

Abstract
Despite the discovery of actinide borohydride complexes over 80 years ago, no plutonium borohydride complexes have been structurally validated using single-crystal X-ray diffraction (XRD). Here we describe Pu2(H3BPtBu2BH3)6, the first example of a Pu(III) borohydride complex authenticated by XRD and NMR spectroscopy. Theoretical calculations (DFT, EDA, and QTAIM) and experimental comparisons of metal–boron distances suggest that metal–borohydride covalency in M2(H3BPtBu2BH3)6 complexes generally decreases in the order M = U(III) > Pu(III) > Ln(III).
Actinide borohydrides, complexes containing An–H–B bonds, were discovered during the Manhattan Project when volatile U(BH4)4 was investigated for isotopically enriching uranium in U-235.1,2 The first crystal structure of an actinide borohydride, again U(BH4)4, was reported several decades later.3,4 These data revealed for the first time the defining structural characteristics of these complexes with uranium coordinated exclusively by hydrogen atoms.5
Though many structurally determined Th and U borohydride complexes have been identified since the discovery of U(BH4)4,5−7 there are very few examples beyond uranium. Np(BH4)4 and Np(MeBH3)4 are the only two transuranium borohydride complexes to be characterized by single-crystal X-ray diffraction (Figure 1).8−14 Pu(BH4)4, the only known plutonium borohydride complex,15 is an unstable liquid at room temperature.11 Powder X-ray diffraction (XRD) data showed that Pu(BH4)4 crystallizes in the same space group and has similar unit cell parameters as Np(BH4)4,11,16 but no data indicating atomic positions were reported.
Figure 1.

Transuranium borohydride complexes structurally characterized by single-crystal XRD.
The dearth of transuranium borohydride structures has come into focus recently because there has been growing evidence of how metal–borohydride covalency can influence the structures and properties of trivalent actinide complexes, a phenomenon commonly associated with more conventional soft donor ligands.17−19 These effects have been observed in U(III) borohydride complexes,20−22 but little is known about how they manifest as the 5f-block is traversed into the transuranium elemental realm.
One set of difficulties in preparing molecular borohydride complexes with Pu concerns radiological safety and isotope availability limitations, as well as the known pyrophoricity of borohydrides when complexed with actinides.10,11,23−25 Another challenge is the instability of Pu(IV) in the presence of reducing borohydride ligands. The aforementioned Pu(BH4)4, for example, decomposes via reduction to form Pu(III) products that have yet to be characterized.11,16 Once reduced, traditional borohydrides like BH41– and BH3Me1– are too small to saturate the relatively large coordination sphere of Pu(III) to form homoleptic complexes soluble in organic solvents.5,7 Thus, the investigation of homoleptic Pu(III) borohydride complexes requires the development of borohydride ligands that can saturate the coordination sphere of this relatively large trivalent ion.26
We recently demonstrated how a class of larger borohydride ligands called phosphinodiboranates, which have the general formula H3BPR2BH31–, can be used to prepare U2(H3BPtBu2BH3)6 and Ln2(H3BPtBu2BH3)6.22,27−30 These dinuclear complexes are isostructural regardless of metal size, which permitted structural comparisons that revealed shorter than expected U–B distances.22 Subsequent calculations corroborated the structural observations and converged to suggest that the uranium–borohydride bonds have increased covalency compared to those with lanthanides.22 Aside from its larger size, H3BPtBu2BH31– was an ideal choice for extension to transuranium elements (that pose greater radiological hazards) because the U and Ln complexes have shown no appreciable volatility and they appear less susceptible to enflaming in air.28
Herein, we report how H3BPtBu2BH31– (tBu-PDB) was used to prepare the first Pu borohydride complex to be characterized by single-crystal XRD and NMR spectroscopy. Given the logistical and safety constraints associated with synthetic Pu chemistry in a fundamental research laboratory setting, we first had to develop procedures to prepare and crystallize the Pu complex on milligram scales. Using 9.0 mg of UI3(THF)424,31,32 as a test surrogate, we showed that reactions with 6.3 mg of K(H3BPtBu2BH3)33 in chlorobenzene, followed by crystallization from pentane, afforded red crystals of U2(H3BPtBu2BH3)6 (1) in yields as high as 79% (5.5 mg; Figure 2). Chlorobenzene, a solvent shown by Edelstein and co-workers to be compatible with the synthesis of actinide borohydrides,10 was used because we previously showed that metathesis reactions with several phosphinodiboranate salts are low yielding and often irreproducible in Et2O and THF.27,28 Moreover, mechanochemical methods used to prepare other tBu-PDB complexes22,29,30 were not amenable to reactions with powdered Pu salts because of the contamination risk.
Figure 2.
