

Abstract
Owing to their high safety and reversibility, aqueous microbatteries using zinc anodes and an acid electrolyte have emerged as promising candidates for wearable electronics. However, a critical limitation that prevents implementing zinc chemistry at the microscale lies in its spontaneous corrosion in an acidic electrolyte that causes a capacity loss of 40% after a ten‐hour rest. Widespread anti‐corrosion techniques, such as polymer coating, often retard the kinetics of zinc plating/stripping and lack spatial control at the microscale. Here, a polyimide coating that resolves this dilemma is reported. The coating prevents corrosion and hence reduces the capacity loss of a standby microbattery to 10%. The coordination of carbonyl oxygen in the polyimide with zinc ions builds up over cycling, creating a zinc blanket that minimizes the concentration gradient through the electrode/electrolyte interface and thus allows for fast kinetics and low plating/stripping overpotential. The polyimide's patternable feature energizes microbatteries in both aqueous and hydrogel electrolytes, delivering a supercapacitor‐level rate performance and 400 stable cycles in the hydrogel electrolyte. Moreover, the microbattery is able to be attached to human skin and offers strong resistance to deformations, splashing, and external shock. The skin‐mountable microbattery demonstrates an excellent combination of anti‐corrosion, reversibility, and durability in wearables.
Keywords: microbatteries, patternable polymers, wearables, Zn anodes, Zn‐ion coordination
A zinc blanket created by the coordination between carbonyl oxygen atoms in a polyimide coating and zinc ions overcomes the thermodynamic instability and facilitates plating/stripping kinetics. Patterning this zinc blanket for a Zn microanode allows for the development of a skin‐mountable microbattery with an outstanding rate capability and stable cycling performance, providing promise for microbatteries integrable for electronic skin.
Advances in the miniaturization of electronic devices have allowed the rapid development of wearable technologies and envisioned seamless and user‐friendly smart systems with excellent wearability.[ 1 , 2 , 3 ] These advanced devices require electrical power to support their smart functions. Unfortunately, general power supplies for miniaturized electronic devices are coin cells and thin‐film batteries, which are rigid, thick, and bulky, and therefore frustrate the design for wearable electronic devices. To circumvent these disadvantages, wireless power generators have been widely integrated into wearables.[ 4 , 5 , 6 , 7 ] However, they suffer from temporal and spatial restrictions, which limit continuous use and human activities. To match up the size and flexibility of miniaturized wearable electronic devices, flexible and microscale energy storage devices, for instance flexible microbatteries, are of great importance.[ 8 , 9 ] Moreover, a prerequisite of next‐generation wearable electronic devices is the high safety standard, even under severe mechanical deformations. As such, despite high energy density, safety concerns about toxic organic solvent‐based lithium ion batteries impede their application in wearable technologies. Aqueous batteries are safe alternatives that offer low cost, environmental benignity, and high tolerance against mishandling.[ 10 , 11 ] Among different aqueous batteries, zinc (Zn) ion batteries utilizing zinc electrochemistry in mildly acidic electrolytes show high energy density and relatively good cycling stability, making them ideal candidates for safe microbatteries.[ 12 , 13 , 14 , 15 ]
Although a Zn anode's reversibility in a mildly acidic electrolyte is better than in alkaline electrolytes, the thermodynamical instability can still cause corrosion. As shown in the Pourbaix diagram (Figure 1a), Zn tends to dissolve along with hydrogen evolution. For an electrolyte containing 2 mol L−1 Zn ions (the typical molarity of Zn ions in the electrolytes of Zn ion batteries), a hydroxide film can form on and passivate the Zn surface when pH increases to larger than approximately 5. As hydrogen evolution is inevitable, the local increase of pH facilitates the formation of passivation films consisting of complexes of hydroxide and anions. Figure 1b directly shows the formation of a passivation film and its impact on the impedance of a Zn anode. The Nyquist plots are fitted by the equivalent circuit (inset in Figure 1b; the fitted plots are shown in Figure S1, Supporting Information). The first semicircle (R dl/Q dl) represents the electric double layer, where R dl is the double layer resistance and Q dl is the corresponding constant phase element (CPE). The n value of a CPE element can be used to determine a resistive (n = 0), a capacitive (n = 1), or a diffusion (n = 0.5) behavior. The R cl and Q cl represent the resistance and CPE of the corrosion layer or diffusion layer. For a fresh Zn anode, only one semicircle is observed, agreeing with the n value of approximately 1 and 0.5 for the Q dl and Q cl, respectively (Table S1, Supporting Information), which corresponds to the formation of the typical electric double layer and the diffusion layer. By contrast, the n value of Q cl of the aged Zn anode decreases to 0.37, indicating the formation of a resistive passivation layer. Meanwhile, R cl increases by almost one order of magnitude (from 845 Ω to 7.6 kΩ), confirming the passivation effect due to the thermodynamical instability of Zn in a mildly acidic electrolyte. In addition to the exacerbation of the corrosion, energy recovery after the rest of a battery in the charged state, a typical usage scenario in practical applications, is also debilitated by the instability of the Zn anode. These problems become more fatal for Zn ion microbatteries compared to bulky batteries because the small amount of Zn in microbatteries can be completely corroded or passivated in a short time. Figure 1c shows the dramatic degradation of the capacity after keeping the Zn ion microbattery at the fully charged state for 10 h. More than 40% of the charged capacity is lost. It therefore seems that the eventual success of Zn ion microbatteries with high practicability for miniaturized electronic devices relies on electrochemically stable Zn microanodes.
Figure 1.
