

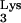
Abstract
Cellular tRNA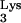 serves as the primer for reverse transcription of human immunodeficiency virus type 1 (HIV-1). tRNA
serves as the primer for reverse transcription of human immunodeficiency virus type 1 (HIV-1). tRNA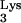 interacts directly with HIV-1 reverse transcriptase (RT), is packaged into viral particles, and anneals to the primer-binding site (PBS) of the HIV-1 genome in order to initiate reverse transcription. Residue A58 of tRNA
interacts directly with HIV-1 reverse transcriptase (RT), is packaged into viral particles, and anneals to the primer-binding site (PBS) of the HIV-1 genome in order to initiate reverse transcription. Residue A58 of tRNA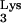 , which lies outside the PBS-complementary region, is posttranscriptionally methylated to form 1-methyladenosine 58 (M1A58). This methylation is thought to serve as a pause signal for plus-strand strong-stop DNA synthesis during reverse transcription. However, formal proof that the methylation is necessary for the pausing of RT has not been obtained in vivo. In the present study, we investigated the role of tRNA
, which lies outside the PBS-complementary region, is posttranscriptionally methylated to form 1-methyladenosine 58 (M1A58). This methylation is thought to serve as a pause signal for plus-strand strong-stop DNA synthesis during reverse transcription. However, formal proof that the methylation is necessary for the pausing of RT has not been obtained in vivo. In the present study, we investigated the role of tRNA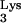 residue A58 in the replication cycle of HIV-1 in living cells. We have developed a mutant tRNA
residue A58 in the replication cycle of HIV-1 in living cells. We have developed a mutant tRNA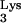 derivative, tRNA
derivative, tRNA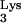 A58U, in which A58 was replaced by U. This mutant tRNA was expressed in CEM cells. We demonstrate that the presence of M1A58 is necessary for the appropriate termination of plus-strand strong-stop DNA synthesis and that the absence of M1A58 allows RT to read the tRNA sequences beyond residue 58. In addition, we show that replacement of M1A58 with U inhibits the replication of HIV-1 in vivo. These results highlight the importance of tRNA primer residue A58 in the reverse transcription process. Inhibition of reverse transcription with mutant tRNA primers constitutes a novel approach for therapeutic intervention against HIV-1.
A58U, in which A58 was replaced by U. This mutant tRNA was expressed in CEM cells. We demonstrate that the presence of M1A58 is necessary for the appropriate termination of plus-strand strong-stop DNA synthesis and that the absence of M1A58 allows RT to read the tRNA sequences beyond residue 58. In addition, we show that replacement of M1A58 with U inhibits the replication of HIV-1 in vivo. These results highlight the importance of tRNA primer residue A58 in the reverse transcription process. Inhibition of reverse transcription with mutant tRNA primers constitutes a novel approach for therapeutic intervention against HIV-1.
Retroviruses contain two copies of an RNA genome but replicate via a DNA intermediate (18). Reverse transcription of the RNA genome into DNA is performed by the viral enzyme reverse transcriptase (RT). The primer for reverse transcription is a cellular tRNA. Retroviruses, long terminal repeat (LTR) retrotransposons, and long interspersed nucleotide element retrotransposons use cellular tRNAs to initiate cDNA synthesis. Different tRNAs are used by different retroviruses and retrotransposons (12, 14).
Lentiviruses, such as feline and simian immunodeficiency viruses and human immunodeficiency virus (HIV), use tRNA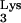 as their primers. tRNA
as their primers. tRNA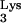 interacts directly with the HIV-1 RT, is packaged into viral particles, and anneals to the PBS of the HIV-1 genome in order to initiate reverse transcription.
interacts directly with the HIV-1 RT, is packaged into viral particles, and anneals to the PBS of the HIV-1 genome in order to initiate reverse transcription.
Early in the viral life cycle, tRNA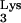 primes minus-strand strong-stop DNA synthesis. Plus-strand strong-stop DNA synthesis is primed by the polypurine tract. During plus-strand strong-stop DNA synthesis, elongation terminates at a 1-methyladenosine at position 58 (M1A58) of tRNA
primes minus-strand strong-stop DNA synthesis. Plus-strand strong-stop DNA synthesis is primed by the polypurine tract. During plus-strand strong-stop DNA synthesis, elongation terminates at a 1-methyladenosine at position 58 (M1A58) of tRNA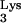 . The precise mechanism of termination at this stage of reverse transcription is not fully characterized. After termination of plus-strand strong-stop DNA synthesis, the tRNA primer is removed by RNase H, allowing the second-strand transfer and subsequent completion of reverse transcription.
. The precise mechanism of termination at this stage of reverse transcription is not fully characterized. After termination of plus-strand strong-stop DNA synthesis, the tRNA primer is removed by RNase H, allowing the second-strand transfer and subsequent completion of reverse transcription.
The role of M1A58 of tRNA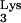 in the retroviral life cycle was first proposed by Gilboa et al. (8), who suggested that termination of plus-strand strong-stop DNA synthesis occurs at base M1A58 in tRNA
in the retroviral life cycle was first proposed by Gilboa et al. (8), who suggested that termination of plus-strand strong-stop DNA synthesis occurs at base M1A58 in tRNA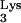 . However, formal proof that the methylation is necessary for the pausing of RT has not been obtained in vivo. In the present study, we investigate the role of tRNA
. However, formal proof that the methylation is necessary for the pausing of RT has not been obtained in vivo. In the present study, we investigate the role of tRNA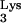 residue A58 in the replication cycle of HIV-1 in living cells. We demonstrate that the presence of M1A58 is necessary for the appropriate termination of plus-strand strong-stop DNA synthesis and that the absence of M1A58 allows RT to read beyond residue 58 during plus-strand strong-stop DNA synthesis. In addition, we show that replacement of M1A58 with U inhibits the replication of HIV-1 in vivo.
residue A58 in the replication cycle of HIV-1 in living cells. We demonstrate that the presence of M1A58 is necessary for the appropriate termination of plus-strand strong-stop DNA synthesis and that the absence of M1A58 allows RT to read beyond residue 58 during plus-strand strong-stop DNA synthesis. In addition, we show that replacement of M1A58 with U inhibits the replication of HIV-1 in vivo.
MATERIALS AND METHODS
Plasmid construction.
Using PCR, a human tRNA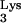 transcriptional unit was obtained and cloned into mutagenesis vector M13mp19 (13). Strand-specific site-directed (11) mutagenesis was employed to generate mutant tRNA
transcriptional unit was obtained and cloned into mutagenesis vector M13mp19 (13). Strand-specific site-directed (11) mutagenesis was employed to generate mutant tRNA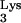 A58U using primer 5′-CCGAACAGGGACATGAACCCTGGAC-3′. The tRNA transcriptional unit from the resultant vector M13-Lys3A58U was cut with SspI and ClaI, generating a 310-bp product containing the tRNA transcriptional unit. This fragment was cloned into N2A using MluI and SnaB1, thus generating N2A-Lys3A58U. HIV-green fluorescent protein (GFP)-ΔEnv was constructed by replacing Thy-1 with GFP in HIV-Thy-ΔEnv (10) using XbaI and XhoI restriction sites.
