

Abstract
SGS1 encodes a DNA helicase whose homologues in human cells include the BLM, WRN, and RECQ4 genes, mutations in which lead to cancer-predisposition syndromes. Clustering of synthetic genetic interactions identified by large-scale genetic network analysis revealed that the genetic interaction profile of the gene RMI1 (RecQ-mediated genome instability, also known as NCE4 and YPL024W) was highly similar to that of SGS1 and TOP3, suggesting a functional relationship between Rmi1 and the Sgs1/Top3 complex. We show that Rmi1 physically interacts with Sgs1 and Top3 and is a third member of this complex. Cells lacking RMI1 activate the Rad53 checkpoint kinase, undergo a mitotic delay, and display increased relocalization of the recombination repair protein Rad52, indicating the presence of spontaneous DNA damage. Consistent with a role for RMI1 in maintaining genome integrity, rmi1Δ cells exhibit increased recombination frequency and increased frequency of gross chromosomal rearrangements. In addition, rmi1Δ strains fail to fully activate Rad53 upon exposure to DNA-damaging agents, suggesting that Rmi1 is also an important part of the Rad53-dependent DNA damage response.
Keywords: checkpoint, genome instability, RecQ helicase, Sgs1, Top3
Introduction
Saccharomyces cerevisiae SGS1 is a member of the recQ family of 3′–5′ DNA helicases, which includes five human homologues (RECQL, BLM, WRN, RECQ4, and RECQ5) (Watt et al, 1995, 1996). Loss-of-function mutations in BLM, WRN, and RECQ4 give rise to Bloom's syndrome (BS), Werner's syndrome (WS), and Rothmund–Thomson syndrome (RTS), respectively (Ellis et al, 1995; Yu et al, 1996; Kitao et al, 1999). Although the spectrum of clinical features of each disease differs, they all result in a predisposition to cancer. The major defects of cells with mutated recQ helicases are hyper-recombination and genomic instability (Hickson, 2003). S. cerevisiae sgs1 mutants show elevated levels of mitotic homologous recombination, illegitimate recombination (Gangloff et al, 1994; Watt et al, 1996; Yamagata et al, 1998), sister chromatid exchanges (Onoda et al, 2000), and gross chromosomal rearrangements (GCRs) (Myung et al, 2001b; Myung and Kolodner, 2002). Cells lacking SGS1 are also moderately sensitive to genotoxic agents such as methyl methanesulfonate (MMS) and hydroxyurea (HU) (Gangloff et al, 1994; Watt et al, 1996; Yamagata et al, 1998; Chang et al, 2002).
A subset of RecQ family members physically interacts with topoisomerase III (Top3) homologues (Gangloff et al, 1994; Goodwin et al, 1999; Johnson et al, 2000; Wu et al, 2000). Escherichia coli RecQ stimulates Top3 to catenate and decatenate covalently closed duplex DNA (Harmon et al, 1999) and BLM is able to stimulate the DNA strand passage activity of Top3α (Oakley and Hickson, 2002). Furthermore, BLM and Top3α can work together to resolve a recombination intermediate containing a double Holliday junction (Wu and Hickson, 2003). S. cerevisiae strains lacking TOP3 exhibit a severe growth defect, sensitivity to DNA-damaging agents, and hyper-recombination (Wallis et al, 1989; Gangloff et al, 1994; Chang et al, 2002). Most of the defects exhibited by top3 mutants can be suppressed by mutation of SGS1 (Gangloff et al, 1994; Chakraverty et al, 2001), a relationship that is conserved in Schizosaccharomyces pombe where mutations in the recQ homologue rqh1+ can suppress the lethality of top3Δ mutants (Maftahi et al, 1999). These data support models in which RecQ helicase action produces a toxic DNA structure that is resolved by Top3 (Gangloff et al, 1994; Ira et al, 2003; Wu and Hickson, 2003).
Two-dimensional hierarchical clustering of synthetic genetic interactions determined by large-scale genetic network analysis in S. cerevisiae has proven useful for identifying genes whose products function within the same pathway or complex (Tong et al, 2004). Such clustering analysis revealed that the genetic interaction profile of the poorly characterized gene RMI1 (RecQ-mediated genome instability) was highly similar to that of SGS1 and TOP3. We show that Rmi1 associates with Sgs1 and Top3 and that strains lacking RMI1 accumulate DNA damage in the absence of exogenous genotoxic agents. Our results indicate that the actions of Sgs1, Top3, and Rmi1 are required in concert in order to maintain genome integrity.
Results
Mutations in SGS1 can suppress the growth defects of an rmi1Δ mutant
Two-dimensional hierarchical clustering of large-scale synthetic genetic array (SGA) data revealed that the set of genes that genetically interact with the uncharacterized gene RMI1 was highly similar to that associated with SGS1, TOP3, and YLR235C (which overlaps the TOP3 open reading frame (ORF) such that a deletion of this ORF likely results in a TOP3 hypomorph) (Tong et al, 2004; Supplementary Figure S1). Synthetic genetic interactions are usually orthogonal to protein–protein interactions, but the products of genes with similar patterns of genetic interactions are often found in the same cellular pathway or protein complex (Tong et al, 2004), suggesting that RMI1 might function in the SGS1/TOP3 pathway.
Crossing the rmi1Δ strain from the Saccharomyces gene deletion collection with a wild-type strain revealed the presence of an extragenic suppressor in the rmi1Δ strain. Tetrads from this cross were dissected to analyze the products of individual meioses (Figure 1A). The resultant colonies were screened to identify those carrying the rmi1Δ mutation. We found that roughly half (10 of 24) of the rmi1Δ isolates exhibited a slow growth phenotype, whereas the other half (14 of 24) grew relatively normally, indicating that the original strain did indeed carry a single extragenic suppressor mutation. To identify the suppressor (supX), we employed synthetic genetic array mapping (SGAM) methodology, in which an rmi1Δ supX query strain was crossed to an array of ∼4600 viable gene deletion mutants. This method maps a group of genes that are tightly linked to the suppressor (Jorgensen et al, 2002). Indeed, we identified a group of linked genes on chromosome XIII (Figure 1B), indicating that the suppressor was in this region. The SGS1 gene was located in the middle of this linkage group, and the sgs1Δ strain was not identified in the SGAM experiment, suggesting that the suppressor might be a loss-of-function allele of SGS1. We crossed the rmi1Δ strain lacking the suppressor with an sgs1Δ strain and found that the double mutants had a normal growth phenotype (Figure 1C). We also sequenced the SGS1 allele from the rmi1Δ supX strain and found that it carried a frame-shift mutation 691 nucleotides into the ORF and so encoded a truncated protein lacking the helicase catalytic domain of Sgs1. Therefore, deletion of RMI1 causes a slow growth phenotype that can be suppressed by deletion of SGS1. This is reminiscent of the TOP3 gene, deletion of which causes slow growth that is suppressed by mutation of SGS1 (Wallis et al, 1989; Gangloff et al, 1994).
Figure 1.
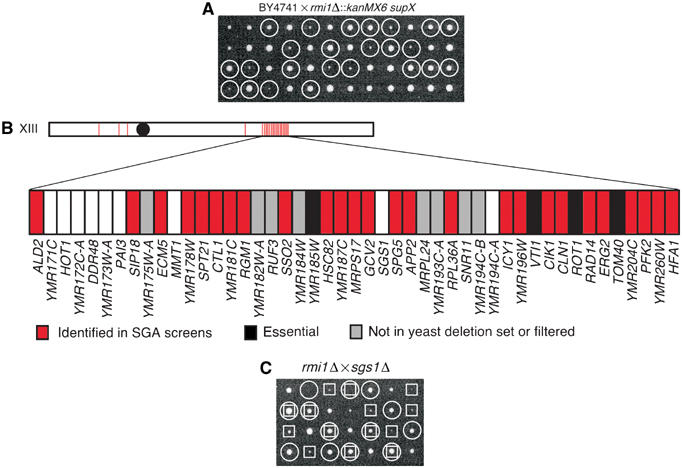
rmi1Δ mutants exhibit a growth defect that can be suppressed by mutation of SGS1. (A) The rmi1Δ∷kanMX6 strain was backcrossed to a wild-type strain (BY4741). The resulting diploids were sporulated and tetrads were dissected on YPD. Each column represents the four spores from a single tetrad. The genotypes of the resulting colonies are indicated with circles (○) for rmi1Δ∷kanMX6. (B) SGAM analysis using an rmi1Δ∷natMX6 query strain (which contains supX) revealed a set of colinear synthetic genetic interactions on chromosome XIII. A red bar indicates that deletion of the corresponding gene resulted in a genetic interaction. Black bars represent essential genes, which are not a part of the gene deletion collection. Gray bars indicate ORFs for which no deletion mutant was made as part of the Saccharomyces Gene Deletion Project (Winzeler et al, 1999) and genes that are often found in control screens using a wild-type query strain, and therefore are filtered from the results of SGA analyses. (C) An rmi1Δ∷natMX6 strain lacking supX was crossed to an sgs1Δ∷kanMX6 strain. The resulting diploids were sporulated for tetrad analysis as in panel A. The genotypes of the resulting colonies are indicated with boxes (□) for sgs1Δ∷kanMX6 and circles (○) for rmi1Δ∷natMX6.
