

Abstract
Owing to advantageous properties attributed to well‐organized structures, multifunctional materials with reversible hierarchical and highly ordered arrangement in solid‐state assembled structures have drawn tremendous interest. However, such materials rarely exist. Based on the reversible phase transition of phase‐change materials (PCMs), phase‐change nanocrystals (C18‐UCNCs) are presented herein, which are capable of self‐assembling into well‐ordered hierarchical structures. C18‐UCNCs have a core–shell structure consisting of a cellulose crystalline core that retains the basic structure and a soft shell containing octadecyl chains that allow phase transition. The distinct core–shell structure and phase transition of octadecyl chains allow C18‐UCNCs to self‐assemble into flaky nano/microstructures. These self‐assembled C18‐UCNCs exhibit efficient thermal transport and light‐to‐thermal energy conversion, and thus are promising for thermosensitive imaging. Specifically, flaky self‐assembled nano/microstructures with manipulable surface morphology, surface wetting, and optical properties are thermoreversible and show thermally induced self‐healing properties. By using phase‐change nanocrystals as a novel group of PCMs, reversible self‐assembled multifunctional materials can be engineered. This study proposes a promising approach for constructing self‐assembled hierarchical structures by using phase‐change nanocrystals and thereby significantly expands the application of PCMs.
Keywords: cellulose, core–shell structure, phase‐change nanocrystals, thermalimaging, thermal reversibility
Phase‐change nanocrystals (C18‐UCNCs) are prepared via surface esterification and further thiol–ene reaction on cellulose nanocrystals. These nanocrystals can assemble into flaky structures in the solid‐state, which exhibit excellent thermoinduced imaging property, thermoinduced reversibility, and a self‐healing property. The multifunctional flaky structures can manipulate energy conversion, surface morphology, surface wetting, and optical properties, largely expanding their potential uses in various fields.
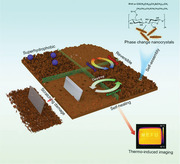
Dry‐state structures engineered using the precise positional arrangement of molecules or nanoparticles at different length scales have attracted great interest for functional materials.[ 1 ] In stimuli‐responsive materials, ordered molecular arrangements can give rise to novel structures and functionalities in response to the environmental changes.[ 2 ] For example, phase‐change materials (PCMs) are temperature‐responsive materials that can reversibly store and release abundant amounts of thermal energy, when they change their molecular arrangements from one physical state to another.[ 3 ] Organic PCMs, such as polyethylene glycol, fatty alcohols, paraffin, and fatty acids, generally exhibit large latent heat and solid–liquid phase transitions, rendering them highly suitable for the preparation of thermal energy storage materials.[ 4 , 5 ] These phase changes are governed by diverse intermolecular interactions, such as van der Waals forces, dipole–dipole interactions, and hydrogen bonding.[ 6 ] Adjusting these interactions during phase transitions allows control not only over thermal energy but self‐assembled structures as well via molecular arrangement. Some solid–liquid PCMs, including stearic acid and alkylated comb‐like polymers, have been used to create distinct functional nanostructures.[ 7 , 8 ] However, the obtained structures are generally irreversible owing to the high melting temperature and ease of deformability.
Substantial efforts have been devoted to stabilizing solid–liquid PCMs to prevent leakage during melting. Conventional techniques include PCM encapsulation in a core material,[ 9 ] porous adsorption,[ 10 ] and physical/chemical bonding in micro‐ or macroscale molecular networks to form composites.[ 11 ] For these stabilized PCMs, the mobility and arrangement of their molecules are generally restricted to a small area, and only crystalline structures can be reversibly changed by varying the temperature.[ 12 ] However, the overall morphologies and shapes of PCMs mainly depend on stabilizing substrates (e.g., silica shells, metal–organic frameworks, porous graphene, and coaxial fibers),[ 13 , 14 ] to obtain PCMs in micro/nanoparticles, porous tubes, foams, bulk sheets, and fibers.
By contrast, the construction of ordered hierarchical nano/microstructures by using nanoparticles still presents a challenge and rarely obtains reversible structures. In the current study, a strategy was developed to prepare phase‐change nanocrystals with self‐stabilized shapes by immobilizing 1‐octadecanethiol onto undecenoated cellulose nanocrystals (UCNCs) (Figure 1a), resulting in core–shell C18‐UCNCs. These phase‐change nanocrystals not only possess efficient thermal transport and conversion properties, but can also self‐assemble into solid‐state thermoreversible hierarchical structures. Different than conventional PCM molecules requiring structuring techniques, immobilized octadecyl chains in the soft shell of C18‐UCNCs can undergo a phase change because of the feasibility of crystallization, while their mobility is strongly confined owing to the presence of CNCs as inert cores. Molecular interactions between the C18‐UCNCs shells ultimately lead to anisotropic flaky nanostructures to which complete reversibility and thermoreversible properties are attributed.
Figure 1.
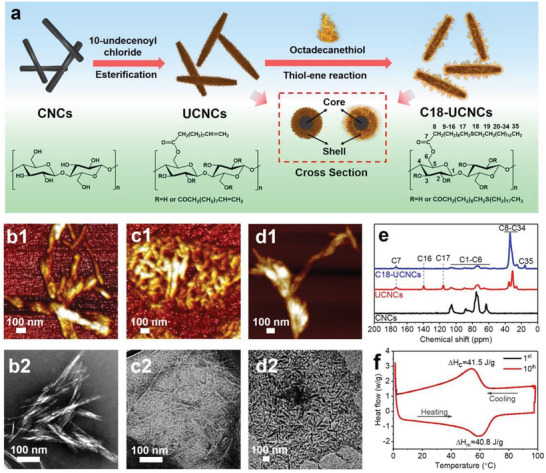
Synthesis and characterization of C18‐UCNCs as phase‐change nanocrystals. a) Schematic of the synthesis of C18‐UCNCs from UCNCs via the thiol–ene reaction with 1‐octadecanethiol. b1–d1) AFM images of CNCs (b1), UCNCs (c1), and C18‐UCNCs (d1). b2–d2) TEM images of CNCs (b2), UCNCs (c2), and C18‐UCNCs (d2). e) Solid‐state 13C NMR spectra of CNCs, UCNCs, and C18‐UCNCs. f) DSC curves of C18‐UCNCs as the 1st and 10th thermal cycle.
