

Abstract
Natural selection and demographic forces can have similar effects on patterns of DNA polymorphism. Therefore, to infer selection from samples of DNA sequences, one must simultaneously account for demographic effects. Here we take a model-based approach to this problem by developing predictions for patterns of polymorphism in the presence of both population size change and natural selection. If data are available from different functional classes of variation, and a priori information suggests that mutations in one of those classes are selectively neutral, then the putatively neutral class can be used to infer demographic parameters, and inferences regarding selection on other classes can be performed given demographic parameter estimates. This procedure is more robust to assumptions regarding the true underlying demography than previous approaches to detecting and analyzing selection. We apply this method to a large polymorphism data set from 301 human genes and find (i) widespread negative selection acting on standing nonsynonymous variation, (ii) that the fitness effects of nonsynonymous mutations are well predicted by several measures of amino acid exchangeability, especially site-specific methods, and (iii) strong evidence for very recent population growth.
Natural selection alters observed patterns of genetic variation within species. For instance, negative selection against slightly deleterious mutations leads to a relative excess of rare variants in a population (1), recurrent positive selection leads to a relative excess of common variants (2), and balancing selection can cause an increase of mutations at intermediate frequencies (3, 4). Because different types of natural selection have different effects on observed genetic variation, it should, in principle, be possible to infer the strength and mode of natural selection from patterns of variation in samples of DNA sequences.
However, a major complicating factor in the effort to infer selection from sequence data is that demographic forces, such as recent population growth, bottlenecks, and subdivision, also affect observed patterns of genetic variation in a manner that can mimic the effects of natural selection (Fig. 3, which is published as supporting information on the PNAS web site). For example, recent population growth, like weak negative selection, leads to a relative excess of rare mutations (5, 6), and certain models of population structure produce an effect identical to that of balancing selection (7). In general, one can only be confident of inferences regarding selection if the “signature” of selection is unique, compared with the effects of other forces. Given that most populations fluctuate in size and do not mate randomly, there is now a growing realization that one must account for demography while inferring selection (8).
Because demographic forces have similar effects over the whole genome, one useful way to account for demography while inferring selection is to compare patterns of variation among different functional classes of mutations. If one has some apriori information that variants in a particular functional class are selectively neutral, then that class can be treated as a neutral standard, to which other classes are compared. The McDonald–Kreitman test (9), for example, contrasts the ratio of polymorphism to divergence at nonsynonymous and synonymous sites. If synonymous mutations are neutral, then this test is a robust test of natural selection at nonsynonymous sites, i.e., it is not sensitive to demographic forces (8). However, the McDonald–Kreitman test and related methods (2, 10–12) do not use allele frequency information; therefore, some power to detect selection is lost (13). An alternative to the McDonald–Kreitman approach is to use nonparametric tests to compare the allele frequency spectrum of segregating sites (hereafter, the site-frequency spectrum) among regions or functional classes (14). This method is quite powerful because it uses the full allele frequency information (13), but, because it is nonparametric, it can be difficult to translate this method into biologically meaningful measures of natural selection, such as estimates of selection parameters, and it is not clear how to compare competing selective models.
Here, we develop a maximum likelihood framework for inferring both selection and demography that contrasts the site-frequency spectrum among functional classes of mutations. We use a population size change model to derive predictions for the site-frequency spectrum both with and without selection. These predictions lead to a method for correcting for the effects of demography while inferring selection. The population size change model is particularly relevant because the species that are the most well studied in population genetics, humans and Drosophila, have probably experienced recent growth. We apply our approach to a large data set composed of DNA sequences from exons, introns, and flanking sequence of 301 human genes sampled in 90 individuals as part of the National Institute of Environmental Health Sciences (NIEHS) Environmental Genome Project (15). Under the assumption that noncoding SNPs are selectively neutral, we find strong evidence for very recent population growth in humans. We correct for this demographic effect to infer selection among nonsynonymous, synonymous, and insertion/deletion (indel) polymorphisms. We find no evidence that natural selection acts on standing synonymous variation, and marginal evidence for selection on noncoding indel polymorphisms. In contrast, we find that negative selection among nonsynonymous polymorphisms is widespread, and that the strength of negative selection on nonsynonymous sites is predicted by several measures of amino acid exchangeability, i.e., “radical” amino acid mutations tend to be more deleterious than “conservative” changes. Several methods have been proposed for quantifying the impact of different types of amino acid changes on protein structure and function (16–22); examples of different criteria that are used include physicochemical properties of the change, phylogenetic patterns of substitution, and experimental measures of the effect of different substitutions on protein structure and activity. However, the relationship between these measures and evolutionary fitness has not been fully explored. We use our methods to compare different measures of exchangeability, finding that the site-specific Polyphen algorithm (22) is the best predictor of the fitness effects of nonsynonymous mutations. Among general (not site-specific) measures of exchangeability, the Miyata (16), EX (20), and PAM-120 (18) matrices all predict the fitness effects of nonsynonymous changes very well. We discuss our results in the context of human genetic disease.
