascites, the range of TC clonal heterogeneity in the TC + CA-MSC group at these sites is included for completion (Figures 1E and 1F). Figure 1G demonstrates that CA-MSC co-injection with TCs resulted in a greater total number of unique TC clones within each tumor-involved organ site. When normalized for total tumor DNA content, the number of unique barcodes per microgram of tumor DNA remained higher in the tumor sites of mice injected with TC + CA-MSCs compared to TC alone (Table S1). Additionally, Figure 1F demonstrates the overlap of clones seeding different organ sites in the TC + CA-MSC group vs. the TC alone group, and displays unique clonal seeding/outgrowth at different organ sites. As an additional quantification of TC clonal diversity, we also compared the number of cells that arise from the top 50 most abundant clones/barcodes at each primary and tumor-involved organ site in the TC and TC + CA-MSC groups (Figure 1I). The TC + CA-MSC sites demonstrated a greater number of unique clones compared to TC alone, indicating that the presence of CA-MSCs improved the survival and propagation of a larger pool of cancer cell clones.
Figure 1. CA-MSCs increase OVCAR3 ovarian TC heterogeneity in vivo.
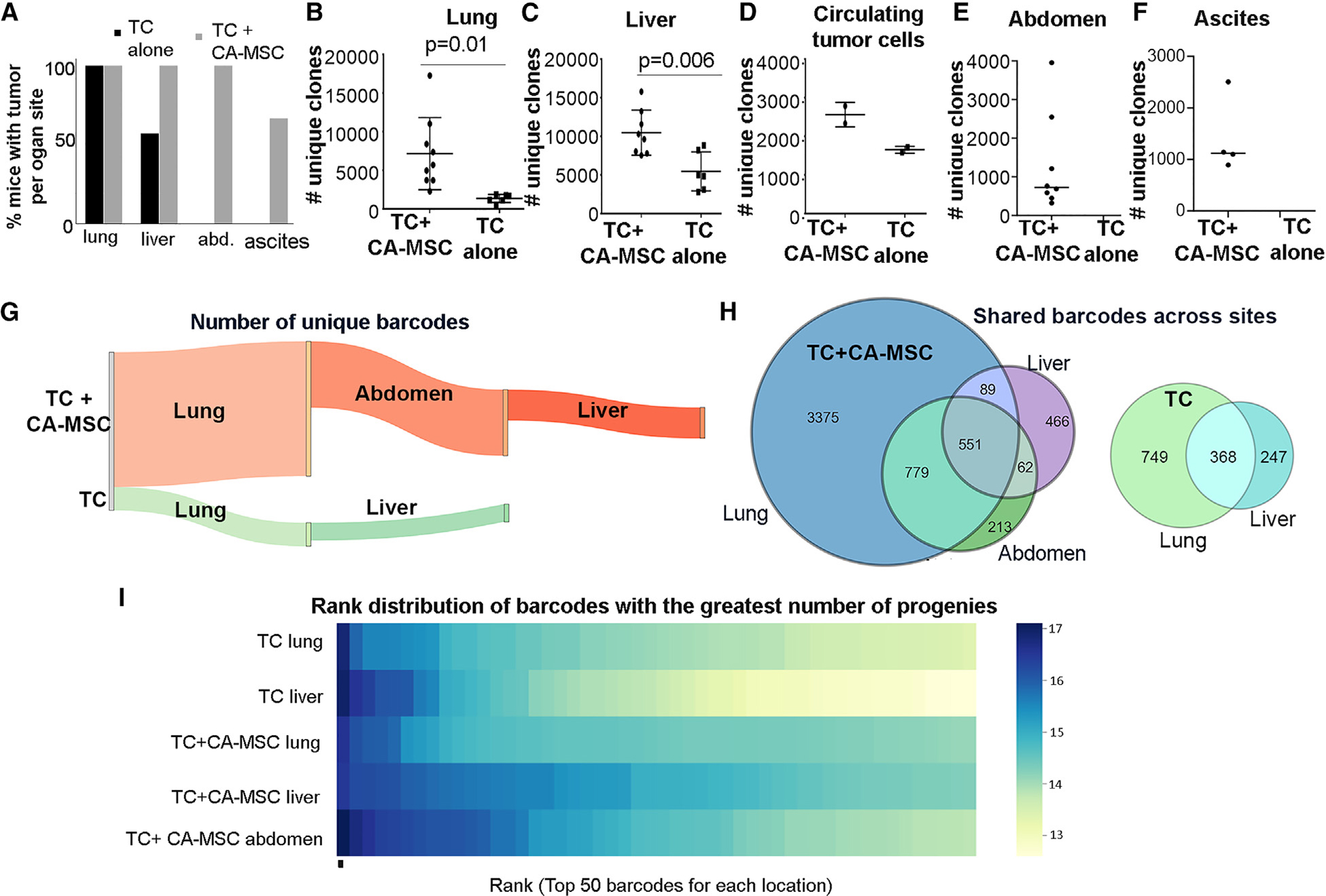
(A) Quantification of organ-specific tumor involvement per mouse group (n = 10 per group): TC alone vs. TC + CA-MSC.
(B–F) Quantification of unique clones at each site of tumor involvement: (B) lung, (C) liver, (D) blood, (E) abdomen, and (F) ascites. n = 8 sites for TC + CA-MSC; n = 6 TC alone; n = 2 blood samples for both groups.
(G) Sankey diagram visually demonstrating the number of unique barcodes at each tumor site in TC + CA-MSC vs. TC-alone mice.
(H) Venn diagrams demonstrating the distribution of TC clones across organ sites per mouse.
(I) Heatmap ranking the log-based total number of unique clones derived from the top 50 most abundant barcodes at each.
For (B) and (C), data are presented as the mean ± SEM; displayed p values obtained using Student’s t test.
CA-MSCs transfer mitochondria to adjacent TCs
Multiple studies show that MSCs utilize direct connections to donate mitochondria to adjacent cells in times of ischemic and metabolic stress, conditions that mirror the hypoxemia and nutrient scarcity of the ovarian tumor microenvironment.15,27–30 As our prior work demonstrated that direct contact between CA-MSCs and TCs was necessary to enhance metastasis, we investigated whether direct interaction also drives TC heterogeneity through mitochondrial donation.11 We generated stable lentiviral-transduced COX8-GFP CA-MSC lines, which incorporate GFP into the subunit of cytochrome c oxidase in mitochondrial complex IV, thereby labeling all endogenous CA-MSC mitochondria. TC mitochondria were tagged with a COX8-dsRed lentivirus. Labeled CA-MSCs and TCs were then grown in coculture and examined using live imaging and fixed confocal microscopy. Within 24 h of coculture, CA-MSCs and TCs readily interacted with multiple points of direct contact, and GFP-labeled CA-MSC mitochondria were transferred to adjacent ovarian TCs (Figure 2A and Video S1). Assessment of the z stacks of the confocal image verifies the intracellular/cytoplasmic location of the CA-MSC-donated GFP-mitochondrial within the TC (Figure 2Aii, iii).
Figure 2. CA-MSCs directly transfer mitochondria to adjacent ovarian TCs.
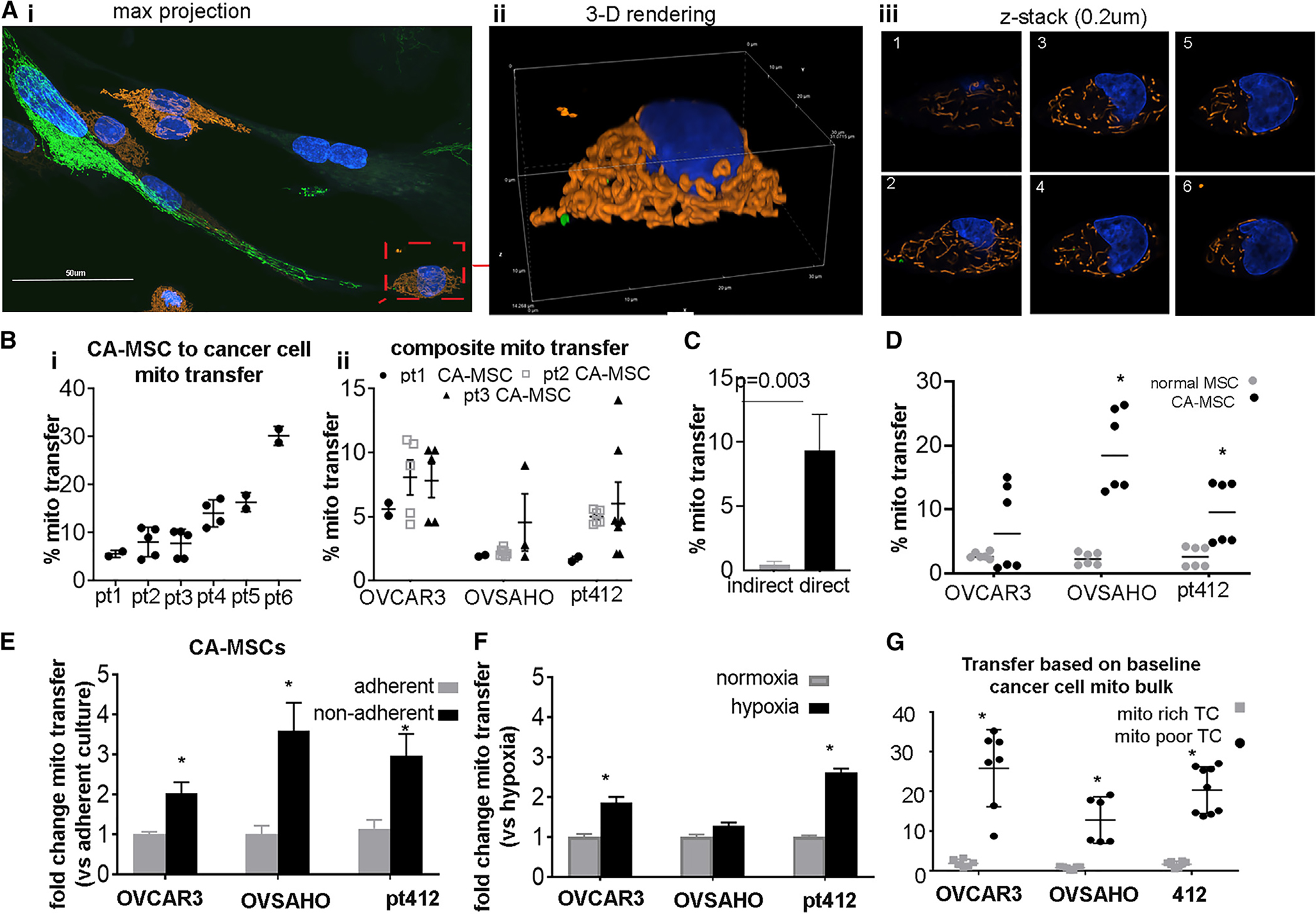
(A) Representative images from coculture demonstrating direct CA-MSC to TC mitochondrial transfer. CA-MSC mitochondria labeled with lentiviral COX8-GFP (green), while endogenous TC mitochondria are COX8-dsRed (orange). (i) Maximum projection image of representative coculture z stack. scale bar, 50 μm. (ii) Three-dimensional (3D) rendering of a subset of (i) showing TC+mitoTrans cell and (iii) representative z slices.
(B) Transfer of GFP-CA-MSC mitochondria to TCs quantified using flow cytometry. Six individual patient (pt)-derived CA-MSCs are represented (i) and three individual patient-derived CA-MSCs donating to three individual ovarian cancer cell lines are represented (ii).
(C) CA-MSC to TC mitochondrial transfer under indirect coculture vs. direct coculture (n = 5 individual experiments).
(D) Comparison of CA-MSCs’ and normal MSCs’ (derived from patients without cancer) ability to transfer mitochondria to TCs (n = 3 individual experiments).
(E) Change in CA-MSC to TC mitochondrial transfer under adherent vs. non-adherent conditions (n = 5 individual experiments).
(F) Change in CA-MSC to TC mitochondrial transfer under normoxic (21% O2) vs. hypoxic (1% O2) conditions (n = 3 individual experiments).
(G) CA-MSC mitochondrial transfer quantified comparing donation to TCs with the highest active mitochondrial content (top 20%, mito rich) and lowest active mitochondrial content (lower 20%, mito poor), as assessed by MitoTracker Deep Red staining.
In (B)–(D), CA-MSCs were cocultured, resulting in a 10:1 TC:CA-MSC ratio. *p < 0.05. Data are presented as the mean ± SEM; displayed p values obtained using Student’s t test.
To determine whether the mitochondrial transfer findings are generalizable to most CA-MSCs, we tested the ability of six independent, patient-derived CA-MSCs to donate mitochondria to three unique ovarian cancer cell lines: two established HGSC lines (OVCAR3 and OVSAHO) and one primary patient-derived HGSC line (PT412). We directly cocultured GFP-COX8 transduced CA-MSCs with CellTrace Blue-labeled cancer cells for 24 h. After coculture, cells were dissociated into single-cell suspension and extensively washed, and flow cytometry was used to quantify the number of TCs that gained CA-MSC mitochondria, based on CellTrace Blue and GFP+ gating. Cell viability was assessed with propidium iodide with both CA-MSC and TCs having over 90% viability at the time of analysis. All six patient-derived CA-MSC lines were capable of donating mitochondria to each cancer cell line tested; however, the amount of mitochondrial transfer varied among the different CA-MSC donors (Figure 2B). To verify that no false signal was present due to mitochondria sticking to the outside of the TC, we directly isolated COX8-GFP-tagged mitochondria from CA-MSCs and incubated them with TCs for 5 min, followed by wash and flow cytometric evaluation. Less than 0.5% of TCs had any green signal, indicating a very low likelihood of exogenous mitochondria on the surface of the TC confounding our flow cytometry results (Figure S1). Mitochondrial transfer between CA-MSCs and TCs requires direct physical contact, as shown by the near-absence of mitochondrial transfer when CA-MSCs and TCs were only allowed to indirectly interact through a transwell system or with CA-MSC conditioned media (Figure 2C). We also examined the capacity of CA-MSCs to donate mitochondria in comparison to non-tumor-educated MSCs derived from patients with benign conditions. While both normal MSCs and CA-MSCs demonstrated an ability to donate mitochondria, CA-MSCs have an increased capacity for donation compared to their tumor-naive counter-parts (Figure 2D). We performed a time course of mitochondrial transfer from CA-MSC to the three different TC lines (OVCAR3, OVSAHO, and PT412) and compared this to the baseline proliferation rates of the TCs. Figure S1 demonstrates peak mitochondrial transfer within 8–24 h (OVCAR at 24 h; OVSAHO and PT412 at 8 h). This did not appear related to the proliferation rates of the TCs (PT412 cells doubled in 24 h; OVSAHO and OVCAR3 cells doubled in 40 h). Again, cell viability remained above 90% for all coculture experiments.