CNCs were extracted from microcrystalline cellulose (MCC) by hydrochloric acid hydrolysis.[ 15 ] Obtained CNCs formed stable suspensions in water (Figure S1, Supporting Information). The morphology of the CNCs was characterized by atomic force microscopy (AFM) and transmission electron microscopy (TEM) (Figure 1b), which showed typically rigid rod‐shaped CNCs. CNCs had an average length of 197 ± 73 nm and an average width of 12 ± 4 nm, as revealed by TEM. UCNCs were synthesized by modifying CNCs with 10‐undecenoyl chloride in pyridine under heterogeneous reaction conditions (Figure 1a).[ 16 ] After surface esterification, obtained UCNCs were well dispersed in tetrahydrofuran (THF) (Figure S1, Supporting Information), retaining their rod‐shaped form, as revealed in the AFM and TEM images (Figure 1c). The solid‐state 13C NMR spectrum of UCNCs showed typical signals at 114.9 ppm (C17) and 138.9 ppm (C16) ascribed to carbons in terminal olefin groups. The signals between 20 and 40 ppm and those near 173 ppm were attributed to alkane and carbonyl carbons in the group with 10‐undecenoyl, respectively (Figure 1e).[ 17 ] The average degree of substitution (DS) ascribed to 10‐undecenoyl groups in UCNCs was ≈0.9, as determined by elemental analysis. Thus, more than two‐thirds of the hydroxyl group on cellulose chains remained unreacted. The heterogeneous esterification of CNCs generally begins from the particle surface, extending to the interior.[ 18 ] The intact hydroxyl groups should mainly be located in the core of the UCNCs. The presence of the unreacted cellulose core within the UCNCs was confirmed by solid‐state 13C NMR spectroscopy, which detected the signal at 84 ppm (C4) attributed to crystalline cellulose (Figure S2, Supporting Information).[ 19 ]
Chemicals containing straight long‐chain alkanes are widely used as organic PCMs.[ 5 ] To prepare C18‐UCNCs as phase‐change nanocrystals with phase‐change properties, 1‐octadecanethiol was grafted in the shell layer of UCNCs containing terminal vinyl groups via thiol–ene reaction (Figure 1a).[ 20 ] Compared with those of UCNCs, the peaks related to vinyl groups at 113.9 and 138.2 ppm in the 13C NMR spectrum of C18‐UCNCs disappeared after the thiol–ene reaction, while a new signal attributed to the CH3‐group appeared at 15.0 ppm (Figure 1e). These results confirmed the successful introduction of 1‐octadecanethiol groups. For C18‐UCNCs with long‐chain alkanes, the nanocrystals tend to assemble into aggregates (Figure 1d). Compared with UCNCs, C18‐UCNCs showed a similar crystalline structure, as observed from the solid‐state 13C NMR spectrum, indicating that the cellulose crystalline core was retained (Figure 1a; Figure S2, Supporting Information). Thus, the C18‐UCNCs consist of a highly esterified phase‐change soft shell and an intact crystalline cellulose core.
As a crucial factor, the phase‐change enthalpy of C18‐UCNCs as phase‐change nanocrystals was investigated using differential scanning calorimetry (DSC) (Figure 1f). In comparison to pristine 1‐octadecanethiol with a melting point (T m) at ≈31 °C and a crystallization temperature (T c) at about 26 °C.[ 21 ] C18‐UCNCs with octadecyl chains in the shell had higher T m and T c at nearly 60 and 54 °C, respectively (Figure 1f). This change was related to the presence of the octadecyl chains with a high density in the shell, which promoted the formation of crystalline regions by restricted chains.[ 22 ] C18‐UCNCs showed excellent thermal properties with a latent heat of melting (H m) and crystallization (H c) of about 40.8 and 41.5 J g−1, respectively (Figure 1f). Notably, C18‐UCNCs show excellent energy storage and release stability, and no obvious change of temperature and latent heat after 10 phase‐change cycles.
Thermal transport and conversion are considered as important properties for PCMs. Based on the enhanced crystallization of surface‐attached octadecyl chains, the thermal transport under heating and the light‐to‐thermal energy conversion under irradiation of C18‐UCNCs were examined. The glass slide coated with C18‐UCNCs (referred to as G+C18‐UCNCs) was compared with the glass slide coated with stearic acid (referred to as G+stearic acid) and a pure glass slide. C18‐UCNCs formed a layer of nanoflakes on the surface exhibiting superhydrophobicity (Figure 2a,b) (further investigated in the subsequent section). By contrast, stearic acid formed no such nanoflakes on the glass surface and instead formed only rough aggregates as crystallized stearic acid (Figure S3, Supporting Information). Figure 2c presents the temperature development curves of the three samples during heating and cooling on the plate (Figure S4a, Supporting Information). When the temperature was set to 80 °C and the heater was turned on, the temperatures of G+C18‐UCNCs, G+stearic acid, and the glass increased from ≈28 °C and remained constant at 79.3, 79.2, and 74.9 °C, respectively (Figure 2c; Movie S1, Supporting Information). Stearic acid containing octadecyl chains is recommended as a promising PCM because of the high latent heat storage density and energy preservation ability, which make it more suitable for thermal energy storage applications.[ 23 ] The temperature of G+C18‐UCNCs increased at almost the same rate as that of G+stearic acid, while faster than that of glass, despite the considerably higher thermal conductivity of the glass slide (Figure 2c; Figure S5, Supporting Information). This finding may be related to the grafted octadecyl chains in the shell of C18‐CNCs. Their presence with a high density formed a phase‐change shell that facilitated thermal transport and conversion performance. During heating or cooling, no thermal storage plateau was observed for G+C18‐UCNCs, unlike stearic acid, which should be in turn attributed to the extremely low absolute amount of the octadecyl chains in the C18‐UCNCs shell.
