

Abstract
Human microbiota communicates with its host by secreting signaling metabolites, enzymes, or structural components. Its homeostasis strongly influences the modulation of human tissue barriers and immune system. Dysbiosis‐induced peripheral immunity response can propagate bacterial and pro‐inflammatory signals to the whole body, including the brain. This immune‐mediated communication may contribute to several neurodegenerative disorders, as Alzheimer's disease. In fact, neurodegeneration is associated with dysbiosis and neuroinflammation. The interplay between the microbial communities and the brain is complex and bidirectional, and a great deal of interest is emerging to define the exact mechanisms. This review focuses on microbiota‐immunity‐central nervous system (CNS) communication and shows how gut and oral microbiota populations trigger immune cells, propagating inflammation from the periphery to the cerebral parenchyma, thus contributing to the onset and progression of neurodegeneration. Moreover, an overview of the technological challenges with in vitro modeling of the microbiota‐immunity‐CNS axis, offering interesting technological hints about the most advanced solutions and current technologies is provided.
Keywords: blood‐brain barrier, immune system, microbiota‐gut‐brain axis, neurodegenerative disorders, organ‐on‐a‐chip
Microbiota‐mediated immunity seems to have a determinant role in neurodegeneration onset and progression. Here, recent evidence and key players of this connection are summarized. The authors' overview brings out the most critical issues for in vitro modeling of multisystem interfaces and communication routes along the microbiota‐immune system‐central nervous system axis.
1. Introduction
More than 100 trillion bacteria inhabit different districts of the human body: gut, mouth, skin, respiratory and urogenital tracts.[ 1 ] These microorganisms as a whole are called microbiota. Bacterial composition and abundance are critical for the environmental homeostasis and an imbalance (dysbiosis) of the microbiota equilibrium has a determinant role in human health.[ 2 , 3 , 4 , 5 , 6 ] Depending on the tissue or organ, dysbiosis may trigger different pathological mechanisms. Despite the interaction between microbes and immunity remains poorly understood at some sites (e.g., lung and vaginal mucosa), recent experimental evidence is consistent with the notion that the health of microbiota communities is strictly related to host immune homeostasis.[ 7 , 8 , 9 ] Indeed, the severity and impact of immune cells activity is dependent on the features of tissue‐associated bacterial communities and related dysbiosis.[ 9 ]
Cancer, autoimmune diseases and neurological disorders are the main pathologies related to microbiota‐mediated peripheral immunity and provide clear examples of the wide spectrum of human tissues involved and the extensive range of immune signal transmission.[ 10 , 11 , 12 ]
In the last decade neurodegenerative disorders have been widely studied in relation to gut and oral microbiota. A literature overview of the topic showed a significant trend in changes on bacterial abundance.[ 13 ] For instance, the microbiota of patients with Parkinson's disease (PD) had a general reduction of Firmicutes genera (e.g., Faecalibacterium, Clostridium, and Blautia), and an increase in Bacteroidetes (Bacteroides and Prevotella). Similarly, patients with Alzheimer's disease (AD) showed changes in relative abundance, with a reduction of several Firmicutes genera (e.g., Clostridium and Enterococcus) and an increase in the Bacteroidetes phylum (e.g., Bacteroides).[ 13 ] In addition to relative abundance change of commensal microbiota, cerebral infection of pathogens had a key role in the process of neurodegeneration. Porphyromonas gingivalis and Helicobacter pylori are examples of infecting bacteria associated with AD and PD, respectively.[ 14 , 15 ]
Recent research has demonstrated that microbiota and its induced immunity are correlated with neurodegenerative pathogenesis and progression.[ 13 ] Throughout the body, microorganisms and secreted molecules widely interact with local immune cells and structures to maintain microenvironment homeostasis and communicate with the distal neuroimmune system.[ 16 ] For instance, gut microbiota is able to stimulate CNS‐resident immune cells through both peripheral immunity and neurotransmission.[ 17 ] Indeed, bacteria can directly release (or stimulate gut‐resident cells to produce) signaling mediators—such as short‐chain fatty acids, lipopolysaccharide, serotonin—relevant for peripheral immune cells or the vagal nerve central transmission.[ 18 ] Despite the increasing evidence of the immunity importance for the microbiota‐brain axis, to what extent the microbiota‐immunity communication contributes in triggering neurodegeneration is still a matter of debate.[ 19 ] Starting from this state‐of‐the‐art, here we discuss the bioengineering challenges of reproducing in vitro the wide immune signaling profile that mediates microbiota‐brain communication, at the basis of neurodegeneration onset and progression. We start with a focus on the peripheral immune response triggered by oral and gut microorganisms, to move on to the immune signal transmission to the central nervous system (CNS) populations and the link between CNS immune activity and neurodegenerative disorders. We finally offer a technological analysis and discussion of advanced in vitro models for the study of the neurological impact of microbiota‐mediated immune responses (Figure 1 ).
Figure 1.
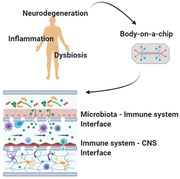
The microbiota‐immunity‐CNS communication. This graphic representation shows the bioengineering approaches used to reproduce the main in vivo immune‐stimulated interfaces during neuroinflammation and neurodegeneration. The most advanced approach used to recapitulate each interface is the organ‐on‐a‐chip technology (OOC) based on microfluidic multi‐cell culture. (Image created with BioRender.com).
2. Microbiota‐Peripheral Immunity Communication
The physiological interaction between microbiota and the immune system is a beneficial two‐way communication and it is the basis of the homeostasis of both systems. Through the release of specific metabolites, bacteria can protect the host from infection and promote body barrier integrity and immune cell maturation.[ 20 ] The microbiota interacts with the host by physical interaction or molecule‐mediated stimulation. Bacteria physically interact with host cells and tissues mostly when a pathological condition leads to impairment of the mucosal barriers and allows bacterial infection.[ 21 ] Otherwise, bacterial molecule‐mediated stimulation constantly contributes to barriers and immune system homeostasis.[ 22 ]
Bacterial molecules able to regulate or influence host mechanisms can be mainly distinguished as: 1) structural components such as lipopolysaccharide (LPS) and flagellin;[ 23 ] 2) functional enzymes such as the P. gingivalis peptidylarginine deiminase (PPAD);[ 24 ] 3) products of bacteria metabolism, like short chain fatty acids (SCFA) and indole‐3‐aldehyde (aryl‐hydrocarbon receptor ligand).[ 7 ] Most of these have an immunomodulatory effect that may lead to immune tolerance or increasing inflammation.[ 25 ]
Depending on the human body tissue, we find different microbiota composition, hence specific bacterial molecules, and metabolites. The principal components—enzymes and metabolites—can be classified according to the microbiota‐associated body tract, the immunomodulatory effect, and the involved host protein mediator.
To better depict the first steps of the microbiota‐mediated immune response that lead to neuroinflammation and neurodegeneration, we now focus on gut and oral microbiota, which are the two bacterial communities mostly linked to neurodegenerative disorders (Table 1 ).
Table 1.
Summary of bacterial molecules and metabolites with immunomodulatory properties. Molecules are classified according to the microbiota‐associated body tract, the related host interacting mediator and the immunomodulatory effects
| Molecules | Mediator | Immunomodulatory effects | Ref. | |
|---|---|---|---|---|
| Gut | PSA | TLR2, TLR4 | Promotion of T cell development and homeostasis; CD4+ T cell‐mediated IL‐10 secretion; immune tolerance increase; | [ 30 , 31 , 32 ] |
| Peptidoglican | Nod1‐2 | Pro‐inflammatory cytokine secretion | [ 176 ] | |
| Flagellin | TLR5 | IL‐22 production by DCs | [ 177 ] | |
| Formyl peptides | FPR | Leukocyte recruitment; pro‐inflammatory cytokine production | [ 178 ] | |
| HBP | TIFA | NF‐κB‐mediated innate inflammation | [ 29 ] | |
| Nucleic acids | TLR3‐7‐8‐9 | NF‐κB‐mediated production of pro‐inflammatory cytokines and chemokines | [ 179 ] | |
| SCFA | GPCRs | GPCR activation; anti‐inflammatory effect by macrophage HDAC inhibition; autophagy regulation; stimulation of serotonin production; promotion of B cell maturation; | [ 19 ] | |
| AhR ligand | AhR | IL‐22 secretion by ILC3 and Th17 cells; AMPs production; T cell differentiation | [ 25 , 180 , 181 , 182 ] | |
| Sphingolipid | CD1b | Stimulation of iNKT cell activity | [ 25 ] | |
| Polyamine | Ion channels | Inhibition of pro‐inflammatory cytokine production; CD8+ and CD4+ T cell maturation | [ 183 ] | |
| Histamine | HR1‐4 | Regulation of cytokine production; regulation of immune cell maturation and activity (e.g., Treg cells, Th1 cells, iNKT cells) | [ 35 ] | |
| Mouth | PPAD | Native arginine residues | Immune response evasion by citrullination of host peptides (e.g., C5a complement component) | [ 24 ] |
| LPS | TLR2‐4 | Co‐activation of TLRs‐ and C5aR1‐mediated pathways induce neutrophils and macrophages to lower phagocytosis, higher pro‐inflammatory cytokines production, and severe inflammation | [ 3 , 12 , 21 , 184 ] | |
| Gingipain | C5aR1 |
AhR, aryl hydrocarbon receptor; AMP, antimicrobial peptide; C5aR1, complement C5a receptor 1; FPR, formyl peptide receptor; GPCR, G protein‐coupled receptor; HBP, D‐glycero‐β‐D‐manno‐heptose‐1,7‐biphosphate; HDAC, histone deacetylase; HR, histamine receptor; IL, interleukin; iNKT cells, invariant natural killer T cells; LPS, lipopolysaccharide; PPAD, P. gingivalis peptidylarginine deiminase; PSA, polysaccharide A; SCFA, short chain fatty acid; Th, helper T cell; TIFA, TRAF‐interacting protein with FHA domain‐containing protein A; TLR, toll‐like receptor.
2.1. Gut Microbiota
The gut mucosal barrier is a key mediator of gut microbiota‐immunity communication, physically separating bacteria from the lamina propria.[ 26 ] The gut barrier is basically composed of a mucosal layer and a cell epithelium. Every component of the gut mucosa has a role in maintaining the homeostasis of the gut barrier or in defending against infection.[ 27 , 28 ] At the basal side of the cell epithelium, immune cells inhabit the lamina mucosa, a connective tissue layer. The immune cells involved in the gut microbiota‐immunity communication are: 1) innate lymphoid cells (ILC); 2) effector T cells; 3) B cells; 4) macrophages; 5) mast cells; and 6) dendritic cells (DC).[ 20 ]
In healthy conditions, bacteria stimulate immune cells, so increasing immune tolerance and inhibiting improper intestinal inflammation.[ 29 ] An example of molecule with these immunomodulatory effects is polysaccharide A (PSA), a structural component of the symbiont Bacteroides fragilis. Luminal PSA is captured by Toll‐like receptors 2 and 5 (TLR2/5) of DCs. After PSA‐TLR interaction, DCs process the antigen and present it to CD4+ T cells, which are then activated.[ 30 , 31 ] CD4+ T cells promote regulatory T (Treg) cell anti‐inflammatory responses and inhibit pro‐inflammatory cytokine production by Th1 and Th17 cells (Figure 2 ).[ 32 ]
Figure 2.
