

Abstract

Many RNA-binding proteins, such as TDP-43 or CELF1, interact multivalently with nucleic acid repetitive elements. The molecular stoichiometry of protein to nucleic acid is often difficult to assess, particularly by standard electrophoretic mobility shift assays (EMSAs). Here, we investigate the use of composition-gradient multiangle light scattering (CG-MALS) for quantifying binding affinity and stoichiometry for two RNA-binding proteins with their nucleic acid partners of varied sequence and length: TDP43’s N-terminal RNA recognition motifs with both TG/GU-repeat ssDNA and ssRNA, respectively, and CELF1’s two N-terminal RNA recognition motifs with (TG/UGUU/GU) repeats and an experimentally defined cognate GU-rich element (GRE). Our CG-MALS data derived from each of these interactions is consistent with expected ranges of binding affinity and stoichiometry for proteins binding to shorter nucleic acid repeats. Furthermore, we conclude that CG-MALS can be an excellent method for obtaining quantitative estimates even for high (>2) protein–nucleic acid stoichiometric ratios.
Introduction
Multivalent interactions occur universally in biological systems. Many homotypic and heterotypic protein–protein interactions and protein–nucleic acid interactions utilize multivalency to facilitate cooperativity and achieve cellular signaling (e.g., refs (1−7)). For example, many RNA-binding proteins (RBPs) such as TDP-43 assemble in multiples upon UG-rich elements of various length, and alterations in the stoichiometric ratios between the protein and its RNA target may have significant consequences regarding normal cellular function.4,8 However, current methods still present challenges in providing robust quantitative estimates of the molecular stoichiometry. Electrophoretic mobility shift assays (EMSAs), a widely used technique in protein–nucleic acid studies, require optimization of gel conditions to generate distinct bands. Even with optimized conditions, the number of bands is not formally indicative of the correct stoichiometry. Other biophysical techniques, such as fluorescence anisotropy, isothermal titration calorimetry (ITC), surface plasmon resonance (SPR), or biolayer interferometry (BLI), are indirect methods (i.e., based on changes in fluorescence, enthalpy heat, or “response units” with complex formation). They are generally best suited for quantifying interactions with a 1:1 stoichiometry and can suffer from artifacts for more complex interactions. For example, ITC may not readily distinguish between 1:1 and 2:2 stoichiometric ratios of molecular complexes. In contrast, multiangle static light scattering (MALS) measurements provide a label-free method of directly accessing the molecule’s molar mass, thus directly measuring the essence of complex formation.9,10
Composition-gradient multiangle light scattering (CG-MALS) leverages the fact that changes in molar mass can be directly related to the reversible formation and dissociation of specific complexes, each with its own equilibrium association constant.10 In a typical experiment, each binding partner is prepared in solution at concentrations near the expected equilibrium constant of the interaction. Solutions of varying compositions are prepared automatically, injected into a multiangle light scattering (MALS) detector, and allowed to come to equilibrium inside the MALS flow cell for a user-specified amount of time. The weight-average molar mass (Mw) of the solution is a first-principles calculation based on the light scattering intensity and total concentration of the solution—either specified manually or measured using an inline concentration detector.11 Association of one species with itself or with a binding partner results in a concentration-dependent increase in Mw; thus, multivalent complex formation is readily evident. The measured Mw as a function of composition is fitted to the appropriate expressions describing the equilibrium association of the analytes, which can include both self-interaction (e.g., dimerization) and heteroassociation. Like AUC or ITC, all interactions occur in solution without the need for immobilization or labeling. CG-MALS can be used to measure interactions with the equilibrium dissociation constant (Kd) from ∼100 pM12 to several mM13,14 and has been used to quantify a wide variety of biomolecular interactions, including the self-association of insulin,15,16 antibody–antigen interactions,17 and multivalent interactions with complex stoichiometries.12,18,19 The measured stoichiometry and affinity by CG-MALS agree well with other techniques, including SPR,20 BLI,17 AUC,12,21,22 ITC,23,24 and CryoEM.17,19 Although often applied to measuring interactions under dilute solution conditions in the absence of molecular crowding or phase separations, as in this study, CG-MALS can also be applied to high-concentration measurements to understand what drives phase separation and other thermodynamic processes.25,26 The automated CG-MALS measurements presented in this study can be applied to interactions among one or two binding partners and can accommodate any combination of self-association and heteroassociation.
To explore the utilization of CG-MALS for the analysis of protein–nucleic acid multivalent interactions, we characterized interactions between two proteins and their nucleic acid partners: TDP43RRM1 with (TG)6 and (GT)12 ssDNA and CELF1 with GU-rich elements (GRE) made up of (UGUU)4 and (GU)22 repeats. For each interaction, CG-MALS data were collected in a three-part experiment, as shown in Figure S1: (1) measurement of the protein alone at 3–5 concentrations, (2) measurement of 8–11 mixtures of protein and oligonucleotide at varying concentrations and stoichiometric ratios to assess, and (3) measurement of the oligonucleotide at 3–5 concentrations. Three technical replicates were performed for each interaction. The change in Mw as a function of composition was then fit to an appropriate equilibrium association model to determine the single set of stoichiometries and corresponding equilibrium association constants (Ka) that fit the data.
We show that our CG-MALS data is consistent with some of the quantitative binding measurements and observations reported elsewhere.8,27,28 Furthermore, even without detailed data analysis and model fitting, CG-MALS can provide minimum estimates of stoichiometry based on the maximum measured weight-average molar mass (Mw,max) and the molar composition of the protein and nucleic acid components where Mw,max occurs. Complete fitting of CG-MALS data can provide not only quantitation of the complexes formed but also the equilibrium association constant for the formation of each complex and the individual affinity at each binding site (equilibrium dissociation constant, Kd).