A58U using primer 5′-CCGAACAGGGACATGAACCCTGGAC-3′. The tRNA transcriptional unit from the resultant vector M13-Lys3A58U was cut with SspI and ClaI, generating a 310-bp product containing the tRNA transcriptional unit. This fragment was cloned into N2A using MluI and SnaB1, thus generating N2A-Lys3A58U. HIV-green fluorescent protein (GFP)-ΔEnv was constructed by replacing Thy-1 with GFP in HIV-Thy-ΔEnv (10) using XbaI and XhoI restriction sites.
Generation of CEM cells expressing mutant tRNA.
Immortalized T-cell line CEM (AIDS Repository, Rockville, Md.) was grown in Iscove's medium (BioWhittaker, Walkersville, Md.) supplemented with 10% fetal calf serum, 100 U of penicillin/ml, 100 μg of streptomycin sulfate/ml, 2.9 mg of l-glutamine/ml, and 0.1 mM sodium citrate in 0.14% sodium chloride (Gibco BRL, Grand Island, N.Y.) and kept at a density of 0.5 to 1 million cells/ml. Ten micrograms of DNA from N2A-Lys3A58U was electroporated into 1 million CEM cells by using 0.2-cm gap cuvettes (Bio-Rad, Hercules, Calif.) and a Bio-Rad Gene PulserII electroporator, 280 V, 975 μF. Bulk-transfected CEM-N2A-Lys3A58U cells were grown under normal conditions (37°C, 5% CO2 and 95% H2O) overnight. One day after transfection, medium was replaced with 10% Iscove's with 0.5 mg of G418/ml for antibiotic selection. Cells containing the retroviral construct were neomycin resistant and were selected using G418. Cells were grown for 14 days at a density of 0.5 to 1 million cells/ml. Fourteen days after selection, single-cell clones were obtained by plating in mini-well plates.
Detection of tRNA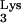 A58U.
A58U.
RNA extracted from cell clones was subjected to RT-PCR amplification of both mutant tRNA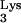 A58U and normal tRNA
A58U and normal tRNA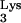 . RT-PCR products from each clone were cloned into the SrfI site of sequencing vector PCR-script (Stratagene, Cedar Creek, Tex.). Ligated product from each cell clone was transformed into Escherichia coli cells and grown into minipreps for each clone. Minipreps from each clone were sequenced using an ABI Prism sequencer (Perkin-Elmer, Norwalk, Conn.) and screened for the presence or absence of mutant tRNA
. RT-PCR products from each clone were cloned into the SrfI site of sequencing vector PCR-script (Stratagene, Cedar Creek, Tex.). Ligated product from each cell clone was transformed into Escherichia coli cells and grown into minipreps for each clone. Minipreps from each clone were sequenced using an ABI Prism sequencer (Perkin-Elmer, Norwalk, Conn.) and screened for the presence or absence of mutant tRNA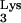 A58U.
A58U.
Infections.
For infections, 5 × 104 CEM cells were added in a volume of 0.5 ml of media into 1.5-ml tubes. Each infection was performed in triplicate tubes. Then, 0.5 ml of virus at the correct dilution with 10 μg of polybrene/ml was added to each tube to achieve multiplicities of infection (MOIs) of 0.01 and 0.1. Infected cultures were rocked at 37°C for 1 h, cells were gently spun out, and infected cell pellets were resuspended in 0.5 ml of 10% Iscove's media and plated in 24-well plates. Cells were cultured for the duration of the experiment at a density of 0.5 to 1 million cells/ml. The percent of infected cells (i.e., percent of Thy-1.2-positive cells) was monitored at regular intervals up to 30 days postinfection. Infections of transduced cell clones were performed in triplicate, and infections of normal CEM cells were performed five times.
Limiting-dilution assay.
Limiting-dilution assay infections were performed using 100,000 cells per well in a volume of 0.25 ml of tissue culture medium. Each well was infected with 0.25 ml of serially diluted (10−1 to 10−10 for HIV-1NL4-3 and 10−1 to 10−7 for HIV-1HXB2-HisAc) virus. Each infection was performed in quadruplicate. Cells were cultured at a density of 0.5 to 1 million cells/ml, and cell-free supernatants were collected at 15 days postinfection for p24 enzyme-linked immunosorbent assay (ELISA) analysis. Viral titers were calculated using the National Center for Biotechnology Information ID50 statistical program.
PCR.
Total DNA from infected cultures was isolated for PCR using urea lysis DNA extraction. DNA was amplified using primer set a-b (5′CCACTGACCTTTGGATGG and 5′GTCCCTGTTCGGGCG, respectively) to test for the presence of plus-strand strong-stop DNA. DNA was amplified with primer set a-c (5′CCACTGACCTTTGGATGG and 5′GCCCGGATAGCTCAGTC, respectively) to test for the presence of plus-strand strong-stop DNA with an attached c-tRNA tail. Products were resolved on a 2% agarose ethidium bromide gel. For sequencing, products were cloned into the sequencing vector, PCR-script (Stratagene), and sequenced using an ABI Prism sequencer (Perkin-Elmer).
Immunological detection of viral antigens.
To assess the level of HIV p24 in culture supernatants, cell-free supernatants were collected from infected cultures at various time points and frozen at −80°C until needed for analysis. Detection of HIV-1 p24 was performed by a capture ELISA with monoclonal antibodies and protein standards obtained from the NIH AIDS Reagent Repository, rabbit polyclonal anti-HIVp24 antibody obtained from Vector Labs (Burlingame, Calif.), and Vector Labs polyclonal anti-rabbit IgG Elite Vectastain ABC Kit. Colorimetric analysis was performed using Vector Labs ABTS substrate and a Microplate Reader Model 550 (Bio-Rad).
Flow cytometry.
To determine the percent of infected cells, flow cytometry was performed with an antibody specific to the murine Thy-1.2 antigen expressed on the surface of infected cells. Analysis was performed with an Epics Elite ESP apparatus (Coulter Corp., Hialeah, Fla.). Gates for detection of Thy-1–fluorescein isothiocyanate were established with mock-infected cells as a background. Because electronic settings varied from experiment to experiment, gates were defined such that the percentage of false-positive events was not higher than 0.3 in the mock-infected population. CD4 was detected using a phycoerythrin (PE)-conjugated monoclonal antibody to human CD4, obtained from Caltag (Burlingame, Calif.), and CXCR4 was detected with PE-conjugated anti-fusin (Pharmingen, San Diego, Calif.).
Production of defective HIV.