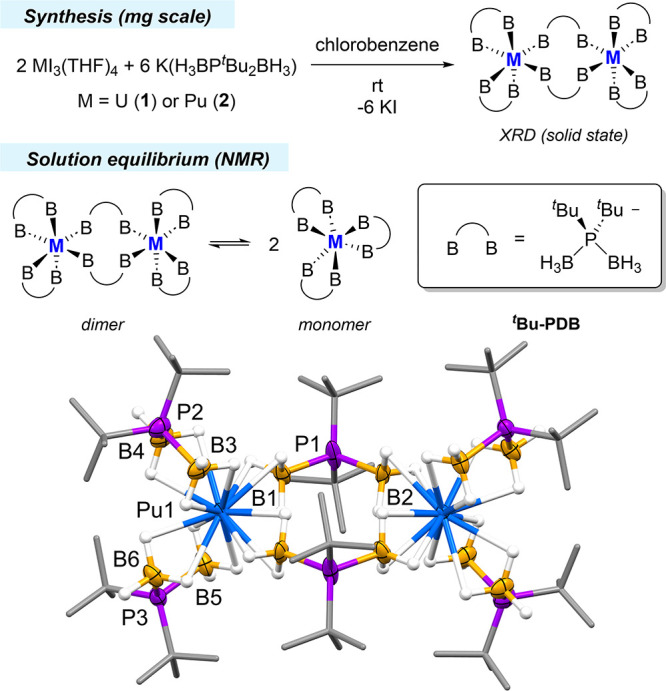
Top – Synthesis of M2(H3BPtBu2BH3)6 and comparison of solution and solid-state structures for M = U (1) and Pu (2). Bottom – Molecular structure of 2. Ellipsoids are drawn at 50%. Carbon atoms are shown as capped sticks, and hydrogen atoms attached to carbon were omitted from the figure.
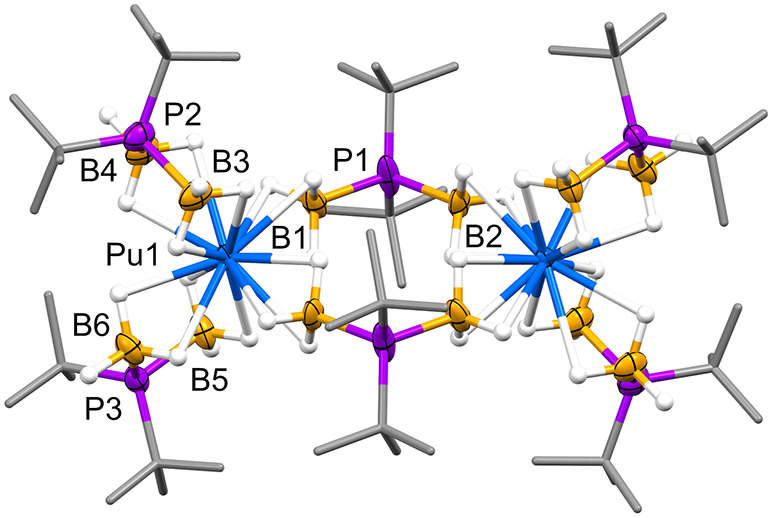
Repeating the same mg-scale procedure using PuI3(THF)4 instead of UI3(THF)4 reproducibly afforded blue crystals of Pu2(H3BPtBu2BH3)6 (2), as confirmed by single-crystal XRD (Figure 2). The structure is dinuclear and isostructural with 1 and homoleptic lanthanide tBu-PDB complexes reported previously.22 The structure has two chelating ligands per metal and two bridging ligands that form the dinuclear core. The metals are coordinated exclusively by hydrogen atoms and are tentatively assigned coordination numbers of 14 based on the Pu–B distances, but this may be lower, as suggested by DFT calculations (vide infra). The chelating Pu–B distances range from 2.848(6) to 2.950(6) Å, indicative of κ2-BH3 groups, whereas the bridging Pu–B distances are shorter at 2.675(6) and 2.678(5) Å and more consistent with κ3-BH3.
The experimental M–B distances in 2 were compared to those in 1 and isostructural lanthanide tBu-PDB complexes (Figure 3).22 Structural assessments of 1 revealed that bridging U–B distances were 0.04 Å shorter than expected when compared to the linear regression afforded by plotting Ln–B distances against Ln ionic radii, whereas the bridging Pu–B distances were 0.02 Å shorter.34 This suggests that the Pu–B bonds are less covalent than U–B bonds, which is consistent with systematic metal–ligand bond comparison studies of trivalent actinide complexes containing ligands with soft chalcogen donor groups.35−37
Figure 3.
