

Abstract
Introduction
Apurinic/apyrimidinic endonuclease 1 (APEX1) is a protein with elevated expression in synovial fluids from rheumatoid arthritis (RA) patients. However, its role in RA pathogenesis remains unexplored. This study investigated the influence of APEX1 on inflammatory pathways in fibroblast-like synoviocytes (FLS) isolated from RA patients.
Material and methods
FLS from RA patients (n = 5) were stimulated with recombinant tumor necrosis factor α (TNF-α) and interleukin (IL)-17. Subsequently, cells were treated with recombinant APEX1, and assessments were made on reactive oxygen species (ROS) production and mitochondrial membrane potential. Additionally, mRNA levels of IL-1 family members were quantified. Cell migration was evaluated through Transwell chamber assays, and levels of key secreted inflammatory cytokines were measured via enzyme-linked immunosorbent assay (ELISA).
Results
The results demonstrated that APEX1 significantly reduced mitochondrial-specific ROS expression and restored mitochondrial membrane potential in TNF-α/IL-17-stimulated RA FLS. Furthermore, APEX1 treatments attenuated TNF-α/IL-17-induced activation of p38 MAPK, NF-κB, and PI3K 110 δ signaling pathways. Similarly, APEX1 significantly diminished TNF-α/IL-17-induced expression of inflammatory cytokines, including IL-1 family members, IL-6, IL-8, and vascular endothelial growth factor (VEGF). Notably, APEX1 downregulated cell migration of TNF-α/IL-17-treated RA FLS via inhibition of matrix metalloproteinase 3 (MMP3).
Conclusions
These findings collectively underscore the role of APEX1 as a key mediator of cytokine-amplified migration, modulating ROS and MMP3 in RA FLS, thus supporting its potential as a therapeutic target in RA treatment.
Keywords: rheumatoid arthritis, inflammation, migration, reactive oxygen species, fibroblast-like synoviocytes
Introduction
Rheumatoid arthritis (RA) is a systemic autoimmune disease characterized by hyperplastic synovial pannus tissue, synovial inflammation, and bone erosion [1, 2]. Fibroblast-like synoviocytes (FLS), residing in the intimal lining of the synovium, play a pivotal role in RA pathogenesis by promoting cartilage destruction [3]. Excessive FLS proliferation, secretion of pro-inflammatory cytokines, and immune cell recruitment into the synovium contribute to RA development and progression [4]. Moreover, activated FLS exhibit heightened secretion of matrix metalloproteinases (MMPs) and possess invasive properties, leading to cartilage and bone degradation [5]. These distinct features, including enhanced migratory capacity and cartilage invasiveness, characterize RA FLS [6].
Reactive oxygen species (ROS) function as signaling molecules regulating cellular processes such as migration, proliferation, and survival [7]. Dysregulated ROS levels can induce inflammation, cancer, aging, and autoimmune diseases, implicating their involvement in RA pathogenesis [8]. Specifically, circulating T cells isolated from RA patients exhibit elevated ROS production compared to healthy controls [9]. Additionally, treatment of RA FLS with MitoTEMPO, a mitochondria-targeted antioxidant, significantly attenuates their migration and invasion capabilities [10], indicating a potential association between ROS and FLS migration. However, the precise factors underlying ROS-mediated migration in RA remain incompletely understood.
Apurinic/apyrimidinic endonuclease 1 (APEX1), also known as APE1 or redox factor-1 (Ref-1), plays a crucial role in DNA damage recognition and repair [11]. It serves as an antioxidant by reducing a redox-sensitive cysteine residue in activator protein-1 (AP-1) in response to oxidative stress [12, 13]. Furthermore, APEX1 regulates the DNA binding affinity and transcription activity of key transcription factors such as p53, nuclear factor-kappa B (NF-κB), and hypoxia-inducible factor-1 (HIF-1), impacting cellular processes including growth, survival, apoptosis, and inflammation [13, 14]. Elevated APEX1 levels are observed in various diseases, such as psoriatic epidermis and serum from patients with coronary artery disease [15, 16]. Similarly, increased APEX1 levels are reported in synovial fluid from RA patients, correlating with increased disease severity [17]. However, the specific roles of APEX1 in RA FLS remain elusive.
Here, we investigated the effects of APEX1 on RA FLS under pro-inflammatory conditions induced by recombinant tumor necrosis factor-alpha (TNF-α) and interleukin (IL)-17 [18, 19]. Our findings revealed that recombinant APEX1 treatment significantly reduced cytokine-induced ROS expression and migratory capacity. Additionally, APEX1 downregulated the activation of key signaling pathways, expression of pro-inflammatory cytokines, and MMP3 expression. These results suggest that APEX1 acts as a critical regulator of the ROS/MMP3-mediated migratory pathway in RA FLS.
Material and methods
Human subjects and ethics statement
Synovial tissues were obtained from five female patients diagnosed with RA and undergoing synovectomy or joint replacement (Table 1). Diagnosis of RA conformed to the College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) 2010 classification criteria [20]. Patients with inflammatory diseases such as septic arthritis, a history of knee surgery, infectious diseases, cancer, multiple sclerosis, stroke, or immune system disorders were excluded from the study.
Table 1.
Baseline characteristics of all patients included in this study
| Variables | Subjects with knee RA (n = 5) |
|
|---|---|---|
| Female (n, %) | 5 (100) | |
| Age (years, mean ±SD) | 53.2 ±11.1 (35-61) | |
| Duration of disease (months, mean ±SD) | 143.6 ±79.3 (40-240) | |
| Rheumatoid factor-positive, n (%) | 5 (100) | |
| Anti CCP antibody-positive, n (%) | 4 (80) | |
| DAS28 (ESR, mean ±SD) | 2.84 ±1.72 (1.75-5.81) | |
| Duration of treatment (months, mean ±SD) | 141 ±85 (28-240) | |
| Treatment (n, %) | ||
| Naive | 0 | |
| Steroid | 4 (80) | |
| Methotrexate | 4 (80) | |
| Hydroxychloroquine | 3 (60) | |
| Sulfasalazine | 1 (20) | |
| Leflunomide | 3 (60) | |
| Tacrolimus | 1 (20) | |
| Biologic DMARD | 1 (20), Golimumab | |
SD – standard deviation, DAS28 – 28-joint disease activity score, DMARD – disease-modifying antirheumatic drug.
