

Abstract
Background
Neuroinflammation reportedly plays a critical role in the pathogenesis of sepsis-associated encephalopathy (SAE). We previously reported that circulating plasma extracellular vesicles (EVs) from septic mice are proinflammatory. In the current study, we tested the role of sepsis plasma EVs in neuroinflammation.
Methods
To track EVs in cells and tissues, HEK293T cell-derived EVs were labeled with the fluorescent dye PKH26. Cecal ligation and puncture (CLP) was conducted to model polymicrobial sepsis in mice. Plasma EVs were isolated by ultracentrifugation and their role in promoting neuronal inflammation was tested following intracerebroventricular (ICV) injection. miRNA inhibitors (anti-miR-146a, -122, -34a, and -145a) were applied to determine the effects of EV cargo miRNAs in the brain. A cytokine array was performed to profile microglia-released protein mediators. TLR7- or MyD88-knockout (KO) mice were utilized to determine the underlying mechanism of EVs-mediated neuroinflammation.
Results
We observed the uptake of fluorescent PKH26-EVs inside the cell bodies of both microglia and neurons. Sepsis plasma EVs led to a dose-dependent cytokine release in cultured microglia, which was partially attenuated by miRNA inhibitors against the target miRNAs and in TLR7-KO cells. When administered via the ICV, sepsis plasma EVs resulted in a marked increase in the accumulation of innate immune cells, including monocyte and neutrophil and cytokine gene expression, in the brain. Although sepsis plasma EVs had no direct effect on cytokine production or neuronal injury in vitro, the conditioned media (CM) of microglia treated with sepsis plasma EVs induced neuronal cell death as evidenced by increased caspase-3 cleavage and Annexin-V staining. Cytokine arrays and bioinformatics analysis of the microglial CM revealed multiple cytokines/chemokines and other factors functionally linked to leukocyte chemotaxis and migration, TLR signaling, and neuronal death. Moreover, sepsis plasma EV-induced brain inflammation in vivo was significantly dependent on MyD88.
Conclusions
Circulating plasma EVs in septic mice cause a microglial proinflammatory response in vitro and a brain innate immune response in vivo, some of which are in part mediated by TLR7 in vitro and MyD88 signaling in vivo. These findings highlight the importance of circulating EVs in brain inflammation during sepsis.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12974-024-03250-0.
Keywords: Sepsis, Sepsis-associated encephalopathy (SAE), Extracellular vesicles (EVs), Extracellular miRNA, Neuroinflammation, Microglia, Neuronal apoptosis, Myeloid differentiation primary response 88 (MyD88), Toll-like receptor 7 (TLR7)
Introduction
Sepsis is a serious clinical syndrome caused by a dysfunctional host response to invading pathogens and is characterized by marked systemic inflammation, hemodynamic instability, coagulopathy, and multiorgan failure [1]. Sepsis-induced brain dysfunction, known as sepsis-associated encephalopathy (SAE), occurs in more than half of septic patients in the intensive care unit (ICU) and is considered one of the predominant causes of encephalopathy in these settings [2]. SAE induces a wide spectrum of brain dysfunctions, from delirium to coma. Cognitive impairment and psychological disorders frequently emerge as long-term consequences among sepsis survivors [2–4]. Moreover, SAE also serves as an independent predictor of mortality in sepsis [5]. Despite the well-documented clinical spectrum of SAE [6], specific treatments are still lacking in part due to the complex molecular mechanism leading to SAE.
Neuroinflammation is reportedly the key pathophysiology of SAE and includes activation of central nervous system (CNS) resident cells such as microglia and astrocytes, blood–brain barrier (BBB) disruption, infiltration of peripheral innate immune cells, and influx of proinflammatory damage molecules [7–9]. Proinflammatory responses by microglia are known to potentiate neuronal apoptosis and influence synaptic function to contribute to neuronal dysfunction and cerebral edema in sepsis [10, 11]. The molecular signaling that controls the CNS innate inflammation during sepsis is the focus of intense investigations in the field. We have previously demonstrated that extracellular microRNAs (ex-miRNAs), including miR-146a, miR-122, miR-34a, and miR-145a, are markedly increased in the plasma of septic humans and animals, where they functions as danger-associated molecular patterns (DAMP)[12]. These ex-miRNAs are capable of inducing cytokine and chemokine production, activating immune cell migration, disrupting cell junctions in the lung [13], and inducing brain inflammation [14]. Most recently, we identified a critical role of miR-146a in brain inflammation and neurological dysfunction in an animal model of SAE, in which it functions as a host-derived damager molecule and innate immune activator that acts through Toll-like receptor 7 (TLR7), a single-stranded RNA (ssRNA) sensor [14]. Moreover, TLR7 has also been shown to contribute to inflammation, coagulopathy, organ injury, and mortality in murine sepsis [13–17]. MyD88, an adaptor molecule essential for the signal transduction of multiple TLRs including TLR7 but not TLR3, is known to mediate systemic inflammation, cardiac dysfunction, and mortality in sepsis [18].
Extracellular vesicles (EVs) are heterogeneous membrane-bound particles secreted into the extracellular space that carry diverse cargo, including biologically active proteins, lipids, and nucleic acids such as miRNAs [19]. Since EVs serve as plasma carriers of ex-miRNAs, it is plausible that EVs may contribute to inflammatory injury both systemically and locally such as in the brain of sepsis mice. Alteration of circulating EVs in sepsis has been suggested to contribute to inflammation, coagulopathy, and delirium [20–24]. We recently characterized plasma EV dynamics at acute, subacute, and chronic time points after cecal ligation and puncture (CLP) in young adult mice and showed that EVs derived from septic mice can cause an inflammatory response [24]. However, the function and mechanism of plasma EVs in neuroinflammation and neuronal injury during sepsis are not well understood.
In the present study, we tested the hypothesis that sepsis-derived plasma EVs induce microglial activation, neuronal injury, and brain neuroinflammation through TLR signaling. To achieve this, we tracked the uptake of fluorescence-labeled EVs by macrophages, microglia and in the intact brain after local and intravenous administration. The effects of sepsis plasma EVs were evaluated in primary cultures of microglia, astrocytes, and neurons. In vivo, the impact of circulating EVs derived from septic mice on the brain was examined in the absence and presence of the TLR7 or MyD88 genes.
Materials and methods
Animals
Male 9–12-week-old WT (C57BL/6J, stock no. 000664) and TLR7−/− (Tlr7tm1Flv/J, stock no. 008380) mice were purchased from the Jackson Laboratories (Bar Harbor, ME), bred, and housed in an animal facility at the University of Maryland School of Medicine (Baltimore, MD). MyD88−/− mice were obtained from Kawai and colleagues [25]. All mice were housed in a specific pathogen-free environment with a 12-h/12-h light–dark cycle and free access to bacteria-free water and food. The use of animals was approved by the Institutional Animal Care and Use Committee of the University of Maryland School of Medicine (Baltimore, MD) and followed the guidelines of the National Institutes of Health (Bethesda, MD). All mice purchased from Jackson Laboratories were housed in the animal facility for at least one week before the experiment.
Labeling and tracking of EVs
EV labeling
To track the localization of EVs in cells and in the brain, EVs derived from HEK293T cell culture media (293EVs) [26] were labeled with PKH26 lipophilic dye (PKH26 red fluorescent cell linker kit, cat. no. NIMI26, Millipore-Sigma, St. Louis, MO) following the manufacturer’s instructions. Free PKH26 was cleared with cold DPBS using a 100 K spin column (Amicon centrifugal filter, Millipore Sigma), as reported previously [27]. In the control group, PKH26 dye was prepared following the same procedure without EVs.
Localization of labeled EVs in cultured cells
The RAW 264.7 (ATCC) macrophages and the primary cultured microglia were seeded in 8-well glass chamber slides (Lab-Tek II, Nunc) and treated with freshly labeled PKH26-EVs at 2.5 × 109 particles/mL at 37 °C with 5% CO2 overnight. The cells were fixed with 4% PFA for 15 min at room temperature. The slides were washed with cold-DPBS three times and mounted with the mounting medium containing DAPI (Shield Medium, Thermo Fisher Scientific). The cellular localization of PKH26-EVs was observed under a confocal microscope (Ti2, Nikon, Japan) and analyzed with the software NIS Element (Nikon, Japan).
Tracking EVs in the brain
PKH26-EVs were administered to naïve mice in vivo either by intravenous (IV) injection via the tail veins at a dose of 2 × 109 particles per mouse based on previous studies [28] or by intra-cerebroventricular (ICV) injection at a dose of 8 × 108 particles per mouse in a volume of 5 μL. Mice were euthanized and perfused with 25 mL of saline followed by 25 mL of 4% paraformaldehyde (Thermo Fisher Scientific) through the left ventricle of the heart 1 h after IV injection or 2 and 18 h after ICV injection. Brains were collected and fixed in paraformaldehyde for 24 h and subsequently soaked in 15% sucrose followed by 30% sucrose solutions at 4 °C until the tissues settled at each step. The dehydrated brains were embedded in a medium, frozen on a cryostat chuck inside of the cryostat, and cryosectioned at 20 µm using a Cryostat CM3050 (Leica biosystems). The brain tissue sections were stained with the following specific markers: Iba1 for microglia (Wako, Japan), GFAP for astrocytes (Thermo Fisher Scientific), neuronal nuclear antigen (NeuN) for neurons (Cell Signaling Technology, Danvers, MA), and DAPI (Fluoshield with DAPI, Sigma-Aldrich) for nuclei. Images were collected using a confocal microscope and analyzed using the NIS-Element (Nikon, Japan).