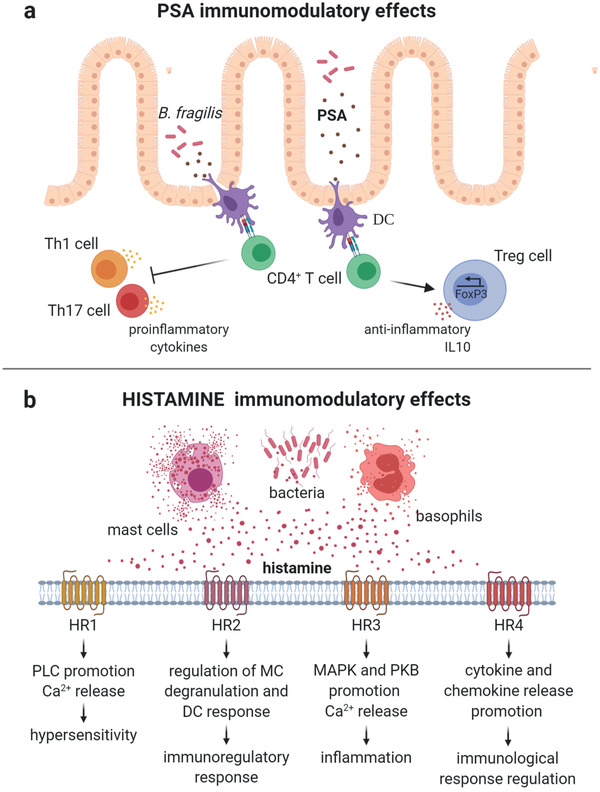
Immunomodulation pathways triggered by bacterial PSA and histamine. a) Bacterial PSA is captured, processed, and presented by DCs. CD4+ T‐cells recognize and bind the DC MHC‐II presenting the antigen. CD4+ T‐cells respond to PSA stimulating Treg‐cell activation and inhibiting Th1 and Th17 cell production of pro‐inflammatory cytokines. b) Histamine is a metabolite of histidine metabolism, released mainly by mast cells, basophils, and bacteria. Depending on the type of histamine receptor (HR), histamine leads to different immunomodulatory responses. PLC, phospholipase C; MAPK, mitogen‐activated protein kinase; PKB, protein kinase B. (Image created with BioRender.com).
Gut dysbiosis can heavily affect the release of signaling molecules and the related immune stimulation.[ 33 ] Histamine is a biogenic amine produced by histidine decarboxylases (HDC) of mammalian cells and gut bacteria (e.g,. Morganella morganii and Citrobacter freundii).[ 34 , 35 , 36 ] Histamine potential to stimulate adaptive and innate immune responses depends on the amounts released not only by bacteria but also by mast cells and basophils, which store and secrete the molecules in response to toxins or immune IgE‐mediated mechanisms (Figure 2).[ 37 ]
2.2. Oral Microbiota
The oral cavity is continuously exposed to external agents such as food, air, and microorganisms. This generates a very dynamic environment with high complexity in its chemical and microbial composition.[ 38 ] As in the gut barrier, the luminal layer of the oral mucosa is predominantly characterized by epithelial cells and the lamina propria is rich in immune cells.[ 39 ]
From a microbiota‐immunity communication viewpoint, the oral cavity is not as characterized as the intestinal compartment. However, it has been demonstrated that the oral microbiota homeostasis is closely related to the immune response, and the presence of oral antigen‐presenting cells (APC) guarantees immune tolerance under physiological conditions.[ 38 ]
Daily oral practices (tooth brushing and mastication) and periodontitis can frequently alter and damage the oral mucosa. Damage of the gingival barrier allows periodontal pathogens (e.g., P. gingivalis and A. actinomycetemcomitans) to enter the circulation. Under this unhealthy condition, the immune system can be strongly stimulated by direct contact with circulating microorganisms and their secreted molecules.[ 40 ]
The keystone pathogens are defined as the oral bacteria fundamental in the initiation and progression of periodontal inflammation and disease (A. actinomycetemcomitans, P. gingivalis, and T. denticola). They subvert the immune system and generate an inflammophilic environment, suitable and protective for other pathogens proliferation.[ 15 ] P. gingivalis seems to be the main influencer of this subverting action and is widely associated with neurodegenerative disorders.[ 21 ] It bypasses the bactericidal immune response mainly through co‐activation of two signaling pathways mediated by LPS, a structural bacterial component, and proteolytic enzymes called gingipains.[ 3 , 41 ]
P. gingivalis LPS (pgLPS) comes in two isoforms characterized by a specific Lipid A region and, depending on the isoform, immune TLRs‐mediated pathways are selectively stimulated or inhibited.[ 3 , 42 ] The cooperation of pgLPS and gingipains leads to a TLR2‐PI3K‐mediated signaling that results in reduction of bactericidal activity, upregulation of pro‐inflammatory cytokines (e.g., IL‐1β, IL‐6, and TNF‐α) and inhibition of phagosome formation and maturation.[ 43 , 44 ] This enables P. gingivalis to regulate immune responses, creating an inflammophilic environment for other pathogens.
3. Immune Communication from Microbiota to CNS‐Resident Cells
The mechanisms that translate the microbiota‐induced peripheral immune response into neuroinflammation are complex and feature two main ways of transmission: the vagus nerve for the gastrointestinal tract and the vascular system in the whole body.[ 19 ]
The vagus nerve innervates most of the digestive tract and allows two‐way gut‐brain signaling mediated by neurotransmitters and hormones such as serotonin, glucagon‐like peptide‐1, and peptide YY.[ 18 , 45 ] Both enteroendocrine cells and vagal sensory ganglia have a receptor system to trigger signaling communication to the CNS and contribute to the effects of gut metabolites on brain function.[ 46 ] Vagal ganglia can use specific receptors—such as pattern‐recognition receptors (PRR), G protein‐coupled receptors (e.g., free fatty acid receptor 3), and the 5‐hydroxytryptamine (5‐HT)‐3 receptor—to sense and process microbial molecules (e.g., LPS), metabolites (e.g., SCFAs), or enterochromaffin cell products (e.g., serotonin).[ 18 , 47 ] Sensory vagal afferent fibers can directly sense the peripheral inflammation and communicate it to the brain and thus, to the neuroimmune system.[ 48 ] Along the vagus nerve, cytokine receptors bind local tissue concentration of cytokines (e.g., IL‐1β) and transmit the signal to the CNS, which responds to regulate and maintain immune homeostasis.[ 49 ] The vagal modulation of the peripheral inflammation severity is a fine‐tuned mechanism called the inflammatory reflex.[ 50 ]
As for the vascular system, it is the direct route for the transport of bacterial molecules, immune signaling peptides, and immune cells, such as APCs. Microbiota molecules can reach the brain directly, crossing the intestinal or oral epithelium, and flowing through the blood vessels.[ 51 ] SCFAs are an example of microbial molecules that can reach the brain and influence CNS‐resident populations under physiological conditions.[ 46 , 52 ] Along the vascular system, SCFAs are captured by the monocarboxylate transporters on blood‐brain barrier (BBB) cells, and imported into the brain. Their effects on neurogenesis, inflammation, or homeostasis change depending on the type of SCFA. For instance propionate, unlike acetate, can reinforce the BBB, promoting tight junction formation and reducing permeability to toxins.[ 52 ] Aryl‐hydrocarbon receptor (AhR) ligands are another microbiota product that can reach the brain in case of barrier disruption and stimulate AhR‐expressing astrocytes. This leads astrocytes to release IL‐33, which in turn stimulates mast cell and microglial maturation.[ 53 ]
Among the molecules transported through the vascular system, cytokines and chemokines are the main factors to transmit inflammation from the periphery to the brain. They stimulate circulatory immune cells and promote their migration and maturation. Furthermore, they can reach CNS‐resident cells and trigger microglial and astrocyte responses.[ 19 ] APCs increase cytokine‐mediated immune signaling, transporting and presenting microbial antigens (e.g., LPS) to the brain.
3.1. The Role of Mast Cells
Mast cells (MCs) are the immune cells with most effect on immune signaling transmission from the periphery to the CNS. Recently, MCs have been widely associated with the neuroinflammation proper of neurodegenerative disorders (Figure 3 ).[ 54 ]
Figure 3.
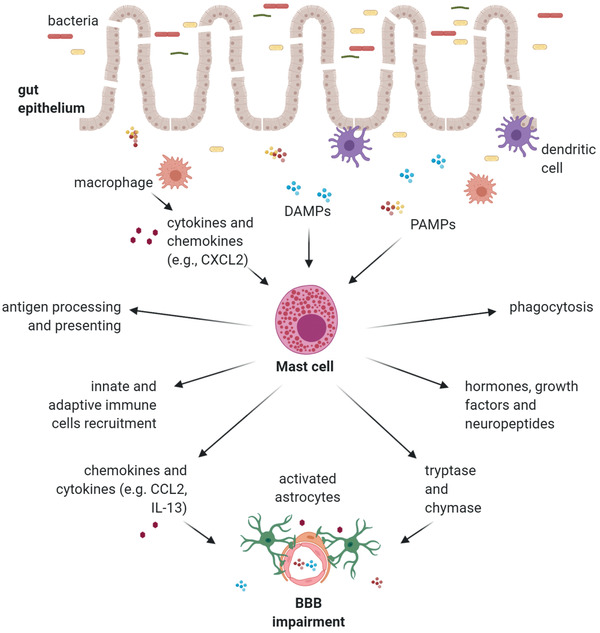
Graphic representation of the mast cells (MCs) response to gut dysbiosis. MCs react to pathogen‐associated molecular pattern (PAMPs), damage‐associated molecular pattern (DAMPs), and macrophage cytokines and chemokines by phagocytosing, processing, and presenting antigens and recruiting adaptive and innate immune cells. The MC release of hormones, growth factors, neuropeptides, inflammatory mediators, and enzymes leads to BBB impairment and stimulation of CNS‐resident cells. (Image created with BioRender.com).
MCs derive from bone marrow hematopoietic cells and migrate to connective tissues rich in blood vessels and nerves, in response to chemokine signaling. Under healthy conditions, MCs do not inhabit the CNS but remain on the abluminal side of BBB.[ 54 ] They are involved in different mechanisms—such as tissue repair and chronic inflammation—due to their ability to: 1) sense and produce hormones, enzymes, cytokines, and chemokines (e.g., tryptase, IL‐1β, and CXCL2); 2) phagocytose, and 3) process and present antigens.[ 55 ]
The recruitment of MCs at microbiota‐interfaced barriers is regulated by the release of chemokines such as CXCR2 (C‐X‐C Motif Chemokine Receptor 2) ligands.[ 56 ] At the gut wall, for instance, macrophages release chemoattractants in response to the activation of TLRs and thus to the presence of bacterial signals.[ 57 ] MCs migrate in proximity of the lamina propria and acts as a connection between connective tissue and the nerves. They contribute to immune transmission via the vagus nerve by releasing hormones, growth factors, and neuropeptides that can be captured by enteric neuron receptors and translated to nerve signal.[ 55 ]
In addition to the MC potential for influencing the nerve response, MCs can directly challenge the pathogen and promote both adaptive and innate immune response.
