

Summary
T cell immune dysfunction is a prominent feature of chronic HIV infection. To evaluate non-specific dysfunction, a method involving both generic activation and T cell receptor (TCR) stimulation is necessary. We created a tunable artificial antigen-presenting cell (aAPC) system. This system consists of lipid bilayers on cytometry-compatible silica microbeads (5 μm). When only anti-CD3 is incorporated, T cell activation is limited. Introducing anti-CD28 agonists significantly elevates the cytokine expression and upregulation of activation-induced markers. CD28 co-stimulation modulates the response profile, preferentially promoting IL-2 expression relative to other cytokines. aAPCs-stimulated CD4+ and CD8+ T cells from untreated HIV-infected individuals exhibit altered effector functions and diminished CD28 dependence. These functions are skewed toward TNFα, IFNγ and CD107a, with reduced IL-2. Antiretroviral therapy partially normalizes this distorted profile in CD4+ T cells, but not in CD8+ T cells. Our findings show T cell intrinsic biases that may contribute to persistent systemic T cell dysfunction associated with HIV pathogenesis.
Subject areas: Immunology, Virology
Graphical abstract
Highlights
-
•
The aAPC system induces robust cytokine production in CD4+ and CD8+ T cells
-
•
CD28 co-stimulation enhances T cell function in a cytokine-dependent manner
-
•
Untreated people with HIV show skewed functions: increased TNF⍺ and reduced IL-2
-
•
Antiviral therapy partially corrects dysfunction in CD4+, but not in CD8+ T cells
Immunology; Virology
Introduction
Optimal T cell activation requires two signals delivered during the interaction between T cells and antigen-presenting cells (APCs). Signal 1 occurs when the T cell receptor (TCR) binds to its cognate antigen presented on a major histocompatibility complex (MHC) class I or class II molecule. Signal 2 is provided by co-stimulatory molecules on T cells, which are engaged by their APC-expressed ligands.1,2 The CD28-B7 axis is the most extensively studied co-stimulatory pathway (reviewed in3,4). The combined effects of signals 1 and 2 activate transcriptional factors governing various “differentiation programs,” each defining a set of functions, such as T cell proliferation and survival, cytokine production, or differentiation.5,6,7,8 It is now evident that the role of CD28 extends beyond enhancing TCR signaling, as it induces distinct transcriptional and epigenetic changes.9,10,11
Systemic chronic activation is a hallmark of progressive HIV infection.12,13 Prolonged exposure to HIV peptides leads to the dysregulation of HIV-specific CD4+ and CD8+ T cells, altering T cell polarization and effector functions.14,15,16,17,18,19,20,21,22 These dysfunctional responses usually fail to control viral replication.22,23 T cell exhaustion is associated with persisting expression of immune checkpoint inhibitory receptors (IR), which exert negative feedback control on the TCR signal.23,24,25 Modulation of CD28 activity plays a pivotal role in this negative feedback loop, as PD-1-associated SHP-2 preferentially dephosphorylates the cytosolic domain of CD28.26 A broader state of T cell dysfunction also exists in non-HIV-specific cells, attributed to systemic activation,27,28 co-infection by other pathogens,29,30,31 and microbial translocation caused by compromised mucosal barrier integrity.32 This generic dysfunction may contribute to co-morbidities and reduced life expectancy in people with HIV (PWH).33 While decreased expression of CD28 on CD8+ T cells is inversely correlated with CD4+ T cell count prior to ART initiation,34 it remains uncertain whether this overall dysfunction is associated with modified responsiveness to TCR and CD28 stimulation.
Stimulation with cognate antigenic peptides loaded onto natural APCs is commonly used to study TCR-mediated T cell functions. This approach provides insights into T cells sharing a defined specificity, but it does not provide information about the overall state of T cells. Furthermore, as natural APCs express an array of other co-stimulatory and inhibitory receptors, simplified model systems are needed to accurately analyze TCR- and CD28-mediated activation. Agonistic anti-CD3 (aCD3) and anti-CD28 (aCD28) antibodies are commonly used to non-selectively activate purified T cells, mimicking TCR and co-stimulation signals, respectively. Commercially available activation systems include crosslinked soluble aCD3 and aCD28 or immobilization of at least one of the antibodies on either plate-, planar-, or bead-based solid supports.35,36,37,38,39,40 Previous studies indicated that having cell-sized constructs (4.5–5 μm)41,42 and a fluid surface allowing the rearrangement of T cell ligands on the artificial APC (aAPC)43,44 leads to enhanced T cell activation. Current embedded agonists on supported lipid membranes were effective for expanding T cells, but required days of stimulation, which complicates phenotyping.45,46,47,48,49
In this study, we developed a customizable artificial antigen-presenting cells (aAPCs) system composed of a fluid lipid bilayer coated onto cytometry-compatible silica microbeads (5 μm) and embedded with agonistic antibodies directed against CD3 and CD28. This system enables strong T cell activation within 15 h. We employed these aAPCs to elucidate the role of CD28 co-stimulation in shaping the CD4+ and CD8+ T cell effector functional profile in uninfected control donors (UD). We investigated how this profile changes in PWH, both before (UNT) and after antiretroviral treatment (ART).
Results
Artificial antigen-presenting cells induce robust cytokine production in CD4+ and CD8+ T cells
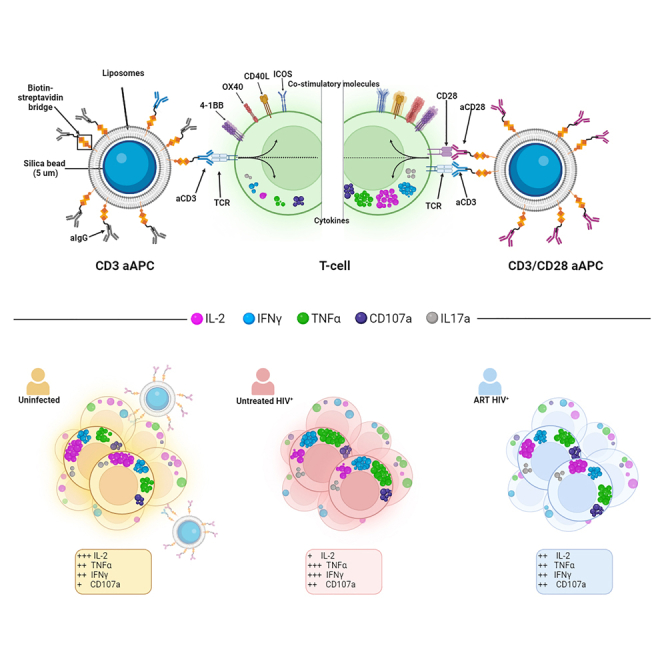
We developed a T cell activation system consisting of silica beads coated with a membrane bilayer to mimic the physiological TCR-mediated T cell activation. This design imitates key features of natural APCs including a similar size (5 μm diameter), a tridimensional structure, and a fluid lipid bilayer that facilitates the clustering of activating ligands. As “artificial antigen-presenting cells (aAPCs)” was a term previously used to describe such a three-dimensional system replicating some aspects of natural APC-mediated activation, we adopted this expression herein for our newly designed activation platform.50,51 The preparation of aAPCs (Figure 1A) involved generating unilamellar liposomes containing biotin acceptor sites through thin film hydration. Bilayered liposomes were formed by extrusion and used to coat the silica beads. Biotin-conjugated anti-CD3 and/or anti-CD28 agonistic antibodies were added via streptavidin-biotin ligation.
Figure 1.

aAPCs induce a robust cytokine production in CD4+ and CD8+ T cells
(A) Schematic representation of aAPCs preparation.
(B) Representative flow cytometry plots of cytokines expression in CD4+ and CD8+ T cells after 15h of aAPCs (aCD3:aCD28:IgG 1:4:0) stimulation of purified T cells from UD. The unstimulated condition represents T cells incubated with control IgG aAPCs (aCD3:aCD28:IgG 0:0:5).
(C) Diagram explaining the OR Boolean gating. For simplicity, only IL-2 and TNFα in CD4+ T cells are shown as a conceptual example.
(D and E) Total cytokine+ (D) CD4+ or (E) CD8+ T cells defined by the ORgate, in 12 UD. Wilcoxon test results are shown. Medians are indicated below the graphs. Error bars represent the interquartile range.
We assessed the potency of aAPCs at stimulating CD4+ and CD8+ T cells isolated from PBMCs of uninfected donors (UD, Table S1). Following purification, T cells were stimulated for 15h with aAPCs coated with aCD3 and aCD28 antibodies at a 4:1 bead-to-cell ratio. Subsequently, an intracellular cytokine staining assay (ICS) was conducted to detect IL-2, IFNγ, TNFα, CD107a, and IL17A. The aAPCs were co-analyzed alongside the T cells (Figure S1A). The successful coating with aCD3 and aCD28 antibodies was verified through anti-IgG staining (Figure S1B). Upon stimulation with aAPCs, a considerable upregulation of cytokines was observed in both CD4+ and CD8+ T cells (Figure 1B). IL17A was not detectable in CD8+ T cells and was consequently excluded from subsequent CD8 analyses (Figure 1B). The extent of activation achieved through aAPCs was close to the level attained through the superantigen staphylococcal enterotoxin B (SEB) (Figure S1C). Of note, SEB-mediated activation was performed in non-purified PBMCs to adequately cross-link MHC-II and TCR molecules and ensure superior activation (Figure S1D).
We assessed the frequencies of CD4+ and CD8+ T cells expressing at least one cytokine. Each cytokine was individually gated, then subjected to ORgate Boolean gating.52 This approach creates a cytokine+ population that includes all cells within IL-2+, IFNγ+, TNFα+, CD107a+ gates. Additionally, the use of the ORgate Boolean gating prevents the redundant counting of cells that simultaneously co-express several effector molecules, which would otherwise occur with a simple summation (Figure 1C). This approach preserved all the possible cytokine combinations, such as single positive cytokine+ cells as well as cell positives for several or all cytokines. Employing this strategy, we determined that a median of 45.8% of CD4+ and 22.9% of CD8+ T cells exerted 1 effector functions upon activation by aAPCs (Figures 1D and 1E). The embedding of antibodies into the lipid-bilayered silica bead was important, given that equivalent concentrations of aCD3 and aCD28 in solution or cross-linked with goat anti-mouse IgG only yielded low activation of CD4+ and CD8+ T cells (Figure S1E). We also compared our system to commercially available magnetic CD3+CD28 beads (Figure S1F). Although magnetic bead-mediated stimulation appeared twice more efficient when gated on valid events, it came at the expense of an alteration of some cell surface marker expression, much higher cell death, and drastically inferior yield of T cell recuperation after stimulation (Figures S1G–S1I). By comparison, aAPCs provided a robust activation signal that nevertheless preserved cell viability and surface CD4 and CD8 expression (Figures S1J and S1K). The engagement of CD3 and CD28 with their agonistic antibodies resulted in their partial downregulation from the surface of both CD4+ and CD8+ T cells owing to strong stimulation cues (Figures S1J and S1K).
These data show that our devised aAPC system, utilizing silica bead-coated bilayers to emulate physiological TCR-mediated activation, robustly induces cytokine expression in both CD4+ and CD8+ T cells while preserving the viability of the cells.
CD28 co-stimulation differentially enhances individual effector functions in CD4+ and CD8+ T cells
We employed aAPCs to analyze the quantitative contributions of aCD3 and aCD28 to T cell activation. To empirically establish the optimal aCD3/aCD28 ratio in the aAPC system, we initially conducted dose-response experiments (Figures S2A and S2B). Increasing the concentration of aCD28 on aAPCs did not compensate for the absence of aCD3. In the absence of aCD28, increasing the aCD3 concentration had little effect, suggesting that CD28 co-stimulation is necessary for optimal activation. In the presence of aCD28, a high concentration of aCD3 proved detrimental. The optimal activation was empirically determined to be at a 1-to-4 aCD3-to-aCD28 molecular ratio. This ratio was selected for subsequent experiments unless otherwise specified.
To evaluate the importance of CD28 co-stimulation for distinct effector functions, we generated 5 sets of aAPCs: 1) negative control lipobeads loaded with non-activating isotype control IgG, and aAPCs containing agonistic antibodies targeting 2) CD3 only, 3) CD28 only, 4) CD3 and CD28 at equivalent but suboptimal molarity, and 5) CD3 and CD28 at the optimal 1-to-4 molecular ratio. Isotype IgG was supplemented onto suboptimal aAPCs to reach an overall antibody density equivalent to that of the optimal 1-to-4 condition. Purified T cells from 12 UD were stimulated with these sets of aAPCs, and total frequencies of cytokine-producing cells were quantified using OR Boolean combination gating (Figures S2C and S2D). Net total responses were calculated after subtracting the background signal (Figures 2A–2D).
Figure 2.