Administration of PKH26-EVs via ICV or IV
The mice were anesthetized with an intraperitoneal (IP) injection of ketamine (120 mg/kg) and xylazine (4 mg/kg). The ICV administration of EVs was conducted using a stereotaxic apparatus, as reported previously [14]. Briefly, the stereotactic coordinates were as follows: from bregma anteroposterior, −0.5 mm; mediolateral, −1.0 mm; and dorsoventral, −2.0 mm. The injection was accomplished using a 30-gauge needle adhered to a Hamilton syringe, with 8 × 108 EV particles per brain in a final volume of 5 μL. IV administration was performed using a 30-gauge needle with 2 × 109 particles per mouse via the tail veins. The EVs were diluted in 200 µL of sterile saline immediately before the IV injection.
Polymicrobial sepsis model
The cecal ligation and puncture (CLP) model was performed in the morning as previously described [13, 29]. In brief, mice were anesthetized by a IP administration of ketamine (120 mg/kg) and xylazine (4 mg/kg). After laparotomy, the cecum was ligated 1.5 cm from the tip and punctured through with an 18-gauge needle. Feces 2–3 mm in height were squeezed out and the cecum returned to the abdominal cavity. Sham-operated mice were subjected to general anesthesia and laparotomy but without CLP. Postoperatively, mice were subcutaneously administered with pre-warmed saline (0.3 mL/10 g). Bupivacaine (3 mg/kg) and buprenorphine (0.1 mg/kg) were given preemptively by subcutaneous injection to alleviate pain. Rectal temperature was taken to measure the sepsis severity, and blood and tissue samples were collected 24 h after surgery after euthanization using injectable anesthetics (Ketamine (80-150 mg/kg)/Xylazine (8–15 mg/kg), IP) followed by exsanguination via cardiac puncture.
Primary cell cultures
Glial cell culture
Cerebral cortex of neonatal mice (1–2 days old) was used to isolate microglia and astrocytes as previously described [30]. After euthanasia, the cerebral cortex was collected in chilled HBSS (Gibco, Waltham, MA) and gently dissociated using papain and DNase I (Roche, Sigma, St. Louis, MO). The dissociated single cells were grown in a T75 flask precoated with poly-D-lysine (PDL) in the complete glia media (Dulbecco’s modified Eagle’s medium (DMEM)/F12 supplemented with 10% FBS and 1% penicillin–streptomycin) at 37 °C with 5% CO2 for 10–14 days. Once cells had grown to confluence, the flasks were shaken at 100 rpm for 1 h at 37 °C, and the floating microglia were collected and cultured for specified experiments.
Neuronal cell culture
Mouse cortical neurons were derived from mouse embryonic (E) 16 to E18 cortices as described previously [31]. Single neuronal cells were dissociated by enzymatic reaction with Collagenase/Dispase (40 μg/mL, Roche, Sigma, St. Louis, MO) and DNase I (0.6 mg/mL, Roche) and by mechanical force using pipettes. The cells were seeded into PDL-coated culture plates in a Neurobasal medium including B-27 supplement (Gibco, Thermo Fisher Scientific). Cell number of 1 ~ 2 × 106 cells/well (6-well plate) were plated for western blotting. One week after the seeding, the neuronal cultures were used for further analysis.
CLP EV isolation and quantification
CLP EVs were isolated from the plasma of sham and CLP mice by ultracentrifugation, as we described previously [24]. In brief, EDTA-anticoagulant plasma was prepared by centrifugation, diluted with an equal volume of sterile, cold DPBS, and then centrifuged at 12,000×g at 4 °C for 30 min. The supernatant was diluted in cold-DPBS and transferred to a polycarbonate ultracentrifuge tube for ultracentrifugation at 110,000×g at 4 °C for 1 h (Optima MAX-XP ultracentrifuge, Beckmann Coulter with MLA-80 rotor). Following centrifugation, the supernatant in the ultracentrifuge tube was discarded carefully, leaving approx. 100 µl of the solution retained. The pellet containing EVs was then resuspended in the remaining solution in the tube. The EVs were quantified using ViewSizer® 3000 nanoparticle tracking analysis system (Horiba Instruments, Irvine, CA), aliquoted, and stored at −80 °C immediately for future experiments. For treatment, EVs were pooled after isolation from the plasma of 5–6 mice within the same group. Notably, the isolation of EVs was conducted using aseptic techniques.
EV treatment in vitro and in vivo
EV treatment in vitro
Primary microglia, astrocytes, and neurons were incubated with sham or CLP plasma EVs at various concentrations at 37 °C with 5% CO2 overnight. Following treatment, the supernatant was collected and ELISAs for CXCL2, TNFα, and IL-6 (Duo set ELISA kit, R&D systems, Minneapolis, MN) were performed following the manufacturer’s instructions. For anti-miRNA treatment in microglial cultures, miRCURY LNA miRNA Power Inhibitors (Qiagen) against miR-146a, -122, -34a, and -145a (200 nM of anti-miRNA mixture with 50 nM each miRNA) or control oligos (200 nM) were preincubated with EVs one hour before being administered to the cell cultures. LPS (50 ng/mL, Sigma) and R837 (100 ng/mL, InvivoGen) were used as positive controls.
EV treatment in vivo
Sham and CLP EVs, each with 8 × 108 particles, were administered to the brain (13) via ICV injection. The operators (JH, ZL) for injection were blinded to the reagents and strain information of the mice. Sixteen to 22 h after injection, the cerebral cortex and hippocampus were harvested for flow cytometry and qRT-PCR analyses. The operators (LZ, FC, JH, JZ, CP, BR) processing the brains and performing the experiments were blinded to both reagents and the mouse strains until all the data analyses were completed.
Flow cytometry
Mice were euthanized as described above and perfused with 25 mL of saline at 16–22 h after the administration of EVs. The isolated cerebral cortex and hippocampus were crushed mechanically using a 1 mL syringe and enzymatically in DMEM containing 10% FBS, collagenase/dispase (1 mg/mL), papain (12 U/mL), and DNase I (1 mg/mL) at 37 °C with shaking at 150 rpm for 50 min. All the enzymes were purchased from Roche, Sigma, St. Louis, MO. The cell suspension was filtered through a 70 μM cell strainer followed by 30 and 70% Percoll gradient isolation (Percoll Plus, Cytiva, Sigma). The isolated single cells were stained with a viability dye, Zombie Red (Invitrogen, Waltham, MA) followed by an Fc receptor blocker, anti-CD16/32 antibody (cone: 93, Cat#: 14-0161-85, Invitrogen), prior to staining for cell surface markers including CD45-efluro 450 (Clone: 30-f11, Cat #: 48-0451-82, Invitrogen), CD11b-BV480 (Clone: M1/70, Cat #: 566149, BD bioscience, San Jose, CA), Ly6G-BV421 (Clone: 1A8, Cat #: 562737, BD bioscience), and Ly6C-PE (Clone:HK1.4, Cat #: 128007, Biolegend, San Diego, CA) for 20 min at 4 °C. Subsequently, the cells were fixed and permeabilized with intracellular fixation and permeabilization buffer (Invitrogen) and subjected to staining with intracellular cytokine antibodies including TNF-α-PE-Cy7 (Clone: MP6-XT22, Cat #: 506323, Biolegend), IL1-β-PerCP-efluro710 (Clone: NJTEN3, Cat #: 46-7114-82, Invitrogen), and IL-6-APC (Clone: MP5-20F3, Cat #: 504508, Biolegend) for 20 min at 4 °C. A fluorescence minus one (FMO) control was used to determine the gating strategy for intracellular cytokines. Data were acquired on a Cytek Aurora (Cytek, Fremont, CA) flow cytometer and analyzed by FlowJo (BD Biosciences).
RNA isolation and qRT-PCR
RNA was isolated from the cerebral cortex and hippocampus using TRIzol (Sigma) and quantified by Nanodrop (Thermo Scientific). One microgram of RNA was transcribed to cDNA using M-MLV reverse transcriptase and random hexamer primers per the manufacturer’s instructions (Promega, Madison, WI). Quantitative real-time PCR was performed using the GoTaq SYBR qPCR master mix (Promega, Madison, WI) in a QuantiStudio3 PCR cycler (Applied Biosystems, ThermoFisher Scientific), as described previously. Following are the PCR primer sequences: GAPDH (Forward: 5′-AACTTTGGCATTGTGGAAGG-3′, Reverse: 5′ GGATGCAGGGATGATGTTCT-3′); TNFα (Forward: 5′CTGGGACA- GTGACCTGGACT-3′, Reverse: 5′-GCACCTCAGGGAAGAGTCT-G-3′), IL-1β (Forward: 5′GCCCATCCTCTGTGACTCAT-3′, Reverse: 5′-AGGCCACAGGTA- TTTTGTCG-3′), IL-6 (Forward: 5′ -AGTTGCCTTCTTGGGACTGA-3′, Reverse: 5′-TCCA- CGATTT- CCCAGAGAAC-3′), CXCL2 (Forward: 5′-CCGCTGTTGTGGCCAGTGAACTGC-G-3′, Reverse: 5′TTAGCCTTGCCTTTGTTCAGTAT3′). Transcript expression was calculated using the comparative Ct method normalized to GPDAH (2−ΔΔCt) and expressed as the fold change in the treatment group over the control.
