

Graphical Abstract

Overview of the study. PCD: primary ciliary dyskinesia; FEV1: forced expiratory volume in 1 s; ODA: outer dynein arm; MCC: multiciliated cell.
Abstract
Background
Primary ciliary dyskinesia is a genetic disorder caused by aberrant motile cilia function that results in defective ciliary airway clearance and subsequently leads to recurrent airway infections and bronchiectasis. We aimed to determine: how many functional multiciliated airway cells are sufficient to maintain ciliary airway clearance?
Methods
To answer this question we exploited the molecular defects of the X-linked recessive primary ciliary dyskinesia variant caused by pathogenic variants in DNAAF6 (PIH1D3), characterised by immotile cilia in affected males. We carefully analysed the clinical phenotype and molecular defect (using immunofluorescence and transmission electron microscopy) and performed in vitro studies (particle tracking in air–liquid interface cultures) and in vivo studies (radiolabelled tracer studies) to assess ciliary clearance of respiratory cells from female individuals with heterozygous and male individuals with hemizygous pathogenic DNAAF6 variants.
Results
Primary ciliary dyskinesia male individuals with hemizygous pathogenic DNAAF6 variants displayed exclusively immotile cilia, absence of ciliary clearance and severe primary ciliary dyskinesia symptoms. Owing to random or skewed X-chromosome inactivation in six female carriers with heterozygous pathogenic DNAAF6 variants, 54.3±10% (range 38–70%) of multiciliated cells were defective. Nevertheless, in vitro and in vivo assessment of the ciliary airway clearance was normal or slightly abnormal. Consistently, heterozygous female individuals showed no or only mild respiratory symptoms.
Conclusions
Our findings indicate that having 30–62% of multiciliated respiratory cells functioning can generate either normal or slightly reduced ciliary clearance. Because heterozygous female carriers displayed either no or subtle respiratory symptoms, complete correction of 30% of cells by precision medicine could improve ciliary airway clearance in individuals with primary ciliary dyskinesia, as well as clinical symptoms.
Shareable abstract
Restoring cilia function in 30–62% of defective respiratory cells is sufficient to improve ciliary clearance in PCD individuals https://bit.ly/45PQQg9
Introduction
The respiratory epithelium consists of different types of cells: non-ciliated cells (e.g. ionocytes, goblet and club cells) and ciliated cells. Coordinated beating of multiple motile cilia is essential for ciliary clearance of the upper and lower airways and represents an effective defence mechanism to prevent airway damage [1, 2]. Respiratory cilia characteristically beat with a forward effective stroke and a backward recovery stroke in the same plane along the cell surface. Primary ciliary dyskinesia (PCD), listed in the Online Mendelian Inheritance in Man catalogue of human genes and genetic disorders under the number 244400, is a rare genetic heterogeneous disorder caused by aberrant motile cilia function. Dysfunction of motile cilia in the airway results in defective ciliary clearance of the upper and lower airways and subsequently leads to recurrent airway infections, chronic inflammation, bronchiectasis and progressive lung failure [2, 3]. Outer dynein arm (ODA) complexes attached at 24 nm intervals to the peripheral microtubule doublets contain molecular motors that drive and regulate ciliary motility, while the inner dynein arms (IDAs) regulate the ciliary and flagellar beating pattern. Dynein axonemal assembly factors (DNAAFs) are responsible for the cytoplasmic preassembly of ODAs and IDAs before transport to the ciliary axoneme [4, 5]. We and others have shown that pathogenic variants in DNAAF6 cause the absence of all ODAs from the ciliary axonemes (shown by the absence of dynein axonemal heavy chain 5 (DNAH5)) as well as IDA defects (shown by the absence of dynein axonemal light intermediate chain 1 (DNALI1)) and therefore result in cilia immotility [6–8]. No causative treatment is thus far available and treatment efforts are aimed at hydration of mucus and early eradication of bacterial airway infections. Azithromycin maintenance therapy currently stands as the only evidence-based medical treatment within this context [9]. Precision medicine approaches in PCD, including gene therapy, mRNA transcript therapy and read-through therapy, are currently being explored [10].