Upon removal of fat and fibrous tissues, the synovium was fragmented into small pieces and incubated with 0.1% collagenase (Sigma-Aldrich, St. Louis, MO, USA) in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, Thermo Fisher Scientific, Waltham, MA, USA) at 37°C for 90 min. Isolated primary FLS with a purity > 95% (CD90+) were cultured and utilized for subsequent experiments after four to six passages. Dissociated cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS; Gibco, Thermo Fisher Scientific), 100 U/ml penicillin, and 100 mg/ml streptomycin, and maintained in a 5% CO2 incubator at 37oC.
This study adhered to the principles outlined in the Declaration of Helsinki and was approved by the Institutional Review Board of Chungnam National University Hospital (CNUH 2019-12-068).
Flow cytometric analysis
FLS were stimulated with human recombinant TNF-α (10 ng/ml; PeproTech, Cranbury, NJ, USA), IL-17 (10 ng/ml; PeproTech), and recombinant human APEX1 (Cat. # MR-EFAPE-100, MediRedox, Daejeon, Korea) for 24 h. Subsequently, cells were washed with phosphate-buffered saline (PBS) and incubated with MitoSOX Red Mitochondrial Superoxide Indicator (Invitrogen, Thermo Fisher Scientific) for ROS detection. To determine the mitochondrial membrane potential, tetramethylrhodamine methyl ester (TMRM; Thermo Fisher Scientific) was used following the manufacturer’s instructions. Flow cytometric analysis was conducted using a FACSCanto II flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA), and data analysis was performed using FlowJo software (Tree Star, Ashland, OR, USA).
Western blot analysis
Cells were lysed on ice using a RIPA lysis kit (ATTO, Co., Tokyo, Japan), containing protease and phosphatase inhibitors. Following centrifugation for clarification, lysates were analyzed using 8% SDS-PAGE. Proteins were transferred onto polyvinylidene (PVDF) membranes (Bio-Rad Laboratories, Inc., Hercules, CA, USA) and blocked with 5% non-fat dry milk (BD Biosciences) in PBS-T (PBS with 0.05% Tween 20). Membranes were incubated overnight at 4°C with antibodies against phospho-p38 mitogen-activated protein kinase (MAPK; Thr180/Tyr182), p38 MAPK, p-NF-κB p65 (Ser536), NF-κB p65, phosphoinositide 3-kinase delta isoform (PI3K p110δ), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). All antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA) and diluted 1:1500 in PBS-T containing 5% non-fat dry milk. Following washing with PBS-T, membranes were incubated with peroxidase-conjugated goat anti-rabbit IgG (1:3000; AbFrontier Co., Ltd., Seoul, Korea). Target proteins were visualized using a chemiluminescent HRP Substrate (Thermo Fisher Scientific).
Reverse transcription (RT)-PCR and quantitative (q)RT-PCR
Total RNA was extracted using TRI Reagent (Molecular Research Center, Cincinnati, OH, USA) following the manufacturer’s instructions. Reverse transcription reactions were performed using ReverTra Ace qPCR RT Master Mix (TOYOBO, Osaka, Japan) to generate cDNA.
qRT-PCR analysis was conducted using SYBR Green Realtime PCR Master Mix (TOYOBO) as per the manufacturer’s instructions. Primers synthesized by Bioneer (Daejeon, Korea) are listed in Table 2. Thermal cycling conditions included initial denaturation at 95°C for 5 min, followed by 40 cycles of 95°C for 10 s, 60°C for 15 s, and 72°C for 20 s. A melting step from 72°C to 95°C was performed after the last cycle. Thermal cycling was performed using a CFX Connect Real-Time PCR Detection System (Bio-Rad Laboratories, Inc.). Expression levels of target genes were normalized to GAPDH levels in the same sample by calculating the cycle threshold (Ct) value. Relative target gene expression was determined using the 2–ΔΔCT relative quantification method.
Table 2.
Primers used for PCR
| Sense primer | Antisense primer | |
|---|---|---|
| IL-1β | GGATATGGAGCAACAAGTGG | ATGTACCAGTTGGGGAACTG |
| IL-18 | CAACAAACTATTTGTCGCAGGA | TGCCACAAAGTTGATGCAAT |
| IL-33 | ATCACGGCAGAATCATCGAG | CTTATGGTGAGGCCAGAACG |
| IL-36α | CATAAATTAGAGGTCCAAAATCG | AAGGGGCTGGGTCAGCTAT |
| MMP3 | GATGCCCACTTTGATGATGATGAA | AGTGTTGGCTGAGTGAAAGAGACC |
| APEX1 | CTGCTCTTGGAATGTGGATGGG | TCCAGGCAGCTCCTGAAGTTCA |
| GAPDH | CACATGGCCTCCAAGGAGTAA | TGAGGGTCTCTCTCTTCCTCTTGT |
For conventional gel-based RT-PCR, synthesized cDNA was mixed with AccuPower RT PreMix (Bioneer) and 10 pmol of each specific PCR primer following the manufacturer’s protocol. Amplified products were separated on 1% agarose gels, stained with Midori Green Advance (NIPPON Genetics, Düren, Germany), and visualized under UV illumination using a GelDoc system (Bio-Rad Laboratories, Inc.).
Wound migration assay
FLS cultures at approximately 90% confluency were in- cubated with serum-free media. FLS monolayers were wounded using pipette tips and treated with recombinant TNF-α (10 ng/ml)/IL-17 (10 ng/ml) alone or in combination with recombinant APEX1 (1, 10, and 100 ng/ml). Wound closure was observed and photographed at 0 and 24 h using an inverted microscope (Olympus, Tokyo, Japan) equipped with a 0.55-numerical aperture dry objective at a magnification of 40× (scale bar, 20 µm). To quantify migrated cells, images of the initial wounded monolayers were compared with corresponding images at the end of the incubation period. Image J software was employed for wound area quantification.