MCs have several receptors to sense not only the pathogen‐associated molecular pattern (PAMP) but also the damage‐associated molecular pattern (DAMP).[ 58 ] DAMPs are molecules produced and secreted during cell damage. Extracellular ATP is an example of DAMPs, since damaged cells, active immune cells and bacteria produce it in unhealthy conditions. MCs can sense and respond to ATP through purinoreceptor P2XT activation and the production of pro‐inflammatory cytokines and chemokines.[ 59 ] In dysbiotic conditions, the bond of PAMPs (e.g., LPS and flagellin) triggers MC activation, which leads to degranulation, release of pro‐inflammatory molecules (e.g., TNFα, IL‐1 and CCL5), phagocytosis, and production of antimicrobial peptides (e.g., cathelicidin). Since MCs can also act as APCs, they can challenge bacterial infection also by processing antigens and presenting them to other immune cells.[ 58 ]
MCs can promote the recruitment, maturation, and activation of innate and adaptive immune cells. MCs stimulate innate immunity mainly by secreting enzymes (tryptase and chymase), antimicrobial peptides and cytokines, while adaptive immune cells are recruited and activated mostly by MC‐produced chemokines and cytokines, such as CCL2, CXCL10, IL‐13, and IFNγ.[ 55 ]
The contribution of MCs during dysbiotic events also comprises their involvement in the regulation of barriers homeostasis (e.g., gut‐blood barrier (GBB) and BBB).[ 54 ]
3.2. Gut‐Blood Barrier Impairment
The GBB is the physical barrier between gut microbiota and blood flow.[ 60 ] It consists of the gut mucosal barrier, the underlying connective tissue, and the endothelium of intestinal capillaries.[ 61 ] The GBB permeability is tightly controlled by both epithelial and endothelial intercellular junctions. The role of the connective tissue in GBB homeostasis is still not clear. Lamina propria fibroblasts seem the key connective tissue players in case of GBB impairment.[ 62 ] Indeed, when stimulated by bacterial endotoxins, lamina propria fibroblasts release inflammatory mediators—such as TNFα—that significantly contribute to transepithelial resistance (TER) reduction, leading to GBB impairment.[ 62 ]
The most important intercellular molecular connections for paracellular permeability regulation are tight junctions (TJ), mainly the transmembrane proteins occludin and claudin, and the cytoplasmic zonulin 1 (ZO‐1).[ 63 ] The structural elements and signaling pathways that influence TJ stability are several and constantly updated. The most determinant factors are: a) inflammatory mediators; b) epithelial or endothelial F‐ actin organization; and c) peripheral inflammatory cell activity.[ 60 , 63 , 64 ]
-
a)
In response to pathological conditions, MCs lead to degranulation and large release of these pro‐inflammatory molecules, such as TNFα, IL‐1β, and IFN‐γ.[ 55 ] TNFα is recognized by epithelial and endothelial tumor necrosis factor receptor, triggering the final expression of the myosin light chain kinase (MLCK).[ 65 ] MLCK influences paracellular permeability mainly by two mechanisms: 1) stimulating myosin light chain phosphorylation with consequent F‐actin reorganization and occludin endocytosis; 2) promoting claudin‐2 expression, which leads to leaky epithelia and increased permeability.[ 66 , 67 , 68 ]
-
b)
F‐actin organization is strictly related to TJ barrier integrity. Both microscopy and pharmacological analysis clearly showed a physical link between TJs and the actin cytoskeleton.[ 64 ] F‐actin directly binds ZO proteins that work as a bridge between the cytoskeletal filaments and other TJ proteins, as occludin, claudin, and the junction adhesion molecule.[ 69 ] Hence, as in the case of the MLCK activity, the alteration of F‐actin arrangement impacts directly on the TJ barrier organization and stability. In addition to cytokine‐mediated mechanisms, immune cells can modulate GBB integrity by inducing F‐actin rearrangement through different signaling pathways. For instance, recruited MCs release tryptase and chymase, two serine proteases targeting the epithelial or endothelial protease‐activated receptor (PAR2). The cleavage of PAR2 leads to cytoskeletal F‐actin redistribution and thus, ZOs delocalization.[ 70 , 71 ]
-
c)
Peripheral inflammatory cells modulate GBB permeability also by the activity of intracellular induced nitric oxide synthase (iNOS).[ 60 ] Under physiological conditions, enterocytes constitutively synthesize nitric oxide (NO) for the maintenance of gut mucosal barrier function. In dysbiotic conditions, endotoxins and cytokines induce inflammatory cell iNOS to overproduce NO.[ 72 ] An excessive NO synthesis leads to GBB leakage by several and multifactorial mechanisms: highly toxic protein oxidation and nitration; S‐nitrosylation; enterocyte apoptosis or necrosis; and impairment of enterocyte migration.[ 60 , 72 ]
On the whole, the GBB impairment significantly increases the flow of bacterial molecules, pro‐inflammatory signals, and APCs along the vascular system into other body tissues. In fact, GBB dysfunction is associated with several pathologies, such as inflammatory diseases and neurodegenerative disorders.[ 73 , 74 ]
3.3. BBB Impairment
Crossing the BBB by bacterial and pro‐inflammatory elements is pivotal in triggering neuroinflammation and neurodegeneration. Under physiological conditions, this barrier is highly selective, avoiding the passage of toxins and noxious biochemical signals. It is composed of cellular and extracellular components: astrocytes, pericytes, endothelial cells, and extracellular matrix (ECM).[ 75 , 76 ] Dysregulation of BBB permeability allows the infiltration of immune signaling molecules and leukocytes.[ 73 ] For instance, MCs can move from the basement membrane of the BBB, penetrate the cerebral parenchyma, and directly stimulate CNS‐resident populations.
In addition to these mechanisms, damage to the BBB is also promoted by interaction between glial cells (astrocytes and microglia) and MCs.[ 77 ] MCs can directly contact glia or communicate by means of exosomes, which can freely pass the BBB. The MC extracellular vesicles can contain signaling molecules and be exploited for two‐way communication between the immune system and the CNS.[ 78 ]
Astrocytes are the most abundant glial component in the CNS and are one of the three fundamental cells of the BBB. Even though astrocytes are not properly considered immune cells,[ 79 ] their complex communication with the immune system makes them pivotal players in neuroinflammation and neurodegeneration. They influence MC and microglia maturation and activation by sensing and releasing immune signaling molecules. In presence of PAMPs and DAMPs (e.g., bacterial LPS and extracellular ATP), BBB astroglia can detect the molecules through specific receptors (e.g., PRR) and respond by releasing cytokines such as IL‐33 and IL‐1β.[ 77 ] These interleukins stimulate MCs to produce IL‐13 that triggers further glial chemokines and cytokine production and increases pro‐inflammatory signaling.
Direct contact between astrocytes and MCs is guided by the CD40L/CD40‐mediated mechanism.[ 80 , 81 ] This interaction leads MCs to degranulation and the release of pro‐inflammatory molecules such as cytokines, histamine, and growth factors. The immediate impact of this pro‐inflammatory secretion is on the integrity of the BBB, since it promotes TJ disruption and increases paracellular permeability.[ 77 , 82 ] In this impaired condition, MCs can abundantly cross the barrier and spread signaling molecules directly into the CNS parenchyma.[ 83 ] For instance, the release of granule‐associated histamine into the brain is easily detected by microglial histamine receptors (HR1‐4). Histamine stimulates the activation of microglia‐triggered neuroinflammation.[ 84 ]
MCs can promote the switch of microglial phenotype to the pro‐inflammatory M1 state also by releasing chemokines such as CCL5.[ 85 ] On the other hand, microglia can stimulate MCs in response to DAMP or PAMP detection. For instance, the microglial receptors P2X and P2Y are activated by ATP sources, stimulating both microglial chemotaxis and pro‐inflammatory cytokine and chemokine secretion, which activate MCs.[ 86 ]
The complement component C5a is another example of MCs‐glia two‐way communication. In severe inflammation, macrophages and neutrophils produce a large amount of C5a that can flow along the vascular system and reach the CNS. Consequently astrocytic, microglial, and MC C5a receptors are activated and upregulated, extending inflammation to the brain.[ 85 ]
A barrier dysfunction induced by PAMPs, DAMPs, and the relative immune response, is common to all the neurodegenerative diseases.[ 16 , 19 ] Indeed, most of them show elevated levels of microbiota molecules reaching the brain.[ 87 , 88 ] In AD and PD the vascular system and the vagus nerve transport peptides and neurotransmitters involved in pathological protein aggregation and neuronal cell degeneration.[ 4 , 12 , 46 ]
3.4. Microbiota‐Induced Gliosis and Neurodegeneration
Activated CNS glial cells are directly involved in the pathogenesis and progression of most neurodegenerative diseases.[ 89 , 90 , 91 , 92 , 93 ] They are activated by bacteria‐derived stimuli and pathological hallmarks, such as protein fibrils and inclusions bodies. Glial cells respond directly to toxic fibrils and inclusions by surrounding them and triggering mechanisms of phagocytosis and recruitment of other immune cells.[ 94 ] The continuous exposure of CNS‐resident cells to inflammatory stimuli induces a continuous glial overresponse. There is evidence that this chronic microglial overstimulation has a detrimental effect on the ability to fight pathological signals.[ 95 , 96 ] They switch to an auto‐aggressive senescent phenotype characterized by dysregulation of phagocytosis and release of pro‐inflammatory molecules.[ 97 ]
Besides these glial responses, astrocytes and microglia make specific contributions to the pathological condition. In Huntington's disease (HD), for instance, the mutated protein huntingtin also affects immune cells. This impairs the immune cells ability to migrate and respond to chemoattractant stimuli. Mutated microglia cannot recruit peripheral immune cells – such as macrophages and monocytes – and are therefore not able to respond properly to toxic events. This causes dysregulation of CNS immunity and increases neuroinflammation.[ 98 ]
In PD, the dysbiotic condition influences not only the peripheral immune response and gut barrier homeostasis, but also the PD hallmark formation and propagation.[ 99 , 100 ] When PD barriers are impaired, for instance, bacterial LPS can reach the brain and promote α‐syn fibrillation.[ 101 ] Together, LPS and LPS‐promoted α‐syn oligomers stimulate neuroinflammation, leading to an immune overresponse.[ 99 ]
AD is the most common neurodegenerative disorder, and its hallmarks are cognitive impairment and intra‐ and extracellular protein aggregates (extracellular senile plaques of Aβ fibrils and intracellular neurofibrillary tangles).[ 102 ] In AD patients, CNS‐resident immune cells concentrate in the proximity of protein aggregates and release pro‐inflammatory mediators (e.g., IL‐6, TNFα, and TGFβ), contributing to pathological neuroinflammation.[ 103 , 104 ] Microbiota play a key role in increasing neuroinflammation and AD hallmark formation.[ 54 ] Several gut bacteria (e.g., Escherichia coli, Staphylococcus aureus, and Bacillus subtilis) are able to produce Aβ fibrils and there is evidence that bacterial LPS further promotes extracellular Aβ fibrillation.[ 88 , 105 ] Dysbiosis not only increases bacterial Aβ fibril formation but also helps Aβ reach the brain by impairing barrier permeability.[ 4 ] Once amyloid plaques appear in the brain, CNS‐resident immune cells respond mainly with microglial clearance of peptides. This process is dependent on the activity of triggering receptor expressed on myeloid cell‐2 (TREM2), which triggers phagocytosis, pro‐inflammatory cytokine secretion, and oxidative stress.[ 106 ] Zhou and colleagues showed that TREM2 immune modulation is negatively regulated by TLR4. In case of dysbiosis, bacterial endotoxins (e.g., LPS) reach the brain, activate TLR4 and indirectly downregulate TREM2 expression. This leads to an overresponse of neuroinflammation and a deficit in Aβ peptide clearance.[ 106 , 107 ]
Amyotrophic lateral sclerosis (ALS) and multiple sclerosis (MS) are two neurodegenerative disorders that affect both muscles and nerves.[ 108 ] ALS consists in a progressive loss of upper and lower motor neurons, leading to paralysis, cognitive impairment, and death.[ 109 ] Since the ALS etiology is complex and multifactorial, the pathogenesis mechanisms are still poorly understood. More than 50 genes are associated with ALS development and code for proteins involved in several molecular and cellular mechanisms, such as dysregulation of protein aggregates degradation, mitochondrial dysfunction, and impairment of axonal signaling.[ 110 ] A common feature of various ALS forms is the systemic inflammation, which leads to neuronal death and ALS motor deficits.[ 111 ] Gut microbial imbalance contributes to increase the pro‐inflammatory state and the severity of the disease.[ 112 , 113 ] For instance, recent works showed that dysbiosis contributes to ALS development by leaking the GBB and increasing the monocyte expression of pro‐inflammatory cytokines and chemokines (e.g., IL‐1β, IL‐8, CXCL1 and ‐2) and the endotoxin levels in ALS serum.[ 114 , 115 ]
MS is an inflammatory demyelinating disease characterized by demyelination of nerve cell fibers, which causes the interruption of nerve signals between the brain and other innervated body tissues, the loss of control of motor functions, and mental and emotional impairment.[ 116 ] The axonal myelin degradation is promoted by a dysregulated immune response of both peripheral and CNS‐resident immune cells.[ 54 , 117 ] In MS, Treg cells are dysregulated leading to Th17 cell proliferation and pro‐inflammatory activity. Th17 cells trigger demyelination by producing pro‐inflammatory cytokines and chemokines, which recruit other immune cells and exacerbate inflammation.[ 118 ] Both gut and oral microbial imbalances are known to deeply influence MS development by promoting a pro‐inflammatory state from the peripheral environment to the brain.[ 119 , 120 ] For instance, dysbiosis‐induced overresponse of microglia and macrophages leads to cerebral oxidative stress and excitotoxicity, which are hallmarks of neurodegeneration in MS.[ 117 ] The interaction between microbiota and neuroinflammation in MS is bidirectional and recent findings demonstrated that it is possible to influence the disease course by targeting microbial composition.[ 54 , 120 ]
Up to now, we have described the biological complexity of microbiota‐immune system‐neurodegeneration connection. The common experimental approach to study the whole microbiota‐immunity‐CNS axis is currently based on animal models.[ 121 , 122 , 123 , 124 ] According to the 3Rs principle (replacement, reduction, and refinement) and taking into account some differences of preclinical models with human physiology (e.g., different microbiota composition), advanced in vitro modeling offers a promising and challenging strategy to reduce the experimental in vivo variability and study the complexity of the axis by analyzing each single microenvironment and biological mediator.[ 125 ] The advent of bioengineering and organ‐on‐a‐chip (OOC) technology in the field of cell culture is providing new technological inputs toward more reliable in vitro tools suitable to model also human immune system‐based mechanisms.