To obtain HIV-GFP-ΔEnv pseudotyped with vesicular stomatitis virus protein G (VSV-G), we cotransfected HIV-GFP-ΔEnv and HCMV–VSV-G (6) into COS-7 cells as previously described (19).
RESULTS
Design and construction of tRNA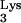 A58U.
A58U.
The cloning of a complete tRNA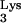 transcriptional unit (Fig. 1A) from human genomic DNA was described earlier (13). The tRNA
transcriptional unit (Fig. 1A) from human genomic DNA was described earlier (13). The tRNA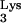 transcriptional unit was modified using site-directed mutagenesis to change residue A58 to U (Fig. 1A). The resulting mutant tRNA transcriptional unit, named tRNA
transcriptional unit was modified using site-directed mutagenesis to change residue A58 to U (Fig. 1A). The resulting mutant tRNA transcriptional unit, named tRNA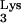 A58U, was then cloned into the retroviral vector N2A (9) to generate N2A-tRNA
A58U, was then cloned into the retroviral vector N2A (9) to generate N2A-tRNA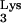 A58U (Fig. 1B). N2A-tRNA
A58U (Fig. 1B). N2A-tRNA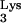 A58U was then stably transfected into CEM cells, and stable transfectants were selected with G418.
A58U was then stably transfected into CEM cells, and stable transfectants were selected with G418.
FIG. 1.

Design of a mutant tRNA and cloning into the retroviral vector, N2A. (A) Schematic diagram depicting the primary and secondary structures of tRNA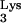 and design of the A58U mutation. Region marked by curved arrow anneals to the HIV-1 PBS. 1-Methyladenosine-58 is shown with a vertical arrow. (B) A complete tRNA
and design of the A58U mutation. Region marked by curved arrow anneals to the HIV-1 PBS. 1-Methyladenosine-58 is shown with a vertical arrow. (B) A complete tRNA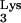 A58U polymerase III transcriptional unit (hatched box) was inserted into the 3′ LTR of the murine leukemia virus-derived retroviral vector, N2A (9), by using a unique SnaBI restriction endonuclease site. S, 5-methoxycarbonylmethyl-2-thiouridine; Ψ, pseudouridine; D, dihydrouridine; Tm, 2′-O-methyl-5-methyluridine; R, 2-methylthio-n-6-threonyl carbomoyladenosine.
A58U polymerase III transcriptional unit (hatched box) was inserted into the 3′ LTR of the murine leukemia virus-derived retroviral vector, N2A (9), by using a unique SnaBI restriction endonuclease site. S, 5-methoxycarbonylmethyl-2-thiouridine; Ψ, pseudouridine; D, dihydrouridine; Tm, 2′-O-methyl-5-methyluridine; R, 2-methylthio-n-6-threonyl carbomoyladenosine.
Detection of tRNA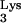 A58U and tRNA
A58U and tRNA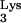 in stable transfectants.
in stable transfectants.
To detect the presence of mutant tRNA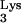 A58U and wild-type tRNA
A58U and wild-type tRNA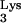 , we performed the following analysis. Bulk RNA from tRNA
, we performed the following analysis. Bulk RNA from tRNA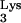 A58U-transfected cell clones 1 and 3 was isolated and amplified by RT-PCR using primers that flanked the tRNA
A58U-transfected cell clones 1 and 3 was isolated and amplified by RT-PCR using primers that flanked the tRNA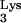 A58U mutation. RT-PCR-amplified products were blunt ended and cloned into the vector, PCR-script (Stratagene). The product of this ligation was then transformed into E. coli cells, and random colonies were used for growing small-scale DNA preparations. DNA minipreps were characterized by DNA sequencing, and the presence of wild-type (A58) versus mutant (U58) residue was verified. tRNA
A58U mutation. RT-PCR-amplified products were blunt ended and cloned into the vector, PCR-script (Stratagene). The product of this ligation was then transformed into E. coli cells, and random colonies were used for growing small-scale DNA preparations. DNA minipreps were characterized by DNA sequencing, and the presence of wild-type (A58) versus mutant (U58) residue was verified. tRNA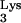 A58U transfectants 1 and 3 produced 4 out of 37 (11%) and 10 out of 46 (22%) bacterial clones containing A58U, respectively, and 89 and 78% containing wild-type tRNA, respectively.
A58U transfectants 1 and 3 produced 4 out of 37 (11%) and 10 out of 46 (22%) bacterial clones containing A58U, respectively, and 89 and 78% containing wild-type tRNA, respectively.
Presence of tRNA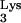 A58U leads to production of chimeric intermediate containing DNA sequences complementary to tRNA.
A58U leads to production of chimeric intermediate containing DNA sequences complementary to tRNA.
Plus-strand strong-stop DNA synthesis normally terminates at base 1-methyladenosine 58 (M1A58) in tRNA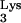 . It was proposed that the presence of a methyl group on the N1 of adenosine blocks the ability of this base to form hydrogen bonds with an incoming nucleotide during reverse transcription (8). The inability of RT to place an incoming nucleotide on the M1A58 residue may prompt RT to pause and terminate plus-strand strong-stop DNA synthesis at this base.
. It was proposed that the presence of a methyl group on the N1 of adenosine blocks the ability of this base to form hydrogen bonds with an incoming nucleotide during reverse transcription (8). The inability of RT to place an incoming nucleotide on the M1A58 residue may prompt RT to pause and terminate plus-strand strong-stop DNA synthesis at this base.
Since 1-adenosine methyltransferase, the enzyme responsible for the posttranscriptional formation of 1-methyladenosine in the tRNA base 58, is nucleotide specific (16), changing base 58 in tRNA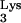 from A to U will abrogate the posttranscriptional methylation (Fig. 1A). We predicted that mutations disrupting methylation of A58 in the tRNA would allow for RT-mediated DNA synthesis residue 58 (Fig. 2).
from A to U will abrogate the posttranscriptional methylation (Fig. 1A). We predicted that mutations disrupting methylation of A58 in the tRNA would allow for RT-mediated DNA synthesis residue 58 (Fig. 2).
FIG. 2.

HIV-1 reverse transcription and the predicted consequences of priming with mutant tRNA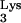 A58U. Plus-strand strong-stop DNA synthesis is initiated from the polypurine tract and continues toward the tRNA PBS-binding region. Normally, synthesis stops at base M1A58 in the tRNA
A58U. Plus-strand strong-stop DNA synthesis is initiated from the polypurine tract and continues toward the tRNA PBS-binding region. Normally, synthesis stops at base M1A58 in the tRNA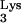 (left). Without the posttranscriptional addition of a methyl group at base 58 (as predicted for the A58U mutant of tRNA
(left). Without the posttranscriptional addition of a methyl group at base 58 (as predicted for the A58U mutant of tRNA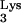 ), RT reads beyond base 58 and continues to copy additional tRNA sequences (right, gray arrow). Since the tRNA-complementary sequences (c-tRNA) are not homologous to the HIV-1 minus strand, plus-strand synthesis cannot be completed.