Transwell migration assay
FLS isolated from RA patients were cultured in serum-free media for 5 h and then stimulated with recombinant TNF-α (10 ng/ml)/IL-17 (10 ng/ml) alone or in combination with recombinant APEX1 (1, 10, and 100 ng/ml) for 24 h. Cells were centrifuged and loaded onto Transwell filters with an 8-µm pore size (Corning, NY, USA) placed on top of the migration chamber. The bottom chambers of the Transwell plates were filled with DMEM containing 10% FBS as a chemoattractant, and cells were incubated for an additional 24 h. Non-migrating cells on the top membrane surface were removed by washing with PBS and cotton swabs. Invaded cells were stained with crystal violet (Sigma-Aldrich) and counted in five random fields per sample under an inverted microscope (Olympus) equipped with a 0.55-numerical aperture dry objective at a magnification of 100× (scale bar, 100 µm).
Enzyme-linked immunosorbent assay (ELISA)
Concentrations of IL-6, IL-8, vascular endothelial growth factor (VEGF), and MMP3 were determined using ELISA kits specific for human IL-6 (Cat. # 555220, BD Biosciences), human IL-8 (Cat. # DY208, R&D Systems, Minneapolis, MN, USA), human VEGF (Cat. # DY293, R&D Systems), and MMP3 (Cat. # DY513, R&D Systems), following the manufacturers’ instructions. Cytokine levels were quantified by interpolation from a standard curve generated using a Sunrise absorbance reader (Tecan, Männedorf, Switzerland) at 450 nm.
siRNA transfection
A specific siRNA duplex targeting APEX1 was purchased from Bioneer with the following sequences: 5´-CAG AUA UAC UGU GCC UUC A-3´ and 5´-UGA AGG CAC AGU AUA UCU G-3´. Control siRNA (Cat. # SN-1003, Bioneer) was utilized as the negative control. Cells were transfected with 100 pmol of each siRNA duplex using Lipofectamine 3000 transfection reagent (Invitrogen, Thermo Fisher Scientific), following the manufacturer’s instructions. Downregulation of target expression was evaluated after 24 h of incubation.
Statistical analysis
Results are presented as mean ± standard deviation. Statistical analysis was conducted using the paired Student’s t-test, with p-values < 0.05 considered statistically significant.
Results
APEX1 decreases TNF-α/IL-17-induced ROS in RA FLS
Previous studies have demonstrated the protective role of APEX1 against oxidative stress and DNA damage during brain development [21]. To assess the direct influence of APEX1 on intracellular ROS levels in RA FLS, cells were treated with recombinant APEX1, and mitochondrial-specific ROS levels were evaluated using MitoSOX staining followed by flow cytometry analysis. The results revealed a significant decrease in ROS levels within RA FLS mitochondria upon APEX1 treatment (Fig. 1A). Furthermore, APEX1 treatment effectively inhibited the ROS levels induced by TNF-α/IL-17 stimulation (Fig. 1B). Additionally, assessment of mitochondrial transmembrane potential using TMRM staining [22] revealed that APEX1 treatment effectively restored the impaired mitochondrial transmembrane potential resulting from TNF-α/IL-17 stimulation (Fig. 1C). These findings indicate that APEX1 acts as a regulator, mitigating ROS-induced damage in FLS derived from patients with RA.
Fig. 1.
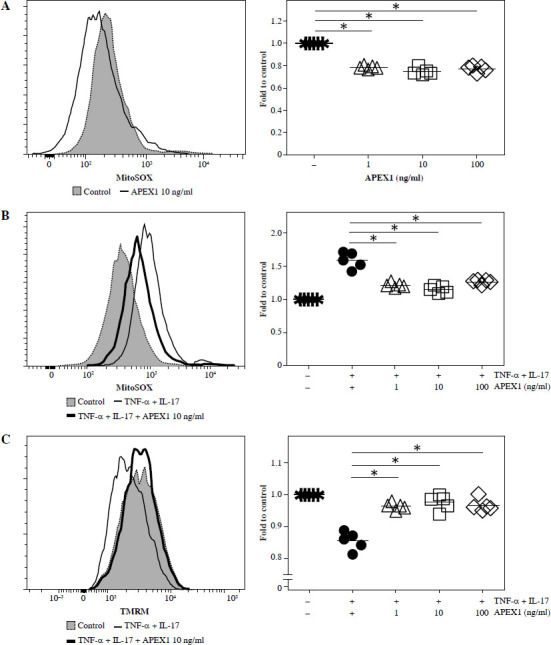
Recombinant APEX1 reduces TNF-α/IL-17-induced ROS in RA FLS. A) RA FLS were treated with recombinant APEX1 (1, 10, and 100 ng/ml) for 24 h, and mitochondrial ROS levels were measured using MitoSOX mitochondrial superoxide indicator. Data represent one median experiment (left panel) with fold change of mean fluorescent intensity (MFI) compared to solvent control (right panel). Statistical analysis between solvent control and APEX1 was performed using the paired Student’s t-test. *p < 0.05. B) RA FLS stimulated with TNF-α (10 ng/ml)/IL-17 (10 ng/ml) alone or with recombinant APEX1 for 24 h were stained with MitoSOX. Data represent one median experiment (left panel), with fold change of MFI for MitoSOX compared to unstimulated control (right panel). Statistical analysis between TNF-α/IL-17 and TNF-α/IL-17/APEX1 was performed using the paired Student’s t-test. *p < 0.05. C) RA FLS stimulated with TNF-α/IL-17 alone or with recombinant APEX1 for 24 h were stained with tetramethylrhodamine methyl ester (TMRM) mitochondrial membrane potential probe. Data represent one median experiment (left panel) with fold change of MFI compared to unstimulated control (right panel). Statistical analysis between TNF-α/IL-17 and TNF-α/IL-17/APEX1 was performed using the paired Student’s t-test. * p < 0.05. Each symbol represents an individual donor (n = 5), and bars represent means
APEX1 inhibits TNF-α/IL-17-induced inflammatory signaling in RA FLS
Excessive ROS stimulation triggers p-38 MAPK, NF-κB, and PI3K signaling, leading to inflammation [23]. Here, stimulation of RA FLS with TNF-α/IL-17 led to increased phosphorylation of p-38 MAPK (1.73-fold) and p-NF-κB p65 (1.79-fold), along with activation of PI3K p100δ (1.82-fold) compared to control conditions (Fig. 2A and p < 0.05). Treatment with APEX1 in the presence of TNF-α/IL-17 significantly attenuated these effects by 36.70% (p-p38 MAPK), 32.42% (p-NF-κB p65), and 33.86% (PI3K p100δ) compared to TNF-α/IL-17 stimulation alone.
Fig. 2.