Theory and Inference
Assume data are available from two functional classes of nucleotide sites (class 1 and class 2), and that apriori knowledge suggests that mutations in class 1 are selectively neutral (e.g., SNPs in noncoding regions, pseudogenes, or at synonymous sites). To estimate demographic parameters and to correct for demography while inferring selection, we follow a two-step procedure. First, assuming that segregating mutations in class 1 are selectively neutral, we estimate the parameters of the population growth model. Second, given these demographic parameter estimates, we infer the strength and mode of natural selection acting on class 2 polymorphisms.
Our inference methods are based on the site-frequency spectrum (SFS), which describes the relative abundances of rare, intermediate, and common polymorphisms in a sample. Let xi represent the number of SNPs at which the derived nucleotide is represented i times in a sample of size n. The SFS is the vector, x, of all xi. To predict the SFS with and without selection, we follow the Poisson Random Field approach (2), i.e., we assume no linkage among sites or interference among mutations. Assuming an infinitely-many sites mutation model, diffusion theory is used to predict the distribution of allele frequencies at a single site and, thereby, the SFS.
The distribution (f) of allele frequency (q) at an arbitrary time (t) is approximated by the general solution to the forward Kolmogorov equation (23–25)
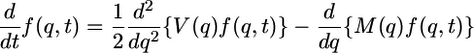 |
[1] |
subject to absorbing boundaries at 0 and 1:
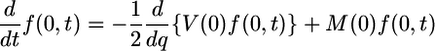 |
[2a] |
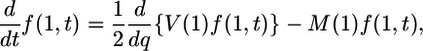 |
[2b] |
where M(q) and V(q) are the mean and variance of the change in allele frequency over 1 unit of time, respectively. Let NC be the current diploid population size. Scaling time in 2NC generations, and assuming Wright–Fisher population structure (random mating, nonoverlapping generations), V(q) = q(1 – q). Also, let s and 2s be the selective advantages of heterozygous and homozygous mutant individuals over wild-type homozygous individuals, so that M(q) = γq(1 – q), where γ = 2NCs.
For inference, Sawyer and Hartl (2) use the stationary solution (1) to the diffusion equation [i.e., the time-independent solution with df(q, t)/dt = 0], subject to irreversible mutation. The stationary solution assumes that the factors affecting changes in allele frequency, such as population size and natural selection, have been constant over recent evolutionary history. To investigate the effect of changes in population size, we require the transient (time-dependent) solution to the forward equation.
Neutral Predictions. To represent population growth or decline, consider a demographic model in which the population experiences two epochs of population size over recent evolutionary history, i.e., it changes instantaneously from an ancestral size, NA, to a contemporary size, NC, at some time τ in the past (Fig. 4a, which is published as supporting information on the PNAS web site). Let the ratio of the two population sizes be ν = NA/NC. Assuming selective neutrality (γ = 0), Kimura (26) found the transient solution to equation (1) for a Wright-Fisher population, conditional on some initial allele frequency p
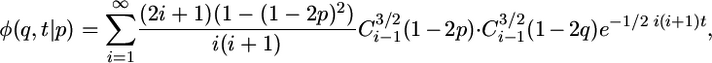 |
[3] |
where C3/2i(z) is the Gegenbauer polynomial with λ = 3/2. To use Kimura's solution in the framework of the two-epoch model, we divide modern variation into two categories: sites that were segregating in the ancestral population (“ancestral sites”) and sites that have mutated since the time of the size change (“modern sites”). Assuming the population was in stationarity before the size change, the distribution of allele frequency at the time of the size change is 4NAμ1/p (2), where μ1 is the mutation rate of the putatively neutral functional class (class 1). The distribution of allele frequency at ancestral sites is then 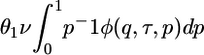 , where θ1 = 4NCμ1. For modern sites, we assume that a fixed number of new mutations, θ1/2, enter the population each generation, and each mutation occurs at a previously unmutated site. Because the frequency of each new mutation is initially 1/(2NC), the distribution of allele frequency at modern sites is equal to the number of new mutations entering the population in a generation, multiplied by the transient distribution given the time difference between that generation and the current population, summed across generations. This sum can be approximated by an integral:
, where θ1 = 4NCμ1. For modern sites, we assume that a fixed number of new mutations, θ1/2, enter the population each generation, and each mutation occurs at a previously unmutated site. Because the frequency of each new mutation is initially 1/(2NC), the distribution of allele frequency at modern sites is equal to the number of new mutations entering the population in a generation, multiplied by the transient distribution given the time difference between that generation and the current population, summed across generations. This sum can be approximated by an integral: 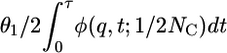 . The neutral prediction for the distribution of allele frequency across sites is then
. The neutral prediction for the distribution of allele frequency across sites is then
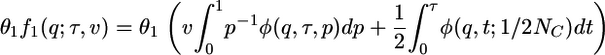 |
[4] |
With this distribution, we can predict the SFS. Define the function F1 to be:
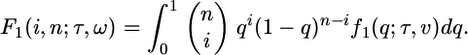 |
[5] |
Then the expected number of polymorphic sites with i derived alleles segregating in a sample of size n is E[xi] = θ1F1(i, n; τ, ν) (Fig. 3), and the probability that a particular SNP is at frequency i out of n is
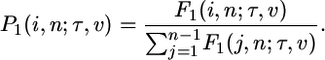 |
[6] |
Note that mutation parameters are absent from this probability, i.e., conditioning on the number of segregating sites, the SFS is independent of the mutation rate, so long as the infinite-sites assumption holds. The above expressions are extensions of Poisson Random Field predictions (2, 4, 27, 28), where the transient distribution of allele frequency (Eq. 4) is substituted for the stationary distribution.
