

Abstract
The matrix (M) proteins of vesicular stomatitis virus (VSV) and rabies virus (RV) play a key role in both assembly and budding of progeny virions. A PPPY motif (PY motif or late-budding domain) is conserved in the M proteins of VSV and RV. These PY motifs are important for virus budding and for mediating interactions with specific cellular proteins containing WW domains. The PY motif and flanking sequences of the M protein of VSV were used as bait to screen a mouse embryo cDNA library for cellular interactors. The mouse Nedd4 protein, a membrane-localized ubiquitin ligase containing multiple WW domains, was identified from this screen. Ubiquitin ligase Rsp5, the yeast homolog of Nedd4, was able to interact both physically and functionally with full-length VSV M protein in a PY-dependent manner. Indeed, the VSV M protein was multiubiquitinated by Rsp5 in an in vitro ubiquitination assay. To demonstrate further that ubiquitin may be involved in the budding process of rhabdoviruses, proteasome inhibitors (e.g., MG132) were used to decrease the level of free ubiquitin in VSV- and RV-infected cells. Viral titers measured from MG132-treated cells were reproducibly 10- to 20-fold lower than those measured from untreated control cells, suggesting that free ubiquitin is important for efficient virus budding. Last, release of a VSV PY mutant was not inhibited in the presence of MG132, signifying that the functional L domain of VSV is required for the inhibitory effect exhibited by MG132. These data suggest that the cellular ubiquitin-proteasome machinery is involved in the budding process of VSV and RV.
The Rhabdoviridae represent a divergent and complex family of negative-sense RNA viruses, of which Vesicular stomatitis virus (VSV) and Rabies virus (RV) are members. VSV maintains a minimal genome encoding five structural proteins: N (nucleoprotein), P (phosphoprotein), M (matrix protein), G (glycoprotein), and L (polymerase protein).
The M protein is an abundant, multifunctional virion protein that plays a role in gene regulation, cellular pathogenesis, and, along with the G protein, virion assembly and budding (2, 6, 7, 10, 14, 20, 23, 29, 30, 41, 43). An important characteristic of the M protein of VSV, shared by the Gag polyprotein of retroviruses (1, 12, 36, 39, 57, 59, 60) and the VP40 protein of Ebola virus (13, 19, 52), is its ability to be released (bud) from cells in the absence of any other viral protein (14, 21, 27). Recent investigations into this budding function exhibited by the M protein revealed that a proline-rich region (PPPY or PY motif) conserved at the N terminus of M was critical for efficient budding (7, 14). Indeed, infectious VSV PY mutants were significantly impaired in their ability to separate completely (pinch off) from the plasma membranes of infected cells (20). The PY motif has been termed a late-budding domain (L domain) for its involvement in a late step of the budding process. The conservation of functional L domains in members of the Rhabdoviridae, Retroviridae, and Filoviridae families is now well documented (1, 7, 13, 14, 20, 33, 36, 39, 45, 49, 57, 59, 60). While the premise that these divergent RNA viruses may utilize common machinery to break out of cells remains intriguing, the mechanism by which these L domains accomplish this task remains unknown.
It has been postulated previously that viral L domains may mediate their function via an interaction with a cellular protein(s). This insight was initiated by Garnier et al. (12), who demonstrated that the PY motif of the Rous sarcoma virus (RSV) Gag mediated interactions in vitro with one of the WW domains present within cellular protein YAP. Unlike SH3 domains, which prefer core consensus sequence PxxP, type I WW domains prefer core consensus sequence PPxY (24, 50, 51). To date, four different types of WW domains have been identified in a wide range of cellular proteins having various functions, and the PY motifs of RSV Gag, VSV M, RV M, and Ebola virus VP40 proteins have been shown to interact with specific, type I WW domain-containing proteins (12–14, 51).
One family of cellular proteins that contain multiple WW domains and that interact strongly with viral PY motifs are E3 ubiquitin ligases (e.g., Nedd4/Rsp5) (13, 14, 28, 58). The mammalian Nedd4 protein and its homolog in yeast, Rsp5, are membrane-localized ubiquitin ligases that play a role in endocytosis (3, 8, 9, 11, 15–18, 22, 25, 26, 38, 42, 47, 54, 55). While ubiquitination often targets a protein for degradation by the 26S proteasome, increasing evidence suggests that ubiquitination, in particular, monoubiquitination, may be a signal for something other than degradation (e.g., endocytosis) (4, 5, 8, 9, 16, 42, 46, 54).
Recent findings have implicated free ubiquitin and ubiquitin ligases as being integral components of the budding machinery of retroviruses and perhaps of filoviruses (13, 37, 45, 49, 53). In this report, we present evidence that the cellular ubiquitin-proteasome machinery is influential in the budding process of VSV and RV. Our results indicate that the VSV M protein can interact both physically and functionally with the Rsp5 ubiquitin ligase in a PY-dependent manner. Moreover, the release of both infectious VSV and RV from infected cells was decreased significantly in the presence of proteasome inhibitors, which reduce the level of free ubiquitin in treated cells (31). In contrast, release of a VSV PY mutant was not affected by proteasome inhibitors.
MATERIALS AND METHODS
Viruses and cells.
VSV (Indiana serotype) was propagated and titered on BHK-21 cells, which were maintained in Dulbecco modified essential medium (DMEM) (Life Technologies, Rockville, Md) supplemented with 10% fetal calf serum (HyClone) and penicillin-streptomycin (Life Technologies). BSR-T7 cells were kindly provided by K.-K. Conzelmann (Max-von-Pettenkofer Institut, Munich, Germany). BSR-T7 cells were maintained as described above with the addition of 1.0 mg of G-418 (Mediatech, Herndon, Va.)/ml every second or third passage.