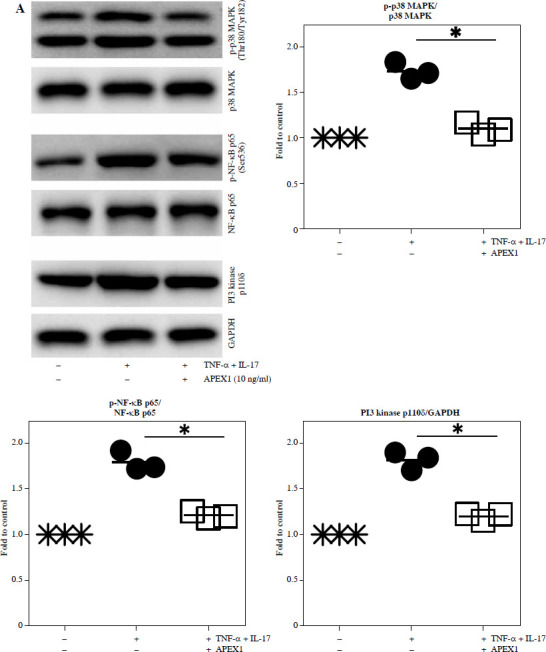
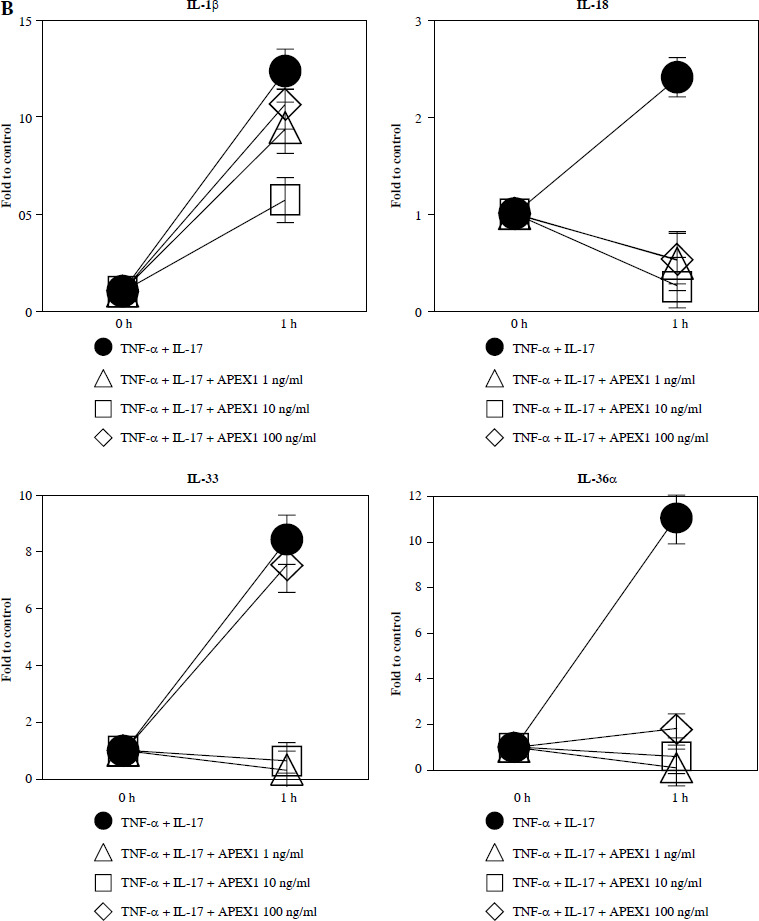
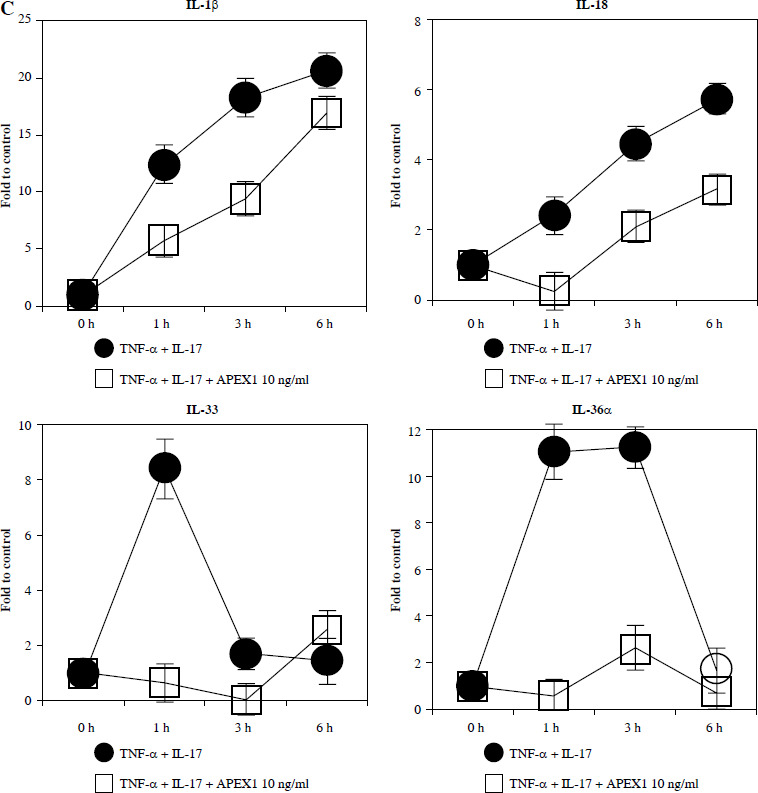
Recombinant APEX1 downregulates TNF-α/IL-17-mediated p38 MAPK and NF-κB phosphorylation, PI3K activation, and IL-1 family production. A) Phosphorylation levels of p-p38 MAPK, p-NF-κB p65, and PI3K delta isoform (110δ) were evaluated by western blot in RA FLS stimulated with TNF-α (10 ng/ml)/IL-17 (10 ng/ml) alone or with recombinant APEX1 (10 ng/ml) for 30 min. Data represent one median experiment (left panel) with fold change of band intensity relative to unstimulated control (right panel). GAPDH served as a loading control. Each symbol represents an individual donor (n = 3), and bars represent means. Statistical analysis between TNF-α/IL-17 and TNF-α/IL-17/APEX1 was performed using the paired Student’s t-test. *p < 0.05 B) RA FLS were treated with TNF-α/IL-17 alone or with recombinant APEX1 (1, 10, and 100 ng/ml) for 1 h, and IL-1β, IL-18, IL-33, and IL-36α mRNA levels were assessed by qRT-PCR. Data represent one experiment conducted in triplicate with similar results C) RA FLS were treated with TNF-α/IL-17 alone or with recombinant APEX1 (10 ng/ml) for 1, 3, and 6 h, and IL-1β, IL-18, IL-33, and IL-36α mRNA levels were analyzed. Data represent one experiment conducted in triplicate with similar results. Data are presented as mean ± standard deviation
NF-κB, p38 MAPK, and PI3K activation is implicated in inflammatory diseases mediated by the IL-1 family [24-26]. Therefore, we investigated whether APEX1 modulates the expression of IL-1 family members in RA FLS. Our results demonstrate that the elevated mRNA expression levels of IL-1β, IL-18, IL-33, and IL-36α induced by TNF-α/IL-17 stimulation were markedly suppressed by APEX1 treatment (Fig. 2B, C). The most effective dose of APEX1 in inhibiting TNF-α/IL-17-induced inflammatory signaling was 10 ng/ml, consistent with the concentration that inhibited ROS generation. Although TNF-α/IL-17 leads to a different increasing trend among IL-1 family members, the overall tendency for levels to be decreased by APEX1 has been consistently confirmed. Further studies are needed to determine why are there such different suppressive effects by APEX1 among IL-1 family members. Additionally, a similar effect of APEX1 was observed for mRNA expression levels of IL-36β and IL-37 (data not shown).