4. Advanced Systems for In Vitro Modeling
OOC technology offers a promising in vitro tool to tackle the above described challenges since conventional 2D cell culture systems cannot recapitulate the complexity of the many interfaces involved along the microbiota‐immunity‐CNS axis.[ 126 , 127 , 128 ] Current in vitro models generally used to mimic immune functions do not include physiologically relevant conditions, such as the fluid flow of their native in vivo microenvironment, 3D architecture of interstitial tissues, molecule passage across multiple barriers, or the interaction between different cell populations.[ 129 ] The main engineering feature in the most advanced models of immune communication is the recapitulation of the immune‐tissue interface. Culturing immune cells in standard wells in presence of tissue specific cellular types is still not representative of the in vivo scenario, where immune cells interact with other cell populations through membranes, matrices, flows in border environments that guide immune cell trafficking, recruitment, and signaling.[ 130 ] Therefore, current OOC systems of immunity‐mediated phenomena employ various technical strategies to recreate an artificial barrier for immune‐organ interface modeling (Table 2 ).
Table 2.
Recent OOC models and related technical features classified according to the immunity‐mediated phenomenon represented
| Immunity‐mediated phenomenon | OOC model | Multi‐system interface | Cell models | Ref. |
|---|---|---|---|---|
| Adipose tissue inflammation | Three concentric compartments in a Si‐based microfluidic chip | Porous barrier formed by a circular capillary channels array | Human pre‐adipocytes; human macrophages | [ 185 ] |
|
Three‐chamber double‐layer PDMS/glass based microfluidic chip |
PET porous membrane (0.4 µm pores) | Human PBMCs; human adipose cells | [ 186 ] | |
| Single circular chamber in a PDMS‐based microfluidic chip | Cell‐to‐cell direct contact | Murine adipocytes; murine macrophages | [ 187 ] | |
| Airway inflammation | Double‐layer PDMS‐based microfluidic chip | PDMS porous membrane (10 µm pores) | Vascular ECs; human alveolar epithelial cells; human neutrophils; E. coli | [ 188 ] |
| Double‐layer PDMS‐based microfluidic chip | Micropore array silicon chip | Human bronchial epithelial cells; human PBMCs | [ 189 ] | |
| Double‐layer PDMS‐based microfluidic chip | PET porous membrane (0.4 µm pores) | Primary human airway epithelial cells; primary human lung microvascular ECs; | [ 190 ] | |
| Allergy | Two‐parallel channels PDMS‐based microfluidic chip | Single side capillary channel | Mast cells; macrophages | [ 191 ] |
|
Double‐layer PMMA‐based microfluidic chip |
PET porous membrane (0.4 µm pores) | Dendritic cells; keratinocytes | [ 192 ] | |
| Single channel PDMS‐based microfluidic chip | Multiple cell layers in a microfluidic channel | Basophils; HUVECs; vascular smooth muscle cells | [ 193 ] | |
| Immune system–endothelia interaction | Double‐layer PDMS‐based microfluidic chip | Porous silicon membrane for cell extravasation | Human microvascular ECs; human promyelocytic leukemia cells | [ 194 ] |
| Double‐layer PDMS‐based microfluidic chip with Y‐channel | ECM‐liquid | HUVECs; smooth muscle cells; leukocytes; platelets | [ 195 ] | |
| Three‐parallel channels microfluidic chip | ECM‐liquid interface with trapezoidal pots | HUVECs; human monocytic leukemia cells | [ 196 ] | |
| Immune system‐cancer communication | Three‐parallel channels microfluidic chip | Capillary channel array | PBMCs; tumor cells | [ 197 ] |
| Three‐parallel channels microfluidic chip | Hydrogel‐liquid with trapezoidal pots | Receptor‐redirected T cells; liver hepatocellular carcinoma; | [ 198 ] | |
| Multi‐compartment microfluidic chip | Porous membranes/wells connected by microfluidic channels | Tumor slice; lymph node slice | [ 199 ] | |
| Intestinal inflammation |
Double‐layer PDMS‐based microfluidic chip |
PDMS porous membrane (10 µm pores) | Intestinal epithelial cells; human capillary ECs; PBMCs; microbial cells | [ 137 ] |
|
PS made four‐layer Biochip (ChipShop GmbH) |
Flow channels and PET porous membrane (8 µm pores) | HUVECs; intestinal epithelial cells | [ 139 ] | |
| Three‐parallel channels microfluidic chip |
ECM‐liquid interface (PhaseGuideTM) |
Human monocyte leukemia cell line; Intestinal epithelial cells | [ 200 ] | |
| Intra‐amniotic inflammation | Three‐parallel channels microfluidic chip | ECM‐liquid interface | Macrophages; HUVECs; human carcinoma | [ 201 ] |
| Three‐parallel channels microfluidic chip | PDMS micropillars array | Differentiated hiPSCs; E. coli | [ 202 ] | |
| Inflammation in gut‐liver axis | Multi organ‐on‐chip in a plate | Wells connected by microfluidic channels | Intestinal epithelial cells; hepatocytes; macrophages | [ 203 ] |
| BBB‐immune system interaction | Hollow polypropylene fibres | Substrate membranes with sub‐micrometric pores | Normal adult human brain microvascular ECs; human adult astrocytes; human monocytic leukemia cells | [ 170 ] |
| Transwell mimetic flow chamber | Nano‐porous SiN membrane (50 nm pores) | CD34+ derived ECs; T cells; pericyte‐conditioned medium | [ 156 ] | |
| Neuroinflammation | PDMS‐based multichamber microfluidic system | Circular micro‐capillary array | Immortalized human microglia; AD neurons; astrocytes | [ 174 ] |
PBMC, peripheral blood mononuclear cell; HUVEC, human umbilical vein endothelial cell; EC, endothelial cell; PET, polyethylene terephtalate; PDMS, polydimethilsiloxane.
In the following paragraphs, we shall discuss the most recent cell culture systems for microbiota‐immunity‐CNS modeling highlighting the fundamental engineering aspects necessary to reproduce physiologically relevant OOCs (Figure 4 ).
Figure 4.
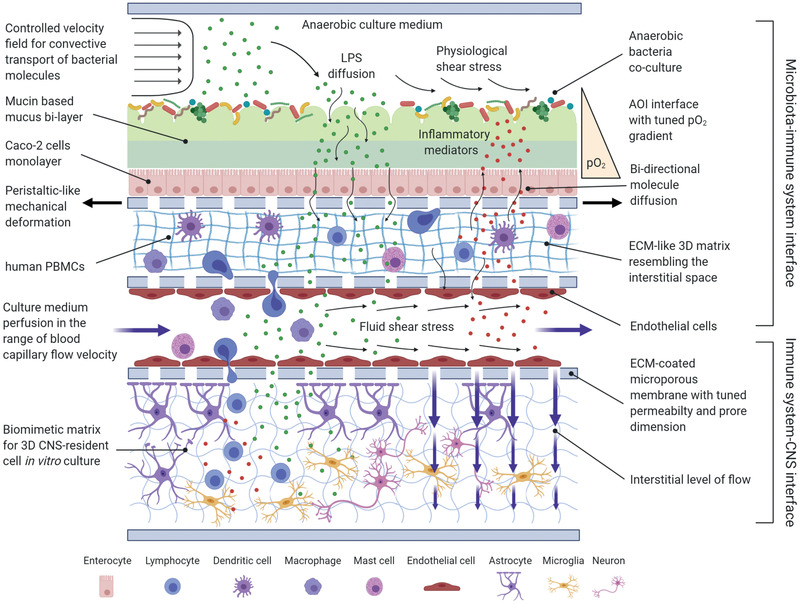
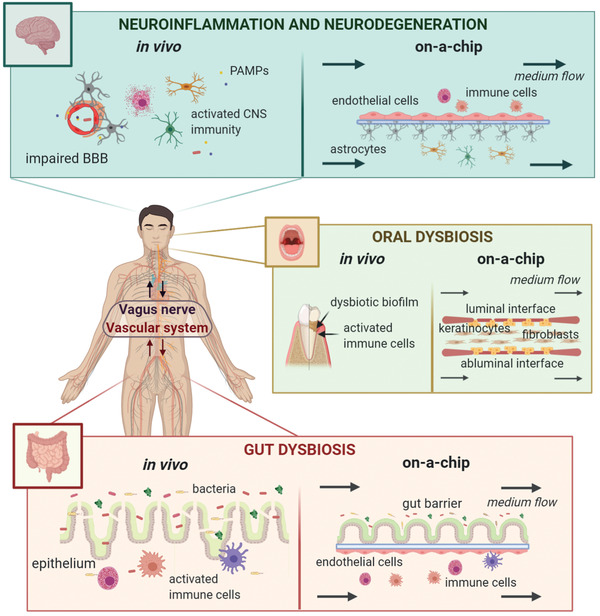
An ideal in vitro model of the microbiota‐immunity‐CNS network crosstalk. This schematic representation highlights the main physical and biological features that may be implemented with the most advanced state‐of‐the‐art engineering systems. From top to bottom: anaerobic gut bacteria are cultured in dynamic conditions, under the effects of physiological levels of shear stress. Controlled anaerobic culture medium flow conveys the secreted bacterial molecules, which diffuse across the intestinal barrier, recreated by a mucin‐based bi‐layered mucus on top of the intestinal epithelium. A tuned oxygen gradient is maintained across the barrier. The microbial activity recruits from the “blood channel” the immune cells in the interstitial space, represented by a perfused micro‐channel. The immune cells in the interstitial space start secreting inflammatory mediators that are free to diffuse back across the intestinal barrier. On the other hand, the bacterial molecules and immune system cytokines can reach the CNS thanks to the bloodstream. At the BBB interface, a selective permeability porous membrane regulates fluid extravasation and molecule diffusion. The final compartment is composed by a multi‐culture of CNS‐resident cells within a biomimetic 3D matrix, resembling the ECM inside the brain. (Image created with BioRender.com.)
4.1. Engineered Microbiota‐Immune System Interface
The intestinal barrier is the first mediator of the inflammatory response in the microbiota‐immunity‐CNS axis.[ 131 ] In vitro modeling of this interface is consolidated in the literature. There are several examples of engineered OOCs, commonly known as gut‐on‐chips (GOCs),[ 132 , 133 , 134 ] which successfully incorporate physiologically relevant features, such as the flow of bacterial molecules, microbial co‐culture in an anoxic–oxic interface (AOI),[ 135 ] a robust mucus bi‐layer with physiological thickness, or physical deformations for peristalsis‐like motion.[ 17 ] However, in the perspective of immune response modeling, most of today's in vitro gut models lack tissue‐resident immune cells.[ 136 ] The inclusion of the typical human microbiota, as well as human immune cells, remains a fundamental need for getting reliable and predictive outcomes.
The preferred cell source for immune system‐on‐chips are human primary immune cells. Ideally, the cell source should present a complete immune function, such as the ability to fight pathogens, secrete cytokines or chemokines, maintain the differentiated state of immune cells, and be available on a large scale.[ 129 ] Isolated human peripheral blood mononuclear cells (PBMCs) have been used in various in vitro models in the past; PBMCs contain a mixed population of innate and adaptive immune cells and can also differentiate into resident dendritic cells and macrophages.[ 137 ]
Kim et al. introduced PBMCs into the basolateral capillary channel of a GOC, to mimic immune cell recruitment into the lamina propria.[ 137 , 138 ] A classic dual‐channel or multilayer configuration was adopted to build the intestinal epithelial layer on the luminal side and the endothelial layer on the other. The inflammatory activation was then mimicked by the inflow of LPS or dextran sulfate sodium (DSS) at the luminal channel, leading to its diffusion across the barrier for recruiting PBMCs.
The most interesting outcome was the induction of intestinal villi injury and alteration of the epithelial barrier function in the presence of PBMCs, with enhanced expression of IL‐8 and other inflammatory cytokines. Furthermore, Maurer and colleagues recently showed how the physiological contact of LPS with intestinal epithelial cells helped DC maturation and triggered their invasion into the epithelial cell layer; DCs formed dendrites through the barrier until they contacted the luminal space.[ 139 ] These examples illustrate the most complete and evolved intestine barrier inflammation‐on‐chip models where all the main physical, biological, and biochemical elements together emulate the full inflammatory scenario at the microbiota‐intestine interface.