Figure 2.
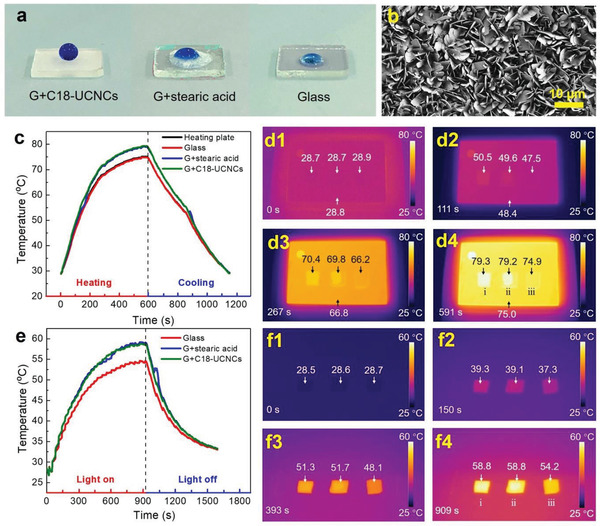
The thermal transport and conversion of C18‐UCNCs. a) Dyed water droplets on the surfaces of G+C18‐UCNCs, G+stearic acid, and glass b) SEM image of the surface morphology of G+C18‐UCNCs. c) Time‐dependent temperature evolution curves of G+C18‐UCNCs, G+stearic acid, and glass during heating and cooling. d) Infrared images showing temperature variations during heating (c). e) Light‐to‐thermal energy conversion curves of G+C18‐UCNCs, G+stearic acid, and glass. f) Infrared images showing the temperature variations of simulated sunlight illumination of (e). Samples i, ii, and iii in (d) and (f) represent the samples of G+C18‐UCNCs, G+stearic acid, and glass, respectively. The numbers above or below the arrows in (b) and (d) denote the temperatures (°C) of the corresponding areas.
To intuitively and visually observe the change in surface transient temperature on the plate during heating and cooling, the thermal infrared images of G+C18‐UCNCs, G+stearic acid, and glass were recorded (Figure 2d). It is notable that the temperature of G+C18‐UCNCs was similar to that of G+stearic acid and consistently higher than those of glass and the plate (Figure 2d; Movie S1, Supporting Information). This difference should be attributed to the efficient thermal conversion and preservation of C18‐UCNCs, which clearly delayed direct energy transport to the surrounding air and thus led to increased temperature in the area of the plate covered by C18‐UCNCs. By contrast, the plate surface without the C18‐UCNCs coverage or covered by glass had a lower temperature due to faster energy transport to the air. Moreover, the leakage test was conducted to evaluate the thermal stability of C18‐UCNCs. Photographs and thermal infrared images of G+C18‐UCNCs and G+stearic acid before and after heat treatment at 80 °C for 5 min are presented in Figures S4 and S6 and Movie S2 of the Supporting Information. The stearic acid on the surface of G+stearic acid completely melted into liquid, acquiring the ability to flow, whereas C18‐UCNCs on the G+C18‐UCNCs surface showed no such leakage. Moreover, C18‐UCNCs also demonstrated considerable light‐to‐thermal energy conversion under light irradiation by using a solar light simulator (Figure S7, Supporting Information). Samples of G+C18‐UCNCs, G+stearic acid, and glass were exposed to the simulated solar light and then cooled to room temperature (RT) after the light was turned off. Time–temperature curves were generated, and thermal infrared images of the samples were obtained using an infrared thermal camera (Figure 2e,f; Movie S3, Supporting Information). Similar to that of G+stearic acid, the temperature of G+C18‐UCNCs increased under light irradiation at a rate faster than that of glass during the illustrating process. G+C18‐UCNCs reached the highest temperature (58.8 °C) at 909 s, which was 4.6 °C higher than that of the glass. This result further indicated the efficient light‐to‐thermal conversion of C18‐UCNCs.
The efficient thermal preservation and light‐to‐thermal conversion of the self‐assembled C18‐UCNCs suggest that C18‐UCNCs show great potential for application in thermosensitive imaging. Imaging via heating or illumination is illustrated in Figure 3a. During heating or under light irradiation, patterned areas formed by C18‐UCNCs should significantly increase their temperatures, which can be captured with a thermal imaging camera. We fabricated patterned letters of “NEFU” with C18‐UCNCs on three substrates as glass, steel, and foil paper (Figure S8a–c, Supporting Information). By heating the patterned glass slide on the plate at different temperatures, the efficient thermal transport and preservation of C18‐UCNCs result in the higher temperatures of the patterned areas and consequently, brighter patterns in the infrared images (Figures S8 and S9, Supporting Information; Figure 3b). In addition, the “NEFU” patterns with higher temperatures than those of the surrounding area on the steel blade and foil paper also became apparent during heating at 50 °C (Figure 3c,d). Under simulated sunlight irradiation, all C18‐UCNCs patterns on glass, steel, and foil paper could be captured with a thermal imaging camera, revealing high‐quality imaging (Figure 3e–g). The C18‐UCNCs showed significantly higher thermal stability than that of molecular PCM stearic acid, as reflected in the patterned letters of “NE” formed by stearic acid. The letters “FU” were formed by C18‐UCNCs (Figure 3h). During the heat treatment at the set temperature (80 °C), the letters “NE” were gradually damaged by the melting of stearic acid. By contrast, the letters “FU” remained unchanged throughout because of the presence of the cellulose crystalline core, indicating excellent thermal stability for C18‐UCNCs (Figure 3h). Therefore, C18‐UCNCs represent promising candidates for thermosensitive imaging.