In line with these findings, we assessed the secretion of IL-1β and IL-36 in culture supernatants of RA FLS. Although APEX1 treatment suppressed the TNF-α/IL-17-induced secretion of IL-1β and IL-36, only trace amounts of the secreted proteins were detected, with levels below 10 pg/ml (Supplementary Fig. 1). Collectively, these findings indicate that APEX1 modulates NF-κB-, p38 MAPK-, and PI3K-mediated inflammation in RA FLS.
APEX1 attenuates TNF-α/IL-17-mediated migration in RA FLS
Activated FLS exhibit heightened migratory capacity, leading to increased invasion into cartilage and bone damage compared to normal FLS [27]. Therefore, we assessed FLS migration in response to TNF-α/IL-17 with or without APEX1 treatment using a scratch assay, where cell movement into a wounded area was evaluated. Our results demonstrate that TNF-α/IL-17-induced cell migration is significantly reduced by APEX1 treatment (Fig. 3A). Additionally, migration of RA patient-derived FLS following stimulation with TNF-α/IL-17 with or without APEX1 was evaluated using a Transwell chamber assay, revealing a substantial decrease in cell migration upon treatment with recombinant APEX1 (Fig. 3B). Quantification of Transwell assay results by eluting the crystal violet dye with 0.1% SDS revealed that cell migration increased by 2.43-fold versus the control in response to cytokine stimulation alone, whereas APEX1 treatment (10 ng/ml) significantly downregulated expression by 44.22% compared to TNF-α/IL-17 stimulation alone (Supplementary Fig. 2 and p < 0.05).
Fig. 3.
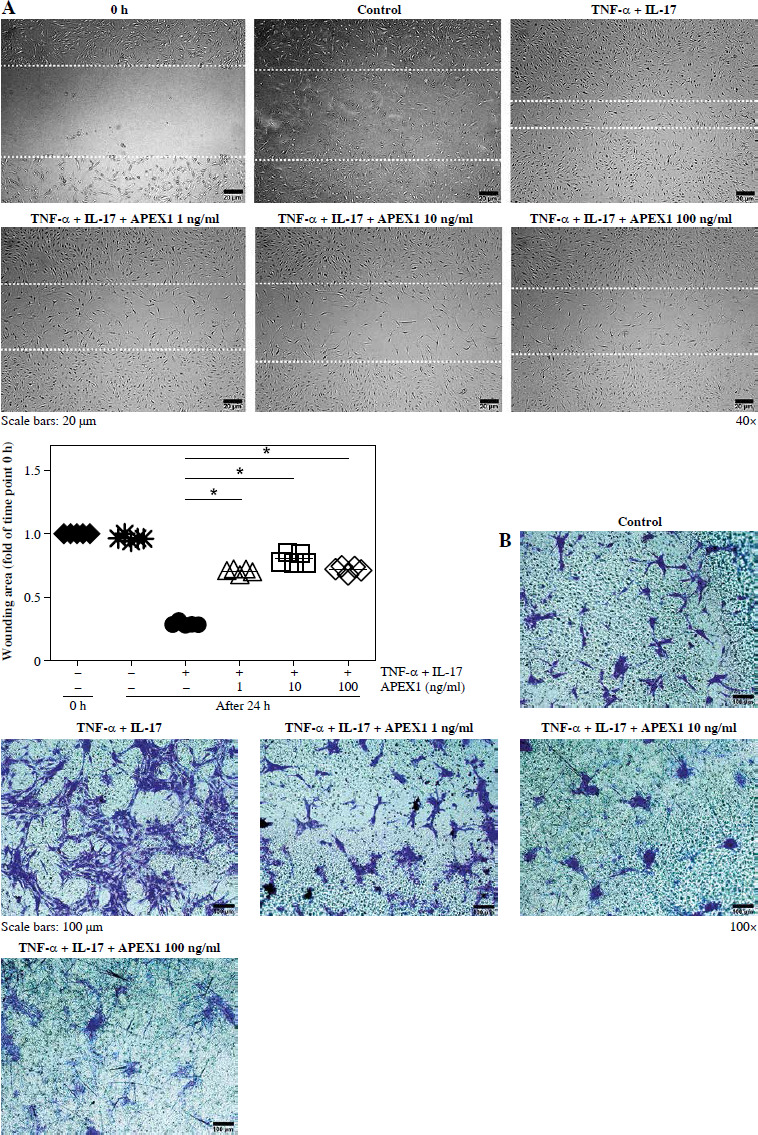
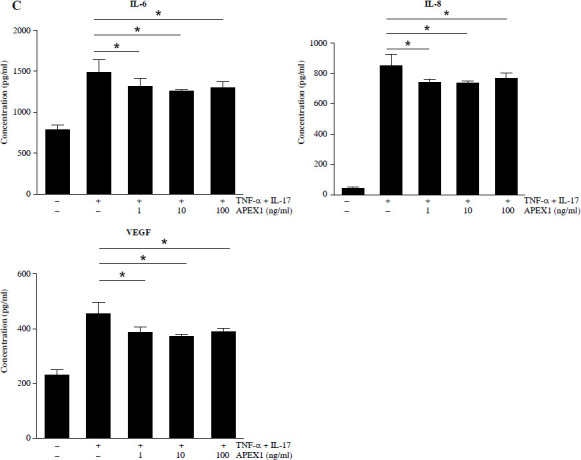
APEX1 mitigates TNF-α/IL-17-induced cell migration in RA FLS. A) RA FLS were subjected to a scratch assay, and migrated cells were photographed after stimulation with TNF-α (10 ng/ml)/IL-17 (10 ng/ml) alone or in combination with recombinant APEX1 (1, 10, and 100 ng/ml) for 24 h. Data represent one median experiment (upper panel), with fold change of wounding area compared to the 0 h time point (lower panel). Each symbol represents an individual donor (n = 5), and bars represent means. Statistical analysis between TNF-α/IL-17 and TNF-α/IL-17/APEX1 was performed using the paired Student’s t-test. *p < 0.05. B) RA FLS were incubated with TNF-α/IL-17 alone or with recombinant APEX1 for 24 h, and cell migration was assessed using a Transwell chamber with crystal violet staining. Data representing one median experiment, with fold change of the optical density, are displayed in Supplementary Fig. 2 (n = 5) C) Culture supernatants of RA FLS were analyzed to measure secreted levels of IL-6, IL-8, and VEGF. Results are expressed as the mean ± SD of five independent donors. Statistical analysis between TNF-α/IL-17 and TNF-α/IL-17/APEX1 was performed using the paired Student’s t-test. *p < 0.05
