

Abstract
The effect of membrane transporters on drug disposition, efficacy and safety is now well recognized. Since the initial publication from the International Transporter Consortium, significant progress has been made in understanding the roles and functions of transporters, as well as in the development of tools and models to assess and predict transporter-mediated activity, toxicity and drug–drug interactions (DDIs). Notable advances include an increased understanding of the effects of intrinsic and extrinsic factors on transporter activity, the application of physiologically based pharmacokinetic modelling in predicting transporter-mediated drug disposition, the identification of endogenous biomarkers to assess transporter-mediated DDIs and the determination of the cryogenic electron microscopy structures of SLC and ABC transporters. This article provides an overview of these key developments, highlighting unanswered questions, regulatory considerations and future directions.
Introduction
Since the original publication of the International Transporter Consortium (ITC, www.itc-transporter.org) in Nature Reviews Drug Discovery1, significant progress has been made in understanding the roles of membrane transporters in drug disposition and response. This first ITC publication identified a subset of transporters of particular clinical interest and outlined decision trees that could be applied to predict the clinical importance of changes in transporter activity. Subsequent ITC publications have highlighted the development of tools and approaches to address the complex and critical issues related to transporters in drug development, evaluated preclinical and clinical data, and provided updated recommendations on decision points for the involvement of transporters and the potential for clinically relevant transporter-mediated drug–drug interactions (DDIs)2–6.
This article is structured in sections that aim to provide an overview of the current status of transporters in drug development, focusing on transporters in two major superfamilies, the solute carrier (SLC) superfamily and the ATP binding cassette (ABC) superfamily. Although there are 65 families in the human SLC superfamily with about 450 genes encoding transport proteins, and 49 genes encoding efflux pumps in the human ABC superfamily, this article will focus on a subset of transporters (Fig. 1) that are involved in DDIs and/or drug toxicity in specific tissues based on a detailed analysis of the recent literature. As a result of our recent analysis, transporter categorization in Fig. 1 differs from earlier assessments by the ITC of transporters relevant in drug development3. As in the past, transport proteins of interest in drug development that play a key role in mediating drug absorption and/or elimination in the liver, kidney and intestine are included. However, Fig. 1 also includes transporters in other specialized blood–tissue barriers such as the blood–brain barrier and placenta. Transporters discussed in this article are multispecific, interact with drugs from diverse pharmacological classes and are associated with DDIs and/or toxicity. Additionally, genetic polymorphisms in several of these transporters have been associated with drug toxicities and/or non-response.
Fig. 1 |. Clinically important uptake and efflux transporters in plasma membranes.
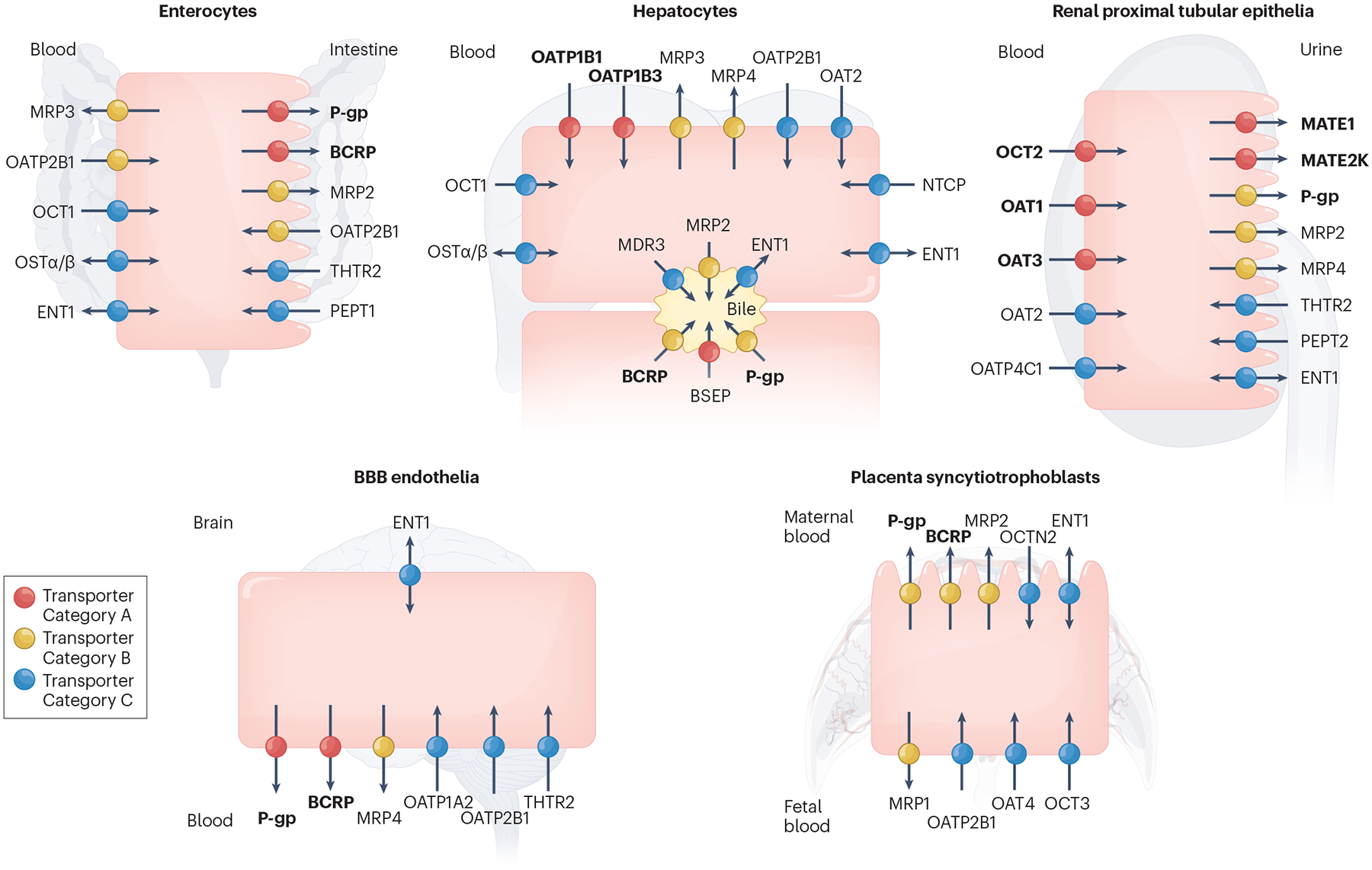
Transporters in the plasma membrane of enterocytes, hepatocytes, renal proximal tubular epithelia, blood–brain barrier (BBB) endothelia and placenta syncytiotrophoblasts are shown. Transporters are only included in Fig. 1 if there is clinical evidence for their involvement in transporter-mediated drug–drug interactions (DDIs) and/or drug toxicity in the specific tissue and are categorized accordingly in each tissue. Thus, the designated colour (categorization) for a transporter may differ across tissues. In some cases, precise transporter categorization is confounded by the absence of specific in vivo inhibitors, the presence of redundant transporters and/or a lack of evidence from knockout models or human polymorphisms. Differences in transporter categorization compared with earlier assessments by the International Transporter Consortium3 are based on our current understanding of the literature, as summarized in Supplementary Table S2. Transporters recommended by regulatory agencies for screening during drug development are highlighted in bold in Fig. 1 (BCRP, MATE1, MATE2K, OAT1, OAT3, OATP1B1, OATP1B3, OCT2, P-gp). Current ICH M12 guidelines recommend evaluation of OCT1, OATP2B1, MRP2 and BSEP on a case-by-case basis. Transporter Category A: Transporters coloured in red transport a wide range of pharmacological drug classes, are critical in drug and/or endogenous substrate disposition in the specific tissue and are the site of clinical DDIs and/or drug-mediated toxicity. Transporter Category B: Transporters coloured in yellow primarily transport a wide range of pharmacological drug classes, but clinical evidence supporting their involvement in the specific tissue in DDIs and/or drug toxicity is limited. Transporter Category C: Transporters coloured in blue primarily transport endogenous substrates and/or fewer drug classes, and there is weak clinical evidence demonstrating their involvement in the specific tissue in DDIs and/or drug toxicity. Transporter Category D: Transporters in this category transport endogenous substrates and a narrow range of drug classes. The significance of these transporters as a target for clinical DDIs in the specific tissue is not well-established, and/or there is limited published data showing that the inhibition of these transporters by a perpetrator leads to abnormal levels of endogenous substrate resulting in negative clinical outcomes. Therefore, these transporters (CNT1–3, ENT2–3, MCT1, MRP5–6, OAT4, OAT7, OCTN1–2, PCFT, PMAT, RFC, THTR1) are not included in Fig. 1. BCRP, breast cancer resistance protein (gene name, ABCG2); BSEP, bile salt export pump (ABCB11); CNT1–3, concentrative nucleoside transporter 1–3 (SLC28A1–3); ENT1–3, equilibrative nucleoside transporter 1 (SLC29A1–3); MATE1, MATE2K, multidrug and toxin extrusion protein (SLC47A1, 2); MCT1, monocarboxylate transporter 1 (SLC16A1); MDR3, multidrug resistance protein 3 (ABCB4); MRP1–6, multidrug resistance-associated protein (ABCC1–6); NTCP, sodium-taurocholate co-transporting polypeptide (SLC10A1); OAT1–3, organic anion transporter 1–3 (SLC22A6–8); OAT4, organic anion transporter 4 (SLC22A11); OAT7, organic anion transporter 7 (SLC22A9); OATP1A2, organic anion transporting polypeptide 1A2 (SLCO1A2); OATP1B1, organic anion transporting polypeptide (SLCO1B1); OATP1B3, organic anion transporting polypeptide 1B3 (SLCO1B3); OATP2B1, organic anion transporting polypeptide 2B1 (SLCO2B1); OATP4C1, organic anion transporting polypeptide 4C1 (SLCO4C1); OCT1–3, organic cation transporter (SLC22A1–3); OCTN1–2, organic cation transporter novel 1–2 (SLC22A4–5); OSTα/β, organic solute transporter alpha/beta (SLC51A/B); PCFT, proton-coupled folate transporter (SLC46A1); PEPT1–2, peptide transporter 1–2 (SLC15A1–2); P-gp, P-glycoprotein (ABCB1); PMAT, plasma membrane monoamine transporter (SLC29A4); RFC, reduced folate carrier (SLC19A1); THTR1–2, thiamine transporter 1–2 (SLC19A2–3).
In the first section of this article, recent research focusing on the role of intrinsic factors (such as genetics, ethnicity, age, sex, physiologic states and organ-based diseases) and extrinsic factors (such as diet, herbal or medication use, the microbiome and environmental exposure) in the modulation of transporter function and abundance is discussed. However, further work is needed to fully understand the mechanisms underlying the effects of some of these factors on transporter function. The second section provides an overview of advances made in the development and application of modelling approaches to predict and understand the role of transporters in drug disposition and DDIs, as well as to predict pharmacokinetic (PK) changes in diseases and in specific populations. The enormous progress since the original ITC publication1, in the discovery and validation of endogenous substrates as biomarkers of transporter function, and their application in assessing risk of transporter-mediated DDIs are discussed in the third section of this article. As reviewed in the fourth section, drug transporters can directly or indirectly contribute to drug-induced organ toxicity. Prominent examples including neurotoxicity and cardiotoxicity, cholestasis and the developing area of environmental toxins such as heavy metals are discussed. The regulatory implications regarding transporter-mediated DDIs are considered in the fifth section. Specifically, different modelling approaches in the regulatory submission process are critically reviewed, together with a consideration of metabolites of drugs as inhibitors of transporters and endogenous biomarkers as additional clinical tools to evaluate transporter-mediated DDIs.
The final two sections summarize key technological advances in transporter research (for example, structural determination of transport proteins and modulation of transport function via genome editing) and future directions in the application of transporter research, including the importance of measurements beyond systemic pharmacokinetics. Information derived from the analysis of tissue-derived small extracellular vesicles (sEVs) and the growing interest in nutrient/endobiotic transport are considered.
The aims of this article are to provide a summary of the current status of transporters in drug development and highlight recent advances in our understanding of the pharmacological roles of transporters in drug development and precision medicine. Critical remaining questions that need to be addressed concerning the role of transport proteins in drug discovery and development, as well as in determining their effects on therapeutic and adverse drug response, are highlighted.
Factors regulating transporter activity
Although the importance of transport proteins in drug disposition and response is now widely recognized, factors such as epigenetics, transcriptional and post-translational regulation that affect the expression, abundance, localization and function of transporters in humans are not well understood7. In this section, current knowledge about the intrinsic and extrinsic factors that affect the abundance and function of transporters is presented, highlighting research needed to fully understand the mechanisms by which transporter activity can be modulated.
Intrinsic factors
A growing body of data support the influence of intrinsic factors such as genetics, ethnicity, age, sex, physiologic states and organ-based diseases on transporter function (Fig. 2).
Fig. 2 |. Intrinsic (pink) and extrinsic (blue) factors affecting the abundance and/or activity of drug transport proteins and mechanisms that may be involved, including specific regulatory pathways and/or inhibitory effects.
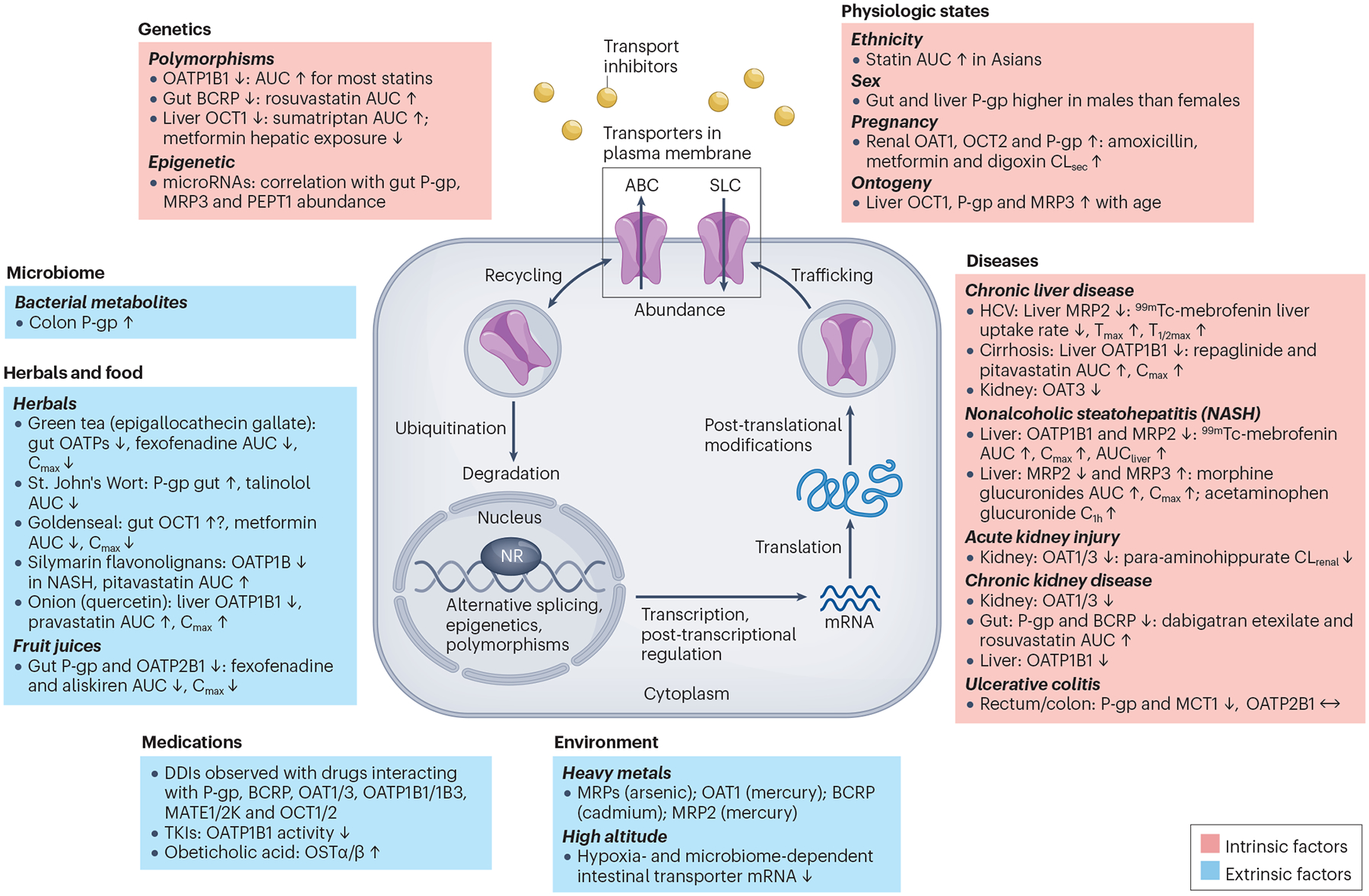
Changes represent protein levels unless noted as mRNA. The examples provided are based on a combination of preclinical and/or clinical data. For details on post-transcriptional and post-translational mechanisms involved in regulation of drug transport proteins, see section ‘Factors regulating transporter activity’ and a 2022 International Transporter Consortium publication7. Polymorphisms: Although some literature suggests that SLCO1B1*37 showed increased transport activity, a review of the literature suggests that the activity of this variant is similar to SLCO1B1*1 (wild type)229. ABC, ATP binding cassette; Asians, defined in ref. 15 as Chinese and Japanese subjects living in the US for at least 12 months; AUC, area under the plasma (serum) concentration-time profile; AUCliver, area under the liver concentration-time profile; BCRP, breast cancer resistance protein; C1h, plasma (serum) concentration at 1 hour; Cmax, maximum plasma concentration; CLrenal, renal clearance; CLsec, renal secretion clearance; Cmax, maximum plasma concentration; DDIs, drug–drug interactions; HCV, hepatitis C virus; MCT1, monocarboxylate transporter 1; MRP, multidrug resistance-associated protein; NASH, nonalcoholic steatohepatitis; NR, nuclear receptor; OAT, organic anion transporter; OATP, organic anion transporting polypeptide; OCT, organic cation transporter; OSTα/β, organic solute transporter alpha/beta; PEPT1, peptide transporter 1; P-gp, P-glycoprotein; SLC, solute carrier; Tmax, time required for maximal hepatic activity; T1/2max, time required for peak activity to decrease by 50%; TKIs, tyrosine kinase inhibitors; ↑, increase, or ↓, decrease, in the protein abundance and/or activity as detailed in the references provided in Supplementary Materials.
Genetics.
Functional genomic studies have revealed that several drug transporters carry common reduced-function polymorphisms, which have been associated with interindividual variation in drug response. Transporter polymorphisms are considered clinically relevant if they result in functional changes and show significant associations in candidate gene studies and genome-wide association studies. For example, the increase in plasma exposure (area under the plasma concentration-time profile (AUC) and/or maximum plasma concentration (Cmax)) of most statins due to hepatic SLCO1B1 variants encoding decreased function of OATP1B1 transporter can lead to myopathy or rhabdomyolysis and is one of the most well-established, clinically relevant examples of the effect of genetics on transporter function8. Additionally, decreased function of intestinal ABCG2 variants increases rosuvastatin exposure9. Recommended dosage adjustments for patients with SLCO1B1 and ABCG2 polymorphisms are available (https://cpicpgx.org/genes-drugs/). Reduced-function variants in SLC22A1 impair hepatic uptake and increase the plasma exposure of drug substrates including sumatriptan, morphine and ondansetron10,11. Advances in transporter science have helped unravel the complexities of some DDIs and provided explanations for unexpected pharmacodynamic effects. In the case of metformin, SLC22A1 variants are associated with decreased hepatic distribution and reduced efficacy; importantly, this change in metformin in the liver and the antihyperglycemic effect is not reflected in plasma exposure12,13. In addition to genetic polymorphisms, an increased understanding of the contribution of epigenetics (for example, microRNAs) and other regulatory mechanisms to transporter function will be important for elucidating the effects of intrinsic and extrinsic factors on transporter-mediated drug disposition7,14. Deep mutational scanning and other technologies should be applied to important drug transporters in the liver, kidney and intestine for a comprehensive understanding of the effects of genetic variants on transporter function, membrane trafficking and overall protein levels.
Ethnicity, sex, pregnancy and ontogeny.
The exposure to several statins, notably rosuvastatin, was higher in Chinese and Japanese subjects living in the US for at least 12 months compared with white individuals15. Although some variability may be explained by a higher frequency of the decreased-function ABCG2 variant (c.421A, p.141K) in Chinese and Japanese populations, additional factors may contribute15. For example, a genotype-independent ethnic variability in OATP1B1-mediated uptake of simvastatin has been suggested in Japanese participants16. Examples of transporter-mediated sex-related differences in pharmacokinetics are rare to date17. Hepatic and intestinal P-gp abundance is slightly higher in males than females18–20, which might contribute to lower saquinavir systemic exposure and higher clearance in males. Interestingly, polyethylene glycol (PEG400) caused a sex-related modulatory effect on P-gp, resulting in up to a 58% increase in urinary excretion of cimetidine in males but not females19. Furthermore, in participants with the c.521TT genotype of SLCO1B1, pravastatin plasma exposure was higher in females than males21. However, OATP1B1 abundance appeared to be similar in males and females18. Although the effects of pregnancy on drug transporters are not as well characterized as for metabolic enzymes, clinical studies revealed increased net renal secretion clearance of amoxicillin (>50%), metformin (~40%) and digoxin (107%) during pregnancy, consistent with increased renal transport likely by OAT1/3, OCT2 and P-gp, respectively22,23.