rmi1Δ synthetic genetic interactions
Since the rmi1Δ mutant readily accumulates mutations in SGS1, we were unable to conduct an RMI1 SGA analysis. Instead, we adopted a candidate approach, analyzing genes with connections to SGS1 and TOP3 function (Klein, 2001; Mullen et al, 2001; Tong et al, 2001, 2004) (Table I). We found that rmi1Δ is synthetic lethal when combined with mutations in genes thought to play roles in restarting stalled replication forks: rrm3Δ, mus81Δ, mms4Δ, slx1Δ, slx4Δ, hex3Δ, slx8Δ, and hpr5Δ. We also found that the slow growth phenotype of rmi1Δ was suppressed by rad51Δ, rad52Δ, and rad54Δ. This is consistent with models in which the presence of the homologous recombination pathway facilitates creation of DNA processing intermediates by Sgs1, which are toxic when Rmi1 is absent. Similar models have been proposed to account for the suppression of top3Δ phenotypes by mutations in recombination repair genes (Oakley et al, 2002; Shor et al, 2002). We also found that rmi1Δ did not display a detectable genetic interaction with top3Δ, consistent with RMI1 and TOP3 functioning in the same pathway. Finally, we found that homozygous rmi1Δ/rmi1Δ diploids are defective in undergoing meiosis to produce four spore asci (Supplementary Figure S2), indicating that like Sgs1 and Top3 (Watt et al, 1995; Gangloff et al, 1999), Rmi1 is essential for proper meiotic cell division.
Table 1.
rmi1Δ genetic interactions
| Gene | Interaction | Proposed function |
|---|---|---|
| sgs1Δ | Suppression | RecQ helicase |
| top3Δ | None | Type I topoisomerase |
| rad53-11 | Lethality | DNA damage checkpoint |
| mrc1Δ | Sickness | S-phase DNA damage checkpoint |
| csm3Δ | Sickness | S-phase DNA damage checkpoint |
| tof1Δ | Sickness | S-phase DNA damage checkpoint |
| rad9Δ | None | G2 DNA damage checkpoint |
| rad24Δ | None | G2 DNA damage checkpoint |
| rrm3Δ | Lethality | DNA helicase; fork restart |
| mus81Δ | Lethality | Nuclease subunit; fork restart |
| mms4Δ | Lethality | Nuclease subunit; fork restart |
| slx1Δ | Lethality | Nuclease subunit; fork restart |
| slx4Δ | Lethality | Nuclease subunit; fork restart |
| hex3Δ | Lethality | Fork restart |
| slx8Δ | Lethality | Fork restart |
| hpr5Δ | Lethality | DNA helicase; fork restart |
| rad51Δ | Suppression | Homologous recombination |
| rad52Δ | Suppression | Homologous recombination |
| rad54Δ | Suppression | Homologous recombination |
Rmi1 physically interacts with Top3 and Sgs1
Genetic analysis placed RMI1 in the SGS1/TOP3 pathway and indicated in several ways that rmi1Δ phenocopies top3Δ. To gain insight into the mechanism underlying these genetic observations, we tested whether Rmi1 physically associates with Sgs1 and Top3. Sgs1 and Top3 interact in vivo and in vitro (Gangloff et al, 1994; Bennett et al, 2000; Fricke et al, 2001); however, the apparent molecular mass of Sgs1/Top3 complexes in yeast extracts suggests that the complexes are not heterodimeric and so may contain other proteins (Fricke et al, 2001). We used strains containing SGS1, TOP3, and RMI1 epitope-tagged at their respective genomic loci to perform co-immunoprecipitations. Rmi1 was found in complex with both Sgs1 and Top3 (Figure 2A and B). This complex was not disrupted in the presence of DNase I, indicating that the interactions are not mediated by DNA (Supplementary Figure S3). When Rmi1-TAP immunoprecipitations were quantified by densitometry, we found that 39% of Rmi1 was depleted from the extract compared with 42% of Sgs1 (data not shown), indicating that a significant fraction of Sgs1 is in complex with Rmi1. We next used gel filtration chromatography to fractionate extract from the tagged strain. We found that Sgs1-HA, Top3-VSV, and Rmi1-TAP co-elute in a high-molecular-weight complex (Figure 2C). Monomeric Rmi1 was not detected. Together, these data suggest that Rmi1 is in a heteromeric complex with both Sgs1 and Top3 and functions as a subunit of the Sgs1/Top3 complex.
Figure 2.
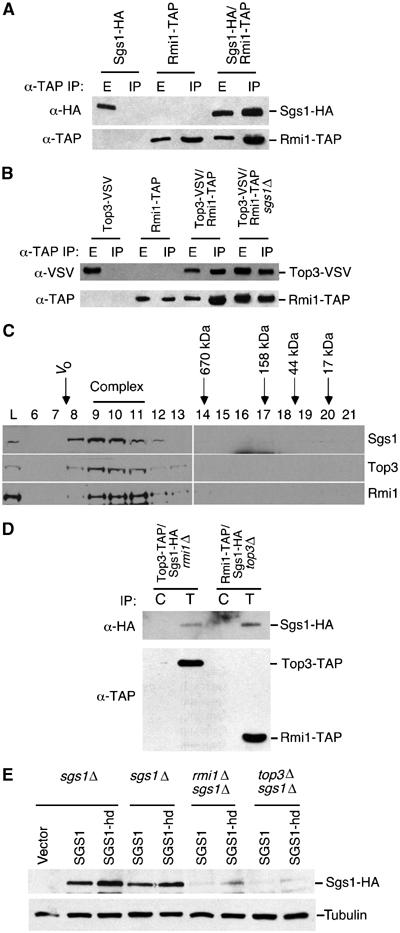
Rmi1 physically associates with the Sgs1/Top3 complex. (A, B) Extracts from yeast strains expressing the indicated epitope-tagged proteins were immunoprecipitated with IgG agarose. In all, 10% of the input extract (E) and the immunoprecipitate (IP) was fractionated by SDS–PAGE. Immunoblots were probed with anti-HA antibody to detect Sgs1, with anti-VSV antibody to detect Top3, or with peroxidase–anti-peroxidase to detect Rmi1-TAP. (C) Extract from a yeast strain expressing Sgs1-HA, Top3-VSV, and Rmi1-TAP was fractionated on a Superose 6 gel filtration column. Fractions were precipitated with TCA and analyzed by immunoblotting. The elution positions of molecular weight standards are indicated, as is the void volume of the column (Vo). (D) Extracts from yeast strains expressing Sgs1-HA and Top3-TAP or Sgs1-HA and Rmi1-TAP in an rmi1Δ or top3Δ background, respectively, were immunoprecipitated with IgG agarose to precipitate the TAP-tagged protein (lanes marked T) or with unconjugated agarose as a control (lanes marked C). The precipitates were immunoblotted and probed with anti-HA antibodies to detect Sgs1-HA (top panel) or with peroxidase–anti-peroxidase to detect the TAP-tagged proteins. (E) sgs1Δ, sgs1Δ rmi1Δ, and sgs1Δ top3Δ strains were transformed with empty vector (vector) or low-copy plasmids expressing HA-tagged Sgs1 (Sgs1) or helicase-dead Sgs1 (Sgs1-hd). TCA-fixed extracts were prepared and fractionated by SGS–PAGE. Immunoblots were probed with anti-HA antibody to detect Sgs1 or Sgs1-hd, and with anti-tubulin antibodies as a loading control.