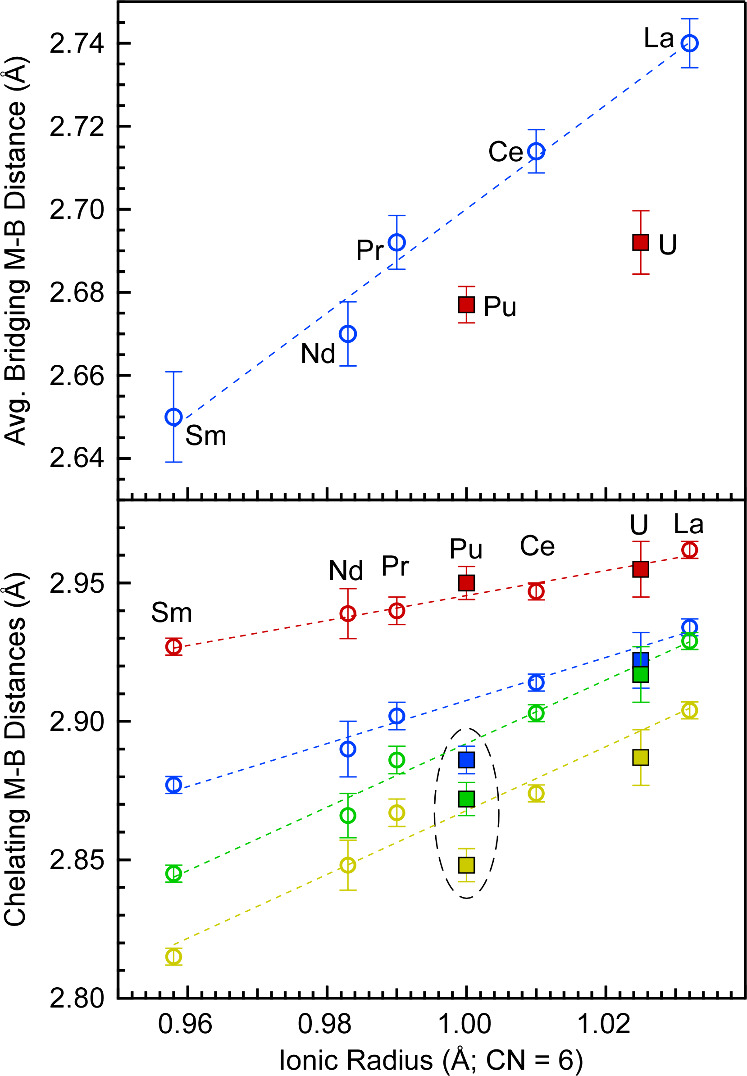
Top – Plot of average bridging M–B distances vs ionic radius26 of the metal for M2(H3BPtBu2BH3)6 complexes (M = actinide or lanthanide). The error bars account for the standard deviation of the averaged M–B distances and the esd’s for the individual M–B distances (see SI for details). Bottom – Plot of chelating M–B distances vs ionic radius of the metal. The shorter chelating Pu–B distances are circled for emphasis. Actinides are represented by solid squares, whereas lanthanides are represented by open circles. The error bars represent esd values from XRD. Dashed lines in both plots represent linear regressions of the lanthanide data points. R2 values are all >0.96.
We next evaluated the chelating M–B distances. It was observed previously that the chelating U–B bonds in 1 show no significant departure from linear regressions obtained when plotting chelating Ln–B distances against their ionic radii (Figure 3). In contrast, data collected for 2 showed that the three shortest chelating Pu–B distances are all 0.02 Å shorter than expected. Only the longest (and presumably most ionic) Pu–B distance at 2.950(6) Å falls close to its respective line.
Before detailing calculations used to assess metal–ligand covalency in 2, we describe its NMR data, the first for a Pu borohydride complex (see SI for solid-state UV–vis–NIR data for 2). 1H, 11B, and 31P NMR data collected on crystals of 2 dissolved in C6D6 revealed that 2 deoligomerizes to form an equilibrium mixture of the dimer and the putative monomer Pu(H3BPtBu2BH3)3 (2a), as described previously for other M2(H3BPtBu2BH3)6 complexes.22 Three paramagnetically shifted and broadened 11B resonances were observed at δ −0.6, 9.7, and 44.7 ppm, and these complemented three 31P{1H} NMR resonances at δ −208.7, −159.2, and 158.9 ppm. Two of the resonances in each set are assigned to the dimer 2 (bridging and chelating tBu-PDB environments), and one of the resonances is assigned to the monomer 2a (chelating tBu-PDB only). Consistent with the 11B and 31P data, the 1H NMR spectrum revealed three major tBu resonances at δ 0.88, 1.01, and 1.26 ppm. Additional 1H resonances assigned to the BH3 groups were more paramagnetically shifted due to their direct binding to Pu(III). A broad multiplet assigned to a single BH3 resonance was observed at δ 12.0 ppm, and overlapping resonances were observed at δ 20.9 ppm, similar to those reported with the lanthanide congener Sm.22
DFT calculations were performed to quantify the thermodynamics of the 2/2a equilibrium for comparison to 1. The structures of 2 and 2a were calculated at the TPSS-D3/def2-TZVP, def-TZVP level of theory,38,39 as previously used for 1.22 The optimized structure of 2 was in good agreement with the experimental data (see SI for details), and bond distances and angles for the calculated structures of 2 and 2a are provided in Tables S1–S3. The ΔG for 1 and 2 are identical within error at 6.3 and 6.7 kcal·mol–1 and ∼2 kcal·mol–1 higher than those for the lanthanide complexes (Table S4). The increase in ΔG of deoligomerization for 1 and 2 is enthalpic in origin, suggesting it is attributable to stronger bridging actinide–borohydride bonds compared to the analogous lanthanide complexes.