However, it is not known how many multiciliated airway cells have to be completely corrected in order to restore ciliary airway clearance. To answer this very important medical and biological question, we exploited the molecular defects caused by pathogenic variants in the DNAAF gene DNAAF6/PIH1D3, responsible for a severe type of PCD characterised by immotile cilia in affected male individuals [6, 7]. Here, aberrant cytoplasmic assembly of axonemal ODAs and IDAs responsible for cilia beat generation and regulation causes complete ciliary immotility. We chose to explore the molecular defects underlying this X-linked model of PCD because X-chromosome inactivation (XCI) has evolved to equalise X-linked gene expression between female XX and male XY individuals to enable dosage compensation in marsupial and placental mammalians [11]. Failure to induce XCI in XX human embryos shows that a double dose of certain X-located genes results in early lethality during development [12]. In somatic tissues, the choice of which X chromosome to inactivate is generally random (figure 1). Because XCI is stably maintained, the same chromosome is inactivated in all progeny cells. Therefore, female individuals are mosaics of two populations of cells that differ in the X-chromosome that is active. Thus, in female individuals with heterozygous pathogenic DNAAF6 variants, half of all ciliated respiratory cells are lined with immotile cilia, whereas male individuals with hemizygous pathogenic DNAAF6 variants display exclusively immotile cilia in the airways (figure 1). However, skewed XCI can occur [13, 14] and even result in disease. For instance, in some female carriers of a dystrophin gene mutation, skewed inactivation (>90%) results in muscular dystrophy [15–17].
FIGURE 1.

Random X-chromosome inactivation (XCI) in female carriers of heterozygous pathogenic DNAAF6 variants results in two populations of respiratory ciliated cells. The differentiated respiratory epithelium consists of different type of cells: non-ciliated cells (e.g. ionocytes, goblet and club cells) and ciliated cells. DNAAF6 is located on the X chromosome. In contrast to female individuals, in male individuals there is no XCI. Male individuals with a hemizygous pathogenic DNAAF6 variant display exclusively immotile cilia in the airways, resulting in primary ciliary dyskinesia. In contrast, due to XCI, female individuals have a mosaic of two populations of cells that differ in the X-chromosome that is active. Thus, in female individuals with a heterozygous pathogenic DNAAF6 variant, it is expected that half of all ciliated respiratory cells are lined with immotile cilia. In this study, we performed detailed in vitro and in vivo studies to assess the impact on ciliary clearance.
Here, we report five male PCD individuals with hemizygous and six female individuals with heterozygous pathogenic DNAAF6 variants from four distinct families (figure 2). We identified three distinct large genomic deletions with variable sizes, a 10 kB deletion (exons 2–4 of DNAAF6), a 28 kB deletion (exon 1 of DNAAF6) and a 400 kB deletion (all DNAAF6 exons), as well as the pathogenic DNA variant c.266c>A resulting in premature stop of translation (p.Trp89*). All male DNAAF6-affected individuals presented with typical PCD symptoms and variant respiratory cilia were immotile due to absence of ODAs and IDAs. Interestingly, we observed in the female heterozygous carriers that, due to random XCI, 38–70% of multiciliated respiratory cells displayed immotile cilia due to aberrant ODAs (absence of DNAH5) and IDAs (absence of DNALI1). We carefully examined the clinical phenotype of the female carriers of the heterozygous pathogenic DNAAF6 variants and performed in vivo functional ciliary clearance analyses using radiolabelled tracer and in vitro ciliary clearance assays in air–liquid interface (ALI) cultures of ciliated respiratory cells. Hemizygous male PCD individuals did not have any in vitro or in vivo ciliary clearance. However, despite 38–70% of the respiratory ciliated cells carrying immotile cilia, female carriers with heterozygous pathogenic DNAAF6 variants showed no or only mild respiratory symptoms. In vitro and in vivo ciliary clearance assays were either normal or showed a subtle reduction in ciliary clearance. We conclude that it is not necessary to have 100% of respiratory ciliated cells functioning to ensure effective ciliary clearance. A range of 30–62% of functioning cells is sufficient to provide either normal or only a subtle reduction in in vitro and in vivo ciliary clearance.
FIGURE 2.