Immunoprecipitates of Rmi1-TAP from extracts of an sgs1Δ strain contain Top3 (Figure 2B), indicating that the interaction of Rmi1 with Top3 does not require Sgs1. When attempting reciprocal experiments, we found that deletion of either TOP3 or RMI1 caused a significant reduction in Sgs1 protein abundance (data not shown). Despite the reduced levels of Sgs1, we detected Sgs1 in both Top3 immunoprecipitates from rmi1Δ cells and in Rmi1 immunoprecipitates from top3Δ cells (Figure 2D). Both wild-type and catalytically inactive helicase-dead mutant Sgs1 were poorly expressed in both rmi1Δ and top3Δ mutants (Figure 2E), indicating that the helicase activity of Sgs1 is not required for the observed reduction in Sgs1 levels.
Cells lacking RMI1 display precocious checkpoint activation
RecQ helicases are thought to play a role in normal DNA replication. The human homologues BLM and WRN are required for normal S-phase progression (Lonn et al, 1990; Poot et al, 1992). Completion of replication in the rDNA array is severely retarded in sgs1Δ mutants (Kaliraman and Brill, 2002; Versini et al, 2003), and in vitro replication in Xenopus egg extracts in the absence of Xblm results in DNA strand breaks (Li et al, 2004). We asked whether Rmi1 was also required for normal S-phase progression. Using cells released synchronously from a G1 arrest, we could not detect a significant defect in bulk DNA synthesis, as assessed by flow cytometry (Supplementary Figure S4). However, asynchronous rmi1Δ cultures exhibited an accumulation of budded cells with one nucleus, suggesting a delay in the late S/G2 phase of the cell cycle (Figure 3A). These observations are similar to those made with top3Δ strains (Gangloff et al, 1994; Chakraverty et al, 2001) and may indicate a checkpoint-dependent mitotic delay. We assayed for activation of the checkpoint kinase Rad53 in these cells, analyzing both the phosphorylation-dependent mobility shift of Rad53 and Rad53 kinase activity (Figure 3B and C). We found that rmi1Δ mutants displayed a modest mobility shift of Rad53 when released from a G1 arrest in the absence of any DNA-damaging agent (Figure 3B). This mobility shift is due to phosphorylation and correlates with activation of Rad53 kinase activity (Pellicioli et al, 1999). We measured Rad53 activation directly using an in situ kinase assay. Activation of Rad53 in rmi1Δ was clearly evident in this assay, even in the sample from the asynchronous culture and from the G1-arrested culture (Figure 3C). Activation of Rad53 was not evident in wild-type cells in either assay. The precocious Rad53 checkpoint activation is likely the cause of the mitotic delay observed in rmi1Δ, suggesting that DNA damage is arising in cells lacking Rmi1 during an unperturbed cell cycle. Consistent with this interpretation, we found that rmi1Δ is synthetic lethal with rad53-11 (Figure 3D), a checkpoint defective allele of RAD53 (Weinert et al, 1994), indicating that an intact checkpoint response is essential for the viability of cells lacking Rmi1.
Figure 3.
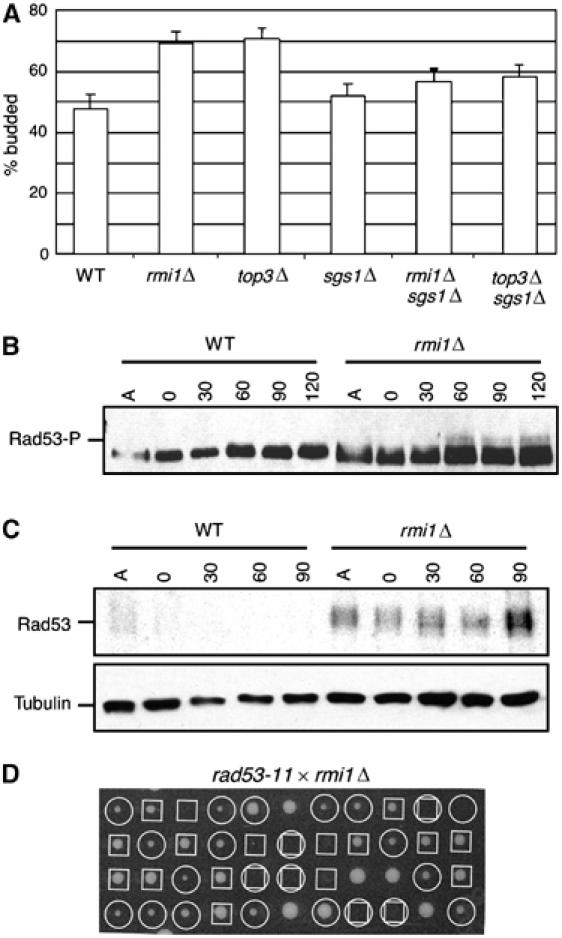
rmi1Δ mutants exhibit Rad53 checkpoint activation during an unperturbed cell cycle. (A) Asynchronous cultures of wild type (WT), rmi1Δ, top3Δ, sgs1Δ, rmi1Δ sgs1Δ, and top3Δ sgs1Δ were examined microscopically to determine the % of cells with a bud. (B) Logarithmically growing cultures were arrested in G1 with alpha factor and released into fresh YPD media. At the indicated times, samples were fixed with TCA, extracts were fractionated on SDS–PAGE, and immunoblotted to detect Rad53. The position of the activated phosphorylated Rad53 is indicated. (C) Samples prepared as in panel B were fractionated on SDS–PAGE for in situ kinase assay of Rad53 (upper panel). A parallel blot was probed with anti-tubulin antibody as a loading control (lower panel). (D) An rmi1Δ∷kanMX6 strain was crossed to a rad53-11∷URA3 strain. The resulting diploids were sporulated and tetrads were dissected on YPD. The genotypes of the resulting colonies are indicated with boxes (□) for rmi1Δ∷kanMX6 and with circles (○) for rad53-11∷URA3. Inferred double mutants are indicated with a box and circle.
We investigated the requirement for other checkpoint proteins in rmi1Δ mutants. We found that deletion of the G1 and G2 DNA damage checkpoint genes RAD24 or RAD9 had no detectable effect on the rmi1Δ mutant. However, deletion of the S-phase checkpoint genes MRC1, TOF1, or CSM3 in the rmi1Δ mutant caused a synthetic sick phenotype (Table I). Therefore, cells lacking RMI1 require the S-phase checkpoint response for optimal growth, suggesting that the DNA damage caused by deletion of RMI1 results from DNA replication defects.
rmi1Δ mutants exhibit increased levels of Rad52 relocalization and genomic instability
RAD52 is essential for efficient homologous recombination. Rad52 relocalizes from a diffuse nuclear localization to distinct subnuclear foci in response to DNA damage, particularly double-strand breaks (DSBs) (Lisby et al, 2001, 2003, 2004). Rad52 tagged with yellow fluorescent protein (Lisby et al, 2003, 2004) was visualized by fluorescence microscopy in asynchronous mitotic haploid cells. As shown in Figure 4A, cells lacking RMI1 display subnuclear Rad52 foci, whereas wild-type cells show infrequent and transient foci. Quantification of the data (Figure 4B) showed that rmi1Δ, sgs1Δ, and top3Δ all have elevated levels of spontaneous Rad52 focus formation, indicating the presence of DNA damage requiring homologous recombination for repair, likely DSBs. Elevated levels of Rad52 foci were observed both in S/G2/M (i.e. budded) cells and in G1 cells. Together with the data indicating that Rad53 is activated in rmi1Δ mutants, these results suggest that DNA replication in the absence of an intact Sgs1/Top3/Rmi1 pathway causes DNA lesions that result in genomic instability (Gangloff et al, 1994; Myung et al, 2001b; Ajima et al, 2002), similar to the effect observed in Xenopus egg extracts, in which replication in the absence of Xblm causes DNA strand breaks (Li et al, 2004).
Figure 4.