Subsequent energy decomposition analysis (EDA) calculations were performed at the PBE/TZP level of theory to determine if the shortened Pu–B distances reflect increased covalency with the bridging and chelating tBu-PDB ligands. Starting with the bridging metal–ligand bonds, the orbitalic energy contribution for the trivalent lanthanide complexes was on average 35.8 ± 0.4% (Table S11). Consistent with the bridging M–B distances, this value was largest in 1 at 39.1%, but decreased to 36.6% in 2. The orbitalic contribution for the chelating ligands in the lanthanide complexes was on average 36.7 ± 1.1% with larger values again obtained for 1 and 2 at 39.1% and 38.0%, respectively (Table S10). While the orbitalic contribution for Pu is closer to the lanthanides (Figure S22), there is a clear difference in the total interaction energies (Figure S23). The actinide complexes had interaction energies that were stronger for the bridging ligands, consistent with the aforementioned deoligomerization energies, but weaker compared to the lanthanides for the chelating ligands. Bond order calculations (PBE/TZP)40,41 showed a much stronger interaction for 1, while 2 had much weaker bond orders for both bridging and chelating ligands consistent with assigning the Pu complex a more lanthanide-like interaction (Tables S5–S9).
To further assess contributions to covalent metal–ligand bonding, quantum theory of atoms in molecules (QTAIM)42 was used to obtain the average electron density (ρ) at bond critical points, as well as delocalization indexes (δ) between metal and hydrogen atoms. Covalency in metal–ligand bonds can be influenced by changes in 1) metal–ligand orbital overlap or 2) frontier orbital energy matching (i.e., energy-degeneracy-driven covalency).43−48 QTAIM has been used to distinguish between these contributions. The ρ values have been used by us and others as a reflection of orbital-driven covalency,44,49 whereas the δ values can be used as a metric for degeneracy-driven covalency.50−52
QTAIM calculations indicate that U and Pu have a larger accumulation of electron density (ρ) at the M–H bond critical points compared to the lanthanides for both the chelating and bridging ligands (Figure 4; Tables S15 and S16). The increased ρ values for 1 and 2 relative to those of lanthanides are consistent with the greater orbital overlap expected due to the larger radial extension of the 6d and 5f orbitals of the actinides. Moreover, the ρ values are slightly larger with Pu than U, but they generally track along the same trend vs radius as observed with lanthanides. By contrast, the delocalization indexes (δ) suggest more favorable orbital energy matching between the bridging ligands and U in 1 compared to Pu in 2 (Figure 4; Figures S27–S29). However, we note that the decrease in coordination number from 14 in 1 to 13 in 2 also contributes to the decrease in δ. Owing to the change in coordination number, the values shown in Figure 4 represent the sum of δ per ligand instead of an average, but the individual values are presented in Figure S30. Unlike the bridging ligands, the sum of the δ values for the chelating ligands are similar for both Pu and U.
Figure 4.
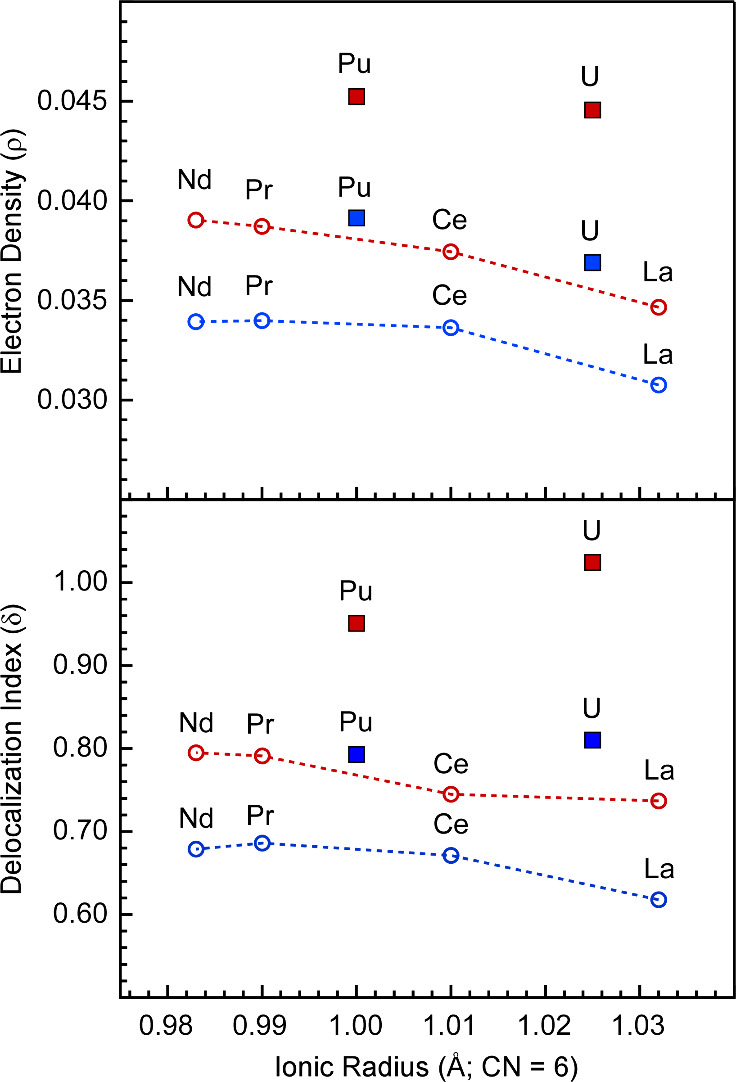
Top – Average QTAIM electron density (ρ) at the M–H bond critical points (PBE/TZP) for the chelating (blue) and bridging (red) ligands in the trivalent lanthanide and actinide dimers plotted as a function of ionic radius.26Bottom – Sum per ligand of the M–H delocalization indices (PBE/TZP) for the chelating (blue) and bridging (red) ligands in the trivalent lanthanide and actinide dimers. Actinides are represented by solid squares, whereas lanthanides are represented by open circles. Data points in red represent average values obtained for bridging ligands, whereas data points shown in blue represent average chelating ligands. Dashed lines between the lanthanide data points are to help guide the eye.