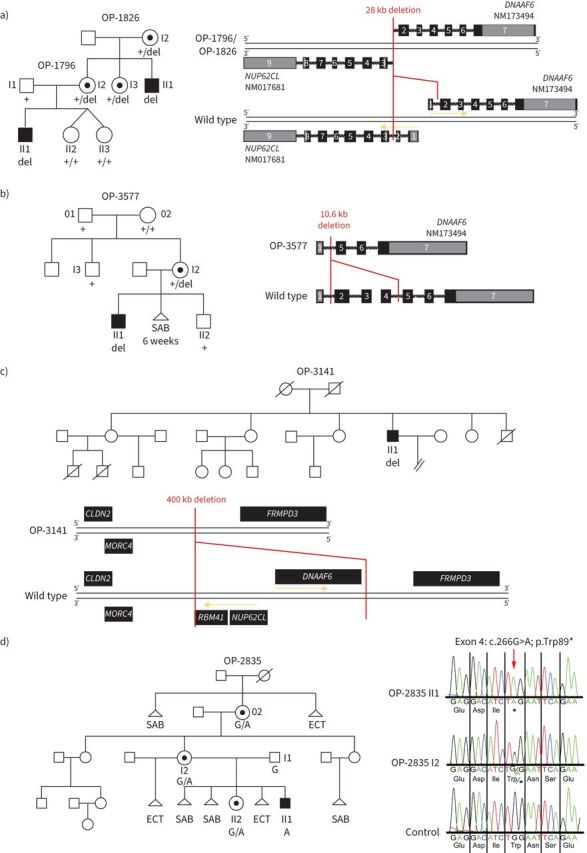
Pathogenic variants in DNAAF6 located on the X chromosome were identified in four families. a) In family OP-1826/OP-1796 we found a 28 kb deletion (del) including the first exon of DNAAF6 and the first two exons of NUP62CL. b) In family OP-3577, a 10.6 kb deletion comprising DNAAF6 exons 2–4 was identified in OP-3577 I2. Segregation analyses revealed that neither her brother (OP-3577 I3) nor grandmother (OP-3577 02) carry the deletion, indicating that this is a de novo variant. OP-3577 I2 reported difficulties in getting pregnant and had one spontaneous abortion in the sixth week of pregnancy. c) In OP-3141 we identified a 400 kb deletion comprising DNAAF6, NUP62CL and RBM41. OP-3141 II1 is the fourth of six children. He has three older and one younger sister. The only brother died 10 days after birth owing to cardiorespiratory failure of unknown cause. His oldest sister gave birth to two male individuals: the first born died 3 months after birth, the second born died 4 months after birth. Both died of unknown causes with cardiorespiratory failure. With another partner, she gave birth to a healthy boy. The second oldest sister has three healthy children, two girls and a boy. The third oldest sister gave birth to a healthy boy. The younger sister has no children. d) In family OP-2835, a transition from G to A (c.266G>A) was identified in DNAAF6 exon 4, predicting premature stop of translation (p.Trp89*). Segregation analyses revealed that the mother (OP-2835 I2) and the grandmother (OP-2835 02) carry this variant in a heterozygous status whereas the father (OP-2835 I1) is unaffected. The great-grandmother of OP-2835 II1 had a spontaneous abortion (SAB) and gave birth to OP-2835 02, who inherited the DNAAF6 variant. The great-grandmother died owing to complications of an ectopic (ECT) pregnancy. OP-2835 02 gave birth to three daughters. The oldest one has two healthy children, a girl, who already has another healthy daughter, and a boy. The second oldest one, OP-2835 I2, is a carrier of the DNAAF6 variant and has two children. She had one ectopic pregnancy with a previous partner and with her current partner two spontaneous abortions and an ectopic pregnancy. OP-2835 II2 was her fourth pregnancy and is also a carrier of the DNAAF6 variant. In addition, she gave birth to the index primary ciliary dyskinesia (PCD) male individual OP-2835 II1, who was her sixth pregnancy and who carries the hemizygous DNAAF6 variant. The younger sister had one abortion and no children. PCD-affected males are shaded black and unaffected siblings are shaded white. Heterozygous female carriers are indicated by a central dot. Genotypes are indicated in the pedigrees.