CD28 co-stimulation differentially enhances effector functions in T cells
Univariate analyses of cytokine expression in CD4+ and CD8+ T cells after 15h of aAPCs stimulation of purified T cells from UD participants. Unless specified otherwise, the 1:4:0 (aCD3:aCD28:IgG) is presented.
(A–D) ORgate-identified net cytokine+ responses after background subtraction. Net ORgate cytokine+ (A) CD4+ and (B) CD8+ T cells responses to aAPCs stimulation with different aCD3:aCD28 ratios in purified T cells. Individual net cytokine production in (C) CD4+ and (D) CD8+ T cells when stimulated with aCD3 aAPCs (aCD3:aCD28:IgG 1:0:4) compared to aCD3+aCD28 aAPCs (aCD3:aCD28:IgG 1:4:0). Wilcoxon test results are shown.
(E and F) Percentage of residual cytokine+ signal, representing the quotient between the 1:0:4 (aCD3:aCD28:IgG) and 1:4:0 (aCD3:aCD28:IgG) conditions in (E) CD4+ and (F) CD8+ T cells.
(G and H) Median fluorescence intensity (MFI) of each cytokine expression in (G) CD4+ and (H) CD8+ T cells when stimulated with aCD3 aAPCs (aCD3:aCD28:IgG 1:0:4) compared to aCD3+aCD28 aAPCs (aCD3:aCD28:IgG 1:4:0). The analyses focus the cytokine+ cells indicated by the subtitles.
(I) Gating strategy defining memory subsets. The populations boxed in colors were further analyzed for CD27 expression, on the right.
(J-K) Percentage of residual ORgate cytokine+ signal in CD4+ and CD8+ memory subsets (as calculated in E,F). In (A–F, J and K), n = 12. Friedman test was used in (A, C, E, F, J, K). In (A–F, J, and K), error bars represent the interquartile range.
In CD4+ T cells, activation with aCD3 alone or aCD28 alone resulted in net responses of low magnitude (Figure 2A), which were still significant compared to negative IgG bead controls (Figure S2C). The signal modestly increased at the suboptimal 1:1 aCD3:aCD28 ratio, and substantially augmented at the optimal 1:4 (aCD3:aCD28) ratio, reaching a 69-fold rise compared to aCD3 alone. Compared to CD4+ T cells, aCD3 alone was slightly more effective in activating CD8+ T cells (Figure 2C). The benefit of co-stimulation was apparent only at the 1:4 M ratio, with a median-fold increase of 9.6 when compared to aCD3 alone. Positive impacts of CD28 co-stimulation were observed for each examined cytokine in both CD4+ and CD8+ T cells (Figures 2B and 2D).
To better illustrate the effect of CD28 co-stimulation on effector functions at the populational level, we calculated the residual signal in the absence of CD28 co-stimulation: ([cytokine+ cells activated by aCD3 alone]/[cytokine+ cells activated by 1:4 (aCD3:aCD28)] X 100) (Figures 2E and 2F). A low residual signal indicates a high dependence on CD28 co-stimulation, whereas a comparatively high residual signal suggests a certain degree of independence from CD28 co-stimulation. These internally controlled side-by-side comparisons allow the ranking of cytokines based on their dependence on CD28. In CD4+ T cells, the expression of IL-2, IFNγ, and TNFα was markedly reduced without CD28. In contrast, IL17A and CD107a were less affected by the lack of CD28 co-stimulation (Figure 2E). CD28 co-stimulation dependency was more heterogeneous in CD8+ T cells. Although a similar hierarchy existed, CD8+ T cells depended less on CD28 co-stimulation than CD4+ T cells for all effector functions, except CD107a (Figure S2E).
CD28 co-stimulation also increased cytokine expression at the single-cell level. Median fluorescence intensities of IL-2, IFNγ, TNFα, and IL17A all increased in the 1:4 (aCD3:aCD28) condition compared to aCD3 alone (Figures 2G, 2H, S2F, and S2G). Notably, only the expression of CD107a remained unchanged in CD4+ and CD8+ T cells. These data show the critical importance of CD28 to fully express cytokines.
Effector functions elicited in CD4+ and CD8+ T cells can vary with memory differentiation.53,54 We, therefore, investigated whether this variability could lead to differing dependence on CD28 co-stimulation. Memory subsets were categorized as follows: naive T cells (TN, CCR7+CD45RA+), central memory (TCM, CCR7+CD45RA−), transitional memory (TTM, CCR7−CD45RA−CD27+), effector memory (TEM, CCR7−CD45RA−CD27−), and terminally differentiated memory (TEMRA, CCR7−CD45RA+) (Figure 2I). There were no disparities in memory subset proportions after stimulation with aCD3+aCD28 aAPCs (Figures S2H and S2I). We evaluated total cytokine+ cells in each subset using OR Boolean combination gating, then calculated the residual signal in the absence of CD28 co-stimulation (Figures 2J and 2K). In CD4+ T cells, TN exhibited the greatest reliance on CD28 co-stimulation. Across CD4+ T memory subsets, we observed a hierarchy for dependence on CD28, wherein the need for co-stimulation diminished as subsets became more differentiated: TN > TCM > TTM> TEM> TEMRA (Figure 2J). CD8+ T cell subsets displayed a three-step hierarchy in which TN and TCM were both strongly reliant on CD28, whereas TTM and TEM and moderately dependent on CD28. As for CD4+ T cells, CD8 TEMRA were the least dependent on CD28. CD8 T cells required less co-stimulation overall than CD4 (Figures 2K and S2J). We tested whether independence from CD28 co-stimulation was linked to decreased CD28 expression in certain memory subsets. We observed that CD28 was expressed in more than 99% of TN, TEM, TTM and TEM CD4 T cell subsets. This proportion dropped to a still high median of 82.5% in TEMRA. There was more variation among subsets in CD8 T cells, with the highest proportion of CD28+ in TCM (median of 100%) and lowest in TEMRA cells (median of 36.6%) (Figure S2K). We next measured the median fluorescence intensity to infer the level of surface CD28 expression per CD28+ cell (Figure S2L). The level of surface CD28 expression was similar across the differentiated subsets (TEM, TTM, TEM and TEMRA), and slightly lower in CD4 and CD8 TN (Figure S2M). Importantly, TCM had the same proportion of CD28+ cells, and expressed the same level of surface CD28 compared to the least CD28 dependent TEM in both CD4 and CD8 T cells. Therefore, independence from CD28 co-stimulation cannot be solely attributed to a lack of CD28 expression in certain differentiated subsets.
These results demonstrate the substantial impact of CD28 co-stimulation on the enhancement of T cell effector functions across CD4+ and CD8+ subsets, underscoring the differential dependence on CD28 co-stimulation across varying memory differentiation stages.
The heightened responsiveness of IL-2 to CD28 co-stimulation shapes the overall effector profile
Given the asymmetric impact of CD28 co-stimulation on cytokine expression, we investigated whether CD28 could act as a determinant shaping the overall effector profile. To avoid predefined marker combinations, we conducted unsupervised analyses utilizing the high-dimensional flow cytometric phenotyping data (Figure 3). We examined cells activated with different aCD3:aCD28:IgG ratios (1:0:4 vs. 1:4:0). We first identified aAPC-reactive cytokine+ cells of both CD4+ and CD8+ T cells using the ORgate method, then downsampled these cells to 1000 per participant per condition, and subsequently utilized the Uniform Manifold Approximation and Projection (UMAP) algorithm55 to visualize the various clusters of CD4+ and CD8+ T populations exerting effector functions. The identification of effector function clusters was performed using Phenograph.56 As the goal of this analysis was to draw an effector function landscape rather than a phenotypic profiling, only cytokine parameters were loaded. The MFI from CD4 and CD8 markers were therefore not included in the UMAP calculation and did not contribute to the clustering. CD4+ and CD8+ cells could be identified post-hoc using categorical labels. Our approach generated 17 effector CD4 and CD8 T cell co-clusters (Figure 3A). The classification of clusters was based on distinct cytokine profiles indicated by fluorescent intensities (Figures 3B and S3A). Further qualification of the identified clusters was based on the percentage of cells expressing the different cytokines (Figure 3C).
Figure 3.

Responsiveness of IL-2 to CD28 co-stimulation shapes the effector profile
Multivariate analyses of cytokine expression in CD4+ and CD8+ T cells after 15h of aAPCs stimulation of purified T cells from UD participants. The 1:4:0 (aCD3:aCD28:IgG) and 1:0:4 (aCD3:aCD28:IgG) are herein qualitatively compared. Cytokine+ CD4 and CD8 T cells are identified by Boolean ORgate gating, downsampled to 1000 cells per participant per condition, concatenated, and then analyzed as follows.
(A) Multiparametric UMAP representation of cytokine+ CD4+ and CD8+ activated T cells from UD, based on IL-2, IFNγ, TNF⍺, CD107a and IL17A expression at two stimulation conditions (aCD3:aCD28:IgG): 1:0:4 and 1:4:0. The categorical CD4+ and CD8+ phenotypic information was reintroduced post-hoc.
(B) Heatmap summarizing the MFI of each parameter for each cluster.
(C) Proportion of cells expressing individual functions in each cluster, using the ORgate as the denominator and combining CD4 and CD8 T cells irrespective of the condition of stimulation; in support of panel B.
(D and E) Proportion of each cytokine+ cluster for (D) CD4+ and (E) CD8+ T cells in uninfected participants. Wilcoxon paired tests are shown to depict the relative changes in cluster composition upon CD28 co-stimulation, compared to aCD3 alone. In (A–E), n = 12.
This analysis revealed six clusters primarily composed of monofunctional cells (C4, C6, C8, C14, C15, C16), while the remaining clusters displayed varying degrees of polyfunctionality (Figure 3C). No cluster could be solely attributed to either the CD4 or the CD8 subset, or the presence or absence or CD28 co-stimulation. The most distinctive clusters between CD4+ and CD8+ T cells were C4, a mostly CD4+ T cell cluster expressing only TNF, and C10, a mostly CD8+ T cell cluster co-expressing IL-2 and IFNγ but not TNF. In CD4+ T cells, IL-2 and IFNγ expression was frequent, and typically associated with TNF co-expression (Figures 3B–3E).
Although the main impact of CD28 co-stimulation was quantitative (Figures S3B and S3C), its relative influence varied among clusters, resulting in three distinct behaviors in CD4+ T cells (Figure 3D): i) clusters whose relative importance declined with CD28 co-stimulation. These clusters (C1, C3, C4, C5, C6, C8, C11, C14) exhibited minimal or no IL-2 expression. These clusters were otherwise characterized by various degrees of TNFα, IFNγ or CD107a co-expression; ii) status quo, with clusters (C2, C7, C10, C15, C17) maintaining consistent proportions with or without CD28 co-stimulation. These clusters were characterized by a high degree of polyfunctionality, excepted for the cluster C15 that exerted only the IL-2 effector function; and iii) clusters enriched after co-stimulation with CD28. These clusters (C9, C12, C13, C16) expressed IL-2 and TNFα, or predominantly IL-2. CD28-mediated co-stimulation did not systematically increase polyfunctionality, as several polyfunctional clusters (C3, C5, C11) proportionally decreased in relative importance with CD28. Overall, similar observations were made on CD8+ T cells, with minor cluster-specific differences (Figures 3E and S3C).
Principal component analyses (PCA) of T cell responses based on the proportions of these clusters in CD4+ (Figure S3D) and CD8+ (Figure S3E) T cells, illustrated the substantial effect of CD28 co-stimulation on the effector function profile. Therefore, CD28 serves a dual role by enhancing the potency of CD3-mediated TCR activation and fine-tuning the nature of the elicited effector functions.
CD28 co-stimulation enhances expression of second wave co-stimulatory molecules
In addition to CD28, a repertoire of other T cell co-stimulatory molecules exists, including CD69, CD40L, OX40, 4-1BB and ICOS. In contrast to the constitutive expression of CD28, these molecules typically undergo upregulation upon activation.57,58,59,60,61 Our investigation aimed to assess the role of CD28 in their upregulation. T cells from uninfected control donors were stimulated with aAPCs for 15h, followed by surface marker staining and subsequent analysis by flow cytometry.