In summary, we have described the synthesis of Pu2(H3BPtBu2BH3)6 (2), the first example of a structurally characterized Pu(III) borohydride complex. The single-crystal XRD data collected for 2 allowed for the first experimental comparison of borohydride complexes with different trivalent actinides. The combined structural and theoretical data suggests that covalent metal–ligand bonding with tBu-PDB generally decreases in the order U > Pu > lanthanides. Evidence of greater metal–ligand covalency with U over Pu was revealed in calculations of the bridging metal–ligand bonds, but the differences between U and Pu were more subtle with chelating tBu-PDB ligands. Efforts to expand these analyses to include Np and transuranium complexes with other borohydride ligands are currently underway.
Acknowledgments
Research presented in this manuscript was supported by the Laboratory Directed Research and Development (LDRD) program of Los Alamos National Laboratory under project number 20220518MFR. DJL and AJG thank the U.S. DOE Office of Science, BES, Heavy Element Chemistry Program at Los Alamos National Laboratory (DE-AC52-06NA25396) for repeat synthesis and spectroscopic characterization of the plutonium complex. RRB, NN, and BV were supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, in the Heavy Element Chemistry program (Grant no. DE-SC0023022). SRD and PAZ also thank the NNSA for support (DE-NA0004151). Computations supporting this project performed on high-performance computing systems were made possible by resources funded by NSF award OAC-1626516.
Data Availability Statement
To ensure reproducibility, the input and output files associated with all calculations are available in a FigShare repository (doi.org/10.6084/m9.figshare.26997388).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.4c09888.
Experimental and theoretical details; crystallographic data and spectra (PDF)
Author Contributions
‡ J.C.Z., D.J.L., and R.B. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Schlesinger H. I.; Brown H. C. Uranium(IV) borohydride. J. Am. Chem. Soc. 1953, 75, 219–221. 10.1021/ja01097a058. [DOI] [Google Scholar]
- Schlesinger H. I.; Brown H. C.; Abraham B.; Bond A. C.; Davidson N.; Finholt A. E.; Gilbreath J. R.; Hoekstra H.; Horvitz L.; Hyde E. K.; Katz J. J.; Knight J.; Lad R. A.; Mayfield D. L.; Rapp L.; Ritter D. M.; Schwartz A. M.; Sheft I.; Tuck L. D.; Walker A. O. New developments in the chemistry of diborane and the borohydrides. General summary. J. Am. Chem. Soc. 1953, 75, 186–190. 10.1021/ja01097a049. [DOI] [Google Scholar]
- Bernstein E. R.; Hamilton W. C.; Keiderling T. A.; La Placa S. J.; Lippard S. J.; Mayerle J. J. 14-Coordinate uranium(IV). Structure of uranium borohydride by single-crystal neutron diffraction. Inorg. Chem. 1972, 11, 3009–3016. 10.1021/ic50118a027. [DOI] [Google Scholar]
- Bernstein E. R.; Keiderling T. A.; Lippard S. J.; Mayerle J. J. Structure of uranium borohydride by single-crystal x-ray diffraction. J. Am. Chem. Soc. 1972, 94, 2552–2553. 10.1021/ja00762a082. [DOI] [Google Scholar]
- Daly S. R.Actinide Borohydrides. In The Heaviest Metals: Science and Technology of the Actinides and Beyond; Evans W. J., Hanusa T. P., Eds.; Major Reference Works, John Wiley & Sons, Ltd., 2018; pp 319–334. [Google Scholar]