Plasmids and antibodies.
The wild-type M gene of VSV (Indiana serotype) was subcloned from vector pSP72 (14) by PCR using primers flanking the entire open reading frame. The PCR product was then inserted into the pYES2 vector (Invitrogen, Carlsbad, Calif.) using SacI/XhoI restriction endonuclease sites to generate pYESMWT. A PY mutant form of VSV M (PPPY changed to AAAA) (14) was also cloned into the SacI/XhoI sites of the pYES2 vector to generate pYESMA4. Plasmids APVSVMWT and APVSVMA4 encoding VSV M and bacterial alkaline phosphatase fusion proteins have been described previously (2). Plasmid GST-Rsp5 encodes a fusion protein consisting of full-length Rsp5 joined to the glutathione S-transferase moiety. Monoclonal antibody 23H12 specific for the M protein of VSV was kindly provided by D. S. Lyles (Bowman-Gray School of Medicine, Winston-Salem, N.C.).
cDNA library screen.
A λExlox cDNA library (Novagen) from a 14-day-old mouse embryo was screened for proteins that interacted with amino acids (aa) 17 to 33 of the VSV M protein in accordance with the protocol of the supplier.
Proteasome inhibitors and VSV budding.
MG132, MG115, lactacystin, and epoxomycin were obtained from Calbiochem and suspended in dimethyl sulfoxide (DMSO) as indicated by the supplier. The suspended inhibitors were stored at −20°C and used within a 2-week period. BSR-T7 cells were infected with VSV at a multiplicity of infection (MOI) of 3.0 for 1 h at 37°C. The inoculum was removed, and the cells were washed with 1× phosphate-buffered saline. Normal medium was added to the cells, and the infection was allowed to proceed for 3 h at 37°C. DMSO alone or proteasome inhibitors at concentrations indicated in Results were added to the appropriate dishes, and the infection was allowed to proceed up to an additional 2.5 h. Supernatants were harvested and stored at −80°C. Serial dilutions of virus-containing supernatants were used to infect fresh monolayers of either BSR-T7 or BHK-21 cells in 35-mm-diameter dishes. Mock-infected and infected cells were overlaid with 1.0% methylcellulose and allowed to incubate up to 48 h at 37°C. VSV titers reported represent averages of six independent experiments. For the time course analysis, the initial MOI was 1.0 PFU/ml and 50 μM MG132 was added to the media at 5.5 h postinfection.
Proteasome inhibitors and RV budding.
BSR-T7 cells were plated in six-well plates and infected at an MOI of 10 with recombinant RV SBN (44). Sixteen hours postinfection, cells were washed with DMEM supplemented with 10% serum. Two milliliters of DMEM plus 10% serum containing 100 μM MG132 diluted in DMSO or DMSO alone was added to the monolayers for 2.5 h. Supernatants were collected, and infectious titers were determined in duplicate on BSR cells. For protein synthesis control studies, BSR cells were plated in 25 cm2 flasks and infected with recombinant RV SBN at an MOI of 10 for 16 h. Monolayers were washed in methionine-free media and then metabolically labeled for 2.5 h with 250 μCi of [35S]methionine in the presence of 100 μM MG132 or DMSO alone. Supernatants were collected, and virions were purified through 10% sucrose for 1 h at 20,000 rpm. The viral pellet was suspended in 50 μl of protein lysis buffer. Cells were washed in 1× phosphate-buffered saline and then lysed in lysis buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 1.0% Nonidet P-40, 0.1% sodium dodecyl sulfate [SDS], 1× protease inhibitor [Sigma]) on ice for 5 min. Proteins from supernatants and cell lysates were analyzed by SDS–10% polyacrylamide gel electrophoresis (PAGE) and visualized by a phosphorimager (Molecular Dynamics).
Immunoprecipitation analysis.
For immunoprecipitation analysis, 100 μCi of 35S-Express label (NEN DuPont) was added to each monolayer concomitant with the addition of the proteasome inhibitor. Cell extract and supernatant samples were immunoprecipitated with monoclonal antibody 23H12 against the VSV M protein and analyzed as described previously (13, 14). Immunoprecipitated proteins were visualized by autoradiography and quantitated using a phosphorimager (Bio-Rad; GS-525 molecular imager system). These experiments were repeated at least three times.
Far-Western blotting.
Plasmids GST-Rsp5, APVSVMWT, and APVSVMA4 were used in this assay. Far-Western blotting was performed as described previously (14).
Ubiquitination assay.
Plasmids pYESMWT and pYESMA4 were employed in this assay. The positive control for this assay was the 52-kDa yeast protein encoded by the YHL002w gene (G. Wang and J. Huibregtse, unpublished data). In vitro ubiquitination assays were performed as described previously (13, 18).
RESULTS
cDNA library screen with the PY motif of VSV M protein.
The PY motifs conserved in the M protein of VSV and RV, the VP40 protein of Ebola virus, and the Gag protein of RSV were shown to mediate interactions with WW domains of several cellular proteins (12–14). To extend these observations, a mouse cDNA library (λExlox) was screened using the PY motif and flanking sequences (aa 17 to 33) from VSV M as the probe. One of the cellular proteins isolated from this screen was identified by DNA sequencing analysis as the mouse Nedd4 gene, which contains three WW domains (GenBank accession no. D85414; Fig. 1). The cDNA insert contained aa 46 to 958 (Fig. 1). The WW domains from the mNedd4 protein were shown previously to interact strongly with the PY motifs of the M proteins of VSV and RV (14). These results lend further support to our hypothesis that the cellular Nedd4 ubiquitin ligase may be an important component of the budding machinery of VSV and RV.