Predictions with Selection. Kimura (29) also found the transient distribution of allele frequency, given some initial frequency p, for the case of directional selection. Unfortunately, his solution is very difficult to calculate accurately, and it is not clear how to adapt his solution to the population growth model we are considering. Instead, we find the distribution of allele frequency by numerically solving Eq. 1 subject to boundary conditions (2). For an initial condition, we again use the stationary distribution of allele frequency for the ancestral population size (1, 2)
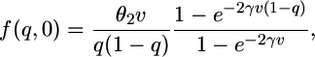 |
[7] |
where θ2 = 4NCμ2, μ2 is the mutation rate in the selected functional class (class 2), and γ = 2NCs. To solve Eq. 1 numerically, we use the Crank–Nicolson finite differencing scheme (ref. 30, pp. 847–851), which is a standard algorithm for solving this sort of advection–diffusion equation. Finite differencing schemes are based on a discrete approximation of the differential equation; we use Δq = 1/2NC as the discrete step size in allele frequency, and Δt = 1/32NC as the discrete step size in time. In addition to imposing the initial condition and boundary conditions (Eq. 2), we model mutation by adding new mutations to the lowest frequency class (1/2NC) at rate θ2/2 per generation. Let f2(x; γ, τ, ν) be the Crank–Nicolson approximation to the distribution of allele frequency at class 2 sites, given selective and demographic parameters. Define the function F2 to be
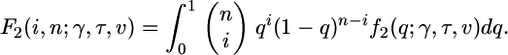 |
[8] |
With selection, the expected number of polymorphic sites segregating at frequency i in a sample of size n is E[xi] = θ2F2(i, n, γ, τ, ν), and the probability that a particular polymorphic site is at frequency i out of n is
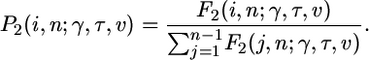 |
[9] |
Likelihood-Based Inference. In the case of perfect information about which nucleotides are ancestral or derived at each SNP, we can evaluate the likelihood of the data under the model by simply taking the product of the probabilities in Eqs. 6 or 9 across all SNPs in the class. To designate ancestral states, one typically “polarizes” a SNP by typing the homologous site in a closely related species. Applying the infinitely-many sites mutation model, the outgroup allelic state at a SNP site should equal the state of the most recent common ancestor (MRCA) of the within-species sample. Unfortunately, minor deviations from the infinitely-many sites model can have a major impact on statistical inference using the SFS. If divergence times are long enough or mutation rates high enough, then there is some nonnegligible probability that a site is both polymorphic in a sample and has experienced a substitution in the lineage connecting the MRCA and the outgroup. If this has occurred, one is likely to misidentify the ancestral state. When ancestral states are occasionally misidentified, the SFS has an overrepresentation of very high-frequency, apparently derived alleles, which, in turn, can cause spurious evidence for positive selection. Visual inspection of observed polarized frequency spectra (Fig. 1) reveals that ancestral misspecification is a nonnegligible problem; i.e., one observes an excess of very high-frequency “derived” alleles (n – 1, n – 2 frequency classes). No realistic evolutionary models can account for such an excess, and it is easily explained by ancestral misspecification.
Fig. 1.

The normalized site-frequency spectra of noncoding, synonymous, and nonsynonymous SNPs discovered in a set of 301 human genes sequenced in 90 individuals, represented as the expected site-frequency spectra of a subsample of size 40. Nonsynonymous SNPs show a relative excess of SNPs with rare alleles. Also note the slight excess of very high-frequency variants, which we explain by ancestral misspecification.
Let a and b be the allelic states for a SNP. Accounting for the possibility of ancestral misspecification, the likelihoods of class 1 and class 2 data are
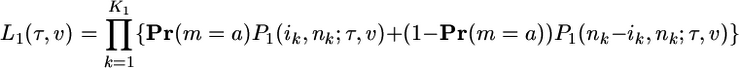 |
[10a] |
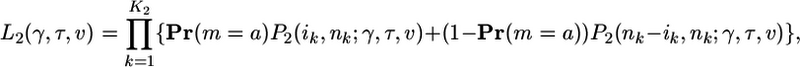 |
[10b] |
where K1 and K2 are the number of SNPs in classes 1 and 2, and ik is the number of b alleles observed at SNP k, nk is the number of chromosomes that were successfully typed at SNP k, and Pr(m = a) is the probability that a is the ancestral state (derived in Supporting Text, which is published as supporting information on the PNAS web site). The above likelihoods assume that the probability that SNP data are missing from a particular individual is independent of the allelic state of that individual; hence, missing data can be treated as a simple reduction in sample size. To estimate the demographic parameters τ and ν, we maximize expression (Eq. 10a) using class 1 (putatively neutral) data. Then, for class 2 data, we fix these demographic parameter estimates and maximize expression (Eq. 10b) to estimate the selection parameter γ. Details of the algorithms used to evaluate and optimize the likelihood functions are given in Supporting Text.