CD69 is a widely employed surface marker to monitor T cell activation due to its rapid and robust upregulation after T cell activation.62,63 Notably, around one-third of CD4+ T cells already expressed low levels of CD69 before stimulation (Figure 4A). This proportion increased to 73% upon exposure to aAPCs conjugated solely with aCD3, with further increase with the optimal 1:4 (aCD3:aCD28) aAPCs, reaching >95% of CD4+ (Figure 4A) and CD8+ (Figure S4A) T cells. To increase the accuracy of activated cell detection following aAPC stimulation, we analyzed CD69 in conjunction with other activation markers using an AND/OR Boolean strategy, an approach validated in previous studies.64,65 Consequently, co-expression of CD40L, OX40, 4-1BB, and ICOS with CD69 was quantified in CD4+ and CD8+ cells. Each activation marker was upregulated after aAPC stimulation, albeit to varying degrees (Figures 4B and S4B). Background signal was negligible (Figures S4C and S4D). We detected only minimal or no induction of T cell activation markers after stimulation with aAPCs incorporating solely aCD3 or aCD28 (Figures 4C–4F). Equimolar addition of CD28 (1:1 ratio of aCD3 to aCD28) marginally increased the signal (Figures 4C and 4D). In contrast, the proportion of activated cells sharply increased at the 1:4 (aCD3:aCD28) ratio, reaching a median of 62.6% in CD4+ (Figure 4C) and 41.1% in CD8+ (Figure 4D). This increase with co-stimulation was observed for all tested activation markers (Figures 4E and 4F).
Figure 4.

CD28 co-stimulation enhances the expression of co-stimulatory molecules
Univariate analyses of co-stimulatory marker expression in CD4+ and CD8+ T cells after 15h of stimulation of purified T cells from UD participants, using aCD3 aAPCs or aCD3 aCD28 aAPCs.
(A and B) Representative flow cytometry plots of purified CD4+ T cells stimulated with indicated aAPCs. (A) CD69 expression. (B) Co-expression of CD69 and other activation molecules.
(C–F) Background-subtracted net responses of CD4+ and CD8+ T cells expressing co-stimulation molecules (CD40L, OX40, 4-1BB, ICOS). Global responses (OR-gate identified cells expressing one or more co-stimulation molecules) for (C) CD4+ and (D) CD8+ T cells are shown, then subdivided individually for each tested co-stimulation molecule for (E) CD4+ and (F) CD8+.
(G and H) Percentage of residual cytokine+ signal, representing the quotient between the 1:0:4 (aCD3:aCD28:IgG; aCD3 aAPCs) and 1:4:0 (aCD3:aCD28:IgG; aCD3+aCD28 aAPCs) conditions in (G) CD4+ and (H) CD8+ T cells. In (C–H), n = 11. Friedman tests are shown below the histograms in (C, D, G, H). In (C–H), error bars represent the interquartile range. Gray borders represent responses <0.1% (in C and D) or <0.01% (in H) that could not be represented on the logarithmic scale and hence were given arbitrary values of 0.1% or 0.01% respectively.
As for the ICS assay, we calculated the residual signal in the absence of aCD28. In CD4+ T cells, CD40L exhibited the highest dependency on CD28 co-stimulation followed by OX40, and ICOS. At the other end of the spectrum, 4-1BB and CD69 displayed greater independence from CD28 (Figure 4G). A comparable hierarchy was observed in CD8+ T cells (Figure 4H). CD8+ T cells exhibited less reliance on CD28 co-stimulation than in CD4+ T cells across all markers except for CD40L, which was poorly expressed on CD8+ T cells (Figure S4E). On a per-cell basis (median FI), CD28 co-stimulation increased the expression levels of CD40L, OX40, ICOS, and CD69, but not 4-1BB (Figures S4F and S4G).
In summary, these results reveal that activation markers have varying levels of dependency on CD28 co-stimulation, from stringent (OX40, CD40L, ICOS), to moderate (4-1BB), and low (CD69).
During viremic human immunodeficiency virus infection, the reliance on CD28 co-stimulation is heightened for IL-2, but diminished for interferon γ and TNFα
Inefficient CD28 co-stimulation has been reported during chronic viral infections.26,66 Therefore, we sought to determine whether the dependence on CD28 co-stimulation was altered in the context of chronic HIV infection. We examined a cohort of 13 people with chronic HIV who had not yet initiated antiretroviral therapy (UNT) (Figure S5A); clinical characteristics are provided in Table S1.
Although not reaching significance, the stimulation with aCD3 aAPCs showed trends for higher levels of cytokine expression in both CD4+ and CD8+ T cells of UNT individuals compared to UD (Figures 5A, 5B, S5B, and S5C). Conversely, UNT’s cytokine expression with CD28 co-stimulation showed trends for lower cytokine expression than those observed in UD (Figures S5B and S5C). We observed consistent results after correcting for the depletion of CD4+ T cells, improving also the significance of the observation (Figures S5D and S5E). Consequently, these trends translated to a lower dependence for CD28 co-stimulation in UNT compared to UD people (Figures 5C and 5D). Notably, these findings were cytokine-dependent; while IFNγ and TNF⍺ were significantly less reliant on CD28 co-stimulation in UNT donors, IL-2 demonstrated an opposing pattern, being more dependent on CD28 co-stimulation in UNT than in UD (Figures 5E and 5F).
Figure 5.

Cytokine-specific dependence on CD28 co-stimulation in untreated HIV infection
Univariate analyses of cytokine expression in CD4+ and CD8+ T cells after 15h of aAPCs stimulation of purified T cells from UD versus UNT participants. Unless specified, the ratios presented are 1:4:0 (aCD3:aCD28:IgG; aCD3 aCD28 aAPCs).
(A and B) ORgate-identified, background subtracted net (A) CD4+ and (B) CD8+ cytokine+ T cell responses from UNT after aAPCs stimulation. Friedman test was used.
(C and D) Comparison of residual signal in the absence of CD28 co-stimulation for cytokine+ (C) CD4+ or (D) CD8+ T cells in UD versus UNT participants.
(E and F) Comparison of residual signal in the absence of CD28 co-stimulation in (E) CD4+ or (F) CD8+ T cells between UD and UNT for each individual cytokine. In (C-F), Mann-Whitney test results are shown. In (A–F), n = 12. In (A-F), error bars represent the interquartile range. In (A) and (E), gray borders represent responses <0.1% that could not be represented on the logarithmic scale and hence were given arbitrary values of 0.1%.
We examined whether alterations in CD28 dependence were linked to changes in memory differentiation subsets (Figures S5F and S5G). In CD4+ T cells from UNT, there were significantly lower frequencies of TTM (Figures S5H). In CD8+ T cells, TEM and TEMRA were elevated in UNT, whereas TCM was concomitantly less frequent (Figures S5I). The increased dependence on CD28 co-stimulation for IL-2 in UNT was observed in all memory subsets. In contrast, the reduced dependence of IFNγ and TNF⍺ on co-stimulation was specifically observed in the TEM and TEMRA subsets of UNT participants (Figures S5J and S5K).
These results reveal altered patterns of the requirement for CD28 co-stimulation in the context of chronic HIV infection, with cytokine-specific variations and memory subset influences.
CD4+ and CD8+ T cells from untreated people with human immunodeficiency virus exhibit effector functions skewed toward TNFα, interferon γ, and CD107a, and diminished IL-2 production
We employed an unsupervised approach to evaluate how the altered dependence on CD28 co-stimulation influenced the overall functional profile of CD4+ and CD8+ T cells during viremic HIV infection. Cytokine+ CD4+ and CD8+ T cells from UNT, as defined in the ORgate analysis, were overlaid onto the UMAP analyses from Figure 3 (Figures 6A and 6B). We next compared the proportions of the aforementioned 17 identified clusters in UD and UNT participants. With regard to UNT vs. UD comparisons, these clusters displayed three distinct patterns (Figure 6C): i) clusters enriched in UNT compared to UD (C1, C2, C3, C4, C5, C6, C8. C11, C13. C14). These clusters expressed IFNγ, TNF⍺ and/or CD107A, but not IL-2; ii) clusters similarly represented in both cohorts (C7, C16, C17); iii) clusters enriched in UD (C9, C10, C12, C15). These clusters were all characterized by IL-2 expression. CD8+ T cell clusters exhibited more clusters enriched in UD, with additional IL-2+ expressing clusters (C13, C16, C17) compared to CD4+ T cells (Figure 6D). Notably, C5, characterized by IL17A expression, was similarly represented between cohorts in CD8+ but higher in UNT’s CD4+ T cells. Globally, the clusters that are enriched in UNT compared to UD (C1, C2, C3, C4, C6, C8, C11, C14 for CD4 and CD8, and C5, C13 for CD4; C2 and C13 are the only exceptions) coincided with those that proportionally declined in the absence of CD28 co-stimulation. The overrepresentation of clusters less dependent on CD28 co-stimulation may thus represent a hallmark of the CD28 co-stimulation defect reported in UNT.
Figure 6.

Untreated PWH’s CD4+ and CD8+ T cells show skewed effector functions
Multivariate analyses of cytokine expression in CD4+ and CD8+ T cells after 15h of aAPCs stimulation of purified T cells at the 1:4:0 (aCD3:aCD28:IgG; aCD3+aCD28 aAPCs) ratio. UD and UNT participants are herein qualitatively compared. Cytokine+ CD4 and CD8 cells are identified by Boolean ORgate gating, downsampled to 1000 cell per participant per condition, concatenated, and then analyzed as follows.
(A) Multiparametric UMAP representation of cytokine+ CD4+ and CD8+ T cells from UD or UNT, based on IL-2, IFNγ, TNF⍺, CD107a, and IL17A expression after stimulation with aAPCs (aCD3:aCD28:IgG 1:4:0). The colors identify the same 17 populations clustered by unsupervised analysis in Figure 3. The CD4+ and CD8+ categorical phenotypic information was reintroduced post-hoc.
(B) Heatmap summarizing the MFI of each parameter for each cluster (replicate of Figure 3B panel).
(C and D) Proportion of each cluster in the total, ORgate-defined cytokine+ population of (C) CD4+ T cells and (D) CD8+ T cells.
(E and F) Global PCA analysis (on the left) was done with the proportion cytokine+ (E) CD4+ or (F) CD8 + activated T cell clusters distinguishing UD (yellow) and UNT (red) participants. The percentage on the x and y axes represents the variance attributed to PC1 and PC2, respectively. On the right, histograms depicted the PC1 and PC2 values between the groups are shown.
(G and H) Spearman correlations between each individual cytokine and the PC1 values (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001) for (G) CD4 and (H) CD8. In (C–F), Mann-Whitney test results are shown. In (A–H), UD n = 12, UNT n = 13. In (C–F), error bars represent the interquartile range.
To further examine the alterations in functional profile during HIV infection, we integrated the proportions of CD4+ and CD8+ T cell clusters from UNT and UD controls in a principal component analysis (PCA) (Figures 6E and 6F). For both CD4+ and CD8+ T cells, we observed a clear and statistically significant separation of the two cohorts along the PC1 axis. To identify the cytokines driving this segregation, we correlated PC1 with the univariate percentage of each cytokine+ T cell response (Figures 6G, 6H, S6A, and S6B). CD107a, IL17A, IFNγ, and TNF⍺ all positively correlated with PC1 (or showed a trend in the case of TNF⍺ in CD4+), showing that the increased expression of these cytokines in UNT drove the observed separation (Figures 6G and 6H). Conversely, IL-2 correlated negatively with PC1, indicating that the diminished expression of this cytokine is a distinctive feature of UNT (Figures 6G and 6H). These findings were corroborated by univariate analyses, as we noted a lower proportion of IL-2+ CD4+ (Figure S6C) and CD8+ (Figure S6D) T cells in UNT samples. Conversely, the proportion of IFNγ+, TNF⍺+ and CD107a+ CD4+ and CD8+ T cells was higher in UNT than UD.
In summary, unsupervised integration of cytokine-positive T cell responses reveals distinct patterns enriched in UNT. Principal component analysis highlights the significant divergence between UNT and controls, driven by elevated IFNγ, TNFα, and CD107a alongside reduced IL-2 expression.
Initiation of antiretroviral treatment in people with human immunodeficiency virus partially normalizes cytokine profiles in CD4+ T cells, with a limited impact on CD8+ T cells
Next, we investigated whether ART could rectify the skewed functional profile observed during viremic HIV infection. We performed pairwise comparisons for participants with longitudinal follow-up and available pre- and post-ART leukapheresis samples (Table S1 and Figures S7A–S7C). The pairwise comparisons did not reveal statistically significant changes in global CD4+ and CD8+ cytokine T cell responses after ART, either with aCD3 alone or with CD28 co-stimulation (Figures 7A and 7B), beside slightly increased cytokine-positive CD4+ T cell response to CD3 and CD28 co-stimulation in ART (Figures S7D and S7E). The residual cytokine response without CD28 was globally comparable before and after treatment (Figures 7C and 7D). This also held true for each individual cytokine assessed (Figures 7E and 7F).
Figure 7.

ART initiation in PWH partially normalizes cytokine profiles in CD4+ T cells, with limited impact on CD8+ T cells
(A–F) Univariate analyses of cytokine expression in CD4+ and CD8+ T cells after 15h of aAPCs stimulation of purified T cells from UNT versus ART participants. (A and B) Comparison of total, ORgate-identified, background-subtracted, net cytokine+ (A) CD4+ or (B) CD8+ T cell responses in the UNT and ART cohorts after activation by aAPCs. Both 1:0:4 (aCD3:aCD28:IgG; aCD3 aAPCs, left panel) and 1:4:0 (aCD3:aCD28:IgG; aCD3+aCD28 aAPCs, right panel) stimulations are shown.