- Marks T. J.; Kolb J. R. Covalent transition metal, lanthanide, and actinide tetrahydroborate complexes. Chem. Rev. 1977, 77, 263–293. 10.1021/cr60306a004. [DOI] [Google Scholar]
- Ephritikhine M. Synthesis, Structure, and Reactions of Hydride, Borohydride, and Aluminohydride Compounds of the f-Elements. Chem. Rev. 1997, 97, 2193–2242. 10.1021/cr960366n. [DOI] [PubMed] [Google Scholar]
- Rajnak K.; Banks R. H.; Gamp E.; Edelstein N. Analysis of the optical spectrum of neptunium borodeuteride (Np(BD4)4) diluted in zirconium borodeuteride (Zr(BD4)4) and the magnetic properties of neptunium borohydride (Np(BH4)4) and neptunium methylborohydride (Np(BH3CH3)4). J. Chem. Phys. 1984, 80, 5951–5962. 10.1063/1.446675. [DOI] [Google Scholar]
- Rajnak K.; Gamp E.; Banks R.; Shinomoto R.; Edelstein N. Optical and magnetic properties of uranium and neptunium borohydrides and tetrakismethylborohydrides. Inorg. Chim. Acta 1984, 95, 29–35. 10.1016/S0020-1693(00)85965-4. [DOI] [Google Scholar]
- Shinomoto R.; Gamp E.; Edelstein N. M.; Templeton D. H.; Zalkin A. Syntheses and crystal structures of the tetrakis(methyltrihydroborato) compounds of zirconium(IV), thorium(IV), uranium(IV), and neptunium(IV). Inorg. Chem. 1983, 22, 2351–2355. 10.1021/ic00159a003. [DOI] [Google Scholar]
- Banks R. H.; Edelstein N. M.; Rietz R. R.; Templeton D. H.; Zalkin A. Preparation and properties of the actinide borohydrides: protactinium(IV), neptunium(IV), and plutonium(IV) borohydrides. J. Am. Chem. Soc. 1978, 100, 1957–1958. 10.1021/ja00474a067. [DOI] [Google Scholar]
- Banks R. H.; Edelstein N. M.; Spencer B.; Templeton D. H.; Zalkin A. Volatility and molecular structure of neptunium(IV) borohydride. J. Am. Chem. Soc. 1980, 102, 620–623. 10.1021/ja00522a029. [DOI] [Google Scholar]
- Banks R. H.; Edelstein N. M. Synthesis and characterization of protactinium(IV), neptunium(IV), and plutonium(IV) borohydrides. ACS Symp. Ser. 1980, 131, 331–348. 10.1021/bk-1980-0131.ch016. [DOI] [Google Scholar]
- Banks R. H.; Edelstein N. Vibrational spectra and normal coordinate analysis of neptunium(IV) borohydride and neptunium(IV) borodeuteride. J. Chem. Phys. 1980, 73, 3589–3599. 10.1063/1.440584. [DOI] [Google Scholar]
- Pu(BH4)4 undergoes thermal decomposition to a product that is presumed to be Pu(BH4)3, but as far as we are aware, the composition of this material has yet to be verified.
- Banks R. H.Preparation and spectroscopic properties of three new actinide(IV) borohydrides, Lawrence Berkeley Lab., Univ. California, Berkeley, CA, 1979. 10.2172/5507647 [DOI]
- Hudson M. J.; Harwood L. M.; Laventine D. M.; Lewis F. W. Use of Soft Heterocyclic N-Donor Ligands To Separate Actinides and Lanthanides. Inorg. Chem. 2013, 52, 3414–3428. 10.1021/ic3008848. [DOI] [PubMed] [Google Scholar]
- Ephritikhine M. Molecular actinide compounds with soft chalcogen ligands. Coord. Chem. Rev. 2016, 319, 35–62. 10.1016/j.ccr.2016.04.020. [DOI] [Google Scholar]
- Bessen N. P.; Jackson J. A.; Jensen M. P.; Shafer J. C. Sulfur donating extractants for the separation of trivalent actinides and lanthanides. Coord. Chem. Rev. 2020, 421, 213446. 10.1016/j.ccr.2020.213446. [DOI] [Google Scholar]
- Arliguie T.; Belkhiri L.; Bouaoud S.-E.; Thuery P.; Villiers C.; Boucekkine A.; Ephritikhine M. Lanthanide(III) and actinide(III) complexes [M(BH4)2(THF)5][BPh4] and [M(BH4)2(18-crown-6)][BPh4] (M = Nd, Ce, U): synthesis, crystal structure, and density functional theory investigation of the covalent contribution to metal-borohydride bonding. Inorg. Chem. 2009, 48, 221–230. 10.1021/ic801685v. [DOI] [PubMed] [Google Scholar]