), RT reads beyond base 58 and continues to copy additional tRNA sequences (right, gray arrow). Since the tRNA-complementary sequences (c-tRNA) are not homologous to the HIV-1 minus strand, plus-strand synthesis cannot be completed.
Reverse transcription of tRNA sequences beyond base 58 would result in DNA sequences complementary to tRNA (c-tRNA; Fig. 2). The c-tRNA sequences would be unable to anneal to the viral minus-strand DNA during second-strand transfer, thus interfering with the completion of reverse transcription. If these predictions were true, a chimeric DNA product containing viral sequences linked to inappropriate tRNA-complementary sequences (c-tRNA) should be detected in cells containing tRNA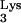 A58U (Fig. 3A).
A58U (Fig. 3A).
FIG. 3.

Analysis of abnormal tRNA-cDNA sequences (c-tRNA). (A) Primer sets a-b and a-c were used to PCR amplify HIV-1 plus-strand strong-stop DNA (537 bp) and plus-strand strong-stop DNA linked to c-tRNA (597 bp), respectively. The 597-bp product corresponds to an intermediate product in Fig. 2 which contains U3–R–U5–c-tRNA. The complementary nucleotides to A58U and Ψ55 are denoted with black arrows. (B) PCR amplified products from infected (+) and uninfected (−) normal CEM and mutant tRNA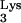 A58U-containing cell lines. Detection of a 597-bp product in infected mutant tRNA cell lines indicated the presence of plus-strand strong-stop DNA linked to c-tRNA sequences (lanes 2 and 3). This aberrant product was not detected in normal, infected CEM cells (lane 4) nor in uninfected cells (lanes 8 and 9). As a control, a 537-bp plus-strand strong-stop DNA product was detected in all infections (lanes 5 to 7) but not in uninfected cells (lanes 8 and 9).
A58U-containing cell lines. Detection of a 597-bp product in infected mutant tRNA cell lines indicated the presence of plus-strand strong-stop DNA linked to c-tRNA sequences (lanes 2 and 3). This aberrant product was not detected in normal, infected CEM cells (lane 4) nor in uninfected cells (lanes 8 and 9). As a control, a 537-bp plus-strand strong-stop DNA product was detected in all infections (lanes 5 to 7) but not in uninfected cells (lanes 8 and 9).
Stable transfectants containing tRNA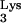 A58U were exposed to HIV-1. We then tested for the presence of c-tRNA by PCR amplification of DNA from infected cells. As a control, infected and uninfected normal CEM cells (untransfected) were also used for this analysis. Cells were infected with HIV-Thy (50 ng of p24), a replication-competent HIV-1 mutant that expresses the murine Thy-1 gene in place of nef (10). At day 25 postinfection, cells were lysed and DNA was extracted and subjected to PCR. Two pairs of primers were used for this analysis (Fig. 3A). Primer pair a-b was designed to amplify a 537-bp product spanning the 3′ LTR and the PBS (plus-strand strong-stop DNA). Primer pair a-c was designed to amplify a 597-bp region comprising the 3′ LTR, PBS, and the c-tRNA (plus-strand strong-stop DNA with attached c-tRNA tail). Because the 537-bp region amplified by primers a-b should be present in all infections regardless of the presence or absence of the mutant tRNA, we used primer pair a-b as a positive control. The region amplified by primer set a-c should be present only if reverse transcription fails to terminate at residue 58 of the tRNA (Fig. 3A).
A58U were exposed to HIV-1. We then tested for the presence of c-tRNA by PCR amplification of DNA from infected cells. As a control, infected and uninfected normal CEM cells (untransfected) were also used for this analysis. Cells were infected with HIV-Thy (50 ng of p24), a replication-competent HIV-1 mutant that expresses the murine Thy-1 gene in place of nef (10). At day 25 postinfection, cells were lysed and DNA was extracted and subjected to PCR. Two pairs of primers were used for this analysis (Fig. 3A). Primer pair a-b was designed to amplify a 537-bp product spanning the 3′ LTR and the PBS (plus-strand strong-stop DNA). Primer pair a-c was designed to amplify a 597-bp region comprising the 3′ LTR, PBS, and the c-tRNA (plus-strand strong-stop DNA with attached c-tRNA tail). Because the 537-bp region amplified by primers a-b should be present in all infections regardless of the presence or absence of the mutant tRNA, we used primer pair a-b as a positive control. The region amplified by primer set a-c should be present only if reverse transcription fails to terminate at residue 58 of the tRNA (Fig. 3A).
The results of this PCR analysis are shown in Fig. 3B. Primer set a-c amplified full-length c-tRNA linked to viral plus-strand strong-stop DNA, generating a product 597 bp in size which was only detected in CEM cells containing tRNA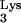 A58U (Fig. 3B). This 597-bp product was not detected in infected, normal CEM cells (Fig. 3B). Primer set a-b, which detects plus-strand strong-stop DNA, amplified a product of 537 bp in size in both normal CEM cells and CEM cells containing tRNA
A58U (Fig. 3B). This 597-bp product was not detected in infected, normal CEM cells (Fig. 3B). Primer set a-b, which detects plus-strand strong-stop DNA, amplified a product of 537 bp in size in both normal CEM cells and CEM cells containing tRNA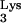 A58U (Fig. 3B). The previous two PCR products could not be detected in uninfected cells (Fig. 3B).
A58U (Fig. 3B). The previous two PCR products could not be detected in uninfected cells (Fig. 3B).
To verify the nature of the 597-bp band, we cloned and sequenced the amplified products shown in Fig. 3, lanes 2 and 3. Sequencing confirmed the presence of full-length c-tRNA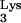 A58U sequences linked to viral plus-strand strong-stop DNA. These data indicate that HIV-1 RT was able to read beyond residue U58 in the mutant tRNA
A58U sequences linked to viral plus-strand strong-stop DNA. These data indicate that HIV-1 RT was able to read beyond residue U58 in the mutant tRNA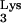 A58U but not beyond A58. The presence of the mutant base A58U was verified by the appearance of a complementary “A” in the c-tRNA (data not shown).
A58U but not beyond A58. The presence of the mutant base A58U was verified by the appearance of a complementary “A” in the c-tRNA (data not shown).
Inhibition of HIV-1 replication by mutant tRNA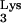 A58U.
A58U.