Material and methods
PCD-affected individuals and families
Signed and informed consent was obtained from individuals fulfilling the diagnostic criteria of PCD [18] and from family members according to protocols approved by the institutional ethics review board of the University Hospital Muenster (Ethik-Kommission Westfalen Lippe in Muenster, reference number 2015-104-f-S) and Copenhagen (Danish Data Protection Agency 2014).
Summary of applied methods
Please see the supplementary file for a detailed method section.
In this study, we performed mutational analyses using a customised PCD gene panel and whole exome sequencing (WES) and whole genome sequencing as well as Sanger sequencing to identify families with male PCD individuals carrying disease-causing hemizygous DNAAF6 DNA variants (figure 2, supplementary figures S1–S4). Genetic variants were evaluated according to American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) guidelines [19]. We only report disease-causing (pathogenic) variants (class 5) according to ACMG/AMP guidelines. Further genetic analyses identified female heterozygous carriers of the DNAAF6 variants (figure 2). Clinical findings such as laterality status and lung function and family history were ascertained (table 1).
TABLE 1.
Clinical findings of female heterozygous pathogenic DNAAF6 variant carriers and PCD males with hemizygous pathogenic DNAAF6 variants
| OP-1796 I2 | OP-1796 I3 | OP-1826 I2 | OP-2835 I2 | OP-2835 II2 | OP-3577 I2 | OP-1796 II1 | OP-1826 II1 | OP-2835 II1 | OP-3141 II1 | OP-3577 II1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gender | F | F | F | F | F | F | M | M | M | M | M |
| Age (years) | 43 | 37 | 67 | 53 | 26 | 34 | 16 | 45 | 19 | 60 | 3 |
| Origin | Denmark | Denmark | Denmark | Germany | Germany | Germany | Denmark | Denmark | Germany | Germany | Germany |
| Mutation in DNAAF6 | 28 kb deletion (het) | 28 kb deletion (het) | 28 kb deletion (het) | c.266G>A; p.Trp89* (het) | c.266G>A; p.Trp89* (het) | 10.6 kb deletion (het) | 28 kb deletion (hem) | 28 kb deletion (hem) | c.266G>A; p.Trp89* (hem) | 400 kb deletion (hem) | 10.6 kb deletion (hem) |
| SI | No | No | No | No | No | No | No | No | No | No | Yes |
| Consanguinity | No | No | No | No | No | No | No | No | No | No | No |
| nNO (nL·min−1) | 459 | 495 | NA | 241.7 | 55 | 122.1 | 9.6 | 19.8 | 29.7 (2017); 3 (2017); 16.1 (2019) | 22.5 | 6.6 |
| Lung function | Obstructive ppFEV1: 87% ppFVC: 110% FEV1/FVC: 64% |
ppFEV1: 101% ppFVC: 116% FEV1/FVC: 72% |
ppFEV1: 115% ppFVC: 121% FEV1/FVC: 73% |
ppFEV1: 123% ppFVC: 130% FEV1/FVC: 81% |
Mild over blow, normal ppFEV1: 111% ppFVC: 117% FEV1/FVC: 83% |
Overblow ppFEV1: 101% ppFVC: 117% FEV1/FVC: 76% |
Obstructive/ restrictive ppFEV1: 74% ppFVC: 86% FEV1/FVC: 72% |
Obstructive ppFEV1: 69% ppFVC: 82% FEV1/FVC: 66% |
Mild over blow, normal ppFEV1 112% |
Obstructive ppFEV1 46.5% |
NA |
| Neonatal RDS | No | No | No | No | No | NA | Yes | Yes | Yes | NA | No |
| Chronic sinusitis | No | No | No | No | Yes | Yes | No | Yes | No | Yes | No |
| Chronic otitis media | No | No | No | No | Yes | No | Yes | Yes | Yes | Yes | No |
| Chronic bronchitis | No | Recurrent pneumonia | No | No | Yes (in childhood) | No | Yes | Yes | Yes | Yes | Yes |
| Chronic wet cough | No | Slight recurrent | No | No | No | No | Yes | Yes | Yes | Yes | Yes |
| Bronchiectasis | NA | No | NA | NA | NA | No | Yes | Yes | Yes | Yes | No |
| TEM | NA | 67% ODA defect and 33% normal | NA | NA | NA | NA | ODA defect | ODA defect | NA | ODA defect | NA |
| IF | 38% ODA defect and 62% normal | 70% ODA defect and 30% normal | 61% ODA defect and 39% normal | 50% ODA defect and 50% normal | 58% ODA defect and 42% normal | 52% ODA defect and 48% normal | ODA/IDA defect | ODA/IDA defect | ODA/IDA defect | ODA/IDA defect | ODA/IDA defect |