Maturation of drug transport, which is often transporter and organ dependent23–26, may cause variability in pharmacokinetics, especially in neonates and infants (for example, lower morphine clearance owing to OCT1 ontogeny)27. Differences in developmental pattern could lead to different contributions of specific transport or metabolic pathways to drug disposition in children versus adults. The available age-dependent protein abundance data for clinically relevant transporters indicate that, in general, age-related changes in transporters are less pronounced than metabolic enzymes23. However, further research regarding the ontogeny of transporter function is needed for successful pharmacokinetic predictions in paediatrics.
Liver disease.
Liver disease has long been associated with reduced clearance of many drugs, although the effects were attributed primarily to a decreased expression of hepatic CYPs and other enzymes involved in drug metabolism28. More recently, the influence of liver disease on hepatic transporters and its effect on hepatobiliary drug disposition have been reviewed29. Changes in hepatic transporters depend on the type and severity of liver disease. For example, in patients with chronic hepatitis C virus and various degrees of fibrosis, 99mTc-mebrofenin hepatic uptake, mediated by OATP1B1/1B3, was impaired relative to the control; increased hepatic exposure of 99mTc-mebrofenin suggested impaired MRP2 function30. Results of a proteomic analysis of hepatitis C virus-infected human liver samples were consistent with downregulation of MRP2, MRP4, NTCP, OATP2B1 and OCT131.
In general, a progressive decrease in OATP1B activity occurs in patients with increasing hepatic impairment. On the basis of plasma concentrations of the OATP1B biomarker coproporphyrin I (CPI) and the systemic exposure of 21 substrate drugs, OATP1B activity was estimated to decrease by as much as ~90% in patients with severe hepatic impairment (Child-Pugh category C)32. Proteomic data for OATP1B1 and other transporters in patients with specific liver diseases have previously been summarized23. The plasma exposure of repaglinide was increased in chronic liver disease patients (Child-Pugh B or C with cirrhosis), relative to controls, consistent with significantly decreased OATP1B1, CYP2C8 and CYP3A4 abundance33,34. Similarly, in cirrhotic patients (Child-Pugh B), pitavastatin plasma exposure was increased ~threefold relative to healthy controls35.
Although obesity and other comorbidities related to nonalcoholic fatty liver disease may confound pharmacokinetic alterations, in patients with noncirrhotic nonalcoholic steatohepatitis (NASH), decreased OATP1B1/1B3 and MRP2 function contributed to increased 99mTc-mebrofenin systemic and hepatic exposure36, consistent with decreased glycosylation of these transporters37. Higher systemic concentrations of glucuronide conjugates of morphine and acetaminophen in noncirrhotic adult and paediatric NASH patients, respectively, are consistent with decreased MRP2 and increased MRP3 efflux38,39.
Renal impairment.
For many years, renal impairment was thought to affect solely renal drug clearance, primarily through the loss of nephrons and an associated decrease in filtration and tubular clearance. Recent studies have revealed the complexity of renal disease on both hepatic and renal drug clearances and, in particular, on drug transporters expressed in these organs. For example, in acute kidney injury, inflammation results in increased plasma concentrations of pro-inflammatory cytokines such as interleukins (for example, IL-1, IL-6, IL-8), tumour necrosis factor alpha (TNF-α) and interferon gamma, and these cytokines can affect membrane transporters40. For instance, in renal allograft patients with ischaemic reperfusion injury-induced acute kidney injury, the total clearance of the OAT1 substrate para-aminohippurate was reduced 8.6-fold41. This correlated with the redistribution of OAT1/3 to the apical plasma membrane in proximal tubule cells and subsequent excretion of the transport proteins in urine42. In chronic kidney disease (CKD), the pharmacokinetics of drugs that undergo renal and non-renal elimination may be altered23,29. For instance, CKD increased systemic exposure of hepatically cleared drugs such as pitavastatin (OATP1B1/1B3 substrate) and fexofenadine (OATP1B1/1B3/2B1 and P-gp substrate)29, and also CPI (OATP1B1/1B3 and MRP2 substrate)43. Interestingly, CKD may also affect intestinal BCRP and/or P-gp based on findings that the inhibitory effect of rifampin was more pronounced on the unbound plasma Cmax than on the unbound AUC0-inf after oral administration of a microdose of dabigatran etexilate, rosuvastatin and atorvastatin in patients with various stages of CKD43. The mechanism by which CKD affects transporters is unclear but may be because of elevated uremic toxins in plasma that inhibit transporters or modulate transporter abundance44. Although no correlation was observed between increased levels of systemic uremic toxins and OATP1B inhibition in patients with various degrees of CKD43, several uremic toxins inhibited OAT1/3 in vitro45.
Extrinsic factors
The function and levels of transport proteins may also be affected by extrinsic factors, such as diet, herbal or medication use, the microbiome and environmental exposure. In some cases, the mechanisms have been elucidated, but in others the exact mechanisms remain unknown (Fig. 2).
Microbiome.
The role of the microbiome in drug disposition and the regulation of drug transporters is an emerging area of research. Many of the uremic toxins that accumulate in CKD and inhibit transporters are produced by the gut microbiome46,47. Short chain fatty acids and secondary bile acids produced by the gut microbiota upregulated P-gp, whereas antibiotic treatment-induced perturbations of the microbiota in mice decreased P-gp levels48. These findings were supported by data from patients with ulcerative colitis48.
Food and herbs.
Food–drug interactions involving grapefruit juice and CYP3A4 are well characterized, but the effect on transporters is now also recognized and extends to other fruit juices. For example, orange and apple juice decreased aliskiren plasma exposure by ~60%, likely through intestinal OATP2B1 inhibition49. Similarly, grapefruit, apple and orange juice reduced fexofenadine exposure up to ~77%50. However, mechanistic data on the role of OATP2B1 versus OATP1A2 or other factors in intestinal DDIs (e.g., fruit juice interactions) are conflicting with the clinical observations because of uncertainty in the localization of OATP2B1 in the intestine51,52, inconsistent effects of polymorphisms in SCLO2B153 and reports on presence/absence of intestinal OATP1A254,55. Flavonoids, which inhibit drug-metabolizing enzymes, are thought to be the constituents in juice that may also inhibit intestinal transporters. More inhibitory compounds continue to be discovered (e.g., avicularin in cranberry juice56). Furthermore, catechins in green tea may inhibit drug transport, as shown by a 70% decrease in fexofenadine exposure following administration of green tea extract57. The flavonoid quercetin, found in many foods (for example, onion), inhibits OATP1B1-mediated statin transport. Although quercetin-mediated increases in pravastatin systemic exposure in healthy participants are modest58, interactions of dietary constituents with transporters, especially those used as herbal supplements, warrant further investigation. The effects of some botanical natural products on P-gp are documented. For instance, the antidepressant hyperforin, the active ingredient in St John’s Wort, induces intestinal P-gp, resulting in reduced oral bioavailability of the P-gp substrate talinolol59. Although components in an extract of the herb Goldenseal were relatively potent inhibitors of several clinically relevant drug transporters in vitro, a significant reduction was observed only in metformin plasma exposure after administration of an oral drug cocktail consisting of furosemide, metformin and rosuvastatin60. In cocktail study design, the selection of probe substrates and their doses is important when establishing a standard approach to study natural product–drug transporter interactions.
Medications.
Since the first ITC publication on transporters1, the mechanisms of transporter inhibition and induction have been increasingly revealed61,62. Initial assessments for transporter (for example, OATP1B) DDI risk typically are based on in vitro substrate and inhibitor studies in recombinant cell lines or membrane vesicles63, assuming competitive inhibition. Preincubation of OATP1B1 inhibitors (for example, cyclosporine) has been shown to increase their inhibitory potency64 and has been recommended for evaluation of OATP1B1/1B365. Several tyrosine kinase inhibitors (TKIs) that are potent inhibitors of LYN kinase reduced OATP1B1 phosphorylation, which correlated with a reduced activity of human OATP1B1 in cell lines, and increased rosuvastatin plasma exposure, at least in mice66. This opens the possibility that the pharmacological activity of TKIs could explain, in part, DDIs caused by this class of drugs. Similarly, the treatment of sandwich-cultured human hepatocytes with the farnesoid X receptor agonist obeticholic acid increased protein levels and activity of OSTα/β67. Clearly, transporter regulation is an emerging field, and more work is needed7.
Environment.
Our understanding of how environmental factors influence transport proteins remains rudimentary. Environmental exposure to heavy metals (e.g., mercury, cadmium) may directly or indirectly inhibit transporters leading to adverse effects, for example, increased fetal exposure to other harmful BCRP substrates such as aflatoxin B1 and heterocyclic amines68. These factors are discussed in more detail in the “Transporters and toxicity” section. Modelling and simulation Physiologically based pharmacokinetic (PBPK) modelling is now the major translational tool in drug development for drugs that are substrates or inhibitors of transporters. PBPK has gained broad acceptance in regulatory submissions69,70 and has been used for a variety of diverse applications, including the characterization of transporter-mediated disposition mechanisms in healthy and other populations23,71,72 (Fig. 3 and below).
Fig. 3 |. Development, validation and applications of physiologically based pharmacokinetic models of transporter-mediated processes.
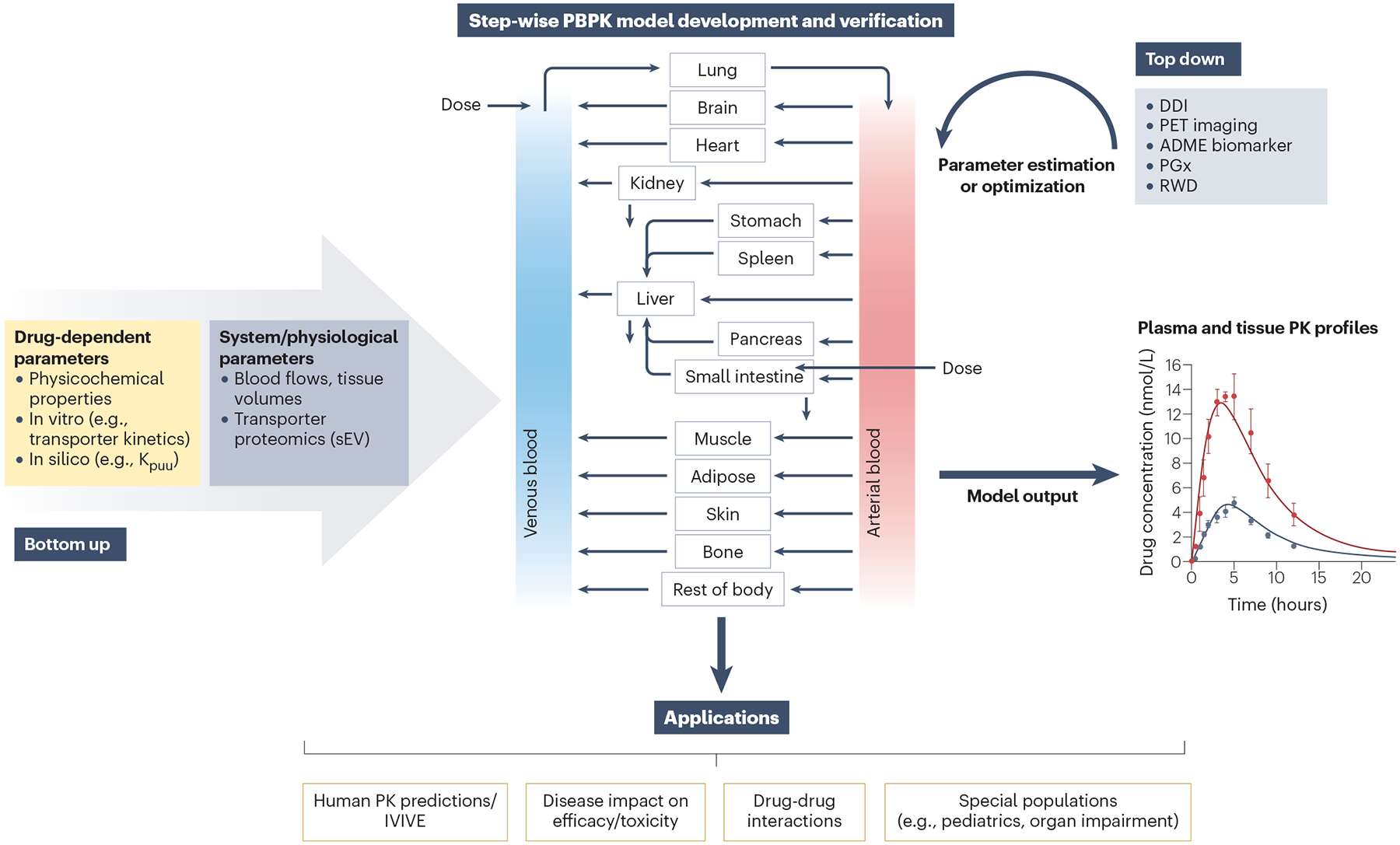
Physiologically based pharmacokinetic (PBPK) modelling is a translational tool that integrates drug-dependent parameters (e.g., in vitro transporter kinetics) with physiological parameters (e.g., transporter/enzyme expression) relevant for a specific patient population for a prospective prediction of transporter-mediated pharmacokinetics (bottom-up approach). PBPK modelling investigates the interplay of multiple processes governing drug distribution and clearance in a mechanistic manner and allows the simulation of plasma and tissue exposure resulting from modulation of enzyme and/or transporter activity. Defining the rate-determining step(s) (uptake, efflux, metabolism or a combination of these processes) in complex transporter-mediated drug disposition is important, and outcomes of the interplay of these multiple processes can be explored by PBPK modelling71. In vitro–in vivo extrapolation (IVIVE) of transporter-mediated clearance considers differences in either individual protein levels between in vitro systems and tissue of interest (REF approach) or implements differences in functional activity between in vitro and in vivo (relative activity factor, RAF); a lack of selective transporter probes hinders wider application of the latter approach. In many instances, direct extrapolation of transporter-mediated clearance using physiological scalars (hepatocellularity) or proteomic-informed IVIVE results in under-estimation of in vivo clearance, thereby requiring additional empirical scaling factors to bridge the IVIVE disconnect noted with animal studies or clinical data230,231. Consequently, a ‘top down’ estimation of missing parameters or ‘middle-out’ approaches to optimize the uncertain system and/or drug-dependent PBPK parameters are favoured. These approaches rely on clinical pharmacokinetic data (generally plasma) to refine PBPK models and ideally should be done for a range of probe drugs to increase confidence in subsequent prospective pharmacokinetic predictions; such examples have been reviewed2,23,71. Following model development and verification, PBPK models can have diverse applications as illustrated here, including prediction of transporter-mediated drug–drug interactions (DDIs; in combination with endogenous biomarkers for drug transporters) and prediction of pharmacokinetics in different patient populations (paediatrics, pregnancy or patients with different diseases, for example, organ impairment, cancer, nonalcoholic steatohepatitis). The current status of these applications and challenges are described in the text. ADME, absorption, distribution, metabolism and excretion; Kpuu, unbound partition coefficient; PET, positron emission tomography; PGx, pharmacogenomics; REF, relative expression factor; RWD, real-world data; sEV, small extracellular vesicles.
Transporter-mediated drug disposition/clearance
Organ clearance of transporter substrates is governed by either transporter–transporter or transporter–enzyme interplay; these individual processes are defined by the extended clearance concept71,73. In vitro– in vivo extrapolation (IVIVE) of transporter-mediated clearance and the ability of PBPK modelling to integrate multiple mechanisms and investigate their interplay mechanistically have proven valuable in predicting pharmacokinetic/DDIs of transporter substrates. Various IVIVE methods have been evaluated, including the relative expression factor (REF) approach. REF is enabled by advances in quantitative proteomics and provides correction for differences in individual protein levels between in vitro systems and tissues71,74,75. Leveraging the IVIVE principles, mechanistic PBPK models have been applied to predict transporter-mediated disposition for certain drugs76,77 and in certain disease populations (e.g., obesity78, Crohn’s disease79). The broader application in disease still needs to be demonstrated, together with establishing correlations between transport protein levels and functional activity for different tissues and disease states, which would further increase confidence in using transporter proteomic data in PBPK modelling. Although for several drug transporters IVIVE is established for cellular systems such as hepatocytes and recombinant cell lines, the translational ability of such data from novel complex cellular models such as microphysiological systems80 remains to be ascertained.
‘Fit-for-purpose’ PBPK models have been extensively described and evaluated for their ability to capture transporter-mediated disposition in the liver and kidney, partly because of the availability of clinical pharmacokinetic data to enable model verification and/or optimization of transporter activity/protein abundance34,81–83. In contrast, IVIVE methods are not as extensively evaluated in areas such as oral absorption or local tissue distributions, due to limitations in the quantitative interpretation of in vitro data for efflux transporters and/or limited availability of in vivo data for model verification. Emerging proteomic or tissue imaging data are critical for the refinement of PBPK-based predictions of tissue exposure (Fig. 3), as demonstrated for the prediction of liver exposure of OATP1B and OCT1 substrates84,85 or brain penetration for P-gp/BCRP substrates86,87. Despite successes, PBPK models to predict changes in transporter-mediated drug disposition and tissue exposure are generally hindered by the presence of multiple transporter orthologues, a lack of substrate specificity, limited selective inhibitors, difficulty in deriving proteomics-based scalars for individual transporters, limitations of in vitro methodologies to delineate multiple transport mechanisms/ratelimiting steps, and a lack of quantitative proteomic methods to differentiate between active and inactive proteins. Further research to address these specific gaps is needed.
Transporter-mediated DDIs
PBPK modelling has been used extensively to predict transportermediated DDIs, to allow study waivers and to inform dosing recommendations in polypharmacy13,23,70,72,83,88,89. With multiple transporters localized in the basolateral membrane of hepatocytes or proximal tubule cells, it is important to characterize the fraction transported (ft) by the individual transporter and the contribution of passive diffusion to the overall uptake. This information is critical for the mechanistic prediction of transporter-mediated DDIs. The ft estimates of OATP1B-mediated hepatic uptake determined using chemical inhibition in primary hepatocytes, or REF approaches using transfected cell data, were shown to reasonably explain the DDIs of statins with OATP1B inhibitors (e.g., rifampin, cyclosporine)90,91. PBPK models enable considerations of multiple interaction mechanisms when evaluating investigational drugs as transporter inhibitors. At this point, there is still some ambiguity in the success of IVIVE of inhibition interaction parameters (IC50/Ki) due to lab-to-lab method variables (cell systems, incubation conditions), substrate-dependent inhibition observed for some transporters and other factors.
Direct use of in vitro interaction parameters has generally resulted in the under-prediction of the magnitude of transporter-mediated DDIs2,72. Therefore, verification of initial ‘bottom-up’ models with relevant clinical data is important before model application to predict DDIs involving specific pathways. To that end, endogenous biomarkers emerged as an alternative to clinical DDI data using probe drug(s), wherein drug-induced changes in biomarker pharmacokinetics or renal clearance can be leveraged to verify and refine the transporter interaction parameters. This concept has been illustrated by using CPI data to estimate in vivo OATP1B Ki for rifampin92 and 4-pyridoxic acid data to refine probenecid OAT1/3 Ki93. Recently, biomarker-informed PBPK modelling using in vivo Ki estimated from CPI data successfully predicted the magnitude of clinical DDIs for multiple OATP1B inhibitors with various magnitudes of inhibition, demonstrating the potential in combining biomarker information and PBPK models to refine/replace clinical transporter-mediated DDI studies94,95 (Fig. 3).
Uptake transporter substrates may exhibit elevated intracellular unbound concentrations relative to plasma and thus pose a higher risk as hepatic CYP/efflux transporter inhibitors or inducers. As such, it is critical to assess the in vitro intracellular concentrations first and then predict the in vivo intracellular concentrations through a PBPK model96. Notably, a PBPK model trained to describe systemic pharmacokinetics may not necessarily predict liver exposure and may need to be further verified with either tissue exposure data obtained by positron-emission tomography imaging or relevant pharmacodynamic data71. Despite the lack of adequate holistic in vitro tools and challenges with the verification of unbound intracellular exposure predictions, consideration should be given in the mechanistic modelling and simulations to understand the elevated CYP modulation risk for uptake transporter substrates. Intestinal efflux transporters, BCRP and P-gp, are important loci for clinically relevant DDIs. Mechanistic models for probe drugs rosuvastatin83,97 and digoxin or dabigatran etexilate88,98,99 have been developed to study the effect of inhibitor/inducer drugs on these mechanisms in vivo, or explore intestinal regional differences/interplay with perpetrator absorption64,88. Recent studies demonstrated adequate predictions of BCRP-mediated DDIs using in vitro inhibition data, implying that a PBPK modelling approach can effectively predict DDI risk involving intestinal efflux in drug development83.
Disease states and specific populations
In recent years, exciting progress has been made in extending PBPK modelling efforts to specific populations, including patients harbouring different diseases (for example organ impairment, NASH) and paediatric and pregnant populations23. In some instances, model development was supported by increased availability of transporter proteomic data in those populations (e.g., cancer100, NASH101). Existing paediatric proteomic data suggest that transporter abundance follows less noticeable age-dependent changes than metabolizing enzymes102. However, the knowledge of transporter developmental biology in extrahepatic organs is still limited103, which needs to be considered when developing paediatric PBPK models23,104.