We next explored whether other physiologic conditions intrinsic to the ovarian tumor microenvironment influence mitochondrial transfer. Tumors exist in three-dimensional orientations, with cells interfacing in multiple planes. Additionally, to effectively metastasize, TCs must detach from the primary tumor and survive under non-adherent conditions. Our previous work demonstrated that CA-MSCs directly interact with TCs to form heterocellular complexes that enhance metastasis and exist within ovarian cancer malignant ascites.4 We expanded on that knowledge by testing whether CA-MSC mitochondrial transfer was impacted by non-adherent, spheroid conditions. We found that TCs cocultured with CA-MSCs (which associate in a 10:1 ratio) under non-adherent conditions exhibited a 2- to 4-fold increase in mitochondrial transfer compared to cocultures in standard adherent conditions (Figure 2E). Given that MSCs have been shown to donate mitochondria under hypoxic stress, we also tested whether hypoxia influenced CA-MSC mitochondrial transfer. CA-MSCs and TCs were grown under non-adherent conditions in normoxic (21% O2) or hypoxic (1% O2) conditions. Hypoxia enhances CA-MSC mitochondrial donation to OVCAR3 and PT412 TC lines 2-fold compared to normoxia (Figure 2F). While these results demonstrate that stressors characteristic of HGSC microenvironments enhance stromal mitochondrial donation, these in vitro assays are limited in their ability to model the complex environmental stressors encountered in vivo and in situ, such as fluid shear stress and peritoneal compression, and they likely underrepresent the amount of transfer that is truly occurring.21,31,32
We next examined whether intrinsic characteristics of the TCs affect mitochondrial donation from CA-MSCs. CA-MSCs from patients 3, 4, and 5, which demonstrated average amounts of mitochondrial donation across the patient samples tested, were used for subsequent experiments. We tested whether the endogenous mitochondrial content within TCs altered CA-MSC mitochondrial gain. TCs were stained with MitoTracker Deep Red, a dye that is taken up by actively respiring mitochondria, and TCs were sorted by flow cytometry into TCs with the highest (top 20%) and lowest (bottom 20%) respiring mitochondrial content. Baseline mitochondrial distribution per cell is represented in Figure S1D. We observed that TCs with the lowest MitoTracker Deep Red staining, referred to as “mito poor” TCs, received significantly more CA-MSC-donated mitochondria compared to TCs with the highest MitoTracker staining, referred to as “mito rich” TCs (Figures 2G and S1). This phenomenon was replicated and consistent across three HGSC cell lines and CA-MSCs derived from three different HGSC patients (Figure 2G).
CA-MSC coculture enhances mito poor TC proliferation and chemotherapy resistance and restores metabolic fitness
To elucidate why CA-MSCs preferentially donate mitochondria to mito poor TCs, we first assessed the baseline phenotypic differences between the mito poor TCs vs. the mito rich TCs. The proliferation rate for each group was quantified through manual cell counting. The mito poor TCs demonstrated remarkably less proliferation compared to the mito rich TCs (Figures 3A–3Ci). We then assessed the proliferation of the mito poor and mito rich TCs following 24 h of CA-MSC coculture. The mito poor and mito rich TC groups were flow sorted by their initial endogenous mitochondrial bulk, not accounting for mitochondria gained from CA-MSCs, through staining TC mitochondria prior to coculture. CA-MSC coculture led to significant increases in proliferation in the mito poor TCs, effectively restoring their proliferative capacity equal to that of the mito rich TCs (Figures 3A–3Cii).
Figure 3. TCs with less endogenous mitochondrial bulk demonstrate decreased proliferation and increased chemotherapy sensitivity compared to TCs with high endogenous mitochondrial bulk.
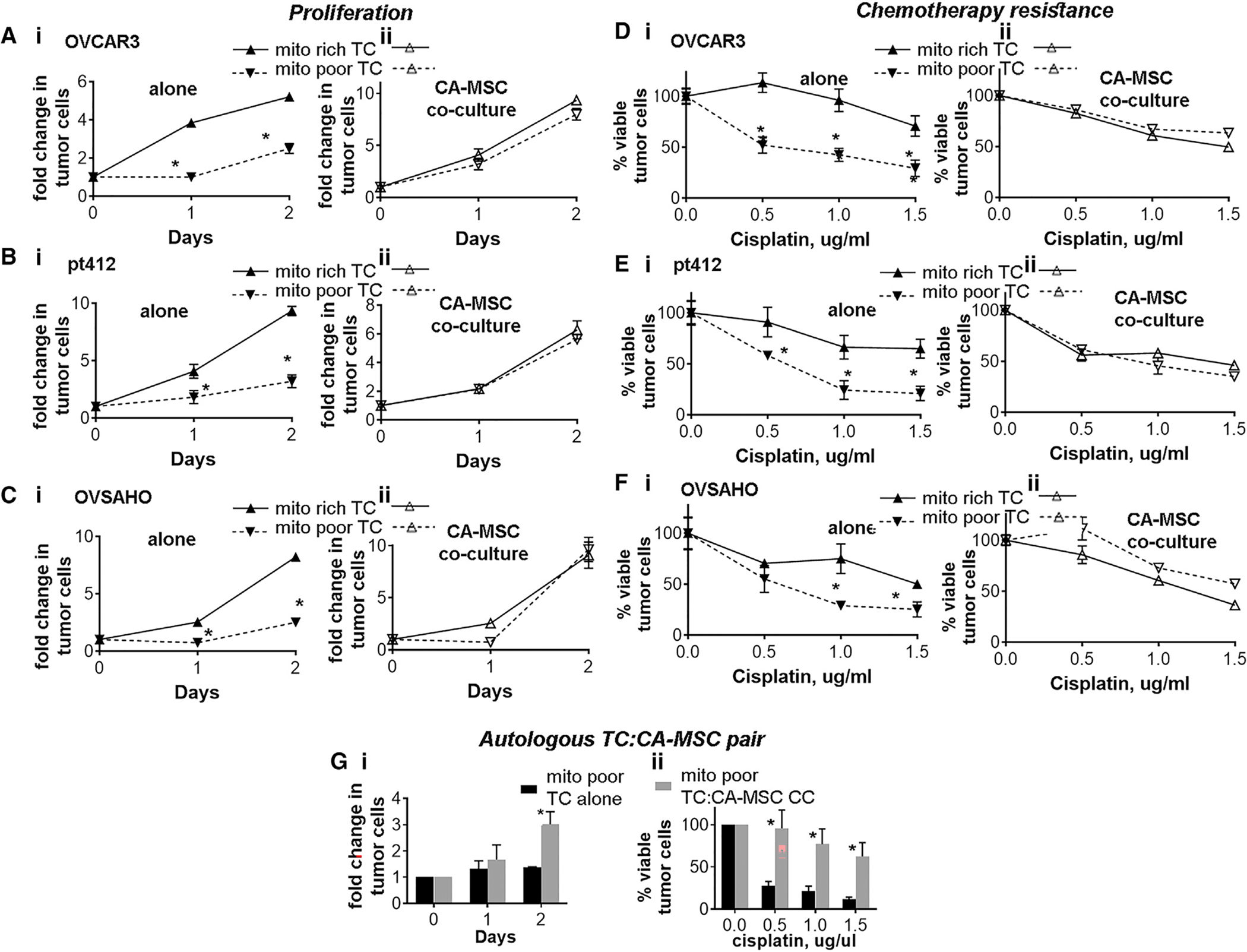
(A–Ci) Mito rich TCs vs. mito poor TCs alone proliferation. (A–Cii) Mito rich vs. mito poor TCs with CA-MSCs co-culture, and TC proliferation was quantified, demonstrating coculture rescues the proliferation of mito poor TCs. Three different TC lines were used: (A) OVCAR3, (B) PT412, and (C) OVSAHO.
(D–F) TC survival to increasing doses of cisplatin in mito rich vs. mito poor (i) TC alone and (ii) with CA-MSC coculture. Three different TC lines were used: (D) OVCAR3, (E) PT412, and (F) OVSAHO.
(G) Mito poor TCs from a paired TC and CA-MSC sample derived from the same patient with HGSC were grown alone or with CA-MSC coculture, and (i) TC proliferation and (ii) chemotherapy sensitivity were compared validating the rescue of mito poor TC growth and chemotherapy survival with CA-MSC coculture in an autologous paired sample.
For all experiments, data are presented as the mean ± SEM; displayed p values obtained using Student’s t test, with *p < 0.05. Three independent experiments were run for each assay.
Surprisingly, despite being less proliferative, the mito poor TCs were also more susceptible to cisplatin treatment at baseline compared to the mito rich TCs. Figures 3D–3Fi demonstrate the survival of mito poor vs. mito rich TCs to increasing doses of cisplatin. To determine the impact of CA-MSC coculture on mito poor vs. mito rich TCs, CA-MSCs and mito poor TC vs. mito rich TCs were cocultured for 24 h. TCs were then separated from CA-MSCs via flow sorting and were treated with cisplatin. Viable TCs were counted after 48 h of cisplatin treatment. CA-MSC coculture enhanced the chemotherapy resistance of the mito poor TCs to a level equal to the mito rich TCs without coculture. The proliferation and chemotherapy resistance assays for the mito poor and mito rich TCs with and without CA-MSC coculture were repeated in triplicate and with three independent ovarian TC lines. Collectively, these data demonstrate that TCs with less endogenous mitochondria preferentially receive CA-MSC mitochondria and that coculture with CA-MSCs enhances the proliferation and cisplatin resistance of mito poor TCs.
We validated the above experiments using an autologous paired patient sample, where TCs and CA-MSCs were isolated from the same patient with HGSC to ensure that the mitochondrial transfer and rescue of mito poor TCs is not influenced by a lack of coevolution of the mtDNA and nuclear genomes.33 We first verified that CA-MSCs donated mitochondria to its autologous TC pair (Figure S2A). We then isolated the mito poor TCs from the paired sample and tested the proliferation and chemotherapy sensitivity of the mito poor TCs alone or after CA-MSC coculture. Figure 3G demonstrates that CA-MSC coculture similarly enhances the proliferation and survival of autologous mito poor TCs.
CA-MSC mitochondrial donation induces ANGPTL3 secretion in recipient ovarian TCs to drive proliferation
While CA-MSC mitochondrial transfer leads to a significant increase in the proliferation of vulnerable ovarian TCs, mitochondrial transfer occurs in only one-third of the mito poor population, suggesting that mito transfer elicits changes in the recipient cells that are propagated to the remaining cell population (Figure 2G). To test whether CA-MSC mitochondrial transfer induced proliferation solely in recipient mito poor ovarian TCs or within the entire mito poor population, we isolated the mito poor TCs that either received or did not receive mitochondria from CA-MSCs (termed TC+mitoTrans and TC−mitoTrans, respectively) via fluorescence-activated cell sorting (FACS). TC+mitoTrans cells demonstrated a significant growth advantage over TC−mitoTrans cells (Figure 4A). However, when we cocultured TC+mitoTrans and TC−mitoTrans cells (labeled with CellTrace Violet and CellTrace Red, respectively), TC−mitoTrans cells demonstrated enhanced proliferation, reaching a growth rate similar to that of TC+mitoTrans cells (Figure 4B). This indicates that TCs that received mitochondria from CA-MSCs act on TCs that did not receive CA-MSC-derived mitochondria to enhance overall growth. To investigate the mechanism of this enhanced proliferation, we first tested whether there was transfer of mitochondria from TC+mitoTrans to TC−mitoTrans cells. TC+mitoTrans cells labeled with MitoTracker Red were cocultured with TC−mitoTrans cells; however, no mitochondrial transfer was observed (Figures S2B and S2C).
Figure 4. Donated mitochondria-to-host nuclear signaling in recipient TCs drives proliferation in non-mitochondria receiving TCs via secretion of ANGPTL3 and MAPK/ERK activation.
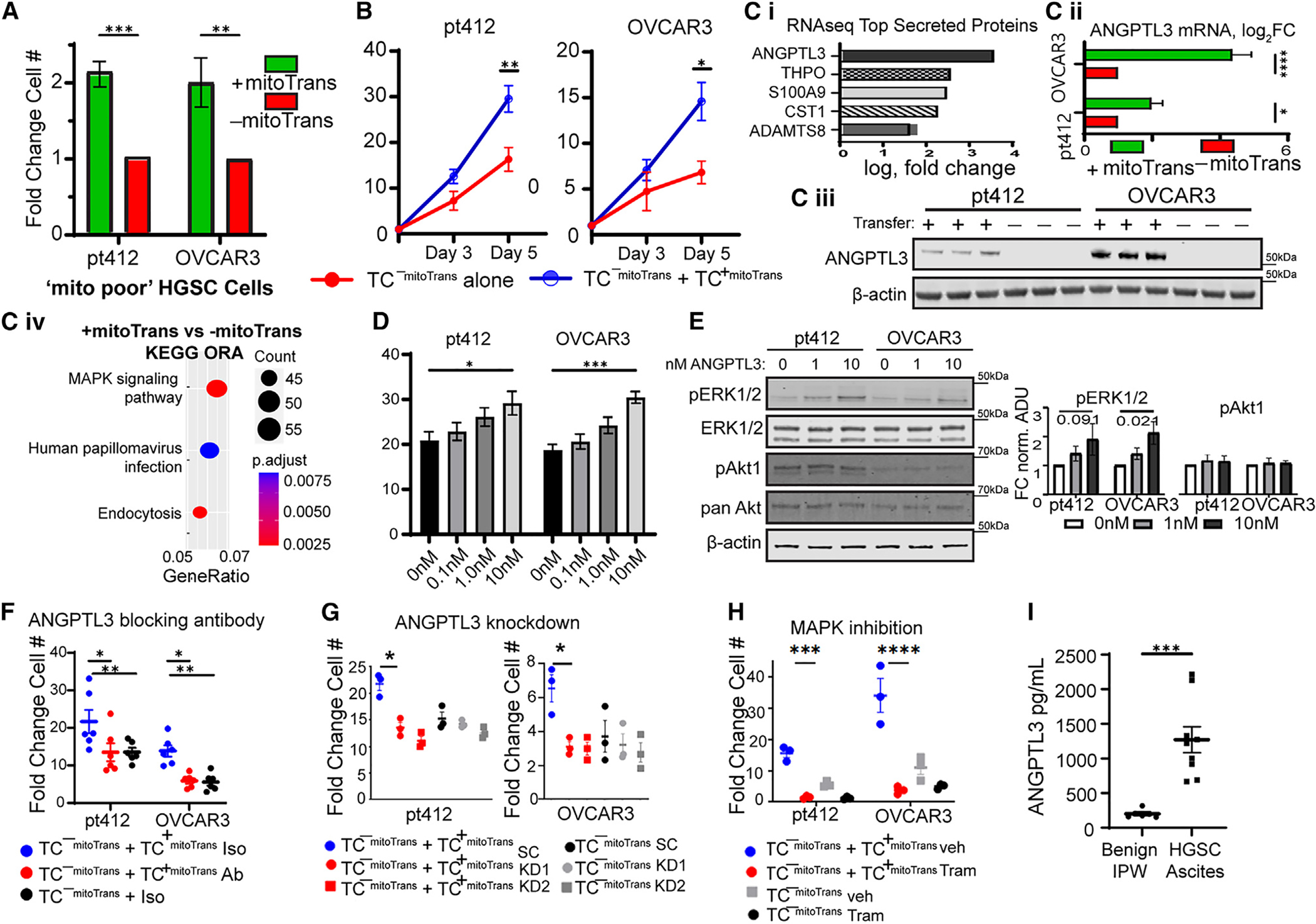
(A) Proliferation of TC+mitoTrans and TC−mitoTrans cells using PT412 and OVCAR3 HGSC cells (n = 6).
