

Abstract
Hemostatic materials have played a significant role in mitigating traumatic injury by controlling bleeding, however, the fabrication of the desirable material's structure to enhance the accumulation of blood cells and platelets for highly efficient hemostasis is still a great challenge. In this work, directed assembly of poly(vinyl alcohol) (PVA) macromolecules covering the rigid Kevlar nanofiber (KNF) network during 3D printing process is utilized to fabricate hydrophilic, biocompatible, and mechanically stable KNF‐PVA aerogel filaments for effective enriching blood components by fast water absorption. As such, KNF‐PVA aerogel gauzes demonstrate remarkable water permeability (338 mL cm−2 s−1 bar−1), water absorption speed (as high as 9.64 g g−1 min−1) and capacity (more than ten times of self‐weight), and ability to enrich micron‐sized particles when contacting aqueous solution. All these properties favor efficient hemostasis and the resulting KNF‐PVA aerogel gauzes significantly outperform the commercial product Quikclot Gauze (Z‐Medica) during in vivo experiments with the rat liver laceration model, reducing the hemostasis time by half (60 ± 4 s) and the blood loss by two thirds (0.07 ± 0.01 g). These results demonstrate a robust strategy to design various aerogel gauzes for hemostasis applications.
Keywords: 3D printing, aerogel gauzes, hemostatic materials, Kevlar nanofibers, poly(vinyl alcohol)
Biocompatible and mechanically stable Kevlar nanofiber‐poly(vinyl alcohol) (KNF‐PVA) hybrid aerogel gauzes demonstrating remarkable water permeability, water absorption speed and capacity, and ability to enrich solution‐suspended micronparticles are fabricated via direct‐ink‐writing‐based 3D printing. A prepared aerogel gauzes significantly outperform the commercial product Quikclot Gauze (Z‐Medica) in rat liver laceration injury model, reducing the hemostasis time by half and the blood loss by two thirds.
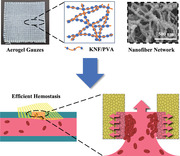
1. Introduction
Uncontrolled hemorrhage is responsible for almost half of the deaths in traumatic accidents before emergency treatments.[ 1 ] The survival rate could substantially increase to 85% by controlling the bleeding within 30 min,[ 2 ] thus making effective hemostatic materials highly desirable. Consisting of woven or non‐woven solid fibers, gauze is by far the most widely applied hemostatic material, which functions by permeating blood while allowing blood cells and platelets to deposit on the fiber surface to form blood clots.[ 3 ] Due to the µm‐scale inter‐fiber spacing of gauzes, however, blood components tend to migrate together with blood fluid instead of settling on fibers, leading to inefficient hemostasis with an undesired large volume of blood loss.[ 3 , 4 ] To address this issue, impregnating gauzes with nanoporous microparticles such as zeolites and clays (e.g., QuikClot Gauze (QCG) from Z‐Medica) are proposed to accumulate blood components by absorbing water.[ 5 , 6 ] Yet the enrichment effect is often limited by the amount of coated microparticles, of which the detachment further reduces the hemostatic efficiency and raises additional safety concerns.[ 3 , 6 ] As such, a proper porous structure that effectively enriches blood cells and platelets while permeating blood liquid is critical for achieving desired hemostatic performance of the gauze.
Aerogel is a well‐known class of nanoporous material characterized by ultrahigh porosity, large surface area, and ultralow density.[ 7 , 8 , 9 ] Particularly, its highly porous structure and nm‐scale pores (2–50 nm) can be designed to quickly absorb and store a large amount of water[ 10 , 11 ] while blocking µm‐sized microparticles,[ 12 , 13 ] both favoring effective enrichment of blood components. In this regard, aerogel gauzes offer a unique strategy to design hemostatic materials, that is, replacing solid fibers in conventional gauzes with highly porous and water‐absorbing aerogels instead of coating them with limited amount of nanoporous microparticles. On the other hand, no such studies have been reported thus far, largely due to the challenge to fabricate common formats of aerogels (e.g., powders, monoliths, and membranes) into the gauze pattern. Meanwhile, although emerging aerogel fibers[ 14 , 15 , 16 ] are compatible with the gauze fabrication, additional weaving procedures not only increase the fabrication time but also require stringent mechanical properties of aerogel fibers.
Here, we develop a Kevlar nanofiber‐poly(vinyl alcohol) (KNF‐PVA) aerogel gauze by designing its chemical compositions and physical structures for hemostatic applications (Scheme 1 ). Microscopically, the composite aerogel involves assembled PVA molecules wrapping around the rigid KNF network, which is designed by utilizing their hydrogen bonding interactions. Kevlar is the common name of para‐aramid and can be properly dissolved to form dispersions of nanoscale fibers. With similar mechanical properties as bulk Kevlar, KNF have been used to prepare a variety of high strength functional materials.[ 17 ] Therefore, the chemical composition of KNF‐PVA and their arrangement simultaneously harness the biocompatibility and hydrophilicity of PVA[ 18 ] and the mechanical performance of KNF network,[ 19 ] which would be impossible for single‐component PVA or KNF aerogels. Macroscopically, the aerogel gauze is conveniently fabricated by 3D printing of KNF‐PVA gel into self‐supporting patterns, which are accurately preserved during subsequent supercritical drying. In the aerogel gauze's hierarchically porous structure for highly efficient hemostasis, nm‐sized pores inside aerogel filaments quickly absorb and store water, which enriches blood components; while the µm‐sized inter‐filament spacing permeates the blood fluid, which induces a substantial flux of blood toward the aerogel surface.
Scheme 1.
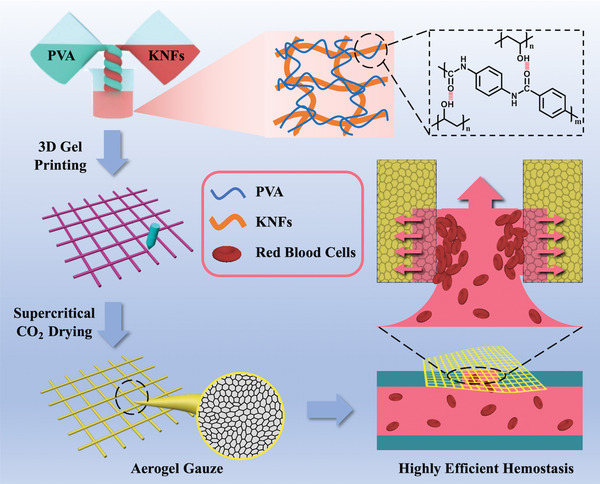
Design of the chemical composition, physical structure, and 3D printing of KNF‐PVA aerogel gauzes for hemostatic application.
Thanks to these material designs, the as‐prepared aerogel gauze is hydrophilic (water contact angle of 42°), flexible (curvature radius of 230 µm), mechanically stable (elastic modulus up to 111 MPa), and possessed unique properties for topical hemostatic applications. First, the large inter‐filament spacing in the aerogel gauze leads to substantially increased water permeation flux of 338 mL cm−2 s−1 bar−1, which is two orders of magnitude higher than values reported with PVA aerogel membranes under similar gravity‐driven flow conditions.[ 20 ] In the meantime, the nanoporous structure of aerogels with specific surface area up to 189 m2 g−1 allows aerogel gauze to absorb water up to ten times of its self‐weight within 1 min, and simultaneously enriches local microparticle concentration by a factor of 3. Altogether, these properties enable aerogel gauze to efficiently concentrate blood cells during in vivo animal experiments, reducing the hemostasis time down to 60 s and blood loss down to 0.07 g with the rat liver laceration model. Notably, these values are only half and one third of the result when using the widely adopted commercial product QCG (Z‐Medica).