In areas where transporter proteomic data are inadequate, an analysis of clinical data for a wide range of substrate drugs in such patients is critical to inform disease-related changes in system parameters in PBPK models. Examples of such approaches include an estimated 50% decrease in the renal OAT1/3 transporter activity, in addition to the decline in glomerular filtration rate in severe stages of CKD105. Alternatively, PBPK modelling of clinical data reported for either transporter probe drugs and/or endogenous biomarkers can be used to gain insight into disease-mediated modulations in transporter function106–108. For instance, PBPK modelling of CPI and several substrate drugs suggested up to ~90% reduction in OATP1B-mediated uptake in patients with hepatic impairment32 and ~40% decrease in CKD106. These examples emphasize the importance of stepwise model development strategies (i.e., initial model verification against pharmacokinetics and DDI data in healthy participants before the extension of the model to specific patient populations), as shown recently for 4-pyridoxic acid109. Despite these advances, prospective PBPK modelling of transporter-mediated processes in specific populations has not yet gained complete confidence in regulatory submissions for exploring untested/’what-if’ scenarios23.
Endogenous biomarkers
The discovery and validation of biomarkers, endogenous substrates of transporters, have significantly advanced the assessment of transporter-mediated DDIs. Monitoring changes in the disposition of these endogenous substances as indicators of altered transporter function in vivo offers the possibility to assess a new molecular entity as a transporter modulator in early drug development. The last decade has witnessed considerable efforts towards the identification, characterization and validation of endogenous biomarkers to monitor transporter activities in vivo and to support the early assessment of DDIs. Quantitative prediction of DDIs involving transporter inhibition is challenging because of the uncertainty in the translatability of in vitro inhibition data (i.e., IC50), and complex interplay among multiple transporters/enzymes2,23,71. Furthermore, current static DDI prediction models based on in vitro data and certain assumptions about the concentrations of inhibitors often result in false-negative and false-positive predictions110,111. As such, measuring biomarkers that are selective for transporter(s) of interest in early-phase clinical investigations (for example, dose escalation studies) has become an attractive approach to facilitate transporter-mediated DDI risk assessments, in conjunction with PBPK modelling (Supplementary Tables S1a and S1b). As a result, there have been numerous academic and industry efforts to discover and validate biomarkers for a variety of drug transporters, including hepatic (OATP1B1, OATP1B3 and OCT1) and renal transporters (OAT1, OAT3, OCT2, MATE1 and MATE2K)112–117. An ITC publication in 2018 summarized the key features of these biomarkers and provided recommended methods to identify and validate biomarkers for the evaluation of DDIs via certain transporters4. Since then, the field has rapidly progressed and resulted in the (1) discovery of additional novel biomarkers with high sensitivity and selectivity for several hepatic/renal transporters115,118–120, (2) generation of rich clinical datasets to further validate selectivity and sensitivity of selected biomarkers (for example, CPI as OATP1B biomarker)95,117,121–123, (3) evaluation of transporter function and DDIs in diseased populations (for example, organ impaired patients)32,43 and (4) development of biomarker-informed modelling approaches to either support their qualification92,93 or to quantitatively translate biomarker data to predict transporter-mediated DDIs94,109,124–126. On the basis of our increased understanding of the in vivo kinetic properties of the various transporter biomarkers, the ITC recommends monitoring several hepatic and renal transporter biomarkers in clinical phase I studies when in vitro studies suggest clinical DDI potential. Figure 4 illustrates the proposed classification of endogenous biomarkers for several hepatic and renal transporters and recommendations for their application in drug development to improve DDI de-risking and management strategies. This classification is based on their selectivity (in vitro transporter phenotyping profile and in vivo pharmacogenomic data), sensitivity (clinical DDI studies with known transporter inhibitors with different inhibition potency and variability in biomarker baseline) and predictability (prediction performance in the clinical DDI studies and/or PBPK modelling and simulation). The data and existing evidence that support our recommendations are summarized in Supplementary Tables S1a and S1b. Measurement of CPI is recommended in early-phase clinical studies, and data for this biomarker can be applied for OATP1B DDI risk assessment given its superior selectivity, sensitivity and prediction performance compared with other biomarkers (i.e., Tier 1 biomarker). Considering existing evidence, we propose collecting data for GCDCA-3G (OATP1B1), GDCA-3G (OATP1B1), GCDCA-S (OATP1B), 4-pyridoxic acid (PDA) (OAT1/3), N1-methylnicotinamide (NMN) and creatinine (OCT2 and MATE1/2K) as Tier 2 biomarkers in early clinical studies. Recent studies have shown that GCDCA-3G and GDCA-3G are more selective for OATP1B1118, whereas GCDCA-S is a more selective biomarker for OATP1B3122, in addition to being an OAT3 substrate. Therefore, monitoring GCDCA-S in both plasma and urine is recommended if the new molecular entity is a dual inhibitor of OATP1B3 and OAT3116,122. These Tier 2 biomarkers should be measured together with a Tier 1 biomarker (CPI) in a multiplexed approach. However, drug-induced changes in these biomarkers should be carefully considered in the DDI risk assessment/decision-making processes, as they are not yet fully validated. In contrast to CPI, all bile acid conjugates show larger baseline diurnal variability and may potentially be affected by food, which needs to be considered in the data interpretation/study design. As clinical studies are time-consuming, costly and associated with some health risks to the trial participants, an important use of a biomarker is to demonstrate no or limited risks of transporter-mediated DDIs (decision tree in Fig. 5), thereby obviating the need for a separate dedicated clinical study. Currently, the Tier 1 biomarker CPI has sufficient clinical data with inhibitors of various potencies as highlighted above and is considered validated for this purpose (Supplementary Table S1a).
Fig. 4 |. Classification of endogenous biomarkers of hepatic and renal transporters and International Transporter Consortium recommendations for their application in drug development.
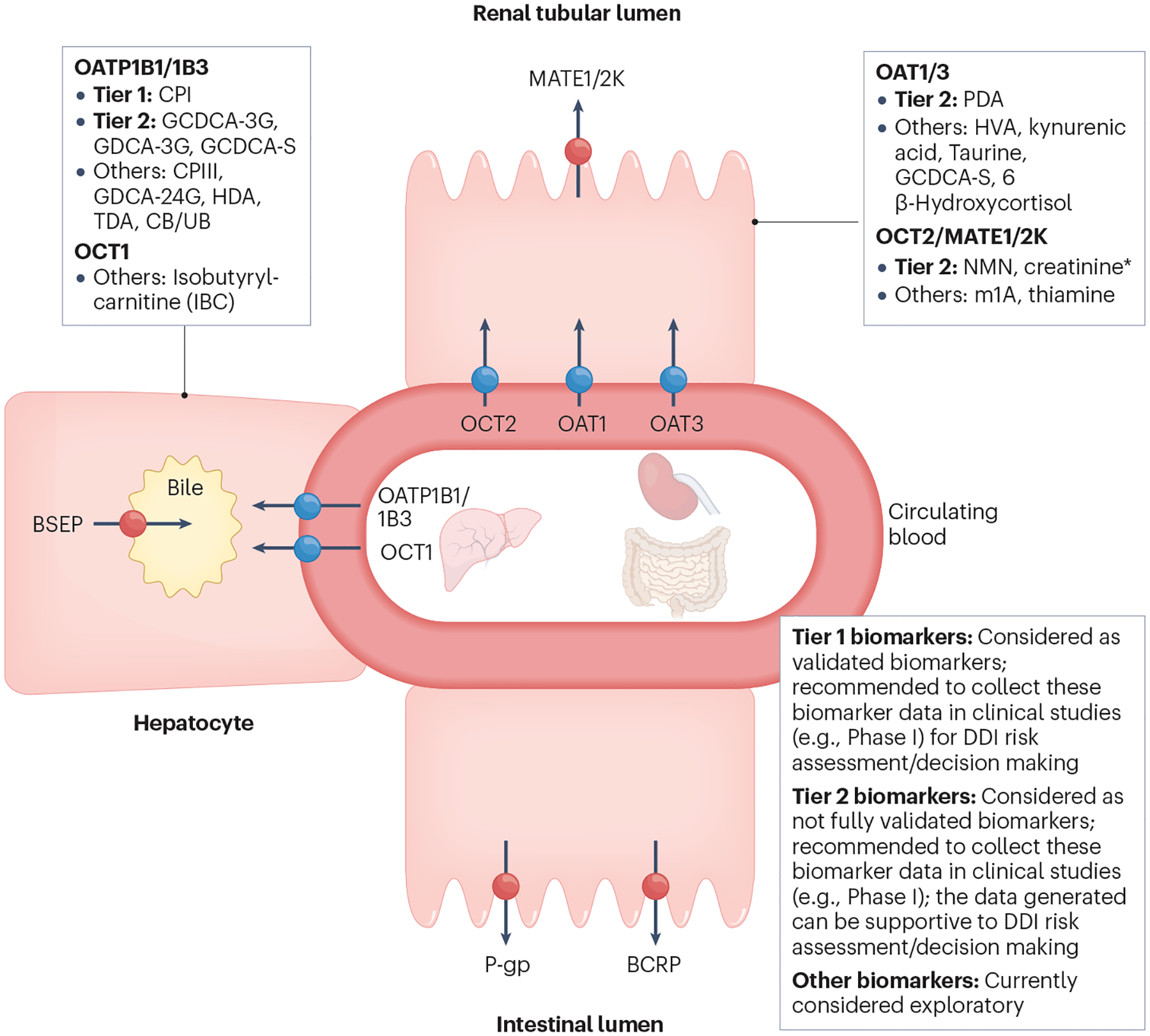
Tier 1 biomarkers: Recommendation to include these biomarkers in clinical Phase I studies when in vitro studies show clinical drug–drug interaction (DDI) potential; considered validated for clinical DDI risk assessment (see Fig. 5 for decision tree). Biomarkers have (1) high sensitivity/selectivity to the transporter of interest (based on in vitro phenotyping or clinical pharmacogenomic data); (2) available clinical DDIs with potent, moderate, weak and non-inhibitors; (3) validated DDI prediction performance with probe drugs; and (4) available mechanistic models. Tier 2 biomarkers: Recommendation to collect data on these biomarkers in clinical Phase I studies when in vitro studies show clinical DDI potential; not considered validated for clinical DDI risk assessment/decision making yet. Biomarkers have (1) high sensitivity/selectivity to the transporter of interest (based on in vitro phenotyping and clinical pharmacogenomic data); (2) limited available clinical DDIs with potent, moderate, weak and non-inhibitors; and (3) models developed for some, but not all Tier 2 biomarkers. Further evaluation is required to understand their DDI prediction performance. * NMN may serve as a more selective and sensitive biomarker than creatinine owing to a higher contribution of active renal secretion clearance to the total clearance (~70% vs. ~30%). Creatinine is included in Tier 2 because of the availability of data and its routine measurement to monitor renal toxicity. Elevation of serum creatinine may also be caused by reduced renal function, and it is important to distinguish the inhibition of OCT2/MATE versus renal toxicity. Other biomarkers: Currently not recommended to collect data on these biomarkers in clinical Phase I studies due to relatively low sensitivity/selectivity or limited data to understand biomarker selectivity/sensitivity or limited clinical reports evaluating DDI predictive performance. BCRP, breast cancer resistance protein; BSEP, bile salt export pump; CB, conjugated bilirubin; CPI, coproporphyrin I; CPIII, coproporphyrin III; GCDCA-3G, glycochenodeoxycholic acid-3-glucuronide; GCDCA-3S, glycochenodeoxycholic acid-3-sulfate; GDCA-3G, glycodeoxycholic acid-3-glucuronide; GDCA-24G, glycodeoxycholic acid-24-glucuronide; HDA, hexadecanedioate; HVA, homovanillic acid; MATE, multidrug and toxin extrusion protein; m1A, N1-methyladenosine; NMN, N1-methylnicotinamide; OAT, organic anion transporter; OATP, organic anion transporting polypeptide; OCT, organic cation transporter; PDA, 4-pyridoxic acid; P-gp, P-glycoprotein; TDA, tetradecanedioate; UB, unconjugated bilirubin. Adapted with permission from ref. 4, Wiley.
Fig. 5 |. Decision tree for organic anion transporting polypeptide (OATP1B)- mediated drug–drug interaction risk assessment with coproporphyrin I.
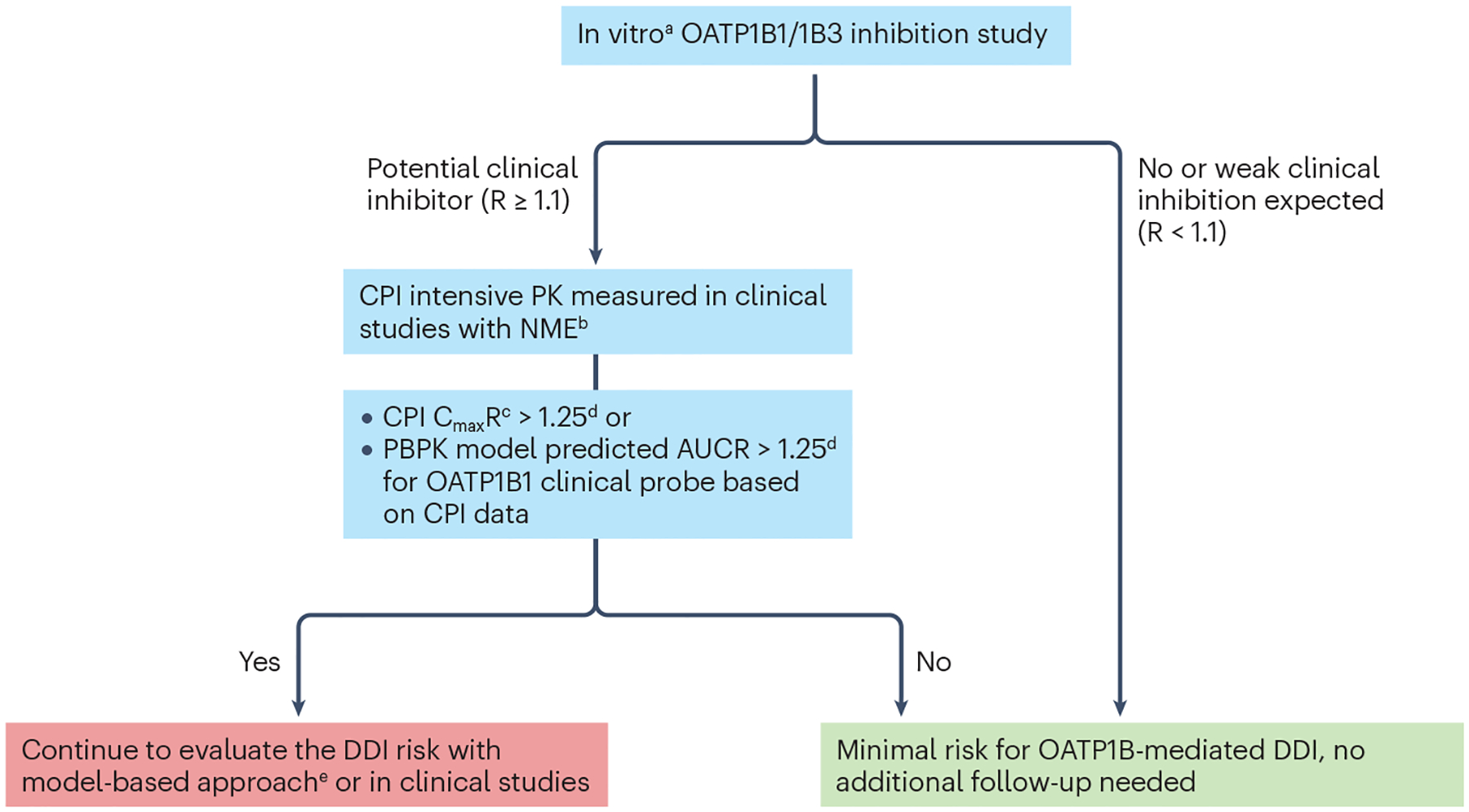
aConsidering substrate-dependent inhibition often seen in the case of OATP1B1, it is recommended to generate in vitro inhibition data with coproporphyrin I (CPI) as a Tier 1 biomarker and a relevant co-medication of interest for a new molecular entity (NME). R is the predicted ratio of the victim drug’s area under the plasma concentration-time profile (AUC) in the presence and absence of the investigational drug as OATP1B inhibitor. bTime-matched biomarker concentrations in the absence of the NME are usually not available from firstin- human or clinical pharmacology studies except for drug–drug interaction (DDI) studies. There are two potential approaches to address this issue, namely, one is to use the pre-dose single time point as the baseline level, and the other is to use data from a separate placebo cohort. The first approach is useful for CPI as there is little to no diurnal variation; however, one must be careful when comparing biomarker kinetics at the steady-state of the NME compared with the pre-dose biomarker data. The second approach is valid except that there is less power to detect an interaction with the parallel, non-crossover comparison. cThe appropriate metrics depends on the kinetic properties of both biomarkers and the NMEs4. Because CPI has a short terminal half-life, AUC is less appropriate, as the ratio of AUC depends on the duration for which AUC is calculated, and this can lead to under-estimation of the magnitude of inhibition compared with CmaxR, as seen in CPI kinetics in the presence of GDC-0810 or cyclosporine A184,232. dOther thresholds can be justified based on the exposure-response relationships of the co-medications of interest for the NME. eFactors that increase confidence in quantitative DDI prediction with model-based approaches: (1) CPI data from dose-ranging trials, especially those including supratherapeutic dose, (2) CPI observations from a sufficiently large number (for example, >10 participants receiving the same dose of the NME as typically seen in a dedicated DDI study) and (3) consistent observations with other biomarkers such as GCDCA-3G. AUCR, AUC of CPI in the presence of an inhibitor relative to the baseline AUC (control); CmaxR, ratio of CPI Cmax in the presence of an inhibitor relative to the baseline Cmax (control); PK, pharmacokinetics.
Recently, a cut-off value for applying CPI data to de-risk OATP1B DDIs has been derived based on a retrospective analysis of clinical CPI and OATP1B DDI data, and recommendations on study design/data interpretation have been provided127. In addition, there are several important considerations regarding clinical development (see the Fig. 5 legend). One important factor is the design of clinical studies from which the biomarker kinetic data are obtained. For example, if a biomarker is monitored in first-in-human studies with a limited number of participants covering the clinical dose, raw observed data might not be sufficient for detecting weak transporter inhibition. In such cases, model-based approaches (for example, population PK modelling) can be used to leverage the entire dataset across wide dose levels128. One way to ensure the robustness of biomarker observations is to not only focus on the point estimate of the observed magnitude of the interaction, but to also provide confidence intervals, either from a statistical summary of the raw observed data or model-based approaches, and to compare that with expected results from clinical studies. The needed level of confidence depends on the stage of clinical development — relatively limited data from firstin- human studies can be sufficient for inclusion/exclusion criteria for Phase 2/3 clinical studies, and more data can be accumulated in parallel with the conduct of confirmatory studies. The final step in biomarker application is to quantitatively predict the magnitude of DDIs based on the biomarker kinetics and inform the co-medication recommendations. This application requires model-based approaches94,95,125,129, as discussed in more detail in the “Modelling and simulation” section and illustrated in Figs. 3 and 5. Transporters and toxicity Given the evidence that drug transporters regulate both systemic and local concentrations of unbound drugs, it is not surprising that drug transporters can directly or indirectly contribute to drug-induced organ toxicity. Over the past decade, several papers130–133 have examined the role of transporters in drug toxicity. Furthermore, recent studies have shown that tissue-specific expression of several transporters can contribute to local drug accumulation and DDIs and that functional alterations in these transporters can directly influence an individual’s susceptibility to drug-induced organ injury. This section will highlight prominent examples of transporter-mediated drug toxicities (Fig. 6a), emerging approaches to navigate these toxic effects and available risk assessment tools. Although SLC transporters play a vital role in the absorption and disposition of essential micronutrients and macronutrients, they can also mediate the uptake of drugs and other xenobiotics, thereby playing unintended and sometimes harmful roles in response to such molecules. For example, injury to neurons and the heart resulting in peripheral neurotoxicity and cardiotoxicity, respectively, are particularly common adverse events of cancer therapeutics. Although the mechanisms underlying these side effects remain incompletely understood, multiple studies have shown that many cytotoxic anticancer drugs accumulate extensively in healthy cells such as peripheral neurons and cardiomyocytes and that this process accounts, at least in part, for selective toxicity to these cells134,135. Several studies have confirmed the expression of certain SLCs in peripheral neurons and cardiomyocytes that are known to transport a broad range of clinically relevant xenobiotics, including anticancer drugs such as oxaliplatin and doxorubicin (Fig. 6). Although still largely unexplored, one strategy that could offer neuroprotection or cardioprotection is to intentionally inhibit this transport process with pharmaceuticals to restrict drug access to the site of injury, thereby preventing drug accumulation that results in the clinical manifestations of toxicity136. Among the class of SLCs, the importance of OCTs as mediators of neuronal and cardiac uptake of drugs has been reasonably well established, and this collective work has demonstrated contributions of OCT2 to oxaliplatin transport137 and of OCT3 to doxorubicin transport138. Similarly, a role for certain OATPs has been implicated in the transport of the neurotoxic chemotherapeutic drug paclitaxel in rodents by a mechanism that is sensitive to pharmacological inhibition by the TKI nilotinib139. In contrast, SLC inhibitors can directly disrupt the uptake and use of nutrients by cells, leading to a range of toxic effects. Fedratinib has been associated with Wernicke’s encephalopathy, which is thought to be because of its ability to inhibit thiamine transporter 2 (THTR2)-mediated uptake in the gut and potentially into the brain140,141. Reduced function of ABC transporters via genetic alteration or inhibition can have toxic consequences. Genetic polymorphisms in BSEP and MDR3 are associated with the cholestatic liver diseases known as progressive familial intrahepatic cholestasis (PFIC) type 2 and 3, in which cholestatic injury occurs from increased intracellular bile acid concentrations due to reduced biliary efflux, and increased free biliary bile acids due to reduced biliary phospholipid translocation from hepatocytes into bile, respectively130. Inhibition of BSEP or MDR3 by xenobiotics has been associated with cholestasis and drug-induced liver injury130. Inhibition of MDR3 has also been associated with bile duct hyperplasia and cholecystitis142,143. Another ABC transporter, P-gp, in the blood–brain barrier can modulate the neurotoxicity of methadone used to treat opioid addiction. Following fatal overdoses with methadone, higher brain-to-blood ratios were detected in patients expressing a polymorphic variant of P-gp associated with reduced function compared to patients expressing the transporter wild type131. Uptake and efflux transporters are also involved in cellular exposure leading to adverse human health effects because of heavy metals, including the drinking-water contaminant arsenic (MRP2/MRP4) and the ubiquitous environmental pollutant mercury (OAT1/OAT3)131. Humans are exposed to the heavy metal cadmium primarily through diet, smoking or industrial use. Cadmium is excreted by BCRP and MRP2, but elimination is not as efficient as uptake/sequestration resulting in adverse health effects in the kidney, liver, bone, lung and cardiovascular system131. Recently, therapeutic approaches aimed at transport pathways to reduce toxicity have been studied. Although further investigations are needed to determine the feasibility of such approaches in the clinic, several caveats exist including the need for pathway-specific transporter inhibitors. Studies to mechanistically link transporters to an underlying toxicity can be challenging because of a lack of selective substrate/inhibitor pairs, robust in vitro and in vivo test systems, profound species differences and lack of clear in vitro-to-in vivo translation. For example, although individuals carrying BSEP gene mutations develop cholestatic injury associated with PFIC2, rodents with Bsep gene knockout are fertile and viable and only develop a mild nonprogressive intrahepatic cholestasis130. Therefore, more comprehensive and physiologically relevant in vitro systems that recapitulate the in vivo functionality (for example, 3D cultures, primary cell co-cultures, microphysiological systems) are in increasing demand. However, the characterization and optimization of advanced cellular systems for transporter function remain at an early stage. Combining in vitro test systems, animal safety data and mathematical modelling (Fig. 6b) approaches is required to de-risk and predict the clinical outcomes of transporter-induced organ toxicity. Assessing in vitro BSEP inhibition for investigational drugs has been increasingly adopted in the pharmaceutical industry130. However, in vitro BSEP IC50 alone cannot accurately predict clinical drug-induced liver injury. Additional mechanisms associated with bile acid accumulation in hepatocytes in addition to BSEP inhibition are investigated in some cases130. Early termination of drug candidates that are associated with toxic findings and the advancement of safe molecules that will most likely succeed are achieved by the introduction of well-defined testing strategies that ensure the implementation of the appropriate in vitro/in vivo test models.