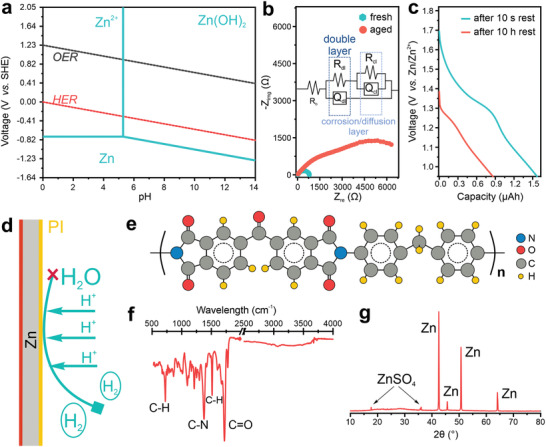
Corrosion of a Zn anode and its suppression by polyimide. a) Pourbaix diagram of Zn in a 2 mol L−1 ZnSO4 solution. b) Nyquist plots of the fresh and aged Zn electrode. The inset shows the equivalent circuit. c) A comparison of the galvanostatic discharge curves of a Zn–MnO2 microbattery at 100 µA and after ten‐hour rest at the fully charged state. d) Schematic of the anti‐corrosion mechanism by a polymeric layer. e) Structure of polyimide. f) FTIR spectrum of the polyimide. g) XRD pattern of piZn after rinsing in 2 mol L−1 ZnSO4 for 24 h.
To improve the stability of Zn anodes, a general strategy is to coat a protective polymer layer onto the Zn anode, which prohibits the contact of Zn with reactive water molecules and corrosive ions in electrolytes.[ 16 , 17 , 18 , 19 ] Despite high efficiency in corrosion inhibition, the polymeric coating, to some extent, impedes the access of cations to the Zn anode, resulting in the large overpotential of the plating/stripping process and the limitation of the reaction kinetics.[ 15 , 18 ] Towards a high‐performance Zn anode, it is of great importance to develop a polymeric protection layer that resolves corrosion‐induced problems and allows for fast kinetics, including low overpotential of plating/stripping and a fast charge/discharge ability. Furthermore, polymers for Zn microanodes may be limited by the lack of spatial control, hence requiring a patternable feature. Photolithography is a well‐established technique for fabricating high‐resolution structures for flexible/wearable microbatteries, and the UV‐induced polymerization should be beneficial to accomplish the goal of developing a patternable protective polymer for Zn microanodes.[ 19 ]
Here, we report a patternable polyimide that can significantly improve the Zn electrodes’ reversibility at the macro‐ and microscale. Although the polyimide coating, as an ion transport barrier, prevents the Zn corrosion in 2 mol L−1 ZnSO4, its high Zn transference number allows for a low Zn plating/stripping overpotential and high reversibility even at a high depth of charge/discharge (85%). We reveal that the carbonyl oxygen atoms coordinate with Zn ions. The coordination becomes stronger as the C—N bonding in the polyimide structure collapses over cycling, which induces the enhancement of the electron‐donating ability of carbonyl oxygen atoms. The in situ formed Zn blanket resolves the common problem of the large overpotential induced by a polymeric layer. We use the polyimide protected Zn electrode in a microbattery (piZn microbattery) coupled with a common MnO2 microcathode and show an overall improvement compared to a bare Zn microanode (Zn microbattery). The capacity loss due to the Zn corrosion during ten‐hour rest is reduced to 10%. The capacity of the piZn microbattery doubles at the same current. More than 60% capacity retention after 1000 cycles is attainable for the piZn microbattery, while the Zn microbattery fails after only 100 cycles. We develop a quasi‐solid‐state piZn microbattery by using polyacrylamide (PAAm) as the electrolyte and demonstrate its capability as a skin‐mountable microbattery with an excellent rate performance, 400 stable charge/discharge cycles, and high resistance to deformations and splashing. As a precedent work for coordination‐enabled, highly reversible Zn electrode, we provide an effective strategy to energize Zn ion microbatteries for flexible/wearable electronic devices.
The corrosion occurs when local charge transfer is available on the Zn surface, where anodic and cathodic sites are coupled. An effective strategy to prevent corrosion is to hamper the migration of ions into the Zn. Figure 1d demonstrates the anti‐corrosion mechanism by a polymeric layer. The polymeric layer separates Zn and the electrolyte (2 m ZnSO4), thus preventing the formation of both anodic and cathodic sites at the interface between Zn and the electrolyte. Pursuing this approach an additional key feature is the possibility to pattern polymeric layer. Polyimide is a patternable polymer and offers advantages over other polymers, such as excellent chemical and thermal stability and low water uptake.[ 20 , 21 ] Besides, carbonyl oxygen atoms in the polyimide chain are electron‐donating sites, forming stable bonds with electron‐accepting sites at the Zn surface upon an annealing process.[ 22 , 23 ] Taking into account these two benefits, polyimide is coated onto the Zn surface. A prepolymerized precursor poly(amic acid) solution made of 4,4′‐methylenedianiline, 3,3′,4,4′‐benzophenonetetracarboxylic dianhydride, 2‐(dimethylamino)ethyl methacrylate, and 5% w/w of photoinitiator (2‐benzyl‐2‐dimethylamino‐1‐(4‐morpholinophenyl)‐butanone‐1) is spin‐coated and crosslinked under ultraviolet (UV) light (Figure S2, Supporting Information). A baking process at 220 °C triggers the imidization to polyimide (Figure 1e) and forms stable adhesion between the polyimide and Zn. Figure 1f shows the Fourier transform infrared spectroscopy (FTIR) of the polyimide. Peaks at 1720 and 1375 cm−1 are assigned to C=O and C—N bonding, respectively. Besides, the spectrum displays C—H peaks associated with benzenes and a methyl group at 720 and 1510 cm−1, respectively.[ 24 ] In comparison to the FTIR spectrum of the precursor poly(amic acid) (Figure S3, Supporting Information), peaks related to O—H, N—H, and carboxylic C=O disappear, indicating the successful imidiazation. The piZn is placed in a 2 m ZnSO4 solution and aged for 24 h. The X‐ray diffraction (XRD) pattern of piZn after aging displays neat and sharp peaks of Zn. The absence of peaks associated with basic Zn sulfate indicates that the polyimide layer can effectively prevent the Zn corrosion in 2 m ZnSO4.