Figure 3.
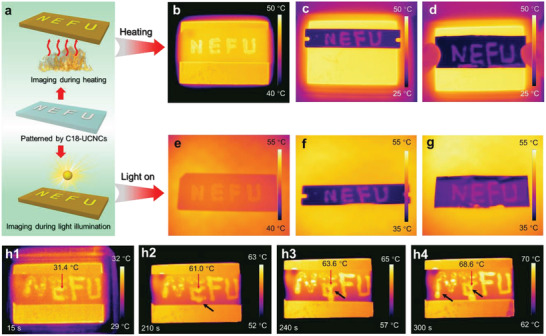
Thermosensitive imaging with C18‐UCNCs. a) Schematic illustration for the thermosensitive imaging of surfaces containing C18‐UCNC patterns of “NEFU” during heating or under light illumination. b–d) Infrared images showing the temperature variations of a patterned glass slide (b), a patterned steel blade (c), and a sheet of patterned foil paper (d) on a heating plate at the set temperature of 50 °C. e–g) Infrared images showing the temperature variations of a patterned glass slide (e), a patterned steel blade (f), and a sheet of patterned foil paper (g) under simulated sunlight illumination. h) Time‐dependent infrared images showing temperature variations of the patterned glass slide on a heating plate with a tilt angle of 45° during heating from RT to the set temperature of 80 °C. The patterned letters “NE” are formed by stearic acid, and the letters “FU” are formed using C18‐UCNCs. The red arrows in (h) indicate the temperature of the corresponding area. The black arrows in (h1), (h2), and (h3) show the flow and leakage of stearic acid caused by melting.
The unique flaky structures were observed on the surface of G+C18‐UCNCs (Figure 2b). To understand the mechanisms underlying the self‐assembly of C18‐UCNCs into flaky structures, self‐standing films were formed by solvent casting from the THF suspensions of C18‐UCNCs on Teflon substrates (Figure 4a; Figure S11a, Supporting Information). The stress–strain curve from the tensile test in Figure S11b of the Supporting Information shows a fracture strength of around 9.71 MPa for C18‐UCNCs films. After the evaporation of THF, peeled‐off C18‐UCNCs films became nontransparent and superhydrophobic, because flaky self‐assembled structures were formed on both surfaces of the films (Figure 4a,b). Irregular flakes with thicknesses of about 150 nm were organized into a flowerlike porous morphology on the film surface (Figure 4a,b; Table S1, Supporting Information). By contrast, the UCNCs suspensions and pure 1‐octadecanethiol solution in THF did not form self‐assembled flaky structures (Figure S12a,b, Supporting Information), but only smooth surface or random aggregates under similar conditions. In addition, polymeric cellulose derivative, which was obtained after the introduction of 1‐octadecanethiol into backbone of cellulose 10‐undecenoyl ester[ 17 ] with the complete derivatization of hydroxy groups (DS = 3) via thiol–ene reaction, also did not form similar self‐assembled structure (Figure S12c, Supporting Information). Furthermore, the density of octadecyl chains in the shell of UCNCs was altered to examine the effect of the octadecyl chains on the self‐assembly process. The surfaces of C180.5‐UCNCs and C180.75‐UCNCs with low densities of octadecyl chain, which were synthesized by partially immobilizing 1‐octadecanethiol (0.5 or 0.75 mol per mol C=C) through the thiol–ene reaction, did not contain fully covered flaky structures, and were only covered by sparsely distributed microlamella clusters (Figure S12d,e, Supporting Information). Resulting surfaces showed a lower hydrophobicity in comparison to those of C18‐UCNCs. Thus, the presence of phase‐change octadecyl chains of a high density within the shell of C18‐UCNCs that are at the same time immobilized in a comfined surrounding on crystalline cellulose cores should be the determining factors for the formation of hierarchical self‐assembled structures. Moreover, the THF suspension of stearoylated CNCs with DS of 1.2, which was obtained after surface esterification of CNCs using stearoyl chloride, only formed transparent films and no self‐assembled flaky structure was observed (Figure S12f, Supporting Information). Therefore, the undecenoyl chains are also critical for the formation of the thermoreversible flaky structures, which guarantee the efficient flexibility and mobility for the ordering assembly of the octadecyl groups in dry‐state.
Figure 4.
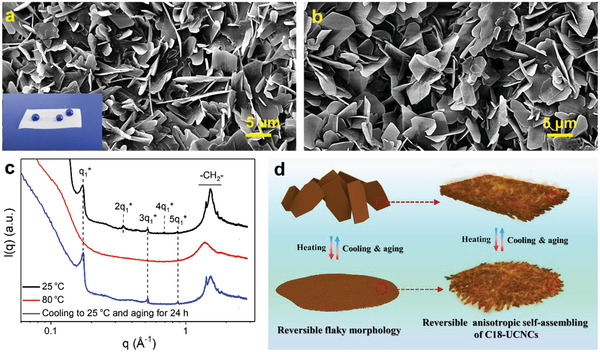
Flaky self‐assembled structures by C18‐UCNCs. a,b) SEM images of flaky self‐assembled structures on the top surface (a) and the bottom surface (the surface in contact with Teflon during film formation) (b) of the as‐prepared C18‐UCNCs film. The inset in (a) shows the dyed water droplets on the surface of the C18‐UCNCs film. c) SAXS curves of a C18‐UCNCs film measured under different conditions: as‐prepared films at 25 °C, heated at 80 °C and cooled from 80 to 25 °C and aging for 24 h. d) Schematic of the formation of a thermoreversible flaky structure.