Intestine‐on‐a‐chip technology can mimic the luminal side of the intestine barrier well, allowing many engineering solutions. However, recapitulation of the deep architecture and functioning of the microenvironment underneath the epithelial monolayer has yet to be fully developed.[ 140 ] The intestinal microvasculature is the direct point of access for inflammatory factors on the route to the CNS and is pivotal for microbiota‐peripheral immunity communication. The dual perfused chambers configuration does not allow mechanistic study of molecule exchange phenomena on the abluminal side. Therefore, the systems need to be made more complex to include the multiple features of the microcirculation architecture.[ 140 ] Multi‐layer micro‐devices have been developed by using innovative rapid prototyping technologies.[ 141 ] In this manner, the micro‐interface of epithelial and endothelial cells (EC) can be added to the GOC model by tuning thickness of the micrometric intercellular spaces,[ 142 ] and introducing ECM‐like biomaterials in the different layers to resemble the outer ECM, which can significantly modulate the endothelial and epithelial activity.[ 143 ]
Of note, 3D architecture reproduction is still challenging. The most advanced model was recently built by recreating a perfused capillary network inside a microfluidic channel after injecting ECs and patient‐derived small intestinal myofibroblasts.[ 144 ] Side micro‐channels allow controlled cell inflows inside the central chamber where angiogenesis might start. A functional microvasculature was obtained, also observing responsiveness to micro‐environmental stimuli including oxygen tension, cell concentration, growth factors, and pharmacotherapy.
The integration of this novel culture technique inside the abluminal compartment of a GOC would make it possible to include the interaction of ECs with the intestinal epithelium and monitor in real‐time the immune system reaction to microbiota influence (e.g., molecule trafficking and leukocytes extravasation). In a more complex scenario, the presence of 3D intestinal villi and internal microcirculation inside a GOC demonstrated the enhancement of the barrier function compared to standardized models.[ 145 , 146 ] In our view, coupling models of systemic circulation with luminal flow would open new routes for modeling intestinal inflammation in vitro.
Including direct neural contacting within intestinal inflammation‐on‐a‐chip models would certainly add a complete representation of the triggering phases in the gut‐brain communication process. Vagus nerve links the biological functions in gut and brain by both direct and indirect interaction with the intestine epithelium.[ 131 ] Only a few examples of innervated OOCs have been reported in literature and these refer to a restricted group of types of tissues that were reported in a recent review by Park and colleagues.[ 147 ] They grouped the current devices in two main categories: the synaptic and the neuroeffector junction (NEJ) innervation‐on‐a‐chip. Here we want to point out the space compartmentalization approach with connecting microchannels: the innervated chips have two synaptic chambers and multiple parallel axon‐passing channels. Such configuration can mimic the neural communication between two distinct compartments as in the case of two brain regions as shown by Virlogeux and colleagues,[ 148 ] or when recreating tissue specific innervation interfaces.[ 149 , 150 ]
The reconfigurable OOC developed by Soucy et al. successfully sustained the sympathetic innervation together with synapsis formation in a functional cardiac tissue‐on‐chip. Precisely, they exploited the meniscus pinning effect by the use of GelPins to compartmentalized 3D cell‐laden materials thus recreating the innervated interface.[ 151 ]
The cited examples represent valuable solutions to be translated into innervated intestinal inflammation‐on‐a‐chip models in order to mimic the vagal root both at the intestinal barrier and CNS levels.
There are no models in the literature addressing the oral microbiota impact on the immune system response that exploit OOC technology. However, one representative example was proposed by Rahimi et al. in 2018.[ 152 ] They developed a mucosa‐on‐a‐chip system to rapidly determine layer‐specific responses of the oral mucosa to influence of the microbial population. The mucosa‐on‐a‐chip recreated the luminal and abluminal interfaces of the oral mucosa, with human keratinocytes co‐cultured with gingival fibroblasts within a collagen‐based matrix inside a microfluidic chamber. This configuration is suitable for simulating sub‐epithelial layer inflammation by adding human neutrophils or lymphocytes to the abluminal channel. This is a promising starting point for improving and adapting OOC existing systems for microbiota‐immunity interface modeling to the oral compartment.[ 152 ]
4.2. Physiologically Relevant Immune System‐CNS Interface
BBB serves as the entrance to the CNS for immune cells and immune mediators, with immune cell traffic intensity changing according to physiological or pathological conditions.[ 153 ]
Currently, the use of microporous track‐etched membranes is consolidated as substrate for the endothelium to study the BBB mechanisms in molecule cross‐diffusion.[ 154 ] Membrane thickness and permeability must be precisely tuned to allow basal transport and barrier cell polarization while ensuring mechanical support and high adhesive strength.[ 155 ]
Mossu et al. developed an in vitro platform for the observation of T‐cells interacting with a BBB model under flow. They proposed a novel ultrathin silicon nanomembrane (50 nm thick) able to guarantee: 1) extraordinary permeability (up to ≈0.1 cm s−1); 2) the co‐culture of ECs with pericytes or pericyte‐conditioned medium recruiting T‐cells, which showed also intense crawling and transmigrating across the TNF‐treated endothelial layer; and 3) high‐quality imaging of immune cell/endothelial interactions.[ 156 ]
As just described, a nanometric thin membrane may mimic the basal lamina in vivo, but Koo et al. went further by recreating a membrane‐free endothelial cell‐perfused structure inside an OOC with the aim to model the diffusion of specific molecules and their neurotoxic effect on the CNS immune system.[ 157 ] Cell‐to‐cell crosstalk between parenchymal cells and the endothelium was maximized without any synthetic membrane in between, creating a more physiological exposure route for chemicals. This introduces a groundbreaking element in cell barrier modeling. The 3D structures adjacent to the endothelial layer were seen to modulate the interaction of the astrocytes with brain‐derived ECs.[ 158 ] 3D‐like interfaces have been successfully integrated by several groups by tuning brain‐like mechanical properties.[ 159 , 160 ]
Besides molecules crosstalk, immune cell trafficking is another fundamental aspect when modeling the immune system‐CNS interface.[ 161 , 162 ] We have already pointed out how BBB impairment causes extravasation of leucocytes and immune signaling molecules, which accumulate inside the brain parenchyma, with a CNS neuroinflammatory response.
Reliable in vitro models of immune cells trafficking into the brain might provide valuable insights into the pathologic immune response and help to understand how to act on it. Immune cell migration is a multistep dynamic process, with a complex spatial‐temporal course: lymphocytes initially become tethered to the ECs, roll along the endothelium and then, after firmly adhering, start crawling and transmigrate. This takes place within a 3D dynamic microenvironment inside the brain micro‐capillaries where leukocytes experience blood flow and interface with the ECM matrix after transendothelial migration.[ 163 , 164 ] Difficulties in recreating such complex steps in vitro have limited the analysis of immune cells crossing the BBB toward inflammatory hubs.
Classical 2D static models such as Transwell or Boyden chambers have been widely used to characterize responses to chemokines signals and measure cell migration across a porous membrane.[ 163 , 165 , 166 , 167 ] While such systems allowed quantitative evaluation of chemokine‐mediated trafficking, the absence of physiological shear stress acting on the EC monolayer conditioned the endothelial interaction with transmigrating cells. To faithfully mimic the in vivo blood flow, microfluidic systems are the most suitable solution and they recapitulate the whole recruitment process, from the “rolling step” to the extravasation.[ 165 ] The most advanced in vitro models included EC stimulation with physiologic levels of shear stress.[ 168 , 169 ] Even though there is general agreement on the importance of incorporating such stimuli to effectively capture transport phenomena, it is still not clear whether that shear stress range needs to be narrowed for more tuned and accurate modulation of the brain microvascular epithelial cells behavior.[ 170 , 171 ]
Some dynamic BBB in vitro models replicated the recruiting and crawling of immune cells very well, but they could not go further because they used substrate membranes with sub‐micrometric pores, which prevented trans‐endothelial migration (TEM) of cells.[ 156 , 170 ] Trans‐capillary pore diameters of 2–4 µm were sufficient to allow cells movement, as demonstrated in a few works.[ 164 , 170 ] More complex fabrication techniques are needed to obtain membrane‐free lumens. The LENS (LumENext‐Stacks) device designed by McMinn et al. guided the formation of perfusable vessels lined by ECs that self‐sustain within an ECM‐like 3D environment. LENS includes all the main features for TEM modeling: 1) flow perfusion; 2) a high cell permeable interface; 3) a 3D microenvironment; and 4) the possibility of detecting real‐time immune cell extravasation process in space and time.[ 172 ]
Another diffused approach to study immune system‐CNS interface transport involves PDMS‐based microfluidic devices made up of three parallel channels separated by micropillars. Micro‐capillary forces determine fluid separation and allow iPSC‐ECs to migrate inside a 3D matrix chamber and form micro‐vessels in a brain‐like environment.[ 166 , 173 ]
Apart from recreating the immune system‐CNS interface in physiology, there is also the need to model the inflammation response inside the brain microenvironment, mediated by neural immune cells. Most of the in vitro AD models do not include neuroinflammatory changes mediated by microglia. The inclusion of neurons and microglia would lead to more complete models of the whole inflammatory scenario while, however, increasing the complexity of the system. There are also few examples of BBB co‐culture models including microglia. As it is challenging to isolate and culture pure populations of microglia, efforts have been made to understand what are the best culture conditions for long‐term phenotypic expression of primary microglia in vitro.[ 167 ] Park et al. introduced the possibility of studying microglial behavior under the effect of chemokines inside an OOC modeling neurodegeneration and neuroinflammation in AD. They designed a micro‐device with specific geometry to permit the study of microglia recruitment and accumulation in a tri‐culture model with neurons and astrocytes.[ 174 ] Another significant example consists in a tetra‐culture system inside a microfluidic device where microglia and neurons were embedded in a collagen‐based gel matrix adjacent to the EC microvessel and astrocytes. This offers the most complete model of the immune system‐CNS interface where it is possible to trace the route of a neurotoxic compound from the luminal compartment, the BBB barrier, and up to the brain.[ 157 ]
5. Conclusion
Human microbiota can profoundly influence several body tissues, and the immune system is a pivotal player in this crosstalk. Recent findings have evidenced several molecular mechanisms that support the involvement of microbiota‐mediated immunity in developing neuroinflammation and neurodegeneration. In neurodegenerative disorders, bacteria play a key role by producing structural components, functional enzymes and metabolites that trigger a spreading immune signaling response, mainly responsible for body barriers impairment and cerebral gliosis. Furthermore, microbiota‐mediated immunity also affects the progress of neurodegeneration by influencing the development of specific pathological hallmarks—such as Aβ fibrils in AD or α‐syn inclusions in PD—and thus, the severity of neuroinflammation.
The whole microbiota‐immunity‐CNS axis is characterized by dynamic and heterogeneous micro‐environments, in a complex integrated network of chemical and cellular mediators, biological structures, and processes. Microfluidic OOCs in vitro modeling offers an extraordinary opportunity to dissect specific axis mechanisms and define the real impact of microbiota‐mediated immunity in the etiology of neurodegenerative disorders. In this review, we went through the main biological and technological features for engineering in vitro models recapitulating the microbiota‐immunity‐CNS interfaces. We explained which are the most advanced technologies, for instance GOC with ECM‐like biomaterials representing the epithelial–endothelial interface for recreating the intestinal microenvironment, side micro‐channels providing a perfused capillary network to mimic microvasculature or the perfusable artificial vessels implemented with an ECM‐like 3D environment offering the possibility to stimulate the extravasation and follow it in real time. Despite all this, much effort is still needed in tuning the engineering parameters to build reliable and physiologically relevant OOCs. Currently, the most challenging aspects calling for research are the co‐culture of multiple cell populations in a 3D microenvironment, the integration, and combination of multi‐physics elements in a dynamic functional ensemble, and connection of the microbiota compartment with the CNS compartment in multi stage OOCs.[ 175 ]
Multi compartment and dynamic in vitro models will open new prospects for understanding microbiota‐immunity‐CNS axis thanks to the possibility of pooling together all the relevant cellular populations within an interconnected environment.
Conflict of Interest
The authors declare no conflict of interest.