Results and Discussion
To evaluate CG-MALS as a method for characterizing multivalent interactions, we chose protein–nucleic acid pairs available to us. To avoid aggregation, we focused on truncated constructs: the first RRM domain of TDP43 (TDP43RRM1) and the first two RRM domains of CELF1 (CELF1RRM1–2). In all cases, at least two proteins are bound to each nucleic acid ligand. For two of the pairs, higher-order binding with four proteins per oligonucleotide was also present. The description and analysis for each of the protein–nucleic acid pair are detailed below.
TDP43RRM1 Binding (TG)6 or (GT)12 ssDNA
TDP43 is an RNA-binding protein, consisting of 2 RRM domains that recognize long GU repeats as well as single-strand DNA (ssDNA) TG repeats.29,30 For this study, we focus on TDP43RRM1 and assess its interaction with multivalent nucleic acid repeats (TG)6 and (GT)12. Both DNA sequences have been previously reported as TDP43 cognate sequences. TG repeat elements were found near the human CFTR exon 9 gene29 where TDP-43 can bind and regulate proper splicing activities to avoid exon skipping and ensure correct CFTR protein translation.31 In other ssDNA studies, TDP43 was demonstrated to form higher-order assemblies (up to four TDP43 RRM1–2) with 24 GT repeats.8 We set out to test how many domains might bind (TG)6 and (GT)12 sequences and with what affinity, i.e., if two molecules of TDP43RRM1 could bind to (TG)6, we expected four molecules to bind to (GT)12 DNA. EMSA provides qualitative evidence that TDP43 binds to both (TG)6 and (GT)12. For (TG)6, we observe the disappearance of the free ssDNA and the consequent smearing of the lanes with faint bands corresponding to bound states (Figure 1a). Similarly, for (GT)12, we observed binding and the appearance of a major distinct band and a minor more shifted band (Figure 1b). The more pronounced bands for (GT)12 suggest stronger binding and a tighter affinity. However, it is difficult to assess stoichiometry from these results. In contrast, the stoichiometry of the interactions is obvious when they are measured by CG-MALS (Figure 1c,d).
Figure 1.

TDP43RRM1 interactions with (TG)6 ssDNA and (GT)12 ssDNA by EMSA (a,b) and CG-MALS (c–f). (a,b) EMSA of TDP43RRM1 and (TG)6 ssDNA (a) and (GT)12 ssDNA (b) showing disappearance of free ssDNA accompanied by band smearing (bracket) and less distinct bands representing complex formation (denoted by *). (c,d) Measured weight-average molar mass (Mw) from three independent CG-MALS experiments as a function of composition. The data was fit assuming 2 or 4 equiv sites on each ssDNA for the TDP43RRM1 protein. Measured molar mass data for all three experiments (blue circles) are shown along with the best fit (blue line), alternative nonfitting model (green line), and “no interaction” reference curve (dashed gray). The best fit for the (TG)6 interaction includes two equivalent binding sites with Kd = 10.4 ± 3.2 μM; the best fit for the (GT)12 interaction includes four equivalent binding sites with Kd = 1.34 ± 0.12 μM (average and standard deviation from three replicates; Table 1). (e,f) Concentration of each complex formed, as determined by the best fit of the CG-MALS data. The fraction of unbound protein and ssDNA has been left off for clarity.
Triplicate experiments were performed to investigate each interaction, each one consisting of three gradients, as described in the Materials and Methods section. Each experiment was fit separately, and we present the average and standard deviations of all measured and fit parameters. The measured Mw for each species (Table 1) was in good agreement with the expected monomer molar mass (Table S1). Concentration gradient data for the protein and nucleic acid species alone (i.e., in the absence of the binding partner) revealed no measurable change in Mw for concentrations up to 20 μM for TDP43RRM1, 30 μM for (TG)6, and 3.5 μM for (GT)12, confirming the lack of reversible self-association under the conditions tested. Although the measured Mw was slightly greater than the expected value, it was invariant with the concentration, indicating the presence of a small fraction of irreversible aggregates. Taken together, these results indicate that any change in Mw upon mixing protein and oligonucleotide resulted from the specific, reversible heteroassociation of the two species, and only these complexes were included when fitting the CG-MALS data.
Table 1. Protein:Nucleic Acid (NA) Stoichiometry (n), Equilibrium Dissociation Constant (Kd), and Monomer Molecular Weight (MW) Determined by Fitting CG-MALS Data.
| interaction | nucleic acid | measured
monomer MW (kDa) |
nprotein:nNA | Kd (μM) | |
|---|---|---|---|---|---|
| protein | NA | ||||
| TDP43RRM1:(TG)6 | ssDNA | 15.2 ± 0.5 | 4.51 ± 0.65 | 2:1a | 10.4 ± 3.2 |
| TDP43RRM1:(GT)12 | ssDNA | 14.3 ± 0.1 | 11.0 ± 0.4 | 4:1a | 1.34 ± 0.12 |
| CELF1RRM1–2:(UGUU)4 | ssRNA | 21.2 ± 0.3 | 4.81 ± 0.26 | 1:1b | 0.02 |
| 2:1b | 4 | ||||
| CELF1RRM1–2:(GU)22 | ssRNA | 21.2 ± 0.4 | 16.8 ± 0.3 | 2:1c | 0.0010 ± 0.0002 |
| 4:1d | 0.39 ± 0.03 | ||||
All values are the average and standard deviation of three replicates, except for CELF1RRM1–2:(UGUU)4, which reports a single fit value across data from three experiments.
All binding sites are equivalent with indicated Kd.
Best fit of concatenated data is consistent with two binding sites—one strong and one weak.
