

Abstract
Electroactive hydrogels can be used to influence cell response and maturation by electrical stimulation. However, hydrogel formulations which are 3D printable, electroactive, cytocompatible, and allow cell adhesion, remain a challenge in the design of such stimuli‐responsive biomaterials for tissue engineering. Here, a combination of pyrrole with a high gelatin‐content oxidized alginate‐gelatin (ADA‐GEL) hydrogel is reported, offering 3D‐printability of hydrogel precursors to prepare cytocompatible and electrically conductive hydrogel scaffolds. By oxidation of pyrrole, electroactive polypyrrole:polystyrenesulfonate (PPy:PSS) is synthesized inside the ADA‐GEL matrix. The hydrogels are assessed regarding their electrical/mechanical properties, 3D‐printability, and cytocompatibility. It is possible to prepare open‐porous scaffolds via bioplotting which are electrically conductive and have a higher cell seeding efficiency in scaffold depth in comparison to flat 2D hydrogels, which is confirmed via multiphoton fluorescence microscopy. The formation of an interpenetrating polypyrrole matrix in the hydrogel matrix increases the conductivity and stiffness of the hydrogels, maintaining the capacity of the gels to promote cell adhesion and proliferation. The results demonstrate that a 3D‐printable ADA‐GEL can be rendered conductive (ADA‐GEL‐PPy:PSS), and that such hydrogel formulations have promise for cell therapies, in vitro cell culture, and electrical‐stimulation assisted tissue engineering.
Keywords: 3D‐printing, biomaterials, electrically conductive hydrogels, oxidized alginate, polypyrrole, tissue engineering
Extrusion‐printed hydrogels are presented, which show enhanced electrical conductivity. Oxidized alginate‐gelatin hydrogel in combination with conductive polypyrrole polymer allows for the fabrication of 3D‐printed cell adhesive and electrically functional biomaterial scaffolds. Enzymatic and ionic crosslinking achieve material stability of 30 days in vitro. Together, this material can be used as a platform to study electrical stimulation assisted tissue engineering.
![]()
1. Introduction
The engineering of hydrogel‐based biomaterials is advancing toward increasingly complex and controllable systems.[ 1 , 2 , 3 , 4 , 5 ] While biocompatibility and active cell–material interactions are important material requirements for tissue engineering, the development of smart‐ and stimuli‐responsive hydrogels, which offer an additional degree of control over hydrogel properties and cell fate, has been emerging in recent years.[ 4 , 6 , 7 , 8 , 9 , 10 , 11 , 12 ] Stimuli‐responsive hydrogels have been developed for various applications,[ 8 , 9 ] enabling controlled drug release,[ 13 , 14 ] cellular attachment[ 12 , 15 , 16 , 17 , 18 ] and differentiation,[ 19 ] degradation kinetics,[ 20 ] conductivity,[ 21 ] and electrical stimulation.[ 11 , 22 ] As one specific type of stimuli‐responsive hydrogel, electroactive hydrogels (EAH) containing electrically conductive polymers (CP) are within the scope of intense current research.[ 23 , 24 , 25 ] Electrically conductive hydrogels have been used to offer electroactive substrates for electrical cell stimulation (ES),[ 26 ] as conductive paths to guide electrical current in stimuli‐responsive soft‐electronics,[ 27 ] for motion sensing,[ 21 , 25 ] or to control drug release of biomolecules upon ES.[ 28 , 29 ] In addition, 3D‐printing has been employed to create open‐porous, 3D, electrically conductive hydrogel scaffolds for cell stimulation,[ 26 , 30 ] electroactive tissue‐supports,[ 31 ] or sensor applications with complex geometry.[ 32 ] While EAH have been used for applications like neural[ 33 , 34 , 35 ] or muscle[ 31 , 36 , 37 , 38 , 39 , 40 ] tissue engineering, they have rarely been applied for cartilage tissue regeneration. Recent studies highlighted the possibility to enhance and control cellular differentiation of chondrocytes using ES.[ 41 , 42 , 43 , 44 , 45 ] The knowledge of the potential positive effect of ES on cartilage regeneration has been gathered for more than a decade.[ 46 , 47 , 48 ] Chondrocytes embedded in hyaluronic acid‐gelatin hydrogels showed increased expression of hyaline cartilage markers SOX‐9 and hyaline extracellular matrix (ECM) glycosaminoglycans like aggrecan (ACAN) upon ES.[ 49 ] As CPs can change their redox state once subjected to ES,[ 4 ] with the possibility to control cell attachment,[ 50 ] migration, and differentiation,[ 33 , 51 , 52 ] the use of hydrogels with tunable electrical conductivity may represent a promising approach to enhance ES‐assisted cartilage regeneration. Oligopolypyrrole‐chitosan hydrogel has been synthesized for intended use in cartilage tissue engineering.[ 53 ] The hydrogels showed suitable toughness and strength to be potentially applied for cartilage regeneration applications.[ 53 ] While the hydrogels were degradable and electroactive, 3D‐printed open‐porous scaffolds for advanced matrix‐associated chondrocyte implantation (MACI) have not been reported. To the best of our knowledge, no study has been dedicated to the engineering of electrically conductive, 3D‐printed hydrogels with the purpose to be applied for advanced MACI applications.
Oxidized alginate‐based hydrogels have shown promising performance in various tissue engineering approaches.[ 54 ] We recently engineered a dually crosslinked ADA‐GEL hydrogel which showed good cell attachment and proliferation capacity while offering stability for up to 30 days of cell culture.[ 55 ] In addition, such hydrogels provided a suitable platform for the 3D‐cultivation of primary human nasal chondrocytes.[ 56 ] CP‐functionalized ADA‐GEL hydrogels have been recently engineered with the focus on biosensor applications.[ 57 ] However, the challenge to provide suitable mechanical properties, biocompatibility, and highly defined structures to engineer advanced open‐porous biomaterial scaffolds for cartilage tissue engineering of such conductive hydrogels still needs to be addressed.
Here we investigate pyrrole (Py)‐modified dually crosslinked ADA‐GEL hydrogels that could be 3D‐printed and subsequently rendered conductive, resulting in ADA‐GEL‐PPy:PSS composite hydrogels for advanced cartilage tissue engineering applications. We show that by the addition of Py to a thermally pre‐treated high gelatin‐content ADA‐GEL precursor (Figure 1A), the hydrogel can be 3D‐printed into open‐porous scaffolds, which allow functionalization by oxidation using FeCl3 to form conductive PPy:PSS inside the ADA‐GEL matrix (Figure 1B). Utilizing the dual‐crosslinking approach (Figure 1C), stable hydrogel scaffolds for long‐term cell culture (>14 days) are presented. We identify the optimal composition of ADA‐GEL‐Py hydrogel, maintaining 3D‐printability while facilitating us to increase the electrical conductivity of the hydrogels once the Py is oxidized to polypyrrole, yielding ADA‐GEL‐PPy:PSS‐based hydrogels. The cytocompatibility of the hydrogels and seeding efficiency on 3D‐printed ADA‐GEL‐PPy:PSS was investigated via multiphoton fluorescence microscopy using mouse teratocarcinoma ATDC‐5 cells, which is a prominent cell‐line model for cartilage engineering. Our results highlight that by 3D‐printing of ADA‐GEL, increased cell‐seeding efficiency for MACI applications can be achieved, while the interpenetrating CP matrix in ADA‐GEL adds valuable functionality with potential for ES‐assisted cartilage tissue engineering applications.
Figure 1.
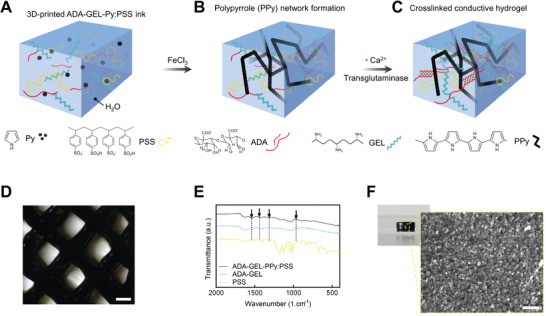
Schematic illustration of the formation of ADA‐GEL‐PPy:PSS conductive hydrogel. A) ADA‐GEL hydrogel precursor containing different molarities of Py and polystyrenesulfonate (PSS) is prepared. B) After 3D‐printing, the formation of polypyrrole (PPy) is triggered by oxidation of Py by immersion in FeCl3 solution. C) Final ADA‐GEL‐PPy:PSS scaffolds are dually crosslinked using Ca2+ and microbial transglutaminase to respectively crosslink the oxidized alginate and gelatin network inside the hydrogel. D) Light microscopy image of 3D‐printed ADA‐GEL‐PPy:PSS in a grit‐like structure. Scale bar: 400 µm. E) Fourier infrared spectroscopy spectra of ADA‐GEL and ADA‐GEL‐PPy:PSS, with characteristic band vibrations at approximately 1540 and 1460 cm−1 indicating symmetric and asymmetric stretching characteristic for Py, verifying the presence of PPy inside ADA‐GEL. F) Scanning electron microscopy (SEM) image of an ADA‐GEL‐PPy:PSS hydrogel cylinder indicating the formation of a dense globular PPy structure on the ADA‐GEL hydrogel surface in comparison to untreated ADA‐GEL (Figure S1, Supporting Information). Scale bar: 400 nm.
2. Results
2.1. Formation of Polypyrrole inside ADA‐GEL Hydrogel
We assessed the formation of PPy inside ADA‐GEL, which was synthesized by a two‐step fabrication process. First, Py was added at different molarities inside ADA‐GEL hydrogel precursor (Figure 1A). PSS was dissolved in the hydrogel precursor to act as a dopant to the PPy network. Next, ADA‐GEL‐Py was 3D‐printed or cast into hydrogel scaffolds, followed by thermal gelation at 22 °C through the thermal solidification of gelatin after printing. To synthesize PPy, the scaffolds were transferred into FeCl3 solution to facilitate oxidation of Py to PPy (Figure 1B), resulting in black hydrogel scaffolds which maintained their 3D‐printed shape (Figure 1D). Following, the scaffolds were crosslinked using Ca2+ and microbial transglutaminase (mTG) (Figure 1C). Fourier transform infrared spectroscopy indicated the formation of PPy by increased absorbance bands at 1530 and 1560 cm−1, which are indicative for C=C stretching and bending vibration of Py, respectively (Figure 1E). Microstructure analysis of the black hydrogel specimens via SEM revealed the formation of a globular and supposedly conductive structure on the hydrogels (Figure 1F), as the hydrogels did not charge by the electron beam during 2.5 kV acceleration voltage imaging in comparison to pristine ADA‐GEL, which would charge and was therefore imaged at 1 kV.
Together, the results confirmed the formation of globular and black PPy inside the ADA‐GEL hydrogel matrix.
2.2. Addition of Pyrrole Changes Hydrogel Precursor Printability
We assessed the printability of ADA‐GEL containing 0.1, 0.2, and 0.4 m Py. Pluronic F127 (38% wt) served as a printability benchmark control. Filament formation tests showed that after 20–30 min of room temperature (22°C, RT) storage, ADA‐GEL and ADA‐GEL‐0.1 m Py allowed for the extrusion of a cohesive hydrogel strut similar to Pluronic F127 hydrogel ink (Figure 2A), with hydrogel filament length of >25 mm without rupture of the extruded filament. It was possible to produce open‐porous, 3D hydrogel scaffolds for all hydrogel inks (Figure 2B). Printability quantification, with a printability factor Pr,[ 58 ] which is reversely proportional to pore circularity, revealed that 0.1 m Py containing ADA‐GEL showed the same capability to form rectangular pores as ADA‐GEL (Pr ≈ 1) (Figure 2C). The 0.2 m Py composition showed a slight drop in Pr with a tendency to have strut fusion and more circular pore morphology (Pr < 1) (Figure 2B). It was possible to print scaffolds with heights exceeding 3 mm (Figure 2D). The uniformity (U) of 3D‐printed filaments was highest for the ADA‐GEL and 0.1 m Py composition, (U ≈ 1[ 59 , 60 ]), while a higher standard deviation of U for the 0.2 m Py composition reflected the tendency for strut non‐uniformities as observed in light microscopy images (Figure 2E, F). To monitor the influence of Py addition on the rheological hydrogel ink characteristics, shear‐rate, time, and temperature sweeps of the hydrogel precursors were performed (Figure 2E, bottom). The results indicated that with increasing Py content dispersed in the ADA‐GEL precursor, the low shear‐rate viscosity (at ≈3 s−1), storage modulus, and complex viscosity decreased (Figure 2E). Py addition reduced the shear‐thinning behavior of the hydrogel precursors (Figure 2E, bottom left), as ADA‐GEL showed a higher extent of shear‐thinning with a decrease of about two orders of magnitude from >100 Pa s to ≈1 Pa s between shear‐rates of 3–1100 s−1. Temperature sweeps indicated a decrease in storage modulus for all hydrogel precursors (Figure 2E, bottom middle). However, ADA‐GEL and ADA‐GEL‐0.1 m Py showed similar storage moduli over all temperatures. A decreased storage modulus from G′ ADA‐GEL ≈ 500–600 Pa to G′ ADA‐GEL‐0.2 m Py ≈ 150 Pa and G′ ADA‐GEL‐0.4 m Py ≈ 20–30 Pa at ≈22 °C was observed for 0.2 and 0.4 m Py containing ADA‐GEL. The complex viscosity increased over storage time at 22 °C for all samples (Figure 2E, bottom right). In addition, increased Py content decreased the sol–gel transition temperature, especially pronounced for 0.4 m Py containing ADA‐GEL, explaining its reduced printability (Figure S3, Supporting Information). ADA‐GEL‐0.1 m Py showed a similar complex viscosity to ADA‐GEL (≈30–100 Pa s) in comparison to ADA‐GEL‐0.2 m Py (≈6–30 Pa s) and ADA‐GEL‐0.4 m Py (≈2–10 Pa s), which had about two orders of magnitude decreased complex viscosity. As ADA‐GEL‐0.4 m Py showed the lowest viscosity among all groups, the main printability assessments were performed for ADA‐GEL, 0.1 m Py, and 0.2 m Py ADA‐GEL. UV–vis assessments of the ADA‐GEL‐Py hydrogel precursors indicated a peak formation at λ ≈ 420 nm and a visible aging behavior over time (Figure S5, Supporting Information), expressed highest for the 0.4 m Py composition, which is in accordance to the changes in rheological behavior over time. After printing, the oxidation process decreased the strut diameter by ≈10% (Figure S12, Supporting Information), due to the formation of the PPy network. Together, the results confirm that it is possible to disperse 0.1 m Py inside ADA‐GEL maintaining the 3D‐printability of the ADA‐GEL hydrogel precursor, while printability decreased at a loading degree of Py > 0.2 m Py inside ADA‐GEL. ADA‐GEL‐0.1 m Py showed the best printability among all Py‐modified ADA‐GEL samples.