Neuronal cell apoptosis
Neuronal treatment and caspase-3 immunoblotting
Primary neuronal cells in 6-well plates were incubated with (1) conditioned media (CM) collected from microglial cultures treated with sham EVs or CLP EVs at 2.5 × 109 particles/mL, (2) sham EVs or CLP EVs (2.5 × 109 particles/mL) alone, and (3) bafilomycin A1 (10 nM, Sigma). Sixteen hours after the treatment, the neuronal cells were washed with chilled DPBS and lysed for western blot with NP-40 lysis buffer, containing 50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 0.25% Na-deoxycholate, and complete protease inhibitor cocktail (Roche). Protein concentration was quantified by a Pierce BCA quantification assay (Thermo Scientific), and 60 μg of protein lysate was fractionated by SDS-PAGE (4–20% TGX stain-free gel, Bio-Rad). Proteins were transferred to a PVDF membrane using a semi-dry transfer unit, Trans-blot Turbo (Bio-Rad), at the default setting for low molecular weight proteins. Membranes were blocked with 10% nonfat milk containing 0.1% Tween 20 in Tris-buffered saline and then blotted with anti-cleaved caspase 3 antibody (Cat. #: 9664, 1:1,000, Cell Signaling, USA) at 4 °C overnight, followed by incubation with a HRP-conjugated secondary antibody (1:1000, Cell Signaling). Membranes were developed with Forte Western HRP substrate (Millipore Sigma), and proteins were visualized using a ChemiDoc imager (Bio-Rad). The intensity of the images was analyzed using Fuji Image J.
Annexin V staining
Primary neuronal cells were seeded in chambered borosilicate cover glass (Lab-Tek, Thermo Scientific) and incubated with conditioned media from microglia treated with sham EVs or CLP EVs as described above, and bafilomycin A1 (10 nM) for 16 h. Annexin-V (Annexin-V-FITC Fluorescence microscopy kit, Cat #: 550911, BD Pharmingen) and Hoechst 33258 (2 µl/mL, Sigma) were stained at room temperature for 15 min while protected from light, followed by washing with cold-PBS and cold-assay buffer provided in the kit. The images were collected using fluorescence microscopy (Ti Eclipse, Nikon) and analyzed with NIS Element (Nikon).
Cytokine array and pathway enrichment analysis
Proteome Profiler, Mouse XL Cytokine Array Kit (Cat #: ARY028, R&D Systems) was used to analyze the media from EVs-treated microglial cultures. The expression levels were measured by blot density and quantified using ImageJ. Proteins with a fold change (CLP-EVs/Sham-EVs) >1.2 were transformed into corresponding genes for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis using SRplot (https://www.bioinformatics.com.cn). Metascape analysis (http://metascape.org) was performed to identify gene types, such as the Entrez Gene ID. Proteins with an expression fold change (CLP-EV/Sham-EVs) >1.5 were selected for GO membership term analysis through Metascape. Gene lists from differentially expressed proteins following the treatment of sham-EVs and CLP-EVs were subjected to custom analysis. GO chord plots were analyzed and visualized using SRplot (https://www.bioinformatics.com.cn). A dot graph ranked by statistical significance was used to display the top enrichment clusters.
Luminex analysis
Media from microglia cultures treated with sham or CLP plasma EVs at 2.5 × 109 EVs/mL were analyzed using a Luminex Multiplex Assay from R&D Systems with Luminex 200 instrument (Luminex xMAP Technology) following the manufacturer’s instructions. Luminex xPONENT software was used for data analysis.
Statistical analysis
All mice were randomly assigned to each group. All in vitro analyses were replicated in duplicate or triplicate with fully independent runs on separate days. The treatment groups in both in vivo and in vitro were blinded. Unblinding occurred only after all data collection and data entry had been completed. Statistical analysis was performed using GraphPad Prism 9 software (GraphPad Software, La Jolla, CA, USA). The normality of all numerical data was assessed using the D’Agostino-Pearson test or Kolmogorov–Smirnov test. A parametric test was used if the hypothesis of normality was not rejected. Nonparametric Mann–Whitney U tests were applied to the data that did not pass the normality test. For multiple group comparisons, one-way ANOVA with Tukey’s post hoc test or two-way ANOVA with Bonferroni’s post hoc test was applied when the data met the two assumptions: normal population distribution and equal variance. If the data did not satisfy the assumptions, we performed an ANOVA analysis after the data were transformed into log format or the Kruskal–Wallis test. Data were presented as mean ± SEM unless stated otherwise. Differences were considered significant at a p-value <0.05 according to two-tailed tests.
Results
EVs uptake by brain cells
To track EVs in cells and tissues, we labeled HEK293T cell-derived EVs with the lipophilic fluorescent dye PKH26 and purified PKH26-EVs by removing free PKH26 dye through filtration (Fig. 1A). The cellular uptake of EVs was then examined by incubating RAW macrophages and microglia with fluorescent PKH26-labeled EVs (Fig. 1A). In both cell types, the PKH26-293EVs were engulfed and localized in the cytoplasm (Fig. 1B, C, top panels). In contrast and as a control, the red fluorescent signals of PKH26 alone were not notably distributed inside the cytoplasm (Fig. 1B, C, bottom panels). In vivo, we tested two approaches of EVs administration—ICV and IV injections—and tested their brain distribution. PKH26-293EVs (8 × 108 particles) were directly injected into naïve mouse brains via the ICV (Fig. 2A, B). EVs signals were assessed at both 2 and 18 h after injection to capture the dynamic changes. At 2 h post-injection, F-Actin (purple, component of cytoskeleton of cell) and PKH26-293EVs (the red fluorescence) were co-detected near the injection site compared to PBS control (Fig. 2A). Eighteen hours after the ICV injection, immunohistochemistry staining observed that the cells colocalizing with the EVs were microglia stained by Iba-1 and neurons stained by NeuN (Fig. 2B). EVs were in close contact with astrocytes marked by GFAP (Fig. 2B). To test if EVs penetrate the brain through BBB and localize in brain cells, the PKH26-293EVs (2 × 109 particles) were systemically injected through tail vein. One hour after the injection, the solid red signals were detected inside the Iba-1-positive microglia and NeuN-positive neurons, as shown in the magnified images (Fig. 2C). Consistent with ICV injection, fluorescence-labeled EVs were found in close contact with GFAP-positive astrocytes but rarely inside of cell body (Fig. 2D). To verify that the red fluorescence of PKH26-293EVs is not a result of spillover from other antibody-associated fluorescence, an unstained brain section following IV injection of PKH26-293EVs was displayed in Figure S1. This figure demonstrates consistent presence of red florescence particles independent of any antibody signaling, thereby confirming the specific labeling and presence of PKH26-labeled EVs in brain tissues. Together, these in vitro and in vivo data suggest that circulating or locally delivered EVs can pass through BBB and enter various brain cells including microglia and neurons.
Fig. 1.

Internalization of PKH26-labeled EVs in RAW macrophages and microglia. A Schematic depicting experimental setup. EVs were isolated from HEK293T cultures using ultracentrifugation and labeled with PKH26 (PKH26-293EVs). Cells were incubated with either PKH26-293EVs (2.5 × 109 particles/mL) or the same concentration of PKH26 alone without EVs at 37 °C and imaged using a confocal microscope (Ti2, Nikon, Japan). The PKH26-labeled EVs (orange-red) were taken up by the RAW macrophages (B) and the primary cultured microglial cells (C). Nuclei were stained with DAPI
Fig. 2.