Binding at first two sites is equivalent with indicated Kd.
Binding at the third and fourth sites is assumed equivalent with indicated Kd.
The heteroassociation gradients, which created 11 different combinations of protein and ssDNA, resulted in an increase in overall weight-average molar mass of the solution and indicated more than one protein bound to each ssDNA (Figure 1c,d). For (TG)6, the maximum measured weight-average molar mass (Mw) occurs when TDP43RRM1 is mixed with (TG)6 at a 2:1 molar ratio, with [TDP43RRM1] ∼ 14 μM and [(GT)6] ∼ 7 μM (Figure 1c), suggesting a 2:1 stoichiometry. The maximum Mw of 21.0 ± 1.8 kDa was consistent with two proteins bound to each oligonucleotide (Figure 1c), and this measured value was 40% higher than the Mw that would result in the absence of an interaction upon mixing these analytes at the same concentration (Figure 1c, gray dashed line). The best fit considers two binding sites with an equivalent affinity. Fitting three experiments independently resulted in an average equilibrium dissociation constant at each binding site of Kd = 10.4 ± 3.2 μM (Table 1). The concentrations of the complexes formed as a function of composition are shown in Figure 1e. Similar studies (TDP43RRM1 with sequence (GT)6) by ITC8 reported tighter affinities with Kd ∼ 100 nM for protein binding to the first site and Kd ∼ 10 nM to the second site, suggesting cooperativity between sites. The discrepancies could be attributed to the differences in sequence and experimental conditions. The ITC experiments were performed with lower buffer and salt concentrations (15 mM phosphate buffer, pH 6.8, containing 25 mM KCl) versus our buffer conditions (25 mM HEPES, 50 mM NaCl, pH 7.3). Also, since the CG-MALS experiments were conducted at concentrations from 3 to 30 μM (TG)6 and most of the protein-oligonucleotide mixtures were made in conditions of excess (TG)6 (Figure 1c,e), higher affinity interactions may have been masked, and the effect of cooperativity may have been negligible.
For (GT)12, the measured Mw suggests higher-order binding and higher affinity compared to the interaction with (TG)6. Even at ∼10 times lower overall concentrations of oligonucleotides (i.e., concentrations up to 3.5 μM (GT)12 compared to concentrations up to 30 μM (GT)6), a significant increase in Mw is observed, reflecting both the increase in affinity and stoichiometry. The maximum measured Mw of 46.3 ± 1.9 occurs where [TDP43RRM1] = 4[(GT)12] = 8.3 μM (Figure 1d) and is significantly higher than the maximum possible Mw that could be achieved if each (GT)12 bound only two TDP43RRM1 at these concentrations (39 kDa), as shown by the green alternative nonfitting model in Figure 1d. Thus, the best fit is unambiguously consistent with four equivalent binding sites each with Kd = 1.34 ± 0.12 μM (Figure 1d, blue line), confirming a higher affinity for TDP43RRM1 as compared to (TG)6. The concentrations of each complex formed are shown in Figure 1f. Since the light scattering intensity measured by MALS is proportional to the weight-average molar mass, even a small amount of high-molecular-weight species can significantly impact the data, giving high confidence to the observation of the 4:1 species even at relative concentrations of 10% mol/mol or less. The RSD <10% on the Kd across three technical replicates further increases confidence in the fit.
CELF1RRM1–2 Binding (UGUU)4 or (GU)22 ssRNA
CELF1 is also an RNA-binding protein, consisting of 3 RRM domains with the N-terminal RRMs (RRM1 and RRM2) separated from the C-terminal RRM3 via a long disordered linker.32,33 For the current studies, we focused on CELF1RRM1–2 and its interactions with its defined GU-rich target elements (GREs). Similar to the investigation of TDP43-binding ssDNA, we observed significant differences in both the stoichiometry and affinity for CELF1RRM1–2 depending on the length of the ssRNA binding partner. Similarly, we observed no significant self-association of either CELF1RRM1–2 or its binding partners.
Initially, we investigated CELF1RRM1–2 binding to (UGUU)4. As expected, EMSA showed disappearance of free ssRNA and appearance of smeary band(s) that migrate higher upon increasing protein concentration but without clear insight into the stoichiometry. Triplicate CG-MALS experiments were performed to assess the interaction. In all three experiments, the maximum Mw (∼30 kDa) was achieved when the concentration of protein was approximately twice the concentration of ssRNA (overall mole fraction of CELF1RRM1–2 = [CELF1]/([CELF1]+[(UGUU)4]) ∼ 0.67), consistent with a 2:1 stoichiometry (Figure 2b). The best fit, considering all three experiments together, suggested two nonequivalent binding sites. CELF1RRM1–2 binds strongly at the first site with Kd ∼ 0.02 μM and more weakly to the second site with Kd ∼ 4 μM (Table 1). The results presented consider a single global fit to all three experiments. Significant experiment-to-experiment variation was observed, as seen in Figure 2b, likely due to the presence of aggregates in CELF1RRM1–2. Although CG-MALS confirmed an overall 2:1 stoichiometry for this interaction, the current data were insufficient to conclude whether the two binding sites are equivalent or nonequivalent, as shown by the comparison of the blue best fit line and the green alternative fit line in Figure 2b. However, a binding model considering two nonequivalent binding sites is consistent with literature studies, suggesting that the secondary site could accommodate only 1 RRM domain, resulting in a weaker affinity,28 and thus is presented as the best fit.
Figure 2.