Fig. 6 |. Drug-induced organ injury.
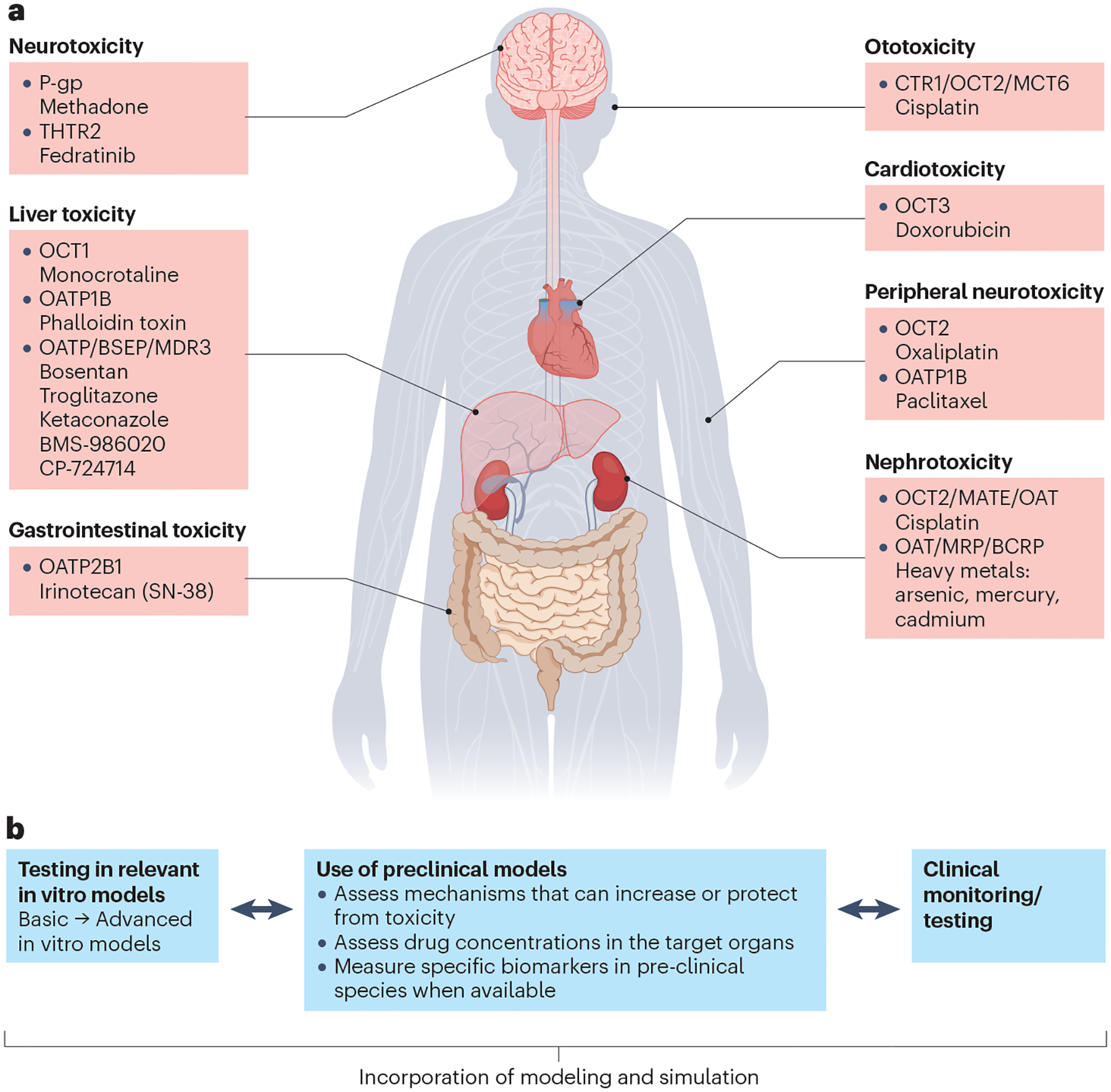
a, Examples of solute carrier superfamily (SLC)/ATP binding cassette superfamily (ABC) transporters involved in drug-induced organ injury. b, Tools to identify/de-risk transporter-mediated drug-induced organ toxicity in humans. The approach to identify or de-risk transporter-mediated toxicity can depend on the site and type of toxicity (or signals observed). Approaches can also differ depending on whether an analysis is conducted prospectively (based on a hypothesis or previous finding) or retrospectively (after a toxicity signal is observed in preclinical or clinical studies). As such, workflows often differ on a case-by-case basis. In vitro, preclinical and/or clinical tools are available to assess the transport mechanism. The following examples showcase the utility and limitations of available tools and highlight different approaches to identify/de-risk transporter-mediated organ toxicity. 1) Bile salt export pump (BSEP) inhibition has been associated with drug-induced liver injury (DILI) and assessing inhibition in vitro has been widely adapted in the pharmaceutical industry130. Often initial assessments are done in a basic in vitro model using membrane vesicles. Follow-up studies in more advanced hepatocyte models, such as sandwich-cultured hepatocytes or micropatterned hepatocyte co-cultures, can be conducted to reduce potential false positives233. Studies in preclinical models are often not conducted because of the need for a specific biomarker and a translational preclinical model. If a perceived liability is found in vitro, establishing structure–activity relationships to screen out inhibition in vitro or powering a phase I study to include markers of cholestasis to assess toxicity potential early in clinical studies could be considered. Further, incorporation of modelling and simulation can also be used for quantitative prediction of mechanistic liabilities, including inhibition of other transport pathways such as multidrug resistance-associated proteins (MRPs), because DILI mechanisms are often multifactorial234. 2) The chemotherapeutic agent oxaliplatin can lead to severe dose-limiting peripheral neurotoxicity because of extensive accumulation in peripheral neurons. In some cases, the toxicity can cause functional impairment lasting well beyond the treatment period. Comparative studies in various transporter knockout mouse models illustrated the role of organic cation transporter 2 (OCT2) in oxaliplatin-mediated neurotoxicity137. Follow-up studies illustrated that pretreatment with OCT2 inhibitors could offer neuroprotection in animal models without affecting drug clearance or efficacy. Dasatinib, an OCT2 inhibitor, is currently being evaluated in phase I/II clinical trials to prevent oxaliplatin-induced neurotoxicity136. 3) Nephrotoxicity is the major dose-limiting toxicity for cidofovir. Probenecid, a pan inhibitor of organic anion transporter 1 (OAT1) and OAT3, can increase the plasma exposure to cidofovir while decreasing its renal clearance by inhibiting the OAT-mediated tubular secretion235,236. The use of cidofovir (VISTIDE®) requires it to be administered with probenecid to reduce renal tubular uptake of cidofovir and subsequent renal toxicity237. BCRP, breast cancer resistance protein; CTR1, copper transporter 1; MATE, multidrug and toxin extrusion protein; MCT6, monocarboxylate transporter 6; MDR3, multidrug resistance protein 3; OATP, organic anion transporting polypeptide; P-gp, P-glycoprotein; THTR2, thiamine transporter 2. Part a adapted with permission from ref. 131, Wiley.
Regulatory science considerations
The assessment of DDIs is an integral part of drug development. These DDIs can involve the modulation of various transporters, which may affect drug concentrations in the systemic circulation and/or specific tissues thereby affecting drug efficacy and/or safety. Here, regulatory considerations are discussed, focusing specifically on transporter-mediated DDIs.
Transporter-mediated DDIs caused by inhibition
The regulatory guidance documents recommend evaluating the inhibition potential of investigational drugs towards several transporters (i.e., BCRP, MATE1, MATE2K, OAT1, OAT3, OATP1B1, OATP1B3, OCT2, P-gp). The EMA guideline also recommends that consideration is given to investigating the inhibitory effect of a drug on OCT1 and preferably also on BSEP. The current guidance documents from several regulatory agencies (for example, FDA, EMA, Pharmaceuticals and Medical Devices Agency) are in line with this general framework, with some differences in terms of the cut-off values or decision criteria used in basic models. Recently, a draft of a globally harmonized DDI guideline, M12, has been published for public consultation by the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH), in which the cut-offs for corresponding transporters are unified144. An initial qualitative assessment of the in vivo potential for a drug to inhibit transporters uses basic models with decision criteria. If the drug interaction potential cannot be ruled out by these basic models, further assessment is warranted either by conducting a clinical DDI study or by quantitative prediction with more sophisticated models (e.g., PBPK models). The cut-offs are generally selected with intention to minimize false negative predictions, but inevitably they lead to false positive predictions. For example, dividing the dose by 250 ml to estimate intestinal luminal concentrations for P-gp and BCRP inhibitors likely overestimates inhibitor concentrations for drugs that have poor solubility, leading to false positive predictions145. For OATP1B1/3 inhibition, various decision criteria were evaluated for prediction performance showing positive prediction error values of 27% to 43%110 (that is, the proportion of studies that were conducted unnecessarily because there was no observed in vivo DDI), with 35% obtained for the current recommended criteria in the FDA guidance. Similar positive prediction error values were observed for the proposed criteria by the FDA to predict DDIs mediated by OCT2/MATEs or OAT1/3 inhibition146. It should be noted that these decision frameworks for the prediction of transporter-mediated DDIs offer a qualitative (yes/no) prediction, primarily to rule out the potential of a drug to inhibit transporters in vivo based on in vitro inhibition data and do not provide a quantitative prediction of the extent of DDIs. In addition, the decision frameworks only consider one transporter at a time, whereas in clinical settings a DDI may occur with multiple enzymes and/or transporters. Mechanistic static models and PBPK models have been used for quantitative predictions of transporter-mediated DDIs; these models are considered at different stages of drug development depending on data availability65,72,147. Although static models require fewer resources and less data than PBPK models, their inability to capture dynamic profiles of perpetrators and substrates at sites relevant for interaction is a significant limitation for the evaluation of transporter-mediated DDIs2. In contrast, PBPK models offer a wider range of capabilities, for example, simulation of systemic and tissue concentration–time profiles at pharmacologically and toxicologically relevant sites, which is critical for the investigation of complex DDI scenarios involving multiple transporters and/or transporter-enzyme interplay (further details in the “Modelling and Simulation” section)2,23,71. The number of regulatory PBPK applications related to transporters has increased significantly in the past 5 years23,69,70; among those submissions, the prediction of transporter-mediated DDIs (mainly for OATP1B1 and BCRP) remains one of the key applications. The analysis of submissions from 2018–2021 showed that 59% of submissions evaluated the investigational drug as a perpetrator, 27% evaluated the investigational drug as a substrate and 14% were intended to evaluate the investigational drug both as a perpetrator and substrate23. Some recent examples of PBPK model applications to inform drug labelling of transporter inhibitors or substrates include cabotegravir (OAT1/3)148, mitapivat (OAT3)149 and atogepant (BCRP)150. Besides the refinement of inhibitor PBPK models, models for transporter substrates (victim drugs) should also be established and verified with appropriate clinical studies. One critical factor for predicting transporter-mediated DDIs is the term ft. In addition to in vitro methods (“Modelling and simulation” section), this value can be derived from clinical DDI studies of a substrate conducted with strong inhibitors that are relatively specific for an individual transporter, similar to the approach used with metabolic DDIs. However, transporter inhibitors are often non-selective and affect multiple transporters and/or enzymes. Equally challenging, transporter substrates often share substrate specificity with other transporters/enzymes4. Hence, it is not straightforward to dissect the contribution of an individual transporter to the observed DDI effect and derive the ft. In certain instances, pharmacogenetic analyses are used to estimate ft values12,64. Although this approach provides useful information, some polymorphisms such as the BCRP c.421C>A and OATP1B1 c.521T>C show decreased transporter activity but do not completely abolish function of BCRP or OATP1B1, and therefore, such data may underestimate ft. Thus, the confidence in the ft of a substrate may need to be considered based on the totality of data. Endogenous biomarkers of drug transporters have received considerable attention as a possible additional clinical tool to support the evaluation and prediction of transporter-mediated DDIs in vivo151. Major advances have been made in recent years to identify various biomarkers for OATP1B1/3 and OAT1/3 (See “Biomarker” section). The evaluation of several biomarkers has now been incorporated into the development process95,152; among these, CPI is the most established biomarker so far (Fig. 4). The strategy of leveraging biomarkers for a drug predicted to have the potential to inhibit a transporter in vivo is that one or multiple biomarkers for that transporter of interest are measured during early clinical studies of the drug in development (for example, single-dose or multiple-dose escalation studies, Fig. 5). If the drug does not significantly alter the level of the biomarker(s), then it is considered less likely to inhibit the transporter in vivo, whereas significant changes in biomarker levels confirm a potential interaction risk and support the conduct of a clinical study as a follow-up. In addition, biomarker data can be used in combination with PBPK models to refine the in vivo inhibitory potency of an investigational inhibitor drug94,109, which is critical for the prediction of the magnitude of the transporter-mediated DDI, but is often not accurately reflected by in vitro measured IC50 or Ki data (further details in “Biomarkers” and “Modelling and simulation”sections).
Metabolites of drugs as inhibitors of transporters
Metabolites of drugs can also contribute to DDIs leading to ‘unexpected’ interactions if only the parent is evaluated as the inhibitor using in vitro methods. Gemfibrozil glucuronide contributes to the OATP1B inhibitory effect of gemfibrozil153. Norverapamil is another example; it has comparable concentrations to verapamil and is a more potent P-gp inhibitor than verapamil154. However, there are generally less data or predictive models with decision criteria available for metabolites compared with parent drugs. The general principles and strategies for the evaluation of parent drugs could be applied to metabolites, when applicable. The draft ICH M12 DDI guideline adopts the same recommendation for transporters as for CYPs. From a pragmatic perspective, it recommends the conduct of in vitro experiments to evaluate the transporter inhibitory potential of metabolites that have AUCmetabolite/AUCparent ≥ 25% and that are also major metabolites (i.e., account for at least 10% of drug-related material in the circulation based on radioactivity data from a mass balance study). This approach may stimulate the generation of more data to fill the knowledge gap, and the recommendation could be revisited when more data become available.
Transporter-mediated DDIs caused by induction
Compared with the induction of CYP enzymes, there are far fewer studies assessing the induction of transporters. P-gp is the most studied transporter and can be induced by activators of the nuclear receptor, pregnane X receptor (PXR), which also regulates the expression of CYP3A4. Because P-gp appears to be less inducible than CYP3A461,155, the draft ICH M12 DDI guideline provides recommendations on whether a clinical DDI study with P-gp substrates may be needed by considering multiple factors including the magnitude of CYP3A induction by an investigational drug144. On the contrary, there is less consensus on the induction of OATP1B61,62. One challenge with interpreting clinical transporter-mediated DDI data is that the substrates are often nonspecific and the potential involvement of other transporters or enzymes confounds the interpretation of the clinical data. Further research is warranted to investigate the clinical relevance and mechanism(s) of OATP1B1/3 induction. Emerging technologies and advances In the past decade, new technologies have been applied to advance basic, translational and clinical research in SLC and ABC transporters, as described in a recent ITC publication156. Here, two major areas that have not been extensively covered previously are focused on: new structures available through cryogenic electron microscopy (cryo-EM), especially ligand-bound structures, and modulation of transporter function via CRISPR–Cas9. Transporters mediate complex molecular events involved in substrate recognition, binding, translocation and release157. Furthermore, many transporters harness ion gradients to bind and translocate their substrates against a concentration gradient. Experimental structures of transporters, along with functional studies and molecular/computational simulation, have helped us to understand these complex mechanisms156 (Table 1). New and ligand-bound structures of SLC and ABC transporters In the past few years, enormous advances have been made in the determination of protein structures for transporters in both the SLC and ABC superfamilies156–159. Notably, structural information is now available for approximately 50% of the transporters in the human ABC superfamily, which has led to a new understanding of substrate binding and transport mechanisms of many members of this important superfamily. In contrast, although progress has been made, structural data are available only for ~15% of the human SLC superfamily members. Since the ITC review in 2022156, several new structures of SLC and ABC transporters have been published (Table 1). Those structures that are particularly relevant to drug development include SLC19A1160, the reduced folate carrier, which plays a role in methotrexate disposition; the organic anion transporting polypeptides, SLCO1B1 and SLCO1B3161,162; and the organic cation transporters, SLC22A1, SLC22A2 and SLC22A3163,164, which play critical roles in the disposition of a variety of drugs and endogenous molecules. Cryo-EM structures of transporters that are bound with ligands that are inhibitors, substrates or inducers allow us to identify the precise residues bound to the ligands and, therefore, to understand the structural determinants of transporter function (see examples in Table 1). Such information is critical in the design and development of drugs that target transporters. With more available structures, de novo prediction by AlphaFold2 and/or comparative modelling, rational drug design and ligand discovery for transporters have been enabled. Modulation of transporter function via CRISPR–Cas9 genome editing CRISPR–Cas9 and related techniques enable fine-tuned genome editing, without the need for time-consuming and costly protein engineering steps that were limitations of earlier techniques. These advances have paved the way for the development of more precise in vitro and in vivo tools for transporter research, as well as for genome-wide approaches to deconvolute transport mechanisms. Cell lines overexpressing heterologous transporters have been used extensively in transporter research. Delineating transport mechanisms is, however, often complicated by the background expression of endogenous transporters in the host cell line, and inhibitors selective for individual transporters are rare. CRISPR–Cas9 genome editing has been used successfully to address such limitations. For example, the expression and function of canine Mdr1/P-gp (Abcb1) was completely ablated in Madine–Darby canine kidney cells, which are commonly used to host human ABC efflux transporters, for example in the assessment of central nervous system exposure165. Canine knockout cells transfected with human P-gp (ABCB1) or BCRP (ABCG2) resulted in improved classification of efflux substrates, reconciling several earlier inter-assay discrepancies, and demonstrating species differences in substrate efflux166–168. Similarly, the selective knockout of P-gp, BCRP or MRP2 in the widely used model of intestinal drug absorption, Caco-2, enabled the deconvolution of efflux pathways in a more complex and in vivo-like system, in which multiple transporters contribute to drug permeability169. Knockout of MRP1 (ABCC1) in NCI-H441 cells via a targeted CRISPR–Cas9 approach identified 5(6)-carboxyfluorescein as a suitable probe to study MRP1 functional activity170. The CRISPR–Cas9 system has also been used in elegant studies to knock out transporters in a human-induced pluripotent stem cell line to validate findings from genome-wide association studies171,172. Mouse knockout models have been used widely for several decades to assess the effects of various transporters in vivo, although data must be interpreted cautiously owing to the potential for compensatory changes in other transport or metabolic pathways. Initially, Zn-finger technology and, more recently, CRISPR–Cas9 have been applied to establish similar models in other species commonly used in nonclinical pharmacokinetics and safety studies. For example, CRISPR–Cas9 knockout models of rat Mdr1a/b (Abcb1a/b), Oatp1b2 (Slco1b2) or the combination of Oat1/3 (Slc22a6/Slc22a8) have been reported, demonstrating an altered systemic exposure of the model substrates digoxin, pitavastatin and furosemide, respectively173–175. CRISPR–Cas9 is a valuable tool to generate knockout mice for preclinical drug evaluation. The relative ease of selective editing using CRISPR, in which multiple genomic regions can be targeted using identical setups by varying only the guide RNA, has greatly simplified genome-wide as well as focused screening for genes involved in specific phenotypes. For example, an SLC-focused CRISPR screening approach in haploid HAP1 cells was used to identify previously unknown transporter interactions for cytotoxic drugs, demonstrating functional dependencies on one or more SLC genes for close to 80% of the 60 compounds tested176. Similar approaches were used to identify OATP1A2 and OATP1B3 as mediators of doxorubicin cardiotoxicity177 and the association of ENT3 with remdesivir cytotoxicity178. An alternative approach, using CRISPR–Cas9 to introduce controlled transcriptional activation, identified SLC transporters that allow cells to survive under the depletion of essential nutrients179. Future directions Understanding the role of transporters beyond systemic pharmacokinetics To date, the clinical relevance of many transporters has been established based on the changes in systemic pharmacokinetics of drugs resulting from DDIs and functional pharmacogenetic variants (Fig. 1). As such, the main focus in drug development has been primarily on (1) hepatic and renal uptake transporters (i.e., OATPs, OATs, OCTs), (2) intestinal efflux transporters (i.e., P-gp, BCRP) and (3) renal excretion by MATEs180. Although efflux transporters (for example, P-gp, BCRP) in clearing organs have been discussed, practical recommendations for drug development have been difficult to articulate. Although the inhibition of these efflux mechanisms can lead to drug accumulation in the clearing organ and potentially toxicity, systemic pharmacokinetics may not be affected5,71,181. Modelling and simulation approaches are often used to predict the intracellular concentrations of P-gp or BCRP substrate/inhibitor drugs and establish relationships with their systemic or pharmacodynamic effects71,88,182 (see also “Modelling and simulation” section). Clinical studies may be designed to further verify the model for use in predicting untested scenarios or studies that are challenging to conduct clinically. A number of recent DDI studies have explored the use of a cocktail of clinical probe drugs (at therapeutic or microdose levels), or monitoring of transporter biomarkers together with clinical probes, to gain a mechanistic understanding of complex DDIs involving multiple transporters and enzymes4,43,119,123,152,183,184. An important future direction for understanding the effect of transporters in drug safety and efficacy is to expand the design of DDI studies to evaluate alterations in drug disposition beyond just systemic pharmacokinetics and changes in the exposure ratio end point. Depending on the specific transport pathway(s) involved, this approach may vary in complexity. Transporters in renal DDIs and toxicity Renal transporter-mediated DDIs can be identified by examining urinary excretion data in addition to systemic pharmacokinetic data. Quantitative collection of urine on a predetermined schedule in clinical studies necessitates a paradigm shift in drug development13,185–187. The acquisition of both systemic and urinary pharmacokinetic data enables the determination of whether apical excretion from the renal proximal tubule has been perturbed, a scenario that may not always be apparent based on systemic pharmacokinetic data. Furthermore, knowledge of changes in urinary drug recovery can help inform whether oral bioavailability may have been affected186. Potential increases in kidney drug exposure when renal excretion is impaired is difficult to directly assess without imaging data, or changes in biological response (for example, toxicity) or biomarker levels. Finally, the availability of both systemic and urinary pharmacokinetic data is critical either for the verification or for the refinement of PBPK model simulations of renal drug exposure.