In the subsequent steps of reverse transcription, following the second strand transfer (Fig. 2), annealing of PBS with PBS′ forms the normal primer for continuing DNA synthesis. However, if the initial priming was performed by tRNA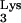 A58U, the additional tRNA-complementary sequences (c-tRNA) linked to plus-strand strong-stop DNA should prevent effective priming after the second-strand transfer (Fig. 2). We hypothesized that the c-tRNA cannot serve as a primer for completion of plus-strand DNA synthesis. If this were true, HIV-1 replication should be delayed or blocked in cells containing tRNA
A58U, the additional tRNA-complementary sequences (c-tRNA) linked to plus-strand strong-stop DNA should prevent effective priming after the second-strand transfer (Fig. 2). We hypothesized that the c-tRNA cannot serve as a primer for completion of plus-strand DNA synthesis. If this were true, HIV-1 replication should be delayed or blocked in cells containing tRNA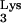 A58U. To test the potential effect of tRNA
A58U. To test the potential effect of tRNA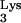 A58U on HIV-1 replication, CEM lymphocytes that were stably transfected with the construct, N2A-tRNA
A58U on HIV-1 replication, CEM lymphocytes that were stably transfected with the construct, N2A-tRNA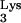 A58U, were tested for their ability to support HIV-1 replication.
A58U, were tested for their ability to support HIV-1 replication.
To examine the kinetics of HIV-1 replication in tRNA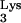 A58U-transfected clones, we utilized a replication-competent HIV-1 recombinant, HIV-Thy, that expresses the murine Thy-1.2 gene in place of nef (10). Cells infected with this virus express the murine Thy-1.2 glycoprotein on their surface and can be detected by flow cytometry. Infection of normal CEM cells with HIV-Thy at an MOI of 0.01 produced infections that reached a level of 30% infected cells or greater between days 16 and 18 postinfection (Fig. 4). In contrast, replication of HIV-Thy in clones of CEM containing tRNA
A58U-transfected clones, we utilized a replication-competent HIV-1 recombinant, HIV-Thy, that expresses the murine Thy-1.2 gene in place of nef (10). Cells infected with this virus express the murine Thy-1.2 glycoprotein on their surface and can be detected by flow cytometry. Infection of normal CEM cells with HIV-Thy at an MOI of 0.01 produced infections that reached a level of 30% infected cells or greater between days 16 and 18 postinfection (Fig. 4). In contrast, replication of HIV-Thy in clones of CEM containing tRNA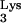 A58U was delayed (Fig. 4, clones 1, 3, and 8). Infection levels in transfected clones remained under 30% during the course of the experiments (30 days).
A58U was delayed (Fig. 4, clones 1, 3, and 8). Infection levels in transfected clones remained under 30% during the course of the experiments (30 days).
FIG. 4.

Replication kinetics of HIV–Thy-1 in mutant tRNA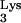 A58U-expressing or normal CEM cells. Cells were grown exponentially and infected with HIV–Thy-1 at the indicated MOI. Infections of CEM containing tRNA
A58U-expressing or normal CEM cells. Cells were grown exponentially and infected with HIV–Thy-1 at the indicated MOI. Infections of CEM containing tRNA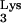 A58U were performed in triplicate. Infection of normal CEM was performed five times. On various days postinfection, cells were analyzed for the expression of the reporter gene Thy-1 using flow cytometry. Results are plotted as the percent of infected cells (those expressing Thy-1 on their cell surface).
A58U were performed in triplicate. Infection of normal CEM was performed five times. On various days postinfection, cells were analyzed for the expression of the reporter gene Thy-1 using flow cytometry. Results are plotted as the percent of infected cells (those expressing Thy-1 on their cell surface).
Antiviral strategies are often overcome when high MOIs are used. To test the ability of tRNA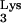 A58U-transfected clones to inhibit viral replication at a high MOI, cells were challenged with a 10-fold-higher amount of virus (Fig. 4, MOI = 0.1). Infection of normal CEM cells at the higher MOI produced infections that reached 30% between days 14 and 16 postinfection. In eight of nine attempts, infection of tRNA
A58U-transfected clones to inhibit viral replication at a high MOI, cells were challenged with a 10-fold-higher amount of virus (Fig. 4, MOI = 0.1). Infection of normal CEM cells at the higher MOI produced infections that reached 30% between days 14 and 16 postinfection. In eight of nine attempts, infection of tRNA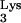 A58U-transfected clones produced either undetected infections (clone 1, experiment 1; clone 3, experiments 1 and 2; clone 3, experiments 2 and 3) or delayed infections (clone 1, experiments 1 and 2; clone 3, experiment 3). One of the infections of clone 3 (experiment 3) produced kinetics of replication which was not significantly different from infection of normal cells. Thus, a significant inhibition or delay of HIV-1 replication was also observed in tRNA
A58U-transfected clones produced either undetected infections (clone 1, experiment 1; clone 3, experiments 1 and 2; clone 3, experiments 2 and 3) or delayed infections (clone 1, experiments 1 and 2; clone 3, experiment 3). One of the infections of clone 3 (experiment 3) produced kinetics of replication which was not significantly different from infection of normal cells. Thus, a significant inhibition or delay of HIV-1 replication was also observed in tRNA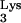 A58U-transfected clones at an MOI of 0.1.
A58U-transfected clones at an MOI of 0.1.
Several experiments produced no detectable levels of infected cells (<0.2%) throughout the duration of the culture. Two explanations could be formulated for the apparent lack of viral replication. First, perhaps infection did not occur or was abortive. Second, perhaps infection occurred, but the levels of viral replication remained very low throughout the experiment. To distinguish between these possibilities, cells from the first set of experiments (MOI of 0.01, clone 1, experiments 1 and 3) were cocultured with wild-type CEM cells. If low levels of virus were present in the cultures, then abundant viral replication should be observed in the cocultures. On the contrary, if infection failed to occur, then the cocultures should demonstrate absence of virus. At 8 days after introduction of wild-type CEM cells, 4.4 and 29.3% of cells were Thy-1.2 positive in cocultures from an MOI of 0.01, clone 1, experiments 1 and 3, respectively.
We conclude from the above observations that cells containing mutant tRNA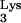 A58U suppress the replication of HIV-1, as demonstrated by the delayed kinetics of HIV-1 infection in cells containing tRNA
A58U suppress the replication of HIV-1, as demonstrated by the delayed kinetics of HIV-1 infection in cells containing tRNA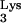 A58U when compared to normal CEM cells.
A58U when compared to normal CEM cells.
Detection of CD4 and CXCR4.
We hypothesized that the inhibition of HIV-1 in tRNA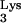 A58U-containing clones is due to expression of tRNA
A58U-containing clones is due to expression of tRNA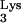 A58U. However, such inhibition could also be explained by the potential loss of viral receptors or coreceptors from the target cells. To rule out this possibility, detection of CD4 and CXCR4 was performed in transfected cell clones by flow cytometry. All tRNA
A58U. However, such inhibition could also be explained by the potential loss of viral receptors or coreceptors from the target cells. To rule out this possibility, detection of CD4 and CXCR4 was performed in transfected cell clones by flow cytometry. All tRNA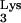 A58U transfectants were found to express levels of CD4 and CXCR4 that were comparable to those of normal cells (Fig. 5). Thus, inhibition of HIV-1 replication in CEM tRNA
A58U transfectants were found to express levels of CD4 and CXCR4 that were comparable to those of normal cells (Fig. 5). Thus, inhibition of HIV-1 replication in CEM tRNA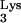 A58U was not due to loss of receptors or coreceptors.