The corrosion behavior is investigated by linear polarization experiments at a sweeping rate of 1 mV s−1. Figure 2a shows the measured Tafel plots of bare Zn and piZn. The cathodic and anodic plots are attributed to the hydrogen evolution reaction and dissolution of Zn, respectively. The Butler–Volmer fit for the bare Zn (Figure S4, Supporting Information) gives a corrosion current of 12 µA cm−2. The cathodic and anodic charge transfer coefficients of 0.56 and 0.44 agree with standard bulk Zn values.[ 25 ] The ohmic resistance‐controlled process is observed for piZn, which is attributed to the insulating polyimide induced IR drop between the local anode and cathode on the Zn surface.[ 26 ] As a result, the corrosion current is reduced by more than one order of magnitude, indicating a lower corrosion rate. The effect of polyimide thickness on the protection efficiency is investigated by a galvanostatic cycling test with a rest period of 1 h for every 5 cycles (Figure S5, Supporting Information). When the thickness decreases from around ≈570 nm to ≈380 nm, the polyimide layer becomes too thin to protect the Zn anode from corrosion. Figure 2b shows the long‐term galvanostatic cycling stability of bare Zn and piZn at a large current density of 4 mA cm−2. A symmetric cell assembled by sandwiching a separator by two identical Zn foils is used. After cycling for only 105 h, a sudden drop of the overpotential resulting from the short‐cut between two bare Zn electrodes is observed. A dense Zn dendrite or a pulverized Zn clusters can penetrate the separator and thus results in an electrical connection between two electrodes. The scanning electron microscopy (SEM) image of the cycled Zn (Figure 2c) does not show a dense dendritic morphology, implying that the pulverized Zn clusters due to the hydrogen evolution is the main cause of the short‐cut, which is further confirmed by the residual Zn clusters on the separator after cycling (Figure S6, Supporting Information). By contrast, piZn stably cycles for 300 h (Figure 2b) and maintains a flat surface (Figure 2d). The basic Zn sulfate due to the corrosion of Zn is found in the XRD spectrum of the bare Zn (Figure 2e), whereas only Zn is observed for the piZn. Both, flat morphology and neat Zn peaks in the XRD spectrum of the cycled piZn demonstrate that the polyimide layer substantially suppresses the Zn corrosion. Given the fact that only a small amount of Zn can be loaded onto a microelectrode, a high coulombic efficiency that allows for high Zn usage is a decisive factor for a high‐capacity microbattery. To evaluate the coulombic efficiency, an asymmetric cell consisting of a Zn foil and a stainless‐steel sheet coated with the polyimide is assembled and cycled at 4 mA cm−2. During the test, Zn deposits onto the coated stainless‐steel sheet and then is stripped off. The coulombic efficiency is determined by the ratio between the charges consumed by the stripped and deposited Zn. As shown in Figure 2f, after a short activation process, the coulombic efficiency is stabilized at around 99.5% (inset in Figure 2f). The high coulombic efficiency allows for cycling at a deep depth of discharge (DOD). The galvanostatic profile with an 85% DOD remains stable over 200 h (Figure S7, Supporting Information). Compared to the state‐of‐the‐art strategies to improve the Zn anode stability, the polyimide shows advantages in simultaneously accomplishing good stability, high coulombic efficiency, and deep charge/discharge ability (Table S2, Supporting Information).
Figure 2.
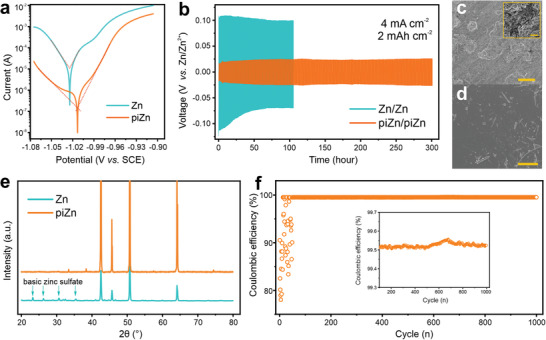
High Zn reversibility. a) Linear polarization curves of Zn and piZn in 2 mol L−1 ZnSO4. b) Long‐term galvanostatic cycling of symmetrical cells with Zn and piZn at a current density of 4 mA cm−2. c,d) SEM images of cycled Zn (c) and cycled piZn (d). Scale bar: 20 µm. e) XRD patterns of cycled Zn and piZn. f) Coulombic efficiency of Zn plating/stripping on stainless steel coated by the polyimide at a current density of 4 mA cm−2. The inset shows the stable region.
Surprisingly, beyond the stable cycling stability, piZn shows a lower overpotential during the Zn plating/stripping process than that for bare Zn albeit the polyimide coating has induced a large IR drop at the surface of Zn. In principle, two factors—ionic conductivity and Zn transference number—of the coating determine the overpotential of the symmetric cell. Figure 3a shows the Nyquist plots of bare Zn and piZn in the symmetrical cells. For bare Zn, we use the equivalent circuit in Figure 1b to fit its Nyquist plot. The n value of Q p is 1, indicating a standard capacitive behavior attributed to the formation of an electrical double layer. The charge transfer resistance is as low as 7.2 Ω, showing the electrolyte's high ionic conductivity. Q z shows the n value of 0.5 and hence indicates a diffusion process. An equivalent circuit including two interfaces established by the coated polyimide layer are used to fit the Nyquist plot of piZn (inset in Figure 3a).[ 27 ] The fitted results are shown in Figure S8a and listed in Table S3, Supporting Information. The n values of two constant phase elements (Q p and Q z) in the circuit demonstrate the electrochemical behaviors at the interfaces. R p, and R z represent the resistance of polyimide and polarization at the Zn/electrolyte interface, respectively. Again, the polyimide layer works as a resistive layer because the n value of the Q z is 0. The polyimide resistance is estimated to be 1500 Ω, thus leading to the ohmic resistance‐controlled process observed in the linear polarization test (Figure 2a). Upon immersion in the electrolyte, Zn ions will migrate into the polyimide layer and reduce charge transfer resistance at the Zn/electrolyte interface (Figure S9, Supporting Information). The ability of polyimide that allows for ion transport is further identified by the n value of Q p of 0.5, which reveals a diffusion behavior. A phase shift of 45° is shown in Figure S8b, Supporting Information, further confirming the diffusion behavior. The resistance of the polyimide coating is around 200 Ω. Considering the polyimide's nano‐thick feature, its ionic conductivity is too low to be able to reduce the overpotential of the Zn plating/stripping process.
Figure 3.