Table 1.
Recently available structures of transporters in the SLC and ABC superfamilies
| Gene | Protein | Endogenous substrate, drugs | Human diseases | PDB entry | Method (resolution) | Ligand (substrate, inhibitor, activator) | Conformation | Findings | Fold | Ref |
|---|---|---|---|---|---|---|---|---|---|---|
| SLC1A2 | EAAT2 | Glutamate | Developmental and epileptic encephalopathy | 7XR4, 7XR6 | Cryo-EM (3.4 Å) | WAY-213613 (inhibitor) | inward-facing | WAY-213613 occupies similar binding pocket with glutamate (substrate). Binding of the inhibitor results in conformational change and stabilizes the inhibitor. | 8 alpha-helical transmembrane domain and 2 hairpins (HP) | 204 |
| SLC1A3 | EAAT1 | Glutamate | Episodic ataxia | 7AWQ, 7AWL, 7AWN, 7AWM | X-ray (3.7 Å) | UCPH101 (allosteric inhibitor) | outward-facing | UCPH101 traps EAAT1 in outward-facing states. | 8 alpha-helical transmembrane domain and 2 hairpins (HP) | 205 |
| SLC5A1 | Sodium glucose co-transporter 1 (SGLT1) | Glucose, galactose, methyl-D-glucopyranoside | Glucose/galactose malabsorption | 7WMV | Cryo-EM (3.2 Å) | LX2761 (Phase 1b trial, SGLT1 and SGLT2 inhibitorl) - for type 2 diabetes | outward-open | LX2761 blocks the putative water permeation pathway of hSGLT1 | LeuT | 206 |
| SLC5A2 | Sodium glucose co-transporter 2 (SGLT2) | Glucose | Renal glucosuria | 7VSI | Cryo-EM (2.95 Å) | empagliflozin - for type 2 diabetes and heart failture | outward-open | Empagliflozin occupies the substrate binding site and external vestibule to lock SGLT2 in outward-open conformation | LeuT | 207 |
| SLC6A1 | Sodium- and chloride-dependent GABA transporter 1 (GAT1) | γ-Aminobutyric acid (GABA) | Myoclonic atomic epilepsy | 7SK2 | Cryo-EM (3.82 Å) | tiagabine (drug inhibitor) - to treat neurological disorders, e.g. epilepsy | inward-open | Two-step mechanism of inhibition by tiagabine | LeuT | 208 |
| SLC6A4 | Sodium- and chloride-dependent serotonin transporter (SERT) | Serotonin, meta-iodobenzylguanidine | Anxiety related trait, Obsessive compulsive disorder | 6VRL, 6W2C | Cryo-EM (3.80 Å), X-Ray (6.3 Å) | paroxetine (drug inhibitor) - antidepressant | outward-open | Paroxetine bind to the substrate-binding site of SERT and stabilize the outward-open conformation | LeuT | 209 |
| 6DZV, 6DZW, 6DZY, 6DZZ | Cryo-EM (3.6–4.2 Å) | ibogaine (inhibitor) - hallucinogenic natural product, psychoactive | outward-open, occluded and inward-open | Ibogaine, a non-competitive inhibitor moves from outward to inward by interacting with transmembrane 1b, 6a and 5. | LeuT | 210 | ||||
| SLC6A9 | Glycine transporter 1 (GLYT1) | Glycine | Glycine encephalopathy with normal serum glycine | 6ZPL | X-Ray (3.94 Å) | Benzoylpiperazine chemotype inhibitor. A drug called Bitopertin, is SLC6A9 inhibitor currently in clinical development for schizophrenia and cognitive impairments | inward-open | Inhibitor locks GLYT1 in an inward-open conformation and binds at the intracellular gate. | LeuT | 211 |
| SLC7A5 | L-type amino acid transporter 1 (LAT1) | Phenylalanine, 3,5-diiodo-L-tyrosine, leucine | Inhibition of SLC7A5 reduce tumor growth | 6IRS, 6IRT, 7DSK, 7DSN, 7DSL | Cryo-EM (3.3–3.5 Å; 2.7–2.8 Å) | BCH, JPH203, JX-075, JX-119, JX-078 (LAT1 inhibitor). JPH203 in Phase II clinical trial for advanced biliary tract cancers | inward open, outward-facing | LAT1 forms a heteromeric amino acid transporter complex with 4F2 cell-surface antigen heavy chain (SLC3A2). LAT1 interacts extensively with 4F2hc on the extracellular side and 4F2hc is essential for LAT1 transport activity. BCH bound to Phe252 and in the center of transport path. | LeuT | 212,213 |
| SLC12A2 | Cation-chloride cotransporters (CCCs) NKCC1 | Symport of Na+, K+, Cl− | Deafness, Kilquist syndrome, Delpire-McNeill Syndrome | 7S1X, 7S1Y, 7S1Z | Cryo-EM (2.9–3.3 Å) | bumetanide (inhibitor) - diuretic for hypertension | inward-facing | Structural and experimental studies suggest that bumetanide wedged into a pocket in the extracellular ion translocation pathway | LeuT | 214 |
| SLC19A1 | Reduced folate carrier (RFC) | Folic acid, thiamine pyrophosphate, cGMP, cyclic di-AMP | Megaloblastic anemia (folate responsive) | 7TX7 | Cryo-EM (3.8 Å) | methotrexate (drug substrate) - anticancer, anti-inflammatory agent | inward-facing | Structure, molecular dynamic simulation and mutagenesis studies reveal a spacious, polarized cavity to allow folates and methotrexate to bind. | MFS, rocker switch | 215 |
| 8GOF, 8GOE | Cryo-EM (3.0 Å) | 5-MTHF, pemetrexed (drug substrate) - anticancer, anti-inflammatory agent | inward-open | The cavity within SLC19A1 is deep, cyclic dinucleotides are located at positively charged section approximately in the middle of the transmembrane region. | 160 | |||||
| 7XQ1, 7XQ2, 78ET7XQ0 | Cryo-EM (3.0–3.4 Å) | various cyclic dinucleotides (endogenous substrates) | inward-open | 160 | ||||||
| SLC22A1 | Organic cation transporter 1 (OCT1) | Thiamine, serotonin | 8ET7 8ET8 |
Cryo-EM (3.5 – 3.8 Å) | Diphenhydramine, verapamil | Outward facing, outward occluded | Structure-based in silico drug docking, coupled with molecular dynamics simulation, provides valuable insights into the key aspects of ligand recognition and transport mechanisms of OCT1 and OCT2. | MFS, alternating access mechanism | 163 | |
| SLC22A2 | Organic cation transporter 2 (OCT2) | Thiamine, serotonin, creatinine | 8ET9 | Cryo-EM (3.61 Å) | MPP+ | Outward facing, outward occluded | MFS, alternating access mechanism | 163 | ||
| SLC22A3 | Organic cation transporter 3 (OCT3) | Norepinephrine, histamine | 7ZH0 7ZH6 7ZHA |
Cryo-EM (3.2–3.7 Å) | Corticosterone, decynium-22 | Outward facing | By binding to two inhibitors, corticosterone and decynium-22, OCT3 structure elucidates the ligand binding pocket and reveals shared characteristics of inhibitors for major facilitator transporters. | MFS | 164 | |
| SLC29A1 | Equilibrative nucleoside transporter 1 (ENT1) | adenosine, various anticancer drugs (gemcitabine, cytarabine) | Human absence of ENT1 show rare Augustine-null blood type (macrocytosis, defective erythropoiesis) (33690842) | 6OB6, 6OB7 | X-Ray (2.3–2.9 Å) | NBMPR, dilazep - known SLC29A1 inhibitor | outward-facing | NBMPR and dilazep share the orthosteric site within the central cavity of ENT1 but explore distinct sites for inhibition. | pseudo-symmetric 6 + 5 topology | 216 |
| SLCO1B1 | Organic Anion Transporting Polypeptide (OATP1B1) | Endogenous metabolites (bilirubin, thyroid, coproporphyrin), anti-cancer, statin) | Hyperbilirubinemia, Rotor type, digenic | 8HNB, 8HNC, 8HND, 8HNH, 8PHW | Cryo-EM (2.92–3.73 Å) | Bilirubin, esterone-3-sulfate, 2’,7’-dichlorofluorescein (DCF), simeprevir | Inward- and outward-facing | The architectures display primary and secondary substrate attachment sites and undergo structural alterations during transit. | MFS | 161,162 |
| SLCO1B3 | Organic Anion Transporting Polypeptide (OATP1B3) | Bilirubin, anti-hypertensive drug, statin | Hyperbilirubinemia, Rotor type, digenic | 8PG0 | Cryo-EM (3.0 Å) | None | inward facing | The configuration of OATP1B3 uncovers a bicarbonate entity linked with the characteristic signature pattern and a histidine element commonly found in OATPs that demonstrate activity dependent on pH. | MFS | 162 |
| ABCB1 | Multi-drug resistance 1 (MDR1); P-glycoprotein (P-gp) | digoxin, etoposide, paclitaxel, vinblastine | Clinical resistance to chemotherapy; drug response | 6C0V, 7O9W, 7A6F, 7A69, 7A6C, 7A6E, 6QEX | Cryo-EM (3.2–3.6 Å) | Encequidar, MRK16, zosuquidar, vincristine, tariquidar, elacridar, taxol | outward facing, occluded | Structures of inhibitor and substrate bound to ABCB1 explain how inhibitors can act as substrates at low concentration, but interfere with the early steps of the peristaltic extrusion mechanism at higher concentration. | Type IV ABC transporter | 217–219 |
| ABCC7 | Cystic fibrosis transmembrane conductance regulator (CFTR) | Mg-ATP-gated chloride channel | Cystic fibrosis | 6O2P, 8EIQ | Cryo-EM (3.0, 3.3 Å) | Ivacaftor (potentiator); tezacaftor (VX-661) (Type I corrector), and the dual-function modulator elexacaftor (VX-445) (Type III corrector) | inward-facing, occluded | Ivacaftor stabilizes the open conformation of the CFTR and hence increases CFTR activity. In the case with CFTR delta508, elexacaftor partially rectified interdomain assembly defects in delta508 CFTR and required tezacaftor together to make CFTR fully function. | Type IV ABC transporter | 220,221 |
| ABCC8 | Sulfonylurea receptor 1 (SUR) | None. SUR1 protein is a subunit of the ATP-sensitive potassium channel. | Diabetes mellitus, noninsulin-dependent, Hyperinsulinemic hypoglycemia, familial, 1, Diabetes mellitus, permanent or transient neonatal | Human: 7S5V, 7S60, 7S61, 7S5Z | Other organism: 6JB3, 5YW7, 7WIT | Cryo-EM (3.1–4.4 Å) | repaglinide, glibenclamide, mitiglinide | inward-facing conformation | Structure of SUR1-ATP-sensitive potassium channel bound to sulfonylurea showed that (i) repaglinide binding to the SUR1 subunit to inhibit SUR1 nucleotide binding domain closure (iii) SUR1 structure showed mitiglinide locks SUR1 in the nucleotide binding domain (NBD)-separated inward-facing conformation. | Type IV ABC transporter | 222–225 |
| ABCG2 | Breast Cancer Resistance Protein (BCRP) | uric acid, steroid conjugates, anti-cancer drugs (e.g., mitoxantrone, SN-38) | Clinical resistance to chemotherapy; increase risk of gout; drug response | 6VXF, 6VXJ, 6VXI, 6VXH, 6ETI, 7NFD, 7NEZ, 7NEQ | Cryo-EM (3.5–4.1 Å) | Drug substrates: SN38, mitoxantrone, imatinib, topotecan, tariquidar; Inhibitor: Ko-143 | inward-facing | Structure of anticancer drugs bound to ABCG2 provided additional insight into (i) how ABCG2 differentiates between inhibitors and substrates; (ii) reveal binding mode how drug molecules open and closed conformation of the transporter; (iii) how drug molecule stabilizing the inward-facing conformation of ABCG2. | Type V ABC transporter | 226–228 |
Transporters in hepatic DDIs and toxicity
Clinical studies to assess the effects of transporter-mediated DDIs on hepatic excretion are even more challenging because bile is not as easily accessible as urine. Although studies including human bile collection have been conducted, these approaches have been rarely applied in drug development to date because, logistically and practically, these are not easy studies to execute188. However, imaging approaches have been used to show that when biliary excretion is impaired, a considerable increase in hepatic drug exposure may be observed36,189,190. Clinical imaging of drug tissue distribution, when feasible, could be incorporated into clinical DDI studies when transporter-mediated perturbation of biliary excretion is expected based on in vitro data and/or liver toxicity has been observed in the clinic. The liver is the target organ for statins, so altered hepatic exposure would be expected to result in changes in systemic cholesterol; unfortunately, the time required to observe alterations in statin pharmacodynamics (>2 weeks) is impractical for DDI studies in drug development9. However, for drugs for which the liver is the site of action and pharmacodynamic or toxicodynamic biomarker response is rapid, these alternative approaches should be considered when hepatic drug exposure is expected to be perturbed, as highlighted in the case of metformin13.
Transporters in nutrient deficiencies
In addition to transport pathways involved in DDIs, the ITC has also discussed the inhibition of nutrient/endobiotic transport as a putative mechanism of drug toxicity. The recommendations have been limited to a retrospective understanding of clinical safety observations rather than prospective testing of these transporters in drug development3,180. The challenge in understanding drug perturbations in nutrient/endobiotic homeostasis is the complexity of the multitude of transport and metabolic mechanisms involved, as well as physiological factors (e.g., diet, disease). This challenge has been highlighted for the interpretation of BSEP data as a hepatotoxicity alert, which requires a consideration of other pathways130. A culture shift has occurred in drug development, in which mechanism(s) of drug toxicities are increasingly scrutinized instead of reporting high-level findings with no understanding of the underlying mechanism(s). For example, THTR2 inhibition was elucidated as a contributing factor for fedratinib-induced encephalopathy141, and modulation of folate transport pathways (PCFT, RFC, FRα) was investigated for the entire HIV integrase inhibitor drug class191. Studies elucidating mechanism(s) underlying drug toxicity may ultimately provide evidence for prospective screening of transporters as toxicity alerts, as discussed above for BSEP.
Transporters as drug targets
Transporters are increasingly being studied as drug targets to treat diseases156,192. For example, SGLT2 inhibitors (for example, canagliflozin, dapagliflozin, empagliflozin) have been approved as a new class of antihyperglycemic compounds for the treatment of type 2 diabetes mellitus193–195. URAT1 inhibitors are being developed for the treatment of hyperuricemia and gout196,197. A grey box illustrating a first-in-class drug targeting ASBT (SLC10A2) and approved for the treatment of cholestatic diseases is shown in Box 1. Future transporter research in drug development will advance understanding of transporters from their effect on pharmacokinetics to targets for drug toxicities (including the use of biomarkers), identify transporters that are targets to treat common and rare diseases and elucidate the role of transporters as important determinants of total body homeostasis. Biomarkers and in vivo measures of transporter activity Monitoring of a cocktail of validated Tier 1 and 2 transporter biomarkers, in combination with PBPK modelling, is envisaged to refine the design and guide decision making on prioritization/need for dedicated clinical transporter-mediated DDI studies (Figs. 4–5). Despite significant progress in identifying multiple potential biomarkers for hepatic and renal transporters (Fig. 4), there are no biomarkers for the intestinal efflux transporter P-gp, and limited data are emerging for BCRP198. In addition to de-risking transporter-mediated DDIs, endogenous biomarkers have great potential as tools to evaluate the modulation of transporter function in diseased and specific populations. Data reported so far focus on CPI and the use of this biomarker to study changes in OATP1B activity in mild-to-severe renal and hepatic impairment, hyperlipidemic children, rheumatoid arthritis and some cancer populations23,32. Knowledge gaps remain in reference values for CPI in other patient populations and the availability of data for biomarkers of other transporters (for example, renal). Important consideration in data interpretation is that disease may also alter biomarker synthesis rates, in addition to the potential modulation of transporter activity. Recent modelling of CPI data in severe renal impairment illustrated this complex interplay between disease-related decreases in CPI synthesis and active uptake via OATP1B1, in addition to renal elimination and protein binding106. Emerging data suggest that tissue-derived plasma sEVs may serve as a ‘liquid biopsy’, a noninvasive detection technique for drug-metabolizing enzymes (DMEs) and transporter profiles in absorption-, distribution-, metabolism-, and/or excretion–related organs199,200. However, currently only liver specific sEVs have been identified because of the unique expression of the asialoglycoprotein receptor (ASGPR), whereas similar markers remain to be identified/validated for other organs. This approach could be especially advantageous in diseased and specific populations in which access to tissue samples is not feasible and/or limited. For instance, when plasma exosomes were isolated from the blood of 29 patients with liver cancer, a good correlation was observed between normalized plasma exosome mRNA expression and protein levels in matched liver tissues for OATP1B1, MRP2, P-gp, BCRP and 12 DMEs201. Quantification of protein levels of OATP1B and CYP3A in liver-specific sEVs confirmed the induction of CYP3A4 but not of hepatic OATP1B1 and OATP1B3 following multiple doses of rifampin, a well-known inducer of CYP3A and P-gp202. sEVs hold great promise as a valuable next generation tool to provide unique and rich information on changes in DME and transporter levels in different diseases and physiological states199,203. Although sEVs have unique advantages such as minimal invasiveness, routine accessibility of clinical samples and the quantitative nature of the measurements, sEVs only provide information on protein levels, with limited data on correlations to transporter function203. The comparison of liquid biopsy (transporter levels) and biomarker data (transporter activity) in the plasma samples obtained from the same individuals would help establish whether and to what extent relationships between transporter abundance and function change because of genetic polymorphisms or in disease.
Box 1 |. First-in-class SLC10A2 inhibitor, for the treatment of disorders associated with abnormal bile acid concentrations.