Localization of PKH26-labeled EVs in brain tissue following different routes of administration. EVs were located in the cytoplasm of microglia and neurons following ICV (A, B) and IV (C, D) routes. At 2 h (A) and 18 h (B) after the ICV injection and 1 h (C, D) after IV injection of PKH26-293EVs and control vehicle, brain sections were stained with Iba-1 for microglia, GFAP for astrocytes, NeuN for neurons, and DAPI for the nuclei. The images were taken and analyzed by a Nikon confocal microscope. Yellow solid filled arrow, EVs colocalized with cells. The magnified neuron and microglia show the localization of PKH26-293EVs inside the cells. LV left cerebral ventricle, CA3 cornu ammonis 3, cc corpus callosum
Sepsis plasma EVs cause brain inflammation when administered via ICV injection
To determine whether circulating sepsis EVs have the potential to induce an inflammatory response in the brain, plasma EVs were isolated from sham or CLP mice at 24 h post-surgery as previously described (24) and CD81 and CD9 expression was confirmed using Western blotting (Figure S2). A total of 8 × 108 plasma EVs were injected per mouse via the ICV and brain inflammation was evaluated at 24 h. Flow cytometry analysis revealed a significant increase in innate immune cell number in the brains injected with CLP EVs compared to those with sham EVs injections (Fig. 3A). These immune cells included the CNS resident microglia (50 ± 5.7 vs. 40.4 ± 4.9 × 104 cells, p < 0.05; CLP EVs vs. sham EVs) and infiltrated myeloid-derived cells (21.7 ± 3.7 vs. 11 ± 3 × 104 cells, p < 0.01), such as monocytes (15 ± 3 vs. 7.8 ± 2.6 × 104 cells, p < 0.01) and neutrophils (20.1 ± 4. vs. 9.5 ± 2.3 × 103 cells, p < 0.01) with the gating strategy shown in Figure S3. Similarly, the percentage of infiltrated myeloid cells (CD11b+CD45high) among total brain cells was increased in the CLP EVs group than in the sham EVs group. Of note, the relative percentage of microglia (CD11b+CD45low) was decreased in the CLP EV group compared to that of sham group (Fig. 3B, C) most likely due to the increase in other cell types such as myeloid cells. Among the immune cells, the percentage of monocytes (Ly6C+Ly6G−) was significantly higher in CLP EV group than sham control (17.9 vs. 14.6%, p < 0.05), and the percentage of neutrophils (Ly6C+Ly6G+) also increased in CLP EV group (2.9 vs. 1.4%, p < 0.05) (Fig. 3B, C). To examine cytokine gene expression in the injected brains (Fig. 3D), we extracted total RNA from the cerebral cortex and hippocampus respectively. In the cortex, qRT-PCR analysis showed that ICV injection of CLP EVs significantly increased the proinflammatory cytokine/chemokine gene expressions including TNFα, IL1-β, IL-6, and CXCL2 compared to sham EVs-treated animals. However, the increase in the pro-inflammatory cytokines and chemokines was not observed in the hippocampus (Fig. S3B). Consistent with cytokine gene expression, the protein level of IL-1 β and TNFα exhibited a significant increase in monocyte, neutrophil and microglia in the mice that receiving CLP EVs (Fig. 3E). Together, these data indicate the prominent pro-inflammatory effects of the circulating EVs from septic mice.
Fig. 3.

Sepsis plasma EVs cause neuroinflammation in vivo. CLP EVs were injected into the mouse brain via ICV at a dose of 8 × 108 particles per brain. Brains were processed 24 h after the injection. The infiltrated immune cells were analyzed by flow cytometry. The cell number (A) and the percentage (B) of myeloid cells, monocytes, and neutrophils in the brain were displayed and represenative flow cytometry figures (C) of microglia (CD45low CD11b+), monocyte (Ly6C+Ly6G−) and neutrophil (Ly6C+Ly6G+) were listed. D Brain cytokine gene expression was assessed using qRT-PCR at 24 h after injection. n = 5/group. E Cytokine expression in monocyte, neutrophil, and microglia tested by flow cytometry. n = 4–7/group
Sepsis EVs activate cultured microglia and indirectly induce neuronal apoptosis
To explore the proinflammatory property of plasma EVs, we treated various brain cells including microglia, astrocytes, and neurons with both sham and CLP plasma EVs and tested their cytokine/chemokine responses. The microglia showed a marked dose-dependent increase in CXCL2 and IL-6 production in response to CLP EVs but not sham EVs (1 × 108 ~ 5 × 109 EVs/mL) (Fig. 4A). In contrast, astrocytes exhibited no or very mild cytokine responses when treated with CLP EVs as compared to that of sham control (Fig. 4B). In neurons, the production of CXCL2 and IL-6 was slightly increased upon treatment of CLP EVs at a dose of 2.5 × 109 EVs/mL (Fig. 4C). Notably, this increase observed in neurons was only a small fraction of the increase seen in the microglia treated with the same concentration of CLP EVs (2.5 × 109 EVs/mL). This is not surpising because neurons have immune capacity, but we do not anticipate the inflamamtory response in neurons will be as strong as that in immune cells such as microglia. However, neuronal cell death plays a pivotal role in the pathophysiology of SAE [32]. To investigate if CLP EVs cause any injury to neurons, we treated cultured neurons with sham and CLP EVs and tested for apoptosis. As illustrated in Fig. 5A–C, direct treatment of CLP EVs to the cultured neurons did not affect cleaved caspase-3 expression (left panel of Fig. 5B, C). In contrast, there was a twofold increase in cleaved caspase-3 when incubated with CLP EVs-treated microglial CM as compared with that of sham control (right panel of Fig. 5B, C). Of note, 10 nM of bafilomycin A1 was included as a positive control for apoptosis induction through the activation of caspase-3 [33]. Additionally, we also observed a greater Annexin V-FITC intensity and Annexin V-positive neurons treated with CM from CLP-EV-treated microglia (Fig. 5D), however, the increase did not reach statistical significance (area: 105.9 vs. 60.2, p = 0.138, cell number: 43 ± 13 vs. 103 ± 18, p = 0.0755, Figure S4). These data suggest that CLP EVs can induce microglial activation, inflammation, and neuronal cell death.
Fig. 4.
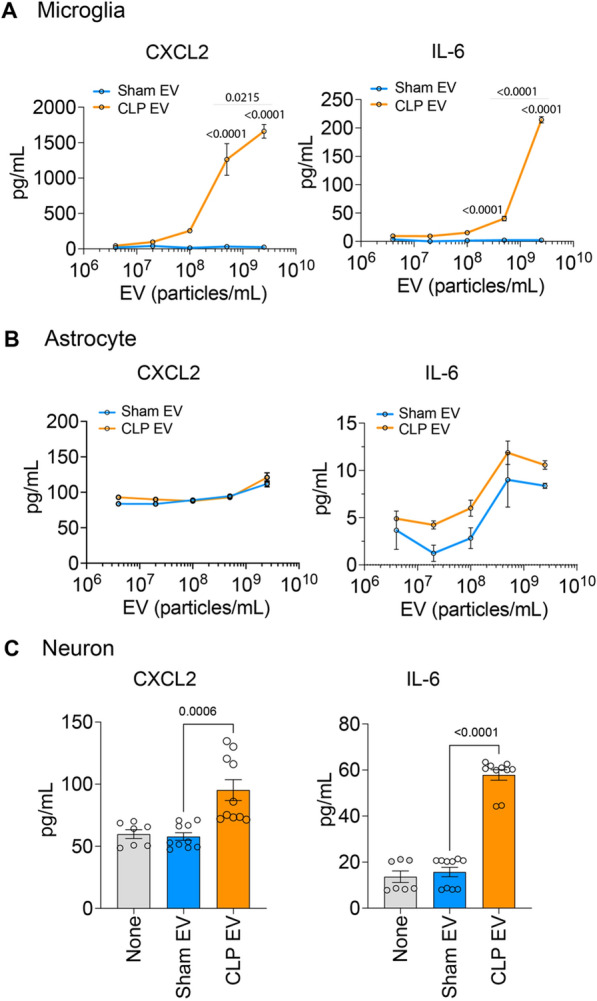
Sepsis plasma EVs selectively activate microglia in vitro. Primarily cutured microglia and astrocytes were treated with sham and CLP plasma EVs in various concentrations (5 × 106 ~ 2.5 × 109 EVs/mL). At 24 h, supernant were collected and cytokine/chemokine were tested using ELISA. A CXCL2 and IL-6 production in microglia. B CXCL2 and IL-6 production in astrocyte. C CXCL2 and IL-6 in the media of neurons treated with sham and CLP plasma EVs (2.5 × 109 EVs/mL). n = 3–10. The experiment was repeated twice in triplicates with independent runs on separate days
Fig. 5 .

Microglial activation by sepsis plasma EVs induces neuronal apoptosis. A Schematic illustration of experimental setup. Conditioned media (CM) were collected at 24 h from microglia treated with plasma CLP- or sham-EVs using aseptic technique. The primarily cultured neurons were incubated with either sham or CLP EVs directly or CM collected above and apoptosis were evaluated at 16 h using western blot and fluoresce imaging. B Conditioned media from CLP EVs treated-microglia but not CLP EVs itself induce apoptosis to neurons. Representative images of blot from three different experiments using different batches of primary neuronal cultures. C Relative intensity of cleaved caspase-3 over caspase-3 precursor. n = 4–10/group D Annexin V-staining in neurons. Neurons were incubated with the conditioned media from EV-treated microglia for 16 h and Annexin V-FITC was stained. Representative images from three different experiments
Sepsis EV-associated miRNAs contribute to microglial activation in part through TLR7
To determine the mechanism by which sepsis-induced EVs drive microglial activation, we focused on EV-cargoed miRNAs. Our previous studies have identified a list of pro-inflammatory miRNAs, such as miR-146a-5p, miR-122-5p, miR-34a-5p, and miR-145a-5p, in sepsis plasma and EVs [34] and their role in the neuroinflammatory responses [14]. To test the role of these miRNAs in sepsis EVs-induced microglial activation, we treated plasma EVs with anti-miR combo targeting the above mentioned 4 miRNAs before incubating the EVs with microglia. As shown in Fig. 6A, treatment of EVs with the anti-miR combo led to a 52% reduction in CXCL2 levels compared to the control oligo (209 ± 17.5 vs. 436 ± 12.4 pg/mL, p < 0.0013) in the CLP EV-induced CXCL2 production. Furthermore, since we previously have demonstrated that miR-146a-5p, miR-122-5p, miR-34a-5p, and miR-145a-5p can induce microglial activation via TLR7 [14], we next explored the role of TLR7 in CLP EVs-activated microglia. As illustrated in Fig. 6B, CLP EVs-induced CXCL2 production was markedly reduced in TLR7-KO microglia as compared to that of WT (173 ± 38.9 vs. 762 ± 91.9 pg/mL, p < 0.0001). In the two important control groups treated with R837 (TLR7 agonist) and LPS (TLR4 agonist), respectively, TLR7-KO microglia exhibited no CXCL2 response to R837, whereas maintained a similar response to LPS as compared with WT microglia, demonstrating the specific functional deficiency of TLR7 in KO microglia. Together, these data demonstrated that sepsis EV-mediated microglial activation is in part via its associated miRNAs and TLR7 signaling.