Figure 2.
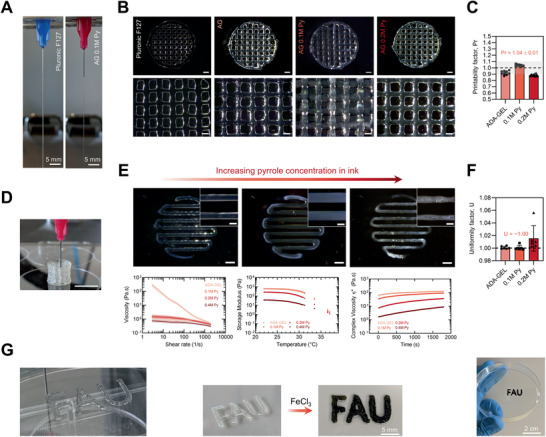
Printability and rheological assessment of ADA‐GEL‐Py:PSS conductive hydrogel precursors. A) Filament formation test indicating proper gelation of the biomaterial‐ink prior to extrusion, 0.1 m Py containing ADA‐GEL, similar to Pluronic F127 40% w/v, which served as biomaterial ink control. B) Optical microscopy images of 3D‐printed 3.75% / 7.5% w/v ADA‐GEL (AG) prepared using temperature pre‐treated gelatin and 0.1 and 0.2 m Py containing modifications. Pluronic F127 (40% wt) served as benchmark ink. Scale bars: 1 mm (top), 500 µm (bottom). C) Printability factor Pr derived from the printed hydrogels, which is inversely proportional to pore‐circularity (n = 9).[ 58 ] The Pr allows an indication of pore circularity and hence printability of the biomaterial ink (Pr = 1 perfectly square pore, Pr < 1 circular pore, and Pr > 1 cluttered, over‐gelated pore) as shown elsewhere.[ 58 , 59 ] The grey region marks the range of sufficient biomaterial‐ink printability, as introduced by Ouyang and co‐workers previously.[ 58 ] D) 3D‐printed ADA‐GEL hydrogel scaffold showing a possible z‐axis height of >5 mm, used as the ADA‐GEL hydrogel basis for the 0.1 m Py and 0.2 m Py containing inks. Scale bar: 10 mm. E) Light microscopy images of one‐layer 3D‐printed ADA‐GEL, 0.1 m Py, and 0.2 m Py, and rheological analysis of Py containing ADA‐GEL. Top: ADA‐GEL and 0.1 m Py show both nearly ideal parallel struts with uniform hydrogel filaments, while 0.2 m Py indicates a non‐homogeneous strut surface. Scale bars: 1 mm, 400 µm (insets). Bottom: Shear‐rate, temperature, and time sweeps. A decrease in viscosity is shown with increasing shear‐rate, indicative for shear thinning behavior. The storage modulus (G′) of the ink decreases with temperature, indicating thermo‐sensitivity. A decrease in G′ with increasing Py molarity is visible. The complex viscosity upon cooling time (s) increases for all compositions, while a drop in complex viscosity η* is shown for ADA‐GEL‐Py compositions >0.1 m Py. F) Hydrogel strut uniformity factor U [ 59 , 60 ] indicating the smoothness of deposited hydrogel struts, which is derived as the strut length in image‐width divided by the theoretical strut length. U = 1 depicts an ideally uniform strut (dashed horizontal line).[ 59 , 60 ] Non‐uniformity of 0.2 m Py is reflected in a higher standard deviation of U in comparison to ADA‐GEL and 0.1 m Py (n = 6). Data are shown as mean ± SD. G) Photographs of 3D‐printed CAD model letters before (left) and after (right) oxidiation of Py to form PPy using FeCl3.
2.3. Polypyrrole Formation Increases Hydrogel Conductivity
The direct‐ and alternating‐current electrical properties of ADA‐GEL and ADA‐GEL‐PPy:PSS formed by the oxidation of 0.1, 0.2, and 0.4 m Py containing ADA‐GEL hydrogels was assessed using electrical impedance spectroscopy (EIS) and four‐point probe measurements (Figure 3 ). ADA‐GEL‐PPy samples oxidized from 0.1, 0.2, and 0.4 m Py containing ADA‐GEL are denoted as 0.1 m PPy, 0.2 m PPy, and 0.4 m PPy, respectively. EIS of the hydrogels confirmed increased alternating current (AC) conductivity of 0.1 m PPy and 0.2 m PPy for frequencies between 100 Hz and 10 MHz (Figure 3A). 0.4 m PPy showed a decreased conductivity in comparison to the other modifications, with a higher standard deviation at higher frequencies (>1 kHz) in comparison to 0.1 m PPy and 0.2 m PPy. The 0.1 m PPy and 0.2 m PPy modifications increased the conductivity of ADA‐GEL at 100 Hz from 0.5 to ≈1.0–1.4 S m−1 (Figure 3B). 0.1 m PPy modification significantly increased the AC conductivity at 100 Hz, while the modifications showed only slight increases of AC conductivity at high frequencies (60 kHz) (Figure 3C). However, the total conductivity (direct current, DC + AC) increased over all frequencies except for the 0.4 m PPy modification, with significant increases in DC conductivity from approximately 0.5 to 1 S m−1 for 0.1 m PPy and 0.2 m PPy in comparison to the pristine ADA‐GEL (Figure 3D). The hydrogel samples were freeze‐dried to assess differences in conductivity in the wet and dry states. While no conductivity was measured for dry ADA‐GEL (sheet resistance > 800 kΩ square, resolution limit of four‐point probe measurement device), the conductivity of the dry specimens increased with increasing molarity of Py during PPy formation (Figure 3E). Dry 0.1 m PPy and 0.2 m PPy showed a conductivity of ≈10−5 to 10−4 S cm−1. It was possible to conduct DC using the PPy modified ADA‐GEL hydrogel to supply a light‐emitting diode (LED) (Figure 3F; Video S1, Supporting Information). Cyclic voltammograms of ADA‐GEL and 0.1 m PPy indicated a broad anodic peak at ≈0.41 V and a cathodic peak at approximately −0.03 V, with half‐wave potential E 1/2 at ≈0.215 V for 0.1 m PPy in comparison to ADA‐GEL. The results confirm that the PPy modification of ADA‐GEL successfully changes its electrical conductivity, adding functionality to the ADA‐GEL hydrogel.
Figure 3.
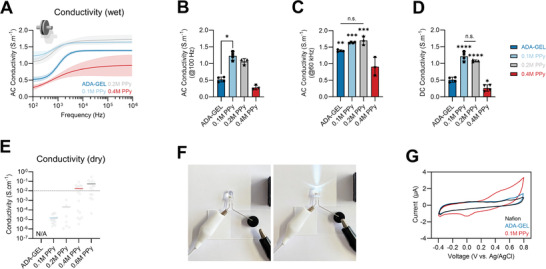
Electrical properties of ADA‐GEL‐PPy:PSS hydrogel. A) EIS of wet ADA‐GEL and ADA‐GEL‐PPy:PSS hydrogels synthesized using 0.1, 0.2, and 0.4 m Py inside ADA‐GEL during oxidation to form PPy. Samples are denoted as 0.1 m PPy, 0.2 m PPy, and 0.4 m PPy. Data are shown as mean curves and SD interval of confidence (n ≤ 4). For low (≈100 Hz) and high (>1 kHz) frequencies, enhanced electrical conductivity is visible for the 0.1 and 0.2 m PPy modifications. B) Corresponding readouts of AC conductivity of the hydrogels at 100 Hz and C) 60 kHz, with a significant increase in AC conductivity at 100 Hz observed for 0.1 m PPy hydrogels. Data are shown as mean ± SD. D) DC conductivity indicating a significant increase in conductivity for 0.1 and 0.2 m PPy (****p < 0.0001) in comparison to pristine ADA‐GEL. Data are shown as mean ± SD. Significant differences of means were analyzed using one‐way ANOVA analysis followed by post‐hoc Bonferroni test (*p < 0.05, **p < 0.01, ***p < 0.001). E) Electrical conductivity of freeze‐dried ADA‐GEL and PPy‐modified ADA‐GEL assessed via four‐point probe measurements (dry samples). While no conductivity could be measured using unmodified ADA‐GEL (N/A), an increase in conductivity was observed with increasing Py content during PPy oxidation. Data are shown as median (colored horizontal) and individual data points (gray dots). F) Light microscopy images of a DC 9V circuit conducted using ADA‐GEL‐PPy:PSS hydrogel to light an LED. G) Cyclic voltammetry measurement of ADA‐GEL and ADA‐GEL‐PPy:PSS (0.1 m Py) modification indicating electrical redox activity of the PPy modified hydrogel.
2.4. Mechanical and Degradation Properties of ADA‐GEL‐PPy:PSS Hydrogels
Uniaxial compression tests were performed to determine the mechanical properties of PPy:PSS functionalized ADA‐GEL. From ADA‐GEL‐PPy:PSS hydrogel cylinders with modification of 0.1, 0.2, 0.4, and 0.6 m PPy (Figure S2, Supporting Information), we only selected 0.1, 0.2, and 0.4 m PPy, as the 0.6 m PPy resulted in highly brittle specimens which were not feasible for further handling and studies.
We observed an increase in hydrogel stiffness for PPy:PSS modified ADA‐GEL (Figure 4 ). 0.1, 0.2, and 0.4 m PPy showed a significantly increased (****p < 0.0001) elastic modulus, exemplarily by approximately one order of magnitude to ≈1270 ± 250 kPa for the 0.1 m Py ADA‐GEL‐PPy:PSS hydrogel modification (Figure 4A,B). The results indicate that the formation of the PPy:PSS matrix inside the ADA‐GEL reinforces the ADA‐GEL matrix by the growth of an interpenetrating network of PPy:PSS (Figure 1C). However, a drop in stiffness and a higher chance of brittle fracture was observed for 0.4 m PPy. Due to the best results for 0.1 m PPy among all PPy‐modifications, it was selected for swelling and degradation studies.
Figure 4.
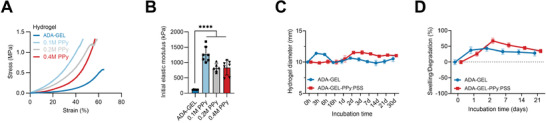
Mechanical properties and degradation behavior of PPy:PSS modified ADA‐GEL. A) Compression stress‐strain curve of ADA‐GEL, 0.1 m PPy, 0.2 m PPy, and 0.4 m PPy hydrogels. A stiffer compression response can be observed for PPy modified hydrogels. All hydrogels show a strain‐stiffening behavior. B) Quantified elastic modulus of the hydrogels. A significant increase of more than tenfold in Young's modulus is observed for all PPy modified ADA‐GELs in comparison to pristine ADA‐GEL (****p < 0.0001). Data are shown as mean ± SD. Statistical significance was assessed using one‐way ANOVA analysis followed by post‐hoc Bonferroni comparison of means. C) Degradation kinetics of ADA‐GEL and ADA‐GEL‐PPy:PSS. Swelling of the hydrogel cylinders measured by the diameter (initial diameter: 10 mm) changing over incubation time in DMEM for 3, 6, and 16 h, as well as 1, 2, 3, 7, 14, 21, and 30 days at 37 °C, 5% CO2. The presence of the PPy:PSS matrix seems to inhibit the initial swelling of the hydrogel cylinders after 3 h of incubation. Data are shown as mean ± SD (n = 6). D) Swelling and degradation of ADA‐GEL and ADA‐GEL‐PPy:PSS measured as the mass of hydrogel cylinders recorded over incubation time. Data are shown as mean ± SD (n = 6). Short‐term swelling behavior (<24 h, Figure S4, Supporting Information) can be found in the Supporting Information.