Data
We obtained human polymorphism data (15) from the NIEHS Environmental Genome Project website (http://egp.gs.washington.edu). Briefly, SNP data were collected by direct sequencing of 301 genes in a sample of 90 individuals representative of populations in the United States (31). The sample was composed of 24 African Americans, 24 Asian Americans, 24 European Americans, 12 Mexican Americans, and 6 Native Americans. Genes were chosen for sequencing on the basis that variation in the genes may be associated with variation in the response to environmental exposures, and most genes contribute to basic cellular processes, such as the cell cycle or DNA repair. We used all genes that were finished as of July 31, 2004; a list of these genes is available from the corresponding author. A full description of the data are found in ref. 15. SNPs are present in several function classes in the data set: synonymous, nonsynonymous, intron, other UTRs of the reading frame, and flanking regions. The site-frequency spectra for different classes of SNPs are shown in Fig. 1. To represent the SFS of all SNPs in a class, including SNPs with missing data, we have plotted the expected SFS in a subsample of the data (32, 33). We assumed that noncoding SNPs (flanking, UTR, intron) are selectively neutral to fit the demographic model, i.e., these SNPs form class 1. We allowed for the possibility of selection on other functional classes of mutation.
Results
Demographic Inference. Maximum likelihood estimates for the time back to the population size change, τ, and the ratio of the ancestral to current population sizes, ν, are given in Table 1. Under the assumption of selective neutrality, noncoding SNPs show evidence for a very recent population growth event. We evaluate the significance of this pattern with a likelihood ratio test (LRT) of stationarity, which contrasts the two-epoch growth model with the equilibrium neutral model. The LRT statistic is 2 log(L1(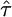 ,
, 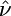 )/L1(∞, 1)); under the null hypothesis, a χ2 distribution with two degrees of freedom provides a conservative critical value for this statistic. This test very strongly rejects the null hypothesis of no growth (P ≪ 10–10).
)/L1(∞, 1)); under the null hypothesis, a χ2 distribution with two degrees of freedom provides a conservative critical value for this statistic. This test very strongly rejects the null hypothesis of no growth (P ≪ 10–10).
Table 1. MLEs of demographic and selective parameters in the two-epoch population growth model, obtained by using different functional classes of human SNPs.
| Functional class | No. of SNPs | MLEs | LRT statistic (df) | P value |
|---|---|---|---|---|
| Demography | ||||
| Noncoding | 36,337 | 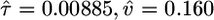 |
9512.2 (2) | «10-10 |
| Selection | ||||
| Nonsynonymous | 880 | 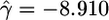 |
66.14 (1) | «10-10 |
| Synonymous | 892 | 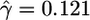 |
0.238 (1) | 0.890 |
| Noncoding indel | 2,710 | 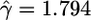 |
5.128 (1) | 0.024 |
τ ≡ time back to population size change; ν ≡ ratio of ancestral to current population size (NA/Nc); γ ≡ selection size parameter (2Ncs).
Our estimate for the time back to the population growth event is in units of 2NC generations. We can use polymorphism and divergence data to estimate the current effective population size, which can be used to convert the time scale to years. Our estimates of the current and ancestral population sizes are N̂C = 51,340 and N̂A = 8,211 (see Supporting Text). Rescaling time, the size change occurred 908 generations ago, or ≈18,200 years. This estimate is more recent than previous estimates (33), probably because of the larger sample size of the NIEHS SNPs data. To detect very recent growth, one must observe mutations that have occurred very recently, which will be rare and only detectable with large sample sizes. The detection of recent mutations will also be complicated by SNP ascertainment schemes with small discovery sample sizes, which is not an issue for the NIEHS SNPs.
Quantifying Selection. Estimates of the selection parameter for nonsynonymous and synonymous SNPs and noncoding indel polymorphisms are given in Table 1. These were obtained by fixing the demographic parameters τ and ν to their maximum likelihood estimates (MLEs) from the noncoding data, then optimizing the likelihood in expression (Eq. 10b). Also shown are the results of LRTs of selective neutrality which compares the demographic model with selection to the demographic model without selection; the LRT static is 2 log(L2(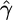 ,
, 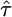 ,
, 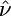 )/L2(0,
)/L2(0, 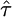 ,
, 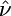 )), which is asymptotically χ2 distributed with one degree of freedom under the null hypothesis. We find strong evidence for negative selection on nonsynonymous sites (P ≪ 10–10), whereas we find no evidence for selection acting on synonymous SNPs (P = 0.89). Interestingly, we find marginal evidence for weak positive selection on noncoding indel polymorphisms (P = 0.024).
)), which is asymptotically χ2 distributed with one degree of freedom under the null hypothesis. We find strong evidence for negative selection on nonsynonymous sites (P ≪ 10–10), whereas we find no evidence for selection acting on synonymous SNPs (P = 0.89). Interestingly, we find marginal evidence for weak positive selection on noncoding indel polymorphisms (P = 0.024).