Fig. 6.

CLP EVs-mediated microglial activation is partially attributed to miRNAs and TLR7 signaling. A Anti-miRs combo (anti-miR-146a, -122, -34a, -145a, 200 nM) paritially attenuated CLP EVs-mediated CXCL2 production in the microglia. Prior to applying to microglia culture, sham EVs and CLP EVs (2.5 × 109 particles/mL) were treated with PBS, control oligonucleotide,or anti-miR combination (combo) at a concentration of 200 nM, including anti-miR-146a, -34a, -miR-122, and miR-145a, for 1 h. Microglial media were then collected 24 h after treatment and assayed for CXCL2 using ELISA. Each bar represents triplicate samples with each experiment repeated twice. n = 3/group B. Plasma EVs from septic mice induce CXCL2 production in part via TLR7 signaling. Cultured microglia from WT and TLR7 KO mice were treated with sham EVs (2.5 × 109 particles/mL), CLP EVs (2.5 × 109 particles/mL), R837 (100 ng/mL), and LPS (50 ng/mL) for 24 h. The supernatant was then collected for CXCL2 analysis using ELISA. n = 3–9/group. Immune blot (C) and quantification of integrated intensity (D) of cytokine array in the conditioned media from EVs-treated microglia. E Enrichment analysis of GO-biological process, molecular function, and KEGG pathway. F GO chord diagram reflects the differential gene expression (shown on the left) and enriched functional categories (shown on the right) including inflammatory response, innate immune response, cell death, and neurons. G Luminex analysis valiated several upregulated molecules in the conditioned media identified from cytokine array and pathway enrichment analysis including Resistin, MMP-9, IL-10, LDLR, CRP, CXCL10, CCL2, and CCL4. n = 3/group
Cytokine profile patterns of conditioned media identified potential effectors responsible for neuronal injury
To identify the mediators responsible for neuronal injury in the conditioned media from sepsis EV-treated microglia, we performed cytokine arrays (Fig. 6C). Among the 111 molecules tested, 39 molecules exhibited a >1.5-fold increase in the conditioned media from CLP EV-treated microglia as compared to sham group. These include various cytokines, chemokines, and growth factors (Fig. 6D). The top 10 upregulated proteins were E-selectin, IL-12 p40, lipocalin2 (neutrophil gelatinase-associated lipocalin; NGAL), MMP9 (matrix metallopeptidase 9), CXCL2 (chemokine C-X-C motif ligand 2), CCL2 (chemokine C–C motif ligand 2), IGF-BP1 (insulin-like growth factor binding protein 1), Myeloperoxidase (MPO), Reg3G (regenerating islet-derived protein 3 gamma), and Resistin.
Pathway enrichment analysis of all upregulated molecules revealed enrichement for Gene Ontology (GO) Biological Process terms including “leukocyte migration,” “granulocyte chemotaxis,” and “myeloid leukocyte migration.” highlighting the activation of microglia by sepsis EVs. In terms of Molecular Function, most of the enriched GO terms, including "cytokine activity,” “receptor ligand activity,” “chemokine activity,” and “complement C1q binding function,” were associated with the proinflammatory response of microglia following the treatment with sepsis EVs. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis indicated an upregulation of cytokine/chemokine signaling pathways, including TNFα and IL-17, and Toll-like receptor signaling pathways with lower counts but statistically significant. To further investigate the functional profile of these mediators, we analyzed the GO membership terms of “cell death” “neuron,” “inflammatory response,” and “innate immune response.” in relation to the upregulated mediators and identified 33 of them as being associated with at least one GO term(Fig. 6F). In the overlapping area of neuron (green) and cell death (dark blue), LCN2 (Lipocalin 2), MMP9 (Matrix metallopeptidase 9), CCL3 (C–C Motif chemokine ligand 3), IL-10 (Interleukin 10), ADIPOQ (Adiponectin), and LEP (Leptin) were identified, suggesting these protein as potential effectors mediating the crosstalk between CLP EV-activated microglia and neuronal cell death. Finally, we validated part of the cytokine array findings using Luminex multiplex assays on 8 selected cytokines and chemokines related to neuronal damage. We found a marked increase of these molecules in the media of sepsis EV-treated microglial cultures (Fig. 6G).
Role of TLR7 and MyD88 signaling in sepsis EV-induced brain inflammation in vivo.
We next examined the signaling pathway by which CLP EVs induce brain inflammation in vivo using a loss-of-function approach. Given that TLR7 contributes to sepsis-EVs-induced microglial activation in vitro (Fig. 6B), we anticipate a similar role of TLR7 in response to ICV injection of sepsis-EVs in vivo. Different from our hypothesis, we found that TLR7 deficiency had no effect on the brain immune cell numbers such as microglia, myeloid cells, monocytes, and neutrophils (Fig. 7A, B) as well as their percentage (data not shown) in the brain following ICV injection of CLP plasma EVs at a dose of 8 × 108/injection. Consistent with this was the lack to difference of cytokine gene expression in the cerebral cortex between the WT and TLR7-KO mice receiving CLP EVs(Fig. 7C). Interestingly, we observed that systemic deficiency of MyD88 reduced CLP EV-induced brain inflammation. As shown in Fig. 8A–C MyD88 KO mice displayed a markedly reduction in the number of brain innate immune cells compared to WT mice after ICV injection of CLP EVs, which include monocytes, and neutrophils. This was applied for both the number and percentage. Regarding brain residential microglia, there was a statistically significant reduction approximately 41% in microglial numbers in MyD88 knockout mice even though the percentage remained unchanged between groups. Moreover, the number of immune cells with positive cytokine expression was also reduced in MyD88-deficient mouse brains (Fig. 8D, E). These include IL-1β-expressing microglia, monocytes and neutrophils and TNFα-expressing neutrophils. Importantly, the cytokine gene expression such as Il-1β, Tnfα, and Cxcl2 in both cortex and hippocampus was markedly attenuated in MyD88KO mice as compared to WT mice that receiving CLP EVs (Fig. 8F).
Fig. 7.

TLR7 is not involved in sepsis plasma EVs-induced brain inflammation in vivo. Plasma EVs (8 × 108 EV particles/brain) were delivered via ICV to both WT and TLR7 KO mice. Cell population in the brain was analyzed at 16–22 h using flow cytometry. A Cell numbers. B Representative figure of flow cytometry. Myeloid cells: CD11b+CD45high, microglia: CD11b+CD45low, monocyte: Ly6C+Ly6Chigh and neutrophils with Ly6C+Ly6Ghigh. MO: monocyte, NE: neutrophil. C. IL-1β, TNFα, and CXCL2 gene expression in the cortex measured by qRT-PCR. n = 4–7/group
Fig. 8.

MyD88 contributes to brain inflammation driven by plasma sepsis EVs. CLP plasma EVs (8 × 108 EV particles/brain) were delivered via ICV to both WT and MyD88 KO mice. Cell population in the brain was analyzed at 16–22 h using flow cytometry. A, B Cell number and percentage of each population. n = 5/group. C. Representative figure of flow cytometry. MO monocyte, NE neutrophil. D Percentage of microglia, monocytes and neutrophils expressing intracellular IL-1β or TNFα. n = 5/group. E Representative histogram of flow cytometry. S-EVs sham plasma EVs, CLP EVs sepsis plasma EVs. FMO fluorescence minus one. F Cytokine gene expression in brain tissues measured by qRT-PCR. n = 4–5/group
Discussion
There are several main findings in the present study. Using fluorescence-labeled EVs and confocal microscopy, we show that EVs enter microglia in vitro and multiple brain cells in vivo via both ICV and IV routes. Direct ICV administration of sepsis EVs causes marked brain inflammation as manifested by multiple innate immune cells accumulations in the brain, such as microglia, monocytes, and neutrophils, as well as cytokine/chemokine gene expression. While sepsis EVs induce a robust and dose-dependent cytokine production in microglia, such an effect was absent in astrocytes and only mild in neurons. Moreover, the proinflammatory effect of sepsis EVs in microglia seems in part dependent on cargo miRNAs and TLR7 signaling as anti-miR inhibitors and genetic deletion of TLR7 attenuate cytokine production. One of the important findings is that conditioned media from the EV-treated microglia causes neuronal cell apoptosis as evidenced by increased positive Annexin-V staining and cleaved caspase-3 in neurons. In contrast, direct treatment with EVs has no effect on neuronal apoptosis. Cytokine array and pathway enrichment analysis of the microglial conditioned media reveals multiple cytokines and chemokines that are linked to cell chemotaxis, leukocyte migration, and neuronal cell death. Finally, and somewhat surprisingly, TLR7 deficiency has no impact on brain inflammation in response to sepsis EV ICV injection, whereas MyD88 KO mice exhibit a significant reduction in brain inflammation following sepsis EV administration.