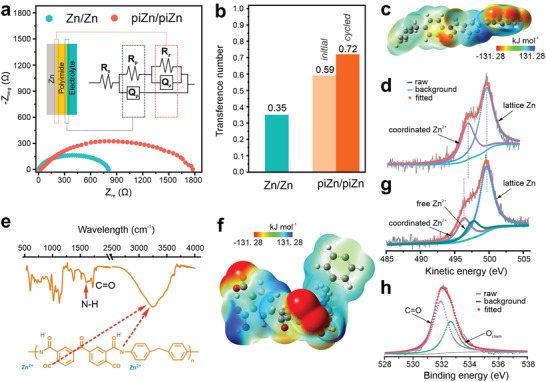
Formation of the polymeric Zn blanket. a) Nyquist plots of symmetrical cells using bare Zn and piZn as electrodes. b) Zn transference number of two symmetrical cells. c) ESP of the polyimide. d) Zn LMM Auger spectrum of piZn before cycling. e) FTIR and chemical structure of the polyimide after cycling. f) ESP of the cycled polyimide. g) Zn LMM Auger spectrum of piZn after cycling. h) XPS spectrum of the O1s core level of piZn after cycling.
The Zn transference number of the polyimide, therefore, plays an important role in reducing the overpotential of Zn plating/stripping. Figure 3b compares the Zn transference number of the symmetrical cells using bare Zn and piZn. A transference number of 0.35 for bare Zn agrees with standard aqueous solutions.[ 18 ] In this case, a large concentration gradient develops during the operation, causing a large overpotential. If coated with a barrier for ion transport, an increase in the overpotential for Zn plating/stripping is observed and has been reported elsewhere.[ 15 , 18 ] On the contrary, an increase in Zn transference number can compensate for the reduced ion transportation by minimizing the depletion of Zn ions during charge/discharge. Moreover, the cation‐controlled ion conduction shows better reversibility at a high rate.[ 28 ] For piZn, the Zn transference number reaches 0.59, indicating cation‐controlled ion conduction. Therefore, a high Zn transference number of the polyimide layer offers better reversibility and reduces the overpotential for Zn plating/stripping compared to the bare Zn.
The cation‐controlled behavior is attributed to the abundant carbonyl oxygen atoms in the polyimide. As mentioned before, these oxygen atoms are electron‐donating sites, which are available for coordinating with Zn ions in the electrolyte.[ 29 , 30 , 31 ] To verify this assumption, electrostatic potential (ESP) calculation of the polyimide, which correlates to the interaction between ions and polymer chains, are conducted and shown in Figure 3c. Negative parts concentrate on carbonyl oxygen atoms, coordinating with Zn ions. Furthermore, the coordinated Zn can be identified in the Zn LMM Auger spectrum measured by X‐ray photoelectron spectroscopy (XPS).[ 32 , 33 ] The coordinated Zn peak centered at 496.7 eV, in addition to the lattice Zn peak centered at 499.8 eV, is evident in Figure 3d.[ 34 ] Likewise, a shoulder peak centered at 532.6 eV in the spectrum of the O1s core level of the piZn confirms the interaction between Zn ions and the carbonyl oxygen atoms (Figure S10, Supporting Information).[ 35 , 36 ]
Figure 3b also shows an increase in the Zn transference number from 0.59 to 0.72 over cycling. Likewise, an activation process is observed in the test for coulombic efficiency (Figure 2f). Both phenomena are the fingerprints of the structural change of the polyimide cycled in the electrolyte. Figure 3e shows the FTIR spectrum of piZn after 300 cycles at 4 mA cm−2. In comparison to the initial structure, the C—N and C=O bonding change substantially, while a new peak correlated to the N—H bonding at 1600 cm−1 occurs. Another change is the occurrence of a broad peak in the range of 3000 to 3500 cm−1. Normally, the O—H stretching from the carboxyl groups and N—H stretching from the amino groups are major sources. Both evidences signify the tendency to form carboxyl and amino groups after the breakage of the bonds between C—N breaks and water uptake over cycling.[ 37 , 38 ] Nevertheless, there is no evidence that the carboxyl and amino groups are formed as the C1s and N1s core level spectra remain identical over cycling (Figure S11, Supporting Information). In comparison to the initial structure, the carbonyl oxygen atoms after the structural change show enhanced interaction with cations as a redder color is observed in the ESP calculation (Figure 3f). These highly concentrated negative sites attract more Zn ions. Examining Figure 3g, we observe a new peak centered at 497.8 eV in the Zn LMM Auger spectrum of cycled piZn, whereas the coordinated Zn peak shifts to a lower energy centered at 496.5 eV. A non‐coordinated Zn has a more positive character and hence higher kinetic energy.[ 34 , 39 ] Therefore, the new peak is attributed to the free Zn ions, which are absorbed in the polymer matrix due to the enhanced electrostatic interaction. The 0.2 eV downshift of the coordinated Zn2+ peak center is attributed to the stronger electron‐donating ability of carbonyl oxygen atoms.[ 34 ] An increasing amount of the Zn ions in the polymer matrix enhances the interaction with oxygen atoms. As a result, the intensity ratio between the shoulder peak centered at 532.6 eV and C=O peak at 531.9 eV in the O1s core level spectrum increases by 24% (Figure 3h). In addition to the strong attraction of cations, the positive sites become stronger than those in raw piZn, hence allowing for an improved ion transport,[ 28 ] which is confirmed by the increased n value for Q p and Q z derived from the impedance profile of cycled piZn (Figure S12 and Table S4, Supporting Information). In short, a strong interaction with Zn ions in the polyimide coating builds up during cycling, forming a Zn blanket in situ with high Zn transference number that reduces the concentration gradient and improves the ion transport. As a result, high Zn reversibility is observed in both galvanostatic cycling tests and coulombic efficiency measurements.