Benzothiazepines have been known to lower LDL cholesterol (US Patent 5998400) since the 1990s, although the mechanism was unclear (see panel a of figure, various doses are illustrated by the different colours). Later it was discovered that benzothiazepines are potent inhibitors of the bile acid transporter, ASBT, encoded by SLC10A2, and that the transporter represents the target for the hypolipidemic effects of the benzothiazepines238. Although rare variants in ASBT lead to primary bile acid malabsorption, which is associated with congenital diarrhoea and steatorrhea239, common variants are associated with lower levels of LDL and increased risk of gallstones240,241, and other sequelae associated with reduced concentrations of bile acids and their conjugates242. As a critical transporter in bile acid–cholesterol balance and enterohepatic circulation of bile acids, ASBT is a promising target for treatment of liver, intestinal and associated metabolic diseases. High-throughput assays using baby hamster kidney cells transfected with human ASBT cDNA (H14 cells) were used to screen a novel series of benzothiazepines, which resulted in the development of the drug candidate, LUM001, also known as SHP625 and maralixibat243–245. Maralixibat was designed to have minimal intestinal absorption246 and was recently approved for cholestatic pruritus in patients with Alagille syndrome (see panel b of figure)247. Clinical development of maralixibat is ongoing for the treatment of cholestatic liver diseases such as Progressive Familial Intrahepatic Cholestasis (PFIC) and biliary atresia (NCT02057718, NCT03905330, NCT04185363, NCT04168385, NCT04729751, NCT04524390)248 (see panel c of figure, the different colours illustrate different indications). The first published clinical trial of maralixibat in 37 children with severe cholestasis-induced pruritis in Alagille syndrome (NCT02057692)249 revealed that it is a safe drug and may potentially reduce pruritus in Alagille syndrome, although no statistically significant effects in the primary analyses of itch-reported outcome were observed. However, a combination of two randomized placebo-controlled trials showed that maralixibat was associated with marked improvement in pruritis and quality of life in 57 children with severe cholestasis secondary to Alagille syndrome250. Another ASBT inhibitor, odevixibat, has been developed to treat children with PFIC251 (US9694018B1). Similar to maralixibat, odevixibat is minimally absorbed after oral administration. Odevixibat was approved in July 2021 in Europe and soon after in the United States by the FDA. Elobixibat, which is a long-acting ASBT inhibitor, is approved in Japan and Thailand for chronic constipation252. Other lead molecules are in development, including GSK2330672 (Linerixibat)253 for the treatment of pruritus in participants with primary biliary cholangitis254.
Conclusions
Major progress has been made over the past two decades in understanding the role of membrane transporters in drug safety and efficacy and in translating this information into practical guidance for drug developers and clinicians. Scientists working together in the academic, regulatory and pharmaceutical industry sectors, with ongoing dialogue facilitated by the ITC, have accelerated discovery and application in the field of transporter science. However, as highlighted in this manuscript, many key questions remain to be answered before we can fully use this knowledge about transporters to optimize drug therapy in individual patients. Rapid technological advances such as single-cell RNA sequencing, CRISPR–Cas9 genome editing, cryo-EM, advanced omics capabilities, exosome-based liquid biopsy, artificial intelligence, machine learning, real-world data, enhanced modelling and simulation tools, and advances in clinical trial design will continue to drive breakthroughs in transporter science and fill existing knowledge gaps. Improved, physiologically relevant in vitro models, state-of-the-art tools to investigate the effect of intrinsic and extrinsic factors on transporter function, identification of novel endogenous biomarkers that may reduce or eliminate the need for clinical DDI studies, and advances in PBPK and quantitative systems pharmacology/toxicology modelling and simulation to more accurately predict transporter-mediated DDIs and pharmacokinetics in different patient populations will aid in unraveling the complexities that currently challenge the interpretation of transporter data. Transport proteins have clearly taken a place alongside DMEs in their relevance to our understanding of drug disposition. In some cases, transport proteins are a key consideration in selecting the appropriate medication for patients and in optimizing drug dosage regimens to enhance efficacy and minimize toxicity. Indeed, this continues to be an exciting era in transporter research, which is advancing at a rapid pace to help achieve the promise of precision medicine.
Supplementary Material
Acknowledgements
The authors thank members of the International Transporter Consortium (ITC) for review of the article prior to submission, in particular Drs. Vikram Arya, Caroline Lee, Micheline Piquette-Miller and Mitchell Taub.
Funding:
A.G. is a deputy director of the Centre for Applied Pharmacokinetic Research, which is a consortium of companies including Genentech, Roche, Merck Serono, Takeda, Janssen, GSK, Servier, Amgen, Eli Lilly and Abbvie. She receives funding from A*STAR (BM/NDR/19/003), Research Council of Norway (15451500) and Asahi Kasei Pharma Corporation for her PhD students and trainees, and she is an unpaid President of the International Society for the Study of Xenobiotics. D.T. owns stock and/or stock options in Merck & Co., Inc. and receives free membership in the International Society for the Study of Xenobiotics. H.S. is an employee of Bristol Myers Squibb and owns stock and/or stock options in Bristol Myers Squibb. K.L.R.B. is co-inventor of the sandwich-cultured hepatocyte technology for quantification of biliary excretion (B-CLEAR) and related technologies, which have been licensed exclusively to Qualyst Transporter Solutions, acquired by BioIVT. She receives funding from a Certara Simcyp Grant and Partnership Scheme to support a PhD student in her laboratory and serves as an unpaid member of the scientific advisory committee for the International Society for the Study of Xenobiotics. K.Y. is an employee of Genentech and owns stock and/or stock options in Genentech and Roche. L.A. serves in an unpaid position for the Society of Toxicology. M.J.H. and X.C. are employees of Merck & Co., Inc. and own stock and/or stock options in Merck & Co., Inc. M.J.Z.-G. is an employee of GlaxoSmithKline and owns stock and/or stock options in GlaxoSmithKline. He also serves in an unpaid position for the American Society for Clinical Pharmacology and Therapeutics. M.V.S.V is an employee of Pfizer and owns stock and/or stock options in Pfizer. N.S. receives funding for graduate student support from the Sigrid Jusélius Foundation. R.E. is an employee of Johnson & Johnson and owns stock and/or stock options in Johnson & Johnson. Y.L. is an employee of Gilead Sciences, Inc. and owns stock and/or stock options in Gilead Sciences, Inc. K.M.G. and S.W.Y. are co-founders of Apricity Therapeutics, Inc., a transporter-based biotechnology company. A.R., J.Y., L.Z., P.M. and X.Y. declare no competing interests. Funding A.R. is supported by the National Institute of General Medical Sciences of the National Institutes of Health (NIH) under award number 5T32GM007546. A.S. is supported, in part, by the NIH under award numbers R01CA238946, R01GM139936 and U24CA247648 and by the Ohio State University Comprehensive Cancer Center Pelotonia Foundation. K.L.R.B. is supported, in part, by the National Institute of General Medical Sciences of the NIH under award number R35GM122576. K.M.G. and S.W.Y. are supported, in part, by the NIH under award numbers R01GM139875, R01GM117163 and UC2HD113474. K.M.G. is also supported, in part by the grant number U01FD004979/U01FD005978 from the FDA, which supports the University of California–San Francisco (UCSF)-Stanford Center of Excellence in Regulatory Sciences and Innovation. L.A is supported, in part, by the NIH under award numbers UC2HD113039, P30ES005022, R01GM123330 and R01ES029275. N.S. is supported, in part, by the Sigrid Jusélius Foundation.
Footnotes
Disclaimer:
The views in this paper are those of the authors and should not be construed to represent FDA’s views or policies.
References
- 1.Giacomini KM et al. Membrane transporters in drug development. Nat. Rev. Drug. Discov 9, 215–236 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; This is a seminal publication from the newly formed ITC that coalesced extensive discussions and output from the first ITC workshop, identified a subset of clinically relevant transporters and outlined decision trees that could be applied in translating in vitro experimental outcomes to likelihood of clinical findings resulting from drug– drug interactions; it formed the basis for regulatory recommendations for drug transporters in guidance documents.
- 2.Zamek-Gliszczynski MJ et al. ITC recommendations for transporter kinetic parameter estimation and translational modeling of transport-mediated PK and DDIs in humans. Clin. Pharmacol. Ther 94, 64–79 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zamek-Gliszczynski MJ et al. Transporters in drug development: 2018 ITC recommendations for transporters of emerging clinical importance. Clin. Pharmacol. Ther 104, 890–899 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chu X et al. Clinical probes and endogenous biomarkers as substrates for transporter drug-drug interaction evaluation: perspectives from the International Transporter Consortium. Clin. Pharmacol. Ther 104, 836–864 (2018). [DOI] [PubMed] [Google Scholar]; This manuscript summarizes challenges and limitations of clinical probe drugs and emerging biomarkers for drug transporters to support clinical development of safe and effective therapeutics; workflows for identification and validation of novel endogenous biomarkers are proposed.
- 5.Chu X et al. Intracellular drug concentrations and transporters: measurement, modeling, and implications for the liver. Clin. Pharmacol. Ther 94, 126–141 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hillgren KM et al. Emerging transporters of clinical importance: an update from the International Transporter Consortium. Clin. Pharmacol. Ther 94, 52–63 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Brouwer KLR et al. Regulation of drug transport proteins-from mechanisms to clinical impact: a white paper on behalf of the International Transporter Consortium. Clin. Pharmacol. Ther 112, 461–484 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper reviews mechanisms of transport protein regulation, which are highly complex and can occur at multiple levels to affect transporter function, and highlights why knowledge about these processes is critical in the development of tools and approaches to improve therapeutic outcomes and accurately predict drug disposition and response.
- 8.Cooper-DeHoff RM et al. The clinical pharmacogenetics implementation consortium guideline for SLCO1B1, ABCG2, and CYP2C9 genotypes and statinassociated musculoskeletal symptoms. Clin. Pharmacol. Ther 111, 1007–1021 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee CA et al. Breast cancer resistance protein (ABCG2) in clinical pharmacokinetics and drug interactions: practical recommendations for clinical victim and perpetrator drug-drug interaction study design. Drug. Metab. Dispos 43, 490–509 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Matthaei J et al. OCT1 mediates hepatic uptake of sumatriptan and loss-of-function OCT1 polymorphisms affect sumatriptan pharmacokinetics. Clin. Pharmacol. Ther 99, 633–641 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Zazuli Z et al. The impact of genetic polymorphisms in organic cation transporters on renal drug disposition. Int. J. Mol. Sci 21, 6627 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yee SW et al. Influence of transporter polymorphisms on drug disposition and response: a perspective from the International Transporter Consortium. Clin. Pharmacol. Ther 104, 803–817 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This review describes actionable polymorphisms in SLC and ABC transporters including SLCO1B1, ABCG2 and SLC22A1, which have a high level of evidence for genotype driven drug or dose selection, and discusses mechanisms responsible for their effects on drug toxicity and response.
- 13.Zamek-Gliszczynski MJ et al. ITC commentary on metformin clinical drug-drug interaction study design that enables an efficacy- and safety-based dose adjustment decision. Clin. Pharmacol. Ther 104, 781–784 (2018). [DOI] [PubMed] [Google Scholar]; This commentary illustrates the complexity of transporter DDIs, which may often result in different effects on the plasma versus tissue exposure of a drug, and proposes a novel clinical DDI study design for metformin to facilitate rational dose adjustment when this drug is co-administered with OCT/MATE inhibitors.
- 14.Hirota T, Tanaka T, Takesue H & Ieiri I Epigenetic regulation of drug transporter expression in human tissues. Expert. Opin. Drug. Metab. Toxicol 13, 19–30 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Birmingham BK et al. Impact of ABCG2 and SLCO1B1 polymorphisms on pharmacokinetics of rosuvastatin, atorvastatin and simvastatin acid in Caucasian and Asian subjects: a class effect. Eur. J. Clin. Pharmacol 71, 341–355 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Tsamandouras N et al. Identification of the effect of multiple polymorphisms on the pharmacokinetics of simvastatin and simvastatin acid using a population-modeling approach. Clin. Pharmacol. Ther 96, 90–100 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Oi Yan Chan J, Moullet M, Williamson B, Arends RH & Pilla Reddy V Harnessing clinical trial and real-world data towards an understanding of sex effects on drug pharmacokinetics, pharmacodynamics and efficacy. Front. Pharmacol 13, 874606 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burt HJ et al. Abundance of hepatic transporters in caucasians: a meta-analysis. Drug. Metab. Dispos 44, 1550–1561 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mai Y et al. Boosting drug bioavailability in men but not women through the action of an excipient. Int. J. Pharm 587, 119678 (2020). [DOI] [PubMed] [Google Scholar]
- 20.Mai Y et al. Quantification of P-glycoprotein in the gastrointestinal tract of humans and rodents: methodology, gut region, sex, and species matter. Mol. Pharm 18, 1895–1904 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Niemi M, Pasanen MK & Neuvonen PJ SLCO1B1 polymorphism and sex affect the pharmacokinetics of pravastatin but not fluvastatin. Clin. Pharmacol. Ther 80, 356–366 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Tasnif Y, Morado J & Hebert MF Pregnancy-related pharmacokinetic changes. Clin. Pharmacol. Ther 100, 53–62, (2016). [DOI] [PubMed] [Google Scholar]
- 23.Chu X et al. Clinical implications of altered drug transporter abundance/function and PBPK modeling in specific populations: an ITC perspective. Clin. Pharmacol. Ther 112, 501–526 (2022). [DOI] [PubMed] [Google Scholar]; This manuscript provides a critical analysis of the clinical implications of altered transporter abundance and/or activity in specific populations using a range of tools, such as quantitative proteomics, pharmacokinetic data for clinical probes/transporter biomarkers, PBPK modelling and integration of these approaches.
- 24.Li CY et al. Optimized renal transporter quantification by using aquaporin 1 and aquaporin 2 as anatomical markers: application in characterizing the ontogeny of renal transporters and its correlation with hepatic transporters in paired human samples. AAPS J 21, 88 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Groen BD et al. Ontogeny of hepatic transporters and drug-metabolizing enzymes in humans and in nonclinical species. Pharmacol. Rev 73, 597–678 (2021). [DOI] [PubMed] [Google Scholar]
- 26.Kiss M et al. Ontogeny of small intestinal drug transporters and metabolizing enzymes based on targeted quantitative proteomics. Drug. Metab. Dispos 49, 1038–1046 (2021). [DOI] [PubMed] [Google Scholar]
- 27.Emoto C et al. PBPK model of morphine incorporating developmental changes in hepatic OCT1 and UGT2B7 proteins to explain the variability in clearances in neonates and small infants. CPT Pharmacomet. Syst. Pharmacol 7, 464–473 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zgheib NK & Branch RA Drug metabolism and liver disease: a drug-geneenvironment interaction. Drug. Metab. Rev 49, 35–55 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Evers R et al. Disease-associated changes in drug transporters may impact the pharmacokinetics and/or toxicity of drugs: a white paper from the International Transporter Consortium. Clin. Pharmacol. Ther 104, 900–915 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper reviews changes in transporter function associated with acute and chronic disease states, describes regulatory pathways affecting transporter expression, discusses potential implications from a drug discovery and development perspective, and identifies opportunities to advance the field.
- 30.Kula M et al. Hepatobiliary function assessed by 99mTc-mebrofenin cholescintigraphy in the evaluation of fibrosis in chronic hepatitis: histopathological correlation. Nucl. Med. Commun 31, 280–285 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Drozdzik M et al. Protein abundance of drug transporters in human hepatitis C livers. Int. J. Mol. Sci 23, 7947 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin J et al. Effect of hepatic impairment on OATP1B activity: quantitative pharmacokinetic analysis of endogenous biomarker and substrate drugs. Clin. Pharmacol. Ther 113, 1058–1069 (2022). [DOI] [PubMed] [Google Scholar]
- 33.Hatorp V, Walther KH, Christensen MS & Haug-Pihale G Single-dose pharmacokinetics of repaglinide in subjects with chronic liver disease. J. Clin. Pharmacol 40, 142–152 (2000). [DOI] [PubMed] [Google Scholar]
- 34.El-Khateeb E, Achour B, Al-Majdoub ZM, Barber J & Rostami-Hodjegan A Non-uniformity of changes in drug-metabolizing enzymes and transporters in liver cirrhosis: implications for drug dosage adjustment. Mol. Pharm 18, 3563–3577 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hui CK, Cheung BM & Lau GK Pharmacokinetics of pitavastatin in subjects with Child-Pugh A and B cirrhosis. Br. J. Clin. Pharmacol 59, 291–297 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ali I et al. Transporter-mediated alterations in patients with NASH increase systemic and hepatic exposure to an OATP and MRP2 substrate. Clin. Pharmacol. Ther 104, 749–756 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clarke JD, Novak P, Lake AD, Hardwick RN & Cherrington NJ Impaired N-linked glycosylation of uptake and efflux transporters in human non-alcoholic fatty liver disease. Liver Int 37, 1074–1081 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferslew BC et al. Altered morphine glucuronide and bile acid disposition in patients with nonalcoholic steatohepatitis. Clin. Pharmacol. Ther 97, 419–427 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Canet MJ et al. Altered regulation of hepatic efflux transporters disrupts acetaminophen disposition in pediatric nonalcoholic steatohepatitis. Drug. Metab. Dispos 43, 829–835 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morgan ET et al. Physiological regulation of drug metabolism and transport: pregnancy, microbiome, inflammation, infection, and fasting. Drug. Metab. Dispos 46, 503–513 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Corrigan G et al. PAH extraction and estimation of plasma flow in human postischemic acute renal failure. Am. J. Physiol 277, F312–F318 (1999). [DOI] [PubMed] [Google Scholar]
- 42.Kunin M, Holtzman EJ, Melnikov S & Dinour D Urinary organic anion transporter protein profiles in AKI. Nephrol. Dial. Transpl 27, 1387–1395 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Tatosian DA et al. A Microdose cocktail to evaluate drug interactions in patients with renal impairment. Clin. Pharmacol. Ther 109, 403–415 (2021). [DOI] [PubMed] [Google Scholar]
- 44.Miners JO, Yang X, Knights KM & Zhang L The role of the kidney in drug elimination: transport, metabolism, and the impact of kidney disease on drug clearance. Clin. Pharmacol. Ther 102, 436–449 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Hsueh CH et al. Identification and quantitative assessment of uremic solutes as inhibitors of renal organic anion transporters, OAT1 and OAT3. Mol. Pharm 13, 3130–3140 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Ramezani A et al. Role of the gut microbiome in uremia: a potential therapeutic target. Am. J. Kidney Dis 67, 483–498 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jarvinen E et al. The role of uptake and efflux transporters in the disposition of glucuronide and sulfate conjugates. Front. Pharmacol 12, 802539 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Foley SE et al. Gut microbiota regulation of P-glycoprotein in the intestinal epithelium in maintenance of homeostasis. Microbiome 9, 183 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen M, Zhou SY, Fabriaga E, Zhang PH & Zhou Q Food-drug interactions precipitated by fruit juices other than grapefruit juice: an update review. J. Food Drug. Anal 26, S61–S71 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dresser GK et al. Fruit juices inhibit organic anion transporting polypeptide-mediated drug uptake to decrease the oral availability of fexofenadine. Clin. Pharmacol. Ther 71, 11–20 (2002). [DOI] [PubMed] [Google Scholar]
- 51.Keiser M et al. The organic anion-transporting peptide 2B1 is localized in the basolateral membrane of the human jejunum and Caco-2 monolayers. J. Pharm. Sci 106, 2657–2663 (2017). [DOI] [PubMed] [Google Scholar]
- 52.Li W et al. Organic anion-transporting polypeptide 2B1 knockout and humanized mice; insights into the handling of bilirubin and drugs. Pharmacol. Res 190, 106724 (2023). [DOI] [PubMed] [Google Scholar]
- 53.McFeely SJ, Wu L, Ritchie TK & Unadkat J Organic anion transporting polypeptide 2B1 - More than a glass-full of drug interactions. Pharmacol. Ther 196, 204–215 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Drozdzik M et al. Protein abundance of clinically relevant multidrug transporters along the entire length of the human intestine. Mol. Pharm 11, 3547–3555 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Couto N et al. Quantitative proteomics of clinically relevant drug-metabolizing enzymes and drug transporters and their intercorrelations in the human small intestine. Drug. Metab. Dispos 48, 245–254 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morita T et al. Inhibitory effects of cranberry juice and its components on intestinal OATP1A2 and OATP2B1: identification of avicularin as a novel inhibitor. J. Agric. Food Chem 70, 3310–3320 (2022). [DOI] [PubMed] [Google Scholar]
- 57.Misaka S et al. Exposure of fexofenadine, but not pseudoephedrine, is markedly decreased by green tea extract in healthy volunteers. Clin. Pharmacol. Ther 112, 627–634 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu LX et al. Inhibition of the organic anion-transporting polypeptide 1B1 by quercetin: an in vitro and in vivo assessment. Br. J. Clin. Pharmacol 73, 750–757 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schwarz UI et al. Induction of intestinal P-glycoprotein by St John’s wort reduces the oral bioavailability of talinolol. Clin. Pharmacol. Ther 81, 669–678 (2007). [DOI] [PubMed] [Google Scholar]
- 60.Nguyen JT et al. Assessing transporter-mediated natural product-drug interactions via in vitro-in vivo extrapolation: clinical evaluation with a probe cocktail. Clin. Pharmacol. Ther 109, 1342–1352 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zamek-Gliszczynski MJ et al. Intestinal P-gp and putative hepatic OATP1B induction: international transporter consortium perspective on drug development implications. Clin. Pharmacol. Ther 109, 55–64 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rodrigues AD et al. Induction of human intestinal and hepatic organic anion transporting polypeptides: where is the evidence for its relevance in drug-drug interactions? Drug. Metab. Dispos 48, 205–216 (2020). [DOI] [PubMed] [Google Scholar]
- 63.Brouwer KLR et al. In vitro methods to support transporter evaluation in drug discovery and development. Clin. Pharmacol. Ther 94, 95–112 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Gertz M et al. Cyclosporine inhibition of hepatic and intestinal CYP3A4, uptake and efflux transporters: application of PBPK modeling in the assessment of drug-drug interaction potential. Pharm. Res 30, 761–780 (2013). [DOI] [PubMed] [Google Scholar]
- 65.US Food and Drug Administration. In vitro Drug Interaction Studies Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry https://www.fda.gov/media/134582/download (2020).