Alkyl side chains with a critical length of longer than 12 carbon atoms can form crystalline structures.[ 24 ] Thus, the formation of the flaky structures should be attributed to the ordered organization of C18‐UCNCs by molecular interactions between octadecyl chains, which led to the crystalline structure within the flaky units as confirmed with small‐angle X‐ray scattering curves (SAXS) (Figure 4c). Within the SAXS curve of dried C18‐UCNCs at RT, the intermediate q range of 0.1–1 Å−1 was dominated by several diffraction peaks, representing the existence of a lamellar morphology with a periodicity of 3.7 nm. Previous studies revealed that the length of stretched octadecyl chains was around 2.0 nm.[ 8 ] Therefore, this periodicity in dried C18‐UCNCs films suggests that the crystalline domains consisted of head‐to‐head packed octadecyl chains belonging to different backbones. Furthermore, the scattering peaks in the wide‐angle region (1 Å−1 < q < 2 Å−1) corresponded to the stacking distance between the octadecyl moieties. The sharp peak at 1.42 Å−1 ascribed to a d‐spacing of 0.44 nm was in accordance with the distance between highly organized octadecyl chains. After heating at 80 °C, C18‐UCNCs became isotropic due to the disassociation of the crystalline domains of packed octadecyl chains (Figure 4d). SAXS analysis exhibited only a broad peak from random scattering of the dissociated octadecyl chains (Figure 4c). By decreasing the temperature to 25 °C and aging for further 24 h, C18‐UCNCs regenerated nanoflakes with the crystalline texture. The SAXS curve exhibited featured peaks reflecting a lamellar structure, revealing the reversibility of this self‐assembled structure (Figure 4c,d).
To elucidate the thermoreversibility of the self‐assembled flaky structures, diverse features were investigated, which include optical properties, wetting ability, and morphology. Owing to the crystalline packing of octadecyl moieties between different C18‐UCNCs, the opaque C18‐UCNCs films at RT exhibited apparent birefringence, as determined from their polarized optical microscopy (POM) spectra (Figure 5a,b). Different from homogenous molecular crystals with uniform crystalline patterns, the long alkane chains only crystallized in the soft shells of C18‐UCNCs, undergoing microphase separation to form flaky structures. Owing to the thermoreversibility of such crystalline packing by octadecyl moieties, these self‐assembled flaky structures endowed obtained C18‐UCNCs films with thermoreversible properties. The nontransparent C18‐UCNCs films at RT became transparent after heating to 80 °C and vice versa after cooling with accompanied aging for several hours at RT (Figure 5a,d). Similarly, the surfaces fully covered with ordered flaky structures turned considerably smoother with uniform morphologies after storage at 80 °C for several seconds (Figure 5c; Figure S13, Supporting Information). This change was accompanied by an increase in visible transparency to ≈80% and a decrease in the static water contact angle to ≈115° (Figure 5e). After cooling down by further aging at RT, flaky structures were regenerated, accompanied by the recovery of opacity, birefringence, and superhydrophobicity (Figure 5a–e). Moreover, the self‐assembled flakes exhibited excellent thermoreversibility and maintained similar morphologies with almost the same thickness (Table S1, Supporting Information). No significant fatigue was observed in the transparency and wetting properties (Figure 5d,e). On the contrary, the density of the porous flaky structures gradually increased after 50 and 100 heating–cooling cycles (Figure 5f–h).
Figure 5.
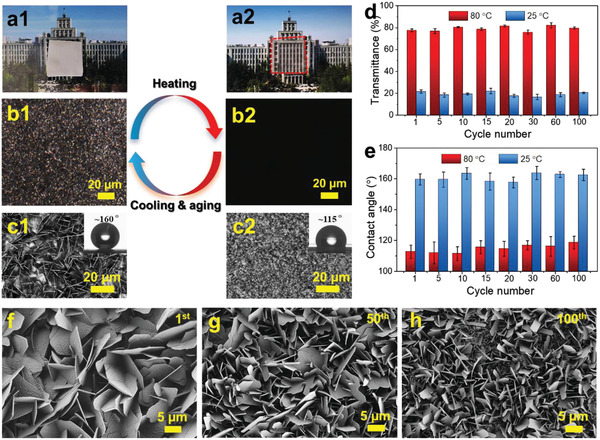
Thermoreversible self‐assembled flaky structures on self‐standing C18‐UCNCs films under repeated heating at 80 °C and cooling with subsequent aging at RT. a) Photos of a self‐standing film of C18‐UCNCs at RT (a1) and 80 °C (a2). b) POM images of a C18‐UCNCs film at RT (b1) and 80 °C (b2). c) Laser scanning microscopy (LSM) images of the surface morphology of the C18‐UCNCs film at RT (c1) and 80 °C (c2). The insets in (c1) and (c2) show the static water contact angles on the corresponding surfaces. d) Transparency of C18‐UCNCs films under repeated heating at 80 °C and cooling with subsequent aging at 25 °C for 100 cycles. e) Water contact angles on the surface of C18‐UCNCs films under repeated heating at 80 °C and cooling with subsequent aging at 25 °C for 100 cycles. f–h) SEM images of flaky structures on self‐standing C18‐UCNCs films after cooling to RT with subsequent aging for >24 h from the first heat treatment at 80 °C (f), the 50th heat treatment at 80 °C (g); and 100th heat treatment at 80 °C (h).