(C and D) Comparison of residual signal in the absence of CD28 co-stimulation of total cytokine+ (C) CD4+ or (D) CD8+ T cells between UNT and ART.
(E and F) Comparison of residual signal in the absence of CD28 co-stimulation in CD4+ (E) or CD8+ (F) T cells between UNT and ART for each individual cytokine.
(G–K) Multivariate analyses of cytokine expression in CD4+ and CD8+ T cells after 15h of 1:4:0 (aCD3:aCD28:IgG; aCD3 aCD28 aAPCs) stimulation of purified T cells from UNT versus ART participants, following the same pipeline analyses as in Figures 3 and 6. The CD4+ and CD8+ categorical phenotypic information was reintroduced post-hoc. (G) Multiparametric UMAP representation of cytokine+ CD4+ and CD8+ T cells from ART, based on IL-2, IFNγ, TNFα, CD107a, and IL17A expression after stimulation with aAPCs (aCD3:aCD28:IgG 1:4:0). The colors identify 17 populations clustered by unsupervised analysis using Phenograph. (H and I) Proportions of clusters among cytokine+ (H) CD4+ and (I) CD8+ T cells in UNT vs. ART.
(J and K) Global PCA analysis (on the left) was done with the proportion of clusters in cytokine+ (J) CD4+ or (K) CD8 + activated T cells. The percentage on the x and y axes presents the variance attributed to PC1 and PC2, respectively. On the right, plots of the PC1 and PC2 between groups are shown. In (A–K), UNT n = 13, ART n = 13. Wilcoxon test was used for paired comparisons between UNT and ART. In (A), gray borders represent responses <0.01% that could not be represented on the logarithmic scale and hence were given arbitrary values of 0.01%.
Given that CD28 expression may influence dependence on co-stimulation, and that UNT have different memory subset compositions compared to UD (Figures S5F and S5G), we assessed CD28 expression in memory subsets from UNT and ART cohorts (Figures S7F–S7I). In CD4+ T cells, almost 100% of TN, TCM, and TTM expressed CD28, but TEM and TEMRA had fewer CD28+ cells in both UNT and ART cohorts. CD8+ T cells showed similar tendencies, as more differentiated memory cells had a lower proportion of CD28+ cells. However, the CD28 expression level per cell was similar between subsets, except for TN, which had slightly lower CD28 expression per CD28+ cell.
We subsequently verified if the unsupervised analysis could reveal subtle qualitative differences between the effector profiles in UNT and ART (Figures 7G–7I). Following ART, some modest yet significant alterations were observed in the proportions of CD4+ T cell clusters, with higher proportion of some IL-2+ clusters (C12 and C15), and lower TNF+ and CD107a+ clusters (C2, C3, C4, C6, and C11) (Figure 7H). These combined alterations reflected a partial yet significant normalization of the CD4+ T cell effector functions, as the ART group is located away from the UNT linearly along the PC1 axis toward the UD group on the PCA analysis plot (Figure 7J). After ART, IL-2+ CD8 clusters were also increased (C12 and C15), and TNF+ and CD107a+ CD8 clusters decreased (C6, C8 and C11) (Figure 7I). These subtle alterations were sufficient to differentiate pre-ART and post-ART samples. However, this separation occurred along the PC2 axis, suggesting that these changes did not represent a normalization toward the UD group (Figure 7K).
These unsupervised analyses reveal that post-ART, global cytokine responses remain consistent, with treatment partially restoring CD4+ T cell cytokine profiles, while CD8+ T cell responses display limited changes.
Discussion
In this study, we developed a versatile and convenient artificial Antigen-Presenting Cell (aAPC) system compatible with flow cytometry. It accommodates brief stimulation periods, fluid interfacing surfaces, and is tunable in terms of agonist antibody densities, as well as aAPC-to-cell ratios. This reductionist model enabled us to characterize in-depth the role of CD28 co-stimulation in primary CD4+ and CD8+ T cells isolated from blood samples, elucidating the varying levels of dependence of individual effector functions and secondary-wave activation molecules on this essential secondary signal. In control donors, our system highlights CD28’s preferential enhancement of IL-2 expression and how this modulation shapes the elicited response profile, favoring it over other cytokines. Additionally, our analyses revealed that CD4+ and CD8+ T cells from viremic UNT prior to ART initiation exhibited a skewed orientation toward TNF⍺, IFNγ, and CD107a expression while diverging from IL-2 production. Expression of TNFα and IFNγ upon sole CD3 stimulation was relatively better preserved in viremic UNT compared to controls, in contrast to a more pronounced dependence on CD28 co-stimulation for IL-2 production. Importantly, this functional imbalance was only partially rectified by ART in CD4+ but not in CD8+ T cells.
Our artificial aAPC system induced robust T cell activation, with over 90% of CD4+ and CD8+ T cells expressing CD69 within just 15 h of stimulation, resulting in stronger activation than that achieved by directly cross-linking the antibodies. Furthermore, both T cell phenotype and viability were preserved following stimulation with aAPCs, unlike the results with magnetic CD3/CD28 beads, which in this experimental setup delivered a strong activation signal but at the cost of surface marker distortion, low cell yield, and high cell mortality.
The ratio of aCD3 to aCD28 antibodies embedded in aAPCs played a pivotal role in optimal cytokine expression. While the agonistic effect of aCD3 was fundamental, it reached a plateau swiftly in the absence of CD28 co-stimulation. In humans, inherited T cell CD28 deficiency leads to severe cutaneous lesions due to human papillomaviruses and moderate impairment of humoral responses, yet appear otherwise usually healthy.67 This underscores the redundant nature of the co-stimulation process in vivo. For instance, murine studies in mice showed that CD27/CD70 signaling can substitute for CD28/B7 in LCMV-specific CD8+ T cell expansion,68 while another study showed that 4-1BBL could replace anti-CD28 as a co-stimulator.69
Our data indeed indicate that co-stimulation is necessary to optimally activate memory CD4+ and CD8+ T cells. This is consistent with murine studies that have demonstrated the essential role of CD28 in the recall responses of CD4+ and CD8+ T cells and subsequent viral clearance.70,71 However, the extent of CD28 co-stimulation required depends on the differentiation state of the cell and was particularly important for naive and central memory cells. These less differentiated cells may require stronger co-activation signal to counter their relative quiescence, whereas effector memory and terminally differentiated cells are likely already poised for reactivation.
Co-stimulation is often understood as a mechanism for TCR signal amplification. Nevertheless, apart from globally increasing the expression of cytokines and activation markers, our data show that CD28 co-stimulation exerts a nuanced influence on the effector functions. Specifically, IL-2, IFNγ, TNFα, CD40L, OX40, and ICOS are preferentially promoted by CD28 compared to CD107a, IL17A, 4-1BB, and CD69. We observed that CD28 co-stimulation enriches certain T cell subpopulations that share IL-2 expression as a common feature. Indeed, CD28 is known to promote IL-2 expression by enhancing its transcription72,73 and stabilizing its mRNA.74 Additionally, CD28 has been found to induce DEC1, a transcription factor controlling IL-2 and IFNγ production.75 Taken together with our findings, these studies suggest that CD28 co-stimulation can selectively promote the transcription of defined effector molecules. Alternatively, CD28 may more broadly impact cells committed to transcriptional programs that include certain functions, such as IL-2 and CD40L expression. The higher dependence on CD28 in the CD4+ T cell lineage and in less differentiated cells is consistent with this interpretation. We cannot exclude that the dependence for CD28 co-stimulation can also be linked to the level of expression CD28. The lower dependence of TEMRA cells for CD28 co-stimulation coincides with their lower expression of surface CD28, which is consistent with this possibility (Figures S2K–S2M and S7F–S7M).76,77 However, TEM and TCM expressed identical levels of CD28 yet were distinctly dependent on CD28 co-stimulation. Moreover, the different dependencies of the tested cytokines for CD28 suggest that other cellular features also play a role, such as the composition of the T cell population and possibly the expression of co-stimulatory and inhibitory receptors.
Compared to uninfected individuals, CD4+ and CD8+ T cells from UNT exhibited a greater overall responsiveness to the first signal given by aCD3, which could be attributed to the higher production of IFNγ and TNFα in response to aCD3. Conversely, we observed an increased dependency on CD28 for IL-2 in UNT. These altered dependencies on CD28 co-stimulation, seen in both naive and memory subsets, resulting in a distorted effector function profile characterized by increased frequencies of IFNγ and TNFα-expressing CD4+ and CD8+ T cell subsets, at the expense of IL-2-expressing subpopulations. The decreased IL-2 expression during HIV infection aligns with earlier reports on mitogen-stimulated PBMCs (such as phytohaemagglutinin PHA or PMA) from UNT.78,79,80,81 Mitogen stimulation circumvents the TCR, which suggests that the altered effector profiles observed may stem from TCR-independent downstream signaling or mechanistic alterations that remain to be identified.
The precise molecular mechanisms underpinning the skewing of non-specific T cell effector functions during HIV infection remain incompletely understood. HIV variably affects CD4+ T cell lineages and anatomic compartments, such as the preferential depletion of Th17 cells in the gut mucosa. This may lead to a preferential representation of other subsets in PWH.82 Conversely, some populations are better restored during ART.83,84 During HIV infection, the upregulation of immune checkpoints such as PD-1, CTLA-4, and Tim-323,85,86 is not restricted to HIV-specific responses but is broadly present in T cell subsets, which may contribute to dysfunction in vivo. Our engineered aAPCs bypass these pathways, as in contrast to natural APCs, they do not contain the corresponding ligands and interact with purified T cells. Therefore, while our data suggest T cell-intrinsic mechanisms, we cannot exclude the possibility that such signals triggered in vivo may have downstream effects in our assays. Consistent with this interpretation would be the epigenetic scars emerging from the persistent engagement of the PD1/PDL1 axis.87,88 Other possible mechanisms include the persistent exposure to microbial products leaking from compromised intestinal barrier,32 co-infection by cytomegalovirus (CMV) and Epstein-Barr virus (EBV),89 and residual expression of HIV products,90 which all contribute to a higher pro-inflammatory environment during ART. This may favor the expansion and maintenance of specific T cell subsets.91
Our data clarify the role of CD28 co-stimulation as a mechanism modulating both the magnitude and nature of effector functions triggered upon TCR-mediated activation in primary human blood cells. We observed distinct CD4+ and CD8+ T effector profiles during chronic HIV infection. Notably, these alterations were not specific to a specific pathogen. This skewing may undermine T cell responses against diverse antigens, thereby potentially contributing to HIV-associated co-morbidities.92,93 Indeed, this non-specific skewing may introduce another layer of dysfunction whose impact, combined with HIV-specific CD4+ and CD8+ T cell impairment, could partially account for the well-documented residual immune dysfunction that persists despite ART (reviewed in94). Deciphering how these defects intertwine will be key to better understanding immune dysfunction during HIV infection.
Limitations of the study
Our experiments do not explore the contribution of other molecules that could compensate for suboptimal CD28 co-stimulation. Through the interaction of other agonistic antibodies or recombinant ligands, our aAPC system could be customized to specifically assess the involvement of other co-stimulatory or co-inhibitory molecules that modulate TCR signaling. This would necessitate the pre-emptive stimulation of T cells, as most co-stimulatory molecules, apart from CD28, are not constitutively expressed on T cells but are expressed following a secondary wave of co-signaling molecules.57,58,59,60,61
Since many non-Th1 cytokine proteins are challenging to detect by flow cytometry, alternative readout approaches should be considered. Further studies should define alterations in the expression of other cytokines and chemokines not measured here, such as those preferentially produced by follicular helper T cells.17 Some factors, such as age, sex, and co-morbidities, are known to influence the immune system,95,96,97 but could not be investigated in this study due to the nature and size of the groups we examined.
The 1:4 (aCD3:aCD28) ratio was empirically determined. No ultrastructural studies were performed to verify the stochiometric implication of this ratio. It should not be interpretated as the necessity of 4 molecules of CD28 to ensure the co-stimulation of 1 molecule of CD3, as other factors such as antibody affinity can explained these empirically determined ratios.
Immune recovery during ART is variable, with a subpopulation of PWH presenting suboptimal CD4 T cell recovery, expansion of the CD8 compartment, and/or heightened inflammation despite optimal viral suppression. The aAPC system presented here could be used to compare immunological responders and non-responders in larger cohorts to identify features of persistent immune dysfunction on ART.
Resource availability
Lead contact
For further information and requests related to resources and reagents, please contact Daniel E. Kaufmann at daniel.kaufmann@chuv.ch.
Materials availability
This study did not produce any new unique reagents.
Data and code availability
-
•
The published article includes all datasets generated and analyzed for this study. For additional information and requests related to resources and reagents, please contact the lead contact Author (daniel.kaufmann@chuv.ch).