Several cytokines are recognized as representative pro-inflammatory factors in RA FLS, including IL-6, IL-8, and VEGF [28]. Therefore, we measured the secreted levels of these cytokines in culture supernatants from RA FLS stimulated with TNF-α/IL-17 and found that all were significantly reduced by APEX1 treatment (Fig. 3C). These findings suggest that recombinant APEX1 alleviates FLS migration-mediated bone damage in RA.
APEX1 suppresses MMP3 expression in RA FLS
MMP3 expression correlates with cell migration and the inflammatory response in RA FLS and is regulated by the MAPK, NF-κB, and PI3K/Akt pathways [29, 30]. To investigate whether MMP3 expression is mediated by APEX1, RA FLS were stimulated with TNF-α/IL-17 with or without APEX1 for 3 h, and cytokine expression was assessed. TNF-α/IL-17 induced a 7.33-fold increase of MMP3 mRNA expression, whereas APEX1 treatment (10 ng/ml) significantly downregulated expression by 67.39% compared to TNF-α/IL-17 stimulation alone (Fig. 4A, B). Additionally, secreted levels of MMP3 protein induced by TNF-α/IL-17 were reduced by APEX1 treatment (Fig. 4C).
Fig. 4.
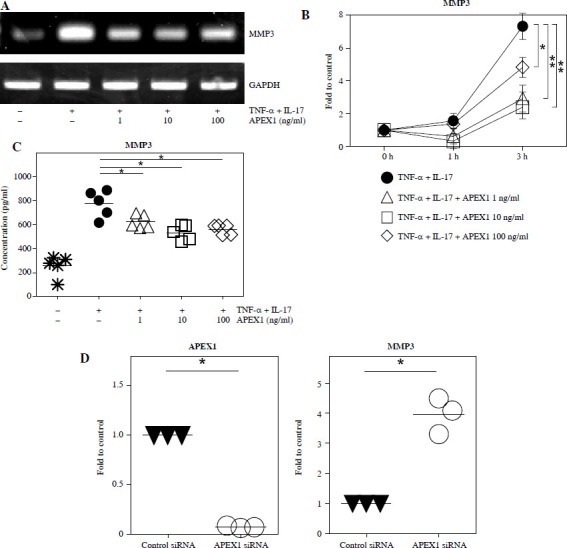
TNF-α/IL-17-activated MMP3 is suppressed by APEX1 in RA FLS. A) RA FLS were stimulated with TNF-α (10 ng/ml)/IL-17 (10 ng/ml) alone or with recombinant APEX1 (1, 10, and 100 ng/ml). After 3 h, mRNA expression of MMP3 was measured. Data represent one experiment, which was performed in triplicate with similar results. B) RA FLS were stimulated with TNF-α/IL-17 alone or with recombinant APEX1 for 1 or 3 h. Data show fold change of MMP3 expression compared to unstimulated control. Data represent one experiment conducted in triplicate with similar results, and bars represent means. Statistical analysis between TNF-α/IL-17 and TNF-α/IL-17/APEX1 was performed using the paired Student’s t-test. *p < 0.05. **p < 0.01. C) RA FLS were incubated with TNF-α/IL-17 alone or with recombinant APEX1 for 24 h, and MMP3 levels in supernatants were measured by ELISA. Each symbol represents an individual donor (n = 5), and bars represent means. Statistical analysis between TNF-α/IL-17 and TNF-α/IL-17/APEX1 was performed using the paired Student’s t-test. *p < 0.05. D) The level of APEX1 was inhibited in RA FLS using siRNA transfection. After 24 h, mRNA expression of APEX1 and MMP3 was assessed. Each symbol represents an individual donor (n = 3), and the bar represents the mean. Statistical analysis between control siRNA and APEX1 siRNA was performed using the paired Student’s t-test. *p < 0.05
To confirm the role of APEX1 in MMP3 expression, APEX1 mRNA levels in RA FLS were reduced using siRNA transfection. After 24 h, APEX1 silencing reduced APEX1 mRNA levels by 0.07-fold compared to control siRNA (Fig. 4D). Upon confirmation of APEX1 silencing, MMP3 expression significantly increased by 4.79-fold in RA FLS. Furthermore, expression of IL-1β (2.47-fold), IL-6 (3.23-fold), and IL-36α (2.03-fold) mRNAs was also significantly increased by APEX1 siRNA (data not shown). These results collectively indicate that recombinant APEX1 suppresses MMP3 production, ultimately reducing cell migration and aiding in alleviating inflammation in RA FLS.