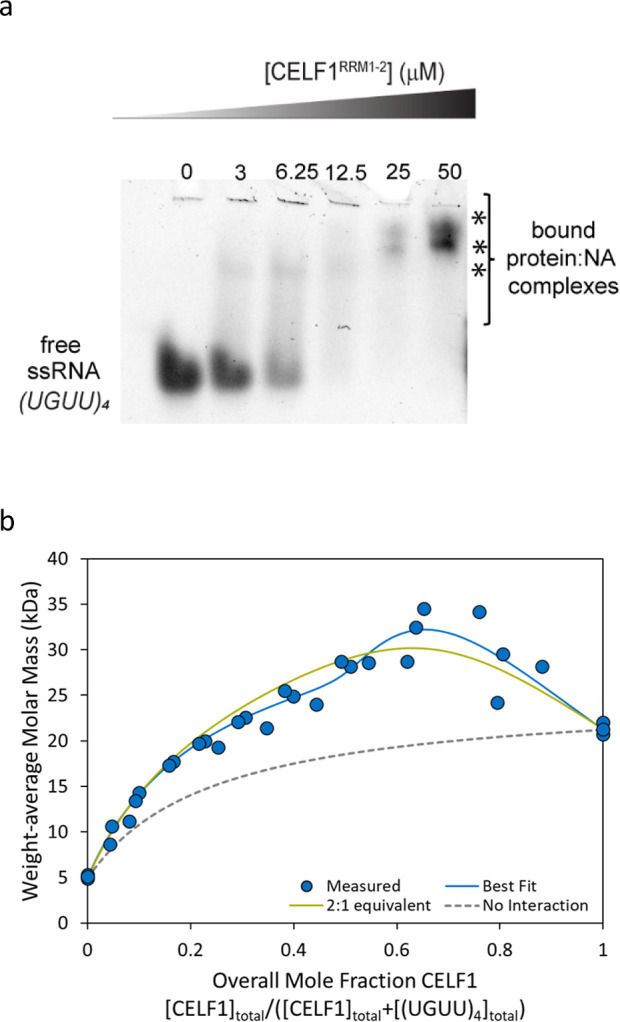
CELF1RRM1–2 and (UGUU)4 ssRNA interactions by EMSA (a) and CG-MALS (b). (a) EMSA of CELF1RRM1–2 and (UGUU)4 ssRNA showing disappearance of free ssRNA accompanied by shifted bands representing complex formation (denoted by *). (b) Mw measured by CG-MALS (blue circles) as a function of composition for three experiments, each with a maximum CELFRRM1–2 concentration of 7–8.5 μM and a maximum concentration of (UGUU)4 of 9–20 μM. The best fit (blue line) considering two nonequivalent binding sites (Table 1) is shown by the blue line. An alternative binding model considering two equivalent binding sites, each with Kd = 3.6 μM, is indicated by the green line. The “no interaction” reference curve (gray dashed line) is shown for reference.
We then investigated CELF1RRM1–2 interactions with the longer RNA (GU)22. With EMSA, we observed binding, which resulted in an upward shifting band. Importantly, the interaction appears to occur at lower concentrations of CELF1, compared with (UGUU)4, suggesting a higher affinity interaction (Figure 3a). With CG-MALS, we can fully quantify this interaction. The maximum measured Mw (86.1 ± 1.2 kDa) suggests that we can see the formation of at least a 4:1 stoichiometric CELF1RRM1–2:(GU)22 complex (Figure 3b). The best fit included the formation of three species with the following protein:ssRNA stoichiometry: 1:1, 2:1, and 4:1 (Table 1). The maximum measured Mw is nearly 50% higher than the maximum possible Mw if only 2:1 binding is considered (Figure 3b, green line), clearly indicating that higher-order assemblies are reversibly formed under these conditions.
Figure 3.
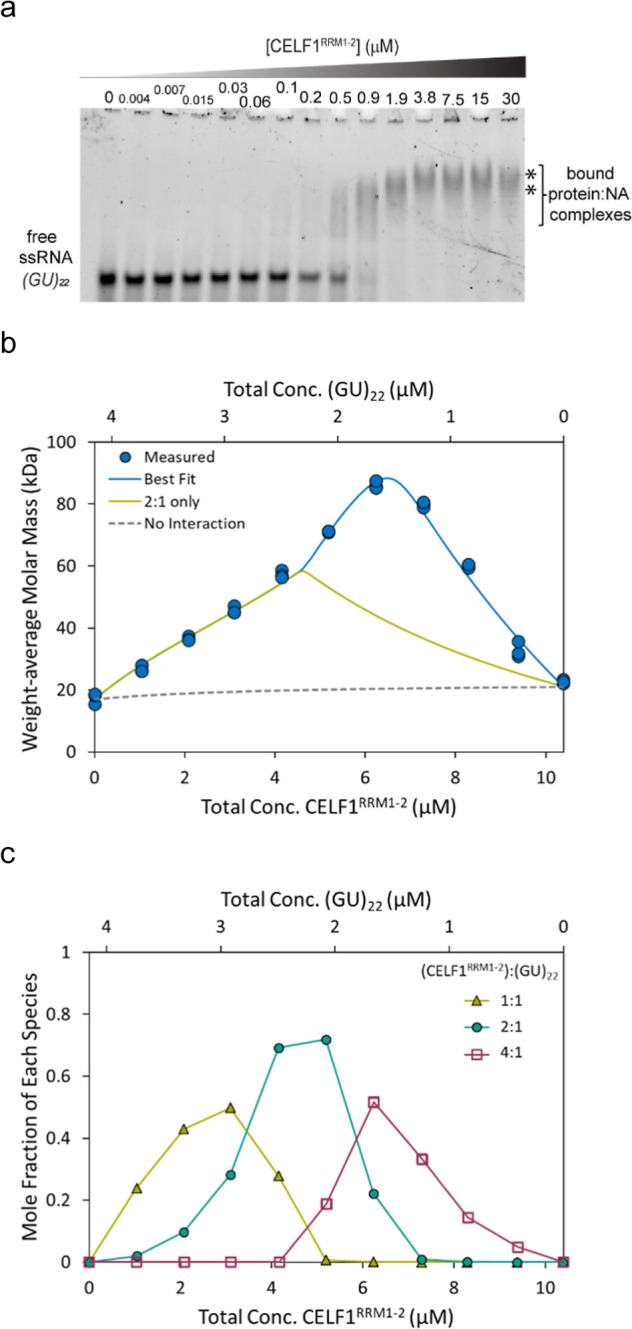
CELF1RRM1–2 and (GU)22 ssRNA interactions by EMSA (a) and CG-MALS data and simulations (b,c). (a) EMSA of CELF1RRM1–2 and (GU)22 ssRNA showing disappearance of free ssRNA accompanied by shifted bands representing complex formation (denoted by *). (b) Measured Mw vs total [CELF1RRM1–2] and total [(GU)22] for triplicate CG-MALS experiments. The best fit (blue line) consists of complexes with 1:1, 2:1, and 4:1 stoichiometry. Including only 1:1 and 2:1 interaction (green line) is not sufficient to capture the measured molar mass. The “no interaction” reference curve (gray dashed line) is shown for reference. c, Calculated concentration of each complex formed across the CG-MALS heteroassociation gradient. The concentration of unbound monomer has been left off for clarity.