(B) Proliferation of TC−mitoTrans cells alone or cocultured with TC +mitoTrans cells (n = 6).
(C) (i) Top upregulated differentially expressed genes coding for secreted proteins in TC+mitoTrans compared to TC−mitoTrans cells (n = 4 paired samples). (ii) qPCR (n = 3 paired samples) and (iii) western blotting for ANGPTL3 in TC+mitoTrans and TC−mitoTrans cells (n = 3). (iv) Over-representation analysis using Kyoto Encyclopedia of Genes and Genomes pathways containing differentially expressed genes in TC+mitoTrans cells.
(D) Proliferation curve showing fold change in TC number in bulk OVCAR3 and PT412 TCs treated with increasing amounts of recombinant ANGPTL3 protein for 5 days (with daily media change/ANGPTL3 re-dosing) (n = 6 per group).
(E) Western blot and corresponding densitometry plot indicating increased phosphorylation of ERK1/2 and unchanged Akt phosphorylation in PT412 and OVCAR3 cells treated with recombinant ANGPTL3 (n = 6 for all groups).
(F) Proliferation on day 5 of TC−mitoTrans cells alone or cocultured with TC+mitoTrans cells in the presence of an ANGPTL3 blocking antibody or IgG control (n = 6 per group).
(G) Proliferation on day 5 of ANGPTL3 KD or scrambled control TC−mitoTrans cells alone or cocultured with KD/scrambled control TC+mitoTrans cells (n = 3 per group).
(H) Proliferation on day 5 of TC−mitoTrans cells alone or cocultured with TC+mitoTrans cells (n = 3 per group) under treatment with either 100 nM trametinib or DMSO vehicle control.
(I) ANGPTL3 ELISA of the non-cellular fraction from patient-derived malignant HGSC ascites (n = 8) or IPWs for benign indications (n = 4).
For (A)–(H), data are presented as the mean ± SEM; displayed p values obtained using Student’s t test (A–Ciii and I) or one-way ANOVA (D–H). For all data, *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
We next hypothesized that the transfer of mitochondria from CA-MSCs induces transcriptional changes in recipient TCs that subsequently drive pro-growth paracrine signaling from TC+mitoTrans to non-recipient TC−mitoTrans cells. RNA sequencing of paired TC+mitoTrans and TC−mitoTrans cells was carried out to determine the factors underlying the enhancement of TC−mitoTrans proliferation by TC+mitoTrans cells (Figures 4C and S3). The secreted cytokine angiopoietin-like protein 3 (ANGPTL3) was identified as one of the most significantly upregulated differentially expressed genes and was the most significantly upregulated secreted protein in TC+mitoTrans cells vs. their paired TC−mitoTrans cells (Figure 4Ci). ANGPTL3 upregulation was independently confirmed via qPCR and western blotting (Figures 4Cii and 4Ciii). Canonically, ANGPTL3 is a multifunctional cytokine with roles in mediating lipid metabolism and angiogenesis; however, recent work has demonstrated an emerging role in promoting tumor progression, including as a driver of cellular proliferation via MAPK/ERK activation.34–36 We observe an enrichment in the MAPK signaling pathway in TC cells (Figure 4Civ). We therefore hypothesized that CA-MSC mitochondrial transfer induced ANGPTL3 secretion in recipient TCs, which then drives proliferation in non-recipient TCs through the activation of MAPK signaling.
The ability of ANGPTL3 to induce proliferation in ovarian TCs was tested by treating bulk OVCAR3 and PT412 cells with increasing amounts of recombinant ANGPTL3 protein for 5 days (Figure 4D). Both lines demonstrated dose-dependent increases in proliferation in response to ANGPTL3 treatment, consistent with the reported literature in other cancer models.35,36 As ANGPTL3 can stimulate both ERK and Akt phosphorylation, we next treated OVCAR3 and PT412 cells with recombinant ANGPTL3 and examined phospho-ERK and phospho-Akt levels via western blot. We observed increasing ERK1/2 phosphorylation at Thr202/Tyr204 in a dose-dependent manner with no notable changes to phosphorylated Akt levels, further suggesting that ANGPTL3 is functioning through MAPK/ERK signaling (Figure 4E).
To further determine whether TC+mitoTrans ANGPTL3 secretion drives TC−mitoTrans cell proliferation, we cultured paired TC−mitoTrans cells with or without TC+mitoTrans cells in the presence of either a commercially available ANGPTL3 blocking antibody (evinacumab) or immunoglobulin G (IgG) matched control (Figure 4F). Evinacumab treatment blocked the growth promotion of TC+mitoTrans cells in cocultured TC−mitoTrans cells compared to cocultures treated with IgG control with the same proliferation rates as TC −mitoTrans cells grown alone (Figure 4F). Likewise, lentiviral small hairpin RNA (shRNA) knockdown (KD) of ANGPTL3 in ovarian TCs similarly replicated this result in coculture, further suggesting that TC+mitoTrans ANGPTL3 secretion drives proliferation in TC−mitoTrans cells (Figures 4G, S4A, and S4C). To further delineate the mechanism, we also used the MAPK/ERK inhibitor trametinib to block downstream ANGPTL3 signaling. The above experiment was repeated with trametinib vs. vehicle control. Trametinib prevented the proliferative effect of TC+mitoTrans on the TC−mitoTrans population. On-target validation of trametinib was performed with western blotting for p-ERK showing a dose-dependent decrease after treatment (Figures 4H and S4B). To validate the clinical relevance of ANGPTL3 in ovarian cancer, the non-cellular component of malignant ascites derived from patients with HGSC or benign intraperitoneal washes (IPWs) from women without cancer was isolated and tested for ANGPTL3 via ELISA. Malignant HGSC ascites contained significantly elevated levels of ANGPTL3 compared with benign IPWs (Figure 4I).
We next assessed whether the mitochondria donated from CA-MSCs to TCs remain actively respiring and alter oxidative metabolism in TCs. We measured the oxygen consumption rate (OCR) using live-cell extracellular flux analysis. We first analyzed the baseline respiratory capacity of the mito rich compared to mito poor TCs. TCs were sorted as above and analyzed with an Agilent XF Seahorse to measure mitochondrial respiration. Figure 5A demonstrates that the mito rich TCs had significantly higher basal respiratory capacity, maximal respiratory capacity, ATP-dependent OCR, and spare respiratory capacity compared to mito poor TCs. However, following coculture with CA-MSCs, the metabolic profile of the mito poor TCs matched that of the mito rich TCs, indicating that CA-MSCs restored the metabolic fitness of the mito poor TCs, possibly through gain of actively respiring CA-MSC mitochondria (Figures 5B–5D). These data provide evidence that the CA-MSC mitochondria gained by TCs are likely metabolically active and may be enhancing the respiratory capacity of TCs that contain less endogenous mitochondria. To further verify the functional significance of mitochondrial transfer, live mitochondria were isolated from CA-MSCs and exogenously given to TCs. Mitochondria were taken up by 5%–10% of TCs after 24 h, and the respiratory capacity of TCs likewise increased (although to a lesser extent than with direct CA-MSC coculture) (Figure S4D).
Figure 5. Respiratory capacity of mito poor TCs improves after CA-MSC coculture.
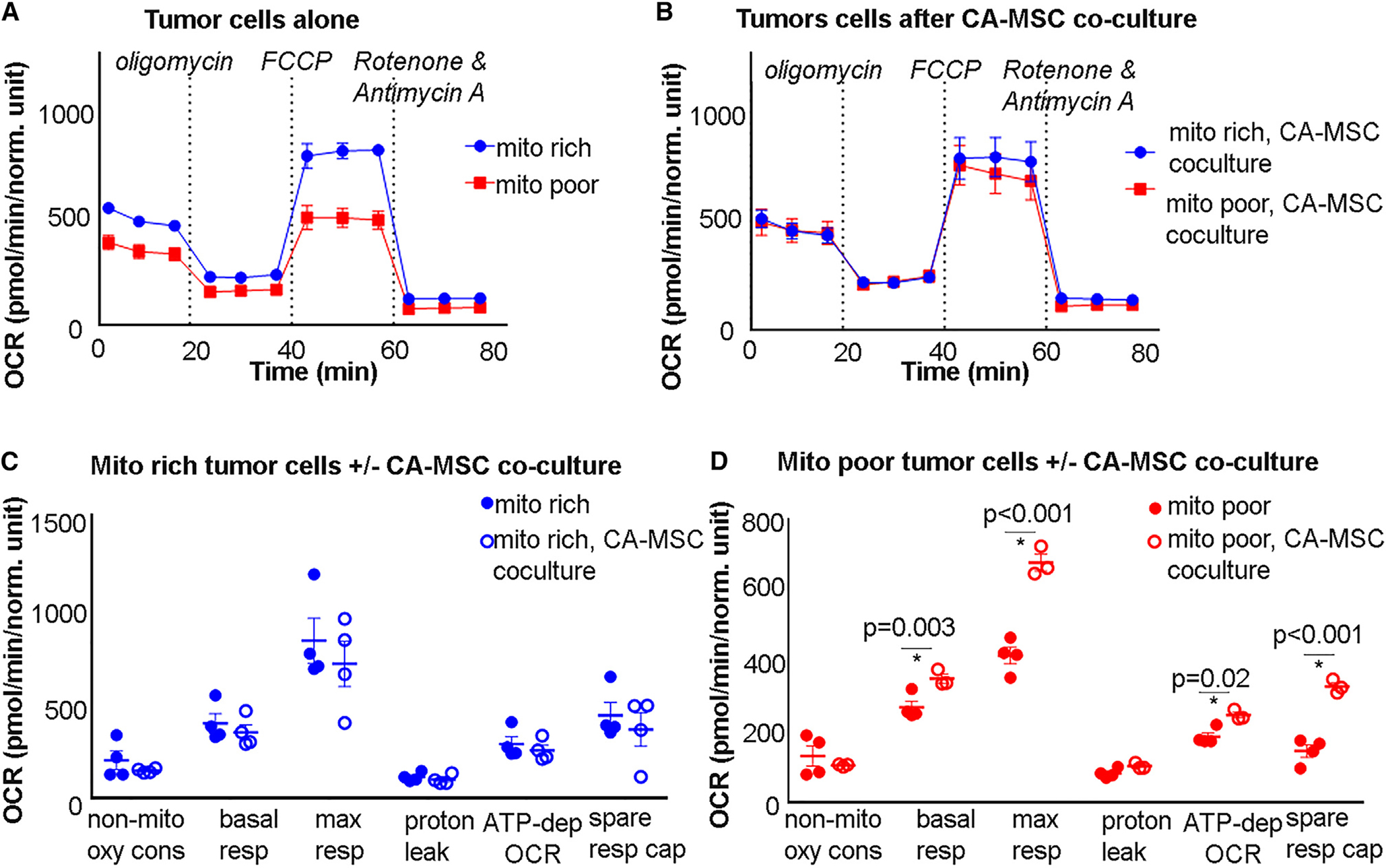
(A and B) Representative extracellular flux assay plots of OCR measurements from mito rich vs. mito poor TCs (A) alone and (B) after CA-MSC coculture.
(C and D) Summary data of metabolic parameters for the mito rich vs. mito poor TCs (C) alone and (D) after CA-MSC coculture. Each point represents the mean OCR value from an independent experiment (n = 4 paired samples; displayed p values obtained from Student’s t test).
MIRO1 KD prevents mitochondrial transfer and decreases the proliferation and chemotherapy resistance benefit of CA-MSC coculture
We demonstrated that mito poor TCs receive CA-MSC mitochondria at higher rates and subsequently have increased proliferation, chemotherapy resistance, and oxidative respiration following coculture with CA-MSCs. To further verify that this impact is due to CA-MSC mitochondrial transfer and not other mediators, we targeted the mitochondrial transfer protein MIRO1. MIRO1 is a mitochondrial transport protein that facilitates intracellular movement of mitochondria via microtubules.37,38 We created MIRO1 KD CA-MSCs, which prevented most mitochondrial transfer while still enabling the CA-MSC to directly interact with the TC. MIRO1 was knocked down in multiple CA-MSC primary cell lines via lentiviral shRNA, with mRNA decreasing 50% and protein expression decreasing 80% (Figure 6A). We validated that MIRO1 KD did not impact CA-MSC identity, viability, growth, mitochondrial bulk/cellular distribution, or ability to interact with TCs (Figures 6B and S5).
Figure 6. Blocking CA-MSC mitochondrial transfer via MIRO1 KD eliminates the proliferation and chemotherapy resistance benefit of TC coculture with CA-MSCs.