- 66.Hayden ER et al. Regulation of OATP1B1 function by tyrosine kinase-mediated phosphorylation. Clin. Cancer Res 27, 4301–4310 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guo C, LaCerte C, Edwards JE, Brouwer KR & Brouwer KLR Farnesoid X receptor agonists obeticholic acid and chenodeoxycholic acid increase bile acid efflux in sandwich-cultured human hepatocytes: functional evidence and mechanisms. J. Pharmacol. Exp. Ther 365, 413–421 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kummu M et al. Cadmium inhibits ABCG2 transporter function in BeWo choriocarcinoma cells and MCF-7 cells overexpressing ABCG2. Placenta 33, 859–865 (2012). [DOI] [PubMed] [Google Scholar]
- 69.Grimstein M et al. Physiologically based pharmacokinetic modeling in regulatory science: an update from the U.S. food and drug administration’s office of clinical pharmacology. J. Pharm. Sci 108, 21–25 (2019). [DOI] [PubMed] [Google Scholar]
- 70.Zhang X et al. Application of PBPK modeling and simulation for regulatory decision making and its impact on US prescribing information: an update on the 2018–2019 submissions to the US FDA’s office of clinical pharmacology. J. Clin. Pharmacol 60, S160–S178 (2020). [DOI] [PubMed] [Google Scholar]
- 71.Guo Y et al. Advancing predictions of tissue and intracellular drug concentrations using in vitro, imaging and physiologically based pharmacokinetic modeling approaches. Clin. Pharmacol. Ther 104, 865–889 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This manuscript illustrates current best practices and outlines practical strategies for selecting appropriate in vitro and in vivo experimental methods to estimate or predict tissue and plasma concentrations resulting from transporter modulation, and discusses the use of these data in the application of PBPK modeling for human pharmacokinetic, efficacy and safety assessment in drug development.
- 72.Taskar KS et al. Physiologically-based pharmacokinetic models for evaluating membrane transporter mediated drug-drug interactions: current capabilities, case studies, future opportunities, and recommendations. Clin. Pharmacol. Ther 107, 1082–1115 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This review provides a critical analysis of the current trends in PBPK modeling of transporter-mediated DDIs and corresponding knowledge gaps, and it provides future directions to improve model predictions and to promote continuous application of PBPK modeling in this challenging area.
- 73.Varma MV et al. Extended Clearance Classification System (ECCS) informed approach for evaluating investigational drugs as substrates of drug transporters. Clin. Pharmacol. Ther 102, 33–36 (2017). [DOI] [PubMed] [Google Scholar]
- 74.Prasad B et al. Toward a consensus on applying quantitative liquid chromatographytandem mass spectrometry proteomics in translational pharmacology research: a white paper. Clin. Pharmacol. Ther 106, 525–543 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Storelli F et al. The next frontier in ADME science: predicting transporter-based drug disposition, tissue concentrations and drug-drug interactions in humans. Pharmacol. Ther 238, 108271 (2022). [DOI] [PubMed] [Google Scholar]
- 76.Sachar M, Kumar V, Gormsen LC, Munk OL & Unadkat JD Successful prediction of positron emission tomography-imaged metformin hepatic uptake clearance in humans using the quantitative proteomics-informed relative expression factor approach. Drug. Metab. Dispos 48, 1210–1216 (2020). [DOI] [PubMed] [Google Scholar]
- 77.Kumar V et al. The importance of incorporating OCT2 plasma membrane expression and membrane potential in IVIVE of metformin renal secretory clearance. Drug. Metab. Dispos 46, 1441–1445 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wegler C et al. Proteomics-informed prediction of rosuvastatin plasma profiles in patients with a wide range of body weight. Clin. Pharmacol. Ther 109, 762–771 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Alrubia S, Al-Majdoub ZM, Achour B, Rostami-Hodjegan A & Barber J Quantitative assessment of the impact of crohn’s disease on protein abundance of human intestinal drug-metabolising enzymes and transporters. J. Pharm. Sci 111, 2917–2929 (2022). [DOI] [PubMed] [Google Scholar]
- 80.van der Made TK et al. Quantitative translation of microfluidic transporter in vitro data to in vivo reveals impaired albumin-facilitated indoxyl sulfate secretion in chronic kidney disease. Mol. Pharm 16, 4551–4562 (2019). [DOI] [PubMed] [Google Scholar]
- 81.Burt HJ et al. Metformin and cimetidine: physiologically based pharmacokinetic modelling to investigate transporter mediated drug-drug interactions. Eur. J. Pharm. Sci 88, 70–82 (2016). [DOI] [PubMed] [Google Scholar]
- 82.Mathialagan S et al. Quantitative prediction of human renal clearance and drug-drug interactions of organic anion transporter substrates using in vitro transport data: a relative activity factor approach. Drug. Metab. Dispos 45, 409–417 (2017). [DOI] [PubMed] [Google Scholar]
- 83.Costales C et al. Quantitative prediction of breast cancer resistant protein mediated drug-drug interactions using physiologically-based pharmacokinetic modeling. CPT Pharmacomet. Syst. Pharmacol 10, 1018–1031 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Scotcher D et al. Physiologically based pharmacokinetic modeling of transporter-mediated hepatic disposition of imaging biomarker gadoxetate in rats. Mol. Pharm 18, 2997–3009 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ito K, Sjostedt N, Malinen MM, Guo C & Brouwer KLR Hepatic transporter alterations by nuclear receptor agonist T0901317 in sandwich-cultured human hepatocytes: proteomic analysis and PBPK modeling to evaluate drug-drug interaction risk. J. Pharmacol. Exp. Ther 373, 261–268 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li J, Jiang J, Wu J, Bao X & Sanai N Physiologically based pharmacokinetic modeling of central nervous system pharmacokinetics of CDK4/6 inhibitors to guide selection of drug and dosing regimen for brain cancer treatment. Clin. Pharmacol. Ther 109, 494–506 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bowman C et al. Evaluation of bottom-up modeling of the blood-brain barrier to improve brain penetration prediction via physiologically based pharmacokinetic modeling. Biopharm. Drug. Dispos 44, 60–70 (2023). [DOI] [PubMed] [Google Scholar]
- 88.Lang J, Vincent L, Chenel M, Ogungbenro K & Galetin A Reduced physiologically-based pharmacokinetic model of dabigatran etexilate-dabigatran and its application for prediction of intestinal P-gp-mediated drug-drug interactions. Eur. J. Pharm. Sci 165, 105932 (2021). [DOI] [PubMed] [Google Scholar]
- 89.Chen Y et al. Physiologically-based pharmacokinetic model-informed drug development for fenebrutinib: understanding complex drug-drug interactions. CPT Pharmacomet. Syst. Pharmacol 9, 332–341 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vildhede A et al. Hepatic uptake of atorvastatin: influence of variability in transporter expression on uptake clearance and drug-drug interactions. Drug. Metab. Dispos 42, 1210–1218 (2014). [DOI] [PubMed] [Google Scholar]
- 91.Bi YA et al. Quantitative contribution of six major transporters to the hepatic uptake of drugs: “SLC-Phenotyping” using primary human hepatocytes. J. Pharmacol. Exp. Ther 370, 72–83 (2019). [DOI] [PubMed] [Google Scholar]
- 92.Barnett S et al. Gaining mechanistic insight into coproporphyrin i as endogenous biomarker for OATP1B-mediated drug-drug interactions using population pharmacokinetic modeling and simulation. Clin. Pharmacol. Ther 104, 564–574 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ahmad A et al. Population pharmacokinetic modeling and simulation to support qualification of pyridoxic acid as endogenous biomarker of OAT1/3 renal transporters. CPT Pharmacomet. Syst. Pharmacol 10, 467–477 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kimoto E et al. Biomarker-Informed model-based risk assessment of organic anion transporting polypeptide 1B mediated drug-drug interactions. Clin. Pharmacol. Ther 111, 404–415 (2022). [DOI] [PubMed] [Google Scholar]
- 95.Tess DA et al. Effect of a ketohexokinase inhibitor (PF-06835919) on in vivo OATP1B activity: integrative risk assessment using endogenous biomarker and a probe drug. Clin. Pharmacol. Ther 112, 605–614 (2022). [DOI] [PubMed] [Google Scholar]
- 96.Pfeifer ND et al. Effect of Ritonavir on (99 m)Technetium-Mebrofenin disposition in humans: a semi-PBPK modeling and in vitro approach to predict transporter-mediated DDIs. CPT Pharmacomet. Syst. Pharmacol 2, e20 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bowman CM, Ma F, Mao J & Chen Y Examination of Physiologically-Based Pharmacokinetic Models of Rosuvastatin. CPT Pharmacomet. Syst. Pharmacol 10, 5–17 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yamazaki S et al. Physiologically-based pharmacokinetic modeling approach to predict rifampin-mediated intestinal P-glycoprotein induction. CPT Pharmacomet. Syst. Pharmacol 8, 634–642 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Moj D et al. A Comprehensive whole-body physiologically based pharmacokinetic model of dabigatran etexilate, dabigatran and dabigatran glucuronide in healthy adults and renally impaired patients. Clin. Pharmacokinet 58, 1577–1593 (2019). [DOI] [PubMed] [Google Scholar]
- 100.Vasilogianni AM et al. Proteomics of colorectal cancer liver metastasis: a quantitative focus on drug elimination and pharmacodynamics effects. Br. J. Clin. Pharmacol 88, 1811–1823 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vildhede A, Kimoto E, Pelis RM, Rodrigues AD & Varma MVS Quantitative proteomics and mechanistic modeling of transporter-mediated disposition in nonalcoholic fatty liver disease. Clin. Pharmacol. Ther 107, 1128–1137 (2020). [DOI] [PubMed] [Google Scholar]
- 102.Prasad B et al. Ontogeny of hepatic drug transporters as quantified by LC-MS/MS proteomics. Clin. Pharmacol. Ther 100, 362–370 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Streekstra EJ, Russel FGM, van de Steeg E & de Wildt SN Application of proteomics to understand maturation of drug metabolizing enzymes and transporters for the optimization of pediatric drug therapy. Drug. Discov. Today Technol 39, 31–48 (2021). [DOI] [PubMed] [Google Scholar]
- 104.Salerno SN, Burckart GJ, Huang SM & Gonzalez D Pediatric drug-drug interaction studies: barriers and opportunities. Clin. Pharmacol. Ther 105, 1067–1070 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tan SPF, Scotcher D, Rostami-Hodjegan A & Galetin A Effect of chronic kidney disease on the renal secretion via organic anion transporters 1/3: implications for physiologically-based pharmacokinetic modeling and dose adjustment. Clin. Pharmacol. Ther 112, 643–652 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Takita H et al. Coproporphyrin I as an endogenous biomarker to detect reduced OATP1B activity and shift in elimination route in chronic kidney disease. Clin. Pharmacol. Ther 112, 615–626 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tan ML et al. Use of physiologically based pharmacokinetic modeling to evaluate the effect of chronic kidney disease on the disposition of hepatic CYP2C8 and OATP1B drug substrates. Clin. Pharmacol. Ther 105, 719–729 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bergman A et al. Effect of hepatic organic anion-transporting polypeptide 1b inhibition and chronic kidney disease on the pharmacokinetics of a liver-targeted glucokinase activator: a model-based evaluation. Clin. Pharmacol. Ther 106, 792–802 (2019). [DOI] [PubMed] [Google Scholar]
- 109.Tan SPF et al. Development of 4-Pyridoxic Acid PBPK model to support biomarker-informed evaluation of OAT1/3 inhibition and effect of chronic kidney disease. Clin. Pharmacol. Ther 114, 1243–1253 (2023). [DOI] [PubMed] [Google Scholar]
- 110.Vaidyanathan J, Yoshida K, Arya V & Zhang L Comparing various in vitro prediction criteria to assess the potential of a new molecular entity to inhibit organic anion transporting polypeptide 1B1. J. Clin. Pharmacol 56, S59–S72 (2016). [DOI] [PubMed] [Google Scholar]
- 111.Mathialagan S, Feng B, Rodrigues AD & Varma MVS Drug-drug interactions involving renal OCT2/MATE transporters: clinical risk assessment may require endogenous biomarker-informed approach. Clin. Pharmacol. Ther 110, 855–859 (2021). [DOI] [PubMed] [Google Scholar]
- 112.Chu X, Chan GH & Evers R Identification of endogenous biomarkers to predict the propensity of drug candidates to cause hepatic or renal transporter-mediated drug-drug interactions. J. Pharm. Sci 106, 2357–2367 (2017). [DOI] [PubMed] [Google Scholar]
- 113.Rodrigues AD, Taskar KS, Kusuhara H & Sugiyama Y Endogenous probes for drug transporters: balancing vision with reality. Clin. Pharmacol. Ther 103, 434–448 (2018). [DOI] [PubMed] [Google Scholar]
- 114.Lai Y et al. Coproporphyrins in plasma and urine can be appropriate clinical biomarkers to recapitulate drug-drug interactions mediated by organic anion transporting polypeptide inhibition. J. Pharmacol. Exp. Ther 358, 397–404 (2016). [DOI] [PubMed] [Google Scholar]
- 115.Shen H et al. Evidence for the validity of pyridoxic acid (PDA) as a plasma-based endogenous probe for OAT1 and OAT3 function in healthy subjects. J. Pharmacol. Exp. Ther 368, 136–145 (2019). [DOI] [PubMed] [Google Scholar]
- 116.Willemin ME et al. Clinical investigation on endogenous biomarkers to predict strong OAT-mediated drug-drug interactions. Clin. Pharmacokinet 60, 1187–1199 (2021). [DOI] [PubMed] [Google Scholar]
- 117.Mori D et al. Dose-dependent inhibition of OATP1B by rifampicin in healthy volunteers: comprehensive evaluation of candidate biomarkers and OATP1B probe drugs. Clin. Pharmacol. Ther 107, 1004–1013 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Neuvonen M et al. Identification of glycochenodeoxycholate 3-O-glucuronide and glycodeoxycholate 3-O-glucuronide as highly sensitive and specific OATP1B1 biomarkers. Clin. Pharmacol. Ther 109, 646–657 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Muller F et al. N(1)-Methylnicotinamide as biomarker for MATE-mediated renal drug-drug interactions: impact of cimetidine, rifampin, verapamil, and probenecid. Clin. Pharmacol. Ther 113, 1070–1079 (2023). [DOI] [PubMed] [Google Scholar]
- 120.Gessner A et al. A Metabolomic analysis of sensitivity and specificity of 23 previously proposed biomarkers for renal transporter-mediated drug-drug interactions. Clin. Pharmacol. Ther 114, 1058–1072 (2023). [DOI] [PubMed] [Google Scholar]
- 121.Neuvonen M, Tornio A, Hirvensalo P, Backman JT & Niemi M Performance of plasma coproporphyrin I and III as OATP1B1 biomarkers in humans. Clin. Pharmacol. Ther 110, 1622–1632 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chan GH et al. Evaluation of the selectivity of several organic anion transporting polypeptide 1B biomarkers using relative activity factor method. Drug. Metab. Dispos 51, 1089–1104 (2023). [DOI] [PubMed] [Google Scholar]
- 123.Huh Y et al. Utilization of rosuvastatin and endogenous biomarkers in evaluating the impact of ritlecitinib on BCRP, OATP1B1, and OAT3 transporter activity. Pharm. Res 40, 2639–2651 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yoshikado T et al. PBPK modeling of coproporphyrin I as an endogenous biomarker for drug interactions involving inhibition of hepatic OATP1B1 and OATP1B3. CPT Pharmacomet. Syst. Pharmacol 7, 739–747 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Turk D et al. Renal transporter-mediated drug-biomarker interactions of the endogenous substrates creatinine and N(1)-methylnicotinamide: a PBPK modeling approach. Clin. Pharmacol. Ther 112, 687–698 (2022). [DOI] [PubMed] [Google Scholar]
- 126.Takita H et al. PBPK model of coproporphyrin i: evaluation of the impact of SLCO1B1 genotype, ethnicity, and sex on its inter-individual variability. CPT Pharmacomet. Syst. Pharmacol 10, 137–147 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kikuchi R et al. Utilization of OATP1B biomarker coproporphyrin-i to guide drug-drug interaction risk assessment: evaluation by the pharmaceutical industry. Clin. Pharmacol. Ther 114, 1170–1183 (2023). [DOI] [PubMed] [Google Scholar]
- 128.Cheung KWK et al. GDC-0810 pharmacokinetics and transporter-mediated drug interaction evaluation with an endogenous biomarker in the first-in-human, dose escalation study. Drug. Metab. Dispos 47, 966–973 (2019). [DOI] [PubMed] [Google Scholar]
- 129.Scotcher D et al. Mechanistic models as framework for understanding biomarker disposition: prediction of creatinine-drug interactions. CPT Pharmacomet. Syst. Pharmacol 9, 282–293 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kenna JG et al. Can bile salt export pump inhibition testing in drug discovery and development reduce liver injury risk? An International Transporter Consortium Perspective. Clin. Pharmacol. Ther 104, 916–932 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This manuscript highlights the importance of inhibition of the bile salt export pump (BSEP) as one of several mechanisms of drug-induced liver injury (DILI) that should be considered, along with other mechanisms, when evaluating potential DILI risk.
- 131.Hafey MJ et al. Transporters and toxicity: insights from the International Transporter Consortium Workshop 4. Clin. Pharmacol. Ther 112, 527–539 (2022). [DOI] [PubMed] [Google Scholar]; This paper highlights multiple ways in which toxicants and drugs interact with transporters to negatively affect therapeutic outcomes and/or cause adverse effects; specific examples focus on anti-cancer drug-induced organ damage, the role of transporters as a barrier in different tissues, and transporters in toxicity of metal ions.
- 132.Pan G Roles of hepatic drug transporters in drug disposition and liver toxicity. Adv. Exp. Med. Biol 1141, 293–340 (2019). [DOI] [PubMed] [Google Scholar]
- 133.Schuetz JD, Swaan PW & Tweedie DJ The role of transporters in toxicity and disease. Drug. Metab. Dispos 42, 541–545 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Anderson JT, Huang KM, Lustberg MB, Sparreboom A & Hu S Solute carrier transportome in chemotherapy-induced adverse drug reactions. Rev. Physiol. Biochem. Pharmacol 183, 177–215 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Uddin ME, Moseley A, Hu S & Sparreboom A Contribution of membrane transporters to chemotherapy-induced cardiotoxicity. Basic. Clin. Pharmacol. Toxicol 130, 36–47 (2022). [DOI] [PubMed] [Google Scholar]
- 136.Hu S, Huang KM, Adams EJ, Loprinzi CL & Lustberg MB Recent developments of novel pharmacologic therapeutics for prevention of chemotherapy-induced peripheral neuropathy. Clin. Cancer Res 25, 6295–6301 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Huang KM et al. Neuronal uptake transporters contribute to oxaliplatin neurotoxicity in mice. J. Clin. Invest 130, 4601–4606 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Huang KM et al. Targeting OCT3 attenuates doxorubicin-induced cardiac injury. Proc. Natl Acad. Sci. USA 118, e2020168118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Leblanc AF et al. OATP1B2 deficiency protects against paclitaxel-induced neurotoxicity. J. Clin. Invest 128, 816–825 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Vora B et al. Drug-nutrient interactions: discovering prescription drug inhibitors of the thiamine transporter ThTR-2 (SLC19A3). Am. J. Clin. Nutr 111, 110–121 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhang Q et al. The Janus kinase 2 inhibitor fedratinib inhibits thiamine uptake: a putative mechanism for the onset of Wernicke’s encephalopathy. Drug. Metab. Dispos 42, 1656–1662 (2014). [DOI] [PubMed] [Google Scholar]
- 142.Yoshikado T et al. Itraconazole-induced cholestasis: involvement of the inhibition of bile canalicular phospholipid translocator MDR3/ABCB4. Mol. Pharmacol 79, 241–250 (2011). [DOI] [PubMed] [Google Scholar]
- 143.Gill MW et al. Mechanism of hepatobiliary toxicity of the LPA(1) antagonist BMS- 986020 developed to treat idiopathic pulmonary fibrosis: contrasts with BMS-986234 and BMS-986278. Toxicol. Appl. Pharmacol 438, 115885 (2022). [DOI] [PubMed] [Google Scholar]
- 144.International Council for Harmonisation. ICH Harmonised Guideline - Drug Interaction Studies M12 (Draft version) https://www.ema.europa.eu/en/documents/scientific-guideline/draft-ich-guideline-m12-drug-interaction-studies-step-2b_en.pdf (2022).
