

Abstract
Stevens-Johnson syndrome and toxic epidermal necrolysis (SJS/TEN) is a rare but life-threatening cutaneous drug reaction mediated by human leukocyte antigen (HLA) class I-restricted CD8+ T cells. For unbiased assessment of cellular immunopathogenesis, here we perform single-cell (sc) transcriptome, surface proteome, and T cell receptor (TCR) sequencing on unaffected skin, affected skin, and blister fluid from 15 SJS/TEN patients. From 109,888 cells, we identify 15 scRNA-defined subsets. Keratinocytes express markers indicating HLA class I-restricted antigen presentation and appear to trigger the proliferation of and killing by cytotoxic CD8+ tissue-resident T cells that express granulysin, granzyme B, perforin, LAG3, CD27, and LINC01871, and signal through the PKM, MIF, TGFβ, and JAK-STAT pathways. In affected tissue, cytotoxic CD8+ T cells express private expanded and unexpanded TCRαβ that are absent or unexpanded in unaffected skin, and mixed populations of macrophages and fibroblasts express pro-inflammatory markers or those favoring repair. This data identifies putative cytotoxic TCRs and therapeutic targets.
Subject terms: Immunological disorders, Allergy, T-cell receptor, Gene regulation in immune cells, Cytotoxic T cells
Stevens-Johnson syndrome and toxic epidermal necrolysis (SJS/TEN) is a rare but life-threatening severe cutaneous drug reaction mediated by CD8+ T cells. Here the authors characterise the immune response in skin samples at the site of tissue damage from patients affected with SJS/TEN and compare this to healthy skin or blister fluid and find populations of CD8+ T cell clonotypes expressing cytotoxic mediator molecules.
Introduction
Stevens-Johnson syndrome and toxic epidermal necrolysis (SJS/TEN) is a rare T cell-mediated severe cutaneous adverse drug reaction characterized by skin and mucous membrane blistering and detachment. The hallmarks of SJS/TEN include a delayed onset of 4–28 days following first drug exposure, keratinocyte death, and mortality up to 50%1. Multiple epithelial surfaces are involved, and long-term complications are significant and include blindness and respiratory, reproductive, and mental health issues. While > 200 culprit drugs may cause SJS/TEN, a narrower range of small molecule drugs, including anti-seizure medications such as carbamazepine, lamotrigine, and phenytoin, sulfonamide antibiotics such as co-trimoxazole, anti-tuberculosis drugs, and the anti-gout medication allopurinol cause the majority of cases globally2. An important hallmark of SJS/TEN is widespread keratinocyte necrosis, which leads to full-thickness epidermal necrosis and detachment of the epidermis from the dermis. Sub-epithelial blisters form due to this separation. A variable perivascular and interstitial infiltrate of lymphocytes and macrophages is seen in the dermis, and it has been proposed that cytotoxic T cells play a crucial role in keratinocyte apoptosis by several different mechanisms: 1. T cell receptor (TCR) recognition of a cognate epitope presented by class I human leukocyte antigen (HLA) triggers cytotoxic T cells to release perforin and granzymes3, 2. Granulysin expressed and released by CD8+ T cells and natural killer (NK) cells is directly cytotoxic4. However, controversy remains about whether the 15kD form of granulysin associated with SJS/TEN is directly cytotoxic to keratinocytes5, 3. Fas ligand (FasL) expressed on T cells interacts with Fas (CD95) on keratinocytes3, 4. TNF and IFN-γ, produced by activated T cells and other immune cells3, contribute to the inflammatory environment and promote keratinocyte apoptosis. Importantly, drug exposure in someone carrying a specific HLA class I risk allele for that drug is necessary for the development of SJS/TEN6. Moreover, for most drugs, with the notable exception of carbamazepine which has a public TCR clonotype, private drug-antigen-expanded TCR clonotypes are identified in the blister fluid of patients with drug-induced SJS/TEN that are rare in the peripheral blood7. In other words, different oligoclonal expanded TCRs are seen in the blister fluid of different patients even if they have been exposed to the same drug and have the same HLA risk allele. These oligoclonal TCRs in the blister fluid are expressed on granulysin-producing CD8+ T cells8. Although therapeutic interventions such as TNF receptor antagonists and cyclosporine have shown promise in one unblinded randomized single-center study and small observational studies or case series, respectively9,10, currently, there are no randomized double-blind controlled studies to inform the best therapeutic intervention for SJS/TEN11. Therefore, the mainstay of clinical management remains drug withdrawal and multidisciplinary supportive care without pharmacological intervention12. We have therefore sought to more deeply phenotype the antigen-driven TCR-expressing T cells and interacting population(s) at the site of tissue damage using unbiased single-cell analyses of normal and diseased skin to provide additional mechanistic understanding and identify potential therapeutic targets. We perform single-cell transcriptome (scRNA-seq), surface protein (scCITE-seq), and TCR-sequencing (scTCR-seq) across unaffected skin, affected skin, and blister fluid from 15 patients with SJS/TEN. Normal skin (n = 1) and burn blister fluid (n = 4) were included as healthy tissue and non-drug-antigen-driven generalized inflammatory blister controls, respectively. Importantly, while patients varied in age, sex, culprit drug, and HLA genotype, single-cell analyses of the cellular response at the site of SJS/TEN tissue damage revealed common features across patients that were unrelated to these different predisposing factors.
Results
CD8+ T cells are enriched at the site of acute SJS/TEN tissue damage
The total scRNA-defined uniform manifold approximation and projection (UMAP, Fig. 1a) includes 109,888 cells, spanning 15 subtypes, including both immune and stromal subsets. Specifically, these include CD8+ and CD4+ T cells, natural killer (NK) cells, hematopoietic stem cells (HSC), B-cells, monocytes, macrophages, dendritic cells (DC), mesenchymal stromal cells (MSC), fibroblasts, endothelial cells, and keratinocytes (Fig. 1b); with the predominant representation of each of these subsets aligning to the same UMAP clusters across disease and sample types (Supplementary Fig. 1). ScCITE-seq confirmed unsupervised transcriptome-based clustering with characteristic protein expression across scRNA-defined CD3+ CD4+ and CD8+ T cells, CD56+ NK cells, CD1c+ DC, CD19+ B-cells, and CD14+ CD11b+ monocytes and macrophages (Fig. 1c). Surface phenotyping identified that few NK or T cells expressed CD3 or CD56, respectively, suggesting an absence of NKT cells, and that myeloid subsets in SJS/TEN were mostly pro-inflammatory, including DC (CD86+, CD112+) but also monocytes and macrophages (CD40+, CD54+, CD31+; Supplementary Fig. 2). Macrophages expressed a mixed pro-inflammatory M1- (CD11c+) and anti-inflammatory M2 (CD11b+, CD163+)-like surface phenotype13,14. As tissue-specific macrophages are recently described, with distinct dermal lineages involved in inflammation or repair and defined by the expression of HLA class II or LYVE1, respectively15, presenting functional resemblances of the traditional M1/M2 classification, we further investigated the signature of these cells. Macrophages in SJS/TEN blister fluid were LYVE1LO HLA class IIHI, as typically seen in inflamed tissue. Indeed, compared to macrophages in burn blister fluid, those in SJS/TEN blister fluid were significantly enriched for HLA class I, HLA class II, STAT1, interferon-induced response (IFITM1-3), and C1Q involved in the phagocytosis of apoptotic cells (Supplementary Fig. 3). However, further transcriptomic analyses confirmed an extended intermediate-like macrophage signature (CD9LO, CD163HI APOELO APOC1HI, MRC1HI)16, supporting a mixed surface phenotype and subpopulations involved in inflammation and others with repair.
Fig. 1. Unbiased multi-focal single-cell sequencing identifies expanded pathogenic CD8+ Tconv in affected skin and blister fluid during Stevens-Johnson syndrome and toxic epidermal necrolysis.

a scRNA-defined uniform manifold approximation and projection (UMAP) of 109,888 live cells from normal skin, burn blister fluid, unaffected skin, affected skin, and/or blister fluid from 15 patients with acute SJS/TEN. b Expression distribution for key cell types, including immune and stromal populations. The majority UMAP location of each subset is labeled with the percentage of each subset within that location indicated. c Integrated scCITE-seq expression of subset-defining cell surface proteins for T cells (CD3, CD4, CD8), NK cells (CD56), DC (CD1c), monocytes and macrophages (CD14, CD11c), and B cells (CD19) on a representative UMAP of 17,404 cells obtained within a single 10x run (SJS/TEN blister fluid from n = 4 patients). Heatmap expression (high to low, red to blue) for each surface protein and average box plot expression is shown between scRNA-defined subsets (n = 11,261 CD8 + T cells, 593 CD4 + T cells, 727 NK cells, 536 DC, 3177 monocytes, 1034 macrophages, 45 B cells). The bounds of the box represent the interquartile range from the 25th to 75th percentile, the center line shows the median expression, and the whiskers identify maximum and minimum values to the 10th and 90th percentile, respectively. Outliers are shown. d Individual scRNA-defined UMAPs for tissue-relevant controls from unrelated donors (burn blister fluid, normal skin) and skin and blister fluid from patients with SJS/TEN. e, f scRNA-defined cell representation. Proportional representation of (e) all subsets in individual samples (culprit drug listed for each patient) and (f) rare, unconventional, and innate-like lymphoid cell (ILC) populations as a proportion of total CD8+ T cells, CD4+ T cells, or NK cells in each sample. The average expression is shown for patients with multiple samples from the same time point, indicated by an asterisk. Figure created using Visual Genomics Analysis Studio (VGAS)89. Source data are provided as a Source Data file. MSC, Mesenchymal stromal cell; HSC, Hematopoietic stem cell; NK, Natural killer; DC, Dendritic cell; Tconv, T conventional cell; ILC, innate-like lymphoid cell; Unaff, Unaffected; Aff, Affected; SJS/TEN, Stevens-Johnson syndrome and toxic epidermal necrolysis.
We compared proportional cell type representation to determine subsets enriched in disease compared to control phenotypes (Fig. 1d). Patient samples varied in culprit drug and time since onset of reaction (Supplementary Table 1), and we theorized this might impact subset representation but were unable to detect any associations (Supplementary Fig. 4). However, we acknowledge we may have been underpowered to do so. The most prevalent immune population in SJS/TEN-affected skin and blister fluid were CD8+ T cells (Fig. 1e), which were the most significantly enriched subset in SJS/TEN blister fluid (n = 15) compared to burn blister fluid (n = 4) (scCODA, p < 0.05)17. On average, SJS/TEN blister fluid contained 70% CD8+ T cells, 12% monocytes, 8% NK cells, 4% macrophages, 4% CD4+ T cells, and 2% DC. Macrophages represented ≥ 0.5% of cells in blister fluids from 6/7 patients with co-trimoxazole-induced SJS/TEN (Supplementary Fig. 4). CD8+ T cells were also the most abundant immune subset in affected skin (18%), with a 2.6-fold increase in CD8+ T cells in affected compared to unaffected skin (7%, Fig. 1e). Consistent with previous studies, CD14+ monocytes were the predominant subset in burn blister fluid18, B-cells represented < 0.5% of cells in SJS/TEN-affected skin and SJS/TEN blister fluid19, and SJS/TEN blister fluid was an immune-rich reservoir devoid of stromal subsets19. Further, while full-thickness skin biopsies also contained immune cells, they also consisted of diverse stromal populations20, including phenotypically similar MSC and fibroblasts21. However, skin digestion methods may result in a variable loss of stromal cells from run to run, and therefore, the relative proportion of stromal cells within all cells cannot be relied upon. In contrast, immune cells are relatively robust, and we, therefore, focused on the relative proportion of immune subsets within the immune cells (Supplementary Fig. 5). These data identified similar proportions of CD4+ and CD8+ T cells in unaffected skin from a patient with SJS/TEN and normal skin from an unrelated donor. While CD4+ T cells were reduced in affected compared to unaffected SJS/TEN patient skin, the relative proportion of the predominant CD8+ T cell population remained the same across healthy, unaffected, and affected biopsies.