The 1:1 and 2:1 complexes appear to form with equivalent affinity at each binding site (Kd = 1.0 ± 0.2 nM), which is over 1000× stronger than the affinity for CELFRRM1–2 for (UGUU)4. This might reflect an increase in cooperative binding affinities as expected for multivalent interactions.34 Under these conditions, a 4:1 complex also appears, and the third and fourth binding events were assumed to have equivalent affinity (Kd = 390 ± 30 nM). In general, we would also expect to observe the formation of the 3:1 complex; however, the model excluded the formation of this species, possibly due to the high concentration at which the measurements were performed (Figure 3c). It is possible that the 3:1 complex would also be evident at lower concentrations closer to Kd, where more binding sites would be unsaturated.
In conclusion, we show that CG-MALS is an excellent technique for obtaining estimates of binding affinities and molecular stoichiometry associated with multivalent interactions. Even when the binding interactions become significantly more complex and involve high stoichiometric ratios (>2), the Mw,max can provide minimum estimates of molecular stoichiometric ratios. Better quantitative estimates can be obtained with CG-MALS data simulations and estimated Kd values from experimental studies for both simple stoichiometry (≤2) and complex interactions by combining data across multiple concentration ranges. Thus, CG-MALS could be a useful technique in investigating the molecular stoichiometry involved in large multivalent protein–nucleic acid complexes. The multivalent interactions of these transcription factors and RNA-binding proteins with nucleic acid repeat sequences are required for their cellular functions. CELF1 is linked to myotonic dystrophy (DM) type 1 disease associated with the toxic effect of (CUG)n expansion.33,35,36 TDP-43 aberrant aggregation with RNA has been linked to amyotrophic lateral sclerosis (ALS) disease.4,37,38 There might be critical protein:RNA stoichiometry thresholds that may distinguish between physiological and pathological complexes and could benefit from the CG-MALS technique.
Materials and Methods
Cloning, Protein Expression, and Purification
TDP43RRM1 (101–192)
The plasmid was prepared by site-directed mutagenesis of TDP43RRM1–2 (101–277), containing both RRM domains, by changing the codon for amino acid V193 for the stop TAG codon. To construct the expression plasmid of TDP-43RRM1–2 tagged with N-terminal His6, DNA fragments encoding TDP-43 (101–277) were amplified from the template plasmid (Addgene 27462) and then ligated into EcoRI/AvrII digested pET302NT-His (Invitrogen).
The TDP43RRM1 plasmid was transformed into Escherichia coli BL21 star-competent cells and grown at 37 °C in Terrific Broth medium containing carbenicillin antibiotic. Cells were induced for expression with 1 mM IPTG when the OD600 reached between 0.8 and 1.0. Growth was allowed to continue overnight at 18 °C, and then cells were harvested by centrifugation at 4,000×g for 20 min. Cells were resuspended in lysis buffer (1.5 M KCl, 25 mM Tris, pH 7.5) containing 1 mM PMSF and the 1X Xpert protease inhibitor (GenDEPOT) and lysed by using a homogenizer (Avestin). The lysate was centrifuged at 4 °C for 1 h at 50,000×g. The supernatant was applied to nickel agarose beads (GoldBio) and washed extensively with lysis buffer containing 20 mM imidazole before elution with lysis buffer containing 200 mM imidazole. The elution was dialyzed to 150 mM NaCl, 25 mM Tris buffer pH 7.5, concentrated to a 2 mL volume, and purified using size exclusion chromatography (SEC) using a Superdex 75 column (Cytiva).
CELF1RRM1–2
The His10-CELF1RRM1–2-expressing plasmid was transformed into E. coli BL21 star-competent cells and grown at 37 °C in LB medium. Cells were induced for expression with 0.4 mM IPTG when the OD600 reached between 0.8 and 1.0. Growth was allowed to continue overnight at 18 °C, and then cells were harvested by centrifugation at 4,000×g for 20 min. Cells were resuspended in lysis buffer (20 mM HEPES, 500 mM NaCl, 1 mM TCEP, 10 mM Imidazole, pH 7.5) supplemented with a protease inhibitor (GoldBio Problock). The cell suspension was lysed using a homogenizer (Avestin). The lysate was centrifuged at 4 °C for 30 min to 1 h at 20,000×g. The cleared lysate is filtered to 0.45 μm using a vacuum filter and applied to Ni-NTA beads equilibrated with the lysis buffer. The beads were washed with 40 column volumes of the same lysis buffer with 1 M NaCl. The protein was eluted with an imidazole step gradient (100, 250, and 500 mM imidazole in 50 mM HEPES, 500 mM NaCl, 1 mM TCEP, pH 7.5).