| 45% IDA defect and 55% normal | 66% IDA defect and 34% normal | 61% IDA defect and 39% normal | 55% IDA defect and 45% normal | 32% IDA defect and 68% normal | 42% IDA defect and 58% normal | ||||||
| HVMA | Patches with motile and immotile cilia | Patches with motile and immotile cilia | Patches with motile and immotile cilia | Patches with motile and immotile cilia | Patches with motile and immotile cilia | Patches with motile and immotile cilia | Immotile | Immotile | Immotile | Immotile | Immotile |
| Fertility problems | No | No | No | Yes | NA | NA | NA | NA | NA | Yes | NA |
F: female; hem: hemizygous; het: heterozygous; HVMA: high-speed video microscopy analyses; IF: high-resolution immunofluorescence microscopy; IDA: inner dynein arm; M: male; nNO: nasal nitric oxide production rate; NA: not available; ODA: outer dynein arm; ppFEV1: % predicted forced expiratory volume in 1 s; ppFVC: % predicted forced vital capacity; RDS: respiratory distress syndrome; SI: situs inversus; TEM: transmission electron microscopy.
To understand the molecular defects in the male hemizygous and female heterozygous DNAAF6 variant individuals we performed various experiments in nasal ciliated airway cells (native cells after brushing), comprising transmission electron microscopy (TEM), high-speed video microscopy analysis (HVMA) and high-resolution immunofluorescence (IF) microscopy. We performed IF using antibodies directed against the ODA components DNAI1, DNAH5, DNAH9 and DNAI2 and the IDA component DNALI1. We counted a total of 500 cells per IF staining for male (n=1) and female (n=2) controls (OP-1796 I2, OP-1796 II1, OP-1826 I2, OP-1826 II1, OP-3577 I2 and OP-3577 II1). For OP-2835 II1, 200 cells and for OP-2835 I2 and OP-2835 II2 a total of 75 cells per IF staining and individual were counted. In addition ALI cultures from male hemizygous and female heterozygous DNAAF6 variant individuals were established and the ciliary beat frequencies, ciliary particle transport, high-resolution IF and immunoblot analyses were performed. For ciliary particle transport measurements (in vitro ciliary clearance assay), fluorescent beads (0.5 and 2.0 μm in diameter) were mixed with pre-warmed cell culture medium and added to the apical compartment of respiratory cells in ALI culture (day 30 after airlift). Transport of fluorescent nanoparticles by ciliary beating was recorded using the Nikon Eclips Ti-S microscope (×20 objective lens) equipped with the NIS-Elements Advanced Research software (20 s; 7.5 frames·s−1). In total, 30 videos per individual were analysed for statistical evaluation and 253 particles were tracked per video on average from three different cell culture inserts from each individual. Because two different bead sizes (0.5 and 2 µm) were used for the analyses, values were normalised against the mean value of the healthy control group. Tracking videos were evaluated using the NIS-Elements Advanced Research software (version 4.51.000) and NIS Advanced 2D Tracking plug-in to generate polargraphs and to determine the speed (μm·s−1). In parallel to each recorded tracking video, a corresponding differential interference contrast (DIC) video was taken by an additionally equipped Basler sc640–120fm monochrome high-speed video camera (recording 125 frames·s−1) to determine the cell condition as well as the ciliary beating pattern. DIC videos were evaluated using SAVA software [20]. Particle transport was measured in 18 healthy female individuals and one healthy male individual (age 25–60 years). The results of the experiments were quantified and statistically analysed using R (www.r-project.org).