Given the widespread nature of negative selection on nonsynonymous variation, it is of great interest to determine what types of nonsynonymous mutations tend to be most deleterious. To do this, we use various measures of amino acid exchangeability to classify nonsynonymous changes as either conservative, moderate, or radical, defining categories such that approximately equal numbers of SNPs appear in each category. We then estimate the selection parameter for each category. Measures for which the selection parameter estimate decreases as nonsynonymous changes become more radical, and which show the greatest difference between conservative and radical changes are the best predictors of the fitness effects of nonsynonymous mutations. Estimates of the selection parameter for conservative, moderate, and radical changes defined by different measures of exchangeability are given in Table 2. Also shown are estimates of the selection parameters for amino acid changes categorized by the program polyphen (22), which uses protein structure and/or sequence conservation information from each gene to predict whether a nonsynonymous mutation is “benign,” “possibly damaging,” or “probably damaging.” Polyphen analyses were conducted as part of the NIEHS environmental genome project, and gene-by-gene results are available on the NIEHS SNPs web site. In general, Polyphen is the best predictor of the fitness effects of nonsynonymous mutations, in that the inferred fitness effect of “probably damaging” mutations is much more negative than the fitness effect of “benign” mutations. Presumably, Polyphen predicts fitness the best because it uses protein- and site-specific information. The strength of selection on “probably damaging” mutations is striking, implying that a large majority of new mutations classified as “probably damaging” are strongly deleterious, relative to other polymorphic amino acid changes. Also note that our method will underestimate the strength of selection: 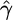 tends to be biased toward 0 if selective effects are variable among new mutations or if deleterious mutations tend to be recessive (34). Among measures of exchangeability that are not site- or protein-specific, the Miyata (16), EX (20), and PAM-120 (18) matrices all predict the fitness consequences of amino acid changes very well.
tends to be biased toward 0 if selective effects are variable among new mutations or if deleterious mutations tend to be recessive (34). Among measures of exchangeability that are not site- or protein-specific, the Miyata (16), EX (20), and PAM-120 (18) matrices all predict the fitness consequences of amino acid changes very well.
Table 2. Estimates of the scaled selection parameter γ(=2NCs) for conservative, moderate, and radical nonsynonymous changes, as defined by different measures of amino acid exchangeability, and for nonsynonymous mutations categorized as “benign,” “possibly damaging,” or “probably damaging” by the polyphen program, which uses site-specific structural and phylogenetic information.
| Conservative
|
Moderate
|
Radical
|
|||||
|---|---|---|---|---|---|---|---|
| Exchangeability measure | Ref. | Range | 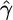 |
Range | 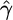 |
Range | 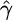 |
| Physicochemical measures | |||||||
| Miyata metric | 16 | x ≤ 0.89 | -5.71* | 0.89 < x ≤ 1.85 | -8.45* | x > 1.85 | -14.06* |
| Grantham's distance | 17 | x ≤ 44 | -7.48* | 44 < x ≤ 82 | -7.77* | x > 82 | -12.04* |
| Experimental measures | |||||||
| Experimental exchangeability | 20 | x > 0.354 | -6.35* | 0.256 < x ≤ 0.354 | -8.79* | x ≤ 0.256 | -12.15* |
| General measures based on structural models | |||||||
| Protein stability matrix | 21 | x > 0.210 | -8.19* | 0.030 < x ≤ 0.210 | -8.55* | x ≤ 0.030 | -10.15* |
| Phylogenetic measures | |||||||
| PAM-120 | 18 | x > 1 | -5.32 | -1 < x ≤ 1 | -8.35* | x ≤ -1 | -12.76* |
| BLOSUM-45 | 19 | x > 0 | -8.41* | -1 < x ≤ 0 | -3.96 | x ≤ -1 | -13.39* |
| BLOSUM-62 | 19 | x > 0 | -8.41* | -1 < x ≤ 0 | -4.09 | x ≤ -1 | -12.75* |
| BLOSUM-80 | 19 | x > 0 | -8.46* | -2 < x ≤ 0 | -4.49 | x ≤ -2 | -13.52* |
| Site-specific structural/phylogenetic measures | |||||||
| Polyphen | 22 | “Benign” (310 SNPs) | -6.072* | “Possibly damaging” (97 SNPs) | -11.732* | “Probably damaging” (56 SNPs) | -23.602* |
Ranges define the values of the exchangeability measure, x, over which the category extends.
Categories for which a likelihood ratio test rejects neutrality (P < 0.01).