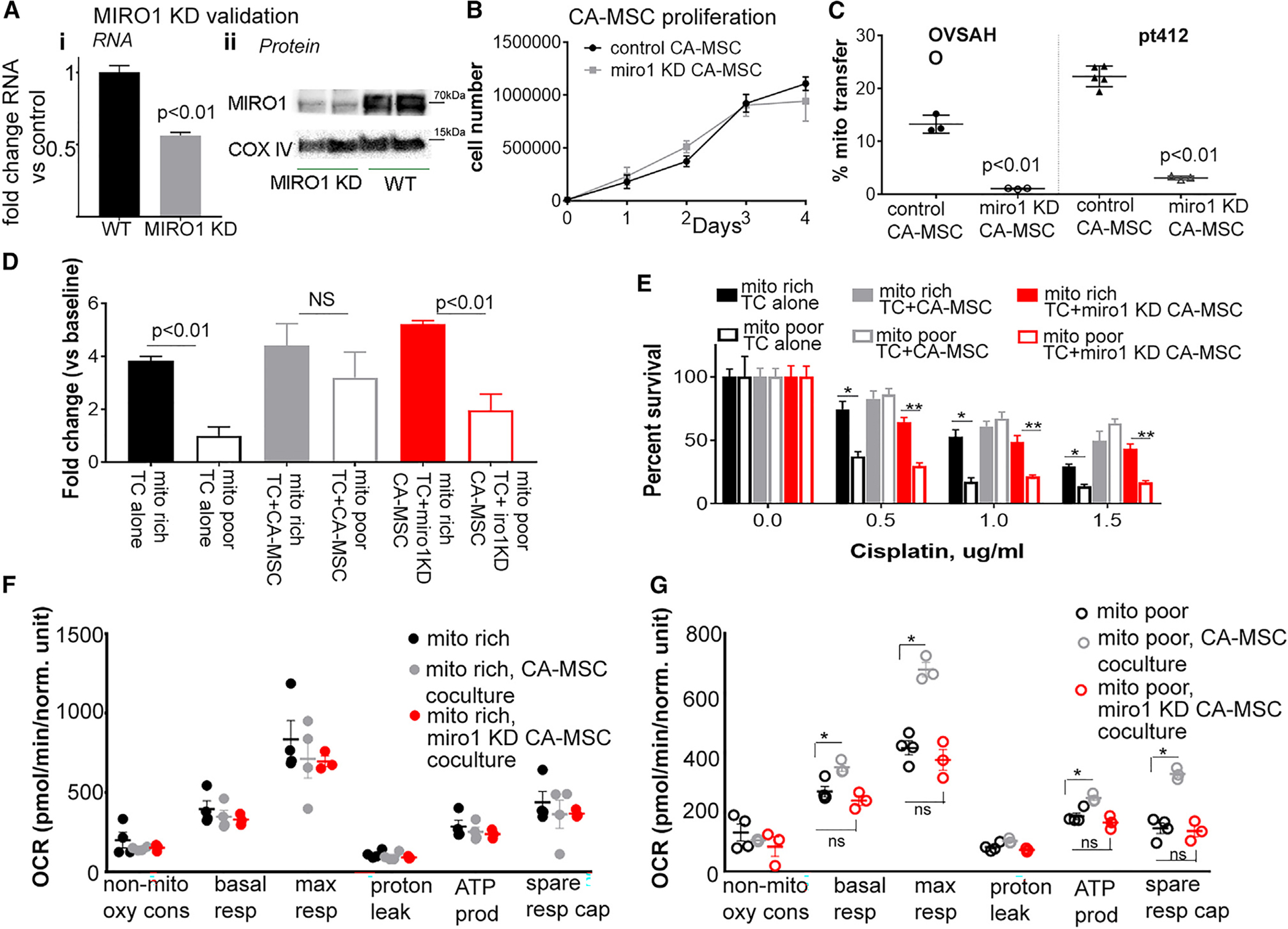
(A) MIRO1 KD validation at the RNA (i) and protein (ii) level in CA-MSCs. WT = wild-type control (n = 3).
(B) Proliferation of MIRO1 KD vs. control CA-MSCs demonstrated that MIRO1 KD did not impact the viability or proliferation of CA-MSCs (n = 3).
(C) Quantification of mitochondrial transfer from control vs. MIRO1 KD CA-MSCs to OVSAHO or PT412 TCs (n = 3).
(D) Quantification of TC proliferation, represented as fold change from baseline at 48 h, in mito rich vs. mito poor TC alone and with control or MIRO1 KD CA-MSC coculture (n = 3 paired samples per coculture condition).
(E) Quantification of TC survival to increasing doses of cisplatin in mito rich vs. mito poor TC alone and with control or MIRO1 KD CA-MSC coculture (n = 3 paired samples per coculture condition).
(F) Composite extracellular flux OCR data in mito rich TC alone and with control vs. MIRO1 KD CA-MSC coculture (n = 3 paired samples per coculture condition).
(G) Composite OCR data in mito poor TC alone and with control vs. MIRO1 KD CA-MSC coculture. Each point is an independent experiment. *p < 0.05, t test. Mean and SEM are represented (n = 3 paired samples per coculture condition).
Data are presented as the mean ± SEM; displayed p values obtained using Student’s t test (A–Ciii and G) or one-way ANOVA (D–F). For all data, ns indicates not significant; *p < 0.05; **p < 0.01.
We next demonstrated that MIRO1 KD effectively prevented CA-MSC-to-TC mitochondrial transfer. MIRO1 KD CA-MSCs with COX8-GFP-labeled mitochondria were cocultured with fluorescently labeled TCs as above, and mitochondrial transfer was quantified via flow cytometry. Figure 6C illustrates that MIRO1 KD CA-MSCs transfer 15- to 20-fold less mitochondria to ovarian TCs vs. CA-MSCs with wild type (WT) MIRO1 expression. We repeated the proliferation, chemotherapy resistance, and metabolomic experiments with mito rich and mito poor TCs with and without control CA-MSC or MIRO1 KD CA-MSC coculture. MIRO1 KD CA-MSCs failed to alter the proliferation or chemotherapy resistance of the mito poor TCs (Figures 6D and 6E). Similarly, MIRO1 KD CA-MSCs did not alter the metabolic profile of the mito rich or mito poor TCs (Figures 6F and 6G), indicating that mitochondrial transfer is critical for the CA-MSC mediated enhancement of mito poor TC proliferation, chemotherapy resistance, and metabolic function.
We next tested whether the donated CA-MSC mitochondria persist after transfer to TCs. TCs that gained mitochondria from cocultured CA-MSCs were FACS isolated and grown alone over time. Immunofluorescence microscopy demonstrated the persistence of the transferred mitochondria after separation of the two cell types for over 24 h (Figure 7A). We visualized transferred mitochondria up to 3–5 days after transfer. However, this method is limited to visualizing only mitochondria that were transferred from the parent CA-MSC, as the COX8-GFP tag is nuclear encoded in the parent CA-MSC. Thus, the COX8-GFP tag cannot account for CA-MSC mitochondria that may have replicated inside TCs and resulted in further amplification of CA-MSC-derived mitochondria. We therefore designed haplotype-specific primers to distinguish CA-MSC-derived mtDNA (using one patient source) from TC-specific mtDNA (OVCAR3, derived from a different patient) to enable the distinction and quantification of all CA-MSC-derived mitochondria within TCs (originally transferred mitochondria plus transferred mitochondria that have replicated). Using qPCR of the haplotype-specific mtDNA with a standard curve method for quantification (Figure 7Bi), we determined the ratio of endogenous TC mitochondria vs. donated CA-MSC mitochondria over 14 days. At each time point, 20,000 TCs were harvested and mtDNA isolated followed by haplotype-specific qPCR (normalized to input DNA amounts). This effectively enables the quantification of CA-MSC-derived mitochondria vs. endogenous TC mitochondria per cell. This method demonstrated that after 24 h there is a 0.63 ratio of CA-MSC/TC-derived mitochondria, which remains relatively stable after 3 days (0.78 ratio) and decreases to 0.19 after 7 days but remains detectable at day 14 (ratio 0.06) (Figure 7Bii). Given that the TC doubling time is ~40 h, this indicates not only that the CA-MSC-derived mitochondria persisted inside the TCs but also that they likely actively replicated and contributed a sizable portion of the total TC mitochondria (10%–30%).
Figure 7. Mitochondria donated from CA-MSCs is retained after TC transfer and in vivo growth, and blockage of CA-MSC mitochondria transfer prolongs mouse survival and decreases TC heterogeneity in a murine ovarian cancer model.
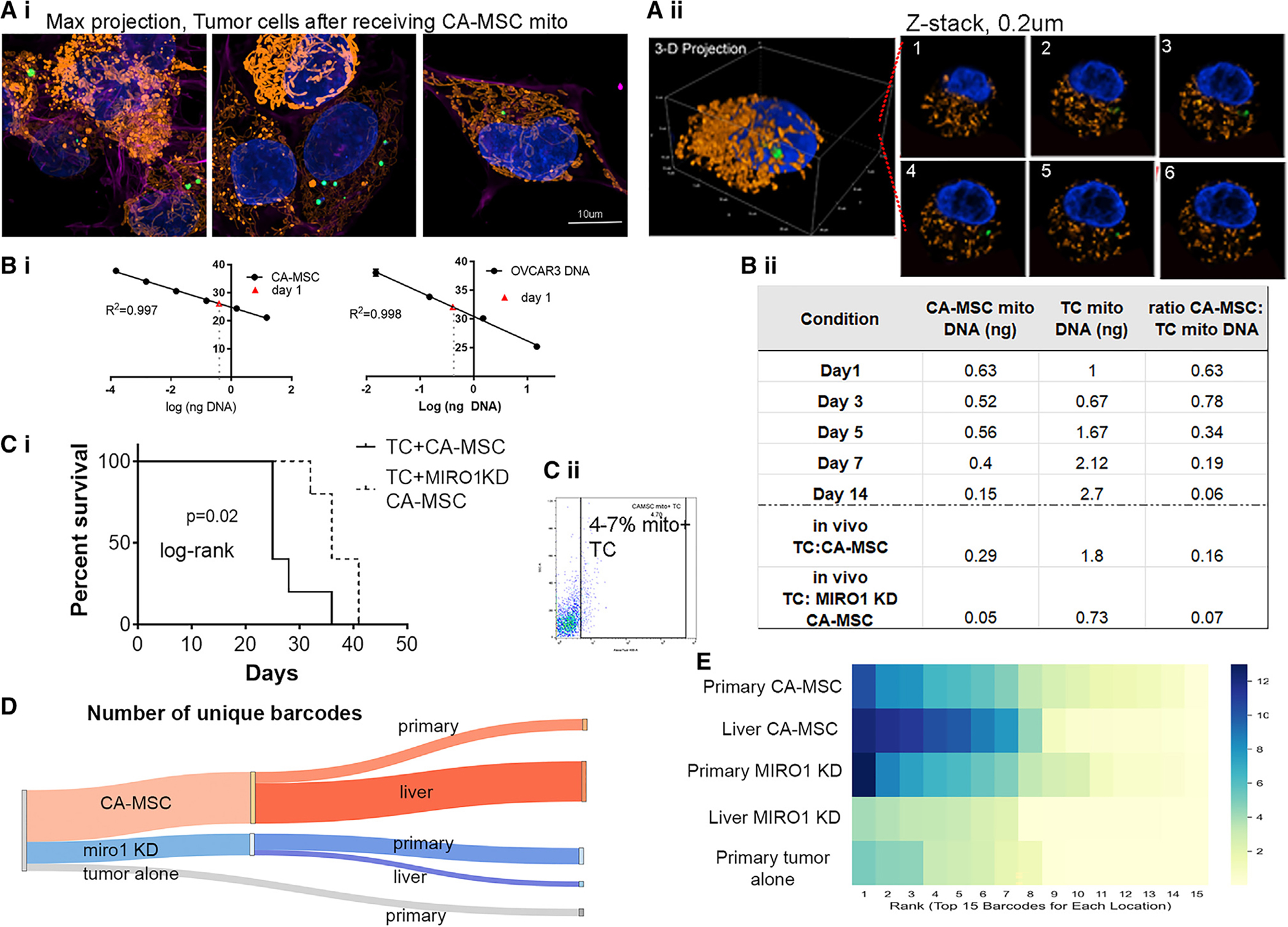
(A) Representative images PT412+mitoTrans cells 24 h post-isolation from coculture with COX8-GFP-tagged CA-MSCs via FACS. (i) Maximum projection images. (ii) 3D rendering of a subset of (i) with (iii) six representative z slices. Scale bar, 10 μm.
(B) (i) qPCR standard curves used to quantify the amount of CA-MSC and endogenous TC mitochondrial DNA in TCs (n = 3). (ii) Ratio of CA-MSC to TC mtDNA in TCs after coculture corresponding to immunofluorescence pictures in (A) and in vivo TCs isolated from TC:CA-MSC xenografts or TC:MIRO1 KD CA-MSC xenografts.
(C) (i) Quantification of TC survival when TCs were cocultured with CA-MSC vs. MIRO1 KD CA-MSC. (ii) Flow cytometry analysis of isolated TCs with CA-MSC-donated mitochondria.
(D) Sankey diagram demonstrating number of unique TC barcodes in the CA-MSC vs. MIRO1 KD CA-MSC xenografts.
(E) Heatmap of the prevalence of the top 15 barcodes at each location in CA-MSC vs. MIRO1 KD CA-MSC mice, indicating a larger diversity of dominate clones consistent with overall increased heterogeneity in the CA-MSC groups. Displayed p value obtained using log rank test.
Ovarian TC proliferation, chemotherapy resistance, and clonal heterogeneity are enhanced by CA-MSC mitochondrial gain in vivo
We next corroborated that CA-MSC mitochondrial transfer is functionally important in vivo. We used an orthotopic ovarian bursal model to more accurately model physiologic ovarian cancer development and metastasis as opposed to the earlier tail vein model, which is a highly permissive metastasis model. TCs were co-injected with either control CA-MSCs or MIRO1 KD CA-MSCs into a unilateral ovarian bursa. Mice implanted with MIRO1 KD CA-MSCs demonstrated decreased metastasis and improved survival compared to mice who received control CA-MSCs (Figure 7Ci). Using flow cytometry and our haplotype-specific qPCR, we validated that TCs coimplanted with control CA-MSCs contained CA-MSC mitochondria at both the primary and metastatic sites (at day 21 post-injection). This was largely absent in the MIRO1 KD CA-MSC group (Figure 7Bii, bottom).
Given the underlying hypothesis that CA-MSCs enhance metastasis through increasing TC heterogeneity by rescuing vulnerable TCs, we also employed our DNA barcoding system in the orthotopic ovarian bursa in vivo model. At time of necropsy, primary and metastatic tumors were removed and sent for DNA barcode sequencing. The barcoding data verified our earlier finding from the tail vein injection model that CA-MSCs enhanced the clonal heterogeneity of TCs compared to TCs grown alone (Figures 7D and 7E). To demonstrate that the enhanced heterogeneity was due to mitochondrial transfer, the same model was used with MIRO 1 KD CA-MSCs, and the tumors in MIRO1 KD mice had significantly less TC heterogeneity compared to the tumors grown in mice injected with control CA-MSCs or with TCs alone (Figure 7D; Table S2).
When we quantified the cells that gave rise to the greatest number of progenies, we found that most cells in the MIRO1 KD CA-MSC tumors originated from fewer uniquely barcoded cells than in the control CA-MSC alone tumors (Figure 7E). This indicates that in MIRO1 KD CA-MSC conditions, fewer unique clones were able to successfully take hold and produce a significant number of progenies as compared to control CA-MSC tumors, thereby demonstrating reduced heterogeneity in MIRO1 KD CA-MSC-containing tumors.