Conventional cytotoxic CD8+ T cells are enriched, and unconventional T cells are rare in SJS/TEN skin and blister fluid
While conventional T cells recognizing protein-derived antigens (Tconv) are implicated as the prevalent cytotoxic subset in SJS/TEN skin19, rare, unconventional, and innate-like lymphoid cell (ILC) populations are important immuno-inflammatory regulators but may not always be detected22. To elucidate effector phenotype, we realigned T- and NK cells to the Azimuth peripheral blood mononuclear cell (PBMC) reference23, which contains T regulatory (Treg) cells, ILC, and unconventional subsets including gamma delta (γδ) T cells and mucosal-associated invariant T (MAIT) cells. Although most cells did not re-align with rare, ILC, or unconventional subsets, small populations were identified (Fig. 1f). The largest of these were Tregs aligned to the CD4+ T cell subset in SJS/TEN blister fluid (16%), unaffected skin (8%), and affected skin (4%). Further, ILC comprised >10% of the NK subset in normal skin, but <5% of those in SJS/TEN-affected skin and blister fluid. In the broadly defined CD8+ T cell subset, ILC and unconventional populations each represented ≤1% of cells in SJS/TEN blister fluid. Interestingly, distinct γδ T cell subsets have recently been characterized, including IFN- (γδT1) and IL-17-responding subsets (γδT17) of particular importance in skin24. In SJS/TEN blister fluid, γδ T cells did not express IL-17-related genes, but CD27, IFITM1, and IFITM2 were highly expressed, as was seen in the CD8+ Tconv subset (Supplementary Fig. 6), suggesting co-stimulation of a limited IFN-responding γδT1-like subset. However, overall, 94% and 98% of CD8+ T cells in SJS/TEN-affected skin and blister fluid, respectively, were defined as Tconv (Fig. 1f).
The same cytotoxic CD8+ Tconv subpopulations are seen in SJS/TEN-affected skin and blister fluid
While the relative proportion of CD8+ T cells remained similar across healthy, unaffected, and affected skin biopsies (Supplementary Fig. 5), we next assessed whether there may be distinct signatures and proportions of CD8+ T cell subsets driving the pathology of SJS/TEN at the site of tissue damage. Within the majority CD8+ Tconv population, eight Seurat-defined clusters (Fig. 2a) shared cluster marker genes (Supplementary Table 2) associated with quiescence/inactivation (clusters 1, 2, 4; ZFP36, BTG1, EEF1A1, TXNIP, AREG and/or IL7R)25, cytotoxicity, activation, and proliferation (cluster 3; GNLY, GZMB, STMN1, PRF1, CD27, GAPDH), migration and post-transcriptional regulation of the inflammasome (cluster 5; NEAT1, MALAT1, FMNL1, CBLB, INPP5D, USF2, MYH9, IKNA, IKZF3)26, and innate-like differentiation (clusters 5–8; TYROBP, FCER1G)27. Notably, ‘innate-like’ clusters were represented by few cells and aligned to UMAP positions associated with other subsets, suggesting that they may be physically interacting cells (PICs)28. ‘Inflammasome-related’ cluster 5 had a high proportion of cells identified as dead or dying during QC filtering (see “Methods”) and was similarly represented in burn (4%) and SJS/TEN blister fluid (5%, Fig. 2a), suggesting a highly-stressed population consistent with generalized inflammation. ‘Inactive’ cluster 1 showed high expression of mitochondrial genes and top genes associated with T cell suppression (TXNIP, BTG1, AREG) without cytotoxic activation, suggesting cell death through basal turnover, and represented < 1.5% of CD8+ Tconv in normal, affected, and unaffected skin. ‘Inactive’ clusters 2 and 4 had high expression of IL7R, which is downregulated upon antigen-induced activation29, indicating populations unstimulated by the drug antigen during SJS/TEN. Indeed cluster 2 was the predominant CD8+ Tconv subset in normal skin, unaffected skin, and burn blister fluid (Fig. 2a). In contrast, cluster 3 expressed cytotoxic markers GNLY, GZMB, and PRF1, but not FASLG, and was the predominant subset in SJS/TEN-affected skin and blister fluid (Fig. 2a).
Fig. 2. Pathogenic CD8+ Tconv are ITGAE +/− cytotoxic tissue-resident memory-like populations.
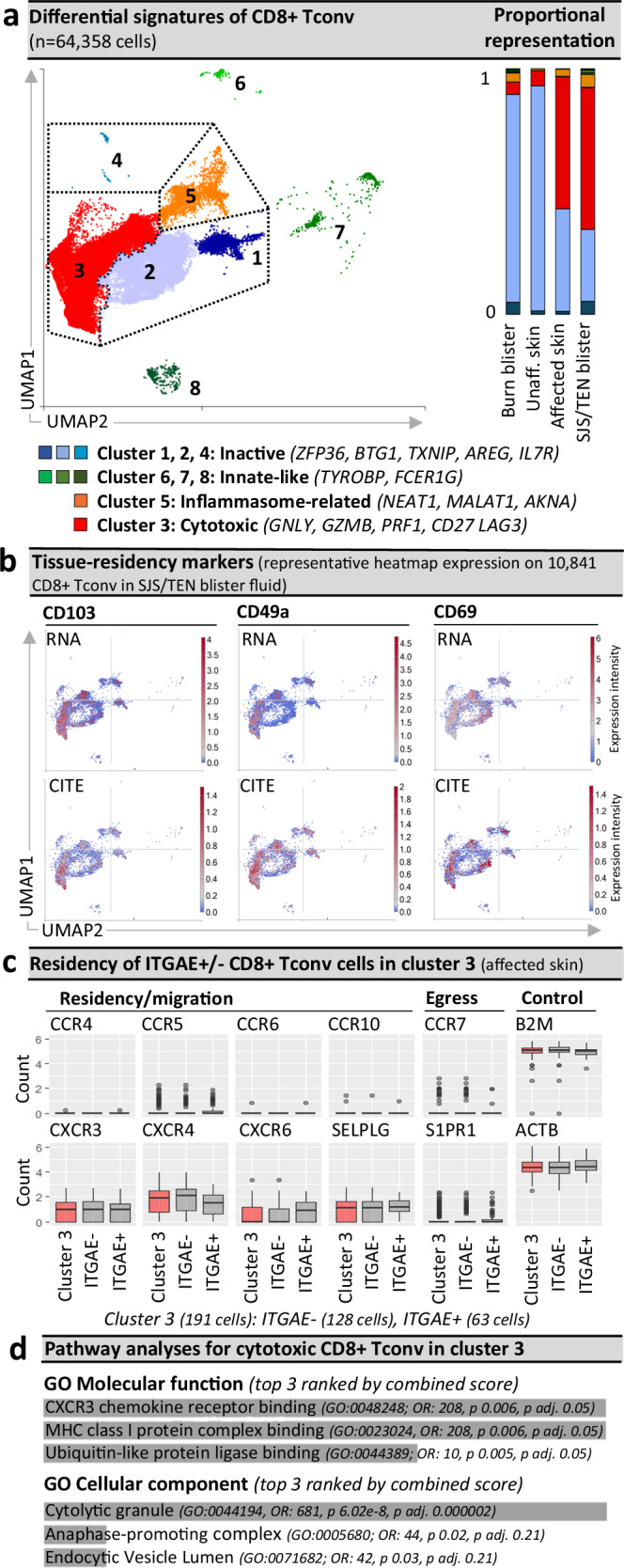
a Seurat-defined CD8+ Tconv clusters 1–8 across cells from normal skin (n = 1 donor), burn blister fluid (n = 4 patients), and unaffected and affected SJS/TEN skin (n = 1 patient) and SJS/TEN blister fluid (n = 15 patients; total n = 64,358 cells: cluster 1, n = 3295 cells; cluster 2, n = 19,761 cells; cluster 3, n = 36,428 cells; cluster 4, 60 cells; cluster 5, 3395 cells; cluster 6, 280 cells; cluster 7, 743 cells; cluster 8, 381 cells) with shared naming according to differential cluster makers. Clusters with < 20 cells were removed from cluster marker analyses (n = 15 cells). A proportional representation of CD8+ Tconv clusters in control and disease sample phenotypes is shown. b Heatmap expression (high to low, red to blue) of RNA and surface protein for ITGAE (CD103), ITGA1 (CD49a), and CLEC2C (CD69) on CD8+ Tconv of SJS/TEN blister fluid. UMAP shows representative 10,841 CD8+ Tconv cells obtained within a single 10x run. c Box plot expression of interest genes aligned with tissue residency, migration, and egress on ITGAE+ and ITGAE- CD8+ Tconv cluster 3 of affected skin (n = 1 patient, 128 ITGAE- cells, 63 ITGAE+ cells). Expression of housekeeping genes B2m and ACTB are shown as a control. For box plots, the bounds of the box represent the interquartile range from the 25th to 75th percentile, the center line shows the median expression, and whiskers identify maximum and minimum values to the 10th and 90th percentile, respectively. Outliers are shown. d Pathway analysis using the significantly enriched cluster markers genes of CD8+ Tconv cluster 3. All cluster marker genes above significance and 0.6 log2 fold change 0.6(log2FC) were included. Analyses were performed using Enrichr90 and Gene Ontology (GO) molecular and cellular terms, which were ranked by combined score. Figure created using Visual Genomics Analysis Studio (VGAS)89. Source data are provided as a Source Data file. Tconv, T conventional cell; GO, Gene Ontology; OR, odds ratio; p adj., adjusted p-value; CITE, cellular indexing of transcriptome and epitopes; SJS/TEN, Stevens-Johnson syndrome and toxic epidermal necrolysis.