Figure 4a illustrates the process of fabricating a piZn microanode for a microbattery. Interdigitated electrodes are patterned and deposited on a substrate, for instance a silicon wafer. The length and width of the microelectrodes are 4.5 mm and 500 µm, respectively. The interspace between two electrodes is 150 µm. Zn is electrodeposited onto the anode side, forming the assembly of Zn nanosheets (Figure S13, Supporting Information). Subsequently, the polyimide precursor solution is spin‐coated onto the substrate and patterned by a described photolithography process. After baking at 220 °C, the Zn microanode is protected by the polyimide. The Bode plot of the protected Zn microanode (Figure S14, Supporting Information) demonstrates the similar interface behavior, indicating that the protection is independent of Zn morphology. In Figure 4b, we compare the galvanostatic cycling performance of the symmetrical microbatteries using bare Zn and piZn with a current density of 4 mA cm−2 and depth of charge/discharge of 80%. It is apparent that the overpotential develops over cycling for the bare Zn microanode due to the corrosion accompanying with the plating/stripping process. The corrosion even occurs when the bare Zn microanode is immersed in the electrolyte as we observe bubbles at the microanode surface (Figure S15, Supporting Information). The corrosion also results in the short‐cut of the symmetrical microbattery as pulverized Zn particles easily precipitate at the interspace between two microelectrodes. A sudden drop in the overpotential during the 60th cycle implies the failure of the microbattery due to the short‐cut. By contrast, more than 800 stable cycles are attainable for the piZn microanode. The polyimide coating, however, covers the surface of the Zn nanosheet assembly that has a high surface area and hence limits the surface kinetics and access of ions to the Zn nanosheets. As a result, the surface overpotential causes a slightly larger overpotential of the piZn microbattery than that for the bare Zn one. Nevertheless, the overpotential of the piZn microbattery is still smaller than that using the bare Zn foil (Figure 2b).
Figure 4.
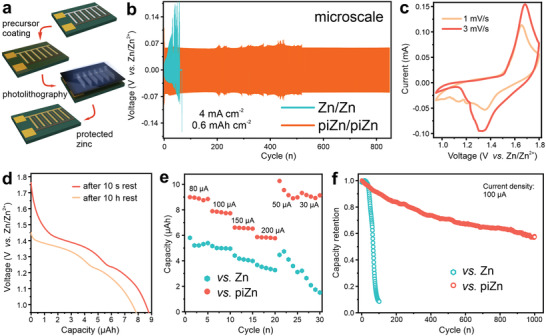
Aqueous microbattery. a) Schematic of the fabrication of a microbattery. b) Galvanostatic cycling performance of Zn and piZn microbattery at a current density of 4 mA cm−2. c) CV curves of the piZn microbattery with a MnO2 microcathode at different scan rates. d) Discharge curves of a fresh piZn microbattery and that after the rest of 10 h at the fully charged state. e,f) Comparisons of rate (e) and cycling (f) performance of the Zn and piZn microbattery.
As we have seen that the piZn offers superior performance at the microscale, it is interesting to investigate a full cell's performance. We deposit a MnO2 nanosphere assembly (Figure S16, Supporting Information) as the microcathode. Figure 4c shows the cyclic voltammetry (CV) curves of the assembled microbattery in a 2 mol L−1 ZnSO4 solution. A pair of redox peaks is observed for various scan rates, correlating to the insertion and extraction of cations (H+ and Zn2+) in MnO2.[ 40 , 41 ] We have presented the capacity loss after resting at the charged state for 10 h due to the degradation of the bare Zn microanode (Figure 1c) and have interpreted that the polyimide coating will address this issue. Figure 4d provides direct evidence that around 90% capacity is attainable after a ten‐hour rest at the charged state. In addition to the anti‐corrosion property, piZn also shows a low overpotential, which is able to allow for a high‐rate capability. Figure 4e compares the rate performance of the microbattery using bare Zn and piZn microanode. The capacity of the microbattery with the piZn microanode is at least 2 times of that for the microbattery with the bare Zn microanode. At high rates (150 and 200 µA), the capacity of the piZn microbattery is more stable than that of the bare Zn microbattery, agreeing with the better Zn reversibility at a high current as seen from Figure 2b. It is natural that the corrosion aggravates at a high rate, which causes an irreversible loss of the capacity. Figure 3e presents a fast drop of the capacity of the bare Zn microbattery after only 5 cycles at 200 µA. By contrast, the capacity of the piZn microbattery recovers after the high‐rate cycling. Combining the advantages of the anti‐corrosion ability and high reversibility of the piZn microanode, the capacity retention of the piZn microbattery after 1000 charge/discharge cycles reaches around 60%, while it rapidly drops to less than 10% in 100 charge/discharge cycles for the bare Zn microbattery.
Besides the good performance in aqueous electrolyte, a quasi‐solid‐state microbattery using the PAAm based hydrogel as the electrolyte is assembled. The contact resistance between the PAAm and the piZn microanode is small enough (23 Ω) to avoid the possible voltage drop (Figure S17, Supporting Information). Charge/discharge profiles of the piZn microbattery at different currents are presented in Figure 5a. At a current of 10 µA, a high capacity of 14 μAh is achieved. Two consecutive plateaus indicating the insertion of H+ and Zn2+ into the MnO2 verify that the charge storage mechanism in the PAAm based hydrogel remains identical to that in the aqueous electrolyte. With increasing currents, the capacity decreases due to the kinetic limitation. The piZn microbattery delivers a capacity of 7 and 5 μAh at a current of 100 and 200 µA, respectively. In comparison to the capacity achieved in the aqueous solution, we do not observe a noticeable capacity loss in the quasi‐solid state. In practical applications, power and energy are two essential parameters to evaluate the performance of a battery. Therefore, Figure 5b shows the energy output of the piZn microbattery with the PAAm electrolyte at different discharging powers. The excellent rate performance seen in the aqueous electrolyte continues here. At a high power of 270 µW, the energy output still reaches 5 μWh, showing the ability to provide electricity for power‐demanding electronic devices. Furthermore, more than 70% energy output can recover after cycling at high power. Generally, a passivation layer of basic Zn sulfate will accumulate rapidly on the cathode's surface at a high rate,[ 42 , 43 ] thus impeding the migration of cations into MnO2. The aqueous electrolyte still can be trapped at the solid interface, thus keeping the performance. Nevertheless, the new solid interface separates the PAAm electrolyte and MnO2. As a result, the piZn microbattery with the PAAm electrolyte shows some energy loss after cycling at high power. The inset in Figure 5b further shows the high‐power capability of the microbattery as a supercapacitor‐like charge/discharge time, ranging from seconds to minutes, is observed. The galvanostatic cycling performance at a current of 100 µA is shown in Figure 5c. 400 stable charge/discharge cycles are attainable. Moreover, the charge/discharge curves at the 50th and 350th cycle show almost identical charge/discharge profile. The well‐kept discharge plateau demonstrates that the piZn microbattery offers stable energy output for a long‐term service time.