DISCUSSION
Long thought of as non-essential in cancer due to the tendency of TCs to undergo aerobic glycolysis, it is now established that mitochondria are indispensable to TC viability and contribute to disease progression.39,40 Depletion of mtDNA in multiple TCs leads to decreased proliferation and tumorigenesis, while the acquisition of mtDNA mutations alters TC phenotypes.41–43 Likewise, cell-to-cell mitochondrial transfer is increasingly recognized as a biological phenomenon with functional implications in cancer, such as promoting TC proliferation and resistance to chemotherapy.19,43–45 Despite its emerging role in tumorigenesis, there remains a lack of understanding in how mitochondrial transfer contributes to malignancy in disease-specific contexts, including ovarian cancer. Here, our findings demonstrate that mitochondrial transfer is a critical mechanism underlying stromal-mediated enhancement of ovarian cancer proliferation, chemotherapy resistance, TC heterogeneity, and metastasis using disease-relevant in vitro and in vivo models.
While mitochondria contain their own genome capable of encoding proteins, they rely on and engage in active bi-directional crosstalk with the nuclear genome for organelle transcription, translation, and respiration.46,47 Likewise, different combinations of mtDNA-to-nuclear DNA promote differing impacts on cellular respiration and metabolism.47,48 Prior studies of mitochondrial transfer largely rely on allogenic sources of mitochondria from multiple origins; given that physiological transfer occurs between autologous sources of mitochondria in situ and some allogenic systems can induce distinct phenotypic changes in recipient cells, more physiological models of transfer should be employed for future studies. This study utilized both allogenic and autogenic cellular pairs to study the impact of stomal mitochondrial transfer on TC proliferation and chemoresistance, demonstrating similar rates of transfer, proliferation, and resistance to cisplatin between both systems, and thereby confirming these findings in a more physiologically appropriate model.
The transfer of mitochondria from the tumor microenvironment to TCs can be both detrimental and beneficial to TCs.49 We find that mitochondrial transfer improves mitochondrial function and decreases chemosensitivity of recipient TCs, as previously reported in other tumor types.17,50–52 However, the exact mechanisms for this remain unclear. Given that mitochondria are central to apoptosis initiation, improved mitochondria function has been linked with chemoresistance. For example, cisplatin-resistant ovarian cancer cells increase their oxidative phosphorylation and maintain a healthy pool of active mitochondria by upregulating their mitophagy-mediated turnover53 and PPAR coactivator-1α-mediated mitogenesis.54 Improved reactive oxygen species (ROS) scavenging following mitochondrial transfer has also been shown to lead to decreased chemosensitivity in TCs.51,52 In glioma cells, the transfer of astrocyte mitochondria alters the metabolism to promote the generation of metabolite precursors necessary for glutathione synthesis, suggesting one mechanism by which recipient TCs increase their ROS scavenging.44 Moreover, since mitochondria are major signaling hubs, given their role as centers for metabolite synthesis, ROS, and calcium homeostasis, it is possible that mitochondrial transfer induces signaling and transcriptional changes that further promote chemoresistance mechanisms,55 which requires further exploration.
Additionally, one hallmark of ovarian cancer, particularly HGSC, is extensive genomic instability and copy-number alterations as well as near-universal mutations to the tumor suppressor gene TP53.3,5,56,57 This genomic instability in turn drives significant intratumor heterogeneity within ovarian cancer, informing disease progression and contributing to patient mortality.3,57 Undoubtedly, the tumor microenvironment plays a critical role in shaping intratumor heterogeneity, as there are a wealth of studies demonstrating that stromal cells can directly influence TC phenotypes, as well as the ability of distinctive microenvironments to provide different selective evolutionary pressures that alter the composition of TC populations.58–60 However, the exact mechanisms underlying how stromal cells alter ovarian cancer heterogeneity remain poorly understood. This study provides further insight into mechanisms of ovarian cancer heterogeneity by demonstrating an important role of CA-MSC-mediated enhancement of ovarian cancer heterogeneity through horizontal mitochondrial transfer.
Beyond the scope of our study, it is possible that CA-MSC mitochondrial donation further promotes disease progression by contributing to TC heteroplasmy. It has long been proposed that alterations to the mitochondrial genome contribute to tumor progression, as dysregulated energy metabolism is a hallmark of malignancy. The recent emergence of single-cell and deep sequencing approaches enabling the quantitative study of subcellular mtDNA populations have since confirmed this connection, with multiple studies indicating the presence of elevated mtDNA copy-number and somatic mutations in ovarian cancer cells and tissues correlated with treatment resistance and tumorigenesis.61–64 We observed elevated TC endogenous mtDNA levels in response to receiving CA-MSC mitochondria using haplotype-specific qPCR, with these mitochondria-receptive TCs displaying enhanced malignant features. While we did not sequence the mtDNA being transferred, other recent studies demonstrate the capacity of stromal cells to transfer mtDNA haplotypes carrying metastasis-enhancing pathogenic mutations, indicating another potential mechanism by which stromal mitochondrial transfer modulates disease progression and clonal heterogeneity.65,66 As somatic mtDNA mutations inform distinct metastatic patterns in ovarian cancer with potential clinical implications, further work is needed to better understand how stromal mitochondrial transfer shapes ovarian TC heteroplasmy.
This work also explored the downstream mechanisms by which exogenously donated mitochondria alter host ovarian TC signaling through the upregulation of ANGPTL3 secretion. To control for paracrine and other stromal signaling factors, we undertook RNA sequencing of paired TCs that either received or did not receive CA-MSC mitochondria from the same population, allowing us to demonstrate that donated mitochondria-to-host nuclear signaling occurs to drive transcriptional changes in the host cell and indicating mitochondrial transfer has a broader impact outside of the receiving cell. This study also explored the role of ANGPTL3 in promoting ovarian cancer proliferation via MAPK/ERK activation; ANGPTL3 plays a contradictory role in several cancers, including ovarian cancer.34–36,67,68 Our findings demonstrate that ANGPTL3 drives proliferation in HGSC cell lines, consistent with Siamakpour-Reihani et al., who found high levels of ANGPTL3 expression in patient-derived tissues correlated with decreased survival, but in contrast to Wu (2023), who found decreased expression in patient tissue and decreased metastatic potential related to elevated ANGPTL3.67,68 Of note is the use of SKOV-3 and the ovarian clear cell line ES-2, which are distinct from the HGSC cell lines employed in our study, potentially explaining this discrepancy. Indeed, differential growth is also seen across mouse models. While OVCAR3 has been reported to have poor growth in nude mice, we demonstrate consistent and robust growth in NSG mice (both in this report and previous papers), which is enhanced by cogrowth with CA-MSCs.11,26 This further highlights the importance of the model system used.
Collectively, this work defines a crucial role for stromal-to-TC mitochondrial transfer in supporting ovarian cancer progression by enhancing TC heterogeneity, survival, and metastasis. These findings contribute to our mechanistic understanding of stromal-mediated metastasis and imply that strategies blocking CA-MSC mitochondrial transfer may have important clinical implications for decreasing the progression of ovarian cancer.
Limitations of the study
The limitations of this work include a relatively small sample size of CA-MSCs and TC lines, and this is unable to account for heterogeneity based on CA-MSC tissue source or TC variables, including exposure to chemotherapy, molecular subclassification (e.g., homologous recombination status), and histologic differences. This study only evaluated treatment-naive HGSC-derived CA-MSCs and HGSC TC lines. Furthermore, while we demonstrate in vivo CA-MSC to TC mitochondrial transfer, our current models do not enable investigations into the kinetics surrounding in vivo transfer. Additionally, both the tail vein model and intrabursal orthotopic models used in this work have limitations. As above, the tail vein model is highly permissive, bypassing critical steps of invasion and intravasation from the primary site of disease; however, it does model organ-seeding differences and the ability to survive in circulation. Furthermore, although lung metastasis is not the most common site of ovarian cancer metastasis, 28% of patients with disease outside of the peritoneum have lung metastasis; therefore, these findings are of physiologic relevance.69,70 The intrabursal model enables orthotopic tumor initiation and models early steps in metastasis, including invasion, intravasation, and extravasation but remains an immune-deficient model, thus not accounting for the important impacts of the immune microenvironment. Conclusions from our work must therefore be tempered by these limitations.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources about this manuscript should be directed to the Lead Contact, Lan G. Coffman (coffmanl@upmc.edu).
Materials availability
Materials unique to this work including cell lines and haplotype specific PCR primers are available upon discussion with the lead contact, Dr. Lan Coffman.
Data and code availability
RNA-seq data have been deposited at Mendeley: https://doi.org/10.17632/25s5258ynf.1 and are publicly available as of the date of publication. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human cancer cell lines
Ovarian cancer cell lines OVCAR3 and OVSAHO were purchased through ATCC and Millipore Sigma, respectively. The primary patient ovarian cancer cell line PT412 was a kind gift from Dr. Ronald Buckanovich (University of Pittsburgh). STR validation was performed on all three cell lines validating the identity of OVCAR3 and OVSAHO while pt412 was consistent with prior in house validation. All cancer cell lines were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. Cancer cells were grown and passaged according to supplier instructions at 37°C in a humidified incubator equilibrated with 5% CO2. Routine mycoplasma testing was carried out and cell lines verified negative for mycoplasma contamination using a commercially available qPCR-based kit (Applied Biological Materials Inc., Richmond, BC) prior to use in experiments.
Human primary cell lines
Cancer associated mesenchymal stem cells (CA-MSCs) were derived from surgical resection of human ovarian cancer involving the fallopian tube, ovary and/or omental metastatic deposits. Normal mesenchymal stem cells (MSCs) were derived from surgical samples (fallopian tube or ovary) of women undergoing surgery for benign indications or propholaytic bilateral salpingo-oophorectomy. Samples were obtained in accordance with protocols approved by the University of Pittsburgh’s IRB (PRO17080326). CA-MSCs and MSCs were grown in Mammary Epithelial cell Basal Medium (MEBM) supplemented as follows: 1% B27, 20 ng/ml epidermal growth factor (EGF), 1% penicillin-streptomycin, 20ug/ml gentamycin, 1 ng/mL hydrocortisone, 5ug/ml insulin, 100um beta-mercaptoethanol, 10 ng/ml recombinant human basic-fibroblast growth factor (bFGF), and 10% fetal bovine serum (FBS). Cells were grown at 37°C in a humidified incubator equilibrated with 5% CO2 and verified negative for mycoplasma contamination.
Mice
All experimental procedures were performed in accordance with protocol approved by the Institutional Animal Care and Use Committee at University of Pittsburgh. Female NSG (NOD scid gamma) mice (6–8 week-old, Jackson Laboratory, ME) were used for both tail vein and orthotopic models. All mice were group housed and kept under 12h:12 h L/D conditions. Food and water were available ad libitum to all mice.
METHOD DETAILS
Tissue harvesting
Patient samples were obtained in accordance with protocols approved by the University of Pittsburgh’s IRB (PRO17080326). CA-MSCs/MSCs were derived from surgical resection of human ovarian cancer involving the fallopian tube, ovary and/or omental deposits, or resection of ovary/fallopian tube under benign/prophylactic indications.10,11,76 Cells derived from these tissues were plated in supplemented MEBM and selected for: plastic adherence, cell surface marker expression (CD105, CD90, CD73 positive; CD45, CD34, CD14, CD19 negative), and trilineage differentiation ability (differentiation into adipocytes, osteocytes and chondrocytes) in accordance with the ISCT criteria for defining multipotent mesenchymal stem/stromal cells77 (Figure S5). Isolated primary lines were grown in complete MEBM.76 CA-MSCs/MSC cell lines failing to meet these criteria were FACS purified, and validated lines were used within 8 passages; all isolated lines were verified negative for mycoplasma contamination.
In vivo tail vein clonal heterogeneity model
Genomically barcoded OVCAR3 cells were generated using Cellecta’s Lentiviral Barcode Library 13k × 13k 30M (Cellecta Inc, Mountain View, CA). 5×105 OVCAR3 cells containing unique genomic barcodes alone or with CA-MSCs (0.5×105 MSCs; 1:10 OV-CAR3: MSC ratio) (n = 10 per group) were injected into the tail vein of NSG mice.11,26 Barcoded clones were stabilized over four passages and sequenced to obtain the baseline barcode pool prior to injection. All mice were euthanized once the first mouse met endpoint criteria (at 21 days post-injection) and metastatic burden assessed at necropsy. Liver, lung and blood were isolated and total DNA extracted using a DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) for subsequent sequencing of genomic barcodes.
Lentiviral Mitochondria labeling of CA-MSCs/MSCs
Early passage (<p4) CA-MSCs/MSCs were transduced with a COX8-GFP lentiviral vector labeling CA-MSC mitochondria with EGFP (Systems Biosciences, Fredrick, MD). Lentiviral labeling was carried out as follows: 5×104 CA-MSCs/MSCs were plated in a 6-well TC plate and allowed to adhere overnight, followed by transduction with the COX8-GFP lentivirus for 48 h using an MOI of 20. Transduced CA-MSCs/MSCs were given a day to recover in fresh complete MEBM followed by 24-h puromycin selection at a concentration of 0.8 μg/mL. A transduction efficiency of >95% was confirmed using fluorescent microscopy.
CA-MSC:Tumor cell mitochondrial ttansfer cocultures
In order to accurately distinguish cell identify in coculture, both ovarian cancer cells and COX8-GFP tagged CA-MSCs/MSCs were dye labeled. Actively respiring mitochondria of OVCAR3, OVSAHO or PT412 tumor cells were stained with MitoTracker Deep Red (Invitrogen, Waltham, MA USA) in PBS suspension at 50nM for 30 min, while CA-MSCs/MSCs were labeled using CellTrace Violet dye (Invitrogen, Waltham, MA USA) in PBS suspension at 5uM for 20 min before neutralizing the dye with serum-containing media. CA-MSCs were grown in coculture with OVCAR3, OVSAHO, or PT412 tumor cells in a 1:1 ratio. A 1:1 ratio was chosen as the more proliferative tumor cells outgrow CA-MSCs, quickly altering the ratio to 1:10 CA-MSC-to-tumor cell. Coculture experiments utilized at least three independently-derived CA-MSC lines per each tumor cell line.
For direct adherent cocultures, 5×105 tumor cells and 5×105 CA-MSCs were mixed in supplemented MEBM media in a T175 cm flask. For direct non-adherent cocultures, 5×105 tumor cells and 5×105 CA-MSCs were combined and mixed with serum-free MEBM in a T75 cm ULA flask (Corning, Corning, NY). After 24-h, adherent cocultures were trypsinized while non-adherent cocultures were pelleted by centrifugation (600 × g, 5 min) and cellular complexes trypsin dissociated for 10 min. Dissociation of cellular complexes and generation of a single cell suspension was confirmed via brightfield microscopy. Cocultures were then neutralized, pelleted (600 × g, 5 min) and resuspended in flow buffer (2% FBS PBS) for flow cytometry analysis with propidium iodide (PI) for live/dead exclusion (all co-cultures resulted in over 90% viability in both CA-MSCs and tumor cells).