FIG. 1.
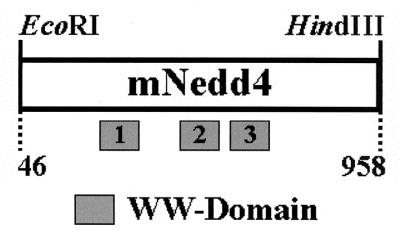
Schematic diagram of mNedd4 cDNA. Shaded boxes, presence and approximate locations of the three WW domains. Amino acids 46 to 958 are present within the cDNA insert.
Functional interaction between full-length VSV M and full-length Rsp5 ubiquitin ligase.
The Nedd4 ubiquitin ligase represents a realistic candidate for a cellular interactor that may facilitate the budding process of VSV for the following reasons. (i) Nedd4 was isolated from a cDNA library screen (above), and its WW domains interact strongly with viral PY motifs (14). (ii) Nedd4 is localized to the plasma membrane of the cell, where it functions in endocytosis and ubiquitination (3, 38, 42). (iii) Recent studies have implicated ubiquitin and ubiquitin ligases as being important in retrovirus and perhaps filovirus budding (13, 37, 45, 49, 53).
The functional homolog of Nedd4 in yeast is Rsp5, a membrane-associated ubiquitin ligase that contains multiple WW domains and that functions in endocytosis. We made use of an established in vitro ubiquitination assay to address whether full-length VSV M could be ubiquitinated by full-length Rsp5 (Fig. 2A). The pYESMWT and pYESMA4 plasmids were used to synthesize wild-type VSV M protein and a mutant M protein containing four alanines in place of PPPY in vitro, respectively. The radiolabeled wild-type M protein was added for the in vitro ubiquitination assays (performed in duplicate) in the presence (Fig. 2A, lanes 5 and 6) or absence of enzymatically active Rsp5 (Fig. 2A, lanes 3 and 4). In the presence of Rsp5, numerous higher-molecular-weight species of multiubiquitinated M protein were markedly evident, along with a concomitant decrease in unmodified VSV M (Fig. 2A, lanes 5 and 6). Ubiquitinated forms of wild-type VSV M were not observed in the absence of Rsp5 (lanes 3 and 4). In stark contrast to what was found for wild-type M, no ubiquitination of the VSV M A4 mutant was detected in the absence (Fig. 2A, lanes 7 and 8) or presence (lanes 9 and 10) of Rsp5. These results demonstrate that full-length VSV M (wild type) can interact both physically and functionally with full-length Rsp5 in vitro.
FIG. 2.

Physical and functional interactions between VSV M and Rsp5. (A) Full-length VSV M was transcribed and translated in vitro in the presence of [35S]methionoine and incubated with ATP, ubiquitin, E1 enzyme, and E2 protein (UBC8 from Arabidopsis thaliana).+, reactions with wild-type (WT) Rsp5 (lanes 2, 5, 6, 9, and 10); −, reactions without WT Rsp5 (lanes 1, 3, 4, 7, and 8). The positions of unmodified WT VSV M (lanes 3 to 6) and of the unmodified VSV A4 mutant (lanes 7 to 10) are indicated. Multiubiquitinated forms of WT VSV M [M-ub(n)] are shown (lanes 5 and 6). A yeast protein of approximately 52 kDa encoded by the YHL002w gene is shown as a positive control for ubiquitination by Rsp5 (lanes 1 and 2). (B) Far-Western binding assay. Two micrograms of GST-Rsp5 (full-length Rsp5) were immobilized onto nitrocellulose filters and probed with BAP-VSV M (WT) or BAP-VSV M (A4 mutant).
To further support the specificity of the physical interaction between VSV M and Rsp5, a far-Western assay was performed (Fig. 2B). Full-length Rsp5 joined to glutathione S-transferase was purified from Escherichia coli and immobilized onto nitrocellulose filters. Identical filters were probed with either full-length wild-type VSV M or the A4 mutant form of VSV M (Fig. 2B). As indicated, the wild-type M protein interacted with Rsp5, whereas the A4 mutant did not (Fig. 2B).
Proteasome inhibitors block release of VSV.
Since the VSV M protein was readily modified by ubiquitin in vitro, we wanted to determine whether alterations of free-ubiquitin levels in the cell would affect the release of progeny VSV. Inhibitors that block the function of the proteasome (e.g., MG132) result in a decrease in the level of free ubiquitin in the cytoplasm (31, 37, 45). BSR-T7 cells were infected with VSV, and, at 4 h postinfection, either DMSO alone or 50 μM MG132 solubilized in DMSO was added to the cells for 2.5 h. Supernatants containing newly released virions were collected and assayed for infectious virus by plaque titration (Fig. 3A). The titers of VSV illustrated represent the averages of six independent experiments (Fig. 3A). In the absence of MG132, newly released VSV achieved an average titer of 2.2 × 108 PFU/ml, while, in the presence of 50 μM MG132, newly released VSV achieved an average titer of 2.0 × 107 PFU/ml (Fig. 3A). This decrease in titer of slightly more than 1 log unit was highly reproducible, and a similar decrease in titer was observed in the presence of other proteasome inhibitors (e.g., MG115, lactacystin, and epoxomycin; data not shown).
FIG. 3.

Inhibition of VSV release by MG132. (A) Titers of VSV present in the supernatant of cells infected in the absence (−) or presence (+) of MG132 (50 μM). VSV titers and standard deviations represent averages of six independent experiments. (B) Time course analysis of VSV replication. Viral titers were determined from supernatant samples harvested at the indicated time points from cells infected in the presence or absence of MG132 (50 μM). MG132 was added to VSV-infected cells at 5.5 h postinfection (arrowhead). (C) Immunoprecipitation of radiolabeled VSV M protein from cell extracts (cells, lanes 1 and 2) and supernatants (media, lanes 1 and 2) of cells infected in the absence (−) or presence (+) of MG132 (50 μM).