To test the role of sepsis plasma EVs in brain inflammation, we first examined if the circulating EVs could penetrate BBB and reach brain tissue. Utilizing PKH26 fluoresences-labled HEK293T cell-derived EVs with an average size of 100 nm, we successfully detected its fluoresence signaling in cortex tissue section when administered intravenously to WT naïve mice, suggesting that EVs can pass through intact BBB. This confirms previous reports that 30–150 nm of small EVs are capable of penetrating BBB via nasal, intratracheal, and intravenous deliveries [35, 36]. When EVs were dispensed directly into intracerebralventrical space, they reaches basal lamina with 2 h of administration and were detected in the microglial and neurons 18 h later. Interestingly, EVs were hardly observed inside of astrocyte after injection of both IV and ICV route. Instead, some EVs were found in close proximity to astrocyte processes, which is consistent with findings from Long et al. [37] that EVs is largely accumulated in microglia and neurons but in close contact with astrocytes within CNS tissue. Similialy, Li et al. revealed that approximately 86.6% of EVs were uptaken by microglia when intravenously administered followed by neurons (12.6%) [38] in the CNS. We speculate that this biased EVs accumulation may be related to different EV-cell interaction mechanism [39]. To facilitate the transfer the EV contents into the cell, mechanisms such as plasma membrane fusion or endocytic uptake of EVs are required. Additionally, EV can also transmit signals through “Kiss-and-Run” mechanism, where they activate receptors on interacting cells or form transient nanometer-size fusion pores before detaching from their target cell [39]. Our observatios of EVs signaling inside the cell body of microglia both in vitro and in vivo implying a potential delivery of cargo molecules such as miRNAs from EVs into the cell, which was later demonstrated by the inhibition of miRNA reducing sepsis EV-associated proinflammatory property in microglia.
Microglia are activated during sepsis in the brain of both mice and human [38, 40] and contribute to neuroinflammation and cognitive impairment [40]. In the current study, we found that microglia but not astrocytes nor neurons, exhibited a marked cytokine production in response to sepsis EVs. In vivo, ICV injection of sepsis EVs promoted infiltration of peripheral monocytes and neutrophils into the brain as well as proinflammatory cytokine TNF-α and IL-1β, two major cytokines that are implicated in brain diseases with inflammation and/or cognitive dysfunction [41, 42]. Chemokine CXCL2, a key attractant for leukocytes was also elevated in sepsis EVs treated mice. Complementary to our ICV approach, Li et al. [38] discovered that intravenously infusion of EVs isolated from serum of LPS-treated mice increased microglial activation and brain proinflammatory cytokines. Together, these facts demonstrate that sepsis plasma EVs are capable of eliciting CNS inflammation in part by activating microglia and promoting migration of blood leukocytes into the brain.
Neuronal dysfunction during sepsis is a significant contributor to cognitive impairment in both survival patients and animals [3, 6, 7]. Post-mortem brain tissues from patients who died from sepsis reveal apoptotic neurons in the amygdala, hypothalamus, and medulla [43]. Additionally, neuronal loss is observed in the hippocampus in animal models of sepsis-like systemic inflammation [44, 45]. Several underlying mechanisms are reported to contribute to neuronal cell damage, including C1q-dependent synaptic pruning by microglia [40] and nitric oxide-mediated neuronal cell death [46]. In our current study, we identified a novel mechanism of EVs-initated microglial activation that induces neuron apoptosis, possibly through released cytokines and chemokines, as identified by cytokine array and bioinformatic analysis. GO term analysis suggests that some of the mediators are related to neuron and neuronal death. For example, Lcn2 is well-studied for its role in the brain, including neuronal death and neuroinflammation [47]. It is reported that neuronal death mediated by Lcn2 relates to oxidative stress and neuroinflammation, including the recruitment of neutrophils [48]. Among the upregulated mediators released from sepsis EVs-activated microglia, IL12-p40, CXCL2, and CCL2 are known for their involvement in peripheral immune cell infiltration and activation [49–51] and Mmp9 contributes to BBB breakdown [52]. Given the ability of small EVs to pass through the BBB, which was demonstrated in the current and previous studies [53], these data support the hypothesis that sepsis plasma EVs contribute to SAE by triggering the microglia release of soluble proteins into the brain to enhance BBB disruption and facilitate the entry of peripheral immune cells into the brain.Among the differential cytokines in the sepsis EV-activated microglia, Reg3G plays various roles in immune defense, including a protective role against bacterial infection [54, 55]. Myeloperoxidase (MPO), known as a major component of neutrophils, is also released in the microglia in our cytokine array. Chen et al. reported that microglial MPO exacerbates brain damage in cerebral ischemia by mediating the neuronal release of HMGB1 [56]. IGF-BP1 participates in metabolic regulation [57], possibly contributing to neuronal dysfunction. The pathway analysis with the differentially expressed cytokines also indicates that the TNFα pathway and the IL-17 pathway are noticeable. TNFα has been reportedly involved in neurodegeneration promoted by activated microglia [58]. IL-17, an essential proinflammatory cytokine secreted by CD4+ helper T cells, has various roles in the brain, including participating in proinflammatory response and regulation of immune cells [59]. Future studies to test these specific molecules will be needed to establish their role in the conditioned media-induced neuron apoptosis and to validate their effects in vivo.
Another important part of this study involves testing the role of EV miRNA cargo and the signaling mechanism. Specifically, the EV cargo miRNAs were targeted based on our previously published EV miRNA array data that show an increased loading of miR-146a-5p, miR-122-5p, miR-34a-5p, and miR-145a-5p in sepsis plasma EVs [24]. The finding that treatment of sepsis EVs with the anti-miRs against the above miRNAs leads to attenuated cytokine production suggests that these cargo miRNAs play a contributory role in sepsis EV-elicited cytokine production in the microglia. Moreover, absence of TLR7 leads to a partical reduction of sepsis EV-meidated inflammation in microglial in vitro but lack of impact on brain inflammation in vivo, suggest the involvement of other yet-to-be identified signaling pathways. In constrast, MyD88 appears to play a major role in mediating the recruitment of cytokine-producing immune cells into the brain following sepsis EV administration as evidenced by a marked reduction in neutrophils and monocytes in MyD88-deficient mice. This may be explained by sepsis EV signaling through other DAMPs-TLR pathways. For instance, EV-encapsulated histones have been shown to promote inflammatory response via TLR4 [60], and exosomes released from infected THP-1 cell have induced robust TNF α production via TLR2 and TLR4 [61]. Given that MyD88 is the key adaptor of all TLRs except TLR3 and part of TLR4 signaling, it is not surprising that absence of MyD88 significantly dampens sepsis EV-induced brain inflammation in vivo. Additionally, study by Papareddy et al. [62] demonstrated a significant impact of EVs fusion on cellular response to inflammatory signaling. Upon fusion, EVs released from inflammatory cells can equip their target cells with the molecular machinery including inflammatory receptors such as IL1R1, a MyD88-dependent receptor for IL-1 α, IL-1β, and the IL-1ra, thereby activing NF-κB pathway in their target cells through MyD88 dependent signaling. This mechanism may also explain our observation that sepsis EVs-mediated neuroinflammation was attenuated in MyD88 KO mice.
The current research has several limitations. EVs in the brain originate from multiple sources, including both local and systemic origins. Our current research focused on the role of plasma-derived EVs in neuronal inflammation. However, reseachers have shown the importance of locally shed EVs by brain cells in inflammation. For example, EVs released into cerebrospinal fluid by the choroid plexus epithelium after systemic-induced inflammation contribute to brain damage [63]. Although astrocytes did not exhibit proinflammatory response to sepsis EVs in the current study, they can release EVs to promote leukocyte recruitment in response to IL-1β, as demonstrated by Dickens et al. [64]. In the present study, we focused primarily on the acute inflammatory response of sepsis EVs, the chronic effects and neurocognitive outcomes in these mice that received sepsis EVs remain unclear and warrant further studies in the future.
Conclusion
We demonstrate that circulating EVs in the blood penetrate the normal mouse brain through BBB and induce neuroinflammation when administered via ICV injection and partially through their cargo ex-miRNAs. The sepsis EV-induced brain inflammation involves not only the infiltration of monocytes and neutrophils but also the activation of the residential microglia. In vitro cell experiments suggest that upon activation by sepsis EVs, microglia release a number of soluble factors that are proinflammatory and capable of inducing neuronal apoptotic death.
Supplementary Information
Acknowledgements
We thank the support from Flow Cytometry Core of University of Maryland School of Medicine Center for Innovative Biomedical Resources, Baltimore, Maryland. Partial funding for open access was provided by the University of Maryland Health Sciences and Human Services Library's Open Access Fund.