Mass loss tests revealed a similar behavior for ADA‐GEL and ADA‐GEL‐PPy:PSS, while the initial swelling in diameter of ADA‐GEL was slightly higher than for ADA‐GEL‐PPy:PSS (Figure S4, Supporting Information). The results confirm that the interpenetrating network of non‐degradable PPy:PSS modification does not alter long term scaffold stability of ADA‐GEL, resulting in stable scaffolds for up to 21 days under in vitro conditions (Figure 4C,D).
Enzyme‐mediated degradation tests by immersion of 3D‐printed ADA‐GEL‐PPy:PSS (0.1 m Py) and ADA‐GEL scaffolds in 1 U mL−1 collagenase II solution at 37 °C demonstrated that the scaffolds maintain stable for three days (Figure S11, Supporting Information), whereas pristine ADA‐GEL scaffolds degraded within one day of incubation. Hence, PPy modification led to increased scaffold stability in comparison to pristine ADA‐GEL.
Based on the printability, conductivity, and mechanical results, we selected 3D‐printed 0.1 m Py for future cell studies as the most promising formulation.
2.5. Cytocompatibility of 3D‐Printed ADA‐GEL‐PPy:PSS Hydrogel
The safety data sheets for the starting materials and products (FeCl3, Py, PPy, and PSS) show FeCl3 to be a corrosive irritant that is toxic to aquatic life, and Py is corrosive and could be toxic if swallowed, however, PPy and PSS are nontoxic. In silico toxicity screening studies of the starting materials and products (FeCl3, Py, PPy, and PSS, Table S1, Supporting Information) using Derek Nexus (Derek Nexus: 6.0.1, Nexus: 2.2.2)[ 61 , 62 ] demonstrated they were nonsensitizers of skin, and in silico mutagenicity screening studies using Sarah Nexus (Sarah Nexus: 3.0.0, Sarah Model: 2.0) demonstrated they were non‐mutagenic. Complementary in vitro studies were undertaken using ATDC‐5 cells seeded on ADA‐GEL and 3D‐printed 0.1 m PPy‐ADA‐GEL (3D AG‐PPy:PSS) scaffolds to determine the cytocompatibility of and cell‐seeding efficiency on the modified ADA‐GEL (Figure 5 ). Fluorescence microscopy images DAPI (nuclei, blue) and rhodamine phalloidin (F‐Actin, red) stained ATDC‐5 cells grown for seven days on the samples confirmed the growth on both, ADA‐GEL and PPy‐modified ADA‐GEL (Figure 5A; Figure S6, Supporting Information). During fluorescence imaging using multiphoton microscopy, absorption of the laser at specific spots on the sample was noticed for 3D AG‐PPy:PSS, resulting in white, high‐intensity spots on the images (Figure 5A, right) which was associated to laser beam absorption on black PPy. Representative X–Z stack projections of z‐cross sections gave an insight into the z‐depth at which cells were visible in optical cross sections of the hydrogel samples (Figure 5A, bottom row). The results show that cell adhesion and growth were possible on 3D AG‐PPy:PSS substrates. Cell quantification indicated fewer cells on 3D AG‐PPy:PSS in comparison to ADA‐GEL and TCPS (Figure 5B). However, we found that due to the 3D‐printing process, cells grown on 3D AG‐PPy:PSS could be detected in greater z‐depth on the hydrogels in comparison to cells seeded on flat TCPS or almost flat ADA‐GEL samples (Figure 5A,C; Figure S7, Supporting Information). This was a result of the 3D surface topography from the 3D‐printing process, allowing cells to grow into scaffold grooves between 3D‐printed hydrogel struts, and indicated increased seeding efficiency in z‐direction in comparison to the non‐printed, flat hydrogel samples (Figure S7, Supporting Information). SEM revealed cell‐material interaction on both, ADA‐GEL and 3D AG‐PPy:PSS samples, while the SEM images indicated a rougher material surface with increased curvature of the AG‐PPy:PSS surface (Figure 5D). The SEM results indicate that this leads to the formation of a less‐dense cell layer and fewer cell–cell contacts in comparison to an almost confluent cell layer, which was grown on the flat ADA‐GEL (Figure 5D; Figure S8, Supporting Information). DNA quantification showed that in comparison to day one, the number of cells increased on all samples after seven days of incubation, which confirmed cell proliferation was highest on TCPS and ADA‐GEL (≈3.5× and 2.8× dsDNA after 7 days, ****p < 0.0001), and by a factor of 1.6× higher on 3D AG‐PPy:PSS in comparison to the day one TCPS control (Figure 5E), however, the latter was not significant (n.s., p = 0.1621). Extracellular lactate dehydrogenase (LDH) release tests after 24 h showed that higher cell necrosis occurred on ADA‐GEL and 3D AG‐PPy:PSS substrates in comparison to TCPS controls (Figure 5F). Cell viability assay WST‐8 indicated a lower viability of cells on ADA‐GEL in comparison to 3D AG‐PPy:PSS and TCPS (Figure 5G). After 14 days of cell culture, a densely confluent cell layer was observed on ADA‐GEL (Figure 5H). Together, the results indicate the cytocompatibility of PPy modified ADA‐GEL, however slightly lower than on pristine ADA‐GEL for ATDC‐5 cells, while 3D‐printed 0.1 m ADA‐GEL increased seeding efficiency in hydrogel z‐direction.
Figure 5.
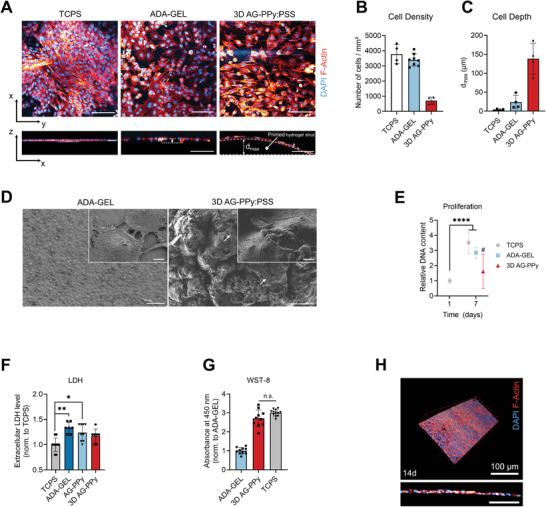
Cytocompatibility study of 3D AG‐PPyhydrogels. A) Multiphoton fluorescence microscopy images of ATDC‐5 cells (Nuclei, DAPI, blue; F‐Actin, Rhodamine Phalloidin, red) grown for seven days on tissue culture polystyrene (TCPS), mTG‐Ca2+ crosslinked ADA‐GEL (ADA‐GEL), and 3D AG‐PPy hydrogels. Bottom: x–z projections of z‐stacks. Scale bars: 100 µm. B) Quantification of the number of cells on each substrate (n = 4). C) Analysis of the depth d max in which cells were found on the hydrogels (cell depth) as a measure of seeding depth and seeding efficiency on the hydrogel scaffolds. D) SEM micrographs of ATDC‐5 cells grown for seven days on the substrates. Cell–material and cell–cell interaction are visible on both materials. Scale bars: 100 µm (left), 50 (right), 4 µm (insets). E) Proliferation study of ATDC‐5 on TCPS, ADA‐GEL, and 3D AG‐PPy. DNA content (dsDNA) after seven days is shown relative to ATDC‐5 cells grown on TCPS for 24 h, which served as control for all samples, and is quantified via PicoGreen assay (n = 4). Data are shown as mean ± SD. ****p < 0.0001, #n.s., relative to control (one‐way ANOVA with Bonferroni post‐hoc test). F) Quantification of extracellular LDH (n ≥ 7) after 24 h of cell culture to detect initial material cytotoxicity. Data are shown as mean ± SD and normalized to the TCPS samples. Statistically significant differences of mean ranks (*p < 0.05, **p < 0.005) were determined by non‐parametric Kruskal–Wallis followed by Dunn's multiple comparisons test. G) Cell viability WST‐8 assay (n ≥ 5) of ATDC‐5 cells on the different substrates, normalized to the ADA‐GEL hydrogel reference. No significant difference (n.s., Student's t‐test) was observed between 3D AG‐PPy samples and the TCPS control. H) ATDC‐5 cells grown for 14 days on ADA‐GEL positive reference. A confluent and dense cell layer is reached on the material.
3. Discussion
In this study, we demonstrate the production of 3D‐printable hydrogels subsequently rendered electrically conductive based on oxidized‐alginate PPy:PSS to create conductive scaffolds for tissue engineering applications. The hydrogels are cell adhesive and show cytocompatibility besides their electrical functionality. Comprehensive literature reviews on CPs[ 23 , 63 , 64 , 65 ] and 3D‐printed conductive hydrogels for biomedical and tissue engineering applications have been reported.[ 32 , 39 , 66 ] However, the main literature on similar alginate‐based materials as used here mainly consists of alginate‐PPy composites,[ 64 , 67 , 68 , 69 , 70 , 71 ] and oxidized alginate‐PPy systems have been explored recently.[ 57 ] The addition of alginate to the PPy synthesis can increase PPy's conductivity, crystallinity, and charge‐carrier mobility,[ 72 ] as it acts as a dopant due to its anionic properties.[ 72 , 73 ] In addition, alginate can act as a template during in situ polymerization synthesis of PPy.[ 67 , 72 ] Consistent with previous studies using an oxidizer combination of ammonium persulfate (APS) and FeCl3 for alginate‐PPy (ALG‐PPy) composite films,[ 67 ] the ADA‐GEL‐PPy:PSS hydrogel introduced here showed a typical globular PPy morphology.[ 67 ] ALG‐carboxymethyl‐chitosan‐PPy hydrogels for peripheral nerve regeneration applications have shown conductivities of up to 8.03 × 10−3 S cm−1 by increasing PPy content, as well as biocompatibility verified using PC‐12 cell adhesion and an in vivo subcutaneous rat model.[ 68 ] The ADA‐GEL‐PPy:PSS hydrogels presented here have a comparable conductivity in the range of 12–16 × 10−3 S cm−1 in comparison to the study[ 68 ] and offer the possibility to be 3D‐printed for scaffold fabrication. While the cytocompatibility and suitability toward PC‐12 nerve‐cell models of our hydrogel remains to be addressed, it is noteworthy that in vivo studies of PPy:PSS implanted subcutaneously or intramuscularly in rats show similar immune response to FDA‐approved poly(lactic acid‐co‐glycolic acid),[ 52 ] or FDA‐approved poly(d,l‐lactide‐co‐glycolide).[ 74 ] The conductivities of ADA‐GEL‐PPy:PSS hydrogel are in the same range as native cartilage tissues (≈1.2 S m−1, bovine[ 75 ]) but also higher than some human cartilage tissue conductivities (≈0.1–0.3 S m−1, human cartilage endplate,[ 76 ] depending on tonicity), which was verified for a broad frequency range in the present study. The conductivity of the hydrogels was lower at frequencies <1 kHz than for higher frequencies and dependent on PPy content. This may allow ADA‐GEL‐PPy:PSS to mimic the cartilage environment regarding its electrical properties. The conductivity could be increased using 0.1 and 0.2 m PPy modifications in comparison to pure ADA‐GEL, while the data suggest a significant contribution of the ionic conductivity of the ADA‐GEL to the overall conductivity of ADA‐GEL‐PPy:PSS. This may further mimic native tissue bulk conductivity which is mainly ionically driven.[ 76 ] In addition, the changes in redox state of PPy upon ES may add functionality to the ADA‐GEL‐PPy:PSS beyond ionic conductivity, as changes in redox state have shown to be able to control, for example, drug release[ 77 ] or cell attachment.[ 50 ] The redox activity may allow to provide electrical stimulus to cells on conductive ADA‐GEL‐PPy:PSS.[ 78 ] From the cyclic voltammogram, a broad anodic peak at ≈0.41 V, shown for PPy coated Pt before,[ 79 ] and a cathodic peak at approximately −0.03 V with a half‐wave potential E 1/2 at ≈0.215 V indicated charge transport in the PPy functionalized hydrogel. This redox activity was absent in pristine ADA‐GEL. Together, the results confirm the successful modification of ADA‐GEL with electroactive conducting polymer.
Collagen‐ALG‐PPy composites with ionic conductivities ranging from ≈2.1–2.8 S m−1 have been shown to be suitable for injectable hydrogel applications.[ 70 ] This hydrogel system has similarities to the ADA‐GEL‐PPy system shown here, as ALG as the unmodified polysaccharide basis of ADA and GEL represents the denatured form of collagen. The authors showed MTT assay data from hydrogel extract tests indicating cell viability of human mesenchymal stem cells.[ 70 ] However, additional data depicting cell–material interaction and comprehensive cytocompatibility analysis were not provided.[ 70 ] As the design of the hydrogel allowed injectability through G21 needles,[ 70 ] the target viscosity may be too low to allow for 3D‐printing via direct extrusion printing and successive scaffold stability. The ADA‐GEL‐PPy system here was developed to feature rheological properties suitable for direct printing and hence, may be advantageous for scaffold fabrication and cell‐seeding applications in contrast to this previous system.[ 70 ] Alternatively, the in situ use of CaSO4 or CaCO3 to tune ADA‐GEL‐Py viscosity prior to 3D‐printing to yield 3D‐printable hydrogel precursor could be assessed in future studies.[ 80 , 81 ] Further, the impact of Ca2+ crosslinker on hydrogel conductivity could be then investigated to check the potential influence of Ca2+ crosslinker concentration on hydrogel conductivity.