-
•
All original code has been deposited at https://github.com/alemi055/scripts-and-data/blob/master/ShaabanKabakiboetal_2024.R and is publicly available as of the publication date. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this article is available from the lead contact upon request.
Acknowledgments
The authors are grateful to the study participants. We thank Gaël Dulude, Philippe St-Onge, and the CRCHUM Flow Cytometry Platform; Olfa Debbeche and the CRCHUM BSL3 platform for technical assistance. We thank Olivier Tastet for his input on unsupervised analyses. This study was supported by the Canadian Institutes of Health Research (CIHR grant #168901 to D.E.K), the US National Institutes of Health (NIH grant HL092565 to D.E.K), an FRQS Merit Research Scholar Award (#268471 to D.E.K), and the Réseau Fonds de la recherche Québec-Santé (FRQ-S) SIDA & Maladies infectieuses and thérapies cellulaires. The Symphony flow cytometer was funded by a John R. Evans Leaders Fund from the Canada Foundation for Innovation (#37521 to D.E.K) and the Fondation Sclérodermie Québec. T.S.K and A.L. are supported by a scholarship from Canada Graduate Scholarships – Master’s (CGS M) program, as well as the FRQS for A.L. T.S.K and E.A was supported by a PREMIER scholarship from Department of Medicine at University of Montreal. J.-P.R. is the holder of the Louis Lowenstein Chair at McGill University. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the article.
Author contributions
T.S.K, M.D., and D.E.K. designed the study; K.P. and E.A. optimized the aAPC system; G.O.D provided assistance with assay protocols; T.S.K. and E.A. performed the experiments; T.S. and A.L. analyzed the data; A.L performed the unsupervised analysis; T.S.K., M.D., N.S. and D.E.K. interpreted the data; T.S.K., M.D., and D.E.K. wrote the initial draft of the article; all co-authors reviewed and edited the article; proofreading was accomplished by M.D., T.S.K., and D.E.K. AI-assisted software was used to help with proofreading. N.S., M.D., and D.E.K. provided supervision.
Declaration of interests
The authors declare no competing interests.
Declaration of generative AI and AI-assisted technologies in the writing process
During the preparation of this work, and after having written a complete version of the text, the authors used GPT 4 from OpenAI and Grammarly in order to improve language and readability, as they are not native English speakers. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| UCHT1 (BUV395) [Human anti-CD3] | BD Bioscience | Cat#563546; Lot: 1214287; RRID:AB_2744387 |
| SK3 (BUV496) [Human anti-CD4] | BD Bioscience | Cat#612936; Lot: 2140305; RRID:AB_2870220 |
| HI100 (BUV563) [Human anti-CD45RA] | BD Bioscience | Cat#612926; Lot: 1078217; RRID:AB_2870211 |
| L128 (BUV661) [Human anti-CD27] | BD Bioscience | Ca#750167; Lot: 3021264; RRID:AB_2874372 |
| NK-1 (BV421) [Human anti-CD57] | Biolegend | Cat#393326; Lot: B361783 ; RRID:AB_2860964 |
| M5E2 (BV480) [Human anti-CD14] | BD Bioscience | Cat#746304; Lot 2286909; RRID:AB_2743629 |
| HIB19 (BV480) [Human anti-CD19] | BD Bioscience | Cat#746457; Lot: 2286911; RRID:AB_2743759 |
| RPA-T8 (BV570) [Human anti-CD8] | Biolegend | Cat#301038; Lot: B346256; RRID:AB_2563213 |
| H4A3 (BV786) [Humananti-CD107a] | BD Bioscience | Cat#563869; Lot: 1207206; RRID:AB_2738458 |
| Mab11 (AF488) [Human anti-TNF⍺] | Biolegend | Cat#502915; Lot: B311742; RRID:AB_493121 |
| FN50 (PerCPeF710) [Human anti-CD69] | ebioscience | Cat#46-0699-42; Lot: 2514279; RRID:AB_2573694 |
| REA1063 (PE) [Human anti- IL17a] | Miltenyi | Cat#130-118-243; Lot: 1323010976; RRID:AB_2751466 |
| MQ1-17H12 (PE-Dazzle594) [Human anti-IL-2] | Biolegend | Cat#500344; Lot:B351592; RRID:AB_2564091 |
| (PE-Cy7) [Human anti-IFNγ] | BD Bioscience | Cat#557643; Lot: 1229911; RRID:AB_396760 |
| 2-L1-A (APC-R700) [Human anti-CCR7] | BD Bioscience | Cat#566766; Lot: 2257402; RRID:AB_2869856 |
| TRAP1 (APC) [Human anti-CD40L] | BD Bioscience | Cat#555702; Lot: 2010706; RRID:AB_398610 |
| LIVE/DEAD Fixable dead cell | BD Bioscience | Cat#L34960 ;Lot:2433781 |
| CD28.2 (BUV737) [Human anti-CD28] | BD Bioscience | Cat#612815; Lot: 2251073; RRID:AB_2870140 |
| EH12.2 (BB515) [Human anti-PD-1] | BD Bioscience | Cat#564494; Lot: 2101875; RRID:AB_2738827 |
| FN50 (BV650) [Human anti-CD69] | Biolegend | Cat#310934; Lot:B360356; RRID:AB_2563158 |
| MBSA43 (PerCPeF710) [Human anti-TIGIT] | Invitrogen ebioscience | Cat#46-9500-42; Lot:2402674; RRID:AB_10853679 |
| 4B4-1 (PE-Dazzle594) [Human anti-4-1BB] | Biolegend | Cat#309826; Lot:B349385; RRID:AB_2566259 |
| ISA-3 (PE-Cy7) [Human anti-ICOS] | Invitrogen ebioscience | Cat#25-9948-42; Lot: 2338678; RRID:AB_1518754 |
| ACT35 (APC) [Human anti-OX40] | BD Bioscience | Cat# 563473; Lot: 2026053; RRID:AB_2738230 |
| TRAP (PE) [Human anti-CD40L] | BD Bioscience | Cat#555700; Lot: 0357889; RRID:AB_396050 |
| CD3 Antibody, anti-human, Biotin functional grade | Miltenyi | Cat#130-093-377; Lot: 5210202091; RRID:AB_1036126 |
| Biotin anti-human CD28 | Biolegend | Cat:302904; Lot: B394472; RRID:AB_314306 |
| Biotin Mouse IgG1, κ Isotype Ctrl Antibody | Biolegend | Cat#400104; Lot: B350305; RRID:AB_326427 |
| Purified Goat anti-mouse IgG (minimal x-reactivity) Antibody | Biolegend | Cat# 405301; Lot: B343022 RRID:AB_315005 |
| Chemicals, peptides, and recombinant proteins | ||
| Staphylococcal Enterotoxin B (SEB) | Toxin technology | Cat#BT202 |
| Silica microspheres | Bangs Laboratoreis | Cat# SS05003 ; Lot: 16497 |
| Srteptavidin | Jackson ImmunoResearch | Cat#016-000-084; Lot: 154826 |
| 18:1 (Δ9-Cis) PC (DOPC) | Avanti polar (via sigma) | Cat# 850375C-200mg |
| 18:1 DGS-NTA(Ni) | Avanti polar (via sigma) | Cat# 790404C-10mg |
| 18:1 Biotinyl Cap PE | Avanti polar (via sigma) | Cat#870273C-25mg |
| RPMI 1640 | GIBCO by Life Technologies | Cat# 11875-119 |
| HEPES | Invitrogen | Cat# 15630-080 |
| Penicillin-Streptomycin | GIBCO by Life Technologies | Cat# 15140-122 |
| Nuclease S7 from Staphylococcus aureus | Roche Diagnostics | Cat# 12344000 |
| Bovine Serum Albumin | Sigma-Aldrich | Cat#A2153-50G CAS#9048-46-8 |
| BD GolgiStop (Monensin) | BD Bioscience | Cat# 554724 |
| BD GolgiPlug (Brefeldin A) | BD Bioscience | Cat#555029 |
| FcR Blocking Reagent, human | Miltenyi | Cat#130-059-901 |
| Invitrogen™ UltraPure™ Tris Buffer (powder format) | Invitrogen | Cat#15504020 |
| Critical commercial assays | ||
| EasySep Human T Cell Enrichment Kit | StemCell | Cat# 19051_C |
| Dynabeads Human T-Activator CD3/CD28 - for physiological activation of human T cells | ThermoFisher | Cat# 11161D; Lot:2860963; RRID:AB_2916088 |
| Deposited data | ||
| Original code is deposited at | N/A | “Original Code : https://github.com/alemi055/scripts-and-data/blob/master/ShaabanKabakiboetal_2024.R” |
| Software and algorithms | ||
| Flow Jo v10.8.0 | Flow Jo | https://www.flowjo.com |
| GraphPad Prism v8.4.1 | GraphPad | https://www.graphpad.com |
Experimental model and study participant details
Ethics statement
All work was conducted in accordance with the Declaration of Helsinki regarding informed consent and approval by an appropriate institutional board. Leukaphereses were obtained from study participants at the Montreal General Hospital, Montreal, Canada and at the Centre Hospitalier de l’Université de Montréal (CHUM) in Montreal, Canada. The study was approved by the respective Institutional Review Boards (IRB CHUM: 17.335) and participants gave written informed consent prior to enrollment. Samples were collected between 2013 and 2019 as part of a multicentric study (MP-37-2018-4029).
Study participants
Subject characteristics are summarized in Table S1. Uninfected donors (UD) are free of HIV-1 infection. Untreated HIV participants had plasma viral loads of at least 5000 copies/ml (except for one at 2700 copies/mL), had been documented by blood testing to live with HIV for at least three months, and were either untreated or off ART for at least 3 months at the time of collection of the “Pre-ART” sample. Longitudinal “Post-ART” samples were collected in these same subjects after at least 6 months on ART and achieving controlled viremia (<50 vRNA copies/mL). PBMCs were isolated from blood samples by Ficoll density gradient centrifugation and cryopreserved in liquid nitrogen until use.
Method details
Liposomes preparation
DOPC, Cap biotin PE and Ni-NTA DOGS lipids were purchased from Avanti Polar Lipids at stock concentrations of 25 mg/mL (DOPC) or 10 mg/mL (biotin-PE and Ni-NTA DOGS) in chloroform layered in argon and stored at -20°C upon receipt (Cat #850375C, #870273C, and #790404C respectively). All lipid solutions were always stored under argon to avoid oxidation. Chloroform solutions of lipids were mixed to obtain a final 4 mM DOPC solution after hydration with DOPC (98%), Cap biotin PE (2%) and Ni-NTA (6.25%) (molar ratios). Lipids were first dried under nitrogen stream and left to completely dry for 2h. The dry lipid film was rehydrated in nitrogen-purged Tris-saline for 30 min at room temperature until the solution became homogenous. The unilamellar liposomes were passed 10 times through a polycarbonate membrane (0.1 μm) in an extruder (Avanti Polar Lipids, Cat #610000) to produce bilayered liposomes (LUVs). The liposome solution was then diluted 10 times to obtain the working stock solution.
Artificial APCs preparation
aAPCs were freshly prepared on the day of stimulation. All centrifuging was done at 600g for 2 min at room temperature. Lipid bilayered aAPCs were obtained by washing 5-μm silica beads (Bangs laboratories, Cat #SS06N) twice with water, then vigorous vortexing with liposome bilayer suspension (2 μL of 1X liposomes solution per 1 μL (1M/μL) of beads) during four sequences of 10 seconds each. Ligands addition and quantities were adapted to experiment requirements and cells number required for flow cytometry each time. Briefly, the lipid-coated beads were washed once with filtered HBS-BSA buffer and incubated with 4 μg/mL streptavidin (Jackson Immunoresearch, Cat #016-000-084) for 30 min at room temperature, then washed again with HBS-BSA buffer. Streptavidin coated beads were incubated 30 min at room temperature with anti-CD3-biotin (0.585 μg/mL) and anti-CD28-biotin (2.344 μg/mL) in HBS-BSA 1% buffer for a aCD3:aCD28 ratio 1:4. Concentrations of aCD3 and aCD28 were adapted for different ratios, and the corresponding Isotype IgG concentration was complemented on suboptimal aAPCs to reach an overall antibody concentration equivalent to that of the aCD3:CD28 1:4 ratio condition. Beads were finally washed with HBS-BSA buffer and resuspended in the proper amount of HBS-BSA.
PBMCs thawing, purification, and stimulation
Frozen PBMCs were thawed in Fetal Bovine Serum (FBS) before total T cells isolation by negative magnetic bead selection (EasySep™ Human T Cell Isolation Kit). Purified T cells were resuspended at 8 × 106/mL in RPMI (GIBCO by Life Technologies) supplemented with penicillin/streptomycin (GIBCO by Life Technologies) and 10% of FBS, and rested for 2 hours. Two million purified T cells were stimulated with aAPCs at a cell:bead ratio of 1:4. As a positive control, 2 million PBMCs were stimulated by 0.5 μL of 1 μg/mL SEB. PBMCs were used for SEB activation instead of purified T cells because SEB activity requires MHC on natural antigen-presenting cells.