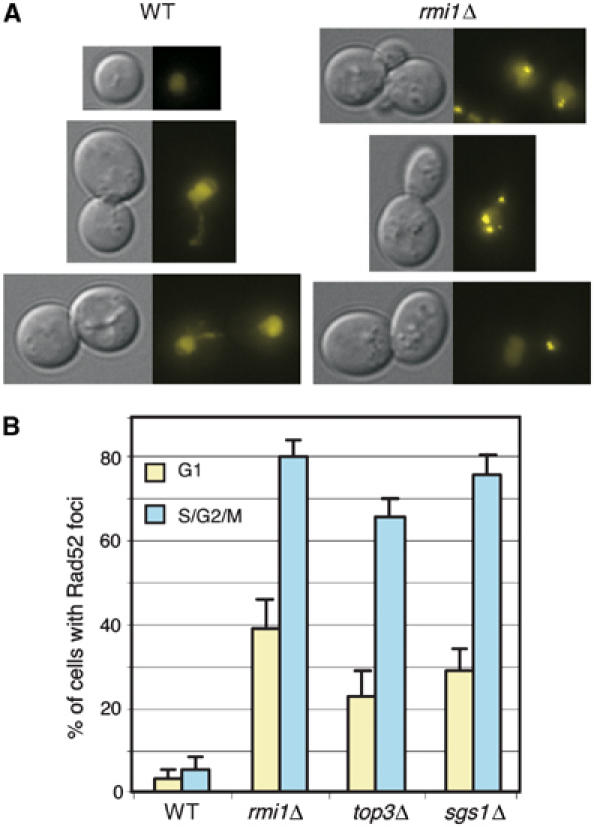
Spontaneous Rad52 focus formation in rmi1Δ cells. (A) Logarithmically growing cells expressing Rad52-YFP were visualized by fluorescence microscopy. For each pair of images, the left panel is a DIC image and the right panel is a fluorescence image showing Rad52-YFP. Representative cells are shown. (B) The percentage of cells with Rad52 foci was determined for the indicated strains. G1 cells with Rad52 foci are represented by the gold bars and S/G2/M cells with Rad52 foci are represented by the blue bars. WT, wild type.
To assess the effect of the DNA damage that arises in rmi1Δ mutants, we applied two assays for genomic instability. Both SGS1 and TOP3 are suppressors of homologous recombination (Shor et al, 2002). We tested the effect of deletion of RMI1 on homologous recombination using a LEU2 direct repeat assay (Smith and Rothstein, 1999). Consistent with the observation that cells lacking RMI1 have high levels of Rad52 foci, we found that rmi1Δ cells have an increased rate of recombination (Figure 5A), approximately six-fold higher than wild type. We also measured the rate of GCRs in rmi1Δ, using an assay that detects large interstitial deletions, translocations, chromosome fusions, and loss of a chromosome arm (Myung et al, 2001a). In this assay (Figure 5B), sgs1Δ and top3Δ showed increased GCR rates of approximately 30-fold over wild type, similar to reported values (Myung et al, 2001b). In contrast, GCR rates in rmi1Δ were more than 150 times wild-type levels. Thus, Rmi1 is a critical suppressor of GCRs.
Figure 5.
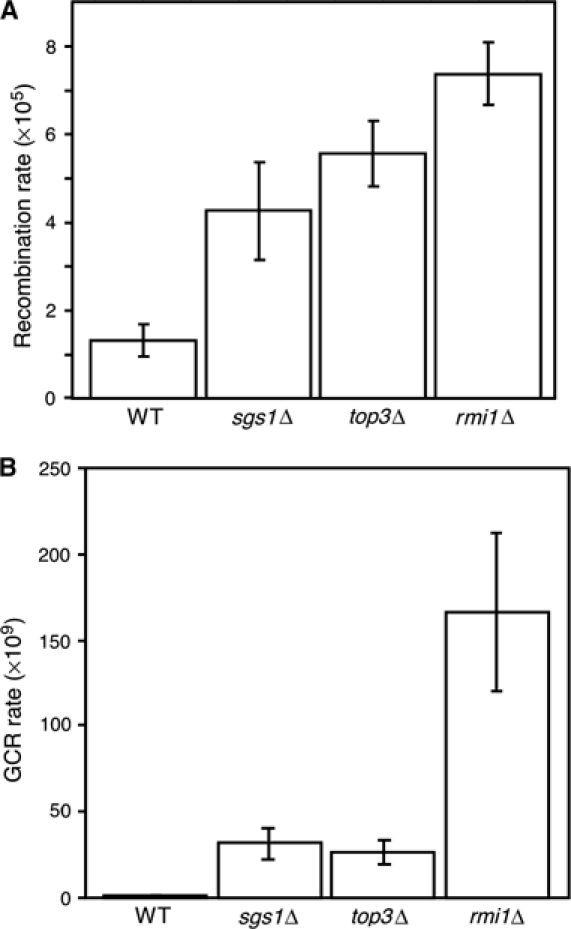
Deletion of RMI1 causes genomic instability. (A) Recombination rate was measured using a direct repeat recombination assay. The average and standard deviation of three fluctuation tests are shown for each strain. (B) GCR rate was measured. The average and standard deviation of four fluctuation tests are shown for each strain. WT, wild type.
RMI1 is required for the response to DNA damage
Both SGS1 and TOP3 are important for the response to DNA damage (Stewart et al, 1997; Davey et al, 1998; Frei and Gasser, 2000; Chakraverty et al, 2001); therefore, we tested whether deletion of RMI1 caused sensitivity to DNA-damaging agents (Figure 6A and B). The rmi1Δ, like top3Δ, displayed slow growth on YPD. The presence of the alkylating agent MMS (at 0.004%) or the replication inhibitor HU (at 10 mM) reduced colony formation by rmi1Δ by at least an order of magnitude, indicating that the rmi1Δ mutant is sensitive to DNA damage and replication stress. Wild-type cells were unaffected by the levels of MMS and HU used. We also tested whether rmi1Δ loses viability during transient exposure to the same concentrations of MMS or HU. The rmi1Δ mutant rapidly lost viability during exposure to MMS. During transient exposure to 10 mM HU, the rmi1Δ mutant displayed little loss of viability, although the growth of rmi1Δ was significantly impaired (Figure 6B). These results are reminiscent of top3Δ, which displays much greater sensitivity to transient MMS exposure than it does to transient HU exposure (Chakraverty et al, 2001; Oakley et al, 2002). The DNA damage sensitivity and loss of viability of rmi1Δ were suppressed by deletion of SGS1, with the double mutant displaying growth similar to that of sgs1Δ.
Figure 6.
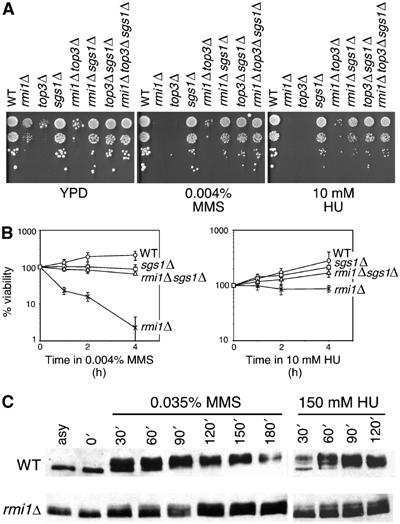
Rmi1 is required for the DNA damage response. (A) Serial dilutions (10-fold) of cultures of the indicated mutants were spotted on YPD, YPD containing 0.004% (v/v) MMS, or YPD containing 10 mM HU. All plates were incubated at 30°C for 2–3 days. (B) Logarithmically growing cultures of the indicated mutants were incubated in YPD containing 0.004% (v/v) MMS or 10 mM HU at 30°C. At the indicated times, samples were withdrawn and plated on YPD to determine the number of viable cells. The percentage of viable cells relative to the number of viable cells at t=0 is shown. Plots represent the average of three experiments, and error bars span 1 s.d. (C) Logarithmically growing cultures were arrested in G1 with alpha factor and released into medium containing either 0.035% (v/v) MMS or 150 mM HU. At the indicated times, samples were fixed and extracts fractionated by SDS–PAGE. Following transfer, the immunoblot was probed with anti-Rad53 antibody. WT, wild type.