These macroscopic thermoreversible hierarchical structures have not been observed in commonly used PCM systems, which generally undergo melting and crystallization within trapped areas by using stable agents and do not affect changes in macroscopic morphologies.[ 13 , 25 ] Compared with conventional PCMs, the immobilization of the phase‐change octadecyl moieties in the soft shell of C18‐UCNCs induced the mobility of the whole shell during the phase change of the octadecyl chains. When the temperature exceeded the melting temperature of crystallized octadecyl chains (e.g., at 80 °C) (Figure 1f), the flakes disintegrated owing to the relaxation and movement of disentangled octadecyl chains in the shell. This process produced a homogeneous nonordered structure in the films (Figures 4d and 5b,c; Movie S4, Supporting Information). Unlike the melting of commonly used solid–liquid PCMs, the intact crystalline cellulose core is considerably more inert and retained the film stability during the phase transitions. The absorbed latent heat during the melting of the octadecyl chains released while cooling down to RT induced the phase separation of the relaxed shell layer and the reorganization of the C18‐UCNCs at RT. The whole transition process in dry‐state caused a slow assembly of the octadecyl chains to form ordered structures. The temperature dropped to RT within 5 min, while the recovery of the flaky structures took much longer time (Figure S14a, Supporting Information). The formation of self‐assembled flaky structures occurred immediately after cooling down and grew gradually, which was visualized by a digital microscope with a large depth of field (Figure S14b and Movie S5, Supporting Information). After 30 min (Figure S14b4, Supporting Information), the surface was already fully covered with the self‐assembled structures with larger populations compared to the image after 1 min in Figure S14b1 of the Supporting Information. With the reassembling of the flaky structures, the static water contact angle on the surface of these films already increased to higher than 150° within 1 h (Figure S14c, Supporting Information). After 3 h aging, the visible transparency of the films decreased to ≈15%. With efficient aging time, the flaky structures gradually reassembled on and fully covered the surface of the C18‐UCNCs films via the crystallization of octadecyl chains.
To investigate the mechanical durability of the assembled structures, an abrasion test was conducted on the surface of C18‐UCNCs films. As shown in Figure 6a and Figure S15 (Supporting Information), the C18‐UCNCs films were abraded using 800 mesh sandpaper with a 50 g loading. After the abrasion, the obvious white powder on the sandpaper indicated significant damage of the flaky structures, revealing limited mechanical stability of the assembled flaky structures. The damage turned the superhydrophobic surface less hydrophobic with a static water contact angle of about 115° (Figure 6b,d). Thanks to the thermoreversibility of these self‐assembled flakes, the C18‐UCNCs film surface was endowed with self‐healing properties. After heating at 80 °C for 30 min, cooling down, and aging at RT for 24 h, the original superhydrophobicity was restored, and distinct hierarchical flaky structures were regenerated on the damaged surfaces (Figure 6c,d). As shown in Figure 6d and Figure S16 (Supporting Information), the C18‐UCNCs films restored their original topographic features with maintained superhydrophobicity even after 10 abrasion–healing cycles. Thus, the self‐assembled flaky structures on C18‐UCNCs films showed excellent self‐healing properties. The crystallization of the octadecyl chains after heating and cooling could induce the exposed C18‐UCNCs to organize and assemble into flaky structures.
Figure 6.
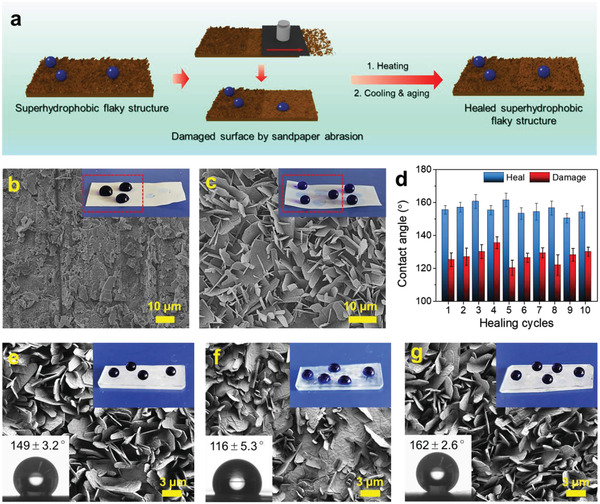
Thermoinduced self‐healing of the flaky structures of self‐assembled C18‐UCNCs. a) Schematic illustration of the self‐healing process. b,c) SEM images of the flaky structures on self‐standing C18‐UCNCs films. b) After the 1st damage by sandpaper abrasion, and c) after the 1st healing by heating at 80 °C for 30 min, cooling down, and aging at RT for 24 h. d) Changes in static water contact angles on the C18‐UCNCs films as the function of the number of abrasion–healing cycles. e–g) SEM images of the surface morphologies of the glass slides coated with C18‐UCNCs under different conditions: e) the as‐prepared sample, f) the sample damaged by scratching, g) the sample healed by heating at 80 °C for 30 min, cooling down and aging at RT for 24 h. The insets in (b) and (c) show dyed water droplets on the C18‐UCNCs film surface, and the red areas show the damaged area and corresponding healed area. The insets in (e–g) at top right show dyed water droplets on the corresponding surfaces, and those at the bottom left show static water contact angles on the corresponding surfaces.