Protein and DNA Concentration Determination
Protein concentrations were calculated based on the UV absorbance extinction coefficient (ε, ml/(mg·cm)) at 280 nm: TDP43RRM1 = 1.16 and CELF1RRM1–2 = 0.49. Oligonucleotides (TG)6, (UGUU)4 were obtained from IDT (Integrated DNA Technologies, Inc., Coralville, IA) and (GU)22, (GT)12 from Sigma. Oligonucleotide concentrations were calculated based on UV absorbance extinction coefficient (ε, ml/(mg·cm)) at 280 nm: (TG)6 = 18.21, (GT)12 = 18.12, (UGUU)4 = 14.03, and (GU)22 = 14.86. The molecular weights of the protein and RNA are summarized in Table S1.
Electrophoretic Mobility Shift Assay (EMSA)
The EMSA binding reactions were prepared from 2-fold dilutions of protein:nucleic acid mix and incubated at room temperature for 10 min prior to gel electrophoresis. For nonfluorescent conjugated nucleic acid, the gel was stained with SYBR Gold (Invitrogen) according to the manufacturer’s instructions. All gels were imaged by using ChemiDoc with appropriate filters. The protein concentrations, nucleic acid concentrations, and gel running conditions are as follows: TDP43RRM1 (1–8 μM); (TG)6 ssDNA (2 μM) or (TG)12 ssDNA (1 μM) in buffer (20 mM HEPES, 50 mM NaCl, 1 mM MgCl2, 10% glycerol, 1 mM TCEP, pH 7.3); 10% tris-glycine (TG, Bio-Rad) gel, 90 min at 100 V, 4 °C, in 0.5× TAE buffer. CELF1RRM1–2 (3–50 μM) and (UGUU)4 ssRNA (5 μM) in buffer (10 mM HEPES, 12.5 mM Tris, 70 mM NaCl, 12.5 mM KCl, 1 mM MgCl2, 10% glycerol, 0.5 mM TCEP, pH 7.4); 10% Tris-Glycine (TG, Bio-Rad) gel, 60 min at 80 V, 4 °C, in 0.5× TAE buffer. CELF1RRM1–2 (4 nM–50 μM) and (GU)22 ssRNA (200 nM) in buffer (20 mM HEPES, 50 mM NaCl, 1 mM MgCl2, 10% glycerol, 1 mM TCEP, pH 7.3); 10% TG (TG, Bio-Rad) gel, 90 min at 80 V, 4 °C, in 0.5× TAE buffer.
Composition Gradient Multiangle Light Scattering (CG-MALS)
Proteins were passed through size exclusion chromatography (Superdex 75) prior to CG-MALS experiments to ensure the removal of high-MW aggregates. Proteins and oligos were diluted in experimental buffer (25 mM HEPES, 50 mM NaCl, and 1 mM TCEP, pH 7.3). CG-MALS data were collected using a fully automated three-syringe pump Calypso II instrumentation (Wyatt Technology, LLC) linked to a DAWN (Wyatt) multiangle light scattering detector equipped with a 661 nm laser and UV/vis detector (Agilent Technologies) collecting absorbance at 280 nm. Each experiment follows a concentration gradient of one species (A) to assess self-association, followed by a “crossover” gradient with varying concentrations of A and species B to characterize the heterointeractions, and finally, a concentration gradient of species B to assess its self-association. An example of one such experiment is shown in Figure S1. Initial concentrations for the different protein:nucleic acid complexes are approximately as follows: TDP43RRM1:(TG)6 are ∼24 μM and ∼14 μM, respectively; TDP43RRM1:(GT)12 are ∼21 μM and ∼5 μM, respectively; CELF1RRM1–2:(UGUU)4 are ∼9 μM and ∼19 μM, respectively; and CELF1RRM1–2:(GU)22 are ∼11 μM and ∼4 μM, respectively. Final protein and oligo concentrations were adjusted based on the UV absorbance signal detected in the CALYPSO software (Wyatt). Three replicates were performed. All analysis, including model fitting and data simulations, was performed using CALYPSO software (Wyatt). Light scattering data were fit to the following equations.9,10,20
where R is the Rayleigh ratio; Kij is the equilibrium association constant of the AiBj complex; A and B are constituent monomers; i and j represent the stoichiometric numbers of A and B in the AiBj complex; MA and MB are the molar masses of monomers A and B, respectively; and K* is a constant defined below
where (dn/dc) is the refractive index increment of a sample–solvent combination; n0 is the refractive index of the solvent; Na is Avogadro’s number; and λ0 is the vacuum wavelength of incident light.
Acknowledgments
We acknowledge the Recombinant Protein Production and Characterization Core (RPPCC) for access to the CG-MALS instrumentation (NIH S10 OD030276). This work was also supported by grants from the NIH R01 GM122763 and Welch Q-2097-20220331 (J.C.F.), the Adrienne Helis Malvin Medical Research Foundation and the Samuel Waxman Cancer Research Foundation (J.R.N), and NIH grant R01 NS105874 (A.C.M.F.).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.4c06358.
Theoretical molecular weights (MW) of protein/nucleic acid components and CG-MALS example method with varying protein and nucleic acid concentrations (PDF)
Author Contributions
Conceptualization, J.C.F.; Methodology, A.C.M.F. and J.C.F.; Software and Formal Analysis, P.S.T., S.K., A.C.M.F., and J.C.F.; Investigation, K.-J.C., P.S.T., A.C.M.F., and J.C.F.; Visualization and Writing—Original Draft, Review, and Editing: A.C.M.F., J.R.N., S.K., and J.C.F.; Supervision, J.R.N., A.C.M.F., and J.C.F.; Funding Acquisition, J.R.N., A.C.M.F., and J.C.F.