For indirect cocultures, 5×104 CA-MSCs were plated on top of polystyrene 0.4um pore Transwell inserts, 5×104 tumor cells were plated in the bottom wells, and each coculture was grown in MEBM +10% FBS. Tumor cells grown alone at equivalent cell densities served as controls. After 24-h, transwells were removed and tumor cells trypsinized and resuspended in flow buffer for flow cytometry analysis PI for live/dead exclusion (all co-cultures resulted in over 90% viability in both CA-MSCs and tumor cells).
For hypoxic cocultures, direct CA-MSC:tumor cell cocultures were incubated for 24-h in a hypoxic incubator workstation (Oxford Optronix, Cambridge, UK) set to 5% CO2 and either 1% O2 for hypoxic experiments or 21% O2 for normoxic controls. Cell culture reagents and media were equilibrated in the hypoxic chamber for 1 h prior to use. After 24-h, cocultures were trypsinized and neutralized in the workstation prior to removal for pelleting, resuspension in flow buffer and flow cytometry analysis DAPI for live/dead exclusion (all co-cultures resulted in over 90% viability in both CA-MSCs and tumor cells).
Quantification of In vitro mitochondrial transfer via flow cytometry
Resuspended cocultures were analyzed and CA-MSC-to-TC mitochondrial transfer quantified using either a Fortessa LSRII (BD Biosciences, Franklin Lakes, NJ) or CytoFLEX S 4L (Beckman Coulter, Pasadena, CA) cytometer. Mitochondrial transfer was quantified as the percentage of CellTrace Violet−, MT-DR+, GFP+ tumor cells amongst all CellTrace Violet−, MT-DR+ tumor cells. Analysis of flow cytometry data was carried out using FlowJo v1.10 (BD Biosciences, Franklin Lakes, NJ). Gating to identify GFP+ tumor cells was done using non-cocultured CellTrace Violet−, MT-DR+ tumor cells.
Confocal imaging
To image isolated tumor cells that had received CA-MSC mitochondria, non-adherent cocultures were set up as described above and transfer-positive and -negative tumor cell populations isolated using FACS at 24-h post coculture. Isolated tumor cells were then seeded at 5,000 cells/plate in 35mm MatTek Poly-D-Lysine glass bottom dishes (No. 1.5 coverglass) and allowed to adhere overnight. For cocultures, 2,000 total cells were plated (1:1 ratio CA-MSC:TC) and allowed to adhere overnight. Cells were then fixed with 2% PFA, 2.5% glutaraldehyde for 20 min at room temperature, followed by 3x washes with PBS and Hoechst staining (1 μg/mL, 5 min), an additional PBS wash and wet mounting with ibidi Mounting Medium (ibidi USA, Inc, Fitchburg, WI). Imaging of fixed cells was carried out using a Nikon AX confocal with a 63× objective (NA 1.42) and a Z-step size of 0.2μm. Image acquisition and processing was done using NIS-Elements AR (v.5.41.02) (Nikon Instruments Inc., Melville, NY).
Proliferation and chemotherapy resistance assays
OVCAR3, OVSAHO or PT412 tumor cells were sorted based on endogenous mitochondrial bulk or were sorted out of coculture with CA-MSCs before use in the assays. Bulk tumor cells that were grown independently and sorted were used as controls. 5 × 103 tumor cells were plated per well in a 96 well dish, 3–5 wells per condition, in DMEM complete media. Cells were incubated overnight to recover, and the following morning cisplatin was spiked into the media to a final concentration of 0, 0.5, 1, or 1.5uM, marking time t = 0. Cells were incubated for an additional 24, 48, or 72 h. For the experiments where tumor cells were sorted out of coculture with CA-MSCs, IncuCyte imaging was used to assess the change in cell density confluence as a proxy for cell viability. For the experiments where tumor cells were sorted based on endogenous mitochondrial bulk, cell viability was counted manually with a hemocytometer and 2:1 Trypan blue exclusion.
Seahorse extracellular flux assay
Tumor cells with top vs. bottom 20% endogenous mitochondria (based on MT-DR staining) were FACS isolated and seeded at a density of 4,000 cells per well overnight. The next day, three measurements of OCR and ECAR were taken at baseline and after each injection of the following mitochondrial stress test compounds in accordance with the Cell Mito Stress Test Protocol using the Seahorse XPf platform (Agilent Technologies, Santa Clara, CA): oligomycin (1μM; complex V inhibitor); FCCP (0.75μM; proton gradient uncoupler); antimycin A (1μM; complex III inhibitor). Basal and maximal respiration were normalized by subtracting non-mitochondrial OCR (i.e., after Antimycin A addition). Respiratory reserve capacity was calculated as the difference between maximal and basal OCR. ATP-linked OCR was derived as the difference between basal and Oligomycin A inhibited OCR. Crystal violet staining was used to normalize data. Analysis of Seahorse data was done using Seahorse Wave Desktop (v2.6.3.5) (Agilent Technologies, Santa Clara, CA).
Transcriptomic sequencing and analysis of transfer +/− tumor cell pairs
Paired PT412 cells that had either received or did not receive CA-MSC mitochondria were FACS isolated from cocultures as described above and total RNA isolated using a Nucleospin RNA isolation kit (Takara Bio USA, San Jose, CA). RNA concentration and quality was assessed via a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA). Samples were then packaged and shipped to Novogene Corporation for further processing and sequencing. mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. After fragmentation, first-strand cDNA synthesis was carried out using random hexamer primers followed by second strand synthesis using dUTP. Qubit (Thermo Fisher Scientific, Waltham, MA) and real-time PCR was used for quantification and bioanalyzer for size distribution detection. Quantified libraries were pooled and sequenced on an Illumina platform (Illumina Inc., San Diego, CA) and paired-end reads were generated.
Raw reads in fastq format were processed through a Novogene in-house perl script for QC. Briefly, reads containing adapters, ploy-N, and low-quality reads were removed and Q20, Q30, and GC content was calculated. The index of the reference genome hg38 was built and paired-end clean reads were aligned to the reference genome using hisat2 (v2.0.5). Quantification of the gene expression level was done via featureCounts (v1.5.0-p3). The FPKM of each gene was calculated based on the length of the gene and read counts mapped to the gene. Differential gene expression analysis was carried out using R (v4.1.3) package DESeq2 (v1.20.0) treating transfer +/− PT412 cells derived from the same coculture as paired samples.72 Specifically, differentially expressed genes were identified based on a p-value less than 0.05, and an absolute log-transformed fold change greater than 1.5. Followed by functional enrichment analysis using R package clusterProfiler,73 with FDR cutoff at 0.05 for KEGG pathway, Gene Ontology, and Gene Set Enrichment Analysis (GSEA).
Quantitative real-time PCR
RNA was isolated using a Nucleospin RNA isolation kit (Takara Bio USA, San Jose, CA) and RNA concentration and quality assessed via a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA). cDNA was synthesized with the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen, Grand Island, NY) using random hexamers per the manufacturer’s protocol. SYBR green-based RT-PCR was performed using the CFX96 or CFX384 Real-Time qPCR Detection System (Bio-Rad, Hercules, CA) and respective primers. The comparative Ct method was used for data analysis with GAPDH as the comparator gene.
Immunoblotting
Cell pellets were homogenized in RIPA buffer (Thermo Fisher Scientific, Waltham, MA) with complete protease inhibitor (Roche, Basel, Switzerland). Insoluble material was removed by centrifugation at 16,000 g at 4°C for 15 min. Protein concentrations were determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA). Equal amounts of protein were separated on 4–12% NuPAGE SDS gel (Invitrogen, Grand Island, NY) and transferred onto a nitrocellulose membrane. α-ANGPTL3, α-pMAPK, α-total MAPK, α-pAKT, α-pan AKT and α-MIRO1 were used at a 1:1000 dilution. α-COXIV and α-β-actin antibodies were used at a 1:2000 dilution. Bands were visualized using an LI-COR near-infrared detection system with a 1:10,000 dilution for secondary antibodies (LI-COR Biosciences, Lincoln, NE).
ANGPTL3 knockdown generation and validation
ANGPTL3 KD OVCAR3 and PT412 tumor cell lines were generated using prepackaged ANGPTL3 Human shRNA Lentiviral Particles (Origene, Rockville, MD). Briefly, 50,000 tumor cells were plated in one well of a 6-well plate (one for each shRNA clone A-D, plus scrambled control) and allowed to adhere overnight. Cells were then transduced using an MOI of 10 for 48 h, followed by removal of the lentivirus and selection with 0.8 μg/mL puromycin for a further 48 h. Knockdowns were validated via qPCR and western blot, and the impact of ANGPTL3 KD on cell proliferation measured. Two shRNA clones (clone A and D) were selected based on their knockdown efficiency and minimal impact to cell proliferation.
Recombinant ANGPTL3/Evinacumab/trametinib treatment
To measure the impact of ANGPTL3 on proliferation, 5,000 cells/well of PT412 and OVCAR3 cells were plated in a 48-well plate and treated with either vehicle control (DMSO) or 0.1, 1, or 10nM of recombinant ANGPTL3 protein (Abcam, Boston, MA) reconstituted per manufacturer’s instructions. Cells were initially plated in DMEM or RPMI containing recombinant protein and media changed daily with fresh protein added. Cells were counted via hemocytometer on days 3 and 5 post plating. To test the inhibition of ANGPTL3 in coculture, the ANGPTL3 blocking antibody Evinacumab (Thermo Fisher Scientific, Waltham, MA) was added to media at a concentration of 1.5 ng/μL. Media was then changed daily with fresh media containing Evinacumab, and proliferation measured across five days (post plating). Trametinib treatment was similarly carried out daily at a concentration of 100nM and cell proliferation measured using a hemocytometer with trypan blue live/dead exclusion.
ANGPTL3 ELISA
ANGPTL3 levels in patient-derived ascites were measured using a commercially available kit, Human ANGPTL3 ELISA Kit (Abcam, Boston, MA). Ascites or intraperitoneal washes from patients with benign indications were centrifuged for 10 min at 1,200 × g to separate out cellular and acellular fractions. The acellular fraction was then assayed per the manufacturer’s protocol.
MIRO1 knockdown CA-MSC lines
shRNA constructs targeting MIRO1/RHOT1 (HSH070029-LVRU6MP, GeneCopoeia, Rockville, MD) were packaged into lentiviral particles using a 3rd Generation Lentiviral Packaging Kit (Applied Biological Materials Inc., Richmond, BC). MIRO1KD patient-derived CA-MSC lines were then generated and validated for MIRO1 knockdown via qPCR and immunoblotting with anti-RHOT1 antibody (Abcam, Boston, MA, USA). The impact of MIRO1KD on CA-MSC viability was determined by comparing proliferative capacity to the wild-type CA-MSC line from which knockdowns were derived; no effect on viability was observed. Additionally, MIRO1KD CA-MSCs expression of MSC surface markers and tri-lineage differentiation potential was confirmed, demonstrating MIRO1KD did not induce or impact CA-MSC differentiation.
mtDNA haplotype-specific qPCR
Mitochondrial DNA (mtDNA) haplotypes were obtained through sequencing of mtDNA from a primary patient-derived CA-MSC line and the OVCAR3 tumor cell line via Sanger sequencing of two mtDNA hypervariable regions.60 Briefly, mitochondria was isolated and DNA was extracted using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) and Sanger sequencing of samples was conducted by the University of Pittsburgh’s Genomics Research Core using previously described primers designed to amplify and sequence the mtDNA hypervariable region.78 CA-MSC and OVCAR3 hypervariable sequences were processed and aligned using SnapGene (Dotmatics, Bishop’s Stortford, UK). Haplotype-specific primer candidates were designed using SnapGene and PrimerQuest (Integrated DNA Technologies, Coralville, IA) to optimize sensitive and specific identification and differentiation between CA-MSC and OVCAR3 derived mtDNA. Primers were validated for specificity using SYBR green- (Applied Biosystems, Waltham, MA) based real time qPCR using the CFX384 Real-Time PCR Detection System (Bio-Rad, Hercules, CA). qPCR standard curves were generated to quantify the amount of CA-MSC and endogenous OVCAR3 mtDNA. All mtDNA haplotype-specific qPCR was normalized based on cell number and into DNA content. To assess retention of CA-MSC mitochondria in tumor cells, GFP-COX8 CA-MSC and OVCAR3 tumor cells were co-cultured for 24 h under non-adherent conditions in a 1:1 cellular ratio. OVCAR3 tumor cells which received CA-MSC mitochondria were isolated using FACS and then plated and grown over 14 days. At each time point (days 1, 3, 5,7 and 14), a subset of 20,000 tumor cells were removed for DNA isolation and used for haplotype-specific qPCR. Results reported in Figure 7B are reported as nanogram (ng) CA-MSC derived mtDNA and ng tumor cell derived mtDNA from 20,000 cells.
Ovarian cancer orthotopic murine model
For the cohort assessing survival and metastatic burden, 5×105 OVCAR3 cells expressing luciferase were injected alone (n = 3), with control CA-MSCs (n = 5) or MIRO1 knockdown CA-MSCs (n = 5) in a 1:1 ratio into the ovarian bursa of NSG mice.11,25 Tumor take and disease progression was monitored via bi-weekly IVIS imaging. On day 7 post injection, mice were treated with cisplatin (0.5 mg/kg every other day × 3 doses). Mice were sacrificed upon meeting endpoint criteria (>10% weight loss, >20% weight gain or poor body condition). For experiments assessing clonal heterogeneity, 5×105 OVCAR3 cells with a unique genomic barcode (Cellecta, Mountain View, CA) expressing luciferase were injected alone (n = 2), with control CA-MSCs (n = 5) or MIRO1 knockdown CA-MSCs (n = 5) ina 1:1 ratio into the ovarian bursa of NSG mice. Bi-weekly IVIS imaging was also used to monitor tumor take and disease progression. All mice were sacrificed when the first mouse met endpoint criteria (day 21). Metastatic burden was assessed during necropsy and both primary and metastatic tumors reisolated. Tumors were then dissociated using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) and isolated DNA used for sequencing of genomic barcodes.
DNA barcode analysis
Sankey diagrams, heat maps, Venn diagrams, and distribution plots were produced in Python version 3.7.3. Barcode data processing for the barcoded tail vein injection model and barcoded in vivo MIRO1 knockdown model was performed in Python version v3.7.3 with pandas v1.2.4 and numpy v1.20.2.
Sankey diagrams
All Sankey diagrams were produced with seaborn v0.11.2 and plotly v5.7.0. In the tail vein injection model, we wanted to understand which barcodes had proliferative activity in each tumor. Therefore, we considered a barcode with reads to be any barcode with >10 reads, indicating that at least ten cells originated from this barcode. Barcoded cells that gave rise to fewer than ten cells have minimal impact on the tumor’s heterogeneity, and the abundance of barcodes with fewer than ten reads obscure the ability to visualize the proportion of barcodes that have a greater contribution to the tumor’s heterogeneity across tumor sites. The total number of reads is the sum of all reads from all barcodes with >10 reads.