VSV release from cells incubated in the presence or absence of MG132 was measured during a time course experiment (Fig. 3B). MG132 (50 μM) or DMSO was added to the media 5.5 h postinfection (Fig. 3B). Release of VSV was inhibited in a time-dependent manner immediately following the addition of MG132 (Fig. 3B), compared to that of VSV in the presence of DMSO alone (Fig. 3B). The difference in VSV titers at 9 h postinfection was approximately 10-fold (5.0 × 107 versus 5.6 × 106 PFU/ml), consistent with results shown in Fig. 3A.
VSV-infected cells were incubated with proteasome inhibitors for no more than 2.5 h since prolonged exposure to these inhibitors can lead to a complete shutdown of protein synthesis. To demonstrate that total protein synthesis in the above experiments was not adversely affected by MG132, radiolabeled viral proteins from infected-cell extracts and supernatants were subjected to immunoprecipitation (Fig. 3C). Equivalent amounts of cell extracts infected with VSV in the absence (Fig. 3C, lane 1 cells) or presence (lane 2 cells) of MG132 were immunoprecipitated with monoclonal antibody 23H12 against the VSV M protein. Virtually identical amounts (<2.0-fold difference) of M protein were present in cells treated with either DMSO or MG132 (Fig. 3C). In contrast, the amount of M protein detected in the supernatant (Fig. 3C, lane 2 media) of MG132-treated cells was reduced by approximately 12-fold compared to that detected in the supernatant of DMSO-treated cells (Fig. 3C, lane 1 media). These data demonstrate that, under these experimental conditions, MG132 did not affect protein synthesis but rather specifically inhibited the release of progeny virions as determined by both virus titration and immunoprecipitation.
Proteasome inhibitors block release of RV.
Since the PY motif is conserved in the M protein of RV and since the RV PY motif also mediates interactions with cellular proteins, we reasoned that release of RV would also be affected by proteasome inhibitors. RV was used to infect monolayers of BSR cells, and MG132 or DMSO was added at 16 h postinfection since the kinetics of RV replication and release are slower that those of VSV. As with VSV, RV-infected cells were incubated with MG132 for no more than 2.5 h. The average RV titers from three independent experiments performed in duplicate were 1.2 × 107 PFU/ml in the presence of DMSO alone (Fig. 4B) and 7.0 × 105 PFU/ml in the presence of 100 μM MG132 (Fig. 4B). This 16-fold reduction in virus titer is similar to those observed for VSV in the presence of 50 μM MG132 (10-fold; shown above) and 100 μM MG132 (>20-fold; data not shown). To ensure that the observed decrease in RV titer was not due to general inhibition of protein synthesis by MG132, radiolabeled proteins from both RV-infected cell extracts and supernatants were examined by SDS-PAGE (Fig. 4A). In two independent experiments, total protein synthesis in cell lysates treated with DMSO alone was shown to be identical to that in cells treated with 100 μM MG132 (Fig. 4A). In contrast, radiolabeled RV proteins (G, N, and M) present in the supernatants from cells treated with DMSO alone were readily detected following SDS-PAGE, whereas those present in supernatants from cells treated with MG132 were not detected in this assay (Fig. 4A). These results are highly consistent with those described above for VSV. These results indicate that inhibitors of the cellular proteasome machinery impair the budding efficiency of VSV and RV.
FIG. 4.

Inhibition of RV release by MG132. (A) Protein gel depicting radiolabeled proteins from both supernatants and cell lysates. Protein patterns from cells infected in the absence (−) or presence (+) of MG132 (100 μM) are shown in duplicate. The G, N, and M proteins of RV are readily visible in supernatant samples from cells infected in the absence of MG132 but are not readily visible in supernatant samples from cells infected in the presence of MG132. (B) Titers of RV present in the supernatants of cells infected in the absence (−) or presence (+) of MG132 (100 μM). RV titers and standard deviations represent averages of three independent experiments.
Release of a VSV PY mutant is not blocked by MG132.
It has been postulated that inhibition of human immunodeficiency virus type 1 (HIV-1) budding by proteasome inhibitors is dependent on the presence of both the functional L domain (PTAPP) within the p6 region of Gag and the protease (PR) function of HIV-1 Gag (45). To determine whether inhibition of VSV budding by proteasome inhibitors is linked to the presence of the PY motif within the VSV M protein, we employed a VSV PY mutant. Recovery of a budding-defective VSV PY mutant containing the sequence AAPA (AAPA virus) in place of PPPY was reported recently (20). We hypothesized that the AAPA virus may be insensitive to the presence of proteasome inhibitors for the following reason. Since the AAPA mutant would likely be impaired in its ability to interact with cellular WW domains of ubiquitin ligases (e.g., Nedd4), it would not be ubiquitinated efficiently. Therefore, the reduction of free ubiquitin in infected cells due to MG132 should have no effect on release of the AAPA virus. To test this hypothesis, the AAPA virus was used to infect monolayers of BHK-21 cells in the presence of DMSO alone or 50 μM MG132 as described above for wild-type VSV. The average titer of AAPA virus from the supernatants of cells infected in the presence of DMSO alone was 3.2 × 106 PFU/ml, while the average titer of AAPA virus from supernatants of cells infected in the presence of MG132 was 2.2 × 106 PFU/ml (Fig. 5A). Furthermore, unlike what was found for wild-type VSV, the amounts of M protein present in both cell extracts and supernatant samples either with or without MG132 were virtually identical (Fig. 5B). Together, these results suggest that inhibition of VSV release by MG132 is dependent on the presence of a functional PY motif within the M protein.