2. Results and Discussion
2.1. Preparation and Characterization of KNF‐PVA Inks
To enable direct‐ink‐writing (DIW) without using a supporting medium or cooling stage, the ink must have specific rheological properties, including appropriate yield stress, large storage modulus (G′), and shear‐thinning behavior.[ 21 , 22 ] Unlike reported strategies by introducing additives or increasing components’ concentration,[ 23 ] KNF‐PVA inks (KP‐I) were prepared by simply mixing PVA and KNF solutions,both in dimethyl sulfoxide (DMSO)[ 17 , 24 ] followed by mechanically stirring at 60 °C, which yielded an orange‐colored mixture that was different from transparent PVA and dark brown KNF solutions (Figure 1a). Upon cooling to 25 °C, KP‐I formed organogel and became viscous liquid again once heated to 60 °C (Figure 1b). In this study, KP‐I involved a fixed mass ratio of PVA to KNF (5:1) to achieve sufficient G′ and the best elongation at break (above 40%) for printed aerogel filaments (Figure S1, Supporting Information).
Figure 1.
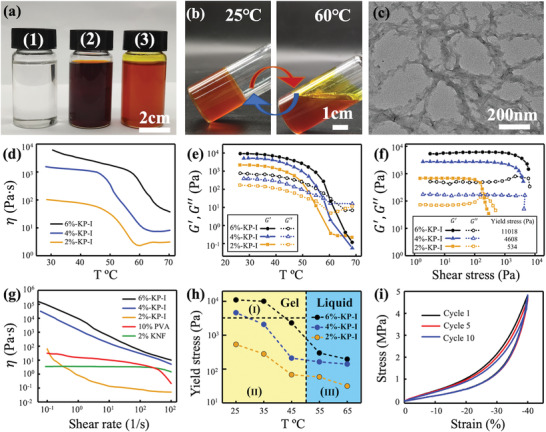
Preparation and characterization of KNF‐PVA inks (KP‐I). a) Mixing transparent 1) PVA/DMSO and dark 2) brown KNF/DMSO solutions yielded 3) orange KNF‐PVA inks. b) Transition of KP‐I between the gel (25 °C) and liquid (60 °C) state. c) TEM image of KP‐I. d) Temperature‐dependence of viscosity for KP‐I with different total concentrations. e) Dynamic modulus of KP‐I measured at different temperatures. f) Yield stress of KP‐I characterized by measuring storage (G′) and loss (G″) modulus at increasing shear stress values. g) Shear‐rate dependent viscosity of KNF‐PVA inks, PVA in DMSO solution (PVA/DMSO) and KNF in DMSO solution (KNF/DMSO). h) Determining printable temperatures (Region II) of KP‐I. Symbols were yield stress values of KP‐I measured at different temperatures, the vertical dashed line indicated the gel‐liquid transition temperature (≈50 °C) of KP‐I, and the horizontal dashed line marked the maximum shear stress generated in the 3D printer. KP‐I was in the gel state in Region I and II, and became liquid in Region III. i) Ten repeated cyclic stress–strain curves for 4%‐KP‐I. All measurements were conducted at 25 °C unless otherwise specified.
Meanwhile, TEM image of KP‐I (Figure 1c) revealed a network of nanofibers similar to that in KNF/DMSO (Figure S2a, Supporting Information), but involving rougher nanofiber surfaces that were likely due to the association of PVA chains. PVA/DMSO, on the other hand, exhibited a different structure arising from the phase separation of PVA polymers during the DMSO evaporation (Figure S2b, Supporting Information). The gel‐liquid transition behavior and TEM characterization suggested physical cross‐linking in KP‐I that directed the assembly of PVA molecules around KNF upon mixing. This interaction was likely driven by the intermolecular hydrogen bonding[ 25 ] between hydroxyl groups of PVA and carbonyl groups of KNF, as indicated by the shift of C=O stretching vibration in FTIR absorption spectra of KNF‐PVA to KNF samples (Figure S3, Supporting Information) and melting points between KNF‐PVA and PVA (Figure S4a, Supporting Information).
Rheological measurements further confirmed that KP‐I, even with total PVA‐KNF concentration as low as 2 wt% (2%‐KP‐I), has occurred during the gel‐liquid transition between 50 and 60 °C. In particular, the viscosity of KP‐I dropped substantially during the transition, where a nearly two orders of magnitude change in viscosity was observed for 4%‐KP‐I (Figure 1d). Similarly, the oscillation rheology measurements indicated a solid‐like behavior of KP‐I at T < 50 °C, as the storage modulus (G′) was at least an order of magnitude larger than the loss modulus (G″). As T increased above 50 °C, however, G′ dropped quickly while G″ increased until complete domination over G′ at T > 60 °C, which indicated the liquid state of KP‐I (Figure 1e). By comparison, both PVA/DMSO and KNF/DMSO showed the liquid property with G″ dominating G′ in the tested temperature range (Figure S5, Supporting Information).
Due to the physical interaction between PVA and KNF, KP‐I at room temperature (25 °C) exhibited large G′, finite yield stress values, and shear‐thinning behavior that are necessary to enable DIW. More specifically, the yield stress of KP‐I increased from 534 Pa to over 10 000 Pa when the concentration increased from 2 to 6 wt% (Figure 1f). In addition, unyielded KP‐I exhibited a solid‐like behavior with nearly constant G′ that was at least an order of magnitude larger than G″ for all concentrations. These properties allow KP‐I to self‐support printed structures and resist deformation caused by self‐weight or external forces. Meanwhile, yielded KP‐I shear thinned to enable smooth extrusion from the printing nozzle. This shear thinning behavior became more profound—nearly exponential reduction in η with —as the total concentration of KP‐I increased above 2 wt% (Figure 1g). By contrast, 10 wt% PVA/DMSO behaved as a Newtonian fluid and 2 wt% KNF/DMSO was only slightly shear‐thinned within the tested range of shear rate.
Because the yield stress of KP‐I spiked by nearly 20 times with the concentration only changing by a factor of 3 (Figure 1f), it is critical to evaluate whether the pressurized air in our printing system can yield KP‐I. To this end, we assume a steady‐state, pressure‐driven laminar flow of KP‐I in the printing nozzle and neglect the gravity effect. This led to
| (1) |
in an axisymmetric cylindrical coordinate, which gave the maximum shear stress as[ 26 ]
| (2) |
where L = 1.3 cm and R = 250 µm were the length and inner radius of the printing nozzle. Using the maximum driving pressure of our printing system ΔP = 0.5 MPa, Pa was estimated to occur at the nozzle wall. While this value seemed to only allow printing of 2%‐KP‐I based on yield stress values measured at 25 °C, we noticed that KP‐I in the gel state showed fairly constant (and large) G′ (Figure 1f) but decreasing τ y with temperature (Figure 1h, symbols). Utilizing this property, the printing temperature of KP‐I can be adjusted to reduce the yield stress for easier extrusion, while preserving large G′ to maintain printed structures without using a cooling stage. To illustrate this, we plot τ y against T for KP‐I of various concentrations and establish an enclosed region of applicable printing temperature for each ink concentration (Region II in Figure 1h). The right bound of Region II (vertical dashed line in Figure 1h) was set by the ink's gel‐liquid transition temperature (≈50 °C), beyond which all inks entered the liquid state (Region III) and hardly possessed fixed shapes after flowing through the printing nozzle (Figure S6, Supporting Information). And the top bound was marked by the horizontal dashed line in Figure 1h located at Pa so Region I indicated unprintable temperature conditions. Depending on how τ y changed with T for KP‐I of different concentrations, distinct options of printing temperatures were available. For example, 2%‐KP‐I can be printed at room temperature (25 °C), whereas 4%‐ KP‐I and 6%‐KP‐I required to be heated to different temperatures (still in the gel state) to fall in Region II.