Top3 is important for full activation of Rad53 in response to DNA damage (Chakraverty et al, 2001) while Sgs1 is necessary for Rad53 activation in the absence of Rad24 (Frei and Gasser, 2000; Bjergbaek et al, 2005). We tested whether RMI1 was also important for Rad53 activation. Rad53 activation was measured after treatment with HU or MMS (Figure 6C). Wild-type cells showed a robust checkpoint response, resulting in phosphorylation-dependent mobility shift of Rad53. By contrast, rmi1Δ mutants showed a defect in Rad53 activation in response to both HU and MMS, as evidenced by incomplete phosphorylation of Rad53. This defect can be suppressed by mutation of SGS1 (data not shown), similar to the suppression of the Rad53 activation defect in a top3Δ mutant by deletion of SGS1 (Chakraverty et al, 2001). Thus, in addition to causing DNA damage during S phase, deletion of RMI1 impedes full checkpoint activation when cells are challenged with exogenous damaging agents, suggesting that like Sgs1 and Top3, Rmi1 is upstream of Rad53 in the S-phase checkpoint response.
Evolutionary conservation of Rmi1
Homologues of Sgs1 and Top3 are found throughout Eucaryota. Using local alignment searches, we identified homologues of Rmi1 in six other yeast species. Sequence alignments of yeast Rmi1 homologues indicated that these proteins share three blocks of high sequence similarity (Figure 7A and Supplementary Figure S5).
Figure 7.
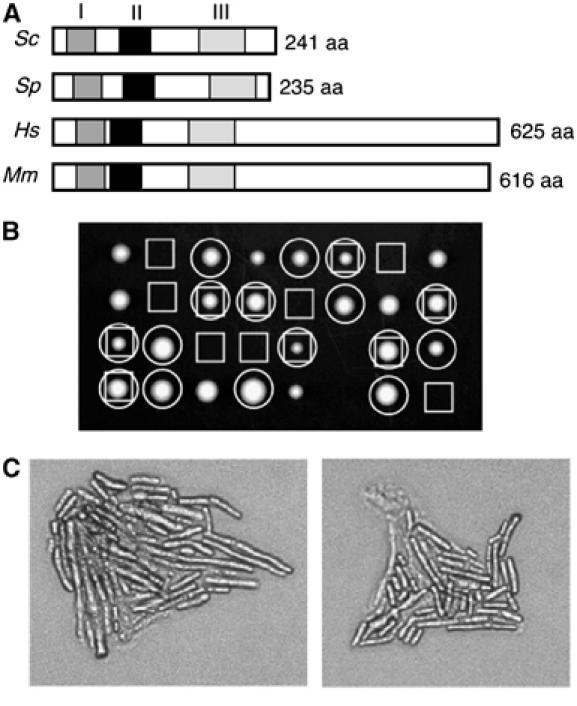
Rmi1 homologues. (A) Schematic diagrams of Rmi1 homologues from S. cerevisiae (Sc), S. pombe (Sp), Homo sapiens (Hs), and Mus musculus (Mm). Regions of high sequence identity are indicated by the three shaded boxes. (B) S. pombe rmi1+ is a functional homologue of RMI1. rmi1Δ∷G418R rqh1Δ∷ura4+ was crossed to rmi1+rqh1+ and tetrads were dissected on YE5S. The genotypes of the resulting colonies are indicated with boxes (□) for (inferred) rmi1Δ∷G418R and with circles (○) for rqh1Δ∷ura4+. (C) Micrographs of rmi1Δ∷G418R rqh1Δ∷ura4+ microcolonies from panel C.
In fission yeast, the top3+ gene is essential for viability (Goodwin et al, 1999; Maftahi et al, 1999), a phenotype that is suppressed by deletion of the fission yeast RecQ homologue rqh1+ (Goodwin et al, 1999; Maftahi et al, 1999). We asked whether the fission yeast rmi1+ gene is, like top3+, essential for viability by replacing the rmi1+ ORF with a G418 resistance gene in a haploid strain carrying a deletion of the rqh1+ gene (rqh1Δ∷ura4+). This strain was viable, indicating that rmi1+ is not essential in an rqh1Δ background. The rmi1Δ∷G418R rqh1Δ∷ura4+ strain was crossed to a wild-type strain and meiotic progeny were examined following tetrad dissection (Figure 7B). All inferred rmi1Δ single mutants failed to form colonies, indicating that rmi1+ is an essential gene. Examination of the resulting microcolonies revealed that the rmi1Δ cells go through several divisions before arresting with an elongated morphology (Figure 7C), a phenotype similar to that found with top3Δ mutants (Maftahi et al, 1999). These results suggest that the fission yeast rmi1+ is the functional homologue of budding yeast RMI1.
We extended our homology search to metazoan species and found that homologues were not readily identified using local alignment searches such as BLAST. We used the three regions of sequence similarity from the yeast analysis to build a hidden Markov model (HMM) for each region. The HMMs were then used to search the NCBI nonredundant protein database, resulting in the identification of homologous proteins in humans and mice. These putative Rmi1 homologues contain the three conserved regions that were evident in the yeast homologues, and also contain a C-terminal extension (Figure 7A and Supplementary Figure S6). The human Rmi1 homologue is identical to the recently described BLAP75, a BLM-associated protein that is important for genome integrity in human cells (Yin et al, 2005).
Discussion
Rmi1 is a novel member of the Sgs1/Top3 complex
We have found that Rmi1 physically associates with both Sgs1 and Top3. Fractionation of cell extracts by gel filtration chromatography and co-immunoprecipitation experiments indicated that Rmi1 is in a high-molecular-weight heteromeric complex that contains both Sgs1 and Top3. In the absence of Rmi1, the levels of Sgs1 decrease, an effect that is also observed in the absence of the Sgs1 binding partner Top3. This suggests that interactions with both Rmi1 and Top3 are important for Sgs1 stability. Finally, rmi1Δ shares many phenotypes with top3Δ, including slow growth and DNA damage sensitivities that are suppressed by deletion of SGS1, indicating that Rmi1 is required for Top3 function in vivo (or vice versa). The simplest interpretation of these data is that Rmi1 is a member of the functional Sgs1/Top3 complex. The exact stoichiometry and architecture of the native cellular Sgs1/Top3/Rmi1 complex remains elusive as Sgs1 is present in a very high-molecular-weight complex of some 1.3 MDa (Fricke et al, 2001), suggesting that other proteins may also be present. Thus, the interaction of Rmi1 with Sgs1/Top3 may be direct or indirect.
The reduction of Sgs1 steady-state protein levels in an rmi1Δ or top3Δ background is especially intriguing given that deleting SGS1 in these backgrounds improves cell viability. Therefore, it appears that even a very low level of Sgs1 is detrimental to cells lacking Rmi1 or Top3. Although abolishing the helicase activity of Sgs1 improves viability of rmi1Δ (data not shown) and top3Δ mutants (Mullen et al, 2001), levels of helicase-dead Sgs1 were still greatly reduced in rmi1Δ and top3Δ compared to wild type (Figure 2E), indicating that the reduced Sgs1 levels are unlikely to be a response to Sgs1 activity. The mechanism by which Sgs1 levels are reduced is currently unknown, but the phenomenon appears to be evolutionarily conserved in that deletion of top3+ in S. pombe results in a reduction in the level of a helicase-inactive Rqh1 (Laursen et al, 2003). Although several models are consistent with our data, the simplest explanation is that absence of either Rmi1 or Top3 from the Sgs1/Top3/Rmi1 complex destabilizes Sgs1 but enough Sgs1 activity remains to cause reduced viability of rmi1Δ or top3Δ cells.
In vitro experiments using purified BLM and TOP3α have demonstrated that together these proteins can resolve a recombination intermediate containing a double Holliday junction (Wu and Hickson, 2003). Deletion of RMI1 results in a phenotype very similar to that displayed by top3Δ mutants, suggesting that Rmi1 may be important for the biochemical activity of Top3. However, deletion of either TOP3 or RMI1 causes a reduction in Sgs1 levels and so is likely to also compromise Sgs1 activity. Additionally, Rmi1 binds to both Top3 and Sgs1, further indicating that Rmi1 may influence the activity of both complex members. It will be of considerable interest to determine if and how the presence of Rmi1 affects the biochemical properties of RecQ/Top3 complexes.