FIG. 5.

Release of a VSV PY mutant is not blocked by MG132. (A) Titers of the AAPA mutant virus present in the supernatant of cells infected in the absence (−) or presence (+) of MG132 (50 μM). Virus titers and standard deviations represent averages of three independent experiments. (B) Immunoprecipitation of radiolabeled VSV M protein from cell extracts (cells, lanes 2 and 3) and media (media, lanes 2 and 3) of cells infected in the absence (−) or presence (+) of MG132 (50 μM). Mock-infected samples are shown in lane 1 in both sections.
DISCUSSION
The L domains of retroviruses, rhabdoviruses, and likely those of filoviruses are important for efficient budding and separation of progeny virions from infected cells. While their role in budding is evident, the mechanism by which these L domains accomplish efficient virus-host separation remains unclear. Initial implications of the possible involvement of host proteins in virus budding were revealed by findings that viral L domains could mediate interactions with cellular proteins (12–14, 39). Recently, a family of cellular proteins known as E3 ubiquitin ligases have been implicated in the budding of retroviruses (34, 35, 37, 45, 49), filoviruses (13), and now rhabdoviruses (this report). One family member, Nedd4/Rsp5, is localized at the plasma membrane, plays a role in ubiquitination and endocytosis, and possesses multiple type I WW domains (3, 8, 9, 38, 42, 54, 55). Although we had demonstrated previously that the L domains of VSV and RV could mediate interactions with WW domains 2 and 3 of Nedd4/Rsp5 (14), results described in this report have augmented the potential significance of this virus-host interaction. For example, the mouse Nedd4 gene was isolated from a cDNA library screen using the L domain of VSV as the bait, and the full-length VSV M protein interacted both physically and functionally with full-length Rsp5 in an in vitro ubiquitination assay. The wild-type VSV M protein was readily modified by the addition of ubiquitin; however, the precise location(s) of ubiquitin addition within the M protein has yet to be identified.
If ubiquitin or ubiquitin modification of the M protein were indeed important for virus budding, then decreasing the level of free ubiquitin in the cell may adversely affect virus budding. Indeed, both VSV and RV release from infected cells was inhibited by 10- to 20-fold in the presence of proteasome inhibitors, compared to that in the absence of proteasome inhibitors. Control experiments clearly demonstrated that inhibition of virus release was not simply due to a global effect of the proteasome inhibitors on protein synthesis. In general, these results are consistent with earlier findings that efficient release of retroviruses is inhibited in the presence of proteasome inhibitors (37, 45, 49); however, the extent of inhibition of rhabdovirus release (10- to 20-fold) was consistently greater than that observed for retrovirus release (3- to 4-fold) (37, 45).
Results from experiments utilizing a VSV PY mutant demonstrated that the effect of proteasome inhibitors on virus release is associated with the presence of a functional L domain. Indeed, release of the VSV AAPA mutant was not significantly blocked in the presence of MG132. These results are consistent with findings reported previously for retrovirus budding in the presence of proteasome inhibitors (45). Indeed, Schubert et al. demonstrated that inhibition of HIV-1 budding by proteasome inhibitors was dependent on both a functional L domain and a functional protease (45).
The fact that PY mutant forms of VSV (e.g., the AAPA mutant) can still bud from cells, albeit at significantly lower levels than wild-type VSV, indicates that additional viral sequences are important for the budding process. One possibility is that there may be redundancy in the mechanism of virus budding at both the virus and cellular levels. We have postulated previously that additional motifs that play a role in budding are likely present within the M protein of VSV. For example, a PSAP motif, similar to the PTAPP motif within the Gag protein of HIV-1, is present just downstream of the PPPY motif in the VSV M protein. Similarly, the VP40 protein of Ebola virus contains overlapping PPxY and PTAPP motifs (13, 49). Although the M protein contains sufficient information to bud from cells independently of other viral proteins, it is certain that the G glycoprotein plays an important role in VSV budding as well (30, 41, 48, 56). The functional interactions between M and G proteins of VSV remain of great interest, and deciphering these interactions will enhance our understanding of the molecular aspects of rhabdovirus budding.
Many questions regarding the precise roles of ubiquitin and L domains in virus budding remain to be answered. For example, whether free ubiquitin or ubiquitin-modified forms of M protein exist in VSV virions and in VSV-infected cells remains uncertain. Interestingly, ubiquitination of the RSV Gag protein has not been detected thus far in virions; however, free ubiquitin has been detected in both RSV and avian leukosis virus (40). Moreover, both free ubiquitin and ubiquitinated Gag molecules have been detected in HIV-1 virions (32, 34, 35, 49). Ubiquitin modification of these viral matrix proteins may function to (i) recruit additional cellular proteins (e.g., proteins involved in endocytosis and/or exocytosis) required to facilitate virus budding or (ii) target virus assembly and budding to specialized regions (e.g., lipid rafts) on the plasma membrane that are active in vesicularization.
The fact that these three diverse families of RNA viruses may utilize a common approach to bud from cells is intriguing. Moreover, the ability to potentially inhibit release of these RNA viruses by targeting this late stage of budding remains an attractive concept. Further analysis of these potential virus-host interactions in vivo is necessary and will serve to expedite our understanding of the role of the ubiquitin-proteasome machinery in virus budding.
ACKNOWLEDGMENTS
We thank D. S. Lyles, J. Paragas, N. T. Wright, and K.-K. Conzelmann for reagents and/or comments.
This work was supported in part by a Formula Fund Award from the USDA to R.N.H. and NIH grant R01 GM-53726 to M.A.W.