To measure pulmonary ciliary clearance in vivo, we performed pulmonary radioaerosol mucociliary clearance (PRMC) measurements in one male PCD individual and three female carriers. For PRMC measurement, insoluble technetium-99m labelled nanocolloids were aerosolised and inhaled by 20 slow inspirations and successive forced expirations. Initial lung deposition and subsequent pulmonary mucociliary clearance immediately after inhalation of the radiolabelled tracer were assessed by repeated dynamic and static scintigraphic acquisitions for 2 h. In addition, a static measurement after 24 h and krypton-81m ventilation imagery of the ventilated lung area was performed. Whole-lung retention 1 h and 2 h after tracer inhalation were compared to predicted values calculated from penetration index, age and sex using previously published reference equations [21]. Tracheobronchial velocity (TBV) (bolus transport) was measured from dynamic acquisitions within the first hour. Involuntary cough was monitored by trained staff and significant coughing rendered the test inconclusive to avoid false positive PRMC results due to cough clearance. If a subject coughed during the test, voluntary cough clearance was assessed by 1 min of rigorous coughing after 2 h by analysing both the central part and the total lung regions [22].
Results
High-resolution IF microscopy analyses
We performed high-resolution IF microscopy to analyse the ciliary defects at the cellular level in females with heterozygous and males with hemizygous pathogenic DNAAF6 variants, respectively (figures 3 and 4). We used antibodies directed against ODA components DNAI1, DNAH5, DNAH9 and DNAI2. Analyses of ciliated respiratory cells from female heterozygous carriers (OP-1796 I2, OP-1796 I3, OP-1826 I2, OP-3577 I2, OP-2835 I2 and OP-2835 II2) revealed that 54.3±10% (range 38–70%, table 1) of ciliated respiratory cells showed abnormal localisation of the ODA components DNAI1, DNAH5, DNAH9 and DNAI2 (table 1, figures 3 and 5, and supplementary figures S5, S9, S11 and S13) when compared to control respiratory cells (1.8±0.67%), consistent with random XCI. However, some female carriers (e.g. OP-1769 I3) had a very low proportion of normal multiciliated cells (30%), whereas others (e.g. OP1796 I2) had a high proportion (62%), indicating that those carriers had a skewed XCI. In accordance with reported findings [6–8], we found no ODAs in analysed ciliated respiratory cells (99.8±0.37%) from male PCD individuals carrying hemizygous variants in DNAAF6 (OP-1796 II1, OP-1826 II1, OP-3577 II1 and OP-2835 II1; figures 4 and 5, and supplementary figures S6, S10 and S12). Next, we analysed the localisation of the IDA component DNALI1. In female carriers, 45.2±13% (range 32–66%, table 1) of the respiratory cells showed abnormal localisation of DNALI1 along the ciliary axoneme when compared to control respiratory cells (2.4±1.6%) (figures 3 and 5, and supplementary figure S7). As expected, we observed an absence of DNALI1 in almost all respiratory cells (99.6±0.4%) from male PCD individuals carrying hemizygous variants in DNAAF6 (figures 4 and 5, and supplementary figure S8). Statistical analysis using the ANOVA two-tailed test showed a significantly increased count of ODA-defective and IDA-defective cells in respiratory cell samples from female carriers with a heterozygous DNAAF6 pathogenic variant when compared to healthy control respiratory cells (p<0.001). In addition, there was a significantly increased count of cells with normal ODA/IDA composition in respiratory cell samples from female carriers with a heterozygous pathogenic DNAAF6 variant when compared to male PCD individuals with a hemizygous pathogenic DNAAF6 variant (p<0.001). For male (n=1) and female (n=2) controls and for OP-1796 I2, OP-1796 I3, OP-1796 II1, OP-1826 I2, OP-1826 II1, OP-3577 I2 and OP-3577 II1, a total of 500 cells per IF staining were counted. For OP-2835II1, 200 cells were counted and for OP-2835 I2 and OP-2835 II2, 75 cells per IF staining were counted.
FIGURE 3.