Besides particle‐based or in situ synthesis approaches, ALG‐PPy composite hydrogels have been prepared by oxidative polymerization[ 34 ] of PPy on existing ALG hydrogels. Specifically, this was done by soaking ALG hydrogels in Py monomer followed by oxidation through immersion in FeCl3 oxidant solution.[ 34 ] In our approach, Py was added to the ADA‐GEL precursor and homogenously distributed prior to 3D‐printing and oxidation, which is similar to the oxidation of Py by immersion of the ALG‐Py hydrogel in FeCl3.[ 34 ] A simultaneous increase in electrical conductivity and stiffness of ALG‐PPy hydrogels by the formation of PPy inside the ALG matrix has been observed,[ 34 ] which is consistent with the results of mechanical compression tests and electrical characterization observed for ADA‐GEL‐PPy:PSS in the present work. The same simultaneous increase in stiffness and electrical conductivity upon PPy synthesis has been shown for hyaluronic acid‐based hydrogels.[ 82 ] Together, the data suggest that PPy matrix formation contributes to both electrical conductivity and stiffness in ALG and hyaluronic acid‐based hydrogels. This observation is in accordance to expectations from classical composite‐reinforcement theory when adding stiffer PPy filler (≈0.6 GPa for pure PPy films[ 83 ]) to the hydrogel matrix, assuming that PPy filler to hydrogel matrix‐interaction occurs. Due to the stiffer response of ADA‐GEL‐PPy:PSS hydrogel in comparison to ADA‐GEL, the data indicate such interaction of PPy filler to the ADA‐GEL matrix and that an interpenetrating polymer network has been formed. Microstructure analysis showed a dense hydrogel structure for as‐prepared ADA‐GEL and ADA‐GEL‐PPy:PSS hydrogels (Figure S1, bottom row, Supporting Information), which was found for 3.75%/7.5% ADA‐GEL before.[ 56 , 84 ] The formation of porosity over incubation time has been demonstrated for the main ADA‐GEL hydrogel component used here for ADA‐GEL‐PPy:PSS hydrogel preparation,[ 56 ] and has been associated to gelatin release and hydrogel degradation in previous studies.[ 56 ] The ADA‐GEL‐PPy:PSS hydrogels here showed a dense PPy:PSS network formation when assessed from the top (Figure S1, top row, Supporting Information). However, we observed a decrease in hydrogel stiffness at PPy modifications using 0.2 and 0.4 m Py in the ADA‐GEL‐PPy:PSS synthesis (Figure 4). After PPy formation in the hydrogels, we saw an increased brittle behavior of the hydrogels with increasing PPy content, especially for the 0.4 m Py‐based synthesis, making them sub‐optimal for further assessments and cell culture. We associate the drop of stiffness of the hydrogels by the abundance of PPy network formed inside the ADA‐GEL for 0.2 and 0.4 m Py. The data suggest that PPy:PSS particles interfere with overall hydrogel cohesion and elasticity. The main elastic matrix contribution to the ADA‐GEL‐PPy:PSS hydrogel is provided by the ionically and enzymatically crosslinked ADA‐GEL network.[ 85 ] As a result, the increased amount of brittle PPy:PSS polymer formation contributes to an increased brittle fracture behavior of the hydrogels, ultimately decreasing bulk hydrogel stiffness due to a lack of gel cohesion. PPy sponges with elastic properties have been introduced via PPy oxidation using deficient amounts of oxidant, resulting in a long‐term aging (≈30 days) synthesis which lead to PPy network formation.[ 86 ] In contrast to this approach, the ADA‐GEL‐PPy:PSS hydrogels here were oxidized over 3 h with n(Py:FeCl3) ratio of 1:4, providing excess FeCl3, which hence leads to a rapid PPy formation. Together, no formation of an elastically behaving PPy network is expected in ADA‐GEL‐PPy:PSS, leading to the hypothesis that the main elastic contribution results from the ADA‐GEL matrix. However, as we qualitatively observed increased strain‐stiffening of ADA‐GEL‐PPy in comparison to pristine ADA‐GEL, PPy at 0.1 and 0.2 m syntheses may contribute to increased non‐linearity of the overall hydrogel response. The formation of PPy in a fixed volume ultimately reduces the water content inside the final hydrogel, which may additionally influence the viscoelastic properties of the bulk hydrogel, and result in increased stiffness of 0.1 m PPy.
Besides the decreased stiffness at high PPy‐content ADA‐GEL, a drop in conductivity for 0.4 m PPy was observed (Figure 3). Previous studies have identified that incorporation of spherical conductive PPy nanoparticle fillers inside ALG hydrogels does not increase but slightly decreases the electrical hydrogel conductivity until forming a percolating PPy network.[ 87 ] An increased PPy formation in the ADA‐GEL network repels water content from the overall hydrogel volume, due to the higher ratio of Py to PSS, and therefore, lowering of the amount of free sulfonates to interact with water. The ionic conductivity of the electrolyte‐swollen hydrogel (≈1 S m−1, Figure 3), which is close to the range of culture medium (≈1.5 S m−1[ 88 , 89 , 90 ] at 37 °C), has a non‐neglectable contribution to the overall hydrogel conductivity. The drop of conductivity for 0.4 m PPy might be a result from electrolyte being expelled and replaced by the PPy:PSS matrix in the ADA‐GEL hydrogel, leading to a significant decrease in overall ionic conductivity (Figure 3D) and ion mobility, overall influencing the electrical performance of the composite.[ 87 ] Others have demonstrated a similar drop of both electrical conductivity and stiffness for high PPy‐content (100 mm Py, in situ oxidized) hyaluronic acid hydrogels,[ 82 ] which was associated to structural instability and non‐uniformity of the final PPy hydrogel.[ 82 ] Consistent with these results, 0.4 m PPy showed similarly decreased electrical conductivity and elastic modulus, suggesting that this modification is not feasible for further investigation. Besides this observation for 0.4 m PPy, our SEM data (Figure 1D, Figure S1, Supporting Information) and impedance spectroscopy measurements showed increased electrical conductivity for 0.1 and 0.2 m PPy hydrogels. This indicates the formation of a dense PPy layer on the ADA‐GEL hydrogel, yielding sufficient percolation to increase the overall electrical conductivity (Figure 3). Cross sections of thick (≈4 mm) ADA‐GEL‐PPy:PSS cylinders were prepared to assess the penetration depth of PPy formation inside ADA‐GEL in order to investigate if the oxidation reaction triggered homogenous formation of PPy inside the hydrogels (Figure S9, Supporting Information). PPy formed throughout the cylinders, exceeding PPy formation depths of ≈1.5 mm. The results verify the homogenous PPy formation inside the thinner 3D‐printed structures prepared in this study, which showed strut thicknesses of <500 µm up to 900 µm, depending on the extrusion needle used (Figure 2B; Figure S12, Supporting Information). As the formation of homogenous conductive polymer in pre‐fabricated hydrogels can be challenging,[ 91 ] our approach of oxidizing the hydrogel precursor containing Py when only the GEL phase is thermally gelled, prior crosslinking the hydrogel using Ca2+ and mTG, demonstrated to be a satisfactory method to yield homogenous penetration of PPy inside the ADA‐GEL hydrogels.
Electroactive oxidized‐alginate based hydrogels have been recently explored for sensor applications.[ 57 , 92 ] Oxidized‐alginate carboxymethyl‐chitosan hydrogels have been reinforced by multi‐walled carbon nanotubes (MWCNT) to form electrically conductive and self‐healing strain sensors.[ 92 ] By this particle‐composite approach, a higher MWCNT content resulted in increased hydrogel stiffness as well as strain‐dependent electrical conduction due to increased particle percolation upon deformation.[ 92 ] The combination of oxidized alginate with chitosan provided self‐healing properties to the hydrogels via Schiff base formation.[ 92 ] Closest to the material combination in the present study, PPy containing oxidized alginate‐gelatin hydrogels viain situ polymerization have been suggested for mechanical sensor applications.[ 57 ] Besides the difference in application, the authors combined ethylenediamine‐modified gelatin, oxidized alginate, Py, and oxidizer in one reaction step in situ.[ 57 ] In contrast to ADA‐GEL‐PPy:PSS, the hydrogel was developed with focus on self‐healing properties and sensor‐application and demonstrated suitability as sensors to detect mechanical deformation.[ 57 ] The hydrogels showed similar conductivities to the ADA‐GEL‐PPy:PSS hydrogels presented here, which were in the range of 1.2–1.6 S m−1.[ 57 ] However, the study did not assess cytocompatibility of the materials nor 3D‐printing capability, as those were not its main scope.[ 57 ] In contrast, we present an approach for 3D‐printable ADA‐GEL‐Py:PSS gels that could be rendered electrically conductive by growth of an interpenetrating network of PPy:PSS, resulting in electroactive ADA‐GEL‐PPy:PSS with cytocompatible properties, focused on tissue engineering applications. The hydrogels may have potential as in vitro systems to study the influence of highly structured conductive hydrogels on cell behavior. Our study used a combination of in situ added Py to ADA‐GEL hydrogel precursor, followed by 3D‐printing of scaffolds and subsequent interfacial polymerization of Py to produce electro‐conductive scaffolds. The scaffolds allowed for cell–material interaction and to be 3D‐printed, which broadens the applicability for ADA‐GEL‐PPy EAH. The classic in situ polymerization of PPy inside ADA‐GEL may limit the processability of the hydrogel composite due to the severe changes in viscosity accompanied by the PPy formation in ADA‐GEL up until the point of forming a fully gelled hydrogel.[ 57 ] As a result, the previous ADA‐GEL‐PPy basedin situ approach may have limited capability to be processed via extrusion.[ 57 ] Other previous approaches suggested the printing of oxidized alginate and oxidizer (ammonium persulfate, APS) simultaneously in combination with amine‐displaying gelatin and Py in one step.[ 57 ] This approach is similar to as shown previously for combining aniline monomers, phytic acid, and oxidizer by ink‐jet printing.[ 93 ] We provide another route to 3D‐print ADA‐GEL‐PPy conductive hydrogels via direct extrusion printing of a Py‐containing precursor and post modification to render it electro‐conductive. To our knowledge, this study is the first showing highly structured, electroactive, and cytocompatible ADA‐GEL‐PPy hydrogels for advanced biomaterial scaffold fabrication and tissue engineering applications.
UV–vis analysis of the hydrogel precursors showed that with increasing Py molarity, absorbance peaks formed at λ ≈ 360 nm and between λ = 400–500 nm (Figure S5, Supporting Information). The absorption peaks indicate the formation of oligopolypyrroles and PPy:PSS[ 94 , 95 ] and are associated with bipolaron absorptions at λ = 460–470 nm, while a peak at 460 nm is related to transitions from the valence band to the antibonding polaron state, indicating a doped‐state PPy.[ 96 ] This suggests the instability of Py inside the ADA‐GEL and indicates the formation of Py dimers, trimers, and oligopolypyrroles, over time, resulting in the observed aging (Figure S5a, Supporting Information) of the biomaterial ink, also seen in the rheological analysis. The 0.4 m Py composition showed a broader absorption between 300 and 800 nm in comparison to all other precursors (Figure S5b, black arrow, Supporting Information). This is indicative for the hydrophobic Py dispersed inside the aqueous ADA‐GEL solution in small droplets which lead to increased turbidity and overall absorption. This may explain the loss of hydrogel cohesiveness and lower viscosity observed in rheological assessments for this precursor. In sum, the data suggest that a critical amount of Py can be added to ADA‐GEL after which printability is sacrificed, while 0.1 m Py identified in this study presents profound printability properties to 3D‐print ADA‐GEL‐Py scaffolds.
In a recent approach without using ALG, gelatin‐methacrylate (GelMA)‐PPy hydrogels have been engineered,[ 97 ] which allowed cell attachment and cytocompatibility with respect to C2C12 myoblasts.[ 97 ] The hydrogel showed similar conductivities (≈0.8–1.6 S m−1) as the ADA‐GEL‐PPy:PSS hydrogels here, but in comparison resulted in softer hydrogels upon mechanical compression. One reason for the higher stiffness of ADA‐GEL‐PPy:PSS may be the higher overall hydrogel concentration of GEL and ADA used in our study. The higher amount of PPy in the system as well as the combination of ionic‐ and enzymatic‐crosslinking in the ADA‐GEL hydrogel may result in the stiffer hydrogels observed.[ 55 , 85 ] In summary, our hydrogels combine the cell‐adhesion properties provided by gelatin[ 97 ] with 3D‐printability, doping,[ 57 , 72 ] and degradation properties of ADA,[ 98 ] which are requirements to fabricate advanced tissue engineering constructs. The results suggest the suitability of the PPy‐modified ADA‐GEL scaffolds in soft‐to‐hard tissue ranges of ≈1–1.5 MPa, which is closer in the range of native cartilage tissue[ 99 , 100 ] than pristine ADA‐GEL. The PPy‐modified ADA‐GEL may potentially be applied as matrices for cartilage, subchondral‐bone, or bone tissue‐engineering. While we demonstrated the 3D‐printing of scaffolds and of a 3D‐CAD model, the capability of the biomaterial ink to create more complex and hierarchically ordered cartilage‐like structures will be addressed in future studies. In situ polymerized ALG‐PPy blended with chitosan has been previously shown to be cytocompatible toward MG‐63 cells and to increase hydrogel mineralization in an 1.5× simulated body fluid (SBF) experiment.[ 69 ] The hydrogel was aimed toward bone engineering applications.[ 69 ] Together, the ADA‐GEL‐PPy:PSS hydrogels may be an attractive system toward further bioactivity and bone engineering investigations.