Abbreviations
- ADIPOQ
Adiponectin
- BBB
Blood Brain Barrier
- CCL3
C-C Motif Chemokine Ligand 3
- CLP
Cecal Ligation and Puncture
- CM
Conditioned Media
- CNS
Central Nervous System
- DAMPs
Damage-associated molecular patterns
- DPBS
Dulbecco’s Phosphate Buffered Saline
- EVs
Extracellular Vesicles
- FMO
Fluorescence minus one
- GFAP
Glial fibrillary acidic protein
- GO
Gene Ontology
- Iba1
Ionized calcium-binding adaptor molecule 1
- ICU
Intensive Care Unit
- ICV
Intra-cerebroventricular injection
- IGF-BP1
Insulin-like Growth Factor Binding Protein 1
- IL10
Interleukin 10
- IP
Intraperitoneal
- IV
Intravenous
- KEGG
Koyto Encyclopedia of Genes and Genomes
- KO
Knock-Out
- LCN2
Lipocalin 2
- LEP
Leptin
- MMP9
Matrix Metallopeptidase 9
- MPO
Myeloperoxidase
- MyD88
Myeloid differentiation primary response 88
- NeuN
Neuronal nuclear antigen
- NTA
Nanoparticle Tracking Analysis
- PAMPs
Pathogen-associated molecular patterns
- PKH26-293EVs
PKH26-labeled EVs from HEK293T cultures
- PRR
Pattern recognition receptors
- REG3G
Regenerating islet-derived protein 3 gamma
- SAE
Sepsis-Associated Encephalopathy
- SOFA
Score of Organ Failure Assessment
- ssRNA
Single-stranded RNA
- TLR
Toll-like Receptor
Author contributions
C.P., Z.L., Y.L., B.R., J.H., F.C., H.L., K.B., and J.Z. performed the in vivo and in vitro experiments. C.P., Z.L., B.R., F.C., B.W., H.H., and L.Z. analyzed the data. S.M.J. provided the HEK293T cell -derived EVs and feedback on the studies. H.H. and L.Z. analyzed the cytokine array and performed pathway enrichment analysis. C.P. and L.Z. organized the figures. C.P. drafted the manuscript. W.C., J.W., and L.Z. contributed to the study conception and design. W.C. J.W., L.Z., C.P., B.W., and K.B. edited the manuscript. All authors have critically read and commented on the final version of the manuscript.
Funding
This work was supported by grants from the NIH R01NS110567 (to W.C./J.W./L.Z.) and RF1NS110637 (to J.W. and S.M.J.).
Availability of data and materials
No datasets were generated or analysed during the current study.
Declarations
Ethics approval and consent to participate
Approved by the IACUC of the University of Maryland School of Medicine.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Junfang Wu, Email: junfang.wu@som.umaryland.edu.
Lin Zou, Email: lzou@som.umaryland.edu.
References
- 1.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA. 2016;315(8):801–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu X, Qin M, Walline JH, Gao Y, Yu S, Ge Z, et al. Clinical Phenotypes of sepsis-associated encephalopathy: a retrospective cohort study. Shock. 2023;59(4):583–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manabe T, Heneka MT. Cerebral dysfunctions caused by sepsis during ageing. Nat Rev Immunol. 2022;22(7):444–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA. 2010;304(16):1787–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feng Q, Ai YH, Gong H, Wu L, Ai ML, Deng SY, et al. Characterization of sepsis and sepsis-associated encephalopathy. J Intensive Care Med. 2019;34(11–12):938–45. [DOI] [PubMed] [Google Scholar]
- 6.Sonneville R, Benghanem S, Jeantin L, de Montmollin E, Doman M, Gaudemer A, et al. The spectrum of sepsis-associated encephalopathy: a clinical perspective. Crit Care. 2023;27(1):386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gofton TE, Young GB. Sepsis-associated encephalopathy. Nat Rev Neurol. 2012;8(10):557–66. [DOI] [PubMed] [Google Scholar]
- 8.Norden DM, Trojanowski PJ, Villanueva E, Navarro E, Godbout JP. Sequential activation of microglia and astrocyte cytokine expression precedes increased Iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia. 2016;64(2):300–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. J Neurochem. 2016;139 Suppl 2(Suppl 2):136–53. [DOI] [PMC free article] [PubMed]
- 10.Pang D, Wu YL, Alcamo AM, Cummings J, Di Caro V, Walko T 3rd, et al. Early axonal injury and delayed cytotoxic cerebral edema are associated with microglial activation in a mouse model of sepsis. Shock. 2020;54(2):256–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luo Y, Yang H, Zhou M, Yang W, Zhang W, Li QQ. Elevated intracranial pressure level is a risk factor for sepsis-associated encephalopathy: a prospective cohort study. In Vivo. 2023;37(6):2585–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang S, Yang Y, Suen A, Zhu J, Williams B, Hu J, et al. Role of extracellular microRNA-146a-5p in host innate immunity and bacterial sepsis. iScience. 2021;24(12):103441. [DOI] [PMC free article] [PubMed]
- 13.Huang H, Zhu J, Gu L, Hu J, Feng X, Huang W, et al. TLR7 mediates acute respiratory distress syndrome in sepsis by sensing extracellular miR-146a. Am J Respir Cell Mol Biol. 2022;67(3):375–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zou L, He J, Gu L, Shahror RA, Li Y, Cao T, et al. Brain innate immune response via miRNA-TLR7 sensing in polymicrobial sepsis. Brain Behav Immun. 2022;100:10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jian W, Gu L, Williams B, Feng Y, Chao W, Zou L. Toll-like receptor 7 contributes to inflammation, organ injury, and mortality in murine sepsis. Anesthesiology. 2019;131(1):105–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams B, Zhu J, Zou L, Chao W. Innate immune TLR7 signaling mediates platelet activation and platelet-leukocyte aggregate formation in murine bacterial sepsis. Platelets. 2022;33(8):1251–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams B, Neder J, Cui P, Suen A, Tanaka K, Zou L, et al. Toll-like receptors 2 and 7 mediate coagulation activation and coagulopathy in murine sepsis. J Thromb Haemost. 2019;17(10):1683–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng Y, Zou L, Zhang M, Li Y, Chen C, Chao W. MyD88 and Trif signaling play distinct roles in cardiac dysfunction and mortality during endotoxin shock and polymicrobial sepsis. Anesthesiology. 2011;115(3):555–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abels ER, Breakefield XO. Introduction to extracellular vesicles: biogenesis, RNA cargo selection, content, release, and uptake. Cell Mol Neurobiol. 2016;36(3):301–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eriksson M, Nelson D, Nordgren A, Larsson A. Increased platelet microvesicle formation is associated with mortality in a porcine model of endotoxemia. Acta Anaesthesiol Scand. 1998;42(5):551–7. [DOI] [PubMed] [Google Scholar]
- 21.Iba T, Ogura H. Role of extracellular vesicles in the development of sepsis-induced coagulopathy. J Intensive Care. 2018;6:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fendl B, Weiss R, Eichhorn T, Linsberger I, Afonyushkin T, Puhm F, et al. Extracellular vesicles are associated with C-reactive protein in sepsis. Sci Rep. 2021;11(1):6996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Plaschke K, Brenner T, Fiedler MO, Holle T, von der Forst M, Wolf RC, et al. Extracellular vesicles as possible plasma markers and mediators in patients with sepsis-associated delirium-a pilot study. Int J Mol Sci. 2023;24(21):15781. [DOI] [PMC free article] [PubMed]
- 24.Xu J, Feng Y, Jeyaram A, Jay SM, Zou L, Chao W. Circulating plasma extracellular vesicles from septic mice induce inflammation via microRNA- and TLR7-dependent mechanisms. J Immunol. 2018;201(11):3392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11(1):115–22. [DOI] [PubMed] [Google Scholar]
- 26.Jeyaram A, Lamichhane TN, Wang S, Zou L, Dahal E, Kronstadt SM, et al. Enhanced loading of functional miRNA cargo via pH gradient modification of extracellular vesicles. Mol Ther. 2020;28(3):975–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puzar Dominkus P, Stenovec M, Sitar S, Lasic E, Zorec R, Plemenitas A, et al. PKH26 labeling of extracellular vesicles: characterization and cellular internalization of contaminating PKH26 nanoparticles. Biochim Biophys Acta Biomembr. 2018;1860(6):1350–61. [DOI] [PubMed] [Google Scholar]
- 28.Kang M, Jordan V, Blenkiron C, Chamley LW. Biodistribution of extracellular vesicles following administration into animals: a systematic review. J Extracell Vesicles. 2021;10(8): e12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zou L, Feng Y, Chen YJ, Si R, Shen S, Zhou Q, et al. Toll-like receptor 2 plays a critical role in cardiac dysfunction during polymicrobial sepsis. Crit Care Med. 2010;38(5):1335–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matyas JJ, O’Driscoll CM, Yu L, Coll-Miro M, Daugherty S, Renn CL, et al. Truncated TrkB.T1-mediated astrocyte dysfunction contributes to impaired motor function and neuropathic pain after spinal cord injury. J Neurosci. 2017;37(14):3956–71. [DOI] [PMC free article] [PubMed]