A58U was not due to loss of receptors or coreceptors.
FIG. 5.

Expression of HIV-1 receptor and coreceptor in mutant tRNA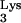 A58U CEM cells. The presence of the CD4 and CXCR4 molecules on the surface of cells was assessed by flow cytometry. CD4 was detected using a phycoerythrin (PE)-conjugated monoclonal antibody to human CD4 (Caltag), and CXCR4 was detected with PE-conjugated anti-fusin (Pharmingen). E and F denote negative and positive gates, respectively.
A58U CEM cells. The presence of the CD4 and CXCR4 molecules on the surface of cells was assessed by flow cytometry. CD4 was detected using a phycoerythrin (PE)-conjugated monoclonal antibody to human CD4 (Caltag), and CXCR4 was detected with PE-conjugated anti-fusin (Pharmingen). E and F denote negative and positive gates, respectively.
Defective HIV-1 pseudotype containing wild-type tRNA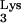 is not inhibited by tRNA
is not inhibited by tRNA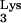 A58U transfectants.
A58U transfectants.
Retroviral particles encapsidate tRNA molecules during virion assembly and budding. Thus, the tRNA molecules that will be utilized as primers during the next infection cycle are not provided by the cell being infected but are present in the virion itself. Based on the previous idea, we predicted that a defective retroviral vector packaged in the presence of wild-type tRNA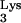 would be able to efficiently produce a single round of infection in cells expressing tRNA
would be able to efficiently produce a single round of infection in cells expressing tRNA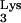 A58U.
A58U.
A defective virus, HIV-GFP-Δenv (Fig. 6A), was packaged in producer cells (COS-7) containing only endogenous, wild-type tRNA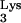 . HIV-GFP-Δenv is a defective HIV-1 that was generated by deleting env and replacing nef with the gene for green fluorescent protein (GFP). The env deficit is complemented in trans by cotransfection of VSV-G (2) to produce a defective virus, HIV-GFP-Δenv/VSV-G. This defective virus is capable of entry, reverse transcription, integration, and expression of the reporter gene, GFP. However, because of the deletion in env, this virus is unable to produce progeny. Cells expressing mutant tRNA
. HIV-GFP-Δenv is a defective HIV-1 that was generated by deleting env and replacing nef with the gene for green fluorescent protein (GFP). The env deficit is complemented in trans by cotransfection of VSV-G (2) to produce a defective virus, HIV-GFP-Δenv/VSV-G. This defective virus is capable of entry, reverse transcription, integration, and expression of the reporter gene, GFP. However, because of the deletion in env, this virus is unable to produce progeny. Cells expressing mutant tRNA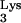 A58U and normal CEM cells were exposed with HIV-GFP-Δenv/VSV-G at an MOI of 1. At 48 h postexposure, cells were analyzed for GFP expression (Fig. 6B). All of the tested CEM tRNA
A58U and normal CEM cells were exposed with HIV-GFP-Δenv/VSV-G at an MOI of 1. At 48 h postexposure, cells were analyzed for GFP expression (Fig. 6B). All of the tested CEM tRNA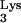 A58U transfectants were infected at levels that were not significantly different from those of wild-type CEM cells (Fig. 6B). These results indicate that tRNA
A58U transfectants were infected at levels that were not significantly different from those of wild-type CEM cells (Fig. 6B). These results indicate that tRNA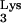 A58U transfectant cell lines are able to support entry, reverse transcription, integration, and gene expression by a pseudotype virus containing normal tRNA
A58U transfectant cell lines are able to support entry, reverse transcription, integration, and gene expression by a pseudotype virus containing normal tRNA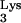 .
.
FIG. 6.

Single-step infection of normal and mutant cells with a replication-defective virus. (A) Schematic representation of the components needed for the defective vector, HIV–GFP–ΔEnv/VSV-G. The defective transfer vector, HIV-GFP-ΔEnv, contains the GFP reporter gene in place of nef and is envelope defective. The VSV-G envelope is supplied in trans by the plasmid, HCMV–VSV-G. (B) GFP expression as a measure of infection with HIV–GFP–ΔEnv/VSV-G in normal CEM and CEM cells transfected with tRNA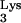 A58U.
A58U.
Quantitation of inhibition of HIV-1 replication by limiting-dilution assay.
To obtain a quantitative measurement of the level of inhibition by tRNA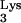 A58U cells, we compared the viral titers of an HIV-1 virus stock in mutant versus normal CEM cells by limiting-dilution assay (Fig. 7). Cells were infected with serially diluted HIV-1NL4-3 (1). At 14 days postinfection, p24 ELISA was used to determine the presence or absence of virus in each well. The titers of HIV-1NL4-3, when used to infect CEM and CEM A58U cells, were 3.6 × 107 and 3.6 × 104 IU/ml, respectively. Thus, the measured titer of HIV in cells containing tRNA
A58U cells, we compared the viral titers of an HIV-1 virus stock in mutant versus normal CEM cells by limiting-dilution assay (Fig. 7). Cells were infected with serially diluted HIV-1NL4-3 (1). At 14 days postinfection, p24 ELISA was used to determine the presence or absence of virus in each well. The titers of HIV-1NL4-3, when used to infect CEM and CEM A58U cells, were 3.6 × 107 and 3.6 × 104 IU/ml, respectively. Thus, the measured titer of HIV in cells containing tRNA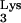 A58U was about 1,000-fold lower than that of normal CEM cells (Fig. 7A).
A58U was about 1,000-fold lower than that of normal CEM cells (Fig. 7A).
FIG. 7.

Measurements of viral infectivity by limiting-dilution assays. Limiting-dilution assays were performed in quadruplicate, using 10-fold dilutions of an initial virus stock and infecting 105 cells of the indicated type per well. (A) Comparison of relative titers of HIV-1NL4-3 (1) in normal and tRNA-expressing CEM cells, clone 1. (B) Comparison of relative titers of HIV-1HXB2-HisAc (17) in the same cell types. HIV-1HXB2-HisAc is a mutant virus that utilizes tRNAHis (17).
Mutant HIV-1 that utilizes tRNAHis as primer is not inhibited by tRNA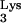 A58U.
A58U.