The cytotoxic CD8+ Tconv enriched in SJS/TEN affected skin and blister fluid are locally proliferating CD8+ TRM T cells
Aside from markers of cytotoxicity, CD8+ Tconv cluster 3 also expressed genes associated with cell remodeling and/or proliferation (STMN1, TUBA1B, TUBB), and expressed RNA and protein for CD103 (ITGAE), CD49a (ITGA1), and CD69 (CLEC2C) (Fig. 2b), suggesting a population of expanded cytotoxic CD8+ tissue-resident memory (TRM) T cells30. Importantly, our data mirror the minor representation of similarly cytotoxic CD8+ TRM T cells reported in published scRNA-seq datasets of full-thickness skin biopsies from healthy individuals31,32, and this population has demonstrated the capacity to proliferate in response to local antigen encounter in the skin33. However, not all cells in the cytotoxic CD8+ Tconv cluster expressed the same TRM-defining markers. While classical TRM markers are not mutually exclusive, with both ITGAE+ and ITGAE- TRM T cells previously defined in the skin and associated with motility or maturation, respectively34, ITGAE- cells can also represent tissue-infiltrating populations from the blood. Thus, to define the origin of this cytotoxic population we performed comparative analyses of ITGAE +/− cells in the cytotoxic CD8+ Tconv cluster of affected skin (Fig. 2c). No significantly differentially expressed genes (DEG) were identified, and all cells co-expressed CXCR3, CXCR4, and SELPLG; associated with both TRM and non-TRM populations35,36. However, while the TRM retention marker CXCR6 was more highly expressed by ITGAE+ cells, we did not identify expression of CCR4 and CCR10, typically co-expressed with SELPLG by tissue-infiltrating skin-homing T cells37, nor tissue egress markers CCR7 or S1PR138 (Fig. 2c). Since the absence of SIPR1 is a requirement for TRM establishment39, these data suggest that all cells of the cytotoxic CD8+ Tconv cluster share a similar and highly cytotoxic TRM-like signature and are retained in the affected tissue. Indeed, immunohistochemistry confirmed that cytotoxic (GNLY+) CD8+ CD103+ TRM T cells line the basement membrane in SJS/TEN-affected skin (Supplementary Fig. 7a). To better understand whether these cells mapped to the cytotoxic cluster, we performed spatial sequencing on a section of SJS/TEN affected skin, and the typical histopathological features of SJS/TEN were seen (Supplementary Fig. 7b). These included a dense lymphocytic population predominating in the dermis, no evidence of eosinophilia, basal keratinocyte disruption with separation of the epidermis from the dermis, and formation of a subepidermal cleft. Importantly, spatially-resolved regions of interest (ROI) nearest the detached epidermis (ROI 1, ROI 9) had the highest transcriptomic expression of CD8+ T cells (CD3E, CD8A; Supplementary Fig. 7c) but also specific markers of the cytotoxic CD8+ Tconv cluster associated with cytotoxicity (GZMB), regulation (LAG3), and activation (CD27, Supplementary Fig. 7c). Pathway analyses of cluster marker genes for the cytotoxic CD8+ Tconv cluster identified processes including HLA class I-binding and the release of cytolytic granules (Fig. 2d). Together these data suggest the pathogenic cytotoxic CD8+ Tconv cluster are a population of locally-proliferating TRM T cells, which are specifically implicated in cytotoxic keratinocyte cell death and blister formation.
The same expanded TCRαβ CD8+ T cell clonotypes are observed in SJS/TEN-affected skin and blister fluid but are unexpanded in unaffected skin
Clonally-expanded HLA class I-restricted TCRαβ CD8+ T cells are implicated as the drug antigen-driven effectors of SJS/TEN7,8. To define signatures of clonally-expanded CD8+ Tconv and whether those in blister fluid represent those in the affected skin, we assessed time-paired samples from a single untreated patient (Fig. 3), including unaffected and affected skin and three blister fluid samples from the arm, face, and foot (Fig. 3a). Initial analyses of total CD8+ Tconv in affected compared to unaffected skin identified downregulated DEG associated with T cell inactivation and quiescence (TSC22D3, ZFP36, ZFP36L2)40, and significantly upregulated DEG (Fig. 3b and Supplementary Table 3) associated with cytotoxicity (GNLY), activation/differentiation (CD27), regulation (LAG3), and the immunoproteasome (PSMB9). In contrast, no genes were significantly enriched in the NK cells of the affected compared to unaffected skin. However, in keratinocytes, downregulated genes matched those similarly downregulated in keratinocytes during psoriasis (GADD45B, MT1X, DUSP1, ZFP36, MYC, BTG2)41 including a regulatory circuit of chronic inflammation (CEBPD, ATF3)42. Moreover, upregulated genes were associated with stress-induced migration (GSTP1, KRT6B), IFN-induced response (IFITM3, IFI6), and HLA class I-associated (HLA-B, HLA-C) proteasomal activation (PSME1, PSME2, PSMB9), suggesting increased capability to present CD8+ T cell-directed epitopes (Fig. 3b and Supplementary Table 3).
Fig. 3. Shared oligoclonal TCRαβ clonotypes on cytotoxic CD8+ Tconv in affected skin and blister fluid are absent from or unexpanded in unaffected skin.

a UMAPs for paired unaffected and affected skin and blister fluid samples from a single patient with SJS/TEN. b Differential gene expression (two-tailed Wilcoxon, Hochberg adj, p < 0.05) between keratinocytes or CD8+ Tconv cells from unaffected and affected skin (n = 1 patient with time-paired samples; unaffected skin keratinocytes, n = 331 cells; unaffected skin CD8+ Tconv, n = 142 cells; affected skin keratinocytes, n = 108 cells; affected skin CD8+ Tconv, 356 cells). Genes colored red are significantly (p < 0.05) increased (light red < 0.6 log2 fold change (log2FC), dark red > 0.6log2FC). The top 10 genes are labeled. c Top functional TCR CDR3αβ pairings and counts (Ct.) in CD8+ Tconv cells. The same top TCR clonotypes across affected samples are highlighted in gray. d Signatures of dominant TCR+ CD8+ Tconv align to the cytotoxic Tconv cluster. Expression of the top 3 dominantly-expanded TCR (black) on CD8+ Tconv cells of each sample. The number of dominant TCR+ cells aligned to CD8+ Tconv cluster 2 (control) and cluster 3 (cytotoxic) are indicated numerically. e Differential gene expression (two-tailed Wilcoxon, Hochberg adj, p < 0.05) between CD8+ Tconv expressing dominant TCRs or other TCRs in affected skin or blister fluids (n = 1 patient with time-paired samples; affected skin DomTCRNEG, 192 cells; affected skin DomTCRPOS, n = 47 cells; blister fluid DomTCRNEG, 2441 cells; blister fluid DomTCRPOS, n = 559 cells). Genes colored red are significantly (p < 0.05) increased (light red < 0.6log2FC, dark red > 0.6log2FC). The top 10 genes are labeled. Figure created using Visual Genomics Analysis Studio (VGAS)89. Source data are provided as a Source Data file. TCR, T cell receptor; Dom TCR, Dominantly-expanded TCR; MSC, Mesenchymal stromal cell; HSC, Hematopoietic stem cell; NK, Natural killer; DC, Dendritic cell; Tconv, T conventional cell; CDR3, Complementary determining region 3; Ct., count; FDR, false discovery rate; FC, fold change; Dom TCR, dominant TCRs; SJS/TEN, Stevens-Johnson syndrome and toxic epidermal necrolysis.
Importantly, while HLA class I was also upregulated in macrophages, endothelial cells, fibroblasts, and MSC of the affected skin (Supplementary Fig. 8a), the checkpoint receptor LAG3 was highly upregulated in cells of the cytotoxic CD8+ Tconv cluster. Thus, as immune checkpoint receptors are known to regulate HLA-restricted drug-induced T cell activation43, we investigated whether decreased expression of LAG3 ligands (HLA class II, FGL1, CLEC4G, LGALS3) may identify disease-susceptible cell populations (Supplementary Fig. 8b). Notably, while CLEC4G and FGL1 were not expressed across samples, and HLA class II was high or upregulated in antigen-presenting cells of affected skin including fibroblasts and MSC, suggesting a role in immune regulation, LGALS3 was among the top 25 most downregulated genes by fold change in keratinocytes of affected skin, which was significant before false discovery rate (FDR) correction (Supplementary Fig. 8b). A protective role for LGALS3-LAG3 has been described to confer cellular resistance to apoptosis44, and the LAG3-LGALS3 interaction is known to be co-inhibitory to CD8+ T cells45. The expression of LGALS3 remained high across other stromal subsets in affected skin. These observations suggest that a decrease in the expression of LGALS3 may facilitate the selective targeting of keratinocytes for LAG3+ cytotoxic CD8+ Tconv-mediated death in SJS/TEN.
Across paired samples from this patient, a productive TCRαβ pair was identified for 66% and 55% of CD4+ and CD8+ T cells, respectively. The most highly expressed TCRαβ variable genes across CD8+ Tconv of affected samples were TRAV19 and TRBV27 (Supplementary Fig. 9), and analyses of the paired complementary-determining region (CDR)3αβ identified the same top three dominantly-expanded TCRs across affected skin and blister fluids (Fig. 3c and Supplementary Fig. 10a, b). These clonotypes were not expanded in unaffected skin (Fig. 3c) nor expressed by CD4+ Tconv cells. The identification of oligoclonal clonotypes in this patient was representative of CD8+ Tconv in blister fluid from all patients with SJS/TEN driven by different drugs (Supplementary Fig. 11a). Interestingly, in this patient, the three dominant clonotypes had similar CDR3 sequences, with two sharing a CDR3β (CASSPDRGGYEQYF) but different CDR3α and these clonotypes were absent from unaffected skin. A third clonotype, expressed on one cell in unaffected skin (Supplementary Fig. 10), had one CDR3β mismatch (CASSQDRGGYEQYF). GLIPH246 predicted that all three TCRs share peptide binding specificity (p = 7.90E-15). Notably, these three dominantly-expanded clonotypes were expressed in the same UMAP location, with the majority of those in affected skin (> 97%) and blister fluid (> 90%) aligned to the cytotoxic CD8+ Tconv cluster (Fig. 3d). This was also the predominant location of dominant TCR+ cells identified in all other patients (Supplementary Fig. 11b), indicating that the cytotoxic CD8+ Tconv cluster represents a common effector phenotype across patients with drug-induced SJS/TEN, even if they use private TCR CDR3αβ clonotypes.
Importantly, compared to CD8+ Tconv cells expressing all other TCR, the same DEG signature (Supplementary Table 4) of dominantly-expanded TCR+ CD8+ Tconv in affected skin and blister fluid (Fig. 3e) aligned with mature cytotoxic effectors (GNLY, GZMB, PRF1, FCGR3A), cytolytic granule exocytosis (RAB27A), and active regulation (TNFRSF9, LAG3). Furthermore, the most downregulated DEG in dominant TCR+ CD8+ Tconv included ZFP36L2 and BTG1, aligned with T cell quiescence/inactivation47, and IL7R, which is downregulated upon antigen-driven TCR-dependent activation29. These data support the model that dominant TCR-expressing CD8+ Tconv are the clonally-expanded antigen-driven cytotoxic cells in the skin and enriched in blister fluid.
Intriguingly, some CD8+ Tconv cells appeared to express more than one TCRαβ chain. For example, the two dominantly-expanded TCRs with the same CASSPDRGGYEQYF CDR3β were predominantly expressed with a second CDR3α on the same CD8+ Tconv cells in blister fluid (221 cells, Supplementary Fig. 12a, b). Cells expressing the third clonotype did not express a second TCR. We considered the possibility that T cells that appeared to express more than one TCRαβ were, in fact, two or more PICs, as suggested by the work of Sun et al. 48. We found that these “dual expressing TCRαβ cells” had higher gene and unique molecular identifier (UMI) counts (Supplementary Fig. 13) than single TCRαβ-expressing T cells, indicating that at least some of these are PICs. These data were mirrored in affected skin (Supplementary Fig. 12b, c); suggesting the possibility that dual TCRαβ+ PIC T cells28,49 play a role in the auto-presentation of drug neoantigen, contributing to the pathogenesis of SJS/TEN.
Expanded oligoclonal and unexpanded polyclonal CD8+ T cells contribute to the acute immunopathogenesis of SJS/TEN
We assessed the total clonality of the cytotoxic CD8+ Tconv cluster, where the three dominantly-expanded TCR showed similar representation among the top 50 clonotypes in affected skin (48%) and blister fluid (average 51%, Fig. 4a). In the affected skin, while up to 32% of cells expressed expanded clonotypes, > 50% expressed unexpanded TCR (n = 1 count, Fig. 4b). Using blister fluid, we investigated whether T cells in the cytotoxic CD8+ Tconv cluster with unexpanded clonotypes had a distinct phenotype. Expanded oligoclonal clonotypes were significantly enriched for cytotoxicity-related genes, including GNLY, PRF1, and FCGR3A, but unexpanded polyclonal clonotypes for GZMA and GZMK (Fig. 4c). Compared to TCRαβ cells from inactive/quiescent cluster 2, unexpanded polyclonal clonotypes in the cytotoxic cluster remained significantly enriched for GNLY, PRF1, and FCGR3A, albeit less than expanded oligoclonal clonotypes, which were uniquely enriched for markers of activation (TNFRSF9), long intergenic non-coding (linc) RNAs (LINC01943), and immunomodulatory genes (TIGIT, KLRC1, KLRD1, Fig. 4d).