Here we have corrected for demography by fitting the two-epoch model to putatively neutral data, then inferring selection at other sites in the context of the fitted model. Even so, the approach makes some assumptions (random mating, only one change in population size) about the underlying demographic history of the population. We have tested the robustness of our approach to violations of the demographic assumptions by simulating neutral data under a variety of demographic models. Data were simulated for different models by using the ms coalescent simulation program (35), then summarized into site-frequency spectra by using the msfreq module of libsequence (36). For each simulated data set, we fit the neutral two-epoch model to a subset of the data (class 1 SNPs), then we fit the two-epoch model with selection to another subset of the data (class 2), given the demographic parameters estimated from the first step. We considered four disparate demographic scenarios (Fig. 4): two-epoch population growth, a single population with a recent bottleneck, a subdivided population of 10 demes with migration (where samples are drawn from only 4 demes), and complex model that is meant to represent a “best guess” of the history of African, Asian, Native American, and European populations. Details of the parameters for all models are given in Table 3, which is published as supporting information on the PNAS web site. Fig. 2a indicates that our estimates of the selection parameter, obtained in the context of the two-epoch model, show very little or no bias under alternative models when new mutations are selectively neutral. Also, in our test of natural selection, the null distribution of the LRT statistic (Fig. 2b) is largely insensitive to the underlying demography. Taken together, these simulations demonstrate that our inference regarding natural selection is robust to the demographic assumptions of the two-epoch population growth model.
Fig. 2.

The sampling distribution of the scaled selection parameter (2NAs)(a) and the likelihood ratio test statistic (b), under selective neutrality (2NAs = 0) for a variety of demographic models. Note that, for ease of comparison among models, the selection parameter is scaled relative to the ancestral, rather than the current, population size. The heavy black line in b represents the χ2 distribution with one degree of freedom.
Coalescent simulations also provide a means of cross-validating the diffusion-based approach. When we simulate data sets with τ = 0.02 and ν = 0.1, then use the diffusion approach to estimate the demographic parameters, the average MLEs are 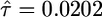 and
and 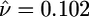 , and the 95% confidence limits for the sampling distribution of τ and ν are (0.0185, 0.0219) and (0.096, 0.108), respectively. Also, asymptotic likelihood theory predicts that the null (selectively neutral) distribution of the LRT of selection should be χ2 distributed with one degree of freedom, and Fig. 2b shows that the simulated null distribution (dotted line) corresponds very closely to the asymptotic prediction (in black). To summarize, the diffusion-based approach very accurately represents the dynamics of allele frequency in the two-epoch population growth model.
, and the 95% confidence limits for the sampling distribution of τ and ν are (0.0185, 0.0219) and (0.096, 0.108), respectively. Also, asymptotic likelihood theory predicts that the null (selectively neutral) distribution of the LRT of selection should be χ2 distributed with one degree of freedom, and Fig. 2b shows that the simulated null distribution (dotted line) corresponds very closely to the asymptotic prediction (in black). To summarize, the diffusion-based approach very accurately represents the dynamics of allele frequency in the two-epoch population growth model.
Coalescent simulations can only be used to investigate the properties of our statistical approach under selective neutrality among all classes of polymorphisms. To explore the properties of the method with selection on class 2 polymorphisms, we have used our predictions from the two-epoch model both with and without selection to simulate data sets. For a given parameter combination, we simulated the site-frequency spectra of class 1 and class 2 data through multinomial sampling using the probabilities defined in Eqs. 6 and 9, respectively. We then estimated the demographic parameters by using the class 1 data, and, fixing those MLEs, we estimated the selection parameter by using class 2 data. The results of these simulations are shown in Table 4, which is published as supporting information on the PNAS web site. The two-step procedure we propose (estimating demographic parameters, then inferring selection in the demographic context) carries essentially no bias in our estimate of the selection or demographic parameters. Likewise, it appears that the asymptotic prediction for the LRT of selection (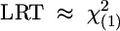 ) holds quite well because only ≈5% of neutral data sets (γ = 0) reject neutrality for all demographic parameters considered. The test also has excellent power for |γ| ≥ 5 for all demographic parameter combinations, rejecting the false null hypothesis of neutrality for selected data >85–90% of the time.
) holds quite well because only ≈5% of neutral data sets (γ = 0) reject neutrality for all demographic parameters considered. The test also has excellent power for |γ| ≥ 5 for all demographic parameter combinations, rejecting the false null hypothesis of neutrality for selected data >85–90% of the time.
Discussion
Here we have developed a method for inferring both selection and demographic parameters from DNA sequence data. We apply this method to a large set of human polymorphism data, and our main results are as follows: (i) negative selection is widespread among nonsynonymous mutations, but that selection is often weak enough that deleterious alleles can reach observable frequencies through genetic drift, an observation that is fully consistent with the nearly neutral theory of molecular evolution (37); (ii) the deleterious effects of nonsynonymous mutations are well predicted by site-specific predictions of the functional consequences of nonsynonymous changes, and by some general measures of amino acid exchangeability; and (iii) the human population has grown very recently. The widespread nature of negative selection on nonsynonymous mutations reflects systematic differences between the site-frequency spectra of nonsynonymous and putatively neutral SNPs. Previous studies have also noted such a systematic difference in site-frequency spectra (38, 39) of different functional classes of human SNPs, but our study quantifies this difference in terms of evolutionary fitness and uses this information to relate amino acid exchangeability with fitness.