Author Contribution
L.B., S.P., and L.I. prepared and wrote the original draft; C.G. and D.A. reviewed and edited the manuscript. All authors listed made a substantial, direct, intellectual contribution to the work, and approved it for publication. All authors have read and agreed to the published version of the manuscript. The authors thank Judith Baggot for English editing.
Acknowledgements
This work was funded by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (Grant agreement No. 724734‐MINERVA). The work reflects only the authors’ views and the Agency is not responsible for any use that may be made of the information contained.
Biographies
Lucia Boeri is a postdoctoral researcher at the Politecnico di Milano (Italy). She received her M.Sc. degree in molecular biology at the University of Parma in 2015 and her Ph.D. in bioengineering in 2018. Since 2014, she has been involved in molecular studies on the genetics of neurodegenerative disorders at the Istituto di Ricerche Farmacologiche Mario Negri IRCCS (Milan, Italy). Her research interests are now in the field of 3D tissue engineering for modeling neurodegeneration and microbiota‐brain communication. She is currently involved in the ERC project “MINERVA” at the TechnoBiology Laboratories‐Politecnico di Milano.

Simone Perottoni is a Ph.D. student in bioengineering. He received his M.Sc. degree in biomedical engineering with specialization on cells and tissue engineering at the Politecnico di Milano (Italy) in 2019. He started working in the field of advanced tissues/organs in vitro modeling during an intern period at the Trinity Centre for Bioengineering, Trinity College Dublin (Ireland) in 2019 and he is now pursuing his doctoral research on multi‐organ microphysiological systems for modeling neurodegeneration and microbiota‐brain communication. He is currently involved in the ERC project “MINERVA” at the TechnoBiology Laboratories‐Politecnico di Milano.

Boeri L., Perottoni S., Izzo L., Giordano C., Albani D., Microbiota‐Host Immunity Communication in Neurodegenerative Disorders: Bioengineering Challenges for In Vitro Modeling. Adv. Healthcare Mater. 2021, 10, 2002043. 10.1002/adhm.202002043
References
- 1. Dekaboruah E., Suryavanshi M. V., Chettri D., Verma A. K., Arch. Microbiol. 2020, 202, 2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gupta S., Kakkar V., Bhushan I., Microb. Pathog. 2019, 136, 103696. [DOI] [PubMed] [Google Scholar]
- 3. Lamont R. J., Koo H., Hajishengallis G., Nat. Rev. Microbiol. 2018, 16, 745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Van Giau V., Wu S. Y., Jamerlan A., An S. S. A., Kim S. Y., Hulme J., Nutrients 2018, 10, 1765.30441866 [Google Scholar]
- 5. Chen Y. E., Fischbach M. A., Belkaid Y., Nature 2018, 553, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boeri L., Izzo L., Sardelli L., Tunesi M., Albani D., Giordano C., Bioengineering 2019, 6, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tomkovich S., Jobin C., Immunology 2016, 147, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Herbst T., Sichelstiel A., Schär C., Yadava K., Bürki K., Cahenzli J., McCoy K., Marsland B. J., Harris N. L., Am. J. Respir. Crit. Care Med. 2011, 184, 198. [DOI] [PubMed] [Google Scholar]
- 9. Belkaid Y., Hand T. W., Cell 2014, 157, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. De Luca F., Shoenfeld Y., Clin. Exp. Immunol. 2019, 195, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gopalakrishnan V., Helmink B. A., Spencer C. N., Reuben A., Wargo J. A., Cancer Cell 2018, 33, 570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Singhrao S. K., Olsen I., J. Oral Microbiol. 2019, 11, 1563405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gerhardt S., Mohajeri M. H., Nutrients 2018, 10, 708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Çamcı G., Oğuz S., J. Clin. Neurol. 2016, 12, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kamer A. R., Fortea J. O., Videla S., Mayoral A., Janal M., Carmona‐Iragui M., Benejam B., Craig R. G., Saxena D., Corby P., Glodzik L., Annam K. R. C., Robbins M., de Leon M. J., Alzheimer's Dementia 2016, 2, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Main B. S., Minter M. R., Front. Neurosci. 2017, 11, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ceppa F. A., Izzo L., Sardelli L., Raimondi I., Tunesi M., Albani D., Giordano C., Front. Cell. Infect. Microbiol. 2020, 10, 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bonaz B., Bazin T., Pellissier S., Front. Neurosci. 2018, 12, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Spielman L. J., Gibson D. L., Klegeris A., Neurochem. Int. 2018, 120, 149. [DOI] [PubMed] [Google Scholar]
- 20. Jandhyala S. M., Talukdar R., Subramanyam C., Vuyyuru H., Sasikala M., Reddy D. N., World J. Gastroenterol. 2015, 21, 8787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dominy S. S., Lynch C., Ermini F., Benedyk M., Marczyk A., Konradi A., Nguyen M., Haditsch U., Raha D., Griffin C., Holsinger L. J., Arastu‐Kapur S., Kaba S., Lee A., Ryder M. I., Potempa B., Mydel P., Hellvard A., Adamowicz K., Hasturk H., 667 Walker G. D., Reynolds E. C., Faull R. L. M., Curtis M. A., Dragunow M., Potempa J., Sci. Adv. 2019, 5, aau3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Antonini M., Lo Conte M., Sorini C., Falcone M., Front. Immunol. 2019, 10, 1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao Y., Jaber V., Lukiw W. J., Front. Cell. Infect. Microbiol. 2017, 7, 318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Olsen I., Singhrao S. K., Potempa J., J. Oral Microbiol. 2018, 10, 1487742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Belkaid Y., Harrison O. J., Immunity 562, 2017, 46, 562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shi N., Li N., Duan X., Niu H., Mil. Med. Res. 2017, 4, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shroyer N. F., Kocoshis S. A., in Pediatric Gastrointestinal and Liver Disease, 4th ed. (Eds: Wyllie R., Hyams J. S.), Elsevier Inc., New York: 2011, Ch. 31, pp. 324. [Google Scholar]
- 28. Allaire J. M., Crowley S. M., Law H. T., Chang S. Y., Ko H. J., Vallance B. A., Trends Immunol. 2018, 39, 677. [DOI] [PubMed] [Google Scholar]
- 29. Rooks M. G., Garrett W. S., Nat. Rev. Immunol. 2016, 16, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Telesford K. M., Yan W., Ochoa‐Reparaz J., Pant A., Kircher C., Christy M. A., Begum‐Haque S., Kasper D. L., Kasper L. H., Gut Microbes 2015, 6, 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cobb B. A., Wang Q., Tzianabos A. O., Kasper D. L., Cell 2004, 117, 677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Erturk‐Hasdemir D., Kasper D. L., Ann. N. Y. Acad. Sci. 2018, 1417, 116. [DOI] [PubMed] [Google Scholar]
- 33. Lobionda S., Sittipo P., Kwon H. Y., Lee Y. K., Microorganisms 2019, 7, 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pugin B., Barcik W., Westermann P., Heider A., Wawrzyniak M., Hellings P., Akdis C. A., O'Mahony L., Microb. Ecol. Health Dis. 2017, 28, 1353881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Barcik W., Wawrzyniak M., Akdis C. A., O'Mahony L., Curr. Opin. Immunol. 2017, 48, 108. [DOI] [PubMed] [Google Scholar]
- 36. Alcañiz L., Vega A., Chacón P., Bekay R. E.l, Ventura I., Aroca R., Blanca M., Bergstralh D. T., Monteseirin J., FASEB J. 2013, 27, 2902. [DOI] [PubMed] [Google Scholar]
- 37. Amin K., Respir. Med. 2012, 106, 9. [DOI] [PubMed] [Google Scholar]
- 38. Idris A., Hasnain S. Z., Huat L. Z., Koh D., Oral Sci. Int. 2017, 14, 27. [Google Scholar]
- 39. Kinikoglu B., Damour O., Hasirci V., J. Artif. Organs 2015, 18, 8. [DOI] [PubMed] [Google Scholar]
- 40. Slocum C., Kramer C., Genco C. A., J. Intern. Med. 2016, 280, 114. [DOI] [PubMed] [Google Scholar]
- 41. De Diego I., Veillard F., Sztukowska M. N., Guevara T., Potempa B., Pomowski A., Huntington J. A., Potempa J., Gomis‐Rüth F. X., J. Biol. Chem. 2014, 289, 32291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Herath T. D. K., Darveau R. P., Seneviratne C. J., Wang C. Y., Wang Y., Jin L., PLoS One 2013, e58496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liang S., Krauss J. L., Domon H., McIntosh M. L., Hosur K. B., Qu H., Li F., Tzekou A., Lambris J. D., Hajishengallis G., J. Immunol. 2011, 186, 869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Makkawi H., Hoch S., Burns E., Hosur K., Hajishengallis G., Kirschning C. J., Nussbaum G., Front. Cell. Infect. Microbiol. 2017, 7, 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kaelberer M. M., Bohórquez D. V., Brain Res. 2018, 1693, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Caspani G., Swann J., Curr. Opin. Pharmacol. 2019, 48, 99. [DOI] [PubMed] [Google Scholar]
- 47. Bohórquez D. V., Shahid R. A., Erdmann A., Kreger A. M., Wang Y., Calakos N., Wang F., Liddle R. A., J. Clin. Invest. 2015, 125, 782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Johnston G. R., Webster N. R., Br. J. Anaesth. 2009, 102, 453. [DOI] [PubMed] [Google Scholar]
- 49. Goehler L. E., Relton J. K., Dripps D., Kiechle R., Tartaglia N., Maier S. F., Watkins L. R., Brain Res. Bull. 1997, 43, 357. [DOI] [PubMed] [Google Scholar]
- 50. Tracey K. J., Nature 2002, 420, 853. [DOI] [PubMed] [Google Scholar]
- 51. Amedei A., Morbidelli L., Molecules 2019, 24, 3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hoyles L., Snelling T., Umlai U. K., Nicholson J. K., Carding S. R., Glen R. C., McArthur S., Microbiome 2018, 6, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fung T. C., Neurobiol. Dis. 2020, 136, 104714. [DOI] [PubMed] [Google Scholar]
- 54. Girolamo F., Coppola C., Ribatti D., Brain, Behav., Immun. 2017, 65, 68. [DOI] [PubMed] [Google Scholar]
- 55. Albert‐Bayo M., Paracuellos I., González‐Castro A. M., Rodríguez‐Urrutia A., Rodríguez‐Lagunas M. J., Alonso‐Cotoner C., Santos J., Vicario M., Cells 2019, 8, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Conti P., Caraffa A., Ronconi G., Kritas S. K., Mastrangelo F., Tettamanti L., Theoharides T. C., Eur. J. Pharmacol. 2018, 818, 294. [DOI] [PubMed] [Google Scholar]
- 57. De Filippo K., Dudeck A., Hasenberg M., Nye E., Van Rooijen N., Hartmann K., Gunzer M., Roers A., Hogg N., Blood 2013, 121, 4930. [DOI] [PubMed] [Google Scholar]
- 58. Urb M., Sheppard D. C., PLoS Pathog. 2012, 8, 10.1371/journal.ppat.1002619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kurashima Y., Amiya T., Nochi T., Fujisawa K., Haraguchi T., Iba H., Tsutsui H., Sato S., Nakajima S., Iijima H., Kubo M., Kunisawa J., Kiyono H., Nat. Commun. 2012, 3, 1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Farhadi A., Banan A., Fields J., Keshavarzian A., J. Gastroenterol. Hepatol. Res. 2003, 18, 479. [DOI] [PubMed] [Google Scholar]
- 61. Ufnal M., Pham K., Med. Hypotheses 2017, 98, 35. [DOI] [PubMed] [Google Scholar]
- 62. Chakravortty D., Kumar K. S. N., Microbiol. Immunol. 1999, 43, 527. [DOI] [PubMed] [Google Scholar]
- 63. Fries W., Muja C., Crisafulli C., Cuzzocrea S., Mazzon E., Am. J. Physiol.: Gastrointest. Liver Physiol. 2008, 294, 938. [DOI] [PubMed] [Google Scholar]