REFERENCES
- 1.Accola M A, Strack B, Gottlinger H G. Efficient particle production by minimal Gag constructs which retain the carboxy-terminal domain of human immunodeficiency virus type 1 capsid-p2 and a late assembly domain. J Virol. 2000;74:5395–5402. doi: 10.1128/jvi.74.12.5395-5402.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmed M, Lyles D S. Effect of vesicular stomatitis virus matrix protein on transcription directed by host RNA polymerases I, II, and III. J Virol. 1998;72:8413–8419. doi: 10.1128/jvi.72.10.8413-8419.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anan T, Nagata Y, Koga H, Honda Y, Yabuki N, Miyamoto C, Kuwano A, Matsuda I, Endo F, Saya H, Nakao M. Human ubiquitin-protein ligase Nedd4: expression, subcellular localization and selective interaction with ubiquitin-conjugating enzymes. Genes Cells. 1998;3:751–763. doi: 10.1046/j.1365-2443.1998.00227.x. [DOI] [PubMed] [Google Scholar]
- 4.Bonifacino J S, Weissman A M. Ubiquitin and the control of protein fate in the secretory and endocytic pathways. Annu Rev Cell Dev Biol. 1998;14:19–57. doi: 10.1146/annurev.cellbio.14.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carthew R W, Xu C. Endocytosis: why not wait to deubiquitinate? Curr Biol. 2000;10:R532–R534. doi: 10.1016/s0960-9822(00)00587-x. [DOI] [PubMed] [Google Scholar]
- 6.Chong L D, Rose J K. Membrane association of functional vesicular stomatitis virus matrix protein in vivo. J Virol. 1993;67:407–414. doi: 10.1128/jvi.67.1.407-414.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Craven R C, Harty R N, Paragas J, Palese P, Wills J W. Late domain function identified in the vesicular stomatitis virus M protein by use of rhabdovirus-retrovirus chimeras. J Virol. 1999;73:3359–3365. doi: 10.1128/jvi.73.4.3359-3365.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dunn R, Hicke L. Domains of the rsp5 ubiquitin-protein ligase required for receptor-mediated and fluid-phase endocytosis. Mol Biol Cell. 2001;12:421–435. doi: 10.1091/mbc.12.2.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunn R, Hicke L. Multiple roles for Rsp5p-dependent ubiquitination at the internalization step of endocytosis. J Biol Chem. 2001;276:25974–25981. doi: 10.1074/jbc.M104113200. [DOI] [PubMed] [Google Scholar]
- 10.Flood E A, Lyles D S. Assembly of nucleocapsids with cytosolic and membrane-derived matrix proteins of vesicular stomatitis virus. Virology. 1999;261:295–308. doi: 10.1006/viro.1999.9856. [DOI] [PubMed] [Google Scholar]
- 11.Gajewska B, Kaminska J, Jesionowska A, Martin N C, Hopper A K, Zoladek T. WW domains of Rsp5p define different functions: determination of roles in fluid phase and uracil permease endocytosis in Saccharomyces cerevisiae. Genetics. 2001;157:91–101. doi: 10.1093/genetics/157.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garnier L, Wills J W, Verderame M F, Sudol M. WW domains and retrovirus budding. Nature. 1996;381:744–745. doi: 10.1038/381744a0. [DOI] [PubMed] [Google Scholar]
- 13.Harty R N, Brown M E, Wang G, Huibregtse J, Hayes F P. A PPxY motif within the VP40 protein of Ebola virus interacts physically and functionally with a ubiquitin ligase: implications for filovirus budding. Proc Natl Acad Sci USA. 2000;97:13871–13876. doi: 10.1073/pnas.250277297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harty R N, Paragas J, Sudol M, Palese P. A proline-rich motif within the matrix protein of vesicular stomatitis virus and rabies virus interacts with WW domains of cellular proteins: implications for viral budding. J Virol. 1999;73:2921–2929. doi: 10.1128/jvi.73.4.2921-2929.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harvey K F, Kumar S. Nedd4-like proteins: an emerging family of ubiquitin-protein ligases implicated in diverse cellular functions. Trends Cell Biol. 1999;9:166–169. doi: 10.1016/s0962-8924(99)01541-x. [DOI] [PubMed] [Google Scholar]
- 16.Hicke L. Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol. 2001;2:195–201. doi: 10.1038/35056583. [DOI] [PubMed] [Google Scholar]
- 17.Huibregtse J M, Scheffner M, Beaudenon S, Howley P M. A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc Natl Acad Sci USA. 1995;92:5249. doi: 10.1073/pnas.92.11.5249-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huibregtse J M, Yang J C, Beaudenon S L. The large subunit of RNA polymerase II is a substrate of the Rsp5 ubiquitin-protein ligase. Proc Natl Acad Sci USA. 1997;94:3656–3661. doi: 10.1073/pnas.94.8.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jasenosky L D, Neumann G, Lukashevich I, Kawaoka Y. Ebola virus vp40-induced particle formation and association with the lipid bilayer. J Virol. 2001;75:5205–5214. doi: 10.1128/JVI.75.11.5205-5214.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jayakar H R, Murti K G, Whitt M A. Mutations in the PPPY motif of vesicular stomatitis virus matrix protein reduce virus budding by inhibiting a late step in virion release. J Virol. 2000;74:9818–9827. doi: 10.1128/jvi.74.21.9818-9827.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Justice P A, Sun W, Li Y, Ye Z, Grigera P R, Wagner R R. Membrane vesiculation function and exocytosis of wild-type and mutant matrix proteins of vesicular stomatitis virus. J Virol. 1995;69:3156–3160. doi: 10.1128/jvi.69.5.3156-3160.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanelis V, Farrow N A, Kay L E, Rotin D, Forman-Kay J D. NMR studies of tandem WW domains of Nedd4 in complex with a PY motif-containing region of the epithelial sodium channel. Biochem Cell Biol. 1998;76:341–350. doi: 10.1139/bcb-76-2-3-341. [DOI] [PubMed] [Google Scholar]