Once extruded and exited the nozzle, KP‐I returned to the gel state and the large G′ allowed printed structures to self‐support. In addition, the mechanical performance of 4% KP‐I in the gel state was characterized by the compression‐relaxation test (Figure 1i), where overlapping strain–stress curves from ten repeated tests with 40% (compression modulus ≈0.007 MPa) strain suggested the long‐term integrity of printed KP‐I (see Figure S7, Supporting Information for the characterization of 6% KP‐I).
2.2. 3D Printing and Fabrication of KNF‐PVA Aerogels
Having demonstrated the appropriate properties of KP‐I for DIW, we now turn to discuss the 3D printing and fabrication of KNF‐PVA aerogels (KP‐A). Given dimensions and moving speed of the printing nozzle, the diameter of printed filament was mainly determined by the extrusion rate of KNF‐PVA inks.[ 27 ] To correlate this rate with the driving pressure of extrusion, the pressure‐driven tubular flow profile of non‐Newtonian KNF‐PVA inks was solved by expressing the shear stress τ rz in Equation (1) with the Herschel‐Bulkley model[ 28 ]
| (3) |
where τ y , K, and n were determined by fitting rheological data (Figure S8, Supporting Information). The extrusion rate of KNF‐PVA inks was calculated by averaging uz over the nozzle's cross‐sectional area (Figure S9, Supporting Information) for different driving pressures (Figure S10, Supporting Information). This correlation was employed to select the driving pressure such that extrusion rate matched with the constant nozzle's moving speed (20 mm s−1). Applying this setting, the variation in the diameter between extruded filaments and the printing nozzle was less than 10% (Figure S6c, Supporting Information). Further combined with computer‐aided design,[ 29 ] this remarkable controlling accuracy enabled layer‐by‐layer printing of delicate patterns (Figure 2a). To prepare final aerogels, printed KNF‐PVA structures were first immersed in water to exchange DMSO, then in ethanol to replace H2O, and finally subject to supercritical CO2 drying to remove all solvents.
Figure 2.
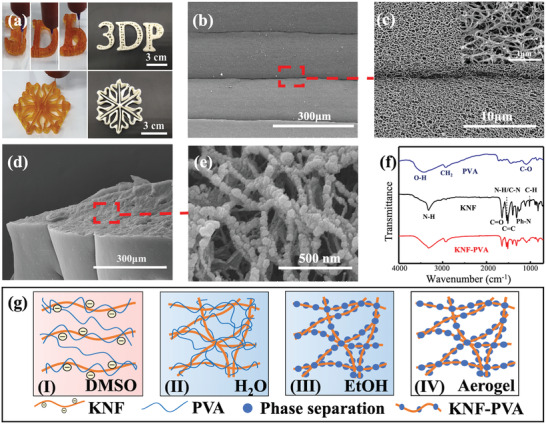
3D printing and fabrication of KNF‐PVA aerogels. a) Digital photos of 3D printed multilayer KNF‐PVA inks and corresponding aerogels after solvent exchange and supercritical drying. b) SEM image of 3D printed KNF‐PVA aerogel showed uniform layer thickness with parallel boundaries. c) Zoomed‐in image of the boxed region in (b) revealed porous surface of KNF‐PVA aerogels. d) SEM image of the cross section and e) structural details of 3D‐printing KNF‐PVA aerogel. f) FTIR spectra of PVA, KNF and KNF‐PVA. g) The process of the formation of the KVF‐PVA aerogel network.
As‐prepared aerogel filaments showed good mechanical strength (elastic modulus up to 111 MPa and elongation at break up to 40%, Table S1, Supporting Information) and flexibility (curvature radius of 230 µm, Figures S11 and S12, Supporting Information). For multi‐layer aerogels, SEM image revealed uniform layer thickness (around 150 µm) with parallel boundaries (Figure 2b), which indicated accurate control of the printing process. More importantly, we identified clear porous structure on the surface (Figure 2c), whereas a dense skin layer (around 1 µm thick) evolved for aerogels printed with liquid KP‐I (Figure S13, Supporting Information). Such observation reflected different flowability of KNFs in liquid and gel KNF‐PVA inks: When liquid KP‐I encountered water, KNFs diffused outward and accumulated at the surface to yield dense skins, whereas such diffusion in the gel KP‐I was restricted and thus prevented the formation of skin layers. Consistent with this explanation, 2%‐KP‐A printed with gel inks showed larger specific area (189 m2 g−1 versus 155 m2 g−1) than aerogels printed with liquid inks (Table S2, Supporting Information). And this difference became negligible for 6% KP‐A as the high viscosity of 6%‐KP‐I also limited the diffusion of KNFs.
Cross‐section SEM image revealed a continuous porous structure that extended through multiple layers without clear boundaries (Figure 2d). Zoomed‐in image further showed nanosized particles covering the continuous backbone (Figure 2e), which was likely caused by the precipitation of PVA molecules on rigid KNF network while ethanol was displacing water. Further measurement of the apparent contact angle of water on aerogels yielded similar values as that of PVA (Figure S14, Supporting Information), suggesting good coverage of PVAs over the KNF network. Therefore, aerogels fabricated by 3D printing KP‐I in the gel state involved homogeneous pore structures and hydrophilic pore surfaces that favored water absorption.
In addition to physical structures, the chemical nature of prepared aerogels was examined with the Fourier transform infrared spectroscopy and thermal analysis. Both the aerogel and KNF involved similar FTIR peaks (Figure 2f),[ 30 ] such as the stretching vibration modes of N—H, C=O, and C=C (3324, 1645, and 1512 cm−1), vibration modes of Ph‐N (1316 cm−1) and in‐plane C—H (1018 cm−1), and the coupling mode of N—H deformation and C‐N stretching vibration (1541 cm−1). Compared with PVA,[ 25 ] prepared aerogel not only exhibited a similar peak corresponding to the stretching vibration of C—H at 2940 cm−1, but also yielded red‐shift in the stretching vibration peaks of O—H and C—OH from 3418 to 3306 cm−1 and from 1092 to 1090 cm−1, respectively. This peak shifting suggested hydrogen bonding between —OH groups from PVA and C=O groups from KNF in the prepared KNF‐PVA aerogel. Because PVA and KNF molecules present a large number of —OH and C=O groups along their polymer chains, this hydrogen bonding enabled considerable intermolecular interaction that was reported between PVA and other molecules.[ 31 , 32 ] Meanwhile, thermogravimetric analysis (Figure S4b, Supporting Information) revealed a 76% weight loss of the aerogel at 500 °C, consistent with the theoretical value calculated with weight loss of pure KNF (6.6%) and PVA (90%) as well as their mass ratio (1:5) in 3D printing inks, that is, . Altogether, these results confirmed the chemical composition of final KNF‐PVA aerogels and suggested that this composition was not affected by the fabrication process.