- 31.Sabirzhanov B, Zhao Z, Stoica BA, Loane DJ, Wu J, Borroto C, et al. Downregulation of miR-23a and miR-27a following experimental traumatic brain injury induces neuronal cell death through activation of proapoptotic Bcl-2 proteins. J Neurosci. 2014;34(30):10055–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu YX, Yu Y, Liu JP, Liu WJ, Cao Y, Yan RM, et al. Neuroimmune regulation in sepsis-associated encephalopathy: the interaction between the brain and peripheral immunity. Front Neurol. 2022;13: 892480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shacka JJ, Klocke BJ, Shibata M, Uchiyama Y, Datta G, Schmidt RE, et al. Bafilomycin A1 inhibits chloroquine-induced death of cerebellar granule neurons. Mol Pharmacol. 2006;69(4):1125–36. [DOI] [PubMed] [Google Scholar]
- 34.Zou L, Feng Y, Xu G, Jian W, Chao W. Splenic RNA and MicroRNA mimics promote complement factor b production and alternative pathway activation via innate immune signaling. J Immunol. 2016;196(6):2788–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhuang X, Xiang X, Grizzle W, Sun D, Zhang S, Axtell RC, et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol Ther. 2011;19(10):1769–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tolomeo AM, Zuccolotto G, Malvicini R, De Lazzari G, Penna A, Franco C, et al. Biodistribution of intratracheal, intranasal, and intravenous injections of human mesenchymal stromal cell-derived extracellular vesicles in a mouse model for drug delivery studies. Pharmaceutics. 2023;15(2):548. [DOI] [PMC free article] [PubMed]
- 37.Long Q, Upadhya D, Hattiangady B, Kim DK, An SY, Shuai B, et al. Intranasal MSC-derived A1-exosomes ease inflammation, and prevent abnormal neurogenesis and memory dysfunction after status epilepticus. Proc Natl Acad Sci U S A. 2017;114(17):E3536–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li JJ, Wang B, Kodali MC, Chen C, Kim E, Patters BJ, et al. In vivo evidence for the contribution of peripheral circulating inflammatory exosomes to neuroinflammation. J Neuroinflammation. 2018;15(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hermann DM, Peruzzotti-Jametti L, Giebel B, Pluchino S. Extracellular vesicles set the stage for brain plasticity and recovery by multimodal signalling. Brain. 2024;147(2):372–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chung HY, Wickel J, Hahn N, Mein N, Schwarzbrunn M, Koch P, et al. Microglia mediate neurocognitive deficits by eliminating C1q-tagged synapses in sepsis-associated encephalopathy. Sci Adv. 2023;9(21):eabq7806. [DOI] [PMC free article] [PubMed]
- 41.Terrando N, Monaco C, Ma D, Foxwell BM, Feldmann M, Maze M. Tumor necrosis factor-alpha triggers a cytokine cascade yielding postoperative cognitive decline. Proc Natl Acad Sci U S A. 2010;107(47):20518–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cibelli M, Fidalgo AR, Terrando N, Ma D, Monaco C, Feldmann M, et al. Role of interleukin-1beta in postoperative cognitive dysfunction. Ann Neurol. 2010;68(3):360–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sharshar T, Gray F, Lorin de la Grandmaison G, Hopkinson NS, Ross E, Dorandeu A, et al. Apoptosis of neurons in cardiovascular autonomic centres triggered by inducible nitric oxide synthase after death from septic shock. Lancet. 2003;362(9398):1799–805. [DOI] [PubMed]
- 44.Semmler A, Hermann S, Mormann F, Weberpals M, Paxian SA, Okulla T, et al. Sepsis causes neuroinflammation and concomitant decrease of cerebral metabolism. J Neuroinflammation. 2008;5:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee JW, Lee YK, Yuk DY, Choi DY, Ban SB, Oh KW, et al. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation. 2008;5:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Semmler A, Okulla T, Sastre M, Dumitrescu-Ozimek L, Heneka MT. Systemic inflammation induces apoptosis with variable vulnerability of different brain regions. J Chem Neuroanat. 2005;30(2–3):144–57. [DOI] [PubMed] [Google Scholar]
- 47.Zhao RY, Wei PJ, Sun X, Zhang DH, He QY, Liu J, et al. Role of lipocalin 2 in stroke. Neurobiol Dis. 2023;179: 106044. [DOI] [PubMed] [Google Scholar]
- 48.Shin HJ, Jeong EA, Lee JY, An HS, Jang HM, Ahn YJ, et al. Lipocalin-2 deficiency reduces oxidative stress and neuroinflammation and results in attenuation of kainic acid-induced hippocampal cell death. Antioxidants (Basel). 2021;10(1):100. [DOI] [PMC free article] [PubMed]
- 49.Hong SO, Kim J, Lee S, Shin J, Choi H, Lee E, et al. Transgenic viral expression of PH-20, IL-12, and sPD1-Fc enhances immune cell infiltration and anti-tumor efficacy of an oncolytic virus. Mol Ther Oncolytics. 2023;30:301–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Y, Xiao J, Cai J, Li R, Sui X, Zhang J, et al. Single-cell immune profiling of mouse liver aging reveals Cxcl2+ macrophages recruit neutrophils to aggravate liver injury. Hepatology. 2024;79(3):589–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang H, Zhang Q, Xu M, Wang L, Chen X, Feng Y, et al. CCL2-CCR2 axis recruits tumor associated macrophages to induce immune evasion through PD-1 signaling in esophageal carcinogenesis. Mol Cancer. 2020;19(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ruan Z, Zhang D, Huang R, Sun W, Hou L, Zhao J, et al. Microglial activation damages dopaminergic neurons through MMP-2/-9-mediated increase of blood-brain barrier permeability in a Parkinson’s disease mouse model. Int J Mol Sci. 2022;23(5):2793. [DOI] [PMC free article] [PubMed]
- 53.Chen CC, Liu L, Ma F, Wong CW, Guo XE, Chacko JV, et al. Elucidation of exosome migration across the blood-brain barrier model in vitro. Cell Mol Bioeng. 2016;9(4):509–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Loonen LM, Stolte EH, Jaklofsky MT, Meijerink M, Dekker J, van Baarlen P, et al. REG3gamma-deficient mice have altered mucus distribution and increased mucosal inflammatory responses to the microbiota and enteric pathogens in the ileum. Mucosal Immunol. 2014;7(4):939–47. [DOI] [PubMed] [Google Scholar]
- 55.Sugisawa E, Kondo T, Kumagai Y, Kato H, Takayama Y, Isohashi K, et al. Nociceptor-derived Reg3gamma prevents endotoxic death by targeting kynurenine pathway in microglia. Cell Rep. 2022;38(10): 110462. [DOI] [PubMed] [Google Scholar]
- 56.Chen S, Pan J, Gong Z, Wu M, Zhang X, Chen H, et al. Hypochlorous acid derived from microglial myeloperoxidase could mediate high-mobility group box 1 release from neurons to amplify brain damage in cerebral ischemia-reperfusion injury. J Neuroinflammation. 2024;21(1):70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nam SY, Lee EJ, Kim KR, Cha BS, Song YD, Lim SK, et al. Effect of obesity on total and free insulin-like growth factor (IGF)-1, and their relationship to IGF-binding protein (BP)-1, IGFBP-2, IGFBP-3, insulin, and growth hormone. Int J Obes Relat Metab Disord. 1997;21(5):355–9. [DOI] [PubMed] [Google Scholar]
- 58.Toyama T, Hoshi T, Noguchi T, Saito Y, Matsuzawa A, Naganuma A, et al. Methylmercury induces neuronal cell death by inducing TNF-alpha expression through the ASK1/p38 signaling pathway in microglia. Sci Rep. 2021;11(1):9832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Milovanovic J, Arsenijevic A, Stojanovic B, Kanjevac T, Arsenijevic D, Radosavljevic G, et al. Interleukin-17 in chronic inflammatory neurological diseases. Front Immunol. 2020;11:947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nair RR, Mazza D, Brambilla F, Gorzanelli A, Agresti A, Bianchi ME. LPS-challenged macrophages release microvesicles coated with histones. Front Immunol. 2018;9:1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bhatnagar S, Shinagawa K, Castellino FJ, Schorey JS. Exosomes released from macrophages infected with intracellular pathogens stimulate a proinflammatory response in vitro and in vivo. Blood. 2007;110(9):3234–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Papareddy P, Tapken I, Kroh K, Varma Bhongir RK, Rahman M, Baumgarten M, et al. The role of extracellular vesicle fusion with target cells in triggering systemic inflammation. Nat Commun. 2024;15(1):1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Balusu S, Van Wonterghem E, De Rycke R, Raemdonck K, Stremersch S, Gevaert K, et al. Identification of a novel mechanism of blood-brain communication during peripheral inflammation via choroid plexus-derived extracellular vesicles. EMBO Mol Med. 2016;8(10):1162–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dickens AM, Tovar YRLB, Yoo SW, Trout AL, Bae M, Kanmogne M, et al. Astrocyte-shed extracellular vesicles regulate the peripheral leukocyte response to inflammatory brain lesions. Sci Signal. 2017;10(473):eaai7696. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No datasets were generated or analysed during the current study.