Fig. 4. Cells in the cytotoxic CD8+ Tconv cluster consist of TCR-dependant expanded and unexpanded clonotypes.

a–c Clonality of cells in the cytotoxic CD8+ Tconv cluster in affected skin and blister fluid samples from a single patient with SJS/TEN. a Clonality of cells in the cytotoxic CD8+ Tconv cluster. The top 50 TCRs are shown to demonstrate a similar expanded representation of the top TCRs. The percentage indicates the proportion of counts aligned to the top 3 dominantly-expanded TCRs. Circos plot segment width is proportionate to dominance (increasing green to red). b Proportional expression of dominantly-expanded TCRαβ (TCR+) cells (red) and unexpanded clonotypes (green, n = 1 count) in all cells of the cytotoxic CD8+ Tconv cluster across affected skin (n = 144 cells) and blister fluid samples (blister fluid 1, n = 919 cells; blister fluid 2, n = 379 cells; blister fluid 3, n = 532 cells). c Differential gene expression signatures (two-tailed Wilcoxon, Hochberg adj, p < 0.05) between expanded or unexpanded TCRαβ (TCR+) cells of the cytotoxic CD8+ Tconv cluster in SJS/TEN blister fluid. Genes colored red are significantly (p < 0.05) increased (light red < 0.6log2FC, dark red > 0.6log2FC). The top 10 genes are labeled. d, e Expression of functionally grouped genes and protein. d Expression of DEG and interest TCR-activation-related genes, and (e) surface scCITE-seq surface protein for CD127 between cells of the cytotoxic Tconv cluster expressing expanded TCR (red, n = 507 cells) or unexpanded TCR (green, n = 652 cells) compared to cells expressing TCR from non-cytotoxic Tconv cluster 2 (control; white, n = 999 cells) in a single patient with SJS/TEN. For box plots, bounds of the box represent the interquartile range from the 25th to 75th percentile, the center line shows the median expression, and whiskers identify maximum and minimum values to the 10th and 90th percentile, respectively. Outliers are shown. *Indicates significant differential expression (two-tailed Wilcoxon, Hochberg adj, p < 0.05 and >0.6 log2FC). Figure created using Visual Genomics Analysis Studio (VGAS)89. Source data are provided as a Source Data file. TCR, T cell receptor; Dom TCR, Dominantly-expanded TCR; Tconv, T conventional cell; CDR3, Complementary determining region 3; FDR, false discovery rate; FC, fold change; LincRNA, long intergenic non-coding RNA; Unaff, Unaffected; Aff, Affected; SJS/TEN, Stevens-Johnson syndrome and toxic epidermal necrolysis.
In the cytotoxic CD8+ Tconv cluster of SJS/TEN blister fluids, while unexpanded T cells expressed diverse CDR3αβ, many expressed the same TRBV27/TRBJ2-7 of dominantly-expanded clonotypes (Supplementary Fig. 14). In fact, several polyclonal unexpanded T cells expressed the dominantly-expanded CDR3β (CASSPDRGGYEQYF) or had one amino acid mismatch (P to H/V/Y/F/L/R). Again, these clonotypes were expressed on single cells expressing two TCRs (Supplementary Table 5), and GLIPH246 predicted shared peptide binding (S_DRGGYE CDR3β motif, fisher p = 7.90E-15). Notably, the dominantly-expanded CASSQDRGGYEQYF CDR3β was not identified on polyclonal T cells, and the polyclonal T cells with a shared CDR3β were only observed in blister fluids and not affected skin. We cannot be sure if this represents a clonal bias between the skin and overlying blister fluid or whether there has been an incomplete capture of the TCR repertoire in the affected skin. Either way, we can be more confident that oligoclonal TCR CDR3αβ expansion that is seen in both blister fluid and affected skin is more likely to be directly relevant to the killing of keratinocytes and most worthy of further investigation. Indeed, between blister fluids, only up to 10% of polyclonal T cells in the cytotoxic CD8+ Tconv cluster shared a TCR, raising the possibility that at least some had not been triggered by the initiating drug-neo-antigen but either by ‘bystander’ cytokine activation50 or a broader array of new antigens produced by the pathological process. However, both oligoclonal and polyclonal T cells significantly downregulated IL7R compared to those in the control cluster 2, indicative of TCR-dependent activation29, and remained KLRK1LO, for which upregulation is associated with TCR-independent activation50 (Fig. 4d). Downregulation of CD127 (IL7R) was mirrored by scCITE-seq expression (Fig. 4e).
Cell communication analyses support the direct HLA class I presentation of epitopes by keratinocytes to cytotoxic CD8+ T cells
To predict interactions between keratinocytes and cells in the cytotoxic CD8+ Tconv cluster we utilized CellChat (Fig. 5a; a bioinformatic cell-cell signaling tool51). Among the highest-scoring interactions were HLA-B and HLA-C on keratinocytes with CD8A on cells of the cytotoxic CD8+ Tconv cluster (Fig. 5a). Further, the interaction for KLRC1/KLRD1 with beta-2-microglobulin (B2M) was specifically predicted for the dominant TCRαβ population, supporting the hypothesis that keratinocytes are directly responsible for HLA class I-mediated antigen presentation to cytotoxic T cells, with the dominantly-expanded population under specific regulation by KLRC1/KLRD1.
Fig. 5. Cell communication and pathway analysis tools predict receptor-ligand interactions and the role of accessory populations during SJS/TEN.

a, b Cellchat communication analyses. a Highly ranked receptor-ligand interaction between keratinocytes and TCRαβ(TCR+)cells of the cytotoxic CD8+ Tconv cluster, including those expressing dominantly-expanded TCR+ cells. LIANA was used to rank and visualize the highest-scoring receptor-ligand interactions from CellChat. Increasing dot size indicates a more significant p-value for the specificity of an interaction to a given pair of cell groups, with coloring blue to yellow indicating higher bioinformatic ligand-receptor (Lr) probability as a measure of the magnitude of interaction strength. p-values are computed from the one-sided permutation test in CellChat. b Box plot expression of key receptors and ligands implicated by Cellchat on keratinocytes and cells of the cytotoxic CD8+ Tconv cluster in unaffected and affected SJS/TEN skin from a single patient with time-paired samples. Cells are shown for unaffected skin (keratinocytes, 331 cells; CD8+ Tconv, 142 cells; cytotoxic CD8+ Tconv, 9 cells) and affected skin (keratinocytes, 108 cells; CD8+ Tconv, 356 cells; cytotoxic CD8+ Tconv, 191 cells, cytotoxic CD8+ DomTCR, 46 cells). *Indicates significant differential expression (two-tailed Wilcoxon, Hochberg adj, p < 0.05 and > 0.6 log2FC). Significantly increased expression in keratinocytes of affected skin was identified for HLA-B (Pc = 5.19E-05), HLA-C (Pc = 8.66E-12), and CD74 (Pc = 1.18E-34). CXCR4 was significantly downregulated in CD8+ Tconv of affected skin (Pc = 1.29e-11). c–e PROGENy pathway analyses. c Comparative heatmap expression (blue to red, low to high) of key cellular pathways in diverse subsets of unaffected skin (CD8+ Tconv, 142 cells; CD4+ Tconv, 118 cells; endothelial cells, 558 cells; fibroblasts, 594 cells; MSC, 673 cells; keratinocytes, 331 cells; macrophages, 18 cells; NK cells, 67 cells; monocytes, 45 cells) and affected skin (CD8+ Tconv, 356 cells; CD4+ Tconv, 155 cells; endothelial cells, 177 cells; fibroblasts, 219 cells; MSC, 578 cells; keratinocytes, 108 cells; macrophages, 21 cells; NK cells, 136 cells; monocytes, 220 cells) from a single SJS/TEN patient. Expression of JAK and STAT genes in (d) keratinocytes and cells of the cytotoxic CD8+ Tconv cluster, including those expressing dominantly-expanded TCR clonotypes, and (e) fibroblasts and MSC of time-paired unaffected and affected skin from a single SJS/TEN patient. *Indicates significant differential expression (two-tailed Wilcoxon, Hochberg adj, p < 0.05 and > 0.6 log2FC). Significantly increased expression in fibroblasts of affected skin was identified for STAT1 (Pc = 4.63E-68), STAT2 (Pc = 1.85E-27), and STAT3 (Pc = 6.41E-09). STAT1 was significantly upregulated in MSC (Pc = 5.55E-10) of affected skin, and significantly upregulated in cytotoxic CD8+ Tconv of affected skin (pc = 0.00073) compared to CD8+ Tconv of unaffected skin. Figure created using Cellchat51, LIANA91, PROGENy56, and Visual Genomics Analysis Studio (VGAS)89. For all box plots, the bounds of the box represent the interquartile range from the 25th to 75th percentile, the center line shows the median expression, and the whiskers identify maximum and minimum values to the 10th and 90th percentile, respectively. Outliers are shown. Cells are shown for unaffected skin (keratinocytes, 331 cells; all CD8+ Tconv, 142 cells; cytotoxic CD8+ Tconv, 9 cells; fibroblasts, 594 cells; MSC, 673 cells) and affected skin (keratinocytes, 108 cells; all CD8+ Tconv, 356 cells; cytotoxic CD8+ Tconv, 191 cells, cytotoxic CD8+ DomTCR, 46 cells; fibroblasts, 219 cells; MSC, 578 cells). Source data are provided as a Source Data file. Pc, corrected p-value; DomTCR, dominantly-expanded T cell receptors; Tconv, T conventional cell; Lr_probs, ligand-receptor probability; Unaff, Unaffected; Affect, Affected; SJS/TEN, Stevens-Johnson syndrome and toxic epidermal necrolysis, MSC, mesenchymal stromal cell.
Of note, although less probable than the interaction with HLA class I, CD8A was also predicted to bind to HLA-E, which like HLA class I was increased (albeit non-significantly) in keratinocytes of affected compared to unaffected skin. These data are consistent with a study by Morel, who reported that HLA-E expression was increased in keratinocytes of SJS/TEN patients and that soluble HLA-E is released into the blister fluid52. However, while the authors suggested that HLA-E bound to the activating CD94/NKG2C receptor to trigger cytotoxic lymphocytes, NKG2C was not expressed by T cells or NK cells in our dataset. Further, the alternate interaction of HLA-E with inhibitory CD94/NKG2A (KLRC1/KLRD1) was deemed less probable than the direct interaction of HLA-E and CD8A. Importantly, while the conventional role of HLA-E is to present signal sequence-derived peptides of HLA class I to inhibit NK and T cell-mediated cell lysis via recognition by CD94/NKG2A, more recently, a second unconventional role is described for the direct HLA-E-mediated presentation of both self- or pathogen-derived peptides to TCRαβ T cells53,54. This interaction has broad primary anchor pocket tolerability, suggesting a potentially diverse peptide repertoire54, and similarities with residues forming the heavily conserved co-receptor-binding flexible loop region in the α3 domain of HLA class I propose the interaction of HLA-E with the CD8 co-receptor53. Thus, alongside the role of HLA class I, these data support the potential for direct recognition between HLA-E and the TCRαβ (near CD8A) and the susceptibility of keratinocytes to lysis by HLA-E-restricted cytotoxic T cells, warranting future studies of both HLA class I and HLA-E-restricted epitopes.