ALG‐PPy hydrogel has recently been engineered to allow 3D‐printability for direct extrusion after PPy in situ formation.[ 101 ] However, as ALG lacks controlled degradation as well as binding sites for cell‐interaction,[ 98 ] we introduce ADA‐GEL‐PPy, with its main advantages of higher degradation and cell attachment potential provided by ADA‐GEL[ 98 ] in comparison to pristine ALG‐based PPy combinations. The degradation of the hydrogels could be tuned by reducing transglutaminase content in the crosslinking step,[ 55 ] which dictates the degradation behavior, as ADA‐GEL‐PPy showed similar swelling and degradation as the ADA‐GEL reference. The stability of the scaffolds in this paper indicated the successful crosslinking of ADA‐GEL‐PPy:PSS using 0.1 m CaCl2 and mTG crosslinking, as insufficiently crosslinked high gelatin‐content ADA‐GEL would quicky dissolve within 24 h under cell culture conditions.[ 84 ] In addition, enzymatic degradation tests confirmed enhanced degradation rate of pristine ADA‐GEL scaffolds in comparison to PPy‐modified ADA‐GEL‐PPy:PSS. Previous studies have shown a fast degradation within 6 h of mTG‐crosslinked gelatin sponges.[ 102 ] The degradation of 5% GelMA scaffolds inside 1 U mL−1 collagenase type II solution has been reported after 24 h of incubation,[ 103 ] similar to what we observed for pristine ADA‐GEL scaffolds here. The enzymatic degradation rate of GelMA has been reduced with increasing CNT concentration[ 103 ] and mitigated by the use of DNA‐functionalized CNTs.[ 30 ] Added CNTs have been described to act as an insoluble backbone‐network hindering collagenase penetration,[ 103 ] optimally providing hydrogel cohesion hindering degradation by DNA‐CNT binding.[ 30 ] We observe a similar effect of PPy:PSS modified ADA‐GEL, mitigating collagenase mediated degradation (Figure S11, Supporting Information), suggesting hindrance of enzymatic degradation by the formation of PPy:PSS, similar to as observed for CNT.[ 103 ] Hence, the data suggest that PPy content could be also used to tailor degradation kinetics of mTG and Ca2+ crosslinked ADA‐GEL, hindering collagenase penetration by the dense PPy layer on top of the hydrogels (Figure S1, top row, Supporting Information), similar to observations for GelMA‐CNT hydrogels.[ 103 ]
In order to assess cytocompatibility, we conducted in vitro studies using ATDC‐5 cells for seven days of cell culture. Our data suggest that ADA‐GEL‐PPy offers cell–material interaction potential, similar to ADA‐GEL. However, we observed that an intense washing step was required to achieve cytocompatibility of ADA‐GEL‐PPy to remove excess H+, potential Py, chlorine, and Fe2+/Fe3+ from the hydrogel after PPy synthesis (Figure S10, Supporting Information). In addition, the attachment and growth of ATDC‐5 on the Ca2+ and mTG‐crosslinked ADA‐GEL reference was similar to tissue culture polystyrene, with a confluent cell layer after 14 days (Video S2, Supporting Information), hence representing a close‐to ideal hydrogel. The PPy modification of ADA‐GEL showed slightly reduced cell attachment and cell number on the overall scaffolds in comparison to this control. While we found a higher cell number after seven days on 3D ADA‐GEL‐PPy in comparison to the one day TCPS control, we could not identify the difference to be statistically significant. However, due to the 3D‐printing, a higher depth of cell seeding in scaffold z‐direction can be achieved due to the strut‐by‐strut structure of the scaffold in comparison to flat 2D cylinders (Figure 5A, bottom right; Figure S7, Supporting Information). Extracellular LDH release data indicated no cell membrane damage after day one, suggesting the washing protocol removed any toxic low molecular weight species (with similar results for ADA‐GEL and ADA‐GEL‐PPy), as supported by pH and UV–vis analysis (Figure S10, Supporting Information). As we found that the PPy results in significantly increased hydrogel stiffness, this may be one reason for the decreased cell number, besides the altered hydrogel surface chemistry caused by the PPy functionalization. As a result, we will assess, in future studies, possible approaches to achieve electrically CP functionalized ADA‐GEL with similar stiffness to pristine ADA‐GEL. Such matrices would be beneficial to allow the study of electrical properties of ADA‐GEL on cell attachment, decoupled from a simultaneous stiffness increase, which might be limited for the here presented hydrogel system having both increased conductivity and stiffness. Ultimately, on hydrogels which allow the independent tuning of their electrical conductivity, gene expression analysis of human stem cells to assess chondrogenic phenotype maintenance and genotype on 3D‐printed samples should be performed, which we will address in the future. In addition, the introduced washing step and the degradation of the hydrogel in this study may alter the overall mechanical and electrical properties of the hydrogels after incubation, which may alter cell response and will be addressed in future research. As we found no significant difference between days 1 and 7 for the group of interest in respect to proliferation, we will conduct further cell viability (e.g., LIVE/DEAD assays) and expanded proliferation studies on multiple time points for PPy functionalized ADA‐GEL with similar stiffness as pristine ADA‐GEL in future work.
In a recent study, in situ polymerized ALG‐PPy hydrogels have been 3D‐printed to create open porous scaffolds for tissue engineering.[ 101 , 104 ] While initial cell adhesion and survival was shown for PC‐12 cells,[ 101 , 104 ] the addition of gelatin in our study as a main hydrogel‐ink component, subsequently crosslinked by a transglutaminase, showed significantly increased cell‐material interaction and growth in comparison to this previous work (Figure 5).[ 101 , 104 ] In comparison to hyaluronic acid‐PPy hydrogels which have been prepared with electrical conductivities of up to 0.73 S m−1 and cytocompatibility with respect for NIH‐3T3 fibroblast cells,[ 82 ] the hydrogels in our study showed increased cell‐material interaction, mostly associated to the presence of gelatin, as well as conductivities in a similar range of up to ≈1.6 S m−1 at high frequencies and >1 S m−1 in DC measurements. In addition, our hydrogels show 3D‐printability, which may be an advantage over previously introduced PPy‐X systems. The results highlight that PPy modified hydrogels prepared via in situ polymerization of PPy may lack in cell‐material interaction potential, while the addition of gelatin crosslinked via mTG as shown here demonstrates a promising route to introduce both, viscosity for 3D‐printing, and cell‐adhesion binding sites for in vitro applications.
PANI nanofibers have been synthesized in the presence of ALG with high electrochemical discharge capacitance,[ 105 , 106 ] however, ALG‐based PANI hydrogels have been rarely explored.[ 107 ] Only recently, PANI‐ALG‐graphene hydrogel hollow fibers have been fabricated as nerve‐conduit materials.[ 108 ] The study demonstrated material conductivities of ≈1.5 S m−1 similar to the hydrogels presented here. The ADA‐GEL used here has been used in former studies to fabricated hydrogel hollow‐fibers.[ 109 , 110 ] Together, the ADA‐GEL‐PPy:PSS hydrogel might be similarly processable into hollow‐fiber vessels, with potential application in nerve conduit engineering. Others have demonstrated that ALG‐conductive‐polymer blends can be used for drug delivery.[ 111 , 112 ] With ALG as the drug carrier matrix, PEDOT was used as the CP to investigate ES triggered drug release.[ 111 ] ALG simultaneously served as carrier matrix for the drug and as a dopant to increase the conductivity of PEDOT by doping.[ 111 ] Similarly, an RGD‐alginate PEDOT system was utilized for drug delivery as cochlea implant coatings.[ 112 ] Consistent with the requirements from these previous results, the ADA‐GEL matrix presented here provides degradation under physiological conditions,[ 55 , 56 ] which is mostly driven by gelatin degradation,[ 55 ] while the PPy:PSS CP could serve as electrical conductor for similar ES‐stimulated drug release applications. By these properties, the ADA‐GEL‐PPy:PSS hydrogels presented here could be potentially applied in the field of drug delivery, broadening the field of application of the here presented hydrogel system. Taking together the results from increased stiffness by PPy modification, altered conductivity, and cytocompatibility, the data suggest that our hydrogels could serve in the future as 3D‐printed hydrogel constructs with increased electrical conductivity for ES‐assisted cartilage engineering applications, drug delivery, or even nerve guidance structures, and could be used to study the effect of substrates with altered conductivity in comparison to solely ionically conductive hydrogels.
4. Conclusions
We identified a 3D‐printable and electroactive oxidized alginate‐gelatin PPy hydrogel which allows for cell‐material interaction and cytocompatibility. Producing Py‐containing ADA‐GEL hydrogel precursors results in decreased hydrogel viscosity, while we achieved to create an ADA‐GEL‐Py precursor with suitable rheological properties to 3D‐print scaffolds via direct extrusion printing. The addition of Py allowed to form ADA‐GEL‐PPy hydrogels with higher electrical conductivity than pristine ADA‐GEL. 3D‐printing of the hydrogel scaffolds can increase seeding efficiency of cells in scaffold depth due to the porosity introduced by 3D‐printing. The finding that PPy formation inside the hydrogel simultaneously increases hydrogel stiffness and electrical conductivity highlights the importance of understanding the effect of CP modification on different hydrogel parameters at once, which together ultimately may affect the cell‐material interaction. Together with 3D‐printability, these properties may enhance the outcome of current cartilage tissue engineering approaches. The hydrogels presented here are candidates for 3D‐printed electroactive hydrogel matrices to study ES assisted tissue cultures and could potentially be employed as drug‐release coatings for electrodes or in advanced MACI applications.
5. Experimental Section
Materials
Sodium alginate (VIVA Pharm, PH176) was purchased from JRS PHARMA GmbH & Co. KG (Germany). All other chemicals were ordered from Sigma‐Aldrich (Germany) if not otherwise specified.
Material Synthesis
Alginate was oxidized by controlled oxidation using sodium metaperiodate as described earlier.[ 98 ] Briefly, 10 g of alginate were dispersed in an ethanol‐water mixture (100 mL, 50:50 v/v) and oxidized in the absence of light under continuous stirring for six hours after addition of 9.375 mmol NaIO4. Next, the reaction was quenched by adding 10 mL of ethylene glycol followed by addition stirring for 30 min. The product was allowed to sediment for 5 min. The ethanol phase was decanted followed by transfer of the oxidized alginate (alginate dialdehyde, ADA) product into dialysis tubings (MWCO: 6000–8000 Da, Spectrum Lab, USA) and dialyzed against ultrapure water (UPW, MilliQ, Germany) for five days with daily water exchange. After dialysis, the product was frozen for a minimum of 24 h at −21 °C and lyophilized using a freeze dryer (LD1‐2 Plus, Martin Christ GmbH, Germany). The degree of oxidation of the final product was determined as ≈13% using a quantification method of residual sodium triperiodate as previously described.[ 55 , 113 ]
Ink Formulation
Gelatin (Typ A, Bloom 300) was pre‐treated by continuous stirring of a 15% w/v solution (in UPW) at 80 °C for 3 h to adapt its rheological properties for increased 3D‐printability.The solution was sterile filtered using 0.22 µm syringe filters (Carl Roth, Germany) and stored at 4 °C until further use. Next, 7.5% w/v ADA was dissolved in phosphate buffered saline (PBS, Thermo Fisher, US) and passed through a 0.45 µm syringe filter (Carl Roth, Germany). Equal amounts of ADA and gelatin solutions were mixed for 10 min (37 °C) to yield a final hydrogel precursor of 3.75%/7.5% ADA‐GEL. Py was purified by passage over a basic alumina column.[ 114 ] PSS was added to the ADA‐GEL mixture until completely dissolved (n(PSS:Py) = 1:16). Next, Py was added to the ADA‐GEL precursor to yield different molarities (0.1, 0.2, 0.4 m Py) inside ADA‐GEL by adding the Py to the stirring ADA‐GEL precursor at high speed (5000 rpm) for 1 min to ensure homogenous dispersion in the precursor, followed by ultrasonication for 5 min. Final ADA‐GEL‐Py:PSS hydrogel precursors of 0.1, 0.2, and 0.4 m Py were prepared, while ADA‐GEL served as the control. 0.1, 0.2, and 0.4 m Py containing ADA‐GEL preparations are denoted as 0.1 m Py, 0.2 m Py, and 0.4 m Py, respectively.
Rheological Characterization
The viscoelastic behavior of ADA‐GEL‐Py:PSS hydrogel precursors was assessed using a HAAKE Mars II Rheometer (ThemoFisher Scientific, Waltham, USA) with a plate‐plate setup (plate‐diameter: 35 mm). The gap size was defined as 0.5 mm, resulting in a sample volume of 0.5 mL precursor ink for each measurement. The dynamic viscosity was determined by a shear‐rate ramp up to a shear‐rate of 2000 s−1 at 22 °C. The viscoelastic properties, the temperature‐dependent, and time‐dependent behavior were measured using dynamic oscillation. A shear‐stress sweep test ranging from 0.1 to 5.0 at a frequency of 1 Hz and a temperature sweep with a shear strain of 0.1 and a varying temperature from 22 to 36 °C over 300 s were performed. To determine the microstructural changes in the polymer framework after the addition of Py, a time sweep over 1800 s at a frequency of 1 Hz and a shear strain of 0.1 was assessed. The dynamic viscosity (ƞ), complex viscosity (ƞ*), storage (G′) and loss modulus (G″), and tan delta were recorded. All measurements were conducted using three individual replicates (n = 3).