The authors declare no competing financial interest.
Supplementary Material
References
- Ruthenburg A. J.; Li H.; Patel D. J.; David Allis C. Multivalent engagement of chromatin modifications by linked binding modules. Nat. Rev. Mol. Cell Biol. 2007, 8 (12), 983–994. 10.1038/nrm2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. W.; Ferreon J. C.; Ferreon A. C. M.; Arai M. A.; Wright P. E. Graded enhancement of p53 binding to CREB-binding protein (CBP) by multisite phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 2010, 107 (45), 19290–19295. 10.1073/pnas.1013078107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P.; Banjade S.; Cheng H. C.; Kim S.; Chen B.; Guo L.; Llaguno M.; Hollingsworth J. V.; King D. S.; Banani S. F.; et al. Phase transitions in the assembly of multivalent signalling proteins. Nature 2012, 483 (7389), 336–340. 10.1038/nature10879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallegger M.; Chakrabarti A. M.; Lee F. C. Y.; Lee B. L.; Amalietti A. G.; Odeh H. M.; Copley K. E.; Rubien J. D.; Portz B.; Kuret K.; et al. TDP-43 condensation properties specify its RNA-binding and regulatory repertoire. Cell 2021, 184 (18), 4680–4696.e22. 10.1016/j.cell.2021.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K. J.; Quan M. D.; Qi C.; Lee J. H.; Tsoi P. S.; Zahabiyon M.; Bajic A.; Hu L.; Prasad B. V. V.; Liao S. C. J.; et al. NANOG prion-like assembly mediates DNA bridging to facilitate chromatin reorganization and activation of pluripotency. Nat. Cell Biol. 2022, 24 (5), 737–747. 10.1038/s41556-022-00896-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoi P. S.; Choi K. J.; Leonard P. G.; Sizovs A.; Moosa M. M.; MacKenzie K. R.; Ferreon J. C.; Ferreon A. C. M. The N-Terminal Domain of ALS-Linked TDP-43 Assembles without Misfolding. Angew. Chem., Int. Ed. Engl. 2017, 56 (41), 12590–12593. 10.1002/anie.201706769. [DOI] [PubMed] [Google Scholar]
- Sharma R.; Choi K. J.; Quan M. D.; Sharma S.; Sankaran B.; Park H.; LaGrone A.; Kim J. J.; MacKenzie K. R.; Ferreon A. C. M.; et al. Liquid condensation of reprogramming factor KLF4 with DNA provides a mechanism for chromatin organization. Nat. Commun. 2021, 12 (1), 5579. 10.1038/s41467-021-25761-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengifo-Gonzalez J. C.; El Hage K.; Clement M. J.; Steiner E.; Joshi V.; Craveur P.; Durand D.; Pastre D.; Bouhss A. The cooperative binding of TDP-43 to GU-rich RNA repeats antagonizes TDP-43 aggregation. Elife 2021, 10, e67605 10.7554/eLife.67605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arisaka F.; Niimura Y.; Minton A. P. Comparison of composition-gradient sedimentation equilibrium and composition-gradient static light scattering as techniques for quantitative characterization of biomolecular interactions: A case study. Anal. Biochem. 2019, 583, 113339. 10.1016/j.ab.2019.06.010. [DOI] [PubMed] [Google Scholar]
- Attri A. K.; Minton A. P. Composition gradient static light scattering: a new technique for rapid detection and quantitative characterization of reversible macromolecular hetero-associations in solution. Anal. Biochem. 2005, 346 (1), 132–138. 10.1016/j.ab.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Wyatt P. J. Light scattering and the absolute characterization of macromolecules. Anal. Chim. Acta 1993, 272, 1–40. 10.1016/0003-2670(93)80373-S. [DOI] [Google Scholar]
- Halling D. B.; Kenrick S. A.; Riggs A. F.; Aldrich R. W. Calcium-dependent stoichiometries of the KCa2.2 (SK) intracellular domain/calmodulin complex in solution. J. Gen. Physiol. 2014, 143 (2), 231–252. 10.1085/jgp.201311007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esfandiary R.; Hayes D. B.; Parupudi A.; Casas-Finet J.; Bai S.; Samra H. S.; Shah A. U.; Sathish H. A. A systematic multitechnique approach for detection and characterization of reversible self-association during formulation development of therapeutic antibodies. J. Pharm. Sci. 2013, 102 (9), 3089–3099. 10.1002/jps.23654. [DOI] [PubMed] [Google Scholar]
- Esfandiary R.; Parupudi A.; Casas-Finet J.; Gadre D.; Sathish H. Mechanism of reversible self-association of a monoclonal antibody: role of electrostatic and hydrophobic interactions. J. Pharm. Sci. 2015, 104 (2), 577–586. 10.1002/jps.24237. [DOI] [PubMed] [Google Scholar]
- Attri A. K.; Fernandez C.; Minton A. P. Self-association of Zn-insulin at neutral pH: investigation by concentration gradient--static and dynamic light scattering. Biophys. Chem. 2010, 148 (1–3), 23–27. 10.1016/j.bpc.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attri A. K.; Fernandez C.; Minton A. P. pH-dependent self-association of zinc-free insulin characterized by concentration-gradient static light scattering. Biophys. Chem. 2010, 148 (1–3), 28–33. 10.1016/j.bpc.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallesen J.; Murin C. D.; de Val N.; Cottrell C. A.; Hastie K. M.; Turner H. L.; Fusco M. L.; Flyak A. I.; Zeitlin L.; Crowe J. E.; et al. Structures of Ebola virus GP and sGP in complex with therapeutic antibodies. Nat. Microbiol. 2016, 1 (9), 16128. 10.1038/nmicrobiol.2016.