The in vivo MIRO1 knockdown model Sankey diagram graphs the number of reads for each location aggregated together, after normalization to control for differences in size between tumors. This experiment used tumors from four mice, each of which had different size tumors at each site. To normalize the number of reads such that we can compare the number of reads from a smaller tumor to that of a larger tumor, the cell counts were divided by each tumor’s mg DNA. After normalizing each sample, the normalized reads were summed by condition, CA-MSC and MIRO1, as well as by site, primary and liver, to produce the total number of reads in each respective category.
Heat maps and distribution plots
All heatmap visualizations and distribution plots were produced with seaborn 0.11.2. The rank of each barcode was determined by ordering the barcodes from greatest number of progenies (1) to fewest number of progenies (n). The value of the upper rank n depends on the number of barcodes that had progenies at a particular site and condition. To account for the large distribution of counts across barcodes and visualize with a heatmap, the negative log of the cell counts associated with each barcode was calculated and plotted in rank order on the heatmap.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as mean ± SEM unless otherwise stated. All experiments preformed on at least three CA-MSCs lines or on multiple mice. Student’s unpaired t test or ANOVA were used to determine the statistical significance. Differences were considered significant at *p < 0.05, **p < 0.01).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENTor RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| α-CD105 | BD Biosciences | Cat#: 561443; RRID: AB_10714629 |
| α-CD90 | BD Biosciences | Cat#: 555596; RRID: AB_395970 |
| α-CD73 | BD Biosciences | Cat#: 560847; RRID: AB_10612019 |
| α-CD45 | BD Biosciences | Cat#: 340664; RRID: AB_400074 |
| α-CD36 | BD Biosciences | Cat#: 561536; RRID: AB_10893348 |
| α-ANGPTL3 | Abcam | Cat#: ab118208; RRID: AB_10933535 |
| α-ANGPTL3 (Evinacumab) | Invitrogen | Cat#: MA5-42049; RRID: AB_2911192 |
| α-p44/42 MAPK | Cell Signaling Technology | Cat#: 4370S; RRID: AB_2315112 |
| α-total MAPK | Cell Signaling Technology | Cat#: 9107S; RRID: AB_10695739 |
| α-pAkt1 | Abcam | Cat#: ab81283; RRID: AB_2224551 |
| α-pan Akt | Abcam | Cat#: ab8805; RRID: AB_306791 |
| α-MIRO1 | Abcam | Cat#: ab188029; RRID: AB_188029 |
| α-COXIV | Proteintech | Cat#: 11242-1-AP; RRID: AB_2085278 |
| α-beta actin | Abcam | Cat#: ab8227; RRID: AB_2305186 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| ANGPTL3 Human shRNA Lentiviral Particle (Locus ID 27329) | Origene | Cat#: TL314795V |
| Human RHOT1 shRNA clones | GeneCopoeia | Cat#: HSH070029-LVRU6MP |
| Mitochondria Cyto-Tracer™, pCT-Mito-GFP (CMV) | System Biosciences | Cat#: CYTO102-VA-1 |
| Lentiviral Barcode Library 13k × 13k 30M | Cellecta, Inc. | Cat#: BC13X13-30M-P |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Recombinant human ANGPTL3 protein | Abcam | Cat#: ab176028 |
| SYBR green master-Mix | Applied Biosystem | Cat#: 4472908 |
| RIPA buffer | Thermo Fisher Scientific | Cat#: 89900 |
| NuPAGE SDS gel | Invitrogen | Cat#: NW04122 |
| Nitrocellulose Membrane | Fisher Scientific | Cat#: PI77012 |
| Cisplatin | Fresenius-Kabi | Cat#: 4015721 |
| Celltrace Violet | Thermo Fisher Scientific | Cat#: C34557 |
| Mitotracker Deep Red | Thermo Fisher Scientific | Cat#: M22426 |
| Trametinib | Selleckchem | Cat#: S2673 |
| Seahorse XF Cell Mito Stress Test | Agilent Technologies | Cat#: 103010-100 |
|
| ||
| Critical commercial assays | ||
|
| ||
| DNeasy Blood & Tissue Kit | Qiagen | Cat#: 69504 |
| Mycoplasma Detection Kit, PCR | Applied Biologic Materials, Inc. | Cat#: G238 |
| Pierce BCA Protein Assay Kit | Fisher Scientific | Cat#: PI23227 |
| SuperScript III First Strand Synthesis System | Fisher Scientific | Cat#: 18080-051 |
| Human ANGPTL3 ELISA Kit | Abcam | Cat#: ab254510 |
| Nucleospin RNA Extraction Kit | Takada Bio | Cat#: 740955.50 |
|
| ||
| Deposited data | ||
|
| ||
| RNA sequencing | Mendeley | https://doi.org/10.17632/25s5258ynl1 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| OVCAR3 | ATCC | Cat#: HTB-161; RRID: CVCL_0465 |
| OVSAHO | Millipore Sigma | Cat#: SCC294; RRID: CVCL_3114 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| NSG mice | The Jackson Laboratory | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| SnapGene Software | Dao et al.71 | www.snapgene.com |
| Rv4.1.3/RStudio 4.3.2 | N/A | www.R-project.org |
| DESeq2 v1.20.0 | Love et al.72 | https://doi.org/10.18129/B9.bioc.DESeq2 |
| clusterProfiler | Yu et al.73 | https://doi.org/10.18129/B9.bioc.clusterProfiler |
| Python v3.7.3 | N/A | https://www.python.org/ |
| pandas v1.2.4 | N/A | https://pandas.pydata.org/ |
| numpy v1.20.2 | Harris et al.74 | https://numpy.org/ |
| seaborn v0.11.2 | Waskom,75 | https://seaborn.pydata.org/archive/0.11/index.html |
| plotly v5.7.0 | N/A | https://plot.ly |
| GraphPad Prism v10 | N/A | www.graphpad.com |
| FlowJo v10.10.0 | N/A | https://www.flowjo.com/ |
| Wave Software | N/A | https://www.agilent.com/en/product/cell-analysis/real-time-cell-metabolic-analysis/xf-software/seahorse-wave-desktop-software-740897 |
Highlights.
CA-MSCs donate mitochondria to ovarian cancer cells with low endogenous mitochondria
Mitochondrial donation enhances cancer cell proliferation, chemoresistance, and OXPHOS
Donated mito-to-nuclear signaling drives ANGPTL3 secretion, which increases tumor cell growth
Mito donation enhances metastasis and clonal heterogeneity in vivo
ACKNOWLEDGMENTS
We thank the University of Pittsburgh Biospecimen Core for access to patient samples, and Francesmary Modugno, MS, PhD, MPH, for use of the Gynecologic Oncology Biospecimen and Data Bank. L.G.C. is supported through the Department of Defense (DoD) Ovarian Cancer Research Program Pilot Award W81XWH-21-1-0371. N.H. is supported by NIH R01CA242021 and DoD Congressionally Directed Medical Research Program – Ovarian Cancer TEAL Award W81XWH2210252.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114551.
REFERENCES
- 1.Siegel RL, Miller KD, and Jemal A (Jan-Feb 2016). Cancer statistics, 2016. CA. Cancer J. Clin. 66, 7–30. 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Dagogo-Jack I, and Shaw AT (2018). Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 15, 81–94. 10.1038/nrclinonc.2017.166. [DOI] [PubMed] [Google Scholar]
- 3.Roberts CM, Cardenas C, and Tedja R (2019). The Role of IntraTumoral Heterogeneity and Its Clinical Relevance in Epithelial Ovarian Cancer Recurrence and Metastasis. Cancers 11, 1083. 10.3390/cancers11081083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schuh A, Becq J, Humphray S, Alexa A, Burns A, Clifford R, Feller SM, Grocock R, Henderson S, Khrebtukova I, et al. (2012). Monitoring chronic lymphocytic leukemia progression by whole genome sequencing reveals heterogeneous clonal evolution patterns. Blood 120, 4191–4196. 10.1182/blood-2012-05-433540. [DOI] [PubMed] [Google Scholar]
- 5.Schwarz RF, Ng CK, Cooke SL, Newman S, Temple J, Piskorz AM, Gale D, Sayal K, Murtaza M, Baldwin PJ, et al. (2015). Spatial and temporal heterogeneity in high-grade serous ovarian cancer: a phylogenetic analysis. PLoS Med. 12, e1001789. 10.1371/journal.pmed.1001789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.El-Sayes N, Vito A, and Mossman K (2021). Tumor Heterogeneity: A Great Barrier in the Age of Cancer Immunotherapy. Cancers 13, 806. 10.3390/cancers13040806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lambrechts S, Smeets D, Moisse M, Braicu EI, Vanderstichele A, Zhao H, Van Nieuwenhuysen E, Berns E, Sehouli J, Zeillinger R, et al. (2016). Genetic heterogeneity after first-line chemotherapy in high-grade serous ovarian cancer. Eur. J. Cancer 53, 51–64. 10.1016/j.ejca.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Atiya HI, Frisbie L, Goldfeld E, Orellana T, Donnellan N, Modugno F, Calderon M, Watkins S, Zhang R, Elishaev E, et al. (2022). Endometriosis-Associated Mesenchymal Stem Cells Support Ovarian Clear Cell Carcinoma through Iron Regulation. Cancer Res. 82, 4680–4693. 10.1158/0008-5472.CAN-22-1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coffman LG, Choi YJ, McLean K, Allen BL, di Magliano MP, and Buckanovich RJ (2016). Human carcinoma-associated mesenchymal stem cells promote ovarian cancer chemotherapy resistance via a BMP4/HH signaling loop. Oncotarget 7, 6916–6932. 10.18632/oncotarget.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coffman LG, Pearson AT, Frisbie LG, Freeman Z, Christie E, Bowtell DD, and Buckanovich RJ (2019). Ovarian Carcinoma-Associated Mesenchymal Stem Cells Arise from Tissue-Specific Normal Stroma. Stem Cell. 37, 257–269. 10.1002/stem.2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fan H, Atiya HI, Wang Y, Pisanic TR, Wang TH, Shih IM, Foy KK, Frisbie L, Buckanovich RJ, Chomiak AA, et al. (2020). Epigenomic Reprogramming toward Mesenchymal-Epithelial Transition in Ovarian-Cancer-Associated Mesenchymal Stem Cells Drives Metastasis. Cell Rep. 33, 108473. 10.1016/j.celrep.2020.108473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wei B, Ji M, Lin Y, Wang S, Liu Y, Geng R, Hu X, Xu L, Li Z, Zhang W, and Lu J (2023). Mitochondrial transfer from bone mesenchymal stem cells protects against tendinopathy both in vitro and in vivo. Stem Cell Res. Ther. 14, 104. 10.1186/s13287-023-03329-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang D, Gao F, Zhang Y, Wong DSH, Li Q, Tse HF, Xu G, Yu Z, and Lian Q (2016). Mitochondrial transfer of mesenchymal stem cells effectively protects corneal epithelial cells from mitochondrial damage. Cell Death Dis. 7, e2467. 10.1038/cddis.2016.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, Rowlands DJ, Quadri SK, Bhattacharya S, and Bhattacharya J (2012). Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 18, 759–765. 10.1038/nm.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Velarde F, Ezquerra S, Delbruyere X, Caicedo A, Hidalgo Y, and Khoury M (2022). Mesenchymal stem cell-mediated transfer of mitochondria: mechanisms and functional impact. Cell. Mol. Life Sci. 79, 177. 10.1007/s00018-022-04207-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dong LF, Rohlena J, Zobalova R, Nahacka Z, Rodriguez AM, Berridge MV, and Neuzil J (2023). Mitochondria on the move: Horizontal mitochondrial transfer in disease and health. J. Cell Biol. 222, e202211044. 10.1083/jcb.202211044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moschoi R, Imbert V, Nebout M, Chiche J, Mary D, Prebet T, Saland E, Castellano R, Pouyet L, Collette Y, et al. (2016). Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 128, 253–264. 10.1182/blood-2015-07-655860. [DOI] [PubMed] [Google Scholar]
- 18.Kidwell CU, Casalini JR, Pradeep S, Scherer SD, Greiner D, Bayik D, Watson DC, Olson GS, Lathia JD, Johnson JS, et al. (2023). Transferred mitochondria accumulate reactive oxygen species, promoting proliferation. Elife 12, e85494. 10.7554/eLife.85494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang W, Zhou H, Li H, Mou H, Yinwang E, Xue Y, Wang S, Zhang Y, Wang Z, Chen T, et al. (2023). Cancer cells reprogram to metastatic state through the acquisition of platelet mitochondria. Cell Rep. 42, 113147. 10.1016/j.celrep.2023.113147. [DOI] [PubMed] [Google Scholar]
- 20.Saha T, Dash C, Jayabalan R, Khiste S, Kulkarni A, Kurmi K, Mondal J, Majumder PK, Bardia A, Jang HL, and Sengupta S (2022). Intercellular nanotubes mediate mitochondrial trafficking between cancer and immune cells. Nat. Nanotechnol. 17, 98–106. 10.1038/s41565-021-01000-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schoutrop E, Moyano-Galceran L, Lheureux S, Mattsson J, Lehti K, Dahlstrand H, and Magalhaes I (2022). Molecular, cellular and systemic aspects of epithelial ovarian cancer and its tumor microenvironment. Semin. Cancer Biol. 86, 207–223. 10.1016/j.semcancer.2022.03.027. [DOI] [PubMed] [Google Scholar]
- 22.Ford CE, Werner B, Hacker NF, and Warton K (2020). The untapped potential of ascites in ovarian cancer research and treatment. Br. J. Cancer 123, 9–16. 10.1038/s41416-020-0875-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szczurek E, Kruger T, Klink B, and Beerenwinkel N (2020). A mathematical model of the metastatic bottleneck predicts patient outcome and response to cancer treatment. PLoS Comput. Biol. 16, e1008056. 10.1371/journal.pcbi.1008056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cascio S, Chandler C, Zhang L, Sinno S, Gao B, Onkar S, Bruno TC, Vignali DAA, Mahdi H, Osmanbeyoglu HU, et al. (2021). Cancer-associated MSC drive tumor immune exclusion and resistance to immunotherapy, which can be overcome by Hedgehog inhibition. Sci. Adv. 7, eabi5790. 10.1126/sciadv.abi5790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Atiya HI, Orellana TJ, Wield A, Frisbie L, and Coffman LG (2021). An Orthotopic Mouse Model of Ovarian Cancer using Human Stroma to Promote Metastasis. J. Vis. Exp, 169. 10.3791/62382. [DOI] [PubMed] [Google Scholar]