Crosslinking antibodies
After resting, two million T cells [8 million/mL] were either left unstimulated or incubated with anti-CD3-biotin (1.5 ug/mL) and anti-CD28-biotin (6 ug/mL) antibodies on ice for 15 minutes, followed by washig with the same culture media. Surface-bound antibodies were subsequently cross-linked using various concentrations of purified goat anti-mouse IgG (Poly4053). Cells were then incubated for 15 hours at 37°C.
CD3/CD28 Dynabeads
Dynabeads were first washed with phosphate buffered saline (PBS 1X) and resuspended in culture media at their original concentration (4x107 beads/mL). After resting, two million T cells [8 million/mL] were either left unstimulated or incubated with Dynabeads at a cell-to-beads ratio of 1:1 for 15h at 37°C. Before flow cytometric analysis, Dynabeads were removed using a magnet.
Intracellular cytokine staining (ICS)
Cell stimulation was carried out for 15h in the presence of mouse anti-human CD107a and anti-human CCR7 at 37°C and 5% CO2. Brefeldin A and monensin (BD Biosciences, San Jose, CA) were added 1h after stimulation. Following stimulation, cells were washed by PBS 1X, and stained for a viability dye (Aquavivid, Thermo Fisher scientific, Waltham, MA) for 20 min at room temperature in PBS. Cells were then stained for surface markers (CD3, CD4, CD45RA, CD27, CD57, CD8, CD19, CD14) for 30 min at 4°C. This was followed by intracellular detection of cytokines (IL-2, IFNγ, TNFα, CD107a, IL17A) using the IC Fixation/Permeabilization kit (Thermo Fisher scientific, Waltham, MA), according to the manufacturer’s protocol. Cells were acquired on a Symphony flow cytometer (BD Biosciences) and analyzed using FlowJo v10.8.1 software. Antibodies used are listed in Table S2. To calculate the degree of dependence on CD28, we used this formula : .
To correct for CD4 counts in UNT and ART in each participant, we used this formula : .
Activation-induced marker (AIM) assay
The AIM assay was performed as previously reported.98 Cells were stimulated for 15h at 37°C and 5% CO2 in the presence of anti-human CCR7. Following stimulation, cells were washed by PBS 1X, and stained for a viability dye (Aquavivid, Thermo Fisher scientific, Waltham, MA) for 20 min at room temperature in PBS. Cells were then stained for surface markers (CD3, CD4, CD45RA, CD27, CD57, CD14, CD19, CD8, CD28, PD-1, CD69, TIGIT, 4-1BB, ICOS, OX40, CD40L) for 30 min at 4°C, and fixed using 2% paraformaldehyde (Sigma-Aldritch, 15min, RT) before acquisition on a Symphony flow cytometer (BD Biosciences) and analyzes using FlowJo BD v10.8.1 software. For total PBMCs conditions, a CD40 blocking antibody (Miltenyi) was added in culture to prevent the interaction of CD40L with CD40 and its subsequent downregulation. Antibodies used are listed in Table S3.
Unsupervised clustering
Unsupervised analyses were performed on CD4+ and CD8+ T cells from UD, UNT, and ART donors that were activated by the aCD3:aCD28:IgG ratios of 1:0:4, 1:1:3, and 1:4:0. These data were first downsampled to an equal number of events (1,000 cells), and FCS files were loaded through the flowCore package (v2.10.0).99 Scaling and logicle transformation of the flow cytometry data was applied using FlowSOM (v2.6.0),100 as done previously.52,101 The uniform manifold approximation and projection (UMAP) algorithm was performed using the R package M3C (v1.20.0),102 while the clustering was achieved using Phenograph (v0.99.1)56 with the k parameter (number of nearest-neighbors) set to 400. Cytokines were the only parameters used for clustering. Therefore, no cells were clustered based on the expression of any surface marker, such as CD4 and CD8. We obtained 17 clusters. All samples of CD4+ and CD8+ activated T cells by aCD3:aCD28:IgG ratios of 1:0:4, 1:1:3, and 1:4:0, from the three cohorts (UD, UNT and ART) were loaded for clustering to form a single Multiparametric UMAP. The identification of T cell clusters relied solely on cytokine expression, and the phenotypic CD4 and CD8 information was reintroduced post-hoc using categorical single-cell phenotypic information. Therefore, CD4+ and CD8+ T cells could be subsequently displayed separately. For example, in the first panel of Figure 3A (CD4 1:0:4), we only visualized CD4+ T cells activated by an aCD3:aCD28:IgG ratio of 1:0:4. Heatmaps were also generated in R (v4.2.2) using the packages gglot2 (v3.4.0)103 and pheatmap (v1.0.12).104 To accommodate cytokines with rare expression, the heatmaps were scaled by individually normalizing the values of each cytokine between 0 and 1, using their respective minimum and maximum values. The R code scripted for this paper is available at “original code: https://github.com/alemi055/scripts-and-data/blob/master/ShaabanKabakiboetal_2024.R”.
Quantification and statistical analysis
Statistical analyses
Symbols represent biologically independent samples from UD, UNT and ART donors. Lines connect data from the same donor. Differences in responses for the same patient before and after ART treatment or from different activation conditions were assessed using Wilcoxon matched-pair tests. Differences in responses between UD and UNT were measured by Mann-Whitney tests. Wilcoxon and Mann-Whitney tests were generated using GraphPad Prism (version 9.2.0). p values < 0.05 were considered significant. p values are indicated for each comparison assessed. For graphical representation on a log scale (but not for statistical tests), null values were arbitrarily set at the minimal values for each assay.
Published: September 13, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.110947.
Contributor Information
Mathieu Dubé, Email: mathieu.dube.chum@ssss.gouv.qc.ca.
Daniel E. Kaufmann, Email: daniel.kaufmann@chuv.ch.
Supplemental information
References
- 1.Bretscher P., Cohn M. A theory of self-nonself discrimination. Science. 1970;169:1042–1049. doi: 10.1126/science.169.3950.1042. [DOI] [PubMed] [Google Scholar]
- 2.Garcia K.C., Degano M., Pease L.R., Huang M., Peterson P.A., Teyton L., Wilson I.A. Structural basis of plasticity in T cell receptor recognition of a self peptide-MHC antigen. Science. 1998;279:1166–1172. doi: 10.1126/science.279.5354.1166. [DOI] [PubMed] [Google Scholar]
- 3.Coyle A.J., Gutierrez-Ramos J.C. The expanding B7 superfamily: increasing complexity in costimulatory signals regulating T cell function. Nat. Immunol. 2001;2:203–209. doi: 10.1038/85251. [DOI] [PubMed] [Google Scholar]
- 4.Linterman M.A., Denton A.E., Divekar D.P., Zvetkova I., Kane L., Ferreira C., Veldhoen M., Clare S., Dougan G., Espéli M., Smith K.G.C. CD28 expression is required after T cell priming for helper T cell responses and protective immunity to infection. Elife. 2014;3 doi: 10.7554/eLife.03180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodríguez-Palmero M., Hara T., Thumbs A., Hünig T. Triggering of T cell proliferation through CD28 induces GATA-3 and promotes T helper type 2 differentiation in vitro and in vivo. Eur. J. Immunol. 1999;29:3914–3924. doi: 10.1002/(sici)1521-4141(199912)29:12<3914::Aid-immu3914>3.0.Co;2-#. [DOI] [PubMed] [Google Scholar]
- 6.Frauwirth K.A., Riley J.L., Harris M.H., Parry R.V., Rathmell J.C., Plas D.R., Elstrom R.L., June C.H., Thompson C.B. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16:769–777. doi: 10.1016/s1074-7613(02)00323-0. [DOI] [PubMed] [Google Scholar]
- 7.Martin P.J., Ledbetter J.A., Morishita Y., June C.H., Beatty P.G., Hansen J.A. A 44 kilodalton cell surface homodimer regulates interleukin 2 production by activated human T lymphocytes. J. Immunol. 1986;136:3282–3287. [PubMed] [Google Scholar]
- 8.Boise L.H., Minn A.J., Noel P.J., June C.H., Accavitti M.A., Lindsten T., Thompson C.B. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity. 1995;3:87–98. doi: 10.1016/1074-7613(95)90161-2. [DOI] [PubMed] [Google Scholar]
- 9.Thomas R.M., Gao L., Wells A.D. Signals from CD28 induce stable epigenetic modification of the IL-2 promoter. J. Immunol. 2005;174:4639–4646. doi: 10.4049/jimmunol.174.8.4639. [DOI] [PubMed] [Google Scholar]
- 10.Butte M.J., Lee S.J., Jesneck J., Keir M.E., Haining W.N., Sharpe A.H. CD28 costimulation regulates genome-wide effects on alternative splicing. PLoS One. 2012;7 doi: 10.1371/journal.pone.0040032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DuPage M., Chopra G., Quiros J., Rosenthal W.L., Morar M.M., Holohan D., Zhang R., Turka L., Marson A., Bluestone J.A. The chromatin-modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity. 2015;42:227–238. doi: 10.1016/j.immuni.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wada N.I., Jacobson L.P., Margolick J.B., Breen E.C., Macatangay B., Penugonda S., Martínez-Maza O., Bream J.H. The effect of HAART-induced HIV suppression on circulating markers of inflammation and immune activation. AIDS. 2015;29:463–471. doi: 10.1097/qad.0000000000000545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.French M.A., King M.S., Tschampa J.M., da Silva B.A., Landay A.L. Serum immune activation markers are persistently increased in patients with HIV infection after 6 years of antiretroviral therapy despite suppression of viral replication and reconstitution of CD4+ T cells. J. Infect. Dis. 2009;200:1212–1215. doi: 10.1086/605890. [DOI] [PubMed] [Google Scholar]
- 14.Lindqvist M., van Lunzen J., Soghoian D.Z., Kuhl B.D., Ranasinghe S., Kranias G., Flanders M.D., Cutler S., Yudanin N., Muller M.I., et al. Expansion of HIV-specific T follicular helper cells in chronic HIV infection. J. Clin. Invest. 2012;122:3271–3280. doi: 10.1172/jci64314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Champagne P., Ogg G.S., King A.S., Knabenhans C., Ellefsen K., Nobile M., Appay V., Rizzardi G.P., Fleury S., Lipp M., et al. Skewed maturation of memory HIV-specific CD8 T lymphocytes. Nature. 2001;410:106–111. doi: 10.1038/35065118. [DOI] [PubMed] [Google Scholar]
- 16.Niessl J., Baxter A.E., Morou A., Brunet-Ratnasingham E., Sannier G., Gendron-Lepage G., Richard J., Delgado G.G., Brassard N., Turcotte I., et al. Persistent expansion and Th1-like skewing of HIV-specific circulating T follicular helper cells during antiretroviral therapy. EBioMedicine. 2020;54 doi: 10.1016/j.ebiom.2020.102727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morou A., Brunet-Ratnasingham E., Dubé M., Charlebois R., Mercier E., Darko S., Brassard N., Nganou-Makamdop K., Arumugam S., Gendron-Lepage G., et al. Altered differentiation is central to HIV-specific CD4(+) T cell dysfunction in progressive disease. Nat. Immunol. 2019;20:1059–1070. doi: 10.1038/s41590-019-0418-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goepfert P.A., Bansal A., Edwards B.H., Ritter G.D., Jr., Tellez I., McPherson S.A., Sabbaj S., Mulligan M.J. A significant number of human immunodeficiency virus epitope-specific cytotoxic T lymphocytes detected by tetramer binding do not produce gamma interferon. J. Virol. 2000;74:10249–10255. doi: 10.1128/jvi.74.21.10249-10255.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kostense S., Vandenberghe K., Joling J., Van Baarle D., Nanlohy N., Manting E., Miedema F. Persistent numbers of tetramer+ CD8(+) T cells, but loss of interferon-gamma+ HIV-specific T cells during progression to AIDS. Blood. 2002;99:2505–2511. doi: 10.1182/blood.v99.7.2505. [DOI] [PubMed] [Google Scholar]
- 20.Shankar P., Russo M., Harnisch B., Patterson M., Skolnik P., Lieberman J. Impaired function of circulating HIV-specific CD8(+) T cells in chronic human immunodeficiency virus infection. Blood. 2000;96:3094–3101. [PubMed] [Google Scholar]