Other interactions included PKM-CD44, a metabolic switch for aerobic glycolysis55, and MIF with each of its receptors: CD74, CXCR4, and CD44 (Fig. 5a). Importantly, while PKM was upregulated by CD8+ Tconv of affected compared to unaffected skin (albeit non-significantly after correction for multiple comparisons), CD44 was not expressed by keratinocytes, suggesting that the relevant interaction is between CD44 on CD8+ Tconv and PKM on keratinocytes (Fig. 5b). In contrast, MIF may drive bidirectional signaling, with high expression across both keratinocytes and cells in the cytotoxic CD8+ Tconv cluster of affected and unaffected skin. While CD74 was the most upregulated DEG in keratinocytes of affected skin, alternate MIF ligands CD44 and CXCR4 were only expressed by CD8+ Tconv, with CXCR4 significantly downregulated on CD8+ Tconv of affected compared to unaffected skin (Fig. 5b). Functional studies are now warranted to ascertain the relevant pathogenic interaction with MIF.
Finally, although the interaction between CD8+ cytotoxic T cells and keratinocytes represents a key immunopathogenic pathway in SJS/TEN, the role of other cells in the skin has not been reported, and we use PROGENy56 to identify common core pathway responsive genes across populations (Fig. 5c). In support of the recently reported efficacy of etanercept10, TNF was upregulated across CD4+ and CD8+ T cells, NK cells, and macrophages of the affected skin, implicating a more general role in the immunopathogenesis of SJS/TEN (Fig. 5c). MSC upregulated TGFβ, which drives tissue residency of T cells and the differentiation of MSC into fibroblasts, and macrophages upregulated PI3K and downregulated androgenic pathways (Fig. 5c), previously associated with M1 to M2-like transition towards cell repair57. This is consistent with our earlier surface and transcriptomic phenotyping of an emerging intermediate-like macrophage population. Hormonal signaling was also modified in keratinocytes, with estrogen and EGFR signaling upregulated in affected skin (Fig. 5c). Although EGFR has various roles in skin homeostasis, wound repair, and innate defense, upregulation of estrogens and EGFR is recently described in keratinocytes of lesional skin during atopic dermatitis and associated with pro‐inflammatory signaling58. Importantly, JAK-STAT signaling was upregulated in CD8+ Tconv of the affected skin, with the highest expression in dominant TCRαβ cells, aligned with activation and clonal expansion. JAK-STAT signaling was also increased in fibroblasts of affected skin (Fig. 5c), mirroring signatures of cancer-associated fibroblasts involved in immune cross-talk and tumor progression59. As selective JAK and STAT inhibitors are recently described, we analyzed individual JAK and STAT expression in keratinocytes, CD8+ Tconv (Fig. 5d), fibroblasts, and MSC (Fig. 5e). On comparing cytotoxic CD8+ Tconv of affected skin compared to CD8+ Tconv of unaffected skin, while JAK1 and STAT3 were also expressed, only STAT1 was significantly increased. Furthermore, in affected skin, STAT1 was significantly upregulated in MSC, and STAT1, STAT2, and STAT3 in fibroblasts.
Discussion
Using a multi-omic single-cell approach, we found clonally-expanded cytotoxic CD8+ TRM T cells at the site of SJS/TEN tissue damage. Specifically, we observed the same cytotoxic CD8+ T cell subpopulation expressing GNLY, GZMB, PRF1, LAG3, CD27, TIGIT, and LINC01871, but not IL7R or FASLG, in blister fluid and affected skin of patients independent of the causal drug or likely HLA risk allele. We propose these cells represent a common pathogenic effector population, which expressed expanded private TCRαβ clonotypes in affected skin and blister fluids but not in unaffected skin. These data are consistent with a previously reported shared drug-expanded TCRαβ polycytotoxic cluster defined across patients with varied drug causality and HLA risk alleles by Villani using a 16-marker flow panel60. Here, we provide an unbiased analysis of this and interacting populations at single-cell resolution to define a shared CD8+ TRM-like cytotoxic cluster with both expanded and unexpanded TCRαβ clonotypes (Fig. 6).
Fig. 6. A common pathogenic population of locally proliferating cytotoxic CD8+ LAG3+ TRM T cells with private expanded and unexpanded TCRαβ clonotypes drives keratinocyte-specific cell death across patients with diverse HLA-restricted drug-induced SJS/TEN.

Predisposition: To develop SJS/TEN, the patient must be exposed to the drug neo-antigen (Step 1: structural risk) and carry a particular HLA risk allele for that drug. The risk HLA allele is necessary but not sufficient for the onset of SJS/TEN, and other currently undefined genetic risk factors may also contribute to disease predisposition (Step 2. Complete genetic risk). The patient must also have cytotoxic CD8+ TRM T cells in the skin and mucous membranes with TCR specificity for the HLA risk-restricted drug-neo-antigen. These cells do not recirculate (Step 3. HLA- and drug neoantigen-specific tissue-resident CD8+ T cells provide the tissue specificity of SJS/TEN). While the drug, HLA, and TRM clonotype may be different between individual patients, the resulting cellular response is shared. Cellular response: Drug-neoantigen is presented by keratinocytes and potentially other stromal cells in the skin by the HLA class I risk allele. HLA class I risk-restricted CD8+ LAG3+ TRM T cells with specificity for the risk HLA-drug neoantigen complex proliferate locally in the skin. In response to local proliferation and inflammation, keratinocytes regulate pathways associated with regulation and death resistance, enabling keratinocyte-specific targeting by cytotoxic LAG3+ CD8+ TRM T cells. The HLA class I-restricted and clonally-expanded cytotoxic CD8+ TRM population initiates keratinocyte death. An unknown antigen or antigens then triggers a similar population of unexpanded cytotoxic CD8+ TRM T cells. We hypothesize that this is either a range of alternate drug neoantigens or a broader array of new antigens produced by the pathological process, and these unexpanded but cytotoxic clonotypes may enhance cytotoxic keratinocyte death. Keratinocyte death leads to epidermal separation and the formation of a sub-epidermal cleft and blister. Both expanded and unexpanded cytotoxic CD8+ TRM T cells enter the cleft and become concentrated in the blister fluid, with eventual separation of the epidermis from the dermis. Repair: Pro-inflammatory M1 and intermediate-like macrophages increase PI3K and androgen signaling towards anti-inflammatory populations associated with re-epithelization and repair. MSC increase TGFβ signaling and differentiate into fibroblasts which express JAK-STAT pathways and are associated with wound repair. TGFβ is also known to promote T cell tissue residency. Created in BioRender. Phillips, E. (2024) BioRender.com/n10c928. APC, antigen-presenting cell; HLA, human leukocyte antigen; TCR, T cell receptor; TRM, tissue-resident memory, MSC, mesenchymal stromal cell.
Expanded clonotypes were enriched for KLRD1, KLRC1, TNFRSF9, and FCGR3A, suggesting that they are terminally-differentiated effectors driving antigen-specific response61–63. Indeed, although KLRC1 and KLRD1 form the inhibitory NKG2A/CD94 receptor, their expression on otherwise activated T cells is shown only to limit excessive activation to sustain the immune response63. Further, expanded clonotypes are expressed (i) under the influence of LINC01871, associated with TCR-dependent cytotoxicity in Sjogren’s disease64, (ii) on a subset of T cells expressing two TCRαβ, associated with auto- and allo-cross-reactivities49, and (iii) in the same pathogenic cluster as diverse polyclonal T cells, with a more limited but nonetheless cytolytic signature consistent with antigen-induced activation50. These data support a ‘selective-signaling’ model of TCR-triggered activation whereby different clonotypes support proliferation and/or cytolytic secretion. These functional differences can be observed by introducing molecular changes in antigens to stimulate the same TCR65, indicating that different TCR-ligand affinities determine how the T cell will respond66.
Importantly, while pathway analyses propose an accessory role for stromal populations, including fibroblasts in immune cross-talk and the culmination of an immunomodulatory microenvironment, our phenotypic, spatial, and cell communication analyses position the cytotoxic CD8+ TRM-like Tconv cluster at the basement membrane in direct contact with keratinocytes. Our data suggest that while keratinocytes and other stromal populations set a basal level of tolerance in the skin controlling HLA-restricted CD8+ T cell activation or killing, loss of such tolerogenic mechanisms may be permissive to or exacerbate the selective targeting of keratinocytes for cell death during SJS/TEN. Specifically, while cells of the cytotoxic CD8+ Tconv cluster and dominant TCR+ cells, in particular, expressed LAG3, keratinocytes were the only stromal cell subset to downregulate the expression of its ligand LGALS3. Previous studies show that high expression of LGALS3 confers resistance to apoptosis in cancer cells44, and the LAG3-LGALS3 interaction is co-inhibitory to CD8+ T cells45. While functional studies are warranted, a similar downregulation of LGALS3 is observed in the lesional skin of patients with atopic dermatitis and psoriasis67, where CD8+ LAG3+ cell-depleting antibody therapies have recently been shown to reduce inflammation and improve lesion severity and barrier integrity, without reported issues surrounding safety or tolerability68. These data are consistent with early studies in non-human primates, which show that the depletion of LAG3+ cells prevents the onset of delayed-type T cell-mediated hypersensitivity reactions69. These data suggest that LAG3+ cell-depleting therapies may have benefits in SJS/TEN and that immune dysregulation represents a conserved defense mechanism enabling epidermal detachment to protect against diverse (drug-, self-) antigens (Fig. 6).
Differential and CellChat analyses also supported a direct CD8-keratinocyte interaction, including antigen presentation via HLA class I and potentially HLA-E but also MIF and its receptors. Intriguingly, MIF was first discovered in 1966 during in vitro studies of delayed hypersensitivity70, and although elevated in the serum of SJS/TEN patients, the expression has not been similarly investigated in the skin. While MIF is pleiotropic, it can recruit and activate T cells, with inhibition shown to suppress autoreactive CD8+ T cells in patients with vitiligo71. Further, MIF signaling is known to counteract the anti-inflammatory action of glucocorticoids72, potentially explaining the limited efficacy of corticosteroids in patients with established SJS/TEN. These data suggest the need for functional studies of MIF and its ligands to investigate their potential as therapeutic targets, given the critical interaction between keratinocytes and cytotoxic CD8+ T cells in SJS/TEN. Our data also suggest that selective targeting of the JAK-STAT pathway may be able to dampen the T cell response during SJS/TEN. However, JAK-STAT signaling is also important in other cell types, and we identify upregulation of STAT1, STAT2, and STAT3 in fibroblasts of affected SJS/TEN skin. As the expression of STAT3 in fibroblasts is important in re-epithelialization and wound repair73, these data highlight the use of single-cell data to understand both the efficacy and potential toxicities of proposed therapeutic interventions, and a need for selective JAK and STAT inhibition to appropriately target pro- and not anti-inflammatory processes. Importantly, specific modulators of MIF74, JAK, STAT, and PKM75 are established in the treatment of cancer and may now provide tissue-specific strategies for therapeutic intervention in SJS/TEN.