- 64. Rodgers L. S., Fanning A. S., Cytoskeleton 2011, 68, 653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Luettig J., Rosenthal R., Barmeyer C., Schulzke J. D., Tissue Barriers 2015, 3, e977176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shen L., Turner J. R., Mol. Biol. Cell 2005, 16, 3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shen L., Black E. D., Witkowski E. D., Lencer W. I., Guerriero V., Schneeberger E. E., Turner J. R., J. Cell Sci. 2006, 119, 2095. [DOI] [PubMed] [Google Scholar]
- 68. Weber C. R., Raleigh D. R., Su L., Shen L., Sullivan E. A., Wang Y., Turner J. R., J. Biol. Chem. 2010, 285, 12037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bazzoni G., Martínez‐Estrada O. M., Orsenigo F., Cordenonsi M., Citi S., Dejana E., J. Biol. Chem. 2000, 275, 20520. [DOI] [PubMed] [Google Scholar]
- 70. Jacob C., Yang P. C., Darmoul D., Amadesi S., Saito T., Cottrell G. S., Coelho A. M., Singh P., Grady E. F., Perdue M., Bunnett N. W., J. Biol. Chem. 2005, 280, 31936. [DOI] [PubMed] [Google Scholar]
- 71. Groschwitz K. R., Wu D., Osterfeld H., Ahrens R., Hogan S. P., Am. J. Physiol.: Gastrointest. Liver Physiol. 2013, 304, G479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chokshi N. K., Guner Y. S., Hunter C. J., Upperman J. S., Grishin A., Ford H. R., Semin. Perinatol. 2008, 32, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Maiuolo J., Gliozzi M., Musolino V., Scicchitano M., Carresi C., Scarano F., Bosco F., Nucera S., Ruga S., Zito M. C., Mollace R., Palma E., Fini M., Muscoli C., Mollace V., Int. J. Mol. Sci. 2018, 19, 2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lee J. Y., Wasinger V. C., Yau Y. Y., Chuang E., Yajnik V., Leong R. W. L., Proteomes 2018, 6, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Xu L., Nirwane A., Yao Y., Stroke Vasc. Neurol. 2019, 4, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Baeten K. M., Akassoglou K., Dev. Neurobiol. 2011, 71, 1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ribatti D., Exp. Cell Res. 2015, 338, 119. [DOI] [PubMed] [Google Scholar]
- 78. Aryani A., Denecke B., Mol. Neurobiol. 2016, 53, 818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zuchero J. B., Barres B. A., Development 2015, 142, 3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Elgueta R., Benson M. J., De Vries V. C., Wasiuk A., Guo Y., Noelle R. J., Immunol. Rev. 2009, 229, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kim D. Y., Hong G. U., Ro J. Y., J. Neuroinflammation 2011, 8, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tran H., Mittal A., Sagi V., Luk K., Nguyen A., Gupta M., Nguyen J., Lamarre Y., Lei J., Guedes A., Gupta K., Front. Cell. Neurosci. 2019, 13, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Dong H., Zhang X., Qian Y., Med. Sci. Monit. Basic Res. 2014, 20, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Barata‐Antunes S., Cristóvão A. C., Pires J., Rocha S. M., Bernardino L., Biochim. Biophys. Acta, Mol. Basis Dis. 2017, 1863, 764. [DOI] [PubMed] [Google Scholar]
- 85. Skaper S. D., Facci L., Giusti P., Immunology 2014, 141, 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hidetoshi T.‐S., Makoto T., Inoue K., Wiley Interdiscip. Rev.: Membr. Transp. Signaling 2012, 1, 493. [Google Scholar]
- 87. Kowalski K., Mulak A., Neurogastroenterol. Motil. 2019, 25, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zhan X., Stamova B., Jin L. W., Decarli C., Phinney B., Sharp F. R., Neurology 2016, 87, 2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Palpagama T. H., Waldvogel H. J., Faull R. L. M., Kwakowsky A., Front. Mol. Neurosci. 2019, 12, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Haukedal H., Freude K., J. Mol. Biol. 2019, 431, 1818. [DOI] [PubMed] [Google Scholar]
- 91. Philips T., Rothstein J. D., Exp. Neurol. 2014, 262, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Fellner L., Jellinger K. A., Wenning G. K., Stefanova N., Acta Neuropathol. 2011, 121, 675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bates K. A., Fonte J., Robertson T. A., Martins R. N., Harvey A. R., Neuroscience 2002, 113, 785. [DOI] [PubMed] [Google Scholar]
- 94. Novellino F., Saccà V., Donato A., Zaffino P., Spadea M. F., Vismara M., Arcidiacono B., Malara N., Presta I., Donato G., Int. J. Mol. Sci. 2020, 21, 1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Giunta B., Fernandez F., Nikolic W. V., Obregon D., Rrapo E., Town T., Tan J., J. Neuroinflammation 2008, 5, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Costantini E., D'Angelo C., Reale M., Mediators Inflammation 2018, 2018, 6039171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Streit W. J., Xue Q. S., Tischer J., Bechmann I., Acta Neuropathol. Commun. 2014, 2, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kwan W., Träger U., Davalos D., Chou A., Bouchard J., Andre R., Miller A., Weiss A., Giorgini F., Cheah C., Möller T., Stella N., Akassoglou K., Tabrizi S. J., Muchowski P. J., J. Clin. Invest. 2012, 122, 4737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Fitzgerald E., Murphy S., Martinson H. A., Front. Neurosci. 2019, 13, 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sampson T. R., Debelius J. W., Thron T., Janssen S., Shastri G. G., Ilhan Z. E., Challis C., Schretter C. E., Rocha S., Gradinaru V., Chesselet M. F., Keshavarzian A., Shannon K. M., Krajmalnik‐Brown R., Wittung‐Stafshede P., Knight R., Mazmanian A. K., Cell 2016, 167, 1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Kim C., Lv G., Lee J. S., Jung B. C., Masuda‐Suzukake M., Hong C. S., Valera E., Lee H. J., Paik S. R., Hasegawa M., Masliah E., Eliezer D., Lee S. J., Sci. Rep. 2016, 6, 30891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Serrano‐Pozo A., Frosch M. P., Masliah E., Hyman B. T., Cold Spring Harbor Perspect. Med. 2011, 1, a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Rea K., Dinan T. G., Cryan J. F., Neurobiol. Stress 2016, 4, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Gentleman S. M., Neuropathol. Appl. Neurobiol. 2013, 39, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Friedland R. P., Chapman M. R., PLoS Pathog. 2017, 13, e1006654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zhou J., Yu W., Zhang M., Tian X., Li Y., Lü Y., Neurochem. Res. 2019, 44, 1138. [DOI] [PubMed] [Google Scholar]
- 107. Gawish R., Martins R., Böhm B., Wimberger T., Sharif O., Lakovits K., Schmidt M., Knapp S., FASEB J. 2015, 29, 1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Trojsi F., Sagnelli A., Cirillo G., Piccirillo G., Femiano C., Izzo F., Monsurrò M. R., Tedeschi G., Case Rep. Med. 2012, 2012, 324685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Zarei S., Carr K., Reiley L., Diaz K., Guerra O., Altamirano P. F., Pagani W., Lodin D., Orozco G., Chinea A., Surg. Neurol. Int. 2015, 6, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Béland L.‐C., Markovinovic A., Jakovac H., De Marchi F., Bilic E., Mazzini L., Kriz J., Munitic I., Brain Commun. 2020, 2, fcaa124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Zhang R., Gascon R., Miller R. G., Gelinas D. F., Mass J., Hadlock K., Jin X., Reis J., Narvaez A., McGrath M. S., J. Neuroimmunol. 2005, 159, 215. [DOI] [PubMed] [Google Scholar]
- 112. Di Gioia D., Bozzi Cionci N., Baffoni L., Amoruso A., Pane M., Mogna L., Gaggìa F., Lucenti M. A., Bersano E., Cantello R., De Marchi F., Mazzini L., BMC Med. 2020, 18, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Rowin J., Xia Y., Jung B., Sun J., Physiol. Rep. 2017, 5, e13443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Zhang R., Miller R. G., Gascon R., Champion S., Katz J., Lancero M., Narvaez A., Honrada R., Ruvalcaba D., McGrath M. S., J. Neuroimmunol. 2009, 206, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Zhao W., Beers D. R., Hooten K. G., Sieglaff D. H., Zhang A., Kalyana‐Sundaram S., Traini C. M., Halsey W. S., Hughes A. M., Sathe G. M., Livi G. P., Fan G. H., Appel S. H., JAMA Neurol. 2017, 74, 677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Dobson R., Giovannoni G., Eur. J. Neurol. 2019, 26, 27. [DOI] [PubMed] [Google Scholar]
- 117. Adamczyk‐Sowa M., Medrek A., Madej P., Michlicka W., Dobrakowski P., J. Immunol. Res. 2017, 2017, 7904821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Danikowski K. M., Jayaraman S., Prabhakar B. S., J. Neuroinflammation 2017, 14, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Sheu J. J., Lin H. C., Eur. J. Neurol. 2013, 20, 1053. [DOI] [PubMed] [Google Scholar]
- 120. Kirby T., Ochoa‐Repáraz J., Med. Sci. 2018, 6, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Erny D., De Angelis A. L. H., Jaitin D., Wieghofer P., Staszewski O., David E., Keren‐Shaul H., Mahlakoiv T., Jakobshagen K., Buch T., Schwierzeck V., Utermöhlen O., Chun E., Garrett W. S., Mccoy K. D., Diefenbach A., Staeheli P., Stecher B., Amit I., Prinz M., Nat. Neurosci. 2015, 18, 965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Minter M. R., Hinterleitner R., Meisel M., Zhang C., Leone V., Zhang X., Oyler‐Castrillo P., Zhang X., Musch M. W., Shen X., Jabri B., Chang E. B., Tanzi R. E., Sisodia S. S., Sci. Rep. 2017, 7, 10411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Cryan J. F., O'riordan K. J., Cowan C. S. M., Sandhu K. V., Bastiaanssen T. F. S., Boehme M., Codagnone M. G., Cussotto S., Fulling C., Golubeva A. V., Guzzetta K. E., Jaggar M., Long‐Smith C. M., Lyte J. M., Martin J. A., Molinero‐Perez A., Moloney G., Morelli E., Morillas E., O'connor R., Cruz‐Pereira J. S., Peterson V. L., Rea K., Ritz N. L., Sherwin E., Spichak S., Teichman E. M., van de Wouw M., Ventura‐Silva A. P., Wallace‐Fitzsimons S. E., Hyland N., Clarke G., Dinan T. G., Physiol. Rev. 2019, 99, 1877. [DOI] [PubMed] [Google Scholar]
- 124. Gotkine M., Kviatcovsky D., Elinav E., Gut Microbes 2020, 11, 1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Raimondi I., Izzo L., Tunesi M., Comar M., Albani D., Giordano C., Front. Bioeng. Biotechnol. 2020, 7, 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Ingber D. E., Cell 2016, 164, 1105. [DOI] [PubMed] [Google Scholar]
- 127. Yesil‐Celiktas O., Hassan S., Miri A. K., Maharjan S., Al‐kharboosh R., Quiñones‐Hinojosa A., Zhang Y. S., Adv. Biosyst. 2018, 2, 1800109. [Google Scholar]
- 128. Wu Q., Liu J., Wang X., Feng L., Wu J., Zhu X., Wen W., Gong X., Biomed. Eng. Online 2020, 19, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Polini A., del Mercato L. L., Barra A., Zhang Y. S., Calabi F., Gigli G., Drug Discovery Today 2019, 24, 517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Shanti A., Teo J., Stefanini C., Pharmaceutics 2018, 10, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Ma Q., Xing C., Long W., Wang H. Y., Liu Q., Wang R. F., J. Neuroinflammation 2019, 16, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Kim H. J., Huh D., Hamilton G., Ingber D. E., Lab Chip 2012, 12, 2165. [DOI] [PubMed] [Google Scholar]
- 133. Shin W., Kim H. J., Methods Cell Biol. 2018, 146, 135. [DOI] [PubMed] [Google Scholar]