Based on characterization results, evolution of KNF‐PVA network's microscopic structure during aerogel fabrication was proposed in Figure 2g. After mixing KNF and PVA solutions in DMSO, PVA molecules assembled around deprotonated KNF due to hydrogen bonding interaction (Figure 2g‐I). In the first solvent‐exchange step, DMSO was displaced by water and re‐protonation of KNF formed the nanofiber network[ 17 ] (Figure 2g‐II). This network greatly enhanced the mechanical strength of subsequent hydrogels (KP‐H) and aerogels (KP‐A), as reflected by the increased compression modulus from 0.024 MPa for 6%‐KP‐I (Figure 1i) to 0.84 MPa for 6%‐KP‐H and 7.47 MPa for 6%‐KP‐A (Figure S15 and Table S3, Supporting Information). Correspondingly, during the second solvent‐exchange step (Figure 2g‐III) and subsequent supercritical drying (Figure 2g‐IV), KNF‐PVA hydrogels resisted deformation from external forces (Figure S16, Supporting Information) and underwent almost negligible volume change and thus ensured accurate printing of aerogel patterns (Figure S17, Supporting Information).
Notably, conventional PVA aerogels were predominantly fabricated at concentration higher than 10 wt% to achieve sufficiently cross‐linked hydrogels, and adopted freeze drying to remove water since solvent exchange with ethanol before supercritical drying would cause significant shrinking of PVA hydrogels.[ 33 ] By contrast, in this work, PVA molecules were anchored on the rigid KNF network by their interaction while undergoing phase separation when water was exchanged for ethanol (Figure 2g‐III), which allowed KP‐A to be fabricated via supercritical drying at lower PVA concentration (5 wt% for 6%‐KP‐A). This advantage led to well preserved porous structure (porosity larger than 90%) and large surface area (up to 111 m2 g−1, Figure S18 and Table S4, Supporting Information) of KP‐A. By achieving mechanical stability and biocompatibility at the same time, the unique core–shell nanofiber network represented in Figure 2g‐IV was particularly desired by aerogel‐based hemostatic materials.
2.3. Characterization of Aerogel Gauzes
Utilizing the accurate control of DIW and aerogel fabrication of KP‐A, we further printed KP‐I into the gauze pattern (Figure 3a) and fabricated KP‐A gauze (Figure 3b). The final aerogel gauze only shrunk by less 20% compared with the pattern after 3D printing and such changes occurred homogeneously in all directions to preserve the original aspect ratio of printed gauzes (Figure S19, Supporting Information). Because of the adhesion between the top and bottom filaments (printed diameter 500 µm), their non‐contacting region experienced extension forces during the printing process and thus showed relatively smaller diameters (450 µm) than that of contacting regions as shown in Figure 3c. The µm‐sized inter‐filament spacing and nm‐sized pores on the filament surface (Figure 3c, inset) thus established hierarchically porous structures.
Figure 3.
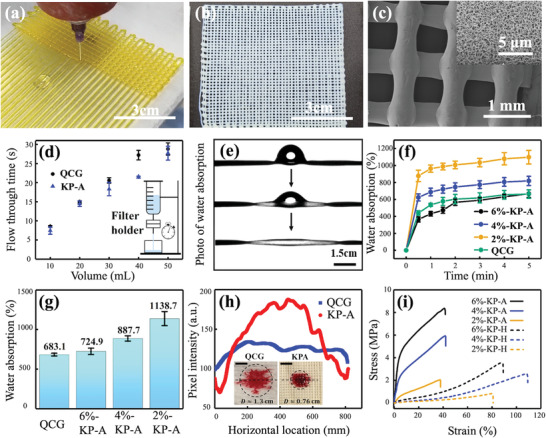
Characterization of aerogel gauzes. a) 3D printing KNF‐PVA ink in the gauze pattern. b) Final KNF‐PVA aerogel gauze after solvent exchange and supercritical drying in sequence. c) SEM image of the aerogel gauze. d) Characterization of water flux for KNF‐PVA aerogel gauze (KP‐A) and QCG. e) Water absorption by the KNF‐PVA aerogel filament. f,g) are dynamic and final water absorption mass scaled by self‐weight for QCG and KNF‐PVA aerogel gauze (KP‐A) with different original concentrations. h) Extracted pixel intensity from images (inset) taken 1 min after depositing drops containing Sudan‐III stained SiO2 particles (diameter ≈ 3 µm) on KP‐A and QCG. i) Tensile curves of KNF‐PVA hydrogel gauzes KP‐H (Figure 2g‐II) and aerogel gauzes (KP‐A). Data in (d), (f), and (g) were shown with mean values and standard deviation acquired from three parallel experiments.
To highlight the advantages of having multiscale pores for hemostatic applications, we compared the performance of water transport and particle enrichment between KP‐A gauze and QCG that only involved µm‐sized porous space between solid fibers (Figure S20, Supporting Information). First of all, both KP‐A gauze and QCG yielded similar water permeation flux of 338 mL cm−2 s−1 bar−1 that was driven by gravity of the water column (Figure 3d). Conversely, water can barely flow through KNF‐PVA aerogel films without driving pressure (Figure S21, Supporting Information), as the confined space of nm‐sized pores significantly restricted the water permeability. In fact, under similar conditions, study[ 20 ] reported PVA aerogel film to yield a water permeation flux close to 3.764 mL cm−2 s−1 bar−1, which was 2 orders of magnitude smaller than that in KP‐A gauze. This contrast demonstrated the important role of µm‐sized pores in aerogel gauze and QCG to ensure high water permeability.
On the other hand, aerogel gauzes presented additional nanoporous structure inside individual filaments that led to enhanced performance of water absorption than QCG. Figure 3e showed the fast water absorption by a single KP‐A filament, where its increasing transparency under transmission light indicated the filling of nm‐sized pores by water. Because of this quick capillary wicking of water,[ 34 ] Figure 3f revealed that the initial water absorption speed (t < 30 s) of aerogel gauzes (9.64 g g−1 min−1) was almost twice of that for QCG (5.37 g g−1 min−1). Additionally, aerogel gauzes exhibited larger capacity for water absorption than QCG, particularly for 2%‐KP‐A gauze that absorbed water more than ten times of its own weight (Figure 3g). By contrast, the water absorption capacity of non‐porous KNF or PVA (less than 60%) was much smaller than KP‐A (Figures S22 and S23, Supporting Information). In fact, the measured water absorption capacity of aerogel gauzes agreed well with the theoretical estimation W ideal = ϕ/ρ (ϕ and ρ is the aerogel's porosity and apparent density, Table S5, Supporting Information), assuming absorbed water was mainly stored inside aerogel filaments.
The quick absorption of water induced a substantial flux of aqueous solution toward the aerogel surface, where water entered the aerogel's porous structure while µm‐sized particles were blocked and accumulated. To estimate the magnitude of this flux, Lucas–Washburn equation[ 34 ] suggested the temporally‐decaying inward flow velocity at the aerogel surface as . Substituting the water‐aerogel contact angle θ ≈ 42° (Figure S14, Supporting Information), aerogel's average pore size r pore ≈ 10 nm (Figure S18, Supporting Information), and water viscosity µ = 0.001 Pa s, the average value of u over the first 30 s reached 96.4 µm s−1. Given the typical inter‐filament spacing less than 1 mm in aerogel gauzes, this large flux could effectively advect and accumulate particles at the aerogel surface. By contrast, the limited water absorption by QCG's solid fibers restricted particle convection toward the fiber surface, which in turn, reduced the particle enrichment effect.