Our two-epoch population growth model is a very rough approximation to the demographic history of human populations, and other demographic forces, such as population bottle-necks or subdivision, might also have a considerable effect on the SFS. For instance, with some sampling schemes, unacknowledged population subdivision may cause spurious evidence for population growth, when the basis of inference is the SFS (40). Therefore, although we find it striking that the time of population growth (18,200 years B.P.) roughly corresponds with events in human history that may have induced population growth, such as the end of the last ice age and the origin of agriculture, we feel that our demographic inferences should be interpreted cautiously until the full range of plausible demographic models has been explored in one coherent framework. However, we argue that our inferences regarding selection are robust to violations of demographic assumptions. By inferring demographic parameters with putatively neutral data, then inferring selection on other types of SNPs given those demographic parameter estimates, we are essentially comparing the site frequency spectra of functional classes in the context of the two-epoch model. That such a comparison is made may be more important than the specifics of the demographic model, especially if the model provides a reasonably good fit to the data. Coalescent simulations support the notion that our inference of selection is robust to violations of demographic assumptions. For instance, estimates of the selection parameter are not strongly biased under a wide range of demographic models. Even if some bias is introduced by more complicated demographic scenarios, we feel that the two-epoch growth model, fit to data, is a much better alternative to the standard (constant population size, random mating) neutral model, which forms the basis of much data analysis in molecular population genetics and cannot be fit to data in the form of the SFS.
We find evidence that negative selection on nonsynonymous mutations is widespread, which implies that deleterious mutations make up a significant proportion of standing nonsynonymous variation. Exactly how this genetic variation contributes to phenotypic variation is a matter of considerable debate, especially for medically interesting phenotypes such as multifactorial genetic disease (41–44). Because deleterious mutations, by definition, have phenotypic effects, and because of the widespread nature of negative selection on nonsynonymous mutations, it seems likely that negatively selected, generally rare nonsynonymous SNPs have some negative impact on human health. If there is a general relationship between nonsynonymous polymorphism and human genetic disease, then our genomic estimates of the fitness effects of different types of mutations contain prior information about the likelihood that a mutation contributes to disease. It may be possible to use this information to aid in identifying SNPs that cause disease. Other studies have suggested this approach (e.g., ref. 15), but it was unclear which of the many measures of exchangeability to use. We feel that the relative fitness of different amino acid changes is the best way to evaluate exchangeability, and we have done that here by using a model that includes demography and selection.
Finally, a common criticism of the Poisson Random Field approach that we have used here is that it assumes SNPs are independent, whereas correlations among SNPs due to limited recombination might alter patterns in the data. However, the presence of limited recombination within genes does not affect the conclusions of this study. First, our method for inferring selection is based on a contrast of functional classes of SNPs; because linkage affects all functional classes within a gene, a contrast among functional classes should be less sensitive to limited recombination. Second, we only observe approximately three nonsynonymous SNPs per gene in the data, and almost all genes are unlinked. Consequently, the vast majority of nonsynonymous SNPs really are unlinked, and our inferences about selection on nonsynonymous sites should not be strongly affected by interaction effects such as Hill-Robertson interference (45). If interference does have an effect, it would bias our estimates of the selection parameter toward 0. And third, our inferences regarding demography should not be affected by linkage. If sites are evolving neutrally, then linkage does not affect the expected SFS, so linkage also should not have a strong effect on the expected values of statistics based on the SFS, such as our demographic parameter estimates.
Supplementary Material
Acknowledgments
We thank B. Payseur, A. Andres, K. Thornton, and A. Clark for many helpful discussions. The comments of two anonymous reviewers greatly improved the manuscript. This research was supported by National Science Foundation Award 0319553 (to M. Purugganan, S. McCouch, R.N., and C.D.B.) and National Institutes of Health Award 1R01HG03229-01 (to A. Clark, T. Mattise, C.D.B., and R.N.).
Author contributions: S.H.W., R.N., and C.D.B. designed research; S.H.W., R.H., and A.F.-A. performed research; S.H.W., R.H., A.F.-A., L.Z., and C.D.B. analyzed data; and S.H.W., R.N., and C.D.B. wrote the paper.
Abbreviations: NIEHS, National Institute of Environmental Health Sciences; SFS, site-frequency spectrum; LRT, likelihood ratio test; MLE, maximum likelihood estimate.