Limitations of this study are that, unlike the blister fluid, the composition of skin is likely to be affected by enzymatic digestion with variable recovery of stromal cell populations. This may explain why the proportion of immune cells appeared to be increased in the normal control skin biopsy compared to unaffected SJS/TEN patient skin. Importantly, there are different methods available for cell preparation, including the laser-dissected isolation of single cells from fixed tissue sections. It is possible that these non-enzymatic methods of sample preparation will differentially affect the results of single-cell transcriptomics and warrant future comparative study. Another limitation is that although single-cell approaches are well-powered to capture expanded clonotypes, the power to detect unexpanded clonotypes is incomplete. Finally, we could not control all patient-specific variables, such as severity and time since the onset of symptoms, and patients were sampled at a single time. Indeed, as the lymphocyte/monocyte ratio has been shown to change over time in samples obtained from a single patient with SJS/TEN19, longitudinal single-cell studies will be required to define the dynamic progression of the disease, including the proposed transition of M1 and intermediate-like macrophages identified in this study toward populations associated with re-epithelization and repair (Fig. 6).
In summary, our data support the hypothesis that drug-expanded oligoclonal CD8+ T cells are highly cytotoxic and are most relevant to initiating and perpetuating keratinocyte death. Such oligoclonal CD8+ T cells have been identified as drug-reactive7,8,76, while polyclonal CD8+ T cells may be driven by distinct drug- or self-antigen. We cannot be sure of the degree to which the polyclonal CD8+ T cells also directly contribute to the effector pathogenesis, which will require further investigation. This more detailed understanding of the complete cytotoxic repertoire will be critical to defining cytotoxic responses in patients with different causal drugs and HLA risk alleles. In contrast to autoimmune diseases with few pathogenic epitopes and TCRs, the epitope(s) driving SJS/TEN remain undefined. Still, drugs have been shown to stimulate T cells through diverse metabolites, drug-modified peptides, and by binding to HLA to alter the self-peptide repertoire77. Thus, as we define private oligoclonal and polyclonal TCRs in the cytotoxic CD8+ LAG3+ Tconv cluster across patients with diverse drug-induced SJS/TEN, this dataset provides a multi-omic atlas of drug-antigen-driven T cells clustered by drug and population that will help define cell-cell and receptor-ligand interactions toward further mechanistic understanding and potential identification of tissue-relevant biological targets for earlier diagnosis and treatment. The identification of private drug-expanded CD8+ LAG3+ Tconv TCR clonotypes8 also prioritizes the most relevant cytotoxic TCR clonotypes to investigate in functional assays to define the epitope-specificities recognized in the context of specific HLA risk allele-drug combinations towards improved genetic and structural prevention strategies.
Methods
Ethical statement
All samples including blister fluid and skin, clinical data, and analyses were obtained with informed consent under institutional review board (IRB) approval from Vanderbilt University Medical Center (IRB 131836, 150754, 171900), the University of Cape Town (HREC R031/2018, 500/2018), Austin Health (HREC 50791/Austin-2019), Murdoch University (HREC 2011/056, 2017/246, 2019/153), and the Western Australian Department of Health (RGS0000001924). All samples and data were de-identified.
Sample acquisition and storage
Burn blister fluid and healthy skin sections from the discarded edges of cutaneous surgeries were obtained from the Western Australian state burns unit. Acute SJS/TEN skin biopsies and aspirated blister fluid were obtained from cryopreserved biorepositories of clinically-confirmed SJS/TEN patients in the US (Vanderbilt University Medical Center), South Africa (University of Cape Town), and Australia (Austin Health and University of Melbourne). All samples were collected from patients either without a prior history of drug allergy or during acute SJS/TEN. Acute disease was defined as the period of symptomatically progressing or non-resolving disease within 10 days of hospitalization (Supplementary Table 1). SJS/TEN patients varied in age (range, 18–74 years; median 38 years), sex (male, n = 5; female, n = 10), underlying disease, culprit drug, reaction timeline, genetic race, and clinical treatment. SJS/TEN has been reported to be associated more commonly with women than men, which aligns with the higher number of female patients identified and included in this study. To consider sex in the design of this study to identify shared signatures of disease, sequencing data were normalized without the inclusion of sex-linked genes to avoid bias due to known expression variability. Data on gender was not collected.
Briefly, upon immediate collection after excision, skin biopsies were placed into cryogenic tubes containing 80% fetal bovine serum (FBS, Atlas Biologicals, Cat. No. F-0500-D) and 20% DMSO (Sigma, Cat. No. D8418) and flash-frozen at − 80 °C before being transferred into liquid nitrogen for long-term storage. Blister fluid samples were strained through a 70 µm mesh filter (Fisher, Cat. No. 229484) and washed twice with PBS. The resulting cell pellet was resuspended in 90% FBS 10% DMSO freeze media and placed into a slow-cooling container (StrataCooler, Agilent, Cat. No. 400005) at − 80 °C before being transferred into liquid nitrogen for long-term storage. Cryopreservation using DMSO is the recommended cell preservation procedure for droplet-based single-cell sequencing, which, after dead cell exclusion, is widely reported to result in minimal batch variation and transcriptomic differences between fresh and cryopreserved human cells78,79.
Enzymatic digestion of skin into a single-cell suspension
Cryopreserved 4 mm punch biopsies from SJS/TEN patients and healthy skin from unrelated donors were quickly thawed in a water bath (37 °C), and full-thickness biopsies were subjected to enzymatic digestion without separation of the epidermis and dermis to obtain a single-cell suspension using published methods optimized for lymphoid cell recovery80 and single-cell sequencing81. Briefly, each sample was washed twice in PBS immediately after thawing to remove freezing media and resuspended in RPMI 1640 medium (Gibco, Cat. No. 11875093) with 10% FBS (Atlas Biologicals, Cat. No. F-0500-D), 1 mg/ml collagenase P (Roche, Cat. No. 11213865001) and 40 µg/ml DNAase I (Sigma, Cat. No. LS002006) with agitation for 90 min. Tissue lysates were washed twice in PBS and passed through a 70 µm Flowmi Cell Strainer (Sigma, Cat. No. BAH136800070). Viable cells were counted using trypan blue (Gibco, Cat. No. 15250061) and an automated cell counter (Countess, Invitrogen). Cells were immediately resuspended in 22.5 µl cell staining buffer (Biolegend, Cat. No. 420201) for single-cell analyses.
Surface antibody staining protocol for scCITE-sequencing
Single-cell suspensions of thawed and enzymatically-digested skin or unprocessed blister fluid were subject to surface antibody staining for a panel of up to 137 functional and surface lineage markers in the TotalSeq-C Human Universal Cocktail, V1.0 (Biolegend, Cat. No. 399905) as per manufacturer’s protocol (https://www.biolegend.com/Files/Images/media_assets/support_protocol/20-0014-00_TotalSeq-UC_8x11.pdf). Briefly, lyophilized antibody panels were resuspended in cell staining buffer (Biolegend Cat. No. 420201), vortexed, and incubated for 5 mins at room temperature. Antibodies were again vortexed and centrifuged (14,000 × g, 10 mins, 4 °C) while cell suspensions were blocked using 2.5 µl Human TruStain FcX Fc blocking reagent (Biolegend, Cat. No. 422301) and incubated for 10 mins at 4 °C. 25 µl of reconstituted antibody cocktail was then added to 25 µl of FcR-blocked cells, and for samples to be pooled into the same well of the 10x chromium chip, 1 µl of the appropriate hashtag antibody was also added, with subsequent incubation for 30 mins at 4 °C. Each cell-antibody cocktail was then resuspended in 3.5 ml cell staining buffer and washed three times in PBS at 400 × g for 5 mins at 4 °C before cells were passed through a 70 µm Flowmi Cell Strainer. Cell count and viability were recorded using trypan blue and an automated cell counter, and cells were diluted as per the manufacturer’s instruction for loading onto the 10X Chromium Next GEM chip K.
Single-cell 5’ RNA-TCR-CITE-sequencing
Cells were loaded onto the chromium Next GEM chip K chip at the manufacturer (10x Genomics) recommended concentration and single-cell libraries made using a chromium controller and Chromium Next GEM Single Cell 5’ Reagent Kit, Version 2.0 (10x Genomics) as per manufacturer instruction (https://www.10xgenomics.com/support/single-cell-immune-profiling/documentation/steps/library-prep/chromium-single-cell-5-reagent-kits-user-guide-v-2-chemistry-dual-index-with-feature-barcoding-technology-for-cell-surface-protein-and-immune-receptor-mapping), with a target of 1000–20,000 cells. 5’ libraries were then sequenced using the Illumina NovaSeq 6000 platform for the capture of 70,000 reads/cell, including 50,000 for cDNA, 5000 for TCR, and 15,000 for the cell surface protein library. Sample sequencing was completed in the VANderbilt Technologies for Advanced GEnomics core (VANTAGE), with subsequent bioinformatic and computational analyses at IIID, Murdoch University.
Computational and statistical analysis
The single-cell sequencing data were processed using CellRanger v6.1.2 (10x Genomics) to demultiplex individual cell barcodes, remove PCR duplicates, and align RNA reads to the reference GRCh38 human transcriptome. The individual hashes within each 10x run were further demultiplexed using Souporcell82 to cluster samples based on SNPs in the RNA-seq reads. The hashtag negatives or doublets, as well as cells with low (< 500) UMIs, were removed. Cells with < 100 genes and > 50% mitochondrial content were removed to filter low-quality, dead, or dying populations for initial QC (QC1, Supplementary Fig. 15), with a second pass filter excluding apoptotic populations characterized by a low percentage of ribosomal genes and a high percentage of mitochondrial genes, using a defined mitochondrial-ribosomal RNA ratio of > 0.4748 (QC step 2, Supplementary Fig. 15). Importantly, a standardized-mitochondrial DNA threshold of 10% is recommended for scRNA-seq of human tissue83, which was met by 98% of ‘live’ cells identified in our dataset. The remaining 2% of cells were excluded from downstream analyses (QC step 3, Supplementary Fig. 15). The median mitochondrial gene content for all ‘live’ cells was 3%, and this matched the immune cell subset-specific median mitochondrial DNA content for CD4+ T cells, CD8+ T cells, NK cells, monocytes, and macrophages. Downstream transcriptome-based graph clustering and principal component analyses (PCA) with cell phenotype consensus calling were performed using R Seurat v4.1.1 package23, without input of TCR, BCR, mitochondrial, ribosomal, or sex-linked genes to avoid bias due to known expression variability. Batch correction for 10x run, sample type (skin or blister fluid), and cell cycle phase were performed using harmony84. While most cell subsets formed distinct clusters, keratinocytes predominantly aligned to a shared cluster, including a minority of T cells, NK cells, and monocytes at the UMAP origin defined by lower UMI; indicative of cell stress. However, these cells met all other criteria for inclusion and were retained for pathogenic importance in the context of drug-induced stress during SJS/TEN. Experimental doublets were identified and removed using a majority consensus benchmark85 of three independent bioinformatic algorithms (DoubletFinder86, scDBLfinder87, scDS88; QC Step 4), and subsets with < 50 scRNA-defined cells were removed (QC step 5, Supplementary Fig. 15). T and NK subsets were re-aligned to the HuBMAP consortium Azimuth PBMC reference multimodal dataset23 for consensus calling of conventional and unconventional subsets. Visualization and differential expression analyses were performed using Visual Genomics Analysis Studio (VGAS)89. Differential gene expression analyses were performed using the two-tailed Wilcoxon rank-sum test with Benjamini-Hochberg adjusted correction (adj. p < 0.05). Pathway analyses were performed using Enrichr90, common core pathway responsive genes identified using PROGENy56, and cell-cell communication analyses using CellChat51. LIANA91 was used to rank and visualize the highest-scoring receptor-ligand interactions. To avoid the bias of testing compositional changes of each cell type independently using univariate models, statistical analysis of single-cell composition was performed using scCODA17 and the software-recommended standard and minimum FDR-corrected significance thresholds of p < 0.05 and p < 0.4, respectively.