Discussion
The synovium in RA exhibits heightened oxygen demand due to immune cell infiltration, synovial hyperplasia, and angiogenesis [31]. FLS play a central role in RA inflammation, characterized by elevated ROS levels and metabolic dysregulation, contributing to RA pathogenesis [32]. Despite being present in the RA synovium, the role of APEX1, a known redox effector in various disorders, remains unexplored in RA. In this study, we investigated the antioxidant function of APEX1 in RA FLS, revealing its ability to diminish ROS levels and restore mitochondrial membrane potential in TNF-α/IL-17-stimulated RA FLS (Fig. 1). Furthermore, we observed significant attenuation of TNF-α/IL-17-induced activation of key inflammation-associated signaling pathways, including p38 MAPK, NF-κB, and PI3K, upon recombinant APEX1 treatment (Fig. 2A). Additionally, APEX1 treatment downregulated the mRNA expression of IL-1 cytokine family members, commonly found in the inflammatory synovial environment during RA pathogenesis (Fig. 2B, C) [33].
In a prior investigation, we found a positive correlation between APEX1 levels and disease activity in RA patients [17]. These findings suggest that elevated APEX1 levels in the RA synovium might serve to mitigate disease progression by regulating redox homeostasis. This notion aligns with previous research indicating that increased APEX1 expression promotes neuronal survival under oxidative stress in a neurodegenerative disorder model [34]. However, contrasting findings by Nishi et al. suggest that APEX1 activates NF-κB signaling by enhancing its DNA binding affinity in vivo [35]. Thus, the role of APEX1 as a regulator of redox homeostasis may be context-dependent, influenced by the specific redox environment. Given the excessive inflammatory state of RA, APEX1 holds promise as a potential therapeutic agent. Further investigation is warranted to evaluate the efficacy of recombinant APEX1 in animal models of RA.
Due to the potential of excessive ROS to directly induce both cell migration and inflammation in RA, we examined the migratory capacity of FLS and cytokine production following recombinant APEX1 treatment [36]. Our findings demonstrate that APEX1 effectively inhibits cytokine-induced cell migration, along with the secretion of IL-6, IL-8, and VEGF secretion in RA FLS (Fig. 3). These results suggest that APEX1 not only suppresses cytokine-induced cell migration but also mitigates inflammation in the synovium. This aligns with a previous study using a vascular inflammation model, where APEX1 attenuated the TNF-α-induced inflammatory response by suppressing ROS, MAPK, and vascular cell adhesion molecule 1 (VCAM-1) [37].
MMP3 functions as a potent inflammatory factor by degrading the extracellular matrix, including the cartilage matrix, and the associated matrix components, and activating several pro-MMPs [38]. Here, we observed significant suppression of MMP3 expression in RA FLS following APEX1 treatment (Fig. 4). Given MMP3’s involvement in the initiation and progression of various disorders, these findings imply that APEX1 may have broader therapeutic potential as a treatment option for inflammation, cancer, and autoimmune diseases [30].
In this study, we investigated the inflammatory response of synovial cells and the activation of associated signaling pathways following the administration of human recombinant APEX1 into a cell culture medium. One potential mechanism through which APEX1 could suppress inflammation in this context is by directly modulating inflammatory signals after endocytosis of the APEX1 protein. Endocytosis serves as the primary pathway for protein uptake into cells. While most cells typically exhibit limited uptake of proteins through this process, FLS, if possessing macrophage-like characteristics, might exhibit a higher propensity for protein endocytosis, including APEX1, during inflammation [39]. Alternatively, APEX1 may regulate the activity of receptors responsible for detecting inflammatory signals in the extracellular fluid or disrupt disulfide bonds in inflammatory receptors via a thiol-exchange mechanism, thereby inhibiting inflammatory signaling [37].
Limitations of this study include the utilization of ex vivo human FLS, which may not fully reflect in vivo responses due to the small size of the sample obtained from RA patients. Therefore, future studies employing animal models of RA are warranted to better understand the role of APEX1 in this disease context.
Conclusions
Our study demonstrates that recombinant APEX1 significantly reduces ROS production, inflammatory cytokine secretion, and activation of the MAPK, NF-κB, and PI3K pathways in RA FLS upon TNF-α/IL-17 stimulation, resulting in inhibition of migration through MMP3 suppression. These findings suggest a novel approach for mitigating pathogenic inflammatory responses and could enhance our comprehension of the interplay between RA pathology and redox homeostasis.
Footnotes
The authors declare no conflict of interest.
Supplementary material is available on the journal website.
Funding
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2020R1F1A1060926, NRF-2022R1A2C1003782, and NRF-2014R1A6A1029617) and Chungnam National University Hospital Research Fund, 2021.
Disclosures
This study was reviewed and approved by the Institutional Review Board of Chungnam National University Hospital, with the approval number: CNUH 2019-12-068. All patients provided informed consent to participate in the study.