Accumulation of DNA damage in cells lacking RMI1
The mitotic cell cycle delay, precocious Rad53 activation, and synthetic genetic interactions with genes required for DNA replication fork stability and the S-phase checkpoint all point to the accumulation of DNA lesions. The genetic suppression data suggest that these lesions are generated from the processing of recombination intermediates by Sgs1. The exact nature of these lesions has yet to be determined but the elevated levels of GCRs, Rad52 foci, and recombination provide insight as to what these lesions may be. GCRs can take the form of nonreciprocal translocations, interstitial deletions, chromosome fusions, and loss of a chromosome arm followed by de novo telomere addition (Chen et al, 1998; Myung et al, 2001a). All of these rearrangements require the creation of DSBs. Thus, we know that at least a significant fraction of the lesions generated in an rmi1Δ mutant are, or result in, DSBs. Consistent with this hypothesis, Rad52 relocalizes into DSB repair foci in rmi1Δ, presumably reflecting the observed increase in recombination frequency. Recent work indicates that abnormal recombination structures accumulate in sgs1Δ and top3Δ mutants when alkylation damage is present (Liberi et al, 2005). Accumulation of these structures was not detected in the absence of DNA damage, however. We found increased levels of Rad52 recombination repair foci in cells lacking Rmi1, Sgs1, or Top3 in an otherwise unperturbed cell cycle, which argues that Sgs1/Top3/Rmi1 function is required to prevent DNA damage from occurring during normal cell cycle progression. Interestingly, we found that the direct repeat recombination rate is higher in an rmi1Δ mutant than in an sgs1Δ mutant; yet, Rad52 foci form to the same extent in both. It has been shown that multiple DSBs can localize to one Rad52 focus; thus, the formation of Rad52 foci may not be directly proportional to the extent of DNA damage (Lisby et al, 2003). As suggested by their slower growth rate and precocious checkpoint activation, it is likely that rmi1Δ mutants accumulate more damage than sgs1Δ mutants, resulting in the higher rate of recombination observed. Alternatively, the DNA lesions present in rmi1Δ cells may simply be more recombinogenic than those present in sgs1Δ cells.
Defects in Rad53 checkpoint activation
Similar to top3Δ mutants (Chakraverty et al, 2001), cells lacking RMI1 are defective in fully activating Rad53 in response to DNA damage induced by HU or MMS, a defect that can be suppressed by the mutation of SGS1. The Sgs1/Top3/Rmi1 complex may be needed to process DNA lesions in order to generate DNA structures that can be recognized by the DNA damage checkpoint machinery, allowing for checkpoint activation. Sgs1 function in the absence of Rmi1 or Top3 could generate a toxic DNA intermediate that is not efficiently recognized by the checkpoint machinery. Alternatively, Rmi1 could be required in a more direct way to facilitate Rad53 activation, perhaps by mediating localization of Rad53 to DNA lesions or stalled replication forks. Either model is consistent with the weak Rad53 activation seen in the Rad53 protein blots of extracts from rmi1Δ mutants treated with MMS or HU (Figure 6A). Although the failure of rmi1Δ mutants to support wild-type checkpoint activation may seem at odds with our data demonstrating precocious checkpoint activation in rmi1Δ in the absence of DNA-damaging agents, it is worth noting that Rad53 is in fact activated in rmi1Δ in response to MMS or HU, but to lower levels than in wild-type cells. Thus, the spontaneous damage present in rmi1Δ might cause more robust checkpoint activation if rmi1Δ mutants were not compromised in checkpoint activation. In this regard, it is interesting that we see evidence of spontaneous DNA damage in G1 rmi1Δ cells (Figure 4B). In wild-type cells, DNA damage accrued during G1 does not induce Rad52 foci formation until cells progress into S phase (Lisby et al, 2004). The presence of Rad52 foci in rmi1Δ G1 cells is likely due to progression through mitosis despite the presence of DNA lesions. Although a single DSB is typically sufficient to prevent passage through mitosis for several cell cycles (Lee et al, 1998), we would expect this checkpoint-mediated mitotic delay to be abrogated in mutants such as rmi1Δ that display compromised checkpoint activation in response to DNA damage. Progression through mitosis in the presence of DNA lesions could be a principal cause of the poor viability of rmi1Δ mutants.
Recent data suggest that top3Δ mutants appear to have a compromised checkpoint due to impaired progression into and through S phase (Bjergbaek et al, 2005). A rad24Δ top3Δ double mutant, which does not exhibit these S-phase defects or the slow growth exhibited by a top3Δ mutant, is fully competent in activating Rad53 upon exposure to HU (Bjergbaek et al, 2005). Flow cytometric analysis of rmi1Δ mutants failed to detect a significant delay in progression into and through S phase, and we found that deletion of RAD24 does not suppress the growth defect of an rmi1Δ mutant. Thus, the underlying mechanism by which the checkpoint is compromised in rmi1Δ may differ from that in top3Δ. Indeed, there are several aspects of the rmi1Δ phenotype that are different from that of top3Δ. rmi1Δ mutants grow slightly better than top3Δ mutants and there is a large difference in their GCR rates. These phenotypic differences are not surprising, given that loss of Top3 from the Rmi1/Sgs1/Top3 complex is likely to be biochemically distinct from loss of Rmi1.
RMI1 function in higher eukaryotes
We have identified homologues of budding yeast Rmi1 in several yeast species, as well as mouse and human. The presence of three conserved regions in diverse species suggests that these regions may constitute functional domains. Human Rmi1 has similarity to nucleic acid binding OB-folds (Koonin et al, 2000) extending through conserved regions II and III, raising the possibility that Rmi1 might bind DNA directly. Of particular interest, the putative human Rmi1 homologue that we identified by sequence similarity is identical to the recently described BLAP75 (Yin et al, 2005). Like Rmi1 in yeast, BLAP75 is an integral component of RecQ/Topo III complexes in human cells, and depletion of BLAP75 results in genome instability in the form of increased sister chromatid exchanges (Yin et al, 2005). Thus, the role of Rmi1 in RecQ/Top3 function appears to be conserved in all eukaryotes. Budding yeast Rmi1 is an important suppressor of DNA damage during S phase, and is also required for a robust checkpoint response to DNA damage and replication stress. It will be of great interest to determine if these functions are conserved in hRmi1/BLAP75, and if hRMI1/BLAP75 polymorphisms are associated with human cancers.
Materials and methods
Yeast strains and media
Yeast strains used in this study are derivatives of BY4741 (Brachmann et al, 1998) and are listed in Supplementary Table S1. Nonessential haploid deletion strains were made by the Saccharomyces Gene Deletion Project (Winzeler et al, 1999). Standard yeast media and growth conditions were used (Moreno et al, 1991; Sherman, 1991). For cell synchrony experiments, cells were arrested in G1 by culturing in the presence of 2 μg/ml alpha mating factor for 2 h at 30°C in YPD, pH 3.9. Cells were released into the cell cycle by harvesting, washing, and resuspending in YPD.
SGAM analysis
SGAM analysis was carried out as described (Tong et al, 2001, 2004; Jorgensen et al, 2002) to map the location of the extragenic suppressor in the rmi1Δ∷natR query strain (Y5646).
Epitope tagging, immunoprecipitation, immunoblotting, and gel filtration
Immunoprecipitation was performed essentially as described (Bellaoui et al, 2003). Purified rabbit IgG agarose (Sigma) was used to immunoprecipitate TAP-tagged proteins, and immunoprecipitates were washed extensively with buffer containing 100 mM NaCl. Proteins were resolved on 7.5% polyacrylamide–SDS gels, transferred to nitrocellulose membranes, and subjected to immunoblot analysis with anti-HA (16B12; Covance), anti-VSV (P5D4; Roche), anti-tubulin (TAT-1) (Woods et al, 1989), or anti-TAP (PAP: peroxidase–anti-peroxidase soluble complex; Sigma) antibodies. Immunoblots were developed using Supersignal ECL (Pierce). For detection of Rad53 and in situ autophosphorylation assays, cells were fixed and extracts were prepared essentially as described (Pellicioli et al, 1999). Proteins were separated on 7.5% polyacrylamide–SDS gels, and immunoblots were probed with anti-RAD53 (yC-19; Santa Cruz). Gel filtration of extracts of GBY635 was carried out on a Superose 6 HR 5/20 column, essentially as described (Fricke et al, 2001).