Scaffold Fabrication
ADA‐GEL‐Py:PSS hydrogel precursor was transferred into 3D‐printing cartridges (Nordson EFD, USA) and centrifuged at 350 rpm (Eppendorf, Germany) for 2 min to remove air bubbles. Hydrogel scaffolds were 3D‐printed using a GeSIM Bioscaffolder (GeSIM GmbH, Germany) equipped with a diameter = 250 µm extrusion nozzle (red colored, Nordson EFD, USA). The hydrogel scaffolds were printed at a speed of 10 mm s−1, extrusion pressure 250–310 kPa, diameter (d) set to 10 mm, height (h) set to 2 mm, and 90° change in printing direction between layers. CAD models of typewritten letters were created using Autodesk TinkerCAD web application tool (Autodesk Inc., USA) and 3D‐printed. After printing, the samples were oxidized using FeCl3 (n(Py:FeCl3 = 1:4))[ 115 ] solution to facilitate PPy formation, as this ratio was found to ensure highest PPy conductivity elsewhere.[ 115 ] Excess FeCl3 was provided to additionally ensure potentially increased oxidant diffusion capacity into the gels by higher FeCl3 concentration as well as to allow for a sufficiently high reaction rate of Py oxidation to reduce the potential of overoxidation and maleimide formation at low reaction rates, which could lead to reduced PPy conductivity.[ 116 ] The samples were subjected to vigorous washing using H2O and Hank's balanced salt solutions (HBSS, Thermo Fisher) to ensure the removal of any oxidation byproducts (FeCl2, H+) until reaching a stable pH of the surrounding HBSS solution (pH ≈ 8.0 ± 0.2, Figure S10, Supporting Information), to render the hydrogel suitable for further cell culture experiments. Washed samples were crosslinked using 0.1 m CaCl2 containing 2.5% w/v mTG (ACTIVA WM, 85–135 U g−1, Ajinomoto Co., Inc., Japan) for 10 min to account for ionic‐crosslinking of the alginate and enzymatic‐crosslinking of the gelatin phase, respectively.[ 55 ]
Printability Assessment
Filament formation tests were performed using 0.1 m Py containing ADA‐GEL. Pristine ADA‐GEL and Pluronic F127 (38% wt) served as biomaterial‐ink controls for the printability assessment. One‐ and two‐layer scaffolds were 3D‐printed using 0.1 and 0.2 m Py containing ADA‐GEL. The printability of the biomaterial‐inks to achieve square‐shaped pores was quantified from light microscopy images (Primovert, Carl Zeiss, Germany) of the scaffolds using a printability factor (Pr) as derived elsewhere,[ 58 ] which was inversely proportional to the pore‐circularity (C), and measured as the ratio of the pore perimeter (P) to the pore area (A) using the following equation[ 58 ]
| (1) |
where Pr < 1 corresponds to under‐gelled hydrogel ink and rounded pore corners, Pr = 1 properly gelled hydrogel ink with ideal square‐shaped pores, and Pr > 1 to over‐gelled hydrogel ink.[ 58 ]
The uniformity of 3D‐printed hydrogel struts was determined using a previously introduced uniformity factor U,[ 59 , 60 ] derived as the measured horizontal length of a deposited hydrogel strut (L) divided by the theoretical horizontal length (corresponding to the width of the image) of a perfectly parallel printed strut (L t)
| (2) |
where U = 1 corresponds to perfectly uniform and parallel hydrogel struts, while U > 1 indicates strut non‐uniformities. All image analyses were performed using Fiji (NIH).
Chemical Characterization
Attenuated total reflectance Fourier transform infrared spectroscopy (ATR‐FTIR) was performed on lyophilized hydrogel specimen using an IRAffinity‐1S FTIR unit (Shimadzu, Japan). UV–vis spectroscopy was used to assess ADA‐GEL hydrogel precursor containing PSS and Py using UV‐transparent plastic cuvettes (Sarstedt, Germany) with an optical path length of 10 mm. UV–vis spectra were recorded using the cuvette‐function on a NanoDrop ONE (ThermoScientific, USA) desktop spectrophotometer.
Electrical Characterization
For electrical testing, hydrogel specimen (d = 10 mm, h = 2 mm) were cast using custom made polydimethylsiloxane (PDMS) molds. All samples were washed and equilibrated in PBS prior to electrical conductivity measurements to assess the electrical properties as close as possible to the hydrogels at in vitro conditions. To assess direct current conductivity, four‐point probe measurements were performed using a Signatone Pro4‐440N test station (Signatone, USA). The bulk‐resistivity of freeze‐dried hydrogel samples (minimum of n = 6) was determined, and the conductivity was calculated by inversion of the measured bulk resistivity.
EIS was performed to assess the frequency dependent conductivity of the hydrogels. Impedance spectra from 100 Hz to 5 MHz of hydrogel samples (diameter = 1.5 cm, height = 2 mm) were measured by an Agilent 4294A impedance analyzer (Keysight Technologies, Inc., USA). The disk sample was rinsed in deionized water within 5 s and the residual water on the surface was removed by blotting paper. Next, impedance measurements for individual samples were performed in capacitive parallel electrodes (16451B dielectric test fixture, Keysight Technologies, Inc., USA). The rinse of sample before measurements with deionized water could wipe out the high conductivity solution on the surface without changing bulk properties of samples, which could reduce the electrode polarization at the interface between the sample surface and electrode. The measured impedance consists of two components, that is, impedance of electrode polarization, ZEP, and impedance of the sample
| (3) |
Z EP is usually represented by a constant phase element (CPE)
| (4) |
where K is a constant describing the amplitude of Z EP, n is a number within 0 < n < 1. A CPE becomes resistive when n → 0 and capacitive when n → 1.
The second term in Equation (3) represents the impedance of the sample, where C 0 is the capacitance of the electrode for air.
Cyclic voltammetry (CV) was performed using a three‐electrode electrochemical cell setup consisting of an Ag/AgCl reference electrode in 3 m NaCl solution (RE‐1BP, ALS, Japan), a Pt‐wire counter electrode (ALS, Japan), and a glassy carbon (inner diameter = 3 mm, ALS, Japan) working electrode (GC) equipped to a Keithley 2450‐EC source meter (Keithley Instruments, USA). The measurement was carried out at a scan rate of 100 mV s−1, three cycles for each sample, and potentials between −0.4 V and 0.8 V with PBS as the electrolyte, similar to as described elsewhere.[ 37 ] Prior to the measurement, the GC was polished using diamond paste (particle size: 1 µm) in an eight‐shape motion using a polishing kit (PK‐3, ALS, Japan; C3 Prozess‐ und Analysetechnik GmbH, Germany) and cleaned by rinsing with distilled water and acetone. Thin hydrogel films were adhered to the GC‐electrode following a procedure described previously.[ 117 ] Briefly, a dilution of 5% Nafion 117 solution (1:10 in EtOH) was prepared and the GC was dipped into the solution and allowed to air‐dry. Next, hydrogel films were dipped into the Nafion solution and laminated on the Nafion‐coated GC, and allowed to air‐dry again until the ethanol was evaporated. The Nafion‐coated GC and pristine ADA‐GEL served as control.
Mechanical Properties
For mechanical testing, cylindrical hydrogel specimen (d = 10 mm, h = 3 mm) were cast using custom‐made PDMS molds. Uniaxial compression tests were performed at 10 mm min−1 using an Instron 5967 testing device equipped with a 100 N load cell (Instron, Germany). Elastic moduli were determined as the slope in the linear elastic deformation region from stress–strain data between 5% and 10% deformation.[ 118 ] The measurement was done using six hydrogel replicates (n = 6).
Degradation and Swelling
To assess hydrogel degradation and swelling, hydrogel cylinders (d = 10 mm, h = 3mm) were fabricated and incubated in Dulbecco's Modified Eagle Medium (DMEM, Gibco, ThermoFisher, USA) at 37 °C, 5% CO2, for 0, 1, 2, 3, 7, 14, 21, and 30 days in an incubator, as well as for short‐time periods of 0, 3, 6, and 16 h to determine initial swelling. The evolution of hydrogel mass and diameter were monitored over time. The swelling and degradation (%) of the hydrogels were determined using the following equation
| (5) |
where mn is the mass after a specific time point and m 0 is the initial mass of the hydrogel specimen as fabricated. All tests were conducted using a minimum of six hydrogel replicates. Image analysis to assess the hydrogel diameter over time was performed using Fiji (NIH).
Enzyme‐driven degradation tests were performed by incubation of hydrogels in 1 U mL−1 collagenase type II (Gibco, Thermo Fisher, USA) solution at 37 °C.
Scanning Electron Microscopy
The microstructure of PPy modified ADA‐GEL was assessed using SEM. ADA‐GEL‐PPy:PSS hydrogel was frozen at −21 °C and freeze‐dried using an LD1‐2 Plus lyophilizer unit (Martin Christ, Germany). SEM images were recorded between 1.5 and 2.5 kV in InLens and secondary electron detection mode using an Auriga CrossBeam Unit (Carl Zeiss GmbH, Germany) scanning electron microscope.
Cell Culture
Mouse teratocarcinoma ATDC‐5 cells were expanded in maintenance medium (MM) consisting of DMEM/HAM's F12 (Gibco, ThermoFisher, USA) containing 5% v/v FCS, 1% penicillin/streptomycin, human transferrin (10 µg mL−1), and 30 nm sodium selenite, at 37 °C, 5.0% CO2, in a humidified atmosphere. Cells were passaged in cell culture T‐75 flasks (Sarstedt, Germany) and counted using Neubauer Chambers (“Neubauer‐improved,” Paul‐Marienfeld, Germany).
Multiphoton Fluorescence Microscopy
Samples (n = 3) were washed using HBSS and fixed using 4% formaldehyde solution (in HBSS) for 5 min. The cells were permeabilized (0.1% TritonX‐100 for 5 min), washed twice using HBSS, and stained using 5 and 1 µL mL−1 Rhodamine‐phalloidin F‐Actin/DAPI (both Life Technologies, Thermo Fisher, USA), in HBSS, for 1 h and 5 min, respectively. The samples were imaged using a multiphoton microscope (TriMScope II, LaVision BioTec, Bielefeld, Germany) equipped with an HC FLUOTAR L 25x/0.95 W VISIR objective. Images were recorded at 810 nm excitation, acquiring DAPI (ET450/70 m) and Rhodamine‐Phalloidin (ET572/35 m) channels using a dichromatic filter (495 nm). The images had a voxel size of 0.85 × 0.85 × 1 µm³ in images with 440 × 440 µm² field of view, where z as the depth in z‐direction of the scaffolds was adjusted to ensure that sample was visible and recorded at all edges of the field of view. As flat (TCPS‐well plate), lightly curved (ADA‐GEL hydrogel cylinder), and strongly curved (3D‐printed) sample substrates were recorded, the z‐value of the stacks required for adjustment to ensure to capture the sample surface.
Image Analysis
All images were processed using Fiji (NIH).[ 119 ] The number of cells on different samples was quantified from maximum intensity z‐projections of image stacks (n = 6) from 512 × 512 pixel images (440 × 440 µm² in x–y). Cell nuclei surrounded by an F‐actin‐positive cytoskeleton were considered as viable and included into the count. The cell depth (d max) at which cells could be detected in z‐direction distance from the hydrogel scaffold surface was determined from y–z cross sections (n = 6). d max was used as a measure of seeding efficiency in z‐direction of the hydrogel scaffolds and counted as an indicator of how deep cells could be seeded and grown into the hydrogel scaffolds. Flat TCPS‐well plate substrates (d max = min) served as controls.
Water Soluble Tetrazolium Salt Viability Assay (WST‐8)
The viability of ATDC‐5 cells grown on ADA‐GEL and 3D‐ADA‐GEL‐PPy:PSS (0.1 m PPy) hydrogels was determined using a WST‐8 viability assay (Cell Counting Kit‐8, Sigma‐Aldrich) according to the manufacturer's instructions. Samples (n = 6) were incubated in 1% WST‐8 stock solution in MM for three hours at 37 °C, 5% CO2, in a humidified atmosphere. Supernatants (100 µL) were transferred into 96‐well plates (Sarstedt, Germany) as technical duplicates, and the absorbance at 450 nm was determined using a multi‐well plate reader (type Phomo, Anthos Mikrosysteme GmbH, Krefeld, Germany) to assess the conversion of water soluble tetrazolium salt into formazan as an indicator for cell viability. Cells grown in tissue culture polystyrene (TCPS) well plates served as control.
In Silico Toxicity Screening
In silico toxicity screening was carried out using Derek Nexus (v. 6.0.1, Nexus: 2.2.2) and Sarah Nexus (Sarah Nexus: 3.0.0, Sarah Model: 2.0) supplied by Lhasa Limited, Leeds, UK.