Figure 5.
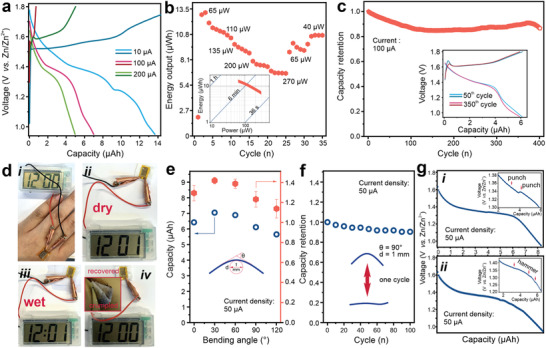
Quasi‐solid‐state microbattery for practical applications. a) Galvanostatic charge/discharge curves of the piZn microbattery with the PAAm‐based electrolyte at different currents. b) Rate performance of the piZn microbattery. The inset shows the Ragone plot of the piZn microbattery. c) Cycling performance of the piZn microbattery. The inset shows the galvanostatic charge/discharge curves of 50th and 350th cycle. d) Optical images of the piZn microbattery in different application scenarios. Panel (i) shows the microbattery attached on skin. Panels (ii and iii) show the microbattery attached to a tissue in dry and wet conditions. Panel (iv) shows the microbattery after being crumpled. e) Capacity of the piZn microbattery at different bending angles. f) Capacity retention of the piZn microbattery as a function of bending cycles. g) Galvanostatic discharge curves of the piZn microbattery under punching (((i)) and hammering (((ii)). The insets show the voltage change.
The fabrication of the piZn microbattery is based on a standard photolithography process and hence compatible with any flexible and flat substrate. As Kapton tape shows high flexibility and resistance to chemicals, we use a Kapton tape as the substrate to fabricate a flexible and skin‐mountable microbattery. Figure 5d presents several application situations for the skin‐mountable microbattery. The microbattery can be easily attached to skin and power an electric timer (Figure 5d, panel (i)). As the microbattery is exposed to environments, it is possible that the microbattery will be splashed. The water may seep into the microbattery from the upper and bottom surface. Therefore, we attach the microbattery onto a tissue and drop water onto it. In this case, the microbattery continues to power the electric timer (Figure 5d, panels (ii and iii)), showing that the adsorbed water has no effect in practical applications. Furthermore, the microbattery on the skin may be severely deformed, while it should keep its performance. Panel (iv) of Figure 5d proves that the microbattery functions well after being heavily crumpled.
Figure 5e shows the capacity of the piZn microbattery under different bending angles. The curvature of the bending is kept at 1 mm, thus imposing sufficient deformation to the microbattery. A small bending angle strengthens the contact between the PAAm electrolyte and the electrode materials. As a result, the capacity increases from 8.2 to around 9 μAh with bending angles of 30° and 60°. Upon further bending, the capacity decreases due to the delamination of the PAAm layer from the electrode materials. Therefore, the capacity decreases to 7.5 μAh, presenting a capacity retention of 85%. Figure 5f shows the capacity retention of the piZn microbattery under multiple bending cycles. In one bending cycle, the microbattery is bent to 90° and recovered. Besides the firm attachment onto the electrode over 100 bending cycles (Figure S18, Supporting Information), the capacity of the microbattery is measured at the recovered state to investigate the potential impact on the energy storage ability. At over 100 bending cycles, 90% capacity can be maintained, showing a good resistance of the skin‐mountable microbattery to the repeated mechanical deformations. In addition to repeated deformations, external impacts that may penetrate the sealing of the microbattery are inevitable for skin‐mountable microbatteries because they are positioned at the outer surface of the human body. Figure 5g presents the galvanostatic discharge profile of the punched ((panel (i)) and hammered (panel (ii)) piZn microbattery at 50 µA. In both situations, a typical discharge plateau is observed and the voltage changes less than 10 mV under the external impacts (insets of Figure 5g), demonstrating the good reliability of the piZn microbattery as an outer interface between a human being and the environment. Based on the stable charge/discharge performance, Video S1, Supporting Information, demonstrates the stability in practical applications. The skin‐mountable microbattery attached to a finger with different directions stably operates a timer under repeated bending. Besides, the stable operation of the timer is observed upon punching and hammering and after being cut by a knife.
In summary, we developed a polyimide coating as a polymeric Zn blanket for a highly reversible Zn anode. As an insulating layer, the polyimide coating induces the ohmic resistance‐controlled suppression of the corrosion. More importantly, the strong polar oxygen atoms in the polyimide coordinate with Zn2+ ions, and the coordination further builds up over cycling. Coordinated Zn ions are able to minimize the concentration gradient through the solid‐electrolyte interface, thus allowing for a small overpotential of the Zn plating/stripping process. As a result, the piZn anode shows a significantly improved lifespan at a high current density. Moreover, the piZn anode offers near 100% coulombic efficiency and a stable cycling performance at 85% DOD, thus guaranteeing the high utilization and reversibility of a microanode with a small mass loading. As the polyimide is patternable by standard photolithography processes, we design a microbattery with the piZn microanode. In the aqueous electrolyte, the corrosion‐induced capacity loss is reduced from 40% to 10%. Compared to the Zn microbattery, the piZn microbattery's capacity at different current densities increases by at least 2 times and recovers after high‐rate charge/discharge cycles. By contrast, irreversible capacity loss is observed for the Zn microbattery. The piZn microbattery shows 60% capacity retention for 1000 cycles, while the Zn microbattery can only run for 60 cycles to attain the same capacity retention. These superior performances in the aqueous electrolyte continue in the hydrogel electrolyte, which shows a high‐rate capability with a charge/discharge time at the supercapacitor level and a long lifespan of 400 stable cycles. Furthermore, a skin‐mountable microbattery is produced, which shows stable performance under deformations, splashing, and external shocks. Therefore, the as‐developed patternable polyimide envisions a reliable and powerful microscale battery with high safety, stability, and wearability to energize the rapid development of wearable electronics.