- Vlaisavljevich B.; Miro P.; Koballa D.; Todorova T. K.; Daly S. R.; Girolami G. S.; Cramer C. J.; Gagliardi L. Volatilities of actinide and lanthanide N,N-dimethylaminodiboranate chemical vapor deposition precursors: a DFT study. J. Phys. Chem. C 2012, 116, 23194–23200. 10.1021/jp305691y. [DOI] [Google Scholar]
- Fetrow T. V.; Zgrabik J.; Bhowmick R.; Eckstrom F. D.; Crull G.; Vlaisavljevich B.; Daly S. R. Quantifying the Influence of Covalent Metal-Ligand Bonding on Differing Reactivity of Trivalent Uranium and Lanthanide Complexes. Angew. Chem., Int. Ed. 2022, 61, e202211145. 10.1002/anie.202211145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly S. R.; Girolami G. S. Synthesis, characterization, and structures of uranium(III) N, N-dimethylaminodiboranates. Inorg. Chem. 2010, 49, 5157–5166. 10.1021/ic100290j. [DOI] [PubMed] [Google Scholar]
- Fetrow T. V.; Grabow J. P.; Leddy J.; Daly S. R. Convenient Syntheses of Trivalent Uranium Halide Starting Materials without Uranium Metal. Inorg. Chem. 2021, 60, 7593–7601. 10.1021/acs.inorgchem.1c00598. [DOI] [PubMed] [Google Scholar]
- Drummond Turnbull R.; Bell N. L. f-Block hydride complexes - synthesis, structure and reactivity. Dalton Trans. 2024, 53, 12814–12836. 10.1039/D4DT00776J. [DOI] [PubMed] [Google Scholar]
- Shannon R. D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr., Sect. A 1976, A32, 751–767. 10.1107/S0567739476001551. [DOI] [Google Scholar]
- Blake A. V.; Fetrow T. V.; Theiler Z. J.; Vlaisavljevich B.; Daly S. R. Homoleptic uranium and lanthanide phosphinodiboranates. Chem. Commun. 2018, 54, 5602–5605. 10.1039/C8CC02862A. [DOI] [PubMed] [Google Scholar]
- Fetrow T. V.; Bhowmick R.; Achazi A. J.; Blake A. V.; Eckstrom F. D.; Vlaisavljevich B.; Daly S. R. Chelating Borohydrides for Lanthanides and Actinides: Structures, Mechanochemistry, and Case Studies with Phosphinodiboranates. Inorg. Chem. 2020, 59, 48–61. 10.1021/acs.inorgchem.9b01628. [DOI] [PubMed] [Google Scholar]
- Fetrow T. V.; Daly S. R. Mechanochemical synthesis and structural analysis of trivalent lanthanide and uranium diphenylphosphinodiboranates. Dalton Trans. 2021, 50, 11472–11484. 10.1039/D1DT01932E. [DOI] [PubMed] [Google Scholar]
- Zgrabik J. C.; Bhowmick R.; Eckstrom F. D.; Harrison A. R.; Fetrow T. V.; Blake A. V.; Vlaisavljevich B.; Daly S. R. The Influence of Phosphorus Substituents on the Structures and Solution Speciation of Trivalent Uranium and Lanthanide Phosphinodiboranates. Inorg. Chem. 2024, 63, 9451–9463. 10.1021/acs.inorgchem.3c02773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark D. L.; Sattelberger A. P.; Bott S. G.; Vrtis R. N. Lewis base adducts of uranium triiodide: a new class of synthetically useful precursors for trivalent uranium chemistry. Inorg. Chem. 1989, 28, 1771–1773. 10.1021/ic00309a004. [DOI] [Google Scholar]
- Clark D. L.; Sattelberger A. P.; Andersen R. A. Lewis base adducts of uranium triiodide and tris[bis(trimethylsilyl)amido]uranium. Inorg. Synth. 1996, 31, 307–315. 10.1002/9780470132623.ch55. [DOI] [Google Scholar]
- Dornhaus F.; Bolte M. (18-Crown-6)potassium di-tert-butylphosphanylbisborohydride. Acta Crystallogr., Sect. E Struct. Rep. Online 2006, 62, m3573–m3575. 10.1107/S1600536806050021. [DOI] [Google Scholar]
- Ionic radii reported for coordination numbers of 6 (CN = 6) were used because these values are known for trivalent U and Pu. The CN = 6 values are the best approximation for bond distance comparisons based on the structures and available radii in the literature.