Fluorescence microscopy
Cells containing the plasmid pWJ1344, which expresses Rad52-YFP, were grown to logarithmic phase at 23°C in SC medium lacking leucine. Microscopy was performed essentially as described (Lisby et al, 2001, 2003, 2004).
Recombination and GCR assays
Recombination assays were performed using a LEU2 direct repeat, as described (Bellaoui et al, 2003). Fluctuation tests of five colonies were repeated three times. GCR assays were performed as described (Myung et al, 2001a). Fluctuation tests of three colonies were repeated at least four times.
MMS and HU sensitivity measurements
Cells were grown in YPD, serially diluted, spotted onto plates, and incubated at 30°C. MMS (Aldrich) plates contained 0.004% (v/v) MMS in YPD and were used within 24 h of preparation. HU plates contained 10 mM HU in YPD. Viability following exposure to 0.004% MMS or 10 mM HU in liquid culture was determined as described (Bellaoui et al, 2003).
Supplementary Material
Supplementary Material
Acknowledgments
We thank Steve Brill for communicating results prior to publication, Igor Stagljar and Steve Brill for strains and plasmids, Dave Alvaro, Jackie Barlow, and Rebecca Burgess for assistance with Rad52 relocalization experiments, and Dan Durocher for constructive comments on the manuscript. MC was supported by an Ontario Graduate Scholarship. This work was supported by the Canadian Institutes of Health Research (to GWB and CB), Genome Canada and Genome Ontario (to CB), and the National Institutes of Health (CA072647 to GAF and GM50237 to RR). GWB is a Research Scientist of the National Cancer Institute of Canada.
References
- Ajima J, Umezu K, Maki H (2002) Elevated incidence of loss of heterozygosity (LOH) in an sgs1 mutant of Saccharomyces cerevisiae: roles of yeast RecQ helicase in suppression of aneuploidy, interchromosomal rearrangement, and the simultaneous incidence of both events during mitotic growth. Mutat Res 504: 157–172 [DOI] [PubMed] [Google Scholar]
- Bellaoui M, Chang M, Ou J, Xu H, Boone C, Brown GW (2003) Elg1 forms an alternative RFC complex important for DNA replication and genome integrity. EMBO J 22: 4304–4313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett RJ, Noirot-Gros MF, Wang JC (2000) Interaction between yeast sgs1 helicase and DNA topoisomerase III. J Biol Chem 275: 26898–26905 [DOI] [PubMed] [Google Scholar]
- Bjergbaek L, Cobb JA, Tsai-Pflugfelder M, Gasser SM (2005) Mechanistically distinct roles for Sgs1p in checkpoint activation and replication fork maintenance. EMBO J 24: 405–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD (1998) Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14: 115–132 [DOI] [PubMed] [Google Scholar]
- Chakraverty RK, Kearsey JM, Oakley TJ, Grenon M, de La Torre Ruiz MA, Lowndes NF, Hickson ID (2001) Topoisomerase III acts upstream of Rad53p in the S-phase DNA damage checkpoint. Mol Cell Biol 21: 7150–7162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M, Bellaoui M, Boone C, Brown GW (2002) A genome-wide screen for methyl methanesulfonate-sensitive mutants reveals genes required for S phase progression in the presence of DNA damage. Proc Natl Acad Sci USA 99: 16934–16939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Umezu K, Kolodner RD (1998) Chromosomal rearrangements occur in S. cerevisiae rfa1 mutator mutants due to mutagenic lesions processed by double-strand-break repair. Mol Cell 2: 9–22 [DOI] [PubMed] [Google Scholar]
- Davey S, Han CS, Ramer SA, Klassen JC, Jacobson A, Eisenberger A, Hopkins KM, Lieberman HB, Freyer GA (1998) Fission yeast rad12+ regulates cell cycle checkpoint control and is homologous to the Bloom's syndrome disease gene. Mol Cell Biol 18: 2721–2728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, Ciocci S, Proytcheva M, German J (1995) The Bloom's syndrome gene product is homologous to RecQ helicases. Cell 83: 655–666 [DOI] [PubMed] [Google Scholar]
- Frei C, Gasser SM (2000) The yeast Sgs1p helicase acts upstream of Rad53p in the DNA replication checkpoint and colocalizes with Rad53p in S-phase-specific foci. Genes Dev 14: 81–96 [PMC free article] [PubMed] [Google Scholar]
- Fricke WM, Kaliraman V, Brill SJ (2001) Mapping the DNA topoisomerase III binding domain of the Sgs1 DNA helicase. J Biol Chem 276: 8848–8855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff S, de Massy B, Arthur L, Rothstein R, Fabre F (1999) The essential role of yeast topoisomerase III in meiosis depends on recombination. EMBO J 18: 1701–1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff S, McDonald JP, Bendixen C, Arthur L, Rothstein R (1994) The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: a potential eukaryotic reverse gyrase. Mol Cell Biol 14: 8391–8398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin A, Wang SW, Toda T, Norbury C, Hickson ID (1999) Topoisomerase III is essential for accurate nuclear division in Schizosaccharomyces pombe. Nucleic Acids Res 27: 4050–4058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmon FG, DiGate RJ, Kowalczykowski SC (1999) RecQ helicase and topoisomerase III comprise a novel DNA strand passage function: a conserved mechanism for control of DNA recombination. Mol Cell 3: 611–620 [DOI] [PubMed] [Google Scholar]
- Hickson ID (2003) RecQ helicases: caretakers of the genome. Nat Rev Cancer 3: 169–178 [DOI] [PubMed] [Google Scholar]
- Ira G, Malkova A, Liberi G, Foiani M, Haber JE (2003) Srs2 and Sgs1–Top3 suppress crossovers during double-strand break repair in yeast. Cell 115: 401–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson FB, Lombard DB, Neff NF, Mastrangelo MA, Dewolf W, Ellis NA, Marciniak RA, Yin Y, Jaenisch R, Guarente L (2000) Association of the Bloom syndrome protein with topoisomerase IIIalpha in somatic and meiotic cells. Cancer Res 60: 1162–1167 [PubMed] [Google Scholar]
- Jorgensen P, Nelson B, Robinson MD, Chen Y, Andrews B, Tyers M, Boone C (2002) High-resolution genetic mapping with ordered arrays of Saccharomyces cerevisiae deletion mutants. Genetics 162: 1091–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaliraman V, Brill SJ (2002) Role of SGS1 and SLX4 in maintaining rDNA structure in Saccharomyces cerevisiae. Curr Genet 41: 389–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitao S, Lindor NM, Shiratori M, Furuichi Y, Shimamoto A (1999) Rothmund–Thomson syndrome responsible gene, RECQL4: genomic structure and products. Genomics 61: 268–276 [DOI] [PubMed] [Google Scholar]
- Klein HL (2001) Mutations in recombinational repair and in checkpoint control genes suppress the lethal combination of srs2Delta with other DNA repair genes in Saccharomyces cerevisiae. Genetics 157: 557–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Wolf YI, Aravind L (2000) Protein fold recognition using sequence profiles and its application in structural genomics. Adv Protein Chem 54: 245–275 [DOI] [PubMed] [Google Scholar]
- Laursen LV, Ampatzidou E, Andersen AH, Murray JM (2003) Role for the fission yeast RecQ helicase in DNA repair in G2. Mol Cell Biol 23: 3692–3705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD, Haber JE (1998) Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 94: 399–409 [DOI] [PubMed] [Google Scholar]
- Li W, Kim SM, Lee J, Dunphy WG (2004) Absence of BLM leads to accumulation of chromosomal DNA breaks during both unperturbed and disrupted S phases. J Cell Biol 165: 801–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberi G, Maffioletti G, Lucca C, Chiolo I, Baryshnikova A, Cotta-Ramusino C, Lopes M, Pellicioli A, Haber JE, Foiani M (2005) Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev 19: 339–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M, Barlow JH, Burgess RC, Rothstein R (2004) Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 118: 699–713 [DOI] [PubMed] [Google Scholar]