- 134. Wang Y., Ahmad A. A., Sims C. E., Magness S. T., Allbritton N. L., Lab Chip 2014, 14, 1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Kim R., Attayek P. J., Wang Y., Furtado K. L., Tamayo R., Sims C. E., Allbritton N. L., Biofabrication 2020, 12, 015006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Ambrosini Y. M., Shin W., Min S., Kim H. J., Immune Network 2020, 20, e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Kim H. J., Li H., Collins J. J., Ingber D. E., Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Shin W., Kim H. J., Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E10539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Maurer M., Gresnigt M. S., Last A., Wollny T., Berlinghof F., Pospich R., Cseresnyes Z., Medyukhina A., Graf K., Gröger M., Raasch M., Siwczak F., Nietzsche S., Jacobsen I. D., Figge M. T., Hube B., Huber O., Mosig A. S., Biomaterials 2019, 220, 119396. [DOI] [PubMed] [Google Scholar]
- 140. Murfee W. L., Peirce S. M., Microcirculation 2017, 24, 23. [DOI] [PubMed] [Google Scholar]
- 141. Hosic S., Puzan M., Lake W., Zhou F., Koppes R., Breault D., Murthy S., Koppes A., bioRxiv, Bioeng. 2018, 400721, 10.1101/400721. [DOI] [Google Scholar]
- 142. Henderson A. R., Choi H., Lee E., Micromachines 2020, 11, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Sharifi F., Htwe S. S., Righi M., Liu H., Pietralunga A., Yesil‐Celiktas O., Maharjan S., Cha B. H., Shin S. R., Dokmeci M. R., Vrana N. E., Ghaemmaghami A. M., Khademhosseini A., Zhang Y. S., Adv. Healthcare Mater. 2019, 8, e1801425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Seiler K. M., Bajinting A., Alvarado D. M., Traore M. A., Binkley M. M., Goo W. H., Lanik W. E., Ou J., Ismail U., Iticovici M., King C. R., VanDussen K. L., Swietlicki E. A., Gazit V., Guo J., Luke C. J., Stappenbeck T., Ciorba M. A., George S. C., Meacham J. M., Rubin D. C., Good M., Warner B. W., Sci. Rep. 2020, 10, 3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Kim W., Kim G., ACS Appl. Mater. Interfaces 2018, 10, 41185. [DOI] [PubMed] [Google Scholar]
- 146. Wang Y., Gunasekara D. B., Reed M. I., DiSalvo M., Bultman S. J., Sims C. E., Magness S. T., Allbritton N. L., Biomaterials 2017, 128, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Park D. Y., Lee J., Chung J. J., Jung Y., Kim S. H., Trends Biotechnol. 2020, 38, 99. [DOI] [PubMed] [Google Scholar]
- 148. Virlogeux A., Moutaux E., Christaller W., Genoux A., Bruyère J., Fino E., Charlot B., Cazorla M., Saudou F., Cell Rep. 2018, 22, 110. [DOI] [PubMed] [Google Scholar]
- 149. Pagella P., Neto E., Jiménez‐Rojo L., Lamghari M., Mitsiadis T. A., Front. Physiol. 2014, 5, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Silva D. I., Dos Santos B. P., Leng J., Oliveira H., Amédée J., Cell Death Dis. 2017, 8, 3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Soucy J. R., Bindas A. J., Brady R., Torregrosa T., Denoncourt C. M., Hosic S., Dai G., Koppes A. N., Koppes R. A., Adv. Biosyst. 2020, 4, e2000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Rahimi C., Rahimi B., Padova D., Rooholghodos S. A., Bienek D. R., Luo X., Kaufman G., Raub C. B., Biomicrofluidics 2018, 12, 054106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Marchetti L., Engelhardt B., Vasc. Biol. 2020, 2, H1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Walter F. R., Valkai S., Kincses A., Petneházi A., Czeller T., Veszelka S., Ormos P., Deli M. A., Dér A., Sens. Actuators, B 2016, 222, 1209. [Google Scholar]
- 155. Sivandzade F., Cucullo L., J. Cereb. Blood Flow Metab. 2018, 38, 1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Mossu A., Rosito M., Khire T., Chung H. L.i, Nishihara H., Gruber I., Luke E., Dehouck L., Sallusto F., Gosselet F., McGrath J. L., Engelhardt B. J., J. Cereb. Blood Flow Metab. 2019, 39, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Koo Y., Hawkins B. T., Yun Y., Sci. Rep. 2018, 8, 2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Hawkins B. T., Grego S., Sellgren K. L., Brain Res. 2015, 1608, 167. [DOI] [PubMed] [Google Scholar]
- 159. Qi D., Wu S., Lin H., Kuss M. A., Lei Y., Krasnoslobodtsev A., Ahmed S., Zhang C., Kim H. J., Jiang P., Duan B., ACS Appl. Mater. Interfaces 2018, 10, 21825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Bischel L. L., Coneski P. N., Lundin J. G., Wu P. K., Giller C. B., Wynne J., Ringeisen B. R., Pirlo R. K., J. Biomed. Mater. Res., Part A 2016, 104, 901. [DOI] [PubMed] [Google Scholar]
- 161. Ransohoff R. M., Schafer D., Vincent A., Blachère N. E., Bar‐Or A., Neurotherapeutics 2015, 12, 896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Prinz M., Priller J., Nat. Neurosci. 2017, 20, 136. [DOI] [PubMed] [Google Scholar]
- 163. Molteni R., Bianchi E., Patete P., Fabbri M., Baroni G., Dubini G., Pardi R., Lab Chip 2015, 15, 195. [DOI] [PubMed] [Google Scholar]
- 164. Schulte‐Mecklenbeck A., Bhatia U., Schneider‐Hohendorf T., Schwab N., Wiendl H., Gross C. C., J. Visualized Exp. 2017, 122, e55390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Goncharova V., Khaldoyanidi S. K., J. Visualized Exp. 2013, 77, e50959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Chen M. B., Whisler J. A., Fröse J., Yu C., Shin Y., Kamm R. D., Nat. Protoc. 2017, 12, 865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Guttenplan K. A., Liddelow S. A., J. Exp. Med. 2019, 216, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Wong J. F., Simmons C. A., Lab Chip 2019, 19, 1060. [DOI] [PubMed] [Google Scholar]
- 169. Dolan J. M., Kolega J., Meng H., Ann. Biomed. Eng. 2013, 41, 1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Cucullo L., Marchi N., Hossain M., Janigro D., J. Cereb. Blood Flow Metab. 2011, 31, 767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Brown J. A., Codreanu S. G., Shi M., Sherrod S. D., Markov D. A., Neely M. D., Britt C. M., Hoilett O. S., Reiserer R. S., Samson P. C., McCawley L. J., Webb D. J., Bowman A. B., McLean J. A., Wikswo J. P., J. Neuroinflammation 2016, 13, 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. McMinn P. H., Hind L. E., Huttenlocher A., Beebe D. J., Lab Chip 2019, 19, 3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Campisi M., Shin Y., Osaki T., Hajal C., Chiono V., Kamm R. D., Biomaterials 2018, 180, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Park J., Wetzel I., Marriott I., Dréau D., D'Avanzo C., Kim D. Y., Tanzi R. E., Cho H., Nat. Neurosci. 2018, 21, 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Raimondi M. T., Albani D., Giordano C., Trends Mol. Med. 2019, 25, 737. [DOI] [PubMed] [Google Scholar]
- 176. Kufer T. A., Banks D. J., Philpott D. J., Ann. N. Y. Acad. Sci. 2006, 1072, 19. [DOI] [PubMed] [Google Scholar]
- 177. Chassaing B., Ley R. E., Gewirtz A. T., Gastroenterology 2014, 147, 1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Bloes D. A., Kretschmer D., Peschel A., Nat. Rev. Microbiol. 2015, 13, 95. [DOI] [PubMed] [Google Scholar]
- 179. Gürtler C., Bowie A. G., Trends Microbiol. 2013, 21, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Zelante T., Iannitti R. G., Cunha C., DeLuca A., Giovannini G., Pieraccini G., Zecchi R., D'Angelo C., Massi‐Benedetti C., Fallarino F., Carvalho A., Puccetti P. P., Romani L., Immunity 2013, 39, 372. [DOI] [PubMed] [Google Scholar]
- 181. Alexeev E. E., Lanis J. M., Kao D. J., Campbell E. L., Kelly C. J., Battista K. D., Gerich M. E., Jenkins B. R., Walk S. T., Kominsky D. J., Colgan S. P., Am. J. Pathol. 2018, 188, 1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Burbach K. M., Poland A., Bradfield C. A., Proc. Natl. Acad. Sci. U. S. A 1992, 89, 8185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Kibe R., Kurihara S., Sakai Y., Suzuki H., Ooga T., Sawaki E., Muramatsu K., Nakamura A., Yamashita A., Kitada Y., Kakeyama M., Benno Y., Matsumoto M., Sci. Rep. 2014, 4, 4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Zhang J., Yu C., Zhang X., Chen H., Dong J., Lu W., Song Z., Zhou W., J. Neuroinflammation 2018, 15, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Liu Y., Kongsuphol P., Chiam S. Y., Zhang Q. X., Gourikutty S. B. N., Saha S., Biswas S. K., Ramadan Q., Lab Chip 2019, 19, 241. [DOI] [PubMed] [Google Scholar]
- 186. Kongsuphol P., Gupta S., Liu Y., Bhuvanendran Nair Gourikutty S., Biswas S. K., Ramadan Q., Sci. Rep. 2019, 9, 4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Zhu J., He J., Verano M., Brimmo A. T., Glia A., Qasaimeh M. A., Chen P., Aleman J. O., Chen W., Lab Chip 2018, 18, 3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Huh D., Matthews B. D., Mammoto A., Montoya‐Zavala M., Yuan Hsin H., Ingber D. E., Science 2010, 328, 1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Punde T. H., Wu W.‐H., Lien P.‐C., Chang Y.‐L., Kuo P.‐H., Chang M. D.‐T., Lee K.‐Y., Huang C.‐D., Kuo H.‐P., Chan Y.‐F., Shih P.‐C., Liu C.‐H., Integr. Biol. 2015, 7, 162. [DOI] [PubMed] [Google Scholar]
- 190. Benam K. H., Villenave R., Lucchesi C., Varone A., Hubeau C., Lee H. H., Alves S. E., Salmon M., Ferrante T. C., Weaver J. C., Bahinski A., Hamilton G. A., Ingber D. E., Nat. Methods 2016, 13, 151. [DOI] [PubMed] [Google Scholar]
- 191. Jiang H., Jiang D., Zhu P., Pi F., Ji J., Sun C., Sun J., Sun X., Biosens. Bioelectron. 2016, 83, 126. [DOI] [PubMed] [Google Scholar]
- 192. Ramadan Q., Ting F. C. W., Lab Chip 2016, 16, 1899. [DOI] [PubMed] [Google Scholar]
- 193. Rothbauer M., Charwat V., Bachmann B., Sticker D., Novak R., Wanzenböck H., Mathies R. A., Ertl P., Lab Chip 2019, 19, 1916. [DOI] [PubMed] [Google Scholar]
- 194. Wu W. H., Punde T. H., Shih P. C., Fu C. Y., Wang T. P., Hsu L., Chang H. Y., Liu C. H., Sens. Actuators, B 2015, 209, 470. [Google Scholar]
- 195. Menon N. V., Tay H. M., Wee S. N., Li K. H. H., Hou H. W., Lab Chip 2017, 17, 2960. [DOI] [PubMed] [Google Scholar]
- 196. Nam U., Kim S., Park J., Jeon J. S., Micromachines 2020, 11, 747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Biselli E., Agliari E., Barra A., Bertani F. R., Gerardino A., De Ninno A., Mencattini A., Di Giuseppe D., Mattei F., Schiavoni G., Lucarini V., Vacchelli E., Kroemer G., Di Natale C., Martinelli E., Businaro L., Sci. Rep. 2017, 7, 12737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Pavesi A., Tan A. T., Koh S., Chia A., Colombo M., Antonecchia E., Miccolis C., Ceccarello E., Adriani G., Raimondi M. T., Kamm R. D., Bertoletti A., JCI Insight 2017, 2, e89762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Shim S., Belanger M. C., Harris A. R., Munson J. M., Pompano R. R., Lab Chip 2019, 19, 1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Gjorevski N., Avignon B., Gérard R., Cabon L., Roth A. B., Bscheider M., Moisan A., Lab Chip 2020, 20, 3365. [DOI] [PubMed] [Google Scholar]
- 201. Yin F., Zhu Y., Zhang M., Yu H., Chen W., Qin J., Toxicol. In Vitro 2019, 54, 105. [DOI] [PubMed] [Google Scholar]
- 202. Yin F., Zhu Y., Wang H., Wang Y., Li D., Qin J., ACS Biomater. Sci. Eng. 2020, 6, 4644. [DOI] [PubMed] [Google Scholar]
- 203. won Jeon J., Choi N., Lee S. H., Sung J. H., Biomed. Microdevices 2020, 22, 65. [DOI] [PubMed] [Google Scholar]