References
- 1.Arnett FC, Edworthy SM, Bloch DA, et al. (1988): The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 31: 315-324. [DOI] [PubMed] [Google Scholar]
- 2.Klareskog L, Catrina AI, Paget S (2009): Rheumatoid arthritis. Lancet 373: 659-672. [DOI] [PubMed] [Google Scholar]
- 3.Bartok B, Firestein GS (2010): Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev 233: 233-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bottini N, Firestein GS (2013): Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol 9: 24-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noss EH, Brenner MB (2008): The role and therapeutic implications of fibroblast-like synoviocytes in inflammation and cartilage erosion in rheumatoid arthritis. Immunol Rev 223: 252-270. [DOI] [PubMed] [Google Scholar]
- 6.Lefevre S, Knedla A, Tennie C, et al. (2009): Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat Med 15: 1414-1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sena LA, Chandel NS (2012): Physiological roles of mitochondrial reactive oxygen species. Mol Cell 48: 158-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin W, Shen P, Song Y, et al. (2021): Reactive oxygen species in autoimmune cells: function, differentiation, and metabolism. Front Immunol 12: 635021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee HR, Yoo SJ, Kim J, et al. (2021): Reduction of oxidative stress in peripheral blood mononuclear cells attenuates the inflammatory response of fibroblast-like synoviocytes in rheumatoid arthritis. Int J Mol Sci 22: 12411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee HR, Yoo SJ, Kim J, et al. (2020): The effect of nicotinamide adenine dinucleotide phosphate oxidase 4 on migration and invasion of fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res Ther 22: 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robson CN, Hickson ID (1991): Isolation of cDNA clones encoding a human apurinic/apyrimidinic endonuclease that corrects DNA repair and mutagenesis defects in E. coli xth (exonuclease III) mutants. Nucleic Acids Res 19: 5519-5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ando K, Hirao S, Kabe Y, et al. (2008): A new APE1/Ref-1-dependent pathway leading to reduction of NF-kappaB and AP-1, and activation of their DNA-binding activity. Nucleic Acids Res 36 4327-4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xanthoudakis S, Curran T (1992): Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J 11: 653-665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaiddon C, Moorthy NC, Prives C (1999): Ref-1 regulates the transactivation and pro-apoptotic functions of p53 in vivo. EMBO J 18: 5609-5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee HM, Yuk JM, Shin DM, et al. (2009): Apurinic/apyrimidinic endonuclease 1 is a key modulator of keratinocyte inflammatory responses. J Immunol 183: 6839-6848. [DOI] [PubMed] [Google Scholar]
- 16.Jin SA, Seo HJ, Kim SK, et al. (2015): Elevation of the serum apurinic/apyrimidinic endonuclease 1/redox factor-1 in coronary artery disease. Korean Circ J 45: 364-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoo IS, Lee YR, Kang SW, et al. (2021): Elevated APE1/Ref-1 levels of synovial fluids in patients with rheumatoid arthritis: reflection of disease activity. J Clin Med 10: 5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chabaud M, Garnero P, Dayer JM, et al. (2000): Contribution of interleukin 17 to synovium matrix destruction in rheumatoid arthritis. Cytokine 12: 1092-1099. [DOI] [PubMed] [Google Scholar]
- 19.Xu T, Ying T, Wang L, et al. (2017): A native-like bispecific antibody suppresses the inflammatory cytokine response by simultaneously neutralizing tumor necrosis factor-alpha and interleukin-17A. Oncotarget 8: 81860-81872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aletaha D, Neogi T, Silman AJ, et al. (2010): 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 62: 2569-2581. [DOI] [PubMed] [Google Scholar]
- 21.Pei DS, Jia PP, Luo JJ, et al. (2019): AP endonuclease 1 (Apex1) influences brain development linking oxidative stress and DNA repair. Cell Death Dis 10: 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farkas DL, Wei MD, Febbroriello P, et al. (1989): Simultaneous imaging of cell and mitochondrial membrane potentials. Biophys J 56: 1053-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yao H, de Boer WI, Rahman I (2008): Targeting lung inflammation: novel therapies for the treatment of COPD. Curr Respir Med Rev 4: 57-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hernandez-Santana YE, Giannoudaki E, Leon G, et al. (2019): Current perspectives on the interleukin-1 family as targets for inflammatory disease. Eur J Immunol 49: 1306-1320. [DOI] [PubMed] [Google Scholar]
- 25.Coulthard LR, White DE, Jones DL, et al. (2009): p38(MAPK): stress responses from molecular mechanisms to therapeutics. Trends Mol Med 15: 369-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bartok B, Boyle DL, Liu Y, et al. (2012): PI3 kinase delta is a key regulator of synoviocyte function in rheumatoid arthritis. Am J Pathol 180: 1906-1916. [DOI] [PubMed] [Google Scholar]
- 27.Pap T, Müller-Ladner U, Gay RE, et al. (2000): Fibroblast biology. Role of synovial fibroblasts in the pathogenesis of rheumatoid arthritis. Arthritis Res 2: 361-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nygaard G, Firestein GS (2020): Restoring synovial homeostasis in rheumatoid arthritis by targeting fibroblast-like synoviocytes. Nat Rev Rheumatol 16: 316-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ribbens C, Andre B, Jaspar JM, et al. (2000): Matrix metalloproteinase-3 serum levels are correlated with disease activity and predict clinical response in rheumatoid arthritis. J Rheumatol 27: 888-893. [PubMed] [Google Scholar]
- 30.Wan J, Zhang G, Li X, et al. (2021): Matrix metalloproteinase 3: a promoting and destabilizing factor in the pathogenesis of disease and cell differentiation. Front Physiol 12: 663978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pucino V, Certo M, Varricchi G, et al. (2020): Metabolic checkpoints in rheumatoid arthritis. Front Physiol 11: 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bustamante MF, Garcia-Carbonell R, Whisenant KDet al. (2017): Fibroblast-like synoviocyte metabolism in the pathogenesis of rheumatoid arthritis. Arthritis Res 19: 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshitomi H (2019): Regulation of immune responses and chronic inflammation by fibroblast-like synoviocytes. Front Immunol 10: 1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mantha AK, Dhiman M, Taglialatela G, et al. (2012): Proteomic study of amyloid beta (25-35) peptide exposure to neuronal cells: impact on APE1/Ref-1’s protein-protein interaction. J Neurosci Res 90: 1230-1239. [DOI] [PubMed] [Google Scholar]
- 35.Nishi T, Shimizu N, Hiramoto M, et al. (2002): Spatial redox regulation of a critical cysteine residue of NF-kappa B in vivo. J Biol Chem 277: 44548-44556. [DOI] [PubMed] [Google Scholar]
- 36.Wang X, Fan D, Cao X, et al. (2022): The role of reactive oxygen species in the rheumatoid arthritis-associated synovial microenvironment. Antioxidants (Basel) 11: 1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park MS, Choi S, Lee YR, et al. (2016): Secreted APE1/Ref-1 inhibits TNF-alpha-stimulated endothelial inflammation via thiol-disulfide exchange in TNF receptor. Sci Rep 6: 23015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramos-DeSimone N, Hahn-Dantona E, Sipley J, et al. (1999): Activation of matrix metalloproteinase-9 (MMP-9) via a converging plasmin/stromelysin-1 cascade enhances tumor cell invasion. J Biol Chem 274: 13066-13076. [DOI] [PubMed] [Google Scholar]
- 39.Lee YK, Kim SW, Park JY, et al. (2017): Suppression of human arthritis synovial fibroblasts inflammation using dexamethasone-carbon nanotubes via increasing caveolin-dependent endocytosis and recovering mitochondrial membrane potential. Int J Nanomedicine 12: 5761-5779. [DOI] [PMC free article] [PubMed] [Google Scholar]