This difference was illustrated by depositing 20 µL aqueous suspensions of silica particles (mean diameter of 3 µm, stained with Sudan III for visual observation, refer to Figure S24, Supporting Information for details of preparation and characterization) on the surface of both materials. Compared with QCG, the wetted area on aerogel gauze was much smaller (0.45 cm2 versus 1.33 cm2, Figure 3h, inset) due to its higher water absorption capacity as discussed previously. Furthermore, quantitative analysis of the pixel intensity profile in wetted area on KP‐A gauze revealed a clear peak arising from particle aggregation (Figure 3h). Comparing the intensity value between the peak and the background yielded an enrichment ratio close to 3, whereas the particle accumulation was almost negligible on QCG as indicated by the flat intensity profile and small enrichment ratio (less than 1.2).
Notably, the water absorption speed and capacity of KP‐A gauzes improved with lower KNF‐PVA concentration (Figure 3f and 3g), likely associated with increased porosity and more smaller pores (Table S4, Supporting Information). On the other hand, as indicated in Figure 3i and Table S1, Supporting Information, the mechanical performance of aerogel (KP‐A) and hydrogel (KP‐H, Figure 2g‐II) filaments enhanced at higher KNF‐PVA concentration due to more developed KNF network, although their elongation at break remained fairly constant. Particularly, when the concentration of KP‐A increased from 2 to 6 wt%, the elastic modulus increased by an order of magnitude (from 14 to 111 MPa) and the mechanical strength by a factor of 4 (from 2 to 8 MPa). To balance both factors for optimal hemostatic function, 4%‐KP‐A gauze was chosen for subsequent in vivo tests.
2.4. In Vivo Hemostasis Experiments with Animal Models
Before in vivo hemostasis experiments, hemolysis rate (Figure S25, Supporting Information) and cell compatibility of QCG and aerogel gauzes were tested to evaluate their biocompatibility. Specifically, KP‐A gauze yielded hemolysis rate of 2.3% and cell viability of 83.2%, which were both better than the result for QCG (10.9% and 73%, respectively), and thus considered as proper hemostatic materials with excellent biocompatibility.[ 35 ] In addition, in vitro degradation study also confirmed the stability of aerogel gauzes in contact with phosphate solutions, during which the 2‐day mass loss ratio of KP‐A (10%) was much smaller than that (17%) of QCG (Figure S26, Supporting Information).
In vivo animal experiments were performed with three Sprague–Dawley (SD) rat's injury models for natural hemostasis involving no topical materials (control) and applying three‐layer QCG or aerogel gauzes. In both rat liver resection and rat tail amputation models, KP‐A achieved efficient hemostasis by reducing the blood loss by an order of magnitude and hemostatic time by half when compared with natural hemostasis (Figure S27, Supporting Information). More importantly, KP‐A outperformed QCG when applied in the rat liver laceration model (Figure 4a). Both QCG and aerogel gauzes achieved hemostasis faster than the control set without blood overflow during the experiment or re‐bleeding after hemostasis materials were removed (Figure S28, Supporting Information). Particularly, the hemostasis time yielded by aerogel gauze (60 ± 4 s) was half of that when using QCG (120 ± 8 s), where the significance parameter p < 0.05 to confirm the statistical accuracy of such results (Figure 4b). The difference in the blood loss during hemostasis was more dramatic (Figure 4c, p < 0.01), with aerogel gauzes reducing the blood loss by a factor of three (0.07 ± 0.01 g) compared with QCG (0.21 ± 0.02 g). Moreover, the blood clotting index (BCI) of aerogel gauze (16 ± 0.2%) was less than half of the value given by QCG (43 ± 3.3%), indicating its better blood clotting performance (Figure 4d). Altogether, these quantitative results suggested aerogel gauzes as more efficient hemostatic materials than commercially available QCG in the rat liver laceration model.
Figure 4.
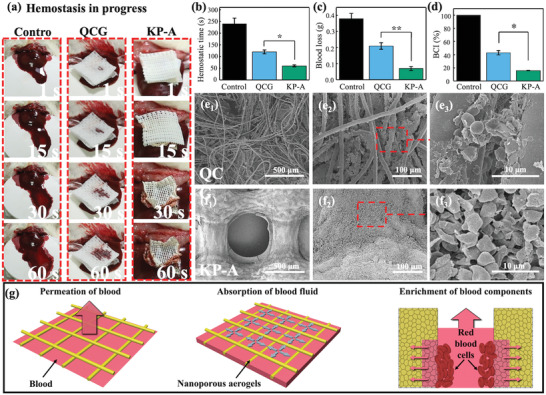
In vivo animal experiments with the rat liver laceration model. a) Images of hemostasis in progress. First column is the natural hemostasis as the control, second and third column correspond to results applying QCG and KNF‐PVA aerogel (KP‐A) gauze. Comparison of b) hemostatic time, c) total blood loss, and d) blood clotting index (BCI) between natural hemostasis and hemostatic materials QCG and aerogel gauze. SEM images and zoomed‐in details of accumulated blood cells on the surface of e) QCG and f) KP‐A gauze. g) Proposed hemostatic mechanism of KP‐A gauze. Data in (b–d) was shown with mean values and standard deviation. The number of experiments in each group is 3 (n = 3). One‐way analysis of variance (ANOVA) by SPSS software (IBM) was applied for statistical analysis. *p < 0.05 and **p < 0.01 were considered significant and greatly significant difference.
To further rationalize why aerogel gauzes much outperformed QCG in hemostasis, their differences in the capability to absorb blood fluid and concentrate blood cells were discussed. First, as shown in Figure 4a, blood appeared on the top layer within 1 s when QCG was pressed against the wound (second column), whereas no obvious blood was observed on the top of aerogel gauze (third column). Since complete hemostasis did not occur within 30 s for either case, QCG and aerogel gauzes would still absorb water at this stage. In particular, the fast water absorption of aerogel gauzes was indicated by its shrinking size (reduction by 25%). Therefore, the contrast shown in Figure 4a indicated that aerogel gauze was able to absorb blood fluid faster than QCG, and also had a larger storage capacity for blood fluid. These properties, consistent with characterization results in Figure 3f,g, prevented blood penetration to the top layer of aerogel gauze and reduced the blood loss during hemostasis.
Meanwhile, SEM images of wound‐contacted surfaces of aerogel gauzes and QCG revealed a clear difference in the density and distribution of blood cells. For QCG, the aggregation of blood cells scattered on individual spots that was likely due to kaolin particles at the fiber's surface,[ 36 ] and many fibers had rather smooth sections without noticeable aggregation of blood cells (Figure 4e). Oppositely, Figure 4f showed that blood cells accumulated more densely and uniformly on the surface of porous fibers in aerogel gauzes, which was consistent with the enrichment effect shown in Figure 3h. This contrast reflected the capability of KP‐A to efficiently accumulate blood cells while absorbing the blood fluid, thus accounting for its fast hemostasis action during in vivo experiments.