Experimental Section
Preparation of piZn
The polyimide precursor solution is synthesized according to the previous work.[ 44 ] The precursor solution is spin coated onto the Zn electrode at a rotation speed of 9000 rpm and dried at 50 °C for 10 min. Subsequently, the poly(amic acid) is polymerized for 300 s using Suss MJB4 mask aligner. The imidization process is conducted at 220 °C.
Preparation of the PAAm‐Based Hydrogel
6 g acrylamide was dissolved in 40 mL 2 mol L−1 ZnSO4, followed by the addition of 30 mg ammonium persulfate and 4 mg N,N′‐methylenebis(acrylamide). The solution was under rigorous stirring until it became clear. Afterward, the prepared solution was injected into rubber molds and covered with a glass plate. The sample was kept at 70 C for 3 h to allow complete polymerization. Subsequently, the samples were taken out of the molds and were directly attached to the microbattery.
Fabrication of the Microbattery
The Cr/Au current collector in the microbattery is deposited by nanoPVD (Moorfield Nanotechnology Ltd.) and patterned by a standard lift‐off process using an AZ‐5214E photoresist (Microchemicals GmbH). The Zn microanode is electrochemically deposited under 1 V versus SCE using the electrolyte of 1 mol L−1 ZnSO4. The piZn microanode is fabricated as described above. The MnO2 microcathode is deposited by a pulse deposition according to the previous work.[ 45 ]
Characterization
FTIR spectroscopy (Nicolet 6700, Thermo Scientific) was carried out in attenuated total reflection mode. The morphologies were characterized by SEM (FESEM, DSM982 Gemini). XRD tests were performed on X'Pert PRO MPD, Philips (Co Kα radiation, λ = 1.78910 Å). XPS tests were performed on ESCALAB 250Xi, ThermoFisher Scientific. Symmetric cells (Zn/Zn and piZn/piZn) and asymmetric cells consisting of Zn/stainless steel coated by the polyimide are used for plating– stripping tests. Plating–stripping profiles and galvanostatic charge–discharge profiles were taken from the battery test system (Arbin 2000). CV curves and impedance spectra were measured by an electrochemical workstation (MULTIAUTOLAB/M101, Metrohm Autolab).The impedance measurement is carried at the open circuit potential.
Calculation
The molecular geometries of the fresh polyimide and the cycled polyimide for the ground states were optimized by DFT at the B3LYP/6‐311G + (d, p) level. ESPs were evaluated. All the DFT calculations were carried out with the Gaussian 09 package.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Supplemental Video 1
Acknowledgements
M.Z. and J.H. contributed equally to this work. The authors appreciate Ronny Engelhard, Barbara Eichler, Sandra Nestler, Carol Schmidt, Martin Bauer, and Lisa Schröder for support in the clean room facilities. The authors acknowledge Kristina Leger for the support on poly(amic acid) synthesis. Q.L., Y.L., H.Y., Z.Q. acknowledge financial support from the China Scholarship Council (CSC). J.H. acknowledges the financial support by the National Natural Science Foundation of China (11904153). D.K. acknowledges financial support by the German Research Foundation SPP 1857 ESSENCE (KA5051/1‐1). O.G.S. acknowledges financial support by the Leibniz Program of the German Research Foundation (SCHM 1298/26‐1). For the experiments shown in Figure 5d, the battery is only placed on the skin firmly with plastic encapsulation and doesn't interact with any skin tissue. The volunteer in this demonstration is the author of this manuscript. No ethical approval was required in this case.
Open access funding enabled and organized by Projekt DEAL.
Zhu M., Hu J., Lu Q., Dong H., Karnaushenko D. D., Becker C., Karnaushenko D., Li Y., Tang H., Qu Z., Ge J., Schmidt O. G., A Patternable and In Situ Formed Polymeric Zinc Blanket for a Reversible Zinc Anode in a Skin‐Mountable Microbattery. Adv. Mater. 2021, 33, 2007497. 10.1002/adma.202007497
References
- 1. Oh J. Y., Bao Z., Adv. Sci. 2019, 6, 1900186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mehrali M., Bagherifard S., Akbari M., Thakur A., Mirani B., Mehrali M., Hasany M., Orive G., Das P., Emneus J., Andresen T. L., Dolatshahi‐Pirouz A., Adv. Sci. 2018, 5, 1700931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu Y., Pharr M., Salvatore G. A., ACS Nano 2017, 11, 9614. [DOI] [PubMed] [Google Scholar]
- 4. García Núñez C., Manjakkal L., Dahiya R., npj Flexible Electron. 2019, 3, 1. [Google Scholar]
- 5. Bandodkar A. J., Gutruf P., Choi J., Lee K. H., Sekine Y., Reeder J. T., Jeang W. J., Aranyosi A. J., Lee S. P., Model J. B., Ghaffari R., Su C. J., Leshock J. P., Ray T., Verrillo A., Thomas K., Krishnamurthi V., Han S., Kim J., Krishnan S., Hang T., Rogers J. A., Sci. Adv. 2019, 5, eaav3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Someya T., Bao Z., Malliaras G. G., Nature 2016, 540, 379. [DOI] [PubMed] [Google Scholar]