Immunohistochemistry and spatial sequencing
Formalin-fixed and paraffin-embedded (FFPE) skin biopsy specimens from healthy control or SJS/TEN patients were sectioned at 5 mm intervals and attached to glass microscope slides. Slides were placed on the Leica Bond Max IHC stainer for immunohistochemistry and deparaffinized. Heat-induced antigen retrieval was performed on the Bond Max using Epitope Retrieval 2 solution for 20 min, before incubation with Ready-To-Use anti-CD8 (Cat. No. MM39-10; clone 144B, StatLab, McKinney, Tex) for 15 min, Ready-To-Use anti-CD103 (Cat. No. PA0374, clone EP206, Leica, Buffalo Grove, IL) for one hour, and polyclonal anti-GNLY (Cat No. HPA058021, Atlas Antibodies, St. Louis, MO) at a dilution of 1:500 for 30 min. The Bond Polymer Refine detection system was used for visualization. Slides were then dehydrated, cleared, and coverslipped. All were performed in the Translational Pathology Shared Resource at VUMC. For GeoMX spatial sequencing, slide-mounted FFPE tissue sections were incubated with high-plex oligo-labeled RNA probes (GeoMX immune-oncology panel) and primary antibodies for the morphology markers: anti-Pan cytokeratin antibody, anti-CD45, and anti-CD3E. Regions of interest (ROIs) were selected, and the ROIs were exposed to UV illumination, releasing the DNA oligos into the layer above the tissue slice. Oligos are hybridized to nCounter optical barcodes to permit ex-situ digital quantification of analyte abundance using the NanoString DSP instrument. Sample processing and sequencing were performed by the Technology Access Program at NanoString. Probe measurements and quality control data were provided by NanoString, and RNA count was normalized using ERCC spike-in controls.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Source data
Acknowledgements
This research was supported by National Institutes of Health awards and grants NIH U01AI154659 (E.J.P.), NIH P50GM115305 (E.J.P. and S.M.), NIH R01HG010863 (E.J.P.), NIH R21AI139021 (E.J.P.), NIH R01AI152183 (E.J.P. and J.P.), NIH P30 AI110527 (S.M., S.K., and C.W.), NIH 2 D43 TW010559 (E.J.P., J.P., and R.L.), and NIH K43 TW011178-01 (J.P.), the Western Australian Future Health Research and Innovation Fund/ Western Australian Department of Health grant WANMA/EL2022/4 (A.G.), and the NHMRC of Australia grants GNT1123499 (E.J.P.) and GNT2028952 (A.G., E.J.P., R.R., and A.C.). We gratefully acknowledge patients and their families, the significant expertize of the VANderbilt Technologies for Advanced GEnomics core (VANTAGE) core for sample sequencing, the Translational Pathology Shared Resource (TPSR) core at Vanderbilt University Medical Center for immunohistochemistry, and researchers and clinicians from the Australasian Registry for Severe Cutaneous Adverse Reaction (AUS-SCAR) and African Registry of Severe Cutaneous Adverse Reactions (AFRiSCAR) for the provision of clinically-curated SJS/TEN tissue samples from Australasia and South Africa, respectively. These thanks are extended but not limited to (AUS-SCAR) Jason A Trubiano, Fiona James, Ar Kar Aung, Michelle S Y Goh, Celia Zubrinich, Douglas Gin, Heather Cleland, Andrew Awad, (AFRiSCAR) Jonny G Peter, Rannakoe J Lehloenya, Phuti Choshi, Sarah Pedretti, Tafadzwa Chimbetete, Rose Selim, and Mireille Porter. Further, for the provision of control samples, we are grateful to and acknowledge Allison Hanlon (Dept of Medicine, Vanderbilt University Medical Center, Nashville, TN, USA).
Author contributions
Concept and design: A.G., R.R., S.A.M., and E.J.P. Sample acquisition and curation: P.C., S.P., J.A.T., J.G.P., S.P., M.W.F., F.M.W., R.J.L., and E.J.P. Acquisition, analysis, or interpretation of data: A.G., R.R., R.G., Y.L., A.M.P., C.N.C., M.T., K.C.K., P.D., S.A.M., and E.J.P. Drafting of the manuscript: A.G. and R.R. Critical revision of the manuscript: A.G., R.R., E.M., R.T.O., C.N.W., S.A.K., S.G., R.J.L., S.S.B., A.C., J.A.T., J.G.P., S.A.M., and E.J.P. Statistical analysis: A.G. and R.R. Supervision: A.G., A.C., and E.J.P. Funding acquisition: E.J.P., J.G.P., and A.G.
Peer review
Peer review information
Nature Communications thanks Juan Quintana, Marta Polak, Joerg Prinz, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Data availability
All raw and normalized data generated by scRNA-TCR-CITE-sequencing and used in this study have been deposited to the NCBI Sequence Read Archive PRJNA1070820 and GEO database under accession code GSE275871. The source data generated in this study, both for main figures and Supplementary Figures are provided in the source data file with this paper. All other data are available in the article and its Supplementary files or from the corresponding author upon request. Source data are provided in this paper.
Competing interests
E.J.P. receives royalties and consulting fees from UpToDate (where she is a Drug Allergy Section Editor) and has received consulting fees from Janssen, Vertex, Verve, Servier, Rapt and Esperion; she is co-director of IIID Pty Ltd, which holds a patent for HLA-B*57:01 testing for abacavir hypersensitivity, and she has a patent pending for detection of HLA-A*32:01 in connection with diagnosing drug reaction with eosinophilia and systemic symptoms (for these patents she does not receive any financial remuneration and neither are related to the submitted work). All other authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A list of authors and their affiliations appears at the end of the paper.
A full list of members and their affiliations appears in the Supplementary Information.
Contributor Information
Elizabeth J. Phillips, Email: elizabeth.j.phillips@vumc.org
On behalf of the AUS-SCAR Consortium:
On behalf of the AFRiSCAR Consortium:
Phuti Choshi, Sarah Pedretti, Rannakoe J. Lehloenya, and Jonny G. Peter
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-52990-3.
References
- 1.Marks, M. E. et al. Updates in SJS/TEN: collaboration, innovation, and community. Front. Med.10, 1213889 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krantz, M., Yoon, B., Stone, C., Yu, R. & Phillips, E. Stevens-Johnson syndrome and toxic epidermal necrolysis in the FDA adverse event reporting system (FAERS) from 1995-2020. J. Allergy Clin. Immunol.149, AB62 (2022). [Google Scholar]
- 3.Posadas, S. J. et al. Delayed reactions to drugs show levels of perforin, granzyme B, and Fas-L to be related to disease severity. J. Allergy Clin. Immunol.109, 155–161 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Chung, W.-H. et al. Granulysin is a key mediator for disseminated keratinocyte death in Stevens-Johnson syndrome and toxic epidermal necrolysis. Nat. Med.14, 1343–1350 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Clayberger, C. et al. 15 kDa granulysin causes differentiation of monocytes to dendritic cells but lacks cytotoxic activity. J. Immunol.188, 6119–6126 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chung, W. H. et al. Medical genetics: a marker for Stevens-Johnson syndrome. Nature428, 486 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Pan, R. Y. et al. Identification of drug-specific public TCR driving severe cutaneous adverse reactions. Nat. Commun.10, 3569 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chung, W.-H. et al. Oxypurinol-specific T cells possess preferential TCR clonotypes and express granulysin in allopurinol-induced severe cutaneous adverse reactions. J. Invest. Dermatol.135, 2237–2248 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Lee, H. Y., Fook-Chong, S., Koh, H. Y., Thirumoorthy, T. & Pang, S. M. Cyclosporine treatment for Stevens-Johnson syndrome/toxic epidermal necrolysis: Retrospective analysis of a cohort treated in a specialized referral center. J. Am. Acad. Dermatol.76, 106–113 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Zhang, J. et al. Evaluation of combination therapy with etanercept and systemic corticosteroids for Stevens-Johnson syndrome and toxic epidermal necrolysis: A multicenter observational study. J. Allergy Clin. Immunology Pract.10, 1295–1304 (2022). [DOI] [PubMed] [Google Scholar]
- 11.Mukherjee, E. M. & Phillips, E. J. Where to place etanercept and combination treatment for Stevens-Johnson syndrome and toxic epidermal necrolysis? Ann. Allergy Asthma Immunol.129, 269–270 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brüggen, M. C. et al. Supportive care in the acute phase of Stevens–Johnson syndrome and toxic epidermal necrolysis: an international, multidisciplinary Delphi‐based consensus. Br. J. Dermatol.185, 616–626 (2021). [DOI] [PubMed] [Google Scholar]
- 13.Chen, S. et al. Macrophages in immunoregulation and therapeutics. Signal Transduct. Target. Ther.8, 207 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morris, D. L. et al. CD40 promotes MHC class II expression on adipose tissue macrophages and regulates adipose tissue CD4+ T cells with obesity. J. Leukoc. Biol.99, 1107–1119 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chakarov, S. et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science363, eaau0964 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Bailin, S. S., et al. Changes in subcutaneous white adipose tissue cellular composition and molecular programs underlie glucose intolerance in persons with HIV. Front. immunol.14, 10.3389/fimmu.2023.1152003 (2023). [DOI] [PMC free article] [PubMed]
- 17.Büttner, M., Ostner, J., Müller, C. L., Theis, F. J. & Schubert, B. scCODA is a Bayesian model for compositional single-cell data analysis. Nat. Commun.12, 6876 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen, S. H. et al. Predominance of CD14+ cells in burn blister fluids. Ann. Plast. Surg.80, S70–s74 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Le Cleach, L. et al. Blister fluid T lymphocytes during toxic epidermal necrolysis are functional cytotoxic cells which express human natural killer (NK) inhibitory receptors. Clin. Exp. Immunol.119, 225–230 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zou, Z. et al. A single-cell transcriptomic atlas of human skin aging. Dev. Cell56, 383–397 (2021). [DOI] [PubMed] [Google Scholar]
- 21.Denu, R. A. et al. Fibroblasts and mesenchymal stromal/stem cells are phenotypically indistinguishable. Acta Haematol.136, 85–97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mayassi, T., Barreiro, L. B., Rossjohn, J. & Jabri, B. A multilayered immune system through the lens of unconventional T cells. Nature595, 501–510 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell184, 3573–3587 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu, Y. et al. γδ T cells: origin and fate, subsets, diseases and immunotherapy. Signal Transduct. Target. Ther.8, 434 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanbar, J. N., et al. The long noncoding RNA Malat1 regulates CD8+ T cell differentiation by mediating epigenetic repression. J. Exp. Med.219, e20211756 (2022). [DOI] [PMC free article] [PubMed]
- 26.Menon, M. P. & Hua, K. F. The long non-coding RNAs: Paramount regulators of the NLRP3 inflammasome. Front. Immunol.11, 569524 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Correia, M. P. et al. Distinct human circulating NKp30(+)FcεRIγ(+)CD8(+) T cell population exhibiting high natural killer-like antitumor potential. Proc. Natl. Acad. Sci. USA115, E5980–e5989 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burel, J. G. et al. Circulating T cell-monocyte complexes are markers of immune perturbations. ELife8, e46045 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schluns, K. S., Kieper, W. C., Jameson, S. C. & Lefrançois, L. Interleukin-7 mediates the homeostasis of naïve and memory CD8 T cells in vivo. Nat. Immunol.1, 426–432 (2000). [DOI] [PubMed] [Google Scholar]
- 30.Cheuk, S. et al. CD49a Expression defines tissue-resident CD8+ T cells poised for cytotoxic function in human skin. Immunity46, 287–300 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He, H. et al. Single-cell transcriptome analysis of human skin identifies novel fibroblast subpopulation and enrichment of immune subsets in atopic dermatitis. J. Allergy Clin. Immunol.145, 1615–1628 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Rojahn, T. B. et al. Single-cell transcriptomics combined with interstitial fluid proteomics defines cell type–specific immune regulation in atopic dermatitis. J. Allergy Clin. Immunol.146, 1056–1069 (2020). [DOI] [PubMed] [Google Scholar]