- 145.Zhou T, Arya V & Zhang L Comparing various in vitro prediction methods to assess the potential of a drug to inhibit P-glycoprotein (P-gp) transporter in vivo. J. Clin. Pharmacol 59, 1049–1060 (2019). [DOI] [PubMed] [Google Scholar]
- 146.Lee SC, Arya V, Yang X, Volpe DA & Zhang L Evaluation of transporters in drug development: Current status and contemporary issues. Adv. Drug. Deliv. Rev 116, 100–118 (2017). [DOI] [PubMed] [Google Scholar]
- 147.Yoshida K et al. In vitro-in vivo extrapolation of metabolism- and transporter-mediated drug-drug interactions-overview of basic prediction methods. J. Pharm. Sci 106, 2209–2213 (2017). [DOI] [PubMed] [Google Scholar]
- 148.US Food and Drug Administration. Food and drug administration integrated review for NDA 212887, Cabotegravir. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/212887Orig1s000,212888Orig1s000IntegratedR.pdf (2021). [Google Scholar]
- 149.US Food and Drug Administration. Food and drug administration integrated review for NDA 216196, Mitapivat. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2022/216196Orig1s000IntegratedR.pdf (2022). [Google Scholar]
- 150.US Food and Drug Administration. Food and drug administration integrated review for NDA 215206, Atogepant. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/215206Orig1s000IntegratedR.pdf (2021). [Google Scholar]
- 151.Arya V, Reynolds KS & Yang X Using endogenous biomarkers to derisk assessment of transporter-mediated drug-drug interactions: a scientific perspective. J. Clin. Pharmacol 62, 1501–1506 (2022). [DOI] [PubMed] [Google Scholar]
- 152.Jones NS et al. Complex DDI by fenebrutinib and the use of transporter endogenous biomarkers to elucidate the mechanism of DDI. Clin. Pharmacol. Ther 107, 269–277 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zamek-Gliszczynski MJ, Chu X, Polli JW, Paine MF & Galetin A Understanding the transport properties of metabolites: case studies and considerations for drug development. Drug. Metab. Dispos 42, 650–664 (2014). [DOI] [PubMed] [Google Scholar]
- 154.Taskar KS et al. Clinical relevance of hepatic and renal P-gp/BCRP inhibition of drugs: an international transporter consortium perspective. Clin. Pharmacol. Ther 112, 573–592 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Elmeliegy M, Vourvahis M, Guo C & Wang DD Effect of P-glycoprotein (P-gp) inducers on exposure of P-gp substrates: review of clinical drug-drug interaction studies. Clin. Pharmacokinet 59, 699–714 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Giacomini KM et al. New and emerging research on solute carrier and ATP binding cassette transporters in drug discovery and development: outlook from the international transporter consortium. Clin. Pharmacol. Ther 112, 540–561 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]; This state-of-the-art manuscript describes a range of technologically driven advances in membrane transporter research including the large number of high-resolution structures enabled by cryo-EM and AlphaFold, new discoveries demonstrating modulation of transporter function by gut microbiome products and pre-clinical and clinical tools such as proteomics, advances in modelling and simulation and biomarkers that provide quantitative estimates of in vivo transporter function.
- 157.Drew D, North RA, Nagarathinam K & Tanabe M Structures and general transport mechanisms by the major facilitator superfamily (MFS). Chem. Rev 121, 5289–5335 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bai X, Moraes TF & Reithmeier RAF Structural biology of solute carrier (SLC) membrane transport proteins. Mol. Membr. Biol 34, 1–32 (2017). [DOI] [PubMed] [Google Scholar]
- 159.Hou WT et al. Plastic structures for diverse substrates: a revisit of human ABC transporters. Proteins 90, 1749–1765 (2022). [DOI] [PubMed] [Google Scholar]
- 160.Zhang Q et al. Recognition of cyclic dinucleotides and folates by human SLC19A1. Nature 612, 170–176 (2022). [DOI] [PubMed] [Google Scholar]
- 161.Shan Z et al. Cryo-EM structures of human organic anion transporting polypeptide OATP1B1. Cell Res 33, 940–951 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ciuta AD et al. Structure of human drug transporters OATP1B1 and OATP1B3. Nat. Commun 14, 5774 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Suo Y et al. Molecular basis of polyspecific drug binding and transport by OCT1 and OCT2. Nat. Struct. Mol. Biol 30, 1001–1011 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Khanppnavar B et al. Structural basis of organic cation transporter-3 inhibition. Nat. Commun 13, 6714 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Simoff I et al. Complete knockout of endogenous Mdr1 (Abcb1) in MDCK cells by CRISPR-Cas9. J. Pharm. Sci 105, 1017–1021 (2016). [DOI] [PubMed] [Google Scholar]
- 166.Chen EC et al. Evaluating the utility of canine Mdr1 knockout madin-darby canine kidney I cells in permeability screening and efflux substrate determination. Mol. Pharm 15, 5103–5113 (2018). [DOI] [PubMed] [Google Scholar]
- 167.Karlgren M et al. A CRISPR-Cas9 generated MDCK cell line expressing human MDR1 without endogenous canine MDR1 (cABCB1): an improved tool for drug efflux studies. J. Pharm. Sci 106, 2909–2913 (2017). [DOI] [PubMed] [Google Scholar]
- 168.Wegler C et al. Expanding the efflux in vitro assay toolbox: a CRISPR-Cas9 edited MDCK cell line with human BCRP and completely lacking canine MDR1. J. Pharm. Sci 110, 388–396 (2021). [DOI] [PubMed] [Google Scholar]
- 169.Feng D et al. Knockout of ABC transporters by CRISPR/Cas9 contributes to reliable and accurate transporter substrate identification for drug discovery. Front. Pharmacol 13, 1015940 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Sake JA et al. Knockout of ABCC1 in NCI-H441 cells reveals CF to be a suboptimal substrate to study MRP1 activity in organotypic in vitro models. Eur. J. Pharm. Sci 181, 106364 (2022). [DOI] [PubMed] [Google Scholar]
- 171.Guan M et al. Generation of a homozygous ABCA7-knockout human iPSC line using the CRISPR/Cas9 system. Stem Cell Res 66, 103000 (2022). [DOI] [PubMed] [Google Scholar]
- 172.Magdy T et al. Identification of drug transporter genomic variants and inhibitors that protect against doxorubicin-induced cardiotoxicity. Circulation 145, 279–294 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ma X et al. Characterization of organic anion transporting polypeptide 1b2 knockout rats generated by CRISPR/Cas9: a novel model for drug transport and hyperbilirubinemia disease. Acta Pharm. Sin. B 10, 850–860 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Gou X, Ran F, Yang J, Ma Y & Wu X Construction and evaluation of a novel organic anion transporter 1/3 CRISPR/Cas9 double-knockout rat model. Pharmaceutics 14, 2307 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Liang C et al. Development and characterization of MDR1 (Mdr1a/b) CRISPR/Cas9 knockout rat model. Drug. Metab. Dispos 47, 71–79 (2019). [DOI] [PubMed] [Google Scholar]
- 176.Girardi E et al. A widespread role for SLC transmembrane transporters in resistance to cytotoxic drugs. Nat. Chem. Biol 16, 469–478 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sapp V et al. Genome-wide CRISPR/Cas9 screening in human iPS derived cardiomyocytes uncovers novel mediators of doxorubicin cardiotoxicity. Sci. Rep 11, 13866 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Akinci E et al. Elucidation of remdesivir cytotoxicity pathways through genome-wide CRISPR-Cas9 screening and transcriptomics. Preprint at bioRxiv 10.1101/2020.08.27.270819 (2020). [DOI] [Google Scholar]
- 179.Rebsamen M et al. Gain-of-function genetic screens in human cells identify SLC transporters overcoming environmental nutrient restrictions. Life Sci. Alliance 5, e202201404 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zamek-Gliszczynski MJ et al. Transporters in drug development: International Transporter Consortium update on emerging transporters of clinical importance. Clin. Pharmacol. Ther 112, 485–500 (2022). [DOI] [PubMed] [Google Scholar]; This manuscript reviews up-to-date clinical evidence for the importance of various transporters in drug–drug interactions and the potential for altered drug efficacy and safety; for the first time, drug–nutrient interactions leading to nutrient deficiencies are also considered.
- 181.Watanabe T, Kusuhara H, Maeda K, Shitara Y & Sugiyama Y Physiologically based pharmacokinetic modeling to predict transporter-mediated clearancse and distribution of pravastatin in humans. J. Pharmacol. Exp. Ther 328, 652–662 (2009). [DOI] [PubMed] [Google Scholar]
- 182.Scotcher D, Jones CR, Galetin A & Rostami-Hodjegan A Delineating the role of various factors in renal disposition of digoxin through application of physiologically based kidney model to renal impairment populations. J. Pharmacol. Exp. Ther 360, 484–495 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wiebe ST et al. Validation of a drug transporter probe cocktail using the prototypical inhibitors rifampin, probenecid, verapamil, and cimetidine. Clin. Pharmacokinet 59, 1627–1639 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Mochizuki T et al. Effect of cyclosporin A and impact of dose staggering on OATP1B1/1B3 endogenous substrates and drug probes for assessing clinical drug interactions. Clin. Pharmacol. Ther 111, 1315–1323 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Barth A et al. Clinical assessment of gepotidacin (GSK2140944) as a victim and perpetrator of drug-drug interactions via CYP3A metabolism and transporters. Clin. Transl. Sci 16, 647–661 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hibma JE et al. The effect of famotidine, a MATE1-selective inhibitor, on the pharmacokinetics and pharmacodynamics of metformin. Clin. Pharmacokinet 55, 711–721 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Song IH et al. The effect of dolutegravir on the pharmacokinetics of metformin in healthy subjects. J. Acquir. Immune Defic. Syndr 72, 400–407 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ghibellini G, Leslie EM & Brouwer KLR Methods to evaluate biliary excretion of drugs in humans: an updated review. Mol. Pharm 3, 198–211 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Nakaoka T et al. Clinical evaluation of [(18)F]pitavastatin for quantitative analysis of hepatobiliary transporter activity. Drug. Metab. Pharmacokinet 44, 100449 (2022). [DOI] [PubMed] [Google Scholar]
- 190.Melillo N et al. Use of in vivo imaging and physiologically-based kinetic modelling to predict hepatic transporter mediated drug-drug interactions in rats. Pharmaceutics 15, 896 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zamek-Gliszczynski MJ et al. Clinical extrapolation of the effects of dolutegravir and other HIV integrase inhibitors on folate transport pathways. Drug. Metab. Dispos 47, 890–898 (2019). [DOI] [PubMed] [Google Scholar]
- 192.Lin L, Yee SW, Kim RB & Giacomini KM SLC transporters as therapeutic targets: emerging opportunities. Nat. Rev. Drug. Discov 14, 543–560 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.US Food and Drug Administration. Food and Drug Administration Drug Label for Farxiga, dapagliflozin (revised 05/2020). https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/202293s020lbl.pdf (2020).
- 194.US Food and Drug Administration. Food and Drug Administration Drug Label for Invokana, canagliflozin (revised 07/2017). https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/204042s026lbl.pdf (2017).
- 195.US Food and Drug Administration. Food and Drug Administration Drug Label for Jardiance, empagliflozin (revised 6/2023). https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/204629s042lbl.pdf (2023).
- 196.Shi X et al. Novel urate transporter 1 (URAT1) inhibitors: a review of recent patent literature (2020-present). Expert. Opin. Ther. Pat 32, 1175–1184 (2022). [DOI] [PubMed] [Google Scholar]
- 197.Dong Y et al. Novel urate transporter 1 (URAT1) inhibitors: a review of recent patent literature (2016–2019). Expert. Opin. Ther. Pat 29, 871–879 (2019). [DOI] [PubMed] [Google Scholar]
- 198.Zhang Y et al. Metabolomic profiling and drug interaction characterization reveal riboflavin as a breast cancer resistance protein -specific endogenous biomarker that demonstrates prediction of transporter activity in vivo. Drug. Metab. Dispos 51, 851–861 (2023). [DOI] [PubMed] [Google Scholar]
- 199.Rodrigues D & Rowland A From endogenous compounds as biomarkers to plasma-derived nanovesicles as liquid biopsy; has the golden age of translational pharmacokinetics-absorption, distribution, metabolism, excretion-drug-drug interaction science finally arrived? Clin. Pharmacol. Ther 105, 1407–1420 (2019). [DOI] [PubMed] [Google Scholar]
- 200.Rowland A et al. Plasma extracellular nanovesicle (exosome)-derived biomarkers for drug metabolism pathways: a novel approach to characterize variability in drug exposure. Br. J. Clin. Pharmacol 85, 216–226 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Achour B et al. Liquid biopsy enables quantification of the abundance and interindividual variability of hepatic enzymes and transporters. Clin. Pharmacol. Ther 109, 222–232 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Rodrigues AD et al. Exploring the use of serum-derived small extracellular vesicles as liquid biopsy to study the induction of hepatic cytochromes P450 and organic anion transporting polypeptides. Clin. Pharmacol. Ther 110, 248–258 (2021). [DOI] [PubMed] [Google Scholar]
- 203.Achour B et al. Liquid biopsy for patient characterization in cardiovascular disease: verification against markers of cytochrome P450 and P-Glycoprotein activities. Clin. Pharmacol. Ther 111, 1268–1277 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Zhang Z et al. Structural basis of ligand binding modes of human EAAT2. Nat. Commun 13, 3329 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Canul-Tec JC et al. The ion-coupling mechanism of human excitatory amino acid transporters. EMBO J 41, e108341 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Niu Y et al. Structural mechanism of SGLT1 inhibitors. Nat. Commun 13, 6440 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Niu Y et al. Structural basis of inhibition of the human SGLT2-MAP17 glucose transporter. Nature 601, 280–284 (2022). [DOI] [PubMed] [Google Scholar]
- 208.Motiwala Z et al. Structural basis of GABA reuptake inhibition. Nature 606, 820–826 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Coleman JA et al. Chemical and structural investigation of the paroxetine-human serotonin transporter complex. eLife 9, e56427 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Coleman JA et al. Serotonin transporter-ibogaine complexes illuminate mechanisms of inhibition and transport. Nature 569, 141–145 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Shahsavar A et al. Structural insights into the inhibition of glycine reuptake. Nature 591, 677–681 (2021). [DOI] [PubMed] [Google Scholar]
- 212.Yan R, Zhao X, Lei J & Zhou Q Structure of the human LAT1–4F2hc heteromeric amino acid transporter complex. Nature 568, 127–130 (2019). [DOI] [PubMed] [Google Scholar]
- 213.Yan R et al. Mechanism of substrate transport and inhibition of the human LAT1–4F2hc amino acid transporter. Cell Discov 7, 16 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Zhao Y et al. Structural basis for inhibition of the Cation-chloride cotransporter NKCC1 by the diuretic drug bumetanide. Nat. Commun 13, 2747 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Wright NJ et al. Methotrexate recognition by the human reduced folate carrier SLC19A1. Nature 609, 1056–1062 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Wright NJ & Lee SY Structures of human ENT1 in complex with adenosine reuptake inhibitors. Nat. Struct. Mol. Biol 26, 599–606 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kim Y & Chen J Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 359, 915–919 (2018). [DOI] [PubMed] [Google Scholar]
- 218.Urgaonkar S et al. Discovery and characterization of potent dual P-Glycoprotein and CYP3A4 inhibitors: design, synthesis, cryo-EM analysis, and biological evaluations. J. Med. Chem 65, 191–216 (2022). [DOI] [PubMed] [Google Scholar]
- 219.Nosol K et al. Cryo-EM structures reveal distinct mechanisms of inhibition of the human multidrug transporter ABCB1. Proc. Natl Acad. Sci. USA 117, 26245–26253 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Liu F et al. Structural identification of a hotspot on CFTR for potentiation. Science 364, 1184–1188 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Fiedorczuk K & Chen J Molecular structures reveal synergistic rescue of Delta508 CFTR by Trikafta modulators. Science 378, 284–290 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Zhao C & MacKinnon R Molecular structure of an open human K(ATP) channel. Proc. Natl Acad. Sci. USA 118, e2112267118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Wu JX et al. Ligand binding and conformational changes of SUR1 subunit in pancreatic ATP-sensitive potassium channels. Protein Cell 9, 553–567 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Ding D, Wang M, Wu JX, Kang Y & Chen L The structural basis for the binding of repaglinide to the pancreatic K(ATP) channel. Cell Rep 27, 1848–1857.e1844 (2019). [DOI] [PubMed] [Google Scholar]
- 225.Wang M, Wu JX & Chen L Structural insights into the high selectivity of the anti-diabetic drug mitiglinide. Front. Pharmacol 13, 929684 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Jackson SM et al. Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat. Struct. Mol. Biol 25, 333–340 (2018). [DOI] [PubMed] [Google Scholar]
- 227.Kowal J et al. Structural basis of drug recognition by the multidrug transporter ABCG2. J. Mol. Biol 433, 166980 (2021). [DOI] [PubMed] [Google Scholar]
- 228.Orlando BJ & Liao M ABCG2 transports anticancer drugs via a closed-to-open switch. Nat. Commun 11, 2264 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Ramsey LB et al. PharmVar geneFocus: SLCO1B1. Clin. Pharmacol. Ther 113, 782–793 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Liang X & Lai Y Overcoming the shortcomings of the extended-clearance concept: a framework for developing a physiologically-based pharmacokinetic (PBPK) model to select drug candidates involving transporter-mediated clearance. Expert. Opin. Drug. Metab. Toxicol 17, 869–886 (2021). [DOI] [PubMed] [Google Scholar]
- 231.Alluri RV, Li R & Varma MVS Transporter-enzyme interplay and the hepatic drug clearance: what have we learned so far? Expert. Opin. Drug. Metab. Toxicol 16, 387–401 (2020). [DOI] [PubMed] [Google Scholar]
- 232.Yoshida K, Jaochico A, Mao J & Sangaraju D Glycochenodeoxycholate and glycodeoxycholate 3-O-glucuronides, but not hexadecanedioate and tetradecanedioate, detected weak inhibition of OATP1B caused by GDC-0810 in humans. Br. J. Clin. Pharmacol 89, 1903–1907 (2023). [DOI] [PubMed] [Google Scholar]
- 233.Hafey MJ et al. A two-tiered in vitro approach to de-risk drug candidates for potential bile salt export pump inhibition liabilities in drug discovery. Drug. Metab. Dispos 48, 1147–1160 (2020). [DOI] [PubMed] [Google Scholar]
- 234.Yang K, Woodhead JL, Watkins PB, Howell BA & Brouwer KLR Systems pharmacology modeling predicts delayed presentation and species differences in bile acid-mediated troglitazone hepatotoxicity. Clin. Pharmacol. Ther 96, 589–598 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Cundy KC, Li ZH, Hitchcock MJ & Lee WA Pharmacokinetics of cidofovir in monkeys. Evidence for a prolonged elimination phase representing phosphorylated drug. Drug. Metab. Dispos 24, 738–744 (1996). [PubMed] [Google Scholar]
- 236.Wolf DL et al. Pharmacokinetics and renal effects of cidofovir with a reduced dose of probenecid in HIV-infected patients with cytomegalovirus retinitis. J. Clin. Pharmacol 43, 43–51 (2003). [DOI] [PubMed] [Google Scholar]
- 237.Gilead Sciences Inc. Cidofovir https://www.gilead.com/~/media/files/pdfs/medicines/other/vistide/vistide.pdf (2010).
- 238.Root C, Smith CD, Winegar DA, Brieaddy LE & Lewis MC Inhibition of ileal sodium-dependent bile acid transport by 2164U90. J. Lipid Res 36, 1106–1115 (1995). [PubMed] [Google Scholar]
- 239.Johns Hopkins University. OMIM Database entry 601295 https://www.omim.org/entry/601295 (2022).
- 240.Ferkingstad E et al. Genome-wide association meta-analysis yields 20 loci associated with gallstone disease. Nat. Commun 9, 5101 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Renner O et al. A variant of the SLC10A2 gene encoding the apical sodium-dependent bile acid transporter is a risk factor for gallstone disease. PLoS ONE 4, e7321 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Yin X et al. Genome-wide association studies of metabolites in Finnish men identify disease-relevant loci. Nat. Commun 13, 1644 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Huang HC et al. Discovery of potent, nonsystemic apical sodium-codependent bile acid transporter inhibitors (Part 2). J. Med. Chem 48, 5853–5868 (2005). [DOI] [PubMed] [Google Scholar]
- 244.Tremont SJ et al. Discovery of potent, nonsystemic apical sodium-codependent bile acid transporter inhibitors (Part 1). J. Med. Chem 48, 5837–5852 (2005). [DOI] [PubMed] [Google Scholar]
- 245.Tollefson MB et al. A novel class of apical sodium co-dependent bile acid transporter inhibitors: the 1,2-benzothiazepines. Bioorg. Med. Chem. Lett 13, 3727–3730 (2003). [DOI] [PubMed] [Google Scholar]
- 246.Shirley M Maralixibat: First Approval. Drugs 82, 71–76 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.US Food and Drug Administration. Food and Drug Administration Integrated Review for NDA 214662, Maralixibat. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/214662Orig1s000IntegratedR.pdf (2021). [Google Scholar]
- 248.Loomes KM et al. Maralixibat for the treatment of PFIC: long-term, IBAT inhibition in an open-label, Phase 2 study. Hepatol. Commun 6, 2379–2390 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Shneider BL et al. Placebo-controlled randomized trial of an intestinal bile salt transport inhibitor for pruritus in alagille syndrome. Hepatol. Commun 2, 1184–1198 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Shneider BL et al. Impact of long-term administration of maralixibat on children with cholestasis secondary to Alagille syndrome. Hepatol. Commun 6, 1922–1933 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Thompson RJ et al. Odevixibat treatment in progressive familial intrahepatic cholestasis: a randomised, placebo-controlled, phase 3 trial. Lancet Gastroenterol. Hepatol 7, 830–842 (2022). [DOI] [PubMed] [Google Scholar]
- 252.Yamamoto A et al. Rationale and design of a multicenter, single-group, open-label trial aiming at investigating the effectiveness of elobixibat for loss of defecation desire in patients with chronic constipation. Contemp. Clin. Trials Commun 28, 100958 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Hegade VS et al. Effect of ileal bile acid transporter inhibitor GSK2330672 on pruritus in primary biliary cholangitis: a double-blind, randomised, placebo-controlled, crossover, phase 2a study. Lancet 389, 1114–1123 (2017). [DOI] [PubMed] [Google Scholar]
- 254.Levy C et al. GLIMMER: a randomized phase 2b dose-ranging trial of linerixibat in primary biliary cholangitis patients with pruritus. Clin. Gastroenterol. Hepatol 21, 1902–1912.e13 (2022). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.