- Gaunt A. J.; Reilly S. D.; Enriquez A. E.; Scott B. L.; Ibers J. A.; Sekar P.; Ingram K. I. M.; Kaltsoyannis N.; Neu M. P. Experimental and Theoretical Comparison of Actinide and Lanthanide Bonding in M[N(EPR2)2]3 Complexes (M = U, Pu, La, Ce; E = S, Se, Te; R = Ph, iPr, H). Inorg. Chem. 2008, 47, 29–41. 10.1021/ic701618a. [DOI] [PubMed] [Google Scholar]
- Gaunt A. J.; Neu M. P. Recent developments in nonaqueous plutonium coordination chemistry. C. R. Chim. 2010, 13, 821–831. 10.1016/j.crci.2010.06.004. [DOI] [Google Scholar]
- Goodwin C. A. P.; Schlimgen A. W.; Albrecht-Schönzart T. E.; Batista E. R.; Gaunt A. J.; Janicke M. T.; Kozimor S. A.; Scott B. L.; Stevens L. M.; White F. D.; Yang P. Structural and Spectroscopic Comparison of Soft-Se vs. Hard-O Donor Bonding in Trivalent Americium/Neodymium Molecules. Angew. Chem., Int. Ed. 2021, 60, 9459–9466. 10.1002/anie.202017186. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Wang Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B Condens. Matter 1992, 45, 13244–13249. 10.1103/PhysRevB.45.13244. [DOI] [PubMed] [Google Scholar]
- Tao J.; Perdew J. P.; Staroverov V. N.; Scuseria G. E. Climbing the density functional ladder: nonempirical meta-generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401. 10.1103/PhysRevLett.91.146401. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized gradient approximation made simple. [Erratum to document cited in CA126:51093]. Phys. Rev. Lett. 1997, 78, 1396. 10.1103/PhysRevLett.78.1396. [DOI] [PubMed] [Google Scholar]
- Bader R. F. W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. 10.1021/cr00005a013. [DOI] [Google Scholar]
- Neidig M. L.; Clark D. L.; Martin R. L. Covalency in f-element complexes. Coord. Chem. Rev. 2013, 257, 394–406. 10.1016/j.ccr.2012.04.029. [DOI] [Google Scholar]
- Kaltsoyannis N. Does Covalency Increase or Decrease across the Actinide Series? Implications for Minor Actinide Partitioning. Inorg. Chem. 2013, 52, 3407–3413. 10.1021/ic3006025. [DOI] [PubMed] [Google Scholar]
- Kelley M. P.; Su J.; Urban M.; Luckey M.; Batista E. R.; Yang P.; Shafer J. C. On the Origin of Covalent Bonding in Heavy Actinides. J. Am. Chem. Soc. 2017, 139, 9901–9908. 10.1021/jacs.7b03251. [DOI] [PubMed] [Google Scholar]
- Su J.; Batista E. R.; Boland K. S.; Bone S. E.; Bradley J. A.; Cary S. K.; Clark D. L.; Conradson S. D.; Ditter A. S.; Kaltsoyannis N.; Keith J. M.; Kerridge A.; Kozimor S. A.; Loble M. W.; Martin R. L.; Minasian S. G.; Mocko V.; La Pierre H. S.; Seidler G. T.; Shuh D. K.; Wilkerson M. P.; Wolfsberg L. E.; Yang P. Energy-Degeneracy-Driven Covalency in Actinide Bonding. J. Am. Chem. Soc. 2018, 140, 17977–17984. 10.1021/jacs.8b09436. [DOI] [PubMed] [Google Scholar]
- Pace K. A.; Klepov V. V.; Berseneva A. A.; zur Loye H.-C. Covalency in Actinide Compounds. Chem. - Eur. J. 2021, 27, 5835–5841. 10.1002/chem.202004632. [DOI] [PubMed] [Google Scholar]
- Pereiro F. A.; Galley S. S.; Jackson J. A.; Shafer J. C. Contemporary Assessment of Energy Degeneracy in Orbital Mixing with Tetravalent f-Block Compounds. Inorg. Chem. 2024, 63, 9687–9700. 10.1021/acs.inorgchem.3c03828. [DOI] [PubMed] [Google Scholar]
- Chowdhury S. R.; Goodwin C. A. P.; Vlaisavljevich B. What is the nature of the uranium(iii)-arene bond?. Chem. Sci. 2024, 15, 1810–1819. 10.1039/D3SC04715F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerridge A. f-orbital covalency in the actinocenes (An = Th-Cm): multiconfigurational studies and topological analysis. RSC Adv. 2014, 4, 12078–12086. 10.1039/C3RA47088A. [DOI] [Google Scholar]
- Kerridge A. Quantification of f-element covalency through analysis of the electron density: insights from simulation. Chem. Commun. 2017, 53, 6685–6695. 10.1039/C7CC00962C. [DOI] [PubMed] [Google Scholar]
- Kohler L.; Patzschke M.; Schmidt M.; Stumpf T.; Marz J. How 5f Electron Polarisability Drives Covalency and Selectivity in Actinide N-Donor Complexes. Chem. - Eur. J. 2021, 27, 18058–18065. 10.1002/chem.202102849. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
To ensure reproducibility, the input and output files associated with all calculations are available in a FigShare repository (doi.org/10.6084/m9.figshare.26997388).