- 21.Appay V., Nixon D.F., Donahoe S.M., Gillespie G.M., Dong T., King A., Ogg G.S., Spiegel H.M., Conlon C., Spina C.A., et al. HIV-specific CD8(+) T cells produce antiviral cytokines but are impaired in cytolytic function. J. Exp. Med. 2000;192:63–75. doi: 10.1084/jem.192.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lichterfeld M., Kaufmann D.E., Yu X.G., Mui S.K., Addo M.M., Johnston M.N., Cohen D., Robbins G.K., Pae E., Alter G., et al. Loss of HIV-1-specific CD8+ T cell proliferation after acute HIV-1 infection and restoration by vaccine-induced HIV-1-specific CD4+ T cells. J. Exp. Med. 2004;200:701–712. doi: 10.1084/jem.20041270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Day C.L., Kaufmann D.E., Kiepiela P., Brown J.A., Moodley E.S., Reddy S., Mackey E.W., Miller J.D., Leslie A.J., DePierres C., et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 24.Barber D.L., Wherry E.J., Masopust D., Zhu B., Allison J.P., Sharpe A.H., Freeman G.J., Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 25.Wherry E.J., Ha S.J., Kaech S.M., Haining W.N., Sarkar S., Kalia V., Subramaniam S., Blattman J.N., Barber D.L., Ahmed R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 26.Hui E., Cheung J., Zhu J., Su X., Taylor M.J., Wallweber H.A., Sasmal D.K., Huang J., Kim J.M., Mellman I., Vale R.D. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017;355:1428–1433. doi: 10.1126/science.aaf1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hazenberg M.D., Stuart J.W., Otto S.A., Borleffs J.C., Boucher C.A., de Boer R.J., Miedema F., Hamann D. T-cell division in human immunodeficiency virus (HIV)-1 infection is mainly due to immune activation: a longitudinal analysis in patients before and during highly active antiretroviral therapy (HAART) Blood. 2000;95:249–255. [PubMed] [Google Scholar]
- 28.Doisne J.M., Urrutia A., Lacabaratz-Porret C., Goujard C., Meyer L., Chaix M.L., Sinet M., Venet A. CD8+ T cells specific for EBV, cytomegalovirus, and influenza virus are activated during primary HIV infection. J. Immunol. 2004;173:2410–2418. doi: 10.4049/jimmunol.173.4.2410. [DOI] [PubMed] [Google Scholar]
- 29.Copeland N.K., Eller M.A., Kim D., Creegan M., Esber A., Eller L.A., Semwogerere M., Kibuuka H., Kiweewa F., Crowell T.A., et al. Brief Report: Increased Inflammation and Liver Disease in HIV/HBV-Coinfected Individuals. J. Acquir. Immune Defic. Syndr. 2021;88:310–313. doi: 10.1097/qai.0000000000002760. [DOI] [PubMed] [Google Scholar]
- 30.Righetti E., Ballon G., Ometto L., Cattelan A.M., Menin C., Zanchetta M., Chieco-Bianchi L., De Rossi A. Dynamics of Epstein-Barr virus in HIV-1-infected subjects on highly active antiretroviral therapy. AIDS. 2002;16:63–73. doi: 10.1097/00002030-200201040-00009. [DOI] [PubMed] [Google Scholar]
- 31.Jabs D.A., Enger C., Bartlett J.G. Cytomegalovirus retinitis and acquired immunodeficiency syndrome. Arch. Ophthalmol. 1989;107:75–80. doi: 10.1001/archopht.1989.01070010077031. [DOI] [PubMed] [Google Scholar]
- 32.Brenchley J.M., Price D.A., Schacker T.W., Asher T.E., Silvestri G., Rao S., Kazzaz Z., Bornstein E., Lambotte O., Altmann D., et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 2006;12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 33.Younas M., Psomas C., Reynes C., Cezar R., Kundura L., Portales P., Merle C., Atoui N., Fernandez C., Le Moing V., et al. Microbial Translocation Is Linked to a Specific Immune Activation Profile in HIV-1-Infected Adults With Suppressed Viremia. Front. Immunol. 2019;10 doi: 10.3389/fimmu.2019.02185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bürgisser P., Hammann C., Kaufmann D., Battegay M., Rutschmann O.T. Expression of CD28 and CD38 by CD8+ T lymphocytes in HIV-1 infection correlates with markers of disease severity and changes towards normalization under treatment. The Swiss HIV Cohort Study. Clin. Exp. Immunol. 1999;115:458–463. doi: 10.1046/j.1365-2249.1999.00818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaizuka Y., Douglass A.D., Varma R., Dustin M.L., Vale R.D. Mechanisms for segregating T cell receptor and adhesion molecules during immunological synapse formation in Jurkat T cells. Proc. Natl. Acad. Sci. USA. 2007;104:20296–20301. doi: 10.1073/pnas.0710258105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grakoui A., Bromley S.K., Sumen C., Davis M.M., Shaw A.S., Allen P.M., Dustin M.L. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285:221–227. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
- 37.Ilani T., Vasiliver-Shamis G., Vardhana S., Bretscher A., Dustin M.L. T cell antigen receptor signaling and immunological synapse stability require myosin IIA. Nat. Immunol. 2009;10:531–539. doi: 10.1038/ni.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yokosuka T., Kobayashi W., Sakata-Sogawa K., Takamatsu M., Hashimoto-Tane A., Dustin M.L., Tokunaga M., Saito T. Spatiotemporal regulation of T cell costimulation by TCR-CD28 microclusters and protein kinase C theta translocation. Immunity. 2008;29:589–601. doi: 10.1016/j.immuni.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bromley S.K., Iaboni A., Davis S.J., Whitty A., Green J.M., Shaw A.S., Weiss A., Dustin M.L. The immunological synapse and CD28-CD80 interactions. Nat. Immunol. 2001;2:1159–1166. doi: 10.1038/ni737. [DOI] [PubMed] [Google Scholar]
- 40.Dustin M.L. Insights into function of the immunological synapse from studies with supported planar bilayers. Curr. Top. Microbiol. Immunol. 2010;340:1–24. doi: 10.1007/978-3-642-03858-7_1. [DOI] [PubMed] [Google Scholar]
- 41.Mescher M.F. Surface contact requirements for activation of cytotoxic T lymphocytes. J. Immunol. 1992;149:2402–2405. [PubMed] [Google Scholar]
- 42.Perica K., Tu A., Richter A., Bieler J.G., Edidin M., Schneck J.P. Magnetic field-induced T cell receptor clustering by nanoparticles enhances T cell activation and stimulates antitumor activity. ACS Nano. 2014;8:2252–2260. doi: 10.1021/nn405520d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hsu C.J., Hsieh W.T., Waldman A., Clarke F., Huseby E.S., Burkhardt J.K., Baumgart T. Ligand mobility modulates immunological synapse formation and T cell activation. PLoS One. 2012;7 doi: 10.1371/journal.pone.0032398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Giannoni F., Barnett J., Bi K., Samodal R., Lanza P., Marchese P., Billetta R., Vita R., Klein M.R., Prakken B., et al. Clustering of T cell ligands on artificial APC membranes influences T cell activation and protein kinase C theta translocation to the T cell plasma membrane. J. Immunol. 2005;174:3204–3211. doi: 10.4049/jimmunol.174.6.3204. [DOI] [PubMed] [Google Scholar]
- 45.Olden B.R., Perez C.R., Wilson A.L., Cardle I.I., Lin Y.S., Kaehr B., Gustafson J.A., Jensen M.C., Pun S.H. Cell-Templated Silica Microparticles with Supported Lipid Bilayers as Artificial Antigen-Presenting Cells for T Cell Activation. Adv. Healthcare Mater. 2019;8 doi: 10.1002/adhm.201801188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen J.Y., Agrawal S., Yi H.P., Vallejo D., Agrawal A., Lee A.P. Cell-Sized Lipid Vesicles as Artificial Antigen-Presenting Cells for Antigen-Specific T Cell Activation. Adv. Healthcare Mater. 2023;12 doi: 10.1002/adhm.202203163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goldstein S.A., Mescher M.F. Cell-sized, supported artificial membranes (pseudocytes): response of precursor cytotoxic T lymphocytes to class I MHC proteins. J. Immunol. 1986;137:3383–3392. [PMC free article] [PubMed] [Google Scholar]
- 48.Cheung A.S., Zhang D.K.Y., Koshy S.T., Mooney D.J. Scaffolds that mimic antigen-presenting cells enable ex vivo expansion of primary T cells. Nat. Biotechnol. 2018;36:160–169. doi: 10.1038/nbt.4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim H.S., Ho T.C., Willner M.J., Becker M.W., Kim H.W., Leong K.W. Dendritic cell-mimicking scaffolds for ex vivo T cell expansion. Bioact. Mater. 2023;21:241–252. doi: 10.1016/j.bioactmat.2022.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wauters A.C., Scheerstra J.F., Vermeijlen I.G., Hammink R., Schluck M., Woythe L., Wu H., Albertazzi L., Figdor C.G., Tel J., et al. Artificial Antigen-Presenting Cell Topology Dictates T Cell Activation. ACS Nano. 2022;16:15072–15085. doi: 10.1021/acsnano.2c06211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang D.K.Y., Cheung A.S., Mooney D.J. Activation and expansion of human T cells using artificial antigen-presenting cell scaffolds. Nat. Protoc. 2020;15:773–798. doi: 10.1038/s41596-019-0249-0. [DOI] [PubMed] [Google Scholar]
- 52.Lemieux A., Sannier G., Nicolas A., Nayrac M., Delgado G.G., Cloutier R., Brassard N., Laporte M., Duchesne M., Sreng Flores A.M., et al. Enhanced detection of antigen-specific T cells by a multiplexed AIM assay. Cell Rep. Methods. 2024;4 doi: 10.1016/j.crmeth.2023.100690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Araújo-Souza P.S., Hanschke S.C.H., Nardy A.F.F.R., Sécca C., Oliveira-Vieira B., Silva K.L., Soares-Lima S.C., Viola J.P.B. Differential interferon-γ production by naive and memory-like CD8 T cells. J. Leukoc. Biol. 2020;108:1329–1337. doi: 10.1002/jlb.2ab0420-646r. [DOI] [PubMed] [Google Scholar]
- 54.Sallusto F., Lenig D., Förster R., Lipp M., Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 55.Becht E., McInnes L., Healy J., Dutertre C.A., Kwok I.W.H., Ng L.G., Ginhoux F., Newell E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2018;37:38–44. doi: 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- 56.Levine J.H., Simonds E.F., Bendall S.C., Davis K.L., Amir E.a.D., Tadmor M.D., Litvin O., Fienberg H.G., Jager A., Zunder E.R., et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell. 2015;162:184–197. doi: 10.1016/j.cell.2015.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Noelle R.J., Roy M., Shepherd D.M., Stamenkovic I., Ledbetter J.A., Aruffo A. A 39-kDa protein on activated helper T cells binds CD40 and transduces the signal for cognate activation of B cells. Proc. Natl. Acad. Sci. USA. 1992;89:6550–6554. doi: 10.1073/pnas.89.14.6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kinnear G., Wood K.J., Fallah-Arani F., Jones N.D. A diametric role for OX40 in the response of effector/memory CD4+ T cells and regulatory T cells to alloantigen. J. Immunol. 2013;191:1465–1475. doi: 10.4049/jimmunol.1300553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gramaglia I., Weinberg A.D., Lemon M., Croft M. Ox-40 ligand: a potent costimulatory molecule for sustaining primary CD4 T cell responses. J. Immunol. 1998;161:6510–6517. [PubMed] [Google Scholar]
- 60.Pollok K.E., Kim Y.J., Zhou Z., Hurtado J., Kim K.K., Pickard R.T., Kwon B.S. Inducible T cell antigen 4-1BB. Analysis of expression and function. J. Immunol. 1993;150:771–781. [PubMed] [Google Scholar]
- 61.Hutloff A., Dittrich A.M., Beier K.C., Eljaschewitsch B., Kraft R., Anagnostopoulos I., Kroczek R.A. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature. 1999;397:263–266. doi: 10.1038/16717. [DOI] [PubMed] [Google Scholar]
- 62.Maino V.C., Suni M.A., Ruitenberg J.J. Rapid flow cytometric method for measuring lymphocyte subset activation. Cytometry. 1995;20:127–133. doi: 10.1002/cyto.990200205. [DOI] [PubMed] [Google Scholar]