- 23.Kaptur P E, McKenzie M O, Wertz G W, Lyles D S. Assembly functions of vesicular stomatitis virus matrix protein are not disrupted by mutations at major sites of phosphorylation. Virology. 1995;206:894–903. doi: 10.1006/viro.1995.1012. [DOI] [PubMed] [Google Scholar]
- 24.Kay B K, Williamson M P, Sudol M. The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 2000;14:231–241. [PubMed] [Google Scholar]
- 25.Kumar S, Harvey K F, Kinoshita M, Copeland N G, Noda M, Jenkins N A. cDNA cloning, expression analysis, and mapping of the mouse Nedd4 gene. Genomics. 1997;40:435–443. doi: 10.1006/geno.1996.4582. [DOI] [PubMed] [Google Scholar]
- 26.Lafont F, Simons K. Raft-partitioning of the ubiquitin ligases Cbl and Nedd4 upon IgE-triggered cell signaling. Proc Natl Acad Sci USA. 2001;98:3180–3184. doi: 10.1073/pnas.051003498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, Luo L, Schubert M, Wagner R R, Kang C Y. Viral liposomes released from insect cells infected with recombinant baculovirus expressing the matrix protein of vesicular stomatitis virus. J Virol. 1993;67:4415–4420. doi: 10.1128/jvi.67.7.4415-4420.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Longnecker R, Merchant M, Brown M E, Fruehling S, Bickford J O, Ikeda M, Harty R N. WW- and SH3-domain interactions with Epstein-Barr virus LMP2A. Exp Cell Res. 2000;257:332–340. doi: 10.1006/excr.2000.4900. [DOI] [PubMed] [Google Scholar]
- 29.Lyles D S, McKenzie M O. Activity of vesicular stomatitis virus M protein mutants in cell rounding is correlated with the ability to inhibit host gene expression and is not correlated with virus assembly function. Virology. 1997;229:77–89. doi: 10.1006/viro.1996.8415. [DOI] [PubMed] [Google Scholar]
- 30.Mebatsion T, Weiland F, Conzelmann K K. Matrix protein of rabies virus is responsible for the assembly and budding of bullet-shaped particles and interacts with the transmembrane spike glycoprotein G. J Virol. 1999;73:242–250. doi: 10.1128/jvi.73.1.242-250.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mimnaugh E G, Chen H Y, Davie J R, Celis J E, Neckers L. Rapid deubiquitination of nucleosomal histones in human tumor cells caused by proteasome inhibitors and stress response inducers: effects on replication, transcription, translation, and the cellular stress response. Biochemistry. 1997;36:14418–14429. doi: 10.1021/bi970998j. [DOI] [PubMed] [Google Scholar]
- 32.Ott D E. Cellular proteins in HIV virions. Rev Med Virol. 1997;7:167–180. doi: 10.1002/(sici)1099-1654(199709)7:3<167::aid-rmv199>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 33.Ott D E, Chertova E N, Busch L K, Coren L V, Gagliardi T D, Johnson D G. Mutational analysis of the hydrophobic tail of the human immunodeficiency virus type 1 p6(Gag) protein produces a mutant that fails to package its envelope protein. J Virol. 1999;73:19–28. doi: 10.1128/jvi.73.1.19-28.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ott D E, Coren L V, Chertova E N, Gagliardi T D, Schubert U. Ubiquitination of HIV-1 and MuLV Gag. Virology. 2000;278:111–121. doi: 10.1006/viro.2000.0648. [DOI] [PubMed] [Google Scholar]
- 35.Ott D E, Coren L V, Copeland T D, Kane B P, Johnson D G, Sowder II R C, Yoshinaka Y, Oroszlan S, Arthur L O, Henderson L E. Ubiquitin is covalently attached to the p6Gag proteins of human immunodeficiency virus type 1 and simian immunodeficiency virus and to the p12Gag protein of Moloney murine leukemia virus. J Virol. 1998;72:2962–2968. doi: 10.1128/jvi.72.4.2962-2968.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parent L J, Bennett R P, Craven R C, Nelle T D, Krishna N K, Bowzard J B, Wilson C B, Puffer B A, Montelaro R C, Wills J W. Positionally independent and exchangeable late budding functions of the Rous sarcoma virus and human immunodeficiency virus Gag proteins. J Virol. 1995;69:5455–5460. doi: 10.1128/jvi.69.9.5455-5460.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patnaik A, Chau V, Wills J W. Ubiquitin is part of the retrovirus budding machinery. Proc Natl Acad Sci USA. 2000;97:13069–13074. doi: 10.1073/pnas.97.24.13069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Plant P J, Yeger H, Staub O, Howard P, Rotin D. The C2 domain of the ubiquitin protein ligase Nedd4 mediates Ca2+-dependent plasma membrane localization. J Biol Chem. 1997;272:32329–32336. doi: 10.1074/jbc.272.51.32329. [DOI] [PubMed] [Google Scholar]
- 39.Puffer B A, Parent L J, Wills J W, Montelaro R C. Equine infectious anemia virus utilizes a YXXL motif within the late assembly domain of the Gag p9 protein. J Virol. 1997;71:6541–6546. doi: 10.1128/jvi.71.9.6541-6546.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Putterman D, Pepinsky R B, Vogt V M. Ubiquitin in avian leukosis virus particles. Virology. 1990;176:633–637. doi: 10.1016/0042-6822(90)90035-p. [DOI] [PubMed] [Google Scholar]