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell S. L.; Ismail A. M.; Kenrick S. A.; Camilli A. The VieB auxiliary protein negatively regulates the VieSA signal transduction system in Vibrio cholerae. BMC Microbiol. 2015, 15, 59. 10.1186/s12866-015-0387-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M.; Wu S.; Zhou Q.; Vivona S.; Cipriano D. J.; Cheng Y.; Brunger A. T. Mechanistic insights into the recycling machine of the SNARE complex. Nature 2015, 518 (7537), 61–67. 10.1038/nature14148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Some D. Light-scattering-based analysis of biomolecular interactions. Biophys. Rev. 2013, 5 (2), 147–158. 10.1007/s12551-013-0107-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monterroso B.; Alfonso C.; Zorrilla S.; Rivas G. Combined analytical ultracentrifugation, light scattering and fluorescence spectroscopy studies on the functional associations of the bacterial division FtsZ. protein. Methods 2013, 59 (3), 349–362. 10.1016/j.ymeth.2012.12.014. [DOI] [PubMed] [Google Scholar]
- del Castillo U.; Alfonso C.; Acebron S. P.; Martos A.; Moro F.; Rivas G.; Muga A. A quantitative analysis of the effect of nucleotides and the M domain on the association equilibrium of ClpB. Biochemistry 2011, 50 (12), 1991–2003. 10.1021/bi101670s. [DOI] [PubMed] [Google Scholar]
- Nishino T.; Rago F.; Hori T.; Tomii K.; Cheeseman I. M.; Fukagawa T. CENP-T provides a structural platform for outer kinetochore assembly. EMBO J. 2013, 32 (3), 424–436. 10.1038/emboj.2012.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H. L.; Fernandez C.; Fan J. B.; Shewmaker F.; Chen J.; Minton A. P.; Liang Y. Quantitative characterization of heparin binding to Tau protein: implication for inducer-mediated Tau filament formation. J. Biol. Chem. 2010, 285 (6), 3592–3599. 10.1074/jbc.M109.035691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voges M.; Herhut M.; Held C.; Brandenbusch C. Light-scattering data of protein and polymer solutions: A new approach for model validation and parameter estimation. Fluid Phase Equilib. 2018, 465, 65–72. 10.1016/j.fluid.2018.02.022. [DOI] [Google Scholar]
- Rakel N.; Bauer K. C.; Galm L.; Hubbuch J. From osmotic second virial coefficient (B22) to phase behavior of a monoclonal antibody. Biotechnol. Prog. 2015, 31 (2), 438–451. 10.1002/btpr.2065. [DOI] [PubMed] [Google Scholar]
- Moosa M. M.; Tsoi P. S.; Choi K. J.; Ferreon A. C. M.; Ferreon J. C. Direct Single-Molecule Observation of Sequential DNA Bending Transitions by the Sox2 HMG Box. Int. J. Mol. Sci. 2018, 19 (12), 3865. 10.3390/ijms19123865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards J.; Malaurie E.; Kondrashov A.; Long J.; de Moor C. H.; Searle M. S.; Emsley J. Sequence determinants for the tandem recognition of UGU and CUG rich RNA elements by the two N--terminal RRMs of CELF1. Nucleic Acids Res. 2011, 39 (19), 8638–8650. 10.1093/nar/gkr510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E.; Baralle F. E. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J. Biol. Chem. 2001, 276 (39), 36337–36343. 10.1074/jbc.M104236200. [DOI] [PubMed] [Google Scholar]
- Lukavsky P. J.; Daujotyte D.; Tollervey J. R.; Ule J.; Stuani C.; Buratti E.; Baralle F. E.; Damberger F. F.; Allain F. H. Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol. 2013, 20 (12), 1443–1449. 10.1038/nsmb.2698. [DOI] [PubMed] [Google Scholar]
- Buratti E.; Dork T.; Zuccato E.; Pagani F.; Romano M.; Baralle F. E. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 2001, 20 (7), 1774–1784. 10.1093/emboj/20.7.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladd A. N.; Charlet N.; Cooper T. A. The CELF family of RNA binding proteins is implicated in cell-specific and developmentally regulated alternative splicing. Mol. Cell. Biol. 2001, 21 (4), 1285–1296. 10.1128/MCB.21.4.1285-1296.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards J. M.; Long J.; de Moor C. H.; Emsley J.; Searle M. S. Structural insights into the targeting of mRNA GU-rich elements by the three RRMs of CELF1. Nucleic Acids Res. 2013, 41 (14), 7153–7166. 10.1093/nar/gkt470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y.; Efremov A. K.; Yan J. Modulating binding affinity, specificity, and configurations by multivalent interactions. Biophys. J. 2022, 121 (10), 1868–1880. 10.1016/j.bpj.2022.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. E.; Cooper T. A. Pathogenic mechanisms of myotonic dystrophy. Biochem. Soc. Trans. 2009, 37 (6), 1281–1286. 10.1042/BST0371281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J. W.; Urbinati C. R.; Teng-Umnuay P.; Stenberg M. G.; Byrne B. J.; Thornton C. A.; Swanson M. S. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000, 19 (17), 4439–4448. 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie I. R.; Rademakers R.; Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010, 9 (10), 995–1007. 10.1016/S1474-4422(10)70195-2. [DOI] [PubMed] [Google Scholar]
- Ling S. C.; Polymenidou M.; Cleveland D. W. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 2013, 79 (3), 416–438. 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.