The formation of hierarchical flaky structures on flat substrates was further explored using C18‐UCNCs. The THF suspension of C18‐UCNCs was coated onto the glass slide and filter paper for surface functionalization. The treated glass slide and filter paper were transformed from being hydrophilic to almost superhydrophobic (Figure 6e; Figure S17a, Supporting Information). The enhanced hydrophobicity was attributed to the formation of the flaky structure consisting of C18‐UCNCs on the surface. This hierarchical structure could trap microscopic air pockets beneath water drops, producing superhydrophobic surfaces.[ 26 ] Specifically, the morphology and superhydrophobicity of the C18‐UCNCs‐coated glass slide and filter paper also exhibited thermoinduced self‐healing properties. The physically damaged flaky structures on the surfaces were healed by heat treatment, together with the recovery of the surface morphology and superhydrophobicity (Figure 6f,g; Figure S17b,c, Supporting Information). Thus, the formation of the C18‐UCNCs flaky structures on substrates via coating represents a facile and effective method of superhydrophobizing materials with self‐healing properties.
In conclusion, phase‐change nanocrystals C18‐UCNCs with core–shell structures as a stabilized PCM was successfully synthesized by immobilizing 1‐octadecanethiol in the shell layer of UCNCs. During the evaporation from its THF suspension, C18‐UCNCs could self‐assemble into well‐ordered flaky hierarchical structures on other substrates or on the surface of self‐standing films formed by themselves. This ability is attributed to the synergistic phase segregation and crystallization of octadecyl chains in the shell of C18‐UCNCs, leading to lamellar crystalline structures with 3.7 nm periodicity. The phase transition of the C18‐UCNCs shell conferred on the flaky structures superior thermoinduced self‐healing and thermoreversible properties, including the morphology, birefringence, superhydrophobicity, and transparency of the material. Moreover, C18‐UCNCs showed efficient thermal transport and light‐to‐thermal energy conversion, largely expanding their potential uses in various fields, such as smart surfaces, temperature‐preserving coatings, and thermosensitive imaging.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Supplemental Movie 1
Supplemental Movie 2
Supplemental Movie 3
Supplemental Movie 4
Supplemental Movie 5
Acknowledgements
Y.W. thanks the National Natural Science Foundation of China (31890774 and 31890770) and China Postdoctoral Science Foundation (2018M640286) for the financial support. K.Z. thanks German Research Foundation (DFG) with Project No. ZH546/2‐1 for the financial support.
Open access funding enabled and organized by Projekt DEAL.
Wang Y., Qiu Z., Lang Z., Xie Y., Xiao Z., Wang H., Liang D., Li J., Zhang K., Multifunctional Reversible Self‐Assembled Structures of Cellulose‐Derived Phase‐Change Nanocrystals. Adv. Mater. 2021, 33, 2005263. 10.1002/adma.202005263
Contributor Information
Yonggui Wang, Email: wangyg@nefu.edu.cn.
Kai Zhang, Email: kai.zhang@uni-goettingen.de.
References
- 1.a) Cui Y., Wei Q., Park H., Lieber C. M., Science 2001, 293, 1289; [DOI] [PubMed] [Google Scholar]; b) Wang Z. L., Song J., Science 2006, 312, 242; [DOI] [PubMed] [Google Scholar]; c) Peer D., Karp J. M., Hong S., Farokhzad O. C., Margalit R., Langer R., Nat. Nanotechnol. 2007, 2, 751; [DOI] [PubMed] [Google Scholar]; d) Tian Q. W., Tang M. H., Sun Y. G., Zou R. J., Chen Z. G., Zhu M. F., Yang S. P., Wang J. L., Wang J. H., Hu J. Q., Adv. Mater. 2011, 23, 3542; [DOI] [PubMed] [Google Scholar]; e) Bao Z., Weatherspoon M. R., Shian S., Cai Y., Graham P. D., Allan S. M., Ahmad G., Dickerson M. B., Church B. C., Kang Z., Abernathy H. W., Summers C. J., Liu M., Sandhage K. H., Nature 2007, 446, 172; [DOI] [PubMed] [Google Scholar]; f) Lim B., Jiang M., Camargo P. H., Cho E. C., Tao J., Lu X., Zhu Y., Xia Y., Science 2009, 324, 1302; [DOI] [PubMed] [Google Scholar]; g) King'ondu C. K., Iyer A., Njagi E. C., Opembe N., Genuino H., Huang H., Ristau R. A., Suib S. L., J. Am. Chem. Soc. 2011, 133, 4186. [DOI] [PubMed] [Google Scholar]
- 2. Zha R. H., Vantomme G., Berrocal J. A., Gosens R., de Waal B., Meskers S., Meijer E. W., Adv. Funct. Mater. 2018, 28, 1703952. [Google Scholar]
- 3. Kenisarin M. M., Sol. Energy 2014, 107, 553. [Google Scholar]