Finally, based on characterization results and in vivo experiments, the advantages of aerogel gauze's hierarchical porous structure for hemostasis were illustrated in Figure 4g. When applied topically to the wound, the hydrophilic nature of KNF‐PVA aerogel filaments and µm‐sized spacing between filaments allowed the permeation of blood that provided sufficient blood components for subsequent accumulation. Meanwhile, the highly nanoporous structure of KNF‐PVA aerogel quickly absorbed and stored blood fluid inside individual filaments, yielding a substantial advection of blood cells toward the aerogel. These µm‐sized blood components cannot enter the nanoporous structure and thus accumulated at the aerogel surface to efficiently promote hemostasis.
3. Conclusion
In conclusion, we have developed 3D printed KNF‐PVA aerogel gauze as a new and effective hemostatic material. The hydrogen bonding interaction between PVA and KNF is critical for directing PVA molecules to cover the rigid KNF backbone, which yields biocompatible, hydrophilic, and mechanically stable KNF‐PVA aerogel filaments for the hemostatic applications. In addition, aerogel's highly nanoporous structure and gauze's large inter‐filament spacing work coherently to permeate and quickly absorb water, inducing substantial fluid advection toward the aerogel filament and promoting accumulation of µm‐sized particles such as blood cells and platelets. In vivo animal experiments with rat liver laceration model confirm that aerogel gauzes reduce the hemostasis time by half (less than 60 s) and blood loss by a factor of three (less than 0.07 g) when compared with the widely applied commercial product QCG. The strategy of designing aerogel gauze's chemical composition and physical structure thus provides important insight for future design of aerogel‐based hemostatic materials.
4. Experimental Section
Chemicals
Poly(vinyl alcohol) (PVA‐117, molecular weight ≈ 145 000 and hydrolysis degree ≈99%) were obtained from Aladdin Co. Kevlar 1000D was obtained from Dongguan SOVETL Co., Ltd. DMSO, KOH, and ethanol were purchased from Sinopharm Chemical Reagent Co. Deionized water (18.2 MΩ·cm−1) was generated in the Millipore Milli‐Q system.
Preparation of the KNF‐PVA Solutions
PVA/DMSO and KNF/DMSO solutions were separately prepared and then mixed to give KNF/PVA/DMSO solutions. PVA was dissolved in DMSO (10 wt%) at 90 °C with constant stirring until the solution became clear. 10 g of Kevlar 1000D and equal weight of KOH were added to 480 g of DMSO and magnetically stirred for 1 week at room temperature to form a 2 wt% Kevlar nanofibers (KNFs) dispersion in DMSO. 6 wt% KNF‐PVA solution was prepared by mixing 10 wt% PVA/DMSO solution and 2 wt% KNF/DMSO dispersion of equal mass at 60 °C followed by constant stirring. This mixing ratio of PVA to KNF (5:1) was selected for optimal stiffness and strength of subsequently fabricated hydrogels and aerogels. Therefore, KNF‐PVA solutions of different total concentrations were obtained by diluting the 6 wt% KNF‐ PVA solution with DMSO.
Preparation of the 3D‐Printed KNF‐PVA Aerogels
KNF‐PVA solutions in DMSO were used as the ink for Direct Ink Writing (DIW) 3D printing. The solution was loaded into the dispensing syringe in the liquid state (at >60 °C) and then both ends of the syringe were sealed to prevent moisture in the air. When the hot dispersion gradually cooled to appropriate temperature, it became gel‐like and ready for direct‐ink 3D printing. The printing process was performed using a customized robotic deposition system involving a 3‐axis desktop controlled by three‐step motors via G‐code commands. The automatic dispensing system (MY3131T, Meiyuan Tech, Shenzhen, China) equipped with a suck‐back dispensing valve was mounted on the x–y–z motor. One end of the dispensing valve was connected to the syringe loaded with inks, and the other end was connected to the oil‐free air compressor (OTS‐550, OTS, Taizhou, China).
To print specific patterns, KNF‐PVA gel‐like ink was squeezed through the syringe needle under a constant extruding pressure of 0.2–0.5 MPa, and then deposited onto the substrate according to the unit‐cell structure designed by computer aided design (CAD). The initial nozzle height from the substrate was optimized at around 400 µm to ensure sufficient adhesion to the substrate, and the moving speed of the nozzle was set to 20 mm s−1. 3D‐printed KNF‐PVA gels then underwent two solvent exchange steps, first replacing DMSO with water and then displacing water with ethanol. Finally, drying with supercritical CO2 removed all solvents in printed structures and yielded 3D‐printed KNF‐PVA aerogels.
Characterization
Rheological properties of inks were measured in the rotational rheometer (Haake RheoStress 6000, Germany) with 20 mm diameter steel plate. Specifically, apparent shear viscosity was obtained in steady‐state flow with a logarithmic sweep of shear rate from 0.1 to 1000 s−1. Viscosity changes at elevated temperature were obtained by steady state flow test at constant shear rate at 1 s−1 and temperature varied between 25 and 70 °C. Yield stress was characterized by oscillation tests with a logarithmic sweep of shear stress from 1 Pa to 10 000 Pa at 1 Hz shear frequency and oscillatory strain of γ = 0.02. Dynamic modulus versus temperature was characterized in oscillation tests with constant shear stress at 100 Pa and temperatures ranging from 25 to 70 °C. All rheological measurements were conducted at fixed temperature (usually 25 °C) with a preliminary equilibration time of 1 min or constant heating rate of 5 °C min−1.
SEM characterization was performed on the S‐4800 (Hitachi, Japan) with the acceleration voltage of 10 kV, with samples coated on Au in a sputter‐coater at the current of 25 mA for 3 min in advance. TEM characterization was carried out on the Tecnai G2 F20 S‐TWIN high‐resolution TEM (FEI, USA) with the acceleration voltage set to 200 kV. Samples were prepared by diluting the dispersion to 1/10 000 mass fraction in DMSO, dipping it onto a copper grid, and drying with infrared light for 2 h before characterization.
The pore size distribution and average diameter of aerogels were analyzed by the Barrett–Joyner–Halenda (BJH) nitrogen adsorption and desorption method (ASAP 2020, Micromeritics, USA). The specific surface area and pore volume were determined following the Brunauer–Emmett–Teller (BET) method using the N2 adsorption data at pressure 0.05 < P/P 0 < 0.3. The porosity of the aerogel fiber ϕ was calculated according to the formula , where ϕ was the porosity, ρ was the density of KNF/PVA aerogel and ρ 0 was the density of KNF/PVA xerogel prepared by drying the hydrogel under ambient pressure.
Fourier transform infrared spectroscopy was performed on the FTIR 5700 (FL, USA) over 64 scans recorded with a resolution wavelength of 4 cm−1. Thermal stability was determined by the thermal gravimetric analyzer (TGA, 209F1, NETZSCH, Germany) with the heating rate of 10 K min−1 in a nitrogen atmosphere. To evaluate the mechanical stability of samples, the stress–strain curves and tensile–strain curves were characterized with the Instron 3365 mechanical testing equipment. The water contact angle measurements were performed at room temperature with the video optical contact angle measuring instrument (OCA15EC, Data physics, Germany).
To measure the water permeability, single‐layer QCG and aerogel gauze were enclosed in a membrane holder and the exposure area of gauze to water permeation was 0.95 cm2. The inlet of the membrane holder was connected to an open syringe holding a column of water and the outlet was open to air. Without applying driving pressure, the drainage time of water passing through the gauze was recorded for different volumes of water. Each experiment was repeated three times.