- 63.Simms P.E., Ellis T.M. Utility of flow cytometric detection of CD69 expression as a rapid method for determining poly- and oligoclonal lymphocyte activation. Clin. Diagn. Lab. Immunol. 1996;3:301–304. doi: 10.1128/cdli.3.3.301-304.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reiss S., Baxter A.E., Cirelli K.M., Dan J.M., Morou A., Daigneault A., Brassard N., Silvestri G., Routy J.P., Havenar-Daughton C., et al. Comparative analysis of activation induced marker (AIM) assays for sensitive identification of antigen-specific CD4 T cells. PLoS One. 2017;12 doi: 10.1371/journal.pone.0186998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Porichis F., Hart M.G., Zupkosky J., Barblu L., Kwon D.S., McMullen A., Brennan T., Ahmed R., Freeman G.J., Kavanagh D.G., Kaufmann D.E. Differential impact of PD-1 and/or interleukin-10 blockade on HIV-1-specific CD4 T cell and antigen-presenting cell functions. J. Virol. 2014;88:2508–2518. doi: 10.1128/jvi.02034-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kamphorst A.O., Wieland A., Nasti T., Yang S., Zhang R., Barber D.L., Konieczny B.T., Daugherty C.Z., Koenig L., Yu K., et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science. 2017;355:1423–1427. doi: 10.1126/science.aaf0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Béziat V., Rapaport F., Hu J., Titeux M., Bonnet des Claustres M., Bourgey M., Griffin H., Bandet É., Ma C.S., Sherkat R., et al. Humans with inherited T cell CD28 deficiency are susceptible to skin papillomaviruses but are otherwise healthy. Cell. 2021;184:3812–3828.e30. doi: 10.1016/j.cell.2021.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Welten S.P.M., Redeker A., Franken K.L.M.C., Oduro J.D., Ossendorp F., Čičin-Šain L., Melief C.J.M., Aichele P., Arens R. The viral context instructs the redundancy of costimulatory pathways in driving CD8(+) T cell expansion. Elife. 2015;4 doi: 10.7554/eLife.07486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Saoulli K., Lee S.Y., Cannons J.L., Yeh W.C., Santana A., Goldstein M.D., Bangia N., DeBenedette M.A., Mak T.W., Choi Y., Watts T.H. CD28-independent, TRAF2-dependent costimulation of resting T cells by 4-1BB ligand. J. Exp. Med. 1998;187:1849–1862. doi: 10.1084/jem.187.11.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Borowski A.B., Boesteanu A.C., Mueller Y.M., Carafides C., Topham D.J., Altman J.D., Jennings S.R., Katsikis P.D. Memory CD8+ T cells require CD28 costimulation. J. Immunol. 2007;179:6494–6503. doi: 10.4049/jimmunol.179.10.6494. [DOI] [PubMed] [Google Scholar]
- 71.Ndejembi M.P., Teijaro J.R., Patke D.S., Bingaman A.W., Chandok M.R., Azimzadeh A., Nadler S.G., Farber D.L. Control of memory CD4 T cell recall by the CD28/B7 costimulatory pathway. J. Immunol. 2006;177:7698–7706. doi: 10.4049/jimmunol.177.11.7698. [DOI] [PubMed] [Google Scholar]
- 72.Fraser J.D., Irving B.A., Crabtree G.R., Weiss A. Regulation of interleukin-2 gene enhancer activity by the T cell accessory molecule CD28. Science. 1991;251:313–316. doi: 10.1126/science.1846244. [DOI] [PubMed] [Google Scholar]
- 73.Verweij C.L., Geerts M., Aarden L.A. Activation of interleukin-2 gene transcription via the T-cell surface molecule CD28 is mediated through an NF-kB-like response element. J. Biol. Chem. 1991;266:14179–14182. [PubMed] [Google Scholar]
- 74.Lindstein T., June C.H., Ledbetter J.A., Stella G., Thompson C.B. Regulation of lymphokine messenger RNA stability by a surface-mediated T cell activation pathway. Science. 1989;244:339–343. doi: 10.1126/science.2540528. [DOI] [PubMed] [Google Scholar]
- 75.Martínez-Llordella M., Esensten J.H., Bailey-Bucktrout S.L., Lipsky R.H., Marini A., Chen J., Mughal M., Mattson M.P., Taub D.D., Bluestone J.A. CD28-inducible transcription factor DEC1 is required for efficient autoreactive CD4+ T cell response. J. Exp. Med. 2013;210:1603–1619. doi: 10.1084/jem.20122387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Salumets A., Tserel L., Rumm A.P., Türk L., Kingo K., Saks K., Oras A., Uibo R., Tamm R., Peterson H., et al. Epigenetic quantification of immunosenescent CD8(+) TEMRA cells in human blood. Aging Cell. 2022;21 doi: 10.1111/acel.13607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Koch S., Larbi A., Derhovanessian E., Ozcelik D., Naumova E., Pawelec G. Multiparameter flow cytometric analysis of CD4 and CD8 T cell subsets in young and old people. Immun. Ageing. 2008;5:1–12. doi: 10.1186/1742-4933-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Clerici M., Hakim F.T., Venzon D.J., Blatt S., Hendrix C.W., Wynn T.A., Shearer G.M. Changes in interleukin-2 and interleukin-4 production in asymptomatic, human immunodeficiency virus-seropositive individuals. J. Clin. Invest. 1993;91:759–765. doi: 10.1172/jci116294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Westby M., Marriott J.B., Guckian M., Cookson S., Hay P., Dalgleish A.G. Abnormal intracellular IL-2 and interferon-gamma (IFN-gamma) production as HIV-1-assocated markers of immune dysfunction. Clin. Exp. Immunol. 1998;111:257–263. doi: 10.1046/j.1365-2249.1998.00505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Clerici M., Shearer G.M. A TH1-->TH2 switch is a critical step in the etiology of HIV infection. Immunol. Today. 1993;14:107–111. doi: 10.1016/0167-5699(93)90208-3. [DOI] [PubMed] [Google Scholar]
- 81.Klein S.A., Dobmeyer J.M., Dobmeyer T.S., Pape M., Ottmann O.G., Helm E.B., Hoelzer D., Rossol R. Demonstration of the Th1 to Th2 cytokine shift during the course of HIV-1 infection using cytoplasmic cytokine detection on single cell level by flow cytometry. AIDS. 1997;11:1111–1118. doi: 10.1097/00002030-199709000-00005. [DOI] [PubMed] [Google Scholar]
- 82.Le Hingrat Q., Sereti I., Landay A.L., Pandrea I., Apetrei C. The Hitchhiker Guide to CD4(+) T-Cell Depletion in Lentiviral Infection. A Critical Review of the Dynamics of the CD4(+) T Cells in SIV and HIV Infection. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.695674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lu L., Li X., Liu X., Qiu Z., Han Y., Song X., Li Y., Li X., Cao W., Lv W., et al. The pattern and magnitude of T cell subsets reconstitution during ten years of ART with viral suppression in HIV-infected patients. Aging (Albany NY) 2022;14:9647–9667. doi: 10.18632/aging.204416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Renault C., Veyrenche N., Mennechet F., Bedin A.S., Routy J.P., Van de Perre P., Reynes J., Tuaillon E. Th17 CD4+ T-Cell as a Preferential Target for HIV Reservoirs. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.822576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kaufmann D.E., Kavanagh D.G., Pereyra F., Zaunders J.J., Mackey E.W., Miura T., Palmer S., Brockman M., Rathod A., Piechocka-Trocha A., et al. Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. Nat. Immunol. 2007;8:1246–1254. doi: 10.1038/ni1515. [DOI] [PubMed] [Google Scholar]
- 86.Jones R.B., Ndhlovu L.C., Barbour J.D., Sheth P.M., Jha A.R., Long B.R., Wong J.C., Satkunarajah M., Schweneker M., Chapman J.M., et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J. Exp. Med. 2008;205:2763–2779. doi: 10.1084/jem.20081398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Youngblood B., Noto A., Porichis F., Akondy R.S., Ndhlovu Z.M., Austin J.W., Bordi R., Procopio F.A., Miura T., Allen T.M., et al. Cutting edge: Prolonged exposure to HIV reinforces a poised epigenetic program for PD-1 expression in virus-specific CD8 T cells. J. Immunol. 2013;191:540–544. doi: 10.4049/jimmunol.1203161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ahn E., Youngblood B., Lee J., Lee J., Sarkar S., Ahmed R. Demethylation of the PD-1 Promoter Is Imprinted during the Effector Phase of CD8 T Cell Exhaustion. J. Virol. 2016;90:8934–8946. doi: 10.1128/jvi.00798-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gianella S., Moser C., Vitomirov A., McKhann A., Layman L., Scott B., Caballero G., Lada S., Bosch R.J., Hoenigl M., et al. Presence of asymptomatic cytomegalovirus and Epstein–Barr virus DNA in blood of persons with HIV starting antiretroviral therapy is associated with non-AIDS clinical events. AIDS. 2020;34:849–857. doi: 10.1097/qad.0000000000002484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sannier G., Dubé M., Dufour C., Richard C., Brassard N., Delgado G.G., Pagliuzza A., Baxter A.E., Niessl J., Brunet-Ratnasingham E., et al. Combined single-cell transcriptional, translational, and genomic profiling reveals HIV-1 reservoir diversity. Cell Rep. 2021;36 doi: 10.1016/j.celrep.2021.109643. [DOI] [PubMed] [Google Scholar]
- 91.Osuji F.N., Onyenekwe C.C., Ahaneku J.E., Ukibe N.R. The effects of highly active antiretroviral therapy on the serum levels of pro-inflammatory and anti-inflammatory cytokines in HIV infected subjects. J. Biomed. Sci. 2018;25:88. doi: 10.1186/s12929-018-0490-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Babu H., Ambikan A.T., Gabriel E.E., Svensson Akusjärvi S., Palaniappan A.N., Sundaraj V., Mupanni N.R., Sperk M., Cheedarla N., Sridhar R., et al. Systemic Inflammation and the Increased Risk of Inflamm-Aging and Age-Associated Diseases in People Living With HIV on Long Term Suppressive Antiretroviral Therapy. Front. Immunol. 2019;10 doi: 10.3389/fimmu.2019.01965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lederman M.M., Funderburg N.T., Sekaly R.P., Klatt N.R., Hunt P.W. Residual immune dysregulation syndrome in treated HIV infection. Adv. Immunol. 2013;119:51–83. doi: 10.1016/b978-0-12-407707-2.00002-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cai C.W., Sereti I. Residual immune dysfunction under antiretroviral therapy. Semin. Immunol. 2021;51 doi: 10.1016/j.smim.2021.101471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Teissier T., Boulanger E., Cox L.S. Interconnections between Inflammageing and Immunosenescence during Ageing. Cells. 2022;11 doi: 10.3390/cells11030359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Klein S.L., Flanagan K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016;16:626–638. doi: 10.1038/nri.2016.90. [DOI] [PubMed] [Google Scholar]
- 97.Poon M.M.L., Byington E., Meng W., Kubota M., Matsumoto R., Grifoni A., Weiskopf D., Dogra P., Lam N., Szabo P.A., et al. Heterogeneity of human anti-viral immunity shaped by virus, tissue, age, and sex. Cell Rep. 2021;37 doi: 10.1016/j.celrep.2021.110071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sannier G., Nicolas A., Dubé M., Marchitto L., Nayrac M., Tastet O., Chatterjee D., Tauzin A., Lima-Barbosa R., Laporte M., et al. A third SARS-CoV-2 mRNA vaccine dose in people receiving hemodialysis overcomes B cell defects but elicits a skewed CD4(+) T cell profile. Cell Rep Med. 2023;4 doi: 10.1016/j.xcrm.2023.100955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hahne F., LeMeur N., Brinkman R.R., Ellis B., Haaland P., Sarkar D., Spidlen J., Strain E., Gentleman R. flowCore: a Bioconductor package for high throughput flow cytometry. BMC Bioinf. 2009;10:106. doi: 10.1186/1471-2105-10-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Quintelier K., Couckuyt A., Emmaneel A., Aerts J., Saeys Y., Van Gassen S. Analyzing high-dimensional cytometry data using FlowSOM. Nat. Protoc. 2021;16:3775–3801. doi: 10.1038/s41596-021-00550-0. [DOI] [PubMed] [Google Scholar]
- 101.Nayrac M., Dubé M., Sannier G., Nicolas A., Marchitto L., Tastet O., Tauzin A., Brassard N., Lima-Barbosa R., Beaudoin-Bussières G., et al. Temporal associations of B and T cell immunity with robust vaccine responsiveness in a 16-week interval BNT162b2 regimen. Cell Rep. 2022;39 doi: 10.1016/j.celrep.2022.111013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.John C.R., Watson D., Russ D., Goldmann K., Ehrenstein M., Pitzalis C., Lewis M., Barnes M. M3C: Monte Carlo reference-based consensus clustering. Sci. Rep. 2020;10 doi: 10.1038/s41598-020-58766-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wickham H. Springer Cham; 2016. ggplot2 Elegant Graphics for Data Analysis. [DOI] [Google Scholar]
- 104.Kolde R. 2019. Pheatmap: Pretty Heatmaps. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The published article includes all datasets generated and analyzed for this study. For additional information and requests related to resources and reagents, please contact the lead contact Author (daniel.kaufmann@chuv.ch).
-
•
All original code has been deposited at https://github.com/alemi055/scripts-and-data/blob/master/ShaabanKabakiboetal_2024.R and is publicly available as of the publication date. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this article is available from the lead contact upon request.