- 41.Robison C S, Whitt M A. The membrane-proximal stem region of vesicular stomatitis virus G protein confers efficient virus assembly. J Virol. 2000;74:2239–2246. doi: 10.1128/jvi.74.5.2239-2246.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rotin D, Staub O, Haguenauer-Tsapis R. Ubiquitination and endocytosis of plasma membrane proteins: role of Nedd4/Rsp5p family of ubiquitin-protein ligases. J Membr Biol. 2000;176:1–17. doi: 10.1007/s00232001079. [DOI] [PubMed] [Google Scholar]
- 43.Schnell M J, Buonocore L, Boritz E, Ghosh H P, Chernish R, Rose J K. Requirement for a non-specific glycoprotein cytoplasmic domain sequence to drive efficient budding of vesicular stomatitis virus. EMBO J. 1998;17:1289–1296. doi: 10.1093/emboj/17.5.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schnell M J, Foley H D, Siler C A, McGettigan J P, Dietzschold B, Pomerantz R J. Recombinant rabies virus as potential live-viral vaccines for HIV-1. Proc Natl Acad Sci USA. 2000;97:3544–3549. doi: 10.1073/pnas.050589197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schubert U, Ott D E, Chertova E N, Welker R, Tessmer U, Princiotta M F, Bennink J R, Krausslich H G, Yewdell J W. Proteasome inhibition interferes with gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. Proc Natl Acad Sci USA. 2000;97:13057–13062. doi: 10.1073/pnas.97.24.13057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Staub O, Abriel H, Plant P, Ishikawa T, Kanelis V, Saleki R, Horisberger J D, Schild L, Rotin D. Regulation of the epithelial Na+ channel by Nedd4 and ubiquitination. Kidney Int. 2000;57:809–815. doi: 10.1046/j.1523-1755.2000.00919.x. [DOI] [PubMed] [Google Scholar]
- 47.Staub O, Dho S, Henry P, Correa J, Ishikawa T, McGlade J, Rotin D. WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle's syndrome. EMBO J. 1996;15:2371–2380. [PMC free article] [PubMed] [Google Scholar]
- 48.Stillman E A, Rose J K, Whitt M A. Replication and amplification of novel vesicular stomatitis virus minigenomes encoding viral structural proteins. J Virol. 1995;69:2946–2953. doi: 10.1128/jvi.69.5.2946-2953.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Strack B, Calistri A, Accola M A, Palu G, Gottlinger H G. A role for ubiquitin ligase recruitment in retrovirus release. Proc Natl Acad Sci USA. 2000;97:13063–13068. doi: 10.1073/pnas.97.24.13063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sudol M. Structure and function of the WW domain. Prog Biophys Mol Biol. 1996;65:113–132. doi: 10.1016/s0079-6107(96)00008-9. [DOI] [PubMed] [Google Scholar]
- 51.Sudol M, Hunter T. NeW wrinkles for an old domain. Cell. 2000;103:1001–1004. doi: 10.1016/s0092-8674(00)00203-8. [DOI] [PubMed] [Google Scholar]
- 52.Timmins J, Scianimanico S, Schoehn G, Weissenhorn W. Vesicular release of ebola virus matrix protein VP40. Virology. 2001;283:1–6. doi: 10.1006/viro.2001.0860. [DOI] [PubMed] [Google Scholar]
- 53.Vogt V M. Ubiquitin in retrovirus assembly: actor or bystander? Proc Natl Acad Sci USA. 2000;97:12945–12947. doi: 10.1073/pnas.97.24.12945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang G, McCaffery J M, Wendland B, Dupre S, Haguenauer-Tsapis R, Huibregtse J M. Localization of the Rsp5p ubiquitin-protein ligase at multiple sites within the endocytic pathway. Mol Cell Biol. 2001;21:3564–3575. doi: 10.1128/MCB.21.10.3564-3575.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang G, Yang J, Huibregtse J M. Functional domains of the Rsp5 ubiquitin-protein ligase. Mol Cell Biol. 1999;19:342–352. doi: 10.1128/mcb.19.1.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Whitt M A, Chong L, Rose J K. Glycoprotein cytoplasmic domain sequences required for rescue of a vesicular stomatitis virus glycoprotein mutant. J Virol. 1989;63:3569–3578. doi: 10.1128/jvi.63.9.3569-3578.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wills J W, Cameron C E, Wilson C B, Xiang Y, Bennett R P, Leis J. An assembly domain of the Rous sarcoma virus Gag protein required late in budding. J Virol. 1994;68:6605–6618. doi: 10.1128/jvi.68.10.6605-6618.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Winberg G, Matskova L, Chen F, Plant P, Rotin D, Gish G, Ingham R, Ernberg I, Pawson T. Latent membrane protein 2A of Epstein-Barr virus binds WW domain E3 protein-ubiquitin ligases that ubiquitinate B-cell tyrosine kinases. Mol Cell Biol. 2000;20:8526–8535. doi: 10.1128/mcb.20.22.8526-8535.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xiang Y, Cameron C E, Wills J W, Leis J. Fine mapping and characterization of the Rous sarcoma virus Pr76gag late assembly domain. J Virol. 1996;70:5695–5700. doi: 10.1128/jvi.70.8.5695-5700.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yasuda J, Hunter E. A proline-rich motif (PPPY) in the Gag polyprotein of Mason-Pfizer monkey virus plays a maturation-independent role in virion release. J Virol. 1998;72:4095–4103. doi: 10.1128/jvi.72.5.4095-4103.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]