In water absorption experiment, single layer of QCG and aerogel gauze (2.5 cm long, 2.5 cm wide, and 1 mm thick) with dry weight m b was soaked in water and taken out at different time points to measure the weight of the gauze m a, and the absorbed mass of water (m a − m b) was scaled by the dry weight of the gauze sample to express the water absorption W abs (%) = (m a − m b)/m b × 100%. The capacity of water absorption by gauze samples was determined by taking the value of W abs (%) measured after 20 min.
Enrichment of particles by gauze samples was performed with SiO2 particles with average diameter of 3 µm (refer to Figure S21, Supporting Information for detailed synthesis and characterization). To visualize the enrichment effect, SiO2 particles were stained with oily dye Sudan III and then dispersed in water at 2% (w/v). 20 µL of this dispersion was deposited on each surface of QCG and aerogel gauze that were both 5 cm by 5 cm and single‐layer. To analyze the particle enrichment ratio, images taken at 1 min after the solution deposition were decomposed into separate red, blue and green channels, and the grayscale intensity of the red channel was used to quantify the distribution of particles on the surface of gauze samples. All image analysis was performed with ImageJ software (National Institutes of Health, USA).
In Vitro Test of Blood Compatibility
The hemolysis rate was determined as follows. The fresh blood of the Sprague–Dawley (SD, provided by the Laboratory Animal Center of Soochow University, P. R. China) rat was centrifuged at 1000 rpm for 10 min at room temperature, rinsed with sterile saline, and further centrifuged until the upper layer was clear. Red blood cells obtained by centrifugation were diluted by phosphate buffer solution (PBS) to a concentration of 5% (v/v) for use. QCG and aerogel gauze were separately cut into squares of 0.5 cm by 0.5 cm, then loaded into 2 mL test tubes involving 500 µL of red blood cell dispersion. After contacting QCG and aerogel gauze at 37 °C for 1‐h, red blood cell dispersions were centrifuged at 1000 rpm for 10 min and the UV absorbance intensity at 540 nm of the supernatant was measured (A s) by the UV–vis Spectrophotometer (V‐660, Jasco, Japan). The positive control group involved 5% (v/v) red blood cells dispersed in deionized water containing 0.1% (v/v) surfactant Triton X‐100 (total volume was 500 µL), and the negative control group was 500 µL 5% (v/v) red blood cells dispersed in PBS. After the same centrifugation process as above, the UV absorbance intensity at 540 nm of the supernatant of the positive (A p,) and negative (A n) control group was measured. The hemolysis rate was then calculated as
| (4) |
To evaluate the blood coagulation index, QCG and aerogel gauze were dried in an oven at 100 °C for 12 h before testing. Three 1 cm2 squares were cut out of the gauze sample and placed in three layers in a 100 mL beaker. The sample and beaker were first held at 37 °C for 10 min, then 100 µL of blood was slowly dropped into the middle of the sample, and then kept at 37 °C for 15 min. 50 mL deionized water was added to the beaker, followed by shaking the beaker slowly to evenly dissolve the uncoagulated blood in the water. The liquid in the beaker was then taken out to measure its absorption peak at 540 nm and recorded as U 1. For the control group, 100 µL of blood was dropped into a blank beaker and kept at 37 °C for 15 min, followed by adding 50 mL deionized water, shaking slowly, and measuring the absorption at 540 nm as U 0. Finally, the blood coagulation index of QCG and aerogel gauze was calculated as
| (5) |
MTT cell counter kit was used to test the cell compatibility of QCG and aerogel gauzes in vitro. Gauze samples were first processed by gamma irradiation sterilization with 25 kGy Cobalt‐60 and extracted with MEM medium (10% fetal bovine serum, Penicillin 100 U mL−1, Streptomycin sulfate 100 µg mL−1). Meanwhile, an L929 murine fibroblast cell was used and cultured in MEM Medium at 37 °C in a humidified atmosphere of 5% CO2 and then digested by 0.25% trypsin containing EDTA and centrifuged (at 200 G for 3 min) to obtain a cell suspension of suitable denseness. The suspended cells were dispensed at 100 µL per well in 96‐well plate, and cultured in cell incubator (5% CO2, 37 °C, >90% humidity). After cells grew to form a monolayer, original culture medium was discarded and the 96 well plates were then treated with 100 µL of extracts of test samples (100%, 50%, 25%, 12.5%), negative control (100%) and positive control (100%), respectively, and incubated at 37 °C in a humidified atmosphere of 5% CO2 for 24 h (five replicates of each extract were tested). A 50 µL aliquot of MTT (1 mg mL−1) was then added to each well and incubated at 37 °C in a humidified atmosphere of 5% CO2 for 2 h. The liquid in each well was tipped out and 100 µL of 99.9% Isopropanol was then added to each well to suspend the cell layer. Finally, the suspension was evaluated with a dual‐wavelength spectrophotometer with the measurement wavelength at 570 nm and reference wavelength at 650 nm. The cell viability was calculated as
| (6) |
where OD570e and OD570b was the measured value of extract and blank sample, respectively.
In vitro degradation of QCG and aerogel gauzes was conducted by measuring the mass loss of samples in Sorensen's phosphate buffer by fixing the sample concentration to 0.01 g mL−1. And the degradation ratio under 70 °C after 1 week was calculated as
| (7) |
where m 0 and m 1 was the vacuumed dried mass of the sample before and after the degradation test.
In Vivo Hemostatic Experiments
SD rat's liver laceration model was conducted according to the standard procedure. All animal experiments were carried out in accordance with the guidelines for the Animal Center of the Suzhou Institute of Nano‐Tech and Nano‐Bionics (SINANO/EC/2022‐069). Rats were randomly selected and anesthetized. Subsequently, the epithelial tissue of the abdomen was cut to expose the liver, and pre‐weighted cotton gauze was placed under the liver. For rat liver laceration model, a scalpel was used to make a linear incision trauma of about 1 cm in length and 0.5 cm in depth on the left lobe of the liver. For rat liver resection model, a 5 mm long, 5 mm wide, and 5 mm deep volume on the left lobe of the rat liver was resected. For the rat tail amputation model, the rat tail was cut off at 1–2 cm from the tail tip. After bleeding for 10 s in each rat injury model, pre‐weighed gauze was applied onto the trauma. Blood permeation in the gauze, bleeding time, and blood loss were recorded (n = 3). QCG and aerogel gauze were both tailored to 2.5 cm × 2.5 cm rectangle swatches (each was 1 mm thick), and three layers of such swatches were stacked together for in vivo hemostasis tests on the rat liver injury model. QCG was the standard military hemostatic agent recommended by the Tactical Combat Injury Care Committee for use as a control. The survived animals were euthanized with 10% chloral hydrate at the end of experiment. Anesthesia was injected intraperitoneally with a 10 wt% chloral hydrate solution (0.4 mL per 100 g). After complete hemostasis, all animals were observed for 2 h or until death.
Statistical Analysis
All data were derived from three parallel experiments (n = 3) without preprocessing and reported with mean values and standard deviation (error bars). One‐way analysis of variance (ANOVA) by SPSS software (IBM) was applied for statistical analysis. *p < 0.05 and **p < 0.01 were considered significant and greatly significant difference, respectively.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
X.Y. and N.S. contributed equally to this work. The authors gratefully acknowledge the support from the Royal Society Newton Advanced Fellowship (NA170184), the National Natural Science Foundation of China (52173052, 52003290), and the Natural Science Foundation of Jiangsu Province (BK20190232).