Extracellular Lactate Dehydrogenase Release Assay (LDH)
To assess potential cytotoxicity of the materials, extracellular LDH release was determined from cell‐culture supernatants using the TOX7 in vitro toxicity kit (Sigma‐Aldrich). Aliquots of 500 µL were removed from ATDC‐5 cells cultured for 24 h on TCPS, ADA‐GEL, 0.1 m PPy, and 3D‐ADA‐GEL‐PPy:PSS (0.1 m PPy) samples and frozen at −21 °C until further use. All samples were treated according to manufacturer's instructions. Briefly, samples were thawed on the day of analysis and 140 µL of the supernatants were transferred into UV–vis cuvettes (path length: 10 mm, Sarstedt, Germany). Next, LDH master‐mix solution (60 µL) containing equal amounts of substrate solution, LDH cofactor solution, and dye solution, were added to the cuvettes and incubated at RT for 30 min in the dark. The samples were diluted using 700 µL UPW and the absorption was immediately measured (λ = 490 nm, 690 nm) using a UV–vis spectrophotometer (NanoDrop One, ThermoFisher, USA).
PicoGreen Proliferation Assay
Cells were grown for seven days on TCPS, ADA‐GEL, and 3D‐printed ADA‐GEL‐PPy:PSS (0.1 m PPy, 3D AG‐PPy). Cells grown on hydrogel samples and TCPS controls (n = 4) were washed using HBSS and frozen at −80 °C to facilitate cell lysis and DNA release. On the day of the experiment, samples were thawed and washed using 1 mL of 1× TE PicoGreen assay buffer solution (Quant‐iT PicoGreen dsDNA Assay‐Kit, Invitrogen, Life Technologies, ThermoFisher, USA). Equal amounts of Quant‐iT PicoGreen working solution and sample (50/50) were mixed and incubated for 3 min at RT in the absence of light. The relative fluorescence (RFU) was recorded using a CFX connect spectrofluorometer (Bio‐Rad, Germany). LambdaDNA assay standard served as calibration curve standard (R² = 0.99). dsDNA data for all samples were normalized to cells grown on TCPS for 24 h as control.
In Vitro Cytocompatibility Assessment
ATDC‐5 cells were culture for seven days on ADA‐GEL and ADA‐GEL‐PPy:PSS samples to assess the cell attachment and proliferation on the different materials. TCPS was used as a control substrate to the hydrogels. Cytocompatibility was assessed by a combination of fluorescence microscopy, cell quantification, proliferation assay, SEM, and indirect cell viability testing (LDH, WST). Multiphoton microscopy was used to quantify cells on the substrates and to assess cell seeding depth. SEM images were taken of cell‐seeded ADA‐GEL and ADA‐GEL‐PPy:PSS samples to assess cell–material interaction. The samples were fixed using two fixing solutions containing 0.1% glutaraldehyde, 2% formaldehyde, 5% sucrose, in distilled water and 0.3% glutaraldehyde, 3% formaldehyde, in distilled water, respectively, for one hour each. Next, samples were subjected to a gradient ethanol series at 30, 50, 70, 75, 80, 85, 90, 95, and 99.6% EtOH for 30 min each step for scaffold dehydration. The samples were critical point dried using an EM CPD300 critical point dryer (Leica, USA) and SEM images were recorded at acceleration voltage of 1 kV using secondary electron detection mode with the same SEM unit used for microstructure imaging. A proliferation assay was performed to assess cell proliferation and growth on the different gels in comparison to cells seeded on TCPS. Extracellular LDH release was investigated after 24 h as an indicator for initial material cytotoxicity. WST‐8 assay was performed as an indicator for cell viability after seven days.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 9.0 software (GraphPad Software, USA). All experiments were performed with at least three replicate samples per group. All data are shown as mean ± standard deviation (SD) if not otherwise noted. Significant differences between means of two groups were determined using two‐tailed Student's t‐tests. Normal distribution of data was assessed using Shapiro–Wilk tests. Differences among means of multiple groups were determined using one‐way analysis of variance (ANOVA) with Bonferroni post‐hoc tests. For non‐normally distributed data, groups were subjected to either Welch's t‐test or non‐parametric Kruskal–Wallis test with Dunn's post‐hoc tests (LDH assay), for two‐group and multiple‐group comparisons, respectively. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 were considered statistically significant. Non‐significant differences (n.s.) were indicated for p ≥ 0.05.
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
T.D., R.D., and A.R.B. designed the study. T.D. conducted main experimentation and data analysis. T.D. performed manuscript formatting, data visualization, and prepared the first draft. T.D., C.P., F.S., D.S., and A.R.B. wrote the manuscript. T.D., C.P., and H.S. designed, conducted, and processed the rheological experiments. F.S. and J.K. designed and conducted electrical impedance spectroscopy analysis. T.D., D.S., and O.F. designed and conducted multiphoton microscopy experiments and image analysis. M.D.A. and J.G.H. performed in silico toxicity screenings. T.D., C.P., F.S., D.S., M.D.A., R.D., J.K., J.G.H., H.S., O.F., and A.R.B. contributed to data interpretation and commented on the manuscript. A.R.B. supervised the overall project. All authors listed contributed directly, substantially, and intellectually to the work. This work was approved by all authors for publication.
Supporting information
Supporting Information
Supplemental Video 1
Supplemental Video 2
Acknowledgements
This research was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—SFB 1270/1−299150580 to J.K., H.S., and A.R.B., and project number 326998133—TRR‐225 (subproject B08) to O.F. The authors would like to additionally thank Jana Papse (Institute of Biomaterials, Friedrich‐Alexander‐University Erlangen‐Nuremberg, Erlangen, Germany) for supporting printability assessment studies. We thank the Engineering and Physical Sciences Research Council (EPSRC) for grants in support of JGH and MDA (EP/R512564/1, 2065445).
Distler T., Polley C., Shi F., Schneidereit D., Ashton M. D., Friedrich O., Kolb J. F., Hardy J. G., Detsch R., Seitz H., Boccaccini A. R., Electrically Conductive and 3D‐Printable Oxidized Alginate‐Gelatin Polypyrrole:PSS Hydrogels for Tissue Engineering. Adv. Healthcare Mater. 2021, 10, 2001876. 10.1002/adhm.202001876
Data Availability Statement
Research data are not shared.
References
- 1. Gaharwar A. K., Detamore M. S., Khademhosseini A., Ann. Biomed. Eng. 2016, 44, 1861. [DOI] [PubMed] [Google Scholar]
- 2. Place E. S., Evans N. D., Stevens M. M., Nat. Mater. 2009, 8, 457. [DOI] [PubMed] [Google Scholar]
- 3. O'Brien F. J., Mater. Today 2011, 14, 88. [Google Scholar]
- 4. Mawad D., Lauto A., Wallace G. G., Polymeric Hydrogels as Smart Biomaterials, Springer, Switzerland: 2016. [Google Scholar]
- 5. Drury J. L., Mooney D. J., Biomaterials 2003, 24, 4337. [DOI] [PubMed] [Google Scholar]
- 6. Liu W. F., Chen C. S., Mater. Today 2005, 8, 28. [Google Scholar]
- 7. Zhang Y. S., Khademhosseini A., Science 2017, 356, eaaf3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Willner I., ACS Chem. Res. 2017, 50, 657. [DOI] [PubMed] [Google Scholar]
- 9. Gholamali I., Regen. Eng. Transl. Med. 2019, 10.1007/s40883-019-00134-1. [DOI] [Google Scholar]
- 10. Jeong B., Gutowska A., Trends Biotechnol. 2002, 20, 305. [DOI] [PubMed] [Google Scholar]
- 11. Municoy S., Á I., Antezana P. E., Galdop J. M., Olivetti C., Mebert A. M., Foglia L., Tuttolomondo V., Alvarez G. S., Hardy J. G., Desimone M. F., Int. J. Mol. Sci. 2020, 21, 4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tanaka M., Nakahata M., Linke P., Kaumann S., Polym. J. 2020, 52, 861. [Google Scholar]
- 13. Culver H. R., Clegg J. R., Peppas N. A., Acc. Chem. Res. 2017, 50, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sawicki L. A., Kloxin A. M., Biomater. Sci. 2014, 2, 1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tibbitt M. W., Kloxin A. M., Dyamenahalli U., Anseth K. S., Soft Matter 2010, 6, 5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen Y., Chung Y., Wang I., Young T., Biomaterials 2012, 33, 1336. [DOI] [PubMed] [Google Scholar]
- 17. Georges P. C., Janmey P. A., J. Appl. Physiol. 2005, 98, 1547. [DOI] [PubMed] [Google Scholar]
- 18. Vats K., Benoit D. S. W., Tissue Eng., Part B 2013, 19, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kloxin A. M., Kasko A. M., Salinas C. N., Anseth K. S., Science 2009, 324, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Konieczynska M. D., Grinsta M. W., Acc. Chem. Res. 2017, 50, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang T., Zhang X., Wang Z., Zhu X., Liu J., Min X., Cao T., Fan X., Polymers 2019, 11, 1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu Y., Yan K., Jiang G., Xiong Y., Du Y., Shi X., Int. J. Polym. Sci. 2014, 2014, 736898. [Google Scholar]
- 23. Balint R., Cassidy N. J., Cartmell S. H., Acta Biomater. 2014, 10, 2341. [DOI] [PubMed] [Google Scholar]
- 24. Visscher P. M., Patel C. J., Lakhani C. M., Tierney B. T., Manrai A. K., Yang J., Physiol. Behav. 2019, 176, 139. [Google Scholar]
- 25. Deng Z., Hu T., Lei Q., He J., Ma P. X., Guo B., ACS Appl. Mater. Interfaces 2019, 11, 6796. [DOI] [PubMed] [Google Scholar]
- 26. Lee S. J., Zhu W., Nowicki M., Lee G., Heo D. N., Kim J., Zuo Y. Y., Zhang L. G., J. Neural Eng. 2018, 15, 016018. [DOI] [PubMed] [Google Scholar]
- 27. Zhu Y., Liu S., Shi X., Han D., Liang F., Mater. Chem. Front. 2018, 2, 2212. [Google Scholar]
- 28. Li Y., Neoh K. G., Kang E. T., J. Biomed. Mater. Res., Part A 2005, 73, 171. [DOI] [PubMed] [Google Scholar]
- 29. Dong R., Zhao X., Guo B., Ma P. X., ACS Appl. Mater. Interfaces 2016, 8, 17138. [DOI] [PubMed] [Google Scholar]
- 30. Shin S. R., Farzad R., Tamayol A., Manoharan V., Mostafalu P., Zhang Y. S., Akbari M., Jung S. M., Kim D., Comotto M., Annabi N., Al‐Hazmi F. E., Dokmeci M. R., Khademhosseini A., Adv. Mater. 2016, 28, 3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shin M., Song K. H., Burrell J. C., Cullen D. K., Burdick J. A., Adv. Sci. 2019, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Distler T., Boccaccini A. R., Acta Biomater. 2020, 101, 1. [DOI] [PubMed] [Google Scholar]
- 33. Heo D. N., Lee S. J., Timsina R., Qiu X., Castro N. J., Zhang L. G., Mater. Sci. Eng., C 2019, 99, 582. [DOI] [PubMed] [Google Scholar]
- 34. Yang S., Jang L. K., Kim S., Yang J., Yang K., Cho S., Lee J. Y., Macromol. Biosci. 2016, 16, 1653. [DOI] [PubMed] [Google Scholar]
- 35. Bagheri B., Zarrintaj P., Surwase S. S., Baheiraei N., Saeb M. R., Mozafari M., Kim Y. C., Park O. O., Colloids Surf., B 2019, 184, 110549. [DOI] [PubMed] [Google Scholar]
- 36. Mawad D., Stewart E., Officer D. L., Romeo T., Wagner P., Wagner K., Wallace G. G., Adv. Funct. Mater. 2012, 22, 2692. [Google Scholar]
- 37. Mawad D., Mansfield C., Lauto A., Perbellini F., Nelson G. W., Tonkin J., Bello S. O., Carrad D. J., Micolich A. P., Mahat M. M., Furman J., Payne D., Lyon A. R., Gooding J. J., Harding S. E., Terracciano C. M., Stevens M. M., Sci. Adv. 2016, 2, e1601007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li M., Guo Y., Wei Y., MacDiarmid A. G., Lelkes P. I., Biomaterials 2006, 27, 2705. [DOI] [PubMed] [Google Scholar]