We hypothesized that the observed inhibition in mutant cells is due to the presence of tRNA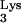 A58U in the mutant cells. Thus, a retrovirus which utilizes a tRNA other than tRNA
A58U in the mutant cells. Thus, a retrovirus which utilizes a tRNA other than tRNA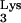 should not be inhibited by tRNA
should not be inhibited by tRNA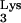 A58U. An HIV-1 mutant that utilizes tRNAHis (HIV-1HXB2-HisAc) was previously described (17). HIV-1HXB2-HisAc was titrated on normal CEM or tRNA
A58U. An HIV-1 mutant that utilizes tRNAHis (HIV-1HXB2-HisAc) was previously described (17). HIV-1HXB2-HisAc was titrated on normal CEM or tRNA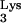 A58U transfectants, using a limiting-dilution assay as described above. The titers of HIV-1HXB2-HisAc on CEM and CEM A58U cells were 9.8 × 104 and 6.1 × 104 IU/ml, respectively (Fig. 7B). These results suggest that tRNA
A58U transfectants, using a limiting-dilution assay as described above. The titers of HIV-1HXB2-HisAc on CEM and CEM A58U cells were 9.8 × 104 and 6.1 × 104 IU/ml, respectively (Fig. 7B). These results suggest that tRNA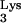 A58U transfectants do not significantly differ from normal cells in their ability to support infection by HIV-1HXB2-HisAc. Thus, we exclude the possibility that inhibition of HIV-1 by tRNA
A58U transfectants do not significantly differ from normal cells in their ability to support infection by HIV-1HXB2-HisAc. Thus, we exclude the possibility that inhibition of HIV-1 by tRNA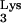 A58U may be due to an intrinsic inability of CEM A58U cells to support HIV-1 replication.
A58U may be due to an intrinsic inability of CEM A58U cells to support HIV-1 replication.
DISCUSSION
We report that cellular expression of mutant tRNA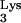 A58U allows HIV-1 plus-strand strong-stop DNA to be elongated beyond the pause site, residue 58 in the tRNA. This event generates DNA sequences complementary to tRNA (c-tRNA), continuous with plus-strand strong-stop DNA. We found c-tRNA product in infected cells containing tRNA
A58U allows HIV-1 plus-strand strong-stop DNA to be elongated beyond the pause site, residue 58 in the tRNA. This event generates DNA sequences complementary to tRNA (c-tRNA), continuous with plus-strand strong-stop DNA. We found c-tRNA product in infected cells containing tRNA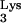 A58U but not in infected cells expressing only wild-type tRNA
A58U but not in infected cells expressing only wild-type tRNA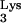 .
.
In vitro studies have suggested that M1A58 is only a minor contributor of termination of plus-strand strong-stop DNA (4). Ben-Artzi et al. (4) found two determinants that may serve as stop signals for plus-strand strong-stop DNA synthesis. One stop signal was the methylated A58 (M1A58) residue in tRNA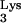 . The second stop signal was the secondary structure of the PBS sequence. However, the second signal appeared to constitute a stronger terminator in vitro (4).
. The second stop signal was the secondary structure of the PBS sequence. However, the second signal appeared to constitute a stronger terminator in vitro (4).
In contrast to observations obtained by Ben-Artzi et al., Burnett et al. (5) found that more than 65% of plus-strand strong-stop DNA terminated at base M1A58 when using natural tRNA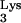 as a primer in an in vitro assay. This suggested that M1A58 is important for correct termination of plus-strand strong-stop DNA synthesis. In addition, plus-strand strong-stop DNA synthesis continued to elongate through tRNA sequences when unmodified, synthetic tRNA
as a primer in an in vitro assay. This suggested that M1A58 is important for correct termination of plus-strand strong-stop DNA synthesis. In addition, plus-strand strong-stop DNA synthesis continued to elongate through tRNA sequences when unmodified, synthetic tRNA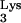 was used as a primer (5).
was used as a primer (5).
Recent work by Wu et al. tested the ability of RT to read beyond M1A58 by using endogenous RT reactions (20). These experiments revealed multiple termination sites for plus-strand strong-stop DNA synthesis. The first termination site observed was M1A58. In addition, a second termination site at pseudouridine 55 in tRNA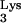 (Ψ55) was found. If the model by Wu et al. is correct, elimination of M1A58 in our experiments should have allowed reverse transcription to proceed beyond residue 58 but not beyond Ψ55. We observed reverse transcription beyond Ψ55 (Fig. 3). Although this may appear to be a discrepancy with the results by Wu et al., we can reconcile these differences if we take into account that the Ψ55 modification does not occur when residue A58 is changed to A58U (3). Thus, the secondary termination site as described by Wu et al. (20) is not a consideration in our model of inhibition. Because substitution of A58 with U abrogates the posttranscriptional formation of Ψ55, our work does not demonstrate whether Ψ55 has a role in RT termination.
(Ψ55) was found. If the model by Wu et al. is correct, elimination of M1A58 in our experiments should have allowed reverse transcription to proceed beyond residue 58 but not beyond Ψ55. We observed reverse transcription beyond Ψ55 (Fig. 3). Although this may appear to be a discrepancy with the results by Wu et al., we can reconcile these differences if we take into account that the Ψ55 modification does not occur when residue A58 is changed to A58U (3). Thus, the secondary termination site as described by Wu et al. (20) is not a consideration in our model of inhibition. Because substitution of A58 with U abrogates the posttranscriptional formation of Ψ55, our work does not demonstrate whether Ψ55 has a role in RT termination.
Knockout mutants of 1-adenosine methyltransferase in mammalian cells are, to our knowledge, not available. A rat adenocarcinoma tumor with diminished 1-adenosine methyltransferase activity has been identified (15); however, infection of this cell line with HIV is not possible due to the species specificity of HIV-1. In an effort to generate a tRNA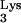 without the methyl group on residue 58, we resorted to mutate A58 to U. Our design is based on the fact that tRNA 1-adenosine methyltransferases are specific for an A in position 58 and not other bases (16). In addition, the mutation A58U was selected for our studies because previous work (7) demonstrated that this mutation does not affect tRNA
without the methyl group on residue 58, we resorted to mutate A58 to U. Our design is based on the fact that tRNA 1-adenosine methyltransferases are specific for an A in position 58 and not other bases (16). In addition, the mutation A58U was selected for our studies because previous work (7) demonstrated that this mutation does not affect tRNA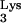 “B-box” transcription and hence does not alter tRNA
“B-box” transcription and hence does not alter tRNA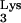 expression levels.
expression levels.
We report that HIV-1 replication is inhibited by a tRNA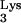 containing the nucleotide substitution A58U. We observed a strong delay of HIV-1 replication kinetics in cells expressing tRNA
containing the nucleotide substitution A58U. We observed a strong delay of HIV-1 replication kinetics in cells expressing tRNA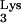 A58U when compared to normal cells. Using a limiting-dilution assay, CEM clones containing tRNA
A58U when compared to normal cells. Using a limiting-dilution assay, CEM clones containing tRNA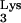 A58U are 1,000 times less infectible than normal cells. Thus, we believe that priming by tRNA may be a potential target for therapeutic inhibition of HIV-1 infection.
A58U are 1,000 times less infectible than normal cells. Thus, we believe that priming by tRNA may be a potential target for therapeutic inhibition of HIV-1 infection.