- 7. Jiang D., Shi B., Ouyang H., Fan Y., Wang Z. L., Li Z., ACS Nano 2020, 14, 6436.. [DOI] [PubMed] [Google Scholar]
- 8. Zheng S., Shi X., Das P., Wu Z. S., Bao X., Adv. Mater. 2019, 31, 1900583. [DOI] [PubMed] [Google Scholar]
- 9. Kyeremateng N. A., Brousse T., Pech D., Nat. Nanotechnol. 2017, 12, 7. [DOI] [PubMed] [Google Scholar]
- 10. Ryu J., Park M., Cho J., Adv. Mater. 2019, 31, 1804784. [DOI] [PubMed] [Google Scholar]
- 11. Liang G., Mo F., Ji X., Zhi C., Nat. Rev. Mater. 2020, 10.1038/s41578-020-00241-4. [DOI] [Google Scholar]
- 12. Tang B., Shan L., Liang S., Zhou J., Energy Environ. Sci. 2019, 12, 3288. [Google Scholar]
- 13. Fang G., Zhou J., Pan A., Liang S., ACS Energy Lett. 2018, 3, 2480. [Google Scholar]
- 14. Konarov A., Voronina N., Jo J. H., Bakenov Z., Sun Y. K., Myung S. T., ACS Energy Lett. 2018, 3, 2620. [Google Scholar]
- 15. Zhang N., Chen X., Yu M., Niu Z., Cheng F., Chen J., Chem. Soc. Rev. 2020, 49, 4203. [DOI] [PubMed] [Google Scholar]
- 16. Jin Y., Han K. S., Shao Y., Sushko M. L., Xiao J., Pan H., Liu J., Adv. Funct. Mater. 2020, 30, 2003932. [Google Scholar]
- 17. Qiu H., Du X., Zhao J., Wang Y., Ju J., Chen Z., Hu Z., Yan D., Zhou X., Cui G., Nat. Commun. 2019, 10, 5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Higashi S., Lee S. W., Lee J. S., Takechi K., Cui Y., Nat. Commun. 2016, 7, 11801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhao Z., Zhao J., Hu Z., Li J., Li J., Zhang Y., Wang C., Cui G., Energy Environ. Sci. 2019, 12, 1938. [Google Scholar]
- 20. Yin Y., Yamada O., Tanaka K., Okamoto K. I., Polym. J. 2006, 38, 197. [Google Scholar]
- 21. Wang J., Bandari V. K., Karnaushenko D., Li Y., Li F., Zhang P., Baunack S., Karnaushenko D. D., Becker C., Faghih M., Kang T., Duan S., Zhu M., Zhuang X., Zhu F., Feng X., Schmidt O. G., ACS Nano 2019, 13, 8067. [DOI] [PubMed] [Google Scholar]
- 22. Muster T. H., Neufeld A. K., Cole I. S., Corros. Sci. 2004, 46, 2337. [Google Scholar]
- 23. Peng J., Chen B., Wang Z., Guo J., Wu B., Hao S., Zhang Q., Gu L., Zhou Q., Liu Z., Hong S., You S., Fu A., Shi Z., Xie H., Cao D., Lin C., Fu G., Zheng L.‐S., Jiang Y., Zheng N., Nature 2020, 586, 390. [DOI] [PubMed] [Google Scholar]
- 24. Snyder R. W., Thomson B., Bartges B., Czerniawski D., Painter P. C., Macromolecules 1989, 22, 4166. [Google Scholar]
- 25. Chamoun M., Hertzberg B. J., Gupta T., Davies D., Bhadra S., Van Tassell B., Erdonmez C., Steingart D. A., NPG Asia Mater 2015, 7, e178. [Google Scholar]
- 26. McCafferty E., in Introduction to Corrosion Science, Springer, New York: 2010, pp. 403–425. [Google Scholar]
- 27. McCafferty E., in Introduction to Corrosion Science, Springer, New York, 2010, pp. 427–451. [Google Scholar]
- 28. Doyle M., Fuller T. F., Newman J., Electrochim. Acta 1994, 39, 2073. [Google Scholar]
- 29. Vallejo E., Pourcelly G., Gavach C., Mercier R., Pineri M., J. Membr. Sci. 1999, 160, 127. [Google Scholar]
- 30. Baran M. J., Braten M. N., Sahu S., Baskin A., Meckler S. M., Li L., Maserati L., Carrington M. E., Chiang Y. M., Prendergast D., Helms B. A., Joule 2019, 3, 2968. [Google Scholar]
- 31. Gracia R., Mecerreyes D., Polym. Chem. 2013, 4, 2206. [Google Scholar]
- 32. Mao C., Fang L., Zhang H., Li W., Wu F., Qin G., Ruan H., Kong C., J. Alloys Compd. 2016, 676, 135. [Google Scholar]
- 33. Li W., Fang L., Qin G., Ruan H., Zhang H., Kong C., Ye L., Zhang P., Wu F., J. Appl. Phys. 2015, 117, 145301. [Google Scholar]
- 34. Cano A., Lartundo‐Rojas L., Shchukarev A., Reguera E., New J. Chem. 2019, 43, 4835. [Google Scholar]
- 35. Oh Y. J., Yoo J. J., Il Kim Y., Yoon J. K., Yoon H. N., Kim J. H., Bin Park S., Electrochim. Acta 2014, 116, 118. [Google Scholar]
- 36. Wu L. Q., Li Y. C., Li S. Q., Li Z. Z., Tang G. D., Qi W. H., Xue L. C., Ge X. S., Ding L. L., AIP Adv. 2015, 5, 097210. [Google Scholar]
- 37. Chen L., Li W., Guo Z., Wang Y., Wang C., Che Y., Xia Y., J. Electrochem. Soc. 2015, 162, A1972. [Google Scholar]
- 38. Yang H., Liu S., Cao L., Jiang S., Hou H., J. Mater. Chem. A 2018, 6, 21216. [Google Scholar]
- 39. Deroubaix G., Marcus P., Surf. Interface Anal. 1992, 18, 39. [Google Scholar]
- 40. Sun W., Wang F., Hou S., Yang C., Fan X., Ma Z., Gao T., Han F., Hu R., Zhu M., Wang C., J. Am. Chem. Soc. 2017, 139, 9775. [DOI] [PubMed] [Google Scholar]
- 41. Wan F., Zhou X., Lu Y., Niu Z., Chen J., ACS Energy Lett. 2020, 12, 3569. [Google Scholar]
- 42. Xue T., Fan H. J., J. Energy Chem. 2021, 54, 194. [Google Scholar]
- 43. Guo X., Zhou J., Bai C., Li X., Fang G., Liang S., Mater. Today Energy 2020, 16, 100396. [Google Scholar]
- 44. Karnaushenko D. D., Karnaushenko D., Grafe H. J., Kataev V., Büchner B., Schmidt O. G., Adv. Electron. Mater. 2018, 4, 1800298. [Google Scholar]
- 45. Zhu M., Wang Z., Li H., Xiong Y., Liu Z., Tang Z., Huang Y., Rogach A. L., Zhi C., Energy Environ. Sci. 2018, 11, 2414. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supplemental Video 1