- 33.Park, S. L. et al. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat. Immunol.19, 183–191 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Mackay, L. K. et al. The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat. Immunol.14, 1294–1301 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Clark, R. A. et al. The vast majority of CLA+ T cells are resident in normal skin. J. Immunol. (Baltim., Md.: 1950)176, 4431–4439 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Tokura, Y., Phadungsaksawasdi, P., Kurihara, K., Fujiyama, T. & Honda, T. Pathophysiology of Skin Resident Memory T Cells. Front. Immunol.11, 618897 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soler, D., Humphreys, T. L., Spinola, S. M. & Campbell, J. J. CCR4 versus CCR10 in human cutaneous TH lymphocyte trafficking. Blood101, 1677–1682 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Mackay, L. K. et al. Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J. Immunol.194, 2059–2063 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Skon, C. N. et al. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol.14, 1285–1293 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.El Hajj, Y. et al. Pregnenolone sulfate induces transcriptional and immunoregulatory effects on T cells. Sci. Rep.14, 6782 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He, Y. et al. Mediators of capillary-to-venule conversion in the chronic inflammatory skin disease psoriasis. J. Invest. Dermatol.142, 3313–3326 (2022). [DOI] [PubMed] [Google Scholar]
- 42.Litvak, V. et al. Function of C/EBPδ in a regulatory circuit that discriminates between transient and persistent TLR4-induced signals. Nat. Immunol.10, 437–443 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hammond, S. et al. Checkpoint inhibition reduces the threshold for drug-specific T-cell priming and increases the incidence of sulfasalazine hypersensitivity. Toxicol. Sci.186, 58–69 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mazurek, N. et al. Cell-surface galectin-3 confers resistance to TRAIL by impeding trafficking of death receptors in metastatic colon adenocarcinoma cells. Cell Death Differ.19, 523–533 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bae, J. et al. Targeting LAG3/GAL-3 to overcome immunosuppression and enhance anti-tumor immune responses in multiple myeloma. Leukemia36, 138–154 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang, H., Wang, C., Rubelt, F., Scriba, T. J. & Davis, M. M. Analyzing the Mycobacterium tuberculosis immune response by T-cell receptor clustering with GLIPH2 and genome-wide antigen screening. Nat. Biotechnol.38, 1194–1202 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hwang, S. S. et al. mRNA destabilization by BTG1 and BTG2 maintains T cell quiescence. Science367, 1255–1260 (2020). [DOI] [PubMed] [Google Scholar]
- 48.Sun, B. et al. Double-jeopardy: scRNA-seq doublet/multiplet detection using multi-omic profiling. Cell Rep. Methods1, 100008 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ji, Q., Perchellet, A. & Goverman, J. M. Viral infection triggers central nervous system autoimmunity via activation of CD8+ T cells expressing dual TCRs. Nat. Immunol.11, 628–634 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee, H., Jeong, S. & Shin, E.-C. Significance of bystander T cell activation in microbial infection. Nat. Immunol.23, 13–22 (2022). [DOI] [PubMed] [Google Scholar]
- 51.Jin, S. et al. Inference and analysis of cell-cell communication using CellChat. Nat. Commun.12, 1088 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morel, E. et al. CD94/NKG2C is a killer effector molecule in patients with Stevens-Johnson syndrome and toxic epidermal necrolysis. J. Allergy Clin. Immunol.125, 703–710 (2010). [DOI] [PubMed] [Google Scholar]
- 53.Grant, E. J. et al. The unconventional role of HLA-E: The road less traveled. Mol. Immunol.120, 101–112 (2020). [DOI] [PubMed] [Google Scholar]
- 54.Walters, L. C. et al. Pathogen-derived HLA-E bound epitopes reveal broad primary anchor pocket tolerability and conformationally malleable peptide binding. Nat. Commun.9, 3137 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tamada, M. et al. Modulation of glucose metabolism by CD44 contributes to antioxidant status and drug resistance in cancer cells. Cancer Res.72, 1438–1448 (2012). [DOI] [PubMed] [Google Scholar]
- 56.Schubert, M. et al. Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat. Commun.9, 20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Salehi, R. et al. Granulosa cell-derived miR-379-5p regulates macrophage polarization in polycystic ovarian syndrome. Front. Immunol.14, 1104550 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Timms, K., Guo, H., Arkwright, P. & Pennock, J. Keratinocyte EGF signalling dominates in atopic dermatitis lesions: A comparative RNAseq analysis. Exp. Dermatol.31, 1373–1384 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu, F. et al. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct. Target. Ther.6, 218 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Villani, A. P. et al. Massive clonal expansion of polycytotoxic skin and blood CD8(+) T cells in patients with toxic epidermal necrolysis. Sci. Adv.7, 10.1126/sciadv.abe0013 (2021). [DOI] [PMC free article] [PubMed]
- 61.Martos, S. N. et al. Single-cell analyses identify dysfunctional CD16(+) CD8 T cells in smokers. Cell Rep. Med.1, 100054 (2020). [DOI] [PMC free article] [PubMed]
- 62.Milner, J. J. et al. Delineation of a molecularly distinct terminally differentiated memory CD8 T cell population. Proc. Natl. Acad. Sci. USA117, 25667–25678 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rapaport, A. S. et al. The inhibitory receptor NKG2A sustains virus-specific CD8+ T cells in response to a lethal poxvirus infection. Immunity43, 1112–1124 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Joachims, M. L. et al. Dysregulated long non-coding RNA in Sjögren’s disease impacts both interferon and adaptive immune responses. RMD Open8, e002672 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kersh, G. J. & Allen, P. M. Structural basis for T cell recognition of altered peptide ligands: a single T cell receptor can productively recognize a large continuum of related ligands. J. Exp. Med.184, 1259–1268 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Evavold, B. D., Sloan-Lancaster, J. & Allen, P. M. Tickling the TCR: selective T-cell functions stimulated by altered peptide ligands. Immunol. Today14, 602–609 (1993). [DOI] [PubMed] [Google Scholar]
- 67.Corrêa, M. P. et al. Expression pattern and immunoregulatory roles of galectin-1 and galectin-3 in atopic dermatitis and psoriasis. Inflammation45, 1133–1145 (2022). [DOI] [PubMed] [Google Scholar]
- 68.Ellis, J. et al. Depletion of LAG-3(+) T cells translated to pharmacology and improvement in psoriasis disease activity: A phase I randomized study of mAb GSK2831781. Clin. Pharmacol. Ther.109, 1293–1303 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Poirier, N. et al. Antibody-mediated depletion of lymphocyte-activation gene-3 (LAG-3+)-activated T lymphocytes prevents delayed-type hypersensitivity in non-human primates. Clin. Exp. Immunol.164, 265–274 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bloom, B. R. & Bennett, B. Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science153, 80–82 (1966). [DOI] [PubMed] [Google Scholar]
- 71.Chen, J. et al. MIF inhibition alleviates vitiligo progression by suppressing CD8(+) T cell activation and proliferation. J. Pathol.260, 84–96 (2023). [DOI] [PubMed] [Google Scholar]
- 72.Mitchell, R. A., Metz, C. N., Peng, T. & Bucala, R. Sustained mitogen-activated protein kinase (MAPK) and cytoplasmic phospholipase A2 activation by macrophage migration inhibitory factor (MIF): REGULATORY ROLE IN CELL PROLIFERATION AND GLUCOCORTICOID ACTION*. J. Biol. Chem.274, 18100–18106 (1999). [DOI] [PubMed] [Google Scholar]
- 73.Huynh, J., Chand, A., Gough, D. & Ernst, M. Therapeutically exploiting STAT3 activity in cancer — using tissue repair as a road map. Nat. Rev. Cancer19, 82–96 (2019). [DOI] [PubMed] [Google Scholar]
- 74.Kok, T. et al. Small-molecule inhibitors of macrophage migration inhibitory factor (MIF) as an emerging class of therapeutics for immune disorders. Drug Discov. Today23, 1910–1918 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rask-Andersen, M., Zhang, J., Fabbro, D. & Schiöth, H. B. Advances in kinase targeting: current clinical use and clinical trials. Trends Pharmacol. Sci.35, 604–620 (2014). [DOI] [PubMed] [Google Scholar]
- 76.Mifsud, N. A. et al. The allopurinol metabolite, oxypurinol, drives oligoclonal expansions of drug-reactive T cells in resolved hypersensitivity cases and drug-naïve healthy donors. Allergy78, 2980–2993 (2023). [DOI] [PMC free article] [PubMed]
- 77.Ostrov, D. A. et al. Drug hypersensitivity caused by alteration of the MHC-presented self-peptide repertoire. Proc. Natl. Acad. Sci. USA109, 9959–9964 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wohnhaas, C. T. et al. DMSO cryopreservation is the method of choice to preserve cells for droplet-based single-cell RNA sequencing. Sci. Rep.9, 10699 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guillaumet-Adkins, A. et al. Single-cell transcriptome conservation in cryopreserved cells and tissues. Genome Biol.18, 45 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Salimi, M. et al. Enhanced isolation of lymphoid cells from human skin. Clin. Exp. Dermatol.41, 552–556 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Waise, S. et al. An optimised tissue disaggregation and data processing pipeline for characterising fibroblast phenotypes using single-cell RNA sequencing. Sci. Rep.9, 9580 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Heaton, H. et al. Souporcell: robust clustering of single-cell RNA-seq data by genotype without reference genotypes. Nat. Methods17, 615–620 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Osorio, D. & Cai, J. J. Systematic determination of the mitochondrial proportion in human and mice tissues for single-cell RNA-sequencing data quality control. Bioinformatics37, 963–967 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xi, N. M. & Li, J. J. Benchmarking computational doublet-detection methods for single-cell RNA sequencing data. Cell Syst.12, 176–194 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McGinnis, C. S., Murrow, L. M. & Gartner, Z. J. Doubletfinder: Doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst.8, 329–337 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Germain, P. L., Lun, A., Garcia Meixide, C., Macnair, W. & Robinson, M. D. Doublet identification in single-cell sequencing data using scDblFinder. F1000Res.10.12688/f1000research.73600.2 (2021). [DOI] [PMC free article] [PubMed]
- 88.Bais, A. S. & Kostka, D. scds: computational annotation of doublets in single-cell RNA sequencing data. Bioinformatics36, 1150–1158 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hertzman, R. J. et al. Visual genomics analysis studio as a tool to analyze multiomic data. Front. Genet.12, 642012 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xie, Z. et al. Gene set knowledge discovery with Enrichr. Curr. Protoc.1, e90 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dimitrov, D. et al. Comparison of methods and resources for cell-cell communication inference from single-cell RNA-Seq data. Nat. Commun.13, 3224 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw and normalized data generated by scRNA-TCR-CITE-sequencing and used in this study have been deposited to the NCBI Sequence Read Archive PRJNA1070820 and GEO database under accession code GSE275871. The source data generated in this study, both for main figures and Supplementary Figures are provided in the source data file with this paper. All other data are available in the article and its Supplementary files or from the corresponding author upon request. Source data are provided in this paper.