Yang X., Shi N., Liu J., Cheng Q., Li G., Lyu J., Ma F., Zhang X., 3D Printed Hybrid Aerogel Gauzes Enable Highly Efficient Hemostasis. Adv. Healthcare Mater. 2022, 12, 2201591. 10.1002/adhm.202201591
Contributor Information
Fengguo Ma, Email: fgma@qust.edu.cn.
Xuetong Zhang, Email: xtzhang2013@sinano.ac.cn.
Data Availability Statement
Research data are not shared.
References
- 1. Moore E. E., Moore H. B., Kornblith L. Z., Neal M. D., Hoffman M., Mutch N. J., Schochl H., Hunt B. J., Sauaia A., Nat. Rev. Dis. Primers 2021, 7, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hickman D. A., Pawlowski C. L., Sekhon U. D. S., Marks J., Gupta A. S., Adv. Mater. 2018, 30, 1804635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. He H., Zhou W., Gao J., Wang F., Wang S., Fang Y., Gao Y., Chen W., Zhang W., Weng Y., Wang Z., Liu H., Nat. Commun. 2022, 13, 552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li Z., Milionis A., Zheng Y., Yee M., Codispoti L., Tan F., Poulikakos D., Yap C. H., Nat. Commun. 2019, 10, 5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pourshahrestani S., Zeimaran E., Djordjevic I., Kadri N. A., Towler M. R., Mater. Sci. Eng., C 2016, 58, 1255. [DOI] [PubMed] [Google Scholar]
- 6. Yu L., Shang X., Chen H., Xiao L., Zhu Y., Fan J., Nat. Commun. 2019, 10, 1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ji X., Du Y., Zhang X., Adv. Mater. 2022, 34, 2107168. [DOI] [PubMed] [Google Scholar]
- 8. Li G., Dong D., Hong G., Yan L., Zhang X., Song W., Adv. Mater. 2019, 31, 1901403. [DOI] [PubMed] [Google Scholar]
- 9. Lyu J., Liu Z., Wu X., Li G., Fang D., Zhang X., ACS Nano 2019, 13, 2236. [DOI] [PubMed] [Google Scholar]
- 10. Jiang F., Hsieh Y.‐L., J. Mater. Chem. A 2014, 2, 350. [Google Scholar]
- 11. Kettunen M., Silvennoinen R. J., Houbenov N., Nykã¤Nen A., Ruokolainen J., Sainio J., Pore V., Kemell M., Ankerfors M., Lindstrom T., Ritala M., Ras R. H. A., Ikkala O., Adv. Funct. Mater. 2011, 21, 510. [Google Scholar]
- 12. Deuber F., Mousavi S., Federer L., Hofer M., Adlhart C., ACS Appl. Mater. Interfaces 2018, 10, 9069. [DOI] [PubMed] [Google Scholar]
- 13. Qiao S., Kang S., Zhu J., Wang Y., Yu J., Hu Z., J. Hazard. Mater. 2021, 415, 125739. [DOI] [PubMed] [Google Scholar]
- 14. Du Y., Zhang X., Wang J., Liu Z., Zhang K., Ji X., You Y., Zhang X., ACS Nano 2020, 14, 11919. [DOI] [PubMed] [Google Scholar]
- 15. Li Y., Zhang X., Adv. Funct. Mater. 2022, 32, 2107767. [Google Scholar]
- 16. Liu Z., Lyu J., Fang D., Zhang X., ACS Nano 2019, 13, 5703. [DOI] [PubMed] [Google Scholar]
- 17. Xu L., Zhao X., Xu C., Kotov N. A., Adv. Mater. 2018, 30, 1703343. [DOI] [PubMed] [Google Scholar]
- 18. Qi M., Gu Y., Sakata N., Kim D., Shirouzu Y., Yamamoto C., Hiura A., Sumi S., Inoue K., Biomaterials 2004, 25, 5885. [DOI] [PubMed] [Google Scholar]
- 19. Yang M., Cao K., Sui L., Qi Y., Zhu J., Waas A., Arruda E. M., Kieffer J., Thouless M. D., Kotov N. A., ACS Nano 2011, 5, 6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu Y., Su Y., Guan J., Cao J., Zhang R., He M., Jiang Z., ACS Appl. Mater. Interfaces 2018, 10, 26546. [DOI] [PubMed] [Google Scholar]
- 21. Cheng Q., Liu Y., Lyu J., Lu Q., Zhang X., Song W., J. Mater. Chem. A 2020, 8, 14243. [Google Scholar]
- 22. Zhu C., Han T. Y.‐J., Duoss E. B., Golobic A. M., Kuntz J. D., Spadaccini C. M., Worsley M. A., Nat. Commun. 2015, 6, 6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jiang Y., Xu Z., Huang T., Liu Y., Guo F., Xi J., Gao W., Gao C., Adv. Funct. Mater. 2018, 28, 1707024. [Google Scholar]
- 24. He H., Li Y., Liu H., Kim Y., Yan A., Xu L., ACS Appl. Mater. Interfaces 2021, 13, 7539. [DOI] [PubMed] [Google Scholar]
- 25. Wang J., Lin Y., Mohamed A., Ji Q., Jia H., J. Mater. Chem. C 2021, 9, 575. [Google Scholar]
- 26. Siqueira G., Kokkinis D., Libanori R., Hausmann M. K., Gladman A. S., Neels A., Tingaut P., Zimmermann T., Lewis J. A., Studart A. R., Adv. Funct. Mater. 2017, 27, 1604619. [Google Scholar]
- 27. Cheng Q., Sheng Z., Wang Y., Lyu J., Zhang X., ACS Nano 2022, 16, 4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lewis J., Adv. Funct. Mater. 2006, 16, 2193. [Google Scholar]
- 29. Yuk H., Zhao X., Adv. Mater. 2018, 30, 1704028. [DOI] [PubMed] [Google Scholar]
- 30. Zhu J., Yang M., Emre A., Bahng J. H., Xu L., Yeom J., Yeom B., Kim Y., Johnson K., Green P., Kotov N. A., Angew. Chem., Int. Ed. 2017, 56, 11744. [DOI] [PubMed] [Google Scholar]
- 31. Liu L., Zhu M., Xu X., Li X., Ma Z., Jiang Z., Pich A., Wang H., Song P., Adv. Mater. 2021, 33, 2105829. [DOI] [PubMed] [Google Scholar]
- 32. Niu W., Zhu Y., Wang R., Lu Z., Liu X., Sun J., ACS Appl. Mater. Interfaces 2020, 12, 30805. [DOI] [PubMed] [Google Scholar]
- 33. Chen C., Zhu Y., Bao H., Shen J., Jiang H., Peng L., Yang X., Li C., Chen G., Chem. Commun. 2011, 47, 5530. [DOI] [PubMed] [Google Scholar]
- 34. Ha J., Kim J., Jung Y., Yun G., Kim D.‐N., Kim H.‐Y., Sci. Adv. 2018, 4, eaao7051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang Y., Fu Y., Li J., Mu Y., Zhang X., Zhang K., Liang M., Feng C., Chen X., Carbohydr. Polym. 2018, 200, 6. [DOI] [PubMed] [Google Scholar]
- 36. Mohamed E., Fitzgerald A., Tsuzuki T., Mat. Today Nano 2021, 16, 100137. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
Research data are not shared.