All known retroviruses utilize tRNAs containing M1A58 as primers for reverse transcription. In addition, all retroviruses have PBS of 18 nucleotides in length, which suggests that residue M1A58 serves as a termination signal for plus-strand strong-stop DNA synthesis of all retroviruses. Thus, it is possible that mutation of M1A58, if made in the appropriate tRNA primer, could have inhibitory effects on any retrovirus.
The effects of tRNA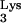 A58U on cells are not known. A rat adenocarcinoma tumor with diminished 1-adenosine methyltransferase activity has been identified (15), suggesting that disruption of M1A58 is not lethal. The growth characteristics and viability of CEM containing tRNA
A58U on cells are not known. A rat adenocarcinoma tumor with diminished 1-adenosine methyltransferase activity has been identified (15), suggesting that disruption of M1A58 is not lethal. The growth characteristics and viability of CEM containing tRNA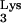 A58U are indistinguishable from those of wild-type CEM cells. However, to carefully examine whether tRNA
A58U are indistinguishable from those of wild-type CEM cells. However, to carefully examine whether tRNA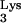 A58U has deleterious consequences for the cell metabolism or division, further experiments will be required.
A58U has deleterious consequences for the cell metabolism or division, further experiments will be required.
ACKNOWLEDGMENTS
We are grateful to Casey Morrow (University of Alabama at Birmingham) for the clone HIV-1HXB2-HisAc.
This work was supported by NIH grants AI41957 to J.D.R. and AI41407 to V.P.
REFERENCES
- 1.Adachi A, Gendelman H E, Koenig S, Folks T W R, Rabson A, Martin M A. Production of acquired immunodeficiency syndrome-associated retrovirus in human and non-human cells transfected with an infectious molecular clone. J Virol. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akkina R K, Walton R M, Chen M L, Li Q X, Planelles V, Chen I S Y. High-efficiency gene transfer into CD34+ cells with a human immunodeficiency virus type 1-based retroviral vector pseudotyped with vesicular stomatitis virus envelope glycoprotein G. J Virol. 1996;70:2581–2585. doi: 10.1128/jvi.70.4.2581-2585.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Becker H F, Motorin Y, Sissler M, Florentz C, Grosjean H. Major identity determinants for enzymatic formation of ribothymidine and pseudouridine in the T psi-loop of yeast tRNAs. J Mol Biol. 1997;274:505–518. doi: 10.1006/jmbi.1997.1417. [DOI] [PubMed] [Google Scholar]
- 4.Ben-Artzi H, Shemesh J, Zeelon E, Amit B, Kleiman L, Gorecki M, Panet A. Molecular analysis of the second template switch during reverse transcription of the HIV RNA template. Biochemistry. 1996;35:10549–10557. doi: 10.1021/bi960439x. [DOI] [PubMed] [Google Scholar]
- 5.Burnett B P, McHenry C S. Posttranscriptional modification of retroviral primers is required for late stages of DNA replication. Proc Natl Acad Sci USA. 1997;94:7210–7215. doi: 10.1073/pnas.94.14.7210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burns J C, Friedmann T, Driever W, Burrascano M, Yee J K. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc Natl Acad Sci USA. 1993;90:8033–8037. doi: 10.1073/pnas.90.17.8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geiduschek E P, Tocchini-Valentini G P. Transcription by RNA polymerase III. Annu Rev Biochem. 1988;57:873–914. doi: 10.1146/annurev.bi.57.070188.004301. [DOI] [PubMed] [Google Scholar]
- 8.Gilboa E, Mitra S W, Goff S, Baltimore D. A detailed model of reverse transcription and tests of crucial aspects. Cell. 1979;18:93–100. doi: 10.1016/0092-8674(79)90357-x. [DOI] [PubMed] [Google Scholar]
- 9.Hantzopoulos P A, Sullenger B A, Ungers G, Gilboa E. Improved gene expression upon transfer of the adenosine deaminase minigene outside the transcriptional unit of a retroviral vector. Proc Natl Acad Sci USA. 1989;86:3519–3523. doi: 10.1073/pnas.86.10.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klimatcheva E, Planelles V, Day S L, Fulreader F, Renda M J, Rosenblatt J D. Defective lentiviral vectors are efficiently trafficked by HIV-1 and inhibit its replication. Mol Ther. 2001;3:928–939. doi: 10.1006/mthe.2001.0344. [DOI] [PubMed] [Google Scholar]
- 11.Kunkel T A. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci USA. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levin H L. It's prime time for reverse transcriptase. Cell. 1997;88:5–8. doi: 10.1016/s0092-8674(00)81851-6. [DOI] [PubMed] [Google Scholar]
- 13.Lu Y, Planelles V, Li X, Palaniappan C, Day B, Challita-Eid P, Amado R, Stephens D, Kohn D B, Bakker A, Fay P, Bambara R A, Rosenblatt J D. Inhibition of HIV-1 replication using a mutated tRNALys-3 primer. J Biol Chem. 1997;272:14523–14531. doi: 10.1074/jbc.272.23.14523. [DOI] [PubMed] [Google Scholar]
- 14.Mak J, Kleiman L. Primer tRNAs for reverse transcription. J Virol. 1997;71:8087–8095. doi: 10.1128/jvi.71.11.8087-8095.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salas C E, Uschmann B D, Leboy P S. Methyl-accepting RNA in 13762 mammary adenocarcinoma correlated with low adenine methyltransferase levels. Cancer Res. 1982;42:5004–5009. [PubMed] [Google Scholar]
- 16.Soll D, RajBhandary U L, editors. tRNA. Washington, D.C.: ASM Press; 1995. [Google Scholar]
- 17.Wakefield J K, Kang S M, Morrow C D. Construction of a type 1 human immunodeficiency virus that maintains a primer binding site complementary to tRNA(His) J Virol. 1996;70:966–975. doi: 10.1128/jvi.70.2.966-975.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weiss R. Experimental biology and assay of RNA tumor viruses. In: Weiss R, Teich N, Varmus H, Coffin J, editors. RNA tumor viruses. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratories; 1982. pp. 209–260. [Google Scholar]
- 19.White S M, Renda M, Nam N Y, Klimatcheva E, Zhu Y, Fisk J, Halterman M, Rimel B J, Federoff H, Pandya S, Rosenblatt J D, Planelles V. Lentivirus vectors using human and simian immunodeficiency virus elements. J Virol. 1999;73:2832–2840. doi: 10.1128/jvi.73.4.2832-2840.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu T, Guo J, Bess J, Henderson L E, Levin J G. Molecular requirements for human immunodeficiency virus type 1 plus-strand transfer: analysis in reconstituted and endogenous reverse transcription systems. J Virol. 1999;73:4794–4805. doi: 10.1128/jvi.73.6.4794-4805.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]