- 4.a) Oro E., de Gracia A., Castell A., Farid M. M., Cabeza L. F., Appl. Energy 2012, 99, 513; [Google Scholar]; b) Sharma A., Tyagi V. V., Chen C. R., Buddhi D., Renewable Sustainable Energy Rev. 2009, 13, 318. [Google Scholar]
- 5. Prajapati D. G., Kandasubramanian B., Ind. Eng. Chem. Res. 2019, 58, 10652. [Google Scholar]
- 6. Han G. G. D., Li H., Grossman J. C., Nat. Commun. 2017, 8, 1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Elschner T., Ludecke C., Kalden D., Roth M., Loffler B., Jandt K. D., Heinze T., Macromol. Biosci. 2016, 16, 522. [DOI] [PubMed] [Google Scholar]
- 8. Zhang K., Geissler A., Chen X., Rosenfeldt S., Yang Y., Förster S., Müller‐Plathe F., ACS Macro Lett. 2015, 4, 214. [DOI] [PubMed] [Google Scholar]
- 9.a) Sari A., Alkan C., Bilgin C., Appl. Energy 2014, 136, 217; [Google Scholar]; b) Li Y., Yu S., Chen P., Rojas R., Hajian A., Berglund L., Nano Energy 2017, 34, 541. [Google Scholar]
- 10.a) Chen L., Zou R., Xia W., Liu Z., Shang Y., Zhu J., Wang Y., Lin J., Xia D., Cao A., ACS Nano 2012, 6, 10884; [DOI] [PubMed] [Google Scholar]; b) Xiao C., Gao H., Mu Y., Dong W., Ge W., Nano Energy 2018, 49, 77. [Google Scholar]
- 11. Wang Y., Liu Z., Zhang T., Zhang Z., Technology E., Royon L., Guiffant G., Flaud P., Energy Convers. Manage. 1997, 38, 517. [Google Scholar]
- 12.a) Alva G., Lin Y. X., Fang G. Y., Sol. Energy Mater. Sol. Cells 2018, 186, 14; [Google Scholar]; b) Lian Q. S., Li K., Sayyed A. A. S., Cheng J., Zhang J. Y., J. Mater. Chem. A 2017, 5, 14562. [Google Scholar]
- 13. Tang J., Chen X., Zhang L., Yang M., Wang P., Dong W., Wang G., Yu F., Tao J., Small 2018, 14, 1801970. [DOI] [PubMed] [Google Scholar]
- 14.a) Zhou M., Lin T., Huang F., Zhong Y., Wang Z., Tang Y., Bi H., Wan D., Lin J., Adv. Funct. Mater. 2013, 23, 2263; [Google Scholar]; b) Li S., Wang H., Mao H., Li J., Shi H., ACS Appl. Mater. Interfaces 2019, 11, 14150. [DOI] [PubMed] [Google Scholar]
- 15. Yu H. Y., Qin Z. Y., Liang B. L., Liu N., Zhou Z., Chen L., J. Mater. Chem. A 2013, 1, 3938. [Google Scholar]
- 16. Wang Y., Groszewicz P. B., Rosenfeldt S., Schmidt H., Volkert C. A., Vana P., Gutmann T., Buntkowsky G., Zhang K., Adv. Mater. 2017, 29, 1702473. [DOI] [PubMed] [Google Scholar]
- 17. Wang Y., Heinze T., Zhang K., Nanoscale 2016, 8, 648. [DOI] [PubMed] [Google Scholar]
- 18. Berlioz S., Molina‐Boisseau S., Nishiyama Y., Heux L., Biomacromolecules 2009, 10, 2144. [DOI] [PubMed] [Google Scholar]
- 19. Fumagalli M., Ouhab D., Boisseau S. M., Heux L., Biomacromolecules 2013, 14, 3246. [DOI] [PubMed] [Google Scholar]
- 20. Wang J., Zhang K., J. Phys. Chem. C 2018, 122, 7474. [Google Scholar]
- 21. Lian Q., Li K., Sayyed A. A. S., Cheng J., Zhang J., J. Mater. Chem. A 2017, 5, 14562. [Google Scholar]
- 22.a) Fu X., Xiao Y., Hu K., Wang J., Lei J., Zhou C., Chem. Eng. J. 2016, 291, 138; [Google Scholar]; b) Sarı A., Alkan C., Biçer A., Karaipekli A., Sol. Energy Mater. Sol. Cells 2011, 95, 3195. [Google Scholar]
- 23. Venkataraman S., Lee A. L. Z., Tan J. P. K., Ng Y. C., Lin A. L. Y., Yong J. Y. K., Yi G. S., Zhang Y. G., Lim I. J., Phan T. T., Yang Y. Y., Polym. Chem. 2019, 10, 412. [Google Scholar]
- 24.a) Chabinyc M. L., Toney M. F., Kline R. J., Mcculloch I., Heeney M., J. Am. Chem. Soc. 2007, 129, 3226; [DOI] [PubMed] [Google Scholar]; b) Sealey J. E., Samaranayake G., Todd J. G., Glasser W. G., J. Polym. Sci., Part B; Polym. Phys. 1996, 34, 1613; [Google Scholar]; c) Vaca‐Garcia C., Gozzelino G., Glasser W. G., Borredon M. E., J. Polym. Sci., Part B: Polym. Phys. 2003, 41, 281; [Google Scholar]; d) Chen X., Zheng N., Wang Q., Liu L. Z., Men Y. F., Carbohydr. Polym. 2017, 162, 28. [DOI] [PubMed] [Google Scholar]
- 25. Liu C., Xu Z., Song Y., Lv P., Zhao J., Liu C., Huo Y., Xu B., Zhu C., Rao Z., J. Mater. Chem. A 2019, 7, 8194. [Google Scholar]
- 26.a) Feng L., Li S. H., Li Y. S., Li H. J., Zhang L. J., Zhai J., Song Y. L., Liu B. Q., Jiang L., Zhu D. B., Adv. Mater. 2002, 14, 1857; [Google Scholar]; b) Geissler A., Chen L. Q., Zhang K., Bonaccurso E., Biesalski M., Chem. Commun. 2013, 49, 4962; [DOI] [PubMed] [Google Scholar]; c) Deng X., Mammen L., Zhao Y., Lellig P., Mullen K., Li C., Butt H. J., Vollmer D., Adv. Mater. 2011, 23, 2962; [DOI] [PubMed] [Google Scholar]; d) Hozumi A., Cheng D. F., Yagihashi M., J. Colloid Interface Sci. 2011, 353, 582. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supplemental Movie 1
Supplemental Movie 2
Supplemental Movie 3
Supplemental Movie 4
Supplemental Movie 5