- Lisby M, Mortensen UH, Rothstein R (2003) Colocalization of multiple DNA double-strand breaks at a single Rad52 repair centre. Nat Cell Biol 5: 572–577 [DOI] [PubMed] [Google Scholar]
- Lisby M, Rothstein R, Mortensen UH (2001) Rad52 forms DNA repair and recombination centers during S phase. Proc Natl Acad Sci USA 98: 8276–8282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonn U, Lonn S, Nylen U, Winblad G, German J (1990) An abnormal profile of DNA replication intermediates in Bloom's syndrome. Cancer Res 50: 3141–3145 [PubMed] [Google Scholar]
- Maftahi M, Han CS, Langston LD, Hope JC, Zigouras N, Freyer GA (1999) The top3(+) gene is essential in Schizosaccharomyces pombe and the lethality associated with its loss is caused by Rad12 helicase activity. Nucleic Acids Res 27: 4715–4724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno S, Klar A, Nurse P (1991) Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol 194: 795–823 [DOI] [PubMed] [Google Scholar]
- Mullen JR, Kaliraman V, Ibrahim SS, Brill SJ (2001) Requirement for three novel protein complexes in the absence of the Sgs1 DNA helicase in Saccharomyces cerevisiae. Genetics 157: 103–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung K, Chen C, Kolodner RD (2001a) Multiple pathways cooperate in the suppression of genome instability in Saccharomyces cerevisiae. Nature 411: 1073–1076 [DOI] [PubMed] [Google Scholar]
- Myung K, Datta A, Chen C, Kolodner RD (2001b) SGS1, the Saccharomyces cerevisiae homologue of BLM and WRN, suppresses genome instability and homeologous recombination. Nat Genet 27: 113–116 [DOI] [PubMed] [Google Scholar]
- Myung K, Kolodner RD (2002) Suppression of genome instability by redundant S-phase checkpoint pathways in Saccharomyces cerevisiae. Proc Natl Acad Sci USA 99: 4500–4507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley TJ, Goodwin A, Chakraverty RK, Hickson ID (2002) Inactivation of homologous recombination suppresses defects in topoisomerase III-deficient mutants. DNA Repair (Amst) 1: 463–482 [DOI] [PubMed] [Google Scholar]
- Oakley TJ, Hickson ID (2002) Defending genome integrity during S-phase: putative roles for RecQ helicases and topoisomerase III. DNA Repair (Amst) 1: 175–207 [DOI] [PubMed] [Google Scholar]
- Onoda F, Seki M, Miyajima A, Enomoto T (2000) Elevation of sister chromatid exchange in Saccharomyces cerevisiae sgs1 disruptants and the relevance of the disruptants as a system to evaluate mutations in Bloom's syndrome gene. Mutat Res 459: 203–209 [DOI] [PubMed] [Google Scholar]
- Pellicioli A, Lucca C, Liberi G, Marini F, Lopes M, Plevani P, Romano A, Di Fiore PP, Foiani M (1999) Activation of Rad53 kinase in response to DNA damage and its effect in modulating phosphorylation of the lagging strand DNA polymerase. EMBO J 18: 6561–6572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poot M, Hoehn H, Runger TM, Martin GM (1992) Impaired S-phase transit of Werner syndrome cells expressed in lymphoblastoid cell lines. Exp Cell Res 202: 267–273 [DOI] [PubMed] [Google Scholar]
- Sherman F (1991) Getting started with yeast. Methods Enzymol 194: 3–21 [DOI] [PubMed] [Google Scholar]
- Shor E, Gangloff S, Wagner M, Weinstein J, Price G, Rothstein R (2002) Mutations in homologous recombination genes rescue top3 slow growth in Saccharomyces cerevisiae. Genetics 162: 647–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J, Rothstein R (1999) An allele of RFA1 suppresses RAD52-dependent double-strand break repair in Saccharomyces cerevisiae. Genetics 151: 447–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart E, Chapman CR, Al-Khodairy F, Carr AM, Enoch T (1997) rqh1+, a fission yeast gene related to the Bloom's and Werner's syndrome genes, is required for reversible S phase arrest. EMBO J 16: 2682–2692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Page N, Robinson M, Raghibizadeh S, Hogue CW, Bussey H, Andrews B, Tyers M, Boone C (2001) Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294: 2364–2368 [DOI] [PubMed] [Google Scholar]
- Tong AH, Lesage G, Bader GD, Ding H, Xu H, Xin X, Young J, Berriz GF, Brost RL, Chang M, Chen Y, Cheng X, Chua G, Friesen H, Goldberg DS, Haynes J, Humphries C, He G, Hussein S, Ke L, Krogan N, Li Z, Levinson JN, Lu H, Menard P, Munyana C, Parsons AB, Ryan O, Tonikian R, Roberts T, Sdicu AM, Shapiro J, Sheikh B, Suter B, Wong SL, Zhang LV, Zhu H, Burd CG, Munro S, Sander C, Rine J, Greenblatt J, Peter M, Bretscher A, Bell G, Roth FP, Brown GW, Andrews B, Bussey H, Boone C (2004) Global mapping of the yeast genetic interaction network. Science 303: 808–813 [DOI] [PubMed] [Google Scholar]
- Versini G, Comet I, Wu M, Hoopes L, Schwob E, Pasero P (2003) The yeast Sgs1 helicase is differentially required for genomic and ribosomal DNA replication. EMBO J 22: 1939–1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis JW, Chrebet G, Brodsky G, Rolfe M, Rothstein R (1989) A hyper-recombination mutation in S. cerevisiae identifies a novel eukaryotic topoisomerase. Cell 58: 409–419 [DOI] [PubMed] [Google Scholar]
- Watt PM, Hickson ID, Borts RH, Louis EJ (1996) SGS1, a homologue of the Bloom's and Werner's syndrome genes, is required for maintenance of genome stability in Saccharomyces cerevisiae. Genetics 144: 935–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt PM, Louis EJ, Borts RH, Hickson ID (1995) Sgs1: a eukaryotic homolog of E. coli RecQ that interacts with topoisomerase II in vivo and is required for faithful chromosome segregation. Cell 81: 253–260 [DOI] [PubMed] [Google Scholar]
- Weinert TA, Kiser GL, Hartwell LH (1994) Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev 8: 652–665 [DOI] [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Liebundguth N, Lockhart DJ, Lucau-Danila A, Lussier M, M'Rabet N, Menard P, Mittmann M, Pai C, Rebischung C, Revuelta JL, Riles L, Roberts CJ, Ross-MacDonald P, Scherens B, Snyder M, Sookhai-Mahadeo S, Storms RK, Veronneau S, Veot M, Volckaert G, Ward TR, Wysocki R, Yen GS, Yu K, Zimmermann K, Philippsen P, Johnston M, Davis RW (1999) Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285: 901–906 [DOI] [PubMed] [Google Scholar]
- Woods A, Sherwin T, Sasse R, MacRae TH, Baines AJ, Gull K (1989) Definition of individual components within the cytoskeleton of Trypanosoma brucei by a library of monoclonal antibodies. J Cell Sci 93: 491–500 [DOI] [PubMed] [Google Scholar]
- Wu L, Davies SL, North PS, Goulaouic H, Riou JF, Turley H, Gatter KC, Hickson ID (2000) The Bloom's syndrome gene product interacts with topoisomerase III. J Biol Chem 275: 9636–9644 [DOI] [PubMed] [Google Scholar]
- Wu L, Hickson ID (2003) The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature 426: 870–874 [DOI] [PubMed] [Google Scholar]
- Yamagata K, Kato J, Shimamoto A, Goto M, Furuichi Y, Ikeda H (1998) Bloom's and Werner's syndrome genes suppress hyperrecombination in yeast sgs1 mutant: implication for genomic instability in human diseases. Proc Natl Acad Sci USA 95: 8733–8738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J, Sobeck A, Xu C, Meetei AR, Hoatlin M, Li L, Wang W (2005) BLAP75, an essential component of Bloom's syndrome protein complexes that maintain genome integrity. EMBO J 24: 1465–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama F, Alisch R, Matthews S, Nakura J, Miki T, Ouais S, Martin GM, Mulligan J, Schellenberg GD (1996) Positional cloning of the Werner's syndrome gene. Science 272: 258–262 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material