- 26.Coffman LG, Burgos-Ojeda D, Wu R, Cho K, Bai S, and Buckanovich RJ (2016). New models of hematogenous ovarian cancer metastasis demonstrate preferential spread to the ovary and a requirement for the ovary for abdominal dissemination. Transl. Res. 175, 92–102.e2. 10.1016/j.trsl.2016.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dong LF, Kovarova J, Bajzikova M, Bezawork-Geleta A, Svec D, Endaya B, Sachaphibulkij K, Coelho AR, Sebkova N, Ruzickova A, et al. (2017). Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. Elife 6, e22187. 10.7554/eLife.22187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Acquistapace A, Bru T, Lesault PF, Figeac F, Coudert AE, le Coz O, Christov C, Baudin X, Auber F, Yiou R, et al. (2011). Human mesenchymal stem cells reprogram adult cardiomyocytes toward a progenitor-like state through partial cell fusion and mitochondria transfer. Stem Cell. 29, 812–824. 10.1002/stem.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang J, Liu L, Oda Y, Wada K, Ago M, Matsuda S, Hattori M, Goto T, Ishibashi S, Kawashima-Sonoyama Y, et al. (2023). Extracellular Vesicles and Cx43-Gap Junction Channels Are the Main Routes for Mitochondrial Transfer from Ultra-Purified Mesenchymal Stem Cells, RECs. Int. J. Mol. Sci. 24, 10294. 10.3390/ijms241210294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Wang C, He T, Zhao T, Chen YY, Shen YL, Zhang X, and Wang LL (2019). Mitochondrial Transfer from Bone Marrow Mesenchymal Stem Cells to Motor Neurons in Spinal Cord Injury Rats via Gap Junction. Theranostics 9, 2017–2035. 10.7150/thno.29400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hyler AR, Baudoin NC, Brown MS, Stremler MA, Cimini D, Davalos RV, and Schmelz EM (2018). Fluid shear stress impacts ovarian cancer cell viability, subcellular organization, and promotes genomic instability. PLoS One 13, e0194170. 10.1371/journal.pone.0194170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asem M, Young A, Oyama C, ClaureDeLaZerda A, Liu Y, Ravosa MJ, Gupta V, Jewell A, Khabele D, and Stack MS (2020). Ascites-induced compression alters the peritoneal microenvironment and promotes metastatic success in ovarian cancer. Sci. Rep. 10, 11913. 10.1038/s41598-020-68639-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bar-Yaacov D, Blumberg A, and Mishmar D (Sep-Oct 2012). Mitochondrial-nuclear co-evolution and its effects on OXPHOS activity and regulation. Biochim. Biophys. Acta 1819, 1107–1111. 10.1016/j.bbagrm.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 34.Camenisch G, Pisabarro MT, Sherman D, Kowalski J, Nagel M, Hass P, Xie MH, Gurney A, Bodary S, Liang XH, et al. (2002). ANGPTL3 stimulates endothelial cell adhesion and migration via integrin alpha vbeta 3 and induces blood vessel formation in vivo. J. Biol. Chem. 277, 17281–17290. 10.1074/jbc.M109768200. [DOI] [PubMed] [Google Scholar]
- 35.Koyama T, Ogawara K, Kasamatsu A, Okamoto A, Kasama H, Minakawa Y, Shimada K, Yokoe H, Shiiba M, Tanzawa H, and Uzawa K (2015). ANGPTL3 is a novel biomarker as it activates ERK/MAPK pathway in oral cancer. Cancer Med. 4, 759–769. 10.1002/cam4.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhong L, Tang L, and He X (2022). Angiopoietin-like 3 (ANGPTL3) drives cell proliferation, migration and angiogenesis in cervical cancer via binding to integrin alpha v beta 3. Bioengineered 13, 2971–2980. 10.1080/21655979.2021.2024951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nguyen TT, Oh SS, Weaver D, Lewandowska A, Maxfield D, Schuler MH, Smith NK, Macfarlane J, Saunders G, Palmer CA, et al. (2014). Loss of Miro1-directed mitochondrial movement results in a novel murine model for neuron disease. Proc. Natl. Acad. Sci. USA 111, E3631–E3640. 10.1073/pnas.1402449111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lopez-Domenech G, Covill-Cooke C, Ivankovic D, Halff EF, Sheehan DF, Norkett R, and Kittler JT (2018). Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. EMBO J. 37, 321–336. 10.15252/embj.201696380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pavlova NN, Zhu J, and Thompson CB (2022). The hallmarks of cancer metabolism: Still emerging. Cell Metab. 34, 355–377. 10.1016/j.cmet.2022.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Warburg O (1956). On the origin of cancer cells. Science 123, 309–314. 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 41.Kim M, Mahmood M, Reznik E, and Gammage PA (2022). Mitochondrial DNA is a major source of driver mutations in cancer. Trends Cancer 8, 1046–1059. 10.1016/j.trecan.2022.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ, Hall J, Lim S, Issa MM, Flanders WD, Hosseini SH, et al. (2005). mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. USA 102, 719–724. 10.1073/pnas.0408894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spees JL, Olson SD, Whitney MJ, and Prockop DJ (2006). Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 103, 1283–1288. 10.1073/pnas.0510511103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watson DC, Bayik D, Storevik S, Moreino SS, Sprowls SA, Han J, Augustsson MT, Lauko A, Sravya P, Røsland GV, et al. (2023). GAP43-dependent mitochondria transfer from astrocytes enhances glioblastoma tumorigenicity. Nat. Cancer 4, 648–664. 10.1038/s43018-023-00556-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Al Amir Dache Z, and Thierry AR (2023). Mitochondria-derived cell-to-cell communication. Cell Rep. 42, 112728. 10.1016/j.celrep.2023.112728. [DOI] [PubMed] [Google Scholar]
- 46.Liu Z, Sun Y, Qi Z, Cao L, and Ding S (2022/05/19 2022). Mitochondrial transfer/transplantation: an emerging therapeutic approach for multiple diseases. Cell Biosci. 12, 66. 10.1186/s13578-022-00805-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dawson ER, Patananan AN, Sercel AJ, and Teitell MA (2020/08/31 2020). Stable retention of chloramphenicol-resistant mtDNA to rescue metabolically impaired cells. Sci. Rep. 10, 14328. 10.1038/s41598-020-71199-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pan J, Wang L, Lu C, Zhu Y, Min Z, Dong X, and Sha H (2019). Matching Mitochondrial DNA Haplotypes for Circumventing Tissue-Specific Segregation Bias. iScience 13, 371–379. 10.1016/j.isci.2019.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cruz-Gregorio A, Aranda-Rivera AK, Amador-Martinez I, and Maycotte P (2023). Mitochondrial transplantation strategies in multifaceted induction of cancer cell death. Life Sci. 332, 122098. 10.1016/j.lfs.2023.122098. [DOI] [PubMed] [Google Scholar]
- 50.Pasquier J, Guerrouahen BS, Al Thawadi H, Ghiabi P, Maleki M, Abu-Kaoud N, Jacob A, Mirshahi M, Galas L, Rafii S, et al. (2013). Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J. Transl. Med. 11, 94. 10.1186/1479-5876-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burt R, Dey A, Aref S, Aguiar M, Akarca A, Bailey K, Day W, Hooper S, Kirkwood A, Kirschner K, et al. (2019). Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood 134, 1415–1429. 10.1182/blood.2019001398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang J, Liu X, Qiu Y, Shi Y, Cai J, Wang B, Wei X, Ke Q, Sui X, Wang Y, et al. (2018). Cell adhesion-mediated mitochondria transfer contributes to mesenchymal stem cell-induced chemoresistance on T cell acute lymphoblastic leukemia cells. J. Hematol. Oncol. 11, 11. 10.1186/s13045-018-0554-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zampieri LX, Grasso D, Bouzin C, Brusa D, Rossignol R, and Sonveaux P (2020). Mitochondria Participate in Chemoresistance to Cisplatin in Human Ovarian Cancer Cells. Mol. Cancer Res. 18, 1379–1391. 10.1158/1541-7786.Mcr-19-1145. [DOI] [PubMed] [Google Scholar]
- 54.Shen L, Zhou L, Xia M, Lin N, Ma J, Dong D, and Sun L (2021). PGC1α regulates mitochondrial oxidative phosphorylation involved in cisplatin resistance in ovarian cancer cells via nucleo-mitochondrial transcriptional feedback. Exp. Cell Res. 398, 112369. 10.1016/j.yexcr.2020.112369. [DOI] [PubMed] [Google Scholar]
- 55.Jin P, Jiang J, Zhou L, Huang Z, Nice EC, Huang C, and Fu L (2022). Mitochondrial adaptation in cancer drug resistance: prevalence, mechanisms, and management. J. Hematol. Oncol. 15, 97. 10.1186/s13045-022-01313-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bakhoum SF, and Cantley LC (2018). The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 174, 1347–1360. 10.1016/j.cell.2018.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Morden CR, Farrell AC, Sliwowski M, Lichtensztejn Z, Altman AD, Nachtigal MW, and McManus KJ (2021). Chromosome instability is prevalent and dynamic in high-grade serous ovarian cancer patient samples. Gynecol. Oncol. 161, 769–778. 10.1016/j.ygyno.2021.02.038. [DOI] [PubMed] [Google Scholar]
- 58.Gillies RJ, Brown JS, Anderson ARA, and Gatenby RA (2018). Eco-evolutionary causes and consequences of temporal changes in intratumoural blood flow. Nat. Rev. Cancer 18, 576–585. 10.1038/s41568-018-0030-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hoefflin R, Lahrmann B, Warsow G, Hübschmann D, Spath C, Walter B, Chen X, Hofer L, Macher-Goeppinger S, Tolstov Y, et al. (2016). Spatial niche formation but not malignant progression is a driving force for intratumoural heterogeneity. Nat. Commun. 7, ncomms11845. 10.1038/ncomms11845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gatenby RA, and Gillies RJ (2008). A microenvironmental model of carcinogenesis. Nat. Rev. Cancer 8, 56–61. 10.1038/nrc2255. [DOI] [PubMed] [Google Scholar]
- 61.Xu Z, Zhou K, Wang Z, Liu Y, Wang X, Gao T, Xie F, Yuan Q, Gu X, Liu S, and Xing J (2023). Metastatic pattern of ovarian cancer delineated by tracing the evolution of mitochondrial DNA mutations. Exp. Mol. Med. 55, 1388–1398. 10.1038/s12276-023-01011-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ni J, Wang Y, Cheng X, Teng F, Wang C, Han S, Chen X, and Guo W (2020). Pathogenic Heteroplasmic Somatic Mitochondrial DNA Mutation Confers Platinum-Resistance and Recurrence of High-Grade Serous Ovarian Cancer. Cancer Manag. Res. 12, 11085–11093. 10.2147/CMAR.S277724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Filograna R, Koolmeister C, Upadhyay M, Pajak A, Clemente P, Wibom R, Simard ML, Wredenberg A, Freyer C, Stewart JB, and Larsson NG (2019). Modulation of mtDNA copy number ameliorates the pathological consequences of a heteroplasmic mtDNA mutation in the mouse. Sci. Adv. 5, eaav9824. 10.1126/sciadv.aav9824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Y, Liu VW, Xue WC, Cheung AN, and Ngan HY (2006). Association of decreased mitochondrial DNA content with ovarian cancer progression. Br. J. Cancer 95, 1087–1091. 10.1038/sj.bjc.6603377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brinker AE, Vivian CJ, Beadnell TC, Koestler DC, Teoh ST, Lunt SY, and Welch DR (2020). Mitochondrial Haplotype of the Host Stromal Microenvironment Alters Metastasis in a Non-cell Autonomous Manner. Cancer Res. 80, 1118–1129. 10.1158/0008-5472.CAN-19-2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Takenaga K, Koshikawa N, and Nagase H (2021). Intercellular transfer of mitochondrial DNA carrying metastasis-enhancing pathogenic mutations from high- to low-metastatic tumor cells and stromal cells via extracellular vesicles. BMC Mol. Cell Biol. 22, 52. 10.1186/s12860-021-00391-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu Y, Zheng Y, and Jin Z (2023/08/01/ 2023). ANGPTL3 affects the metastatic potential and the susceptibility of ovarian cancer cells to natural killer cell-mediated cytotoxicity. Heliyon 9, e18799. 10.1016/j.heliyon.2023.e18799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Siamakpour-Reihani S, Owzar K, Jiang C, Turner T, Deng Y, Bean SM, Horton JK, Berchuck A, Marks JR, Dewhirst MW, and Alvarez Secord A (2015). Prognostic significance of differential expression of angiogenic genes in women with high-grade serous ovarian carcinoma. Gynecol. Oncol. 139, 23–29. 10.1016/j.ygyno.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cao C, and Yang X (Jan-Dec 2020). The Prevalence, Associated Factors for Lung Metastases Development and Prognosis in Ovarian Serous Cancer Based on SEER Database. Technol. Cancer Res. Treat. 19, 1533033820983801. 10.1177/1533033820983801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Deng K, Yang C, Tan Q, Song W, Lu M, Zhao W, Lou G, Li Z, Li K, and Hou Y (2018). Sites of distant metastases and overall survival in ovarian cancer: A study of 1481 patients. Gynecol. Oncol. 150, 460–465. 10.1016/j.ygyno.2018.06.022. [DOI] [PubMed] [Google Scholar]
- 71.Dao VL, Chan S, Zhang J, Ngo RKJ, and Poh CL (2022/03/07 2022). Single 3′-exonuclease-based multifragment DNA assembly method (SENAX). Sci. Rep. 12, 4004. 10.1038/s41598-022-07878-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Love MI, Huber W, and Anders S (2014/12/05 2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Harris CR, Millman KJ, van der Walt SJ, Gommers R, Virtanen P, Cournapeau D, Wieser E, Taylor J, Berg S, Smith NJ, et al. (2020/09/01 2020). Array programming with NumPy. Nature 585, 357–362. 10.1038/s41586-020-2649-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Waskom M (2021). seaborn: statistical data visualization. J. Open Source Softw. 6, 3021. 10.21105/joss.03021. [DOI] [Google Scholar]
- 76.McLean K, Gong Y, Choi Y, Deng N, Yang K, Bai S, Cabrera L, Keller E, McCauley L, Cho KR, and Buckanovich RJ (2011). Human ovarian carcinoma–associated mesenchymal stem cells regulate cancer stem cells and tumorigenesis via altered BMP production. J. Clin. Invest. 121, 3206–3219. 10.1172/jci45273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop D, and Horwitz E (2006). Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8, 315–317. 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 78.. Mita Y, Fukagawa T, Watahiki H, Kitayama T, Fujii K, Mizuno N, and Sekiguchi K. (2020). Developmental validation for sanger sequencing of HV1 and HV2 in mitochondrial DNA. Forensic Sci. Int.: reports 2, 100159. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data have been deposited at Mendeley: https://doi.org/10.17632/25s5258ynf.1 and are publicly available as of the date of publication. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.