References
- 1.Wright, S. (1938) Proc. Natl. Acad. Sci. USA 24, 253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sawyer, S. A. & Hartl, D. L. (1992) Genetics 132, 1161–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaplan, N. L., Darden, T. & Hudson, R. R. (1988) Genetics 120, 819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williamson, S., Fledel-Alon, A. & Bustamante, C. D. (2004) Genetics 168, 463–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Slatkin, M. & Hudson, R. R. (1991) Genetics 129, 555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Griffiths, R. C. & Tavare, S. (1994) Philos. Trans. R. Soc. London Ser. B 344, 403–410. [DOI] [PubMed] [Google Scholar]
- 7.Hudson, R. R. (1990) Oxf. Surv. Evol. Biol. 1, 1–14. [Google Scholar]
- 8.Nielsen, R. (2001) Heredity 86, 641–647. [DOI] [PubMed] [Google Scholar]
- 9.McDonald, J. H. & Kretman, M. (1991) Nature 351, 652–654. [DOI] [PubMed] [Google Scholar]
- 10.Rand, D. M. & Kann, L. M. (1996) Mol. Biol. Evol. 13, 735–748. [DOI] [PubMed] [Google Scholar]
- 11.Bustamante, C. D., Nielsen, R., Sawyer, S. A., Olsen, K. M., Purugganan, M. D. & Hartl, D. L. (2002) Nature 416, 531–534. [DOI] [PubMed] [Google Scholar]
- 12.Smith, N. G. C. & Eyre-Walker, A. (2002) Nature 415, 1022–1024. [DOI] [PubMed] [Google Scholar]
- 13.Akashi, H. (1999) Genetics 151, 221–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akashi, H. & Schaeffer, S. W. (1997) Genetics 146, 295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Livingston, R. J., von Niederhausern, A., Jegga, A. G., Crawford, D. C., Carlson, C. S., Rieder, M. J. Gowrisankar, S., Aronow, B. J., Weiss, R. B. & Nickerson, D. A. (2004) Genome Res. 14, 1821–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyata, T., Miyazawa, S. & Yasunaga, T. (1979) J. Mol. Evol. 12, 219–236. [DOI] [PubMed] [Google Scholar]
- 17.Grantham, R. (1974) Science 185, 862–864. [DOI] [PubMed] [Google Scholar]
- 18.Altshul, S. F. (1991) J. Mol. Biol. 219, 555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henikoff, S. & Henikoff, J. G. (1992) Proc. Natl. Acad. Sci. USA 89, 10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yampolsky, L. Y. & Stoltzfus, A. (2005) Genetics, in press.
- 21.Miyazawa, S. & Jernigan, R. L. (1993) Protein Eng. 6, 267–278. [DOI] [PubMed] [Google Scholar]
- 22.Ramensky, V., Bork, P. & Sunyaev, S. (2002) Nucleic Acids Res. 30, 3894–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kimura, M. (1964) J. Appl. Prob. 1, 177–232. [Google Scholar]
- 24.Crow, J. F. & Kimura M. (1970) An Introduction to Population Genetics Theory (Harper and Row, New York).
- 25.Ewens, W. J. (1979) Mathematical Population Genetics (Springer, New York).
- 26.Kimura, M. (1955) Proc. Natl. Acad. Sci. USA 41, 144–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hartl, D. L., Moriyama, E. N. & Sawyer, S. A. (1994) Genetics 138, 227–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bustamante, C. D., Wakeley, J., Sawyer, S. A. & Hartl, D. L. (2001) Genetics 159, 1779–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kimura, M. (1955) Cold Spring Harbor Symp. Quant. Biol. 20, 33–53. [DOI] [PubMed] [Google Scholar]
- 30.Press, W. H., Flannery, B. P., Teukolsky, S. A. & Vatterling, W. T. (1988) Numerical Recipes in C (Cambridge Univ. Press, Cambridge, U.K.), 2nd Ed.
- 31.Collins, F. S., Brooks, L. D. & Chakravarti, A. (1998) Genome Res. 8, 1229–1231. [DOI] [PubMed] [Google Scholar]
- 32.Nielsen, R., Hubisz, M. J. & Clark, A. G. (2004) Genetics 168, 2373–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marth, G. T., Czabarka, E., Murvai, J. & Sherry, S. T. (2004) Genetics 166, 351–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simmons, M. J. & Crow, J. F. (1977) Annu. Rev. Genet. 11, 49–78. [DOI] [PubMed] [Google Scholar]
- 35.Hudson, R. R. (2002) Bioinformatics 18, 337–338. [DOI] [PubMed] [Google Scholar]
- 36.Thornton, K. (2003) Bioinformatics 19, 2325–2327. [DOI] [PubMed] [Google Scholar]
- 37.Ohta, T. (1973) Nature 246, 96–98. [DOI] [PubMed] [Google Scholar]
- 38.Cargill, M., Altshuler, D., Ireland, J., Sklar, P., Ardlie, K., Patil, N, Shaw, N., Lane, C. R., Lim, E. P., Kalyanaraman, N., et al. (1999) Nat. Genet. 22, 231–238. [DOI] [PubMed] [Google Scholar]
- 39.Sunyaev, S. R., Lathe, W. C., III, Ramensky, V. E. & Bork, P. (2000) Trends Genet. 16, 335–337. [DOI] [PubMed] [Google Scholar]
- 40.Ptak, S. E. & Przeworski, M. (2002) Trends Genet. 18, 559–563. [DOI] [PubMed] [Google Scholar]
- 41.Clark, A. G. (2003) Curr. Opin. Genet. Dev. 13, 1–7. [Google Scholar]
- 42.Pritchard, J. K. & Cox, N. J. (2002) Hum. Mol. Genet. 11, 2417–2423. [DOI] [PubMed] [Google Scholar]
- 43.Chakravarti, A. (1999) Nat. Genet. 21, 56–60. [DOI] [PubMed] [Google Scholar]
- 44.Reich, D. E. & Lander, E. S. (2001) Trends Genet. 17, 502–510. [DOI] [PubMed] [Google Scholar]
- 45.Hill, W. G. & Robertson, A. (1966) Genet. Res. 8, 269–294. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.