- 39. Min J. H., Patel M., Koh W. G., Polymers 2018, 10, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang B., Yao F., Hao T., Fang W., Ye L., Zhang Y., Wang Y., Li J., Wang C., Adv. Healthcare Mater. 2016, 5, 474. [DOI] [PubMed] [Google Scholar]
- 41. Ning T., Zhang K., Heng B. C., Ge Z., Eur. Cells Mater. 2019, 38, 79. [DOI] [PubMed] [Google Scholar]
- 42. Yuan X., Arkonac D. E., Chao P. H. G., Vunjak‐Novakovic G., Sci. Rep. 2015, 4, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hiemer B., Krogull M., Bender T., Ziebart J., Krueger S., Bader R., Jonitz‐Heincke A., Mol. Med. Rep. 2018, 18, 2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Krueger S., Achilles S., Zimmermann J., Tischer T., Bader R., Jonitz‐Heincke A., J. Clin. Med. 2019, 8, 1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brighton C. T., Wang W., Clark C. C., J. Bone Jt. Surg. 2008, 90, 833. [DOI] [PubMed] [Google Scholar]
- 46. Macginitie L. A., Gluzband Y. A., Grodzinsky A. J., J. Orthop. Res. 1994, 12, 151. [DOI] [PubMed] [Google Scholar]
- 47. Baker B., Spadaro J., Marino A., Becker R. O., Ann. N. Y. Acad. Sci. 1974, 238, 491. [DOI] [PubMed] [Google Scholar]
- 48. Rodan G. A., Bourret L. A., Norton L. A., Science 1978, 199, 690. [DOI] [PubMed] [Google Scholar]
- 49. Vaca‐González J. J., Clara‐Trujillo S., Guillot‐Ferriols M., Ródenas‐Rochina J., Sanchis M. J., Ribelles J. L. G., Garzón‐Alvarado D. A., Ferrer G. G., Bioelectrochemistry 2020, 134, 107536. [DOI] [PubMed] [Google Scholar]
- 50. Svennersten K., Bolin M. H., Jager E. W. H., Berggren M., Richter‐Dahlfors A., Biomaterials 2009, 30, 6257. [DOI] [PubMed] [Google Scholar]
- 51. Saigal R., Cimetta E., Tandon N., Zhou J., Langer R., Young M., Vunjak‐Novakovic G., Redenti S., in Proc. Annual Int. Conf. of the IEEE Engineering in Medicine and Biology Society EMBS, IEEE, Piscataway, NJ: 2013, p. 1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schmidt C. E., Shastri V. R., Vacanti J. P., Langer R., Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 8948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kashi M., Baghbani F., Moztarzadeh F., Mobasheri H., Kowsari E., Int. J. Biol. Macromol. 2018, 107, 1567. [DOI] [PubMed] [Google Scholar]
- 54. Reakasame S., Boccaccini A. R., Biomacromolecules 2018, 19, 3. [DOI] [PubMed] [Google Scholar]
- 55. Distler T., McDonald K., Heid S., Karakaya E., Detsch R., Boccaccini A. R., ACS Biomater. Sci. Eng. 2020, 6, 3899. [DOI] [PubMed] [Google Scholar]
- 56. Schwarz S., Kuth S., Distler T., Gögele C., Stölzel K., Detsch R., Bocaccini A. R., Schulze‐Tanzil G., Mater. Sci. Eng., C 2020, 116, 111189. [DOI] [PubMed] [Google Scholar]
- 57. Ren K., Cheng Y., Huang C., Chen R., Wang Z., Wei J., J. Mater. Chem. B 2019, 7, 5704. [DOI] [PubMed] [Google Scholar]
- 58. Ouyang L., Yao R., Zhao Y., Sun W., Biofabrication 2016, 8, 035020. [DOI] [PubMed] [Google Scholar]
- 59. Soltan N., Ning L., Mohabatpour F., Papagerakis P., Chen X., ACS Biomater. Sci. Eng. 2019, 5, 2976. [DOI] [PubMed] [Google Scholar]
- 60. Gao T., Gillispie G. J., Copus J. S., P. R. Anil Kumar, Seol Y.‐J., Atala A., Yoo J. J., Lee S. J., Biofabrication 2018, 10, 034106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Macmillan D. S., Chilton M. L., Regul. Toxicol. Pharmacol. 2019, 101, 35. [DOI] [PubMed] [Google Scholar]
- 62. Ashton M. D., Appen I. C., Firlak M., Stanhope N. E., Schmidt C. E., Eisenstadt W. R., Hur B., Hardy J. G., Polym. Int. 2020, 10.1002/pi.6089. [DOI] [Google Scholar]
- 63. Rahimzadeh Z., Naghib S. M., Zare Y., Rhee K. Y., J. Mater. Sci. 2020, 55, 7575. [Google Scholar]
- 64. Liang Y., Goh J. C.‐H., Bioelectricity 2020, 2, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ares M., Karazehir T., Sezai Sarac A., Curr. Phys. Chem. 2012, 2, 224. [Google Scholar]
- 66. Rogers Z. J., Zeevi M. P., Koppes R., Bencherif S. A., Bioelectricity 2020, 2, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yin J., Liu Q., Zhou J., Zhang L., Zhang Q., Rao R., Liu S., Jiao T., RSC Adv. 2020, 10, 10546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bu Y., Xu H. X., Li X., Xu W. J., Yin Y. X., Dai H. L., Bin Wang X., Huang Z. J., Xu P. H., RSC Adv. 2018, 8, 10806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sajesh K. M., Jayakumar R., Nair S. V., Chennazhi K. P., Int. J. Biol. Macromol. 2013, 62, 465. [DOI] [PubMed] [Google Scholar]
- 70. Ketabat F., Karkhaneh A., Mehdinavaz Aghdam R., Hossein Ahmadi Tafti S., J. Biomater. Sci., Polym. Ed. 2017, 28, 794. [DOI] [PubMed] [Google Scholar]
- 71. Zhang T., Zhang W., Wang B., Liu J., J. Intell. Mater. Syst. Struct. 2018, 29, 232. [Google Scholar]
- 72. Basavaraja C., Jo E. A., Kim B. S., Kim D. G., Huh D. S., Macromol. Res. 2010, 18, 1037. [Google Scholar]
- 73. Chen S., Chen W., Xue G., Macromol. Biosci. 2008, 8, 478. [DOI] [PubMed] [Google Scholar]
- 74. Wang Z., Roberge C., Dao L. H., Wan Y., Shi G., Rouabhia M., Guidoin R., Zhang Z., J. Biomed. Mater. Res., Part A 2004, 70, 28. [DOI] [PubMed] [Google Scholar]
- 75. Binette J. S., Garon M., Savard P., McKee M. D., Buschmann M. D., J. Biomech. Eng. 2004, 126, 475. [DOI] [PubMed] [Google Scholar]
- 76. Wu Y., Cisewski S. E., Sun Y., Damon B. J., Sachs B. L., Pellegrini V. D., Slate E. H., Yao H., Spine (Philadelphia) 2017, 42, E1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Abidian M. R., Kim D.‐H., Martin D. C., Adv. Mater. 2006, 18, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mawad D., Artzy‐Schnirman A., Tonkin J., Ramos J., Inal S., Mahat M. M., Darwish N., Zwi‐Dantsis L., Malliaras G. G., Gooding J. J., Lauto A., Stevens M. M., Chem. Mater. 2016, 28, 6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Brahim S., Guiseppi‐Elie A., Electroanalysis 2005, 17, 556. [Google Scholar]
- 80. Freeman F. E., Kelly D. J., Sci. Rep. 2017, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hazur J., Detsch R., Karakaya E., Kaschta J., Teßmar J., Schneidereit D., Friedrich O., Schubert D. W., Boccaccini A. R., Biofabrication 2020, 12, 045004. [DOI] [PubMed] [Google Scholar]
- 82. Yang J., Choe G., Yang S., Jo H., Lee J. Y., Biomater. Res. 2016, 20, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Foroughi J., Ghorbani S. R., Peleckis G., Spinks G. M., Wallace G. G., Wang X. L., Dou S. X., J. Appl. Phys. 2010, 107, 103712. [Google Scholar]
- 84. Distler T., McDonald K., Heid S., Detsch R., Boccaccini A. R., ACS Biomater. Sci. Eng. 2020, 6, 3899. [DOI] [PubMed] [Google Scholar]
- 85. Weizel A., Distler T., Boccaccini A. R., Budday S., Seitz H., Acta Biomater. 2020, 118, 113. [DOI] [PubMed] [Google Scholar]
- 86. Lu Y., He W., Cao T., Guo H., Zhang Y., Li Q., Shao Z., Cui Y., Zhang X., Sci. Rep. 2014, 4, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Schirmer K., Wright C., Warren H., Thompson B., Quigley A., Kapsa R., Wallace G., Mater. Res. Soc. Symp. Proc. 2015, 1717, 1. [Google Scholar]
- 88. Mazzoleni A. P., Sisken B. F., Kahler R. L., Bioelectromagnetics 1986, 7, 95. [DOI] [PubMed] [Google Scholar]
- 89. Zhadobov M., Augustine R., Sauleau R., Alekseev S., Di Paola A., Quément C. L.e, Mahamoud Y. S., Dréan Y. L.e, Bioelectromagnetics 2012, 33, 346. [DOI] [PubMed] [Google Scholar]
- 90. Taghian T., Narmoneva D. A., Kogan A. B., J. R. Soc. Interface 2015, 12, 20150153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Wu Y., Chen Y. X., Yan J., Quinn D., Dong P., Sawyer S. W., Soman P., Acta Biomater. 2016, 33, 122. [DOI] [PubMed] [Google Scholar]
- 92. Wu L., Li L., Pan L., Wang H., Bin Y., J. Appl. Polym. Sci. 2021, 138, 49800. [Google Scholar]
- 93. Pan L., Yu G., Zhai D., Lee H. R., Zhao W., Liu N., Wang H., Tee B. C.‐K., Shi Y., Cui Y., Bao Z., Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 9287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chougule M. A., Pawar S. G., Godse P. R., Mulik R. N., Sen S., 2011, 2011, 6. [Google Scholar]
- 95. Lei J., Cai Z., Martin C. R., Synth. Met. 1992, 46, 53. [Google Scholar]
- 96. Maruthamuthu S., Chandrasekaran J., Manoharan D., Magesh R., J. Polym. Eng. 2017, 37, 481. [Google Scholar]
- 97. Wang S., Lei J., Yi X., Yuan L., Ge L., Li D., Mu C., ACS Appl. Polym. Mater. 2020, 2, 3016. [Google Scholar]
- 98. Sarker B., Papageorgiou D. G., Silva R., Zehnder T., Gul‐E‐Noor F., Bertmer M., Kaschta J., Chrissafis K., Detsch R., Boccaccini A. R., J. Mater. Chem. B 2014, 2, 1470. [DOI] [PubMed] [Google Scholar]
- 99. Handorf A. M., Zhou Y., Halanski M. A., Li W.‐J., Organogenesis 2015, 11, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Silver F. H., Bradica G., Tria A., Matrix Biol. 2002, 21, 129. [DOI] [PubMed] [Google Scholar]
- 101. Wright C. J., Molino B. Z., Chung J. H. Y., Pannell J. T., Kuester M., Molino P. J., Hanks T. W., Gels 2020, 6, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Yang G., Xiao Z., Long H., Ma K., Zhang J., Ren X., Sci. Rep. 2018, 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Shin S. R., Bae H., Cha J. M., Mun J. Y., Chen Y. C., Tekin H., Shin H., Farshchi S., Dokmeci M. R., Tang S., Khademhosseini A., ACS Nano 2012, 6, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Wright C. J., Zhang B., Kuester M., Molino P. J., Hanks T. W., in Front. Bioeng. Biotechnol. Conference Abstract: 10th World Biomaterials Congress, 2016. 10.3389/conf.FBIOE.2016.01.00314. [DOI]
- 105. Yu Y., Zhihuai S., Chen S., Bian C., Chen W., Xue G., Langmuir 2006, 22, 3899. [DOI] [PubMed] [Google Scholar]
- 106. Li Y., Zhao X., Xu Q., Zhang Q., Chen D., Langmuir 2011, 27, 6458. [DOI] [PubMed] [Google Scholar]
- 107. Pyarasani R. D., Jayaramudu T., John A., J. Mater. Sci. 2019, 54, 974. [Google Scholar]
- 108. Bayat A., Ramazani S.A. A., Int. J. Polym. Mater. Polym. Biomater. 2020, 0, 1. [Google Scholar]
- 109. Ruther F., Distler T., Boccaccini A. R., Detsch R., J. Mater. Sci. Mater. Med. 2019, 30, 8. [DOI] [PubMed] [Google Scholar]
- 110. Distler T., Ruther F., Boccaccini A. R., Detsch R., Macromol. Biosci. 2019, 19, 1900245. [DOI] [PubMed] [Google Scholar]
- 111. Paradee N., Sirivat A., J. Phys. Chem. B 2014, 118, 9263. [DOI] [PubMed] [Google Scholar]
- 112. Chikar J. A., Hendricks J. L., Richardson‐Burns S. M., Raphael Y., Pfingst B. E., Martin D. C., Biomaterials 2012, 33, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Gomez C. G., Rinaudo M., Villar M. A., Carbohydr. Polym. 2007, 67, 296. [Google Scholar]
- 114. Hardy J. G., Cornelison R. C., Sukhavasi R. C., Saballos R. J., Vu P., Kaplan D. L., Schmidt C. E., Bioengineering 2015, 2, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Song H., Li T., Han Y., Wang Y., Zhang C., Wang Q., J. Photopolym. Sci. Technol. 2016, 29, 803. [Google Scholar]
- 116. Thiéblemont J. C., Gabelle J. L., Planche M. F., Synth. Met. 1994, 66, 243. [Google Scholar]
- 117. Ding H., Zhong M., Kim Y. J., Pholpabu P., Balasubramanian A., Hui C. M., He H., Yang H., Matyjaszewski K., Bettinger C. J., ACS Nano 2014, 8, 4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Lee H. P., Gu L., Mooney D. J., Levenston M. E., Chaudhuri O., Nat. Mater. 2017, 16, 1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Schindelin J., Arganda‐Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J.‐Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., Cardona A., Nat. Methods 2012, 9, 676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supplemental Video 1
Supplemental Video 2
Data Availability Statement
Research data are not shared.