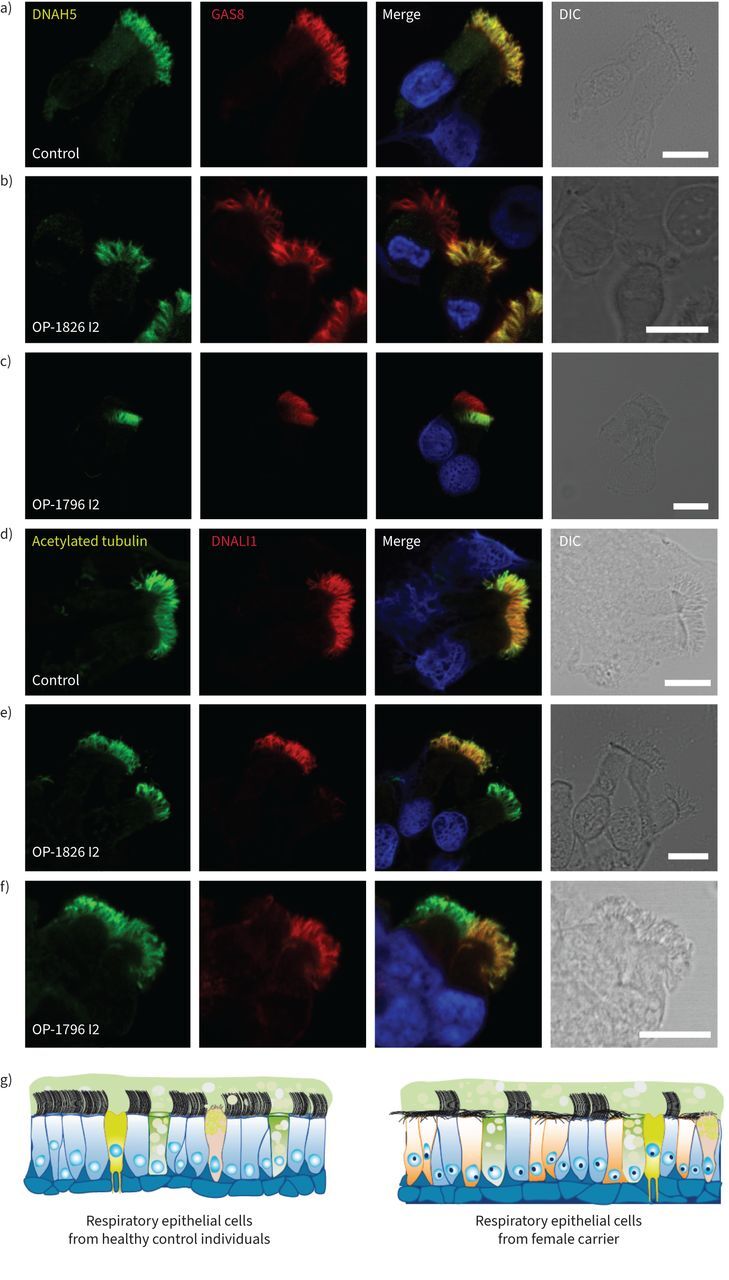
Heterozygous DNAAF6 pathogenic variants in female individuals result in a mosaic of cells with and without axonemal outer and inner dynein arm defects, respectively. a–c) Respiratory epithelial cells from female control individuals and female carriers with a heterozygous pathogenic DNAAF6 variant were double-labelled with antibodies directed against the outer dynein arm protein DNAH5 (green) and GAS8 (red). Both proteins colocalise (yellow) along the ciliary axonemes in all cells from the unaffected control (a). In female carriers with a heterozygous pathogenic DNAAF6 variant, DNAH5 showed a normal localisation in some cells or was completely absent or severely reduced in the ciliary axonemes of other (b, c). d–f) Respiratory epithelial cells from a female control and female carriers with a heterozygous pathogenic DNAAF6 variant were double-labelled with antibodies directed against acetylated tubulin (green) and the inner dynein arm protein DNALI1 (red). Both proteins colocalise (yellow) along the ciliary axonemes in all cells from the unaffected control (d). In female carriers with heterozygous pathogenic DNAAF6 variant, DNALI1 showed a normal localisation in some cells or was completely absent or severely reduced in the ciliary axonemes of other cells (e, f). Nuclei were stained with Hoechst 33342 (blue). g) Schematic summarising the immunofluorescence microscopy results. In female control cells, cytoplasmic preassembly of dynein arms was not altered and all cells showed normal axonemal composition of inner (DNALI1) and outer (DNAH5) dynein arms. In contrast, female carriers with a heterozygous pathogenic DNAAF6 variant show a mosaic of cells with normal axonemal composition of dynein arms (blue) and abnormal composition dynein arms (yellow), respectively. Scale bars: 10 μm. DIC: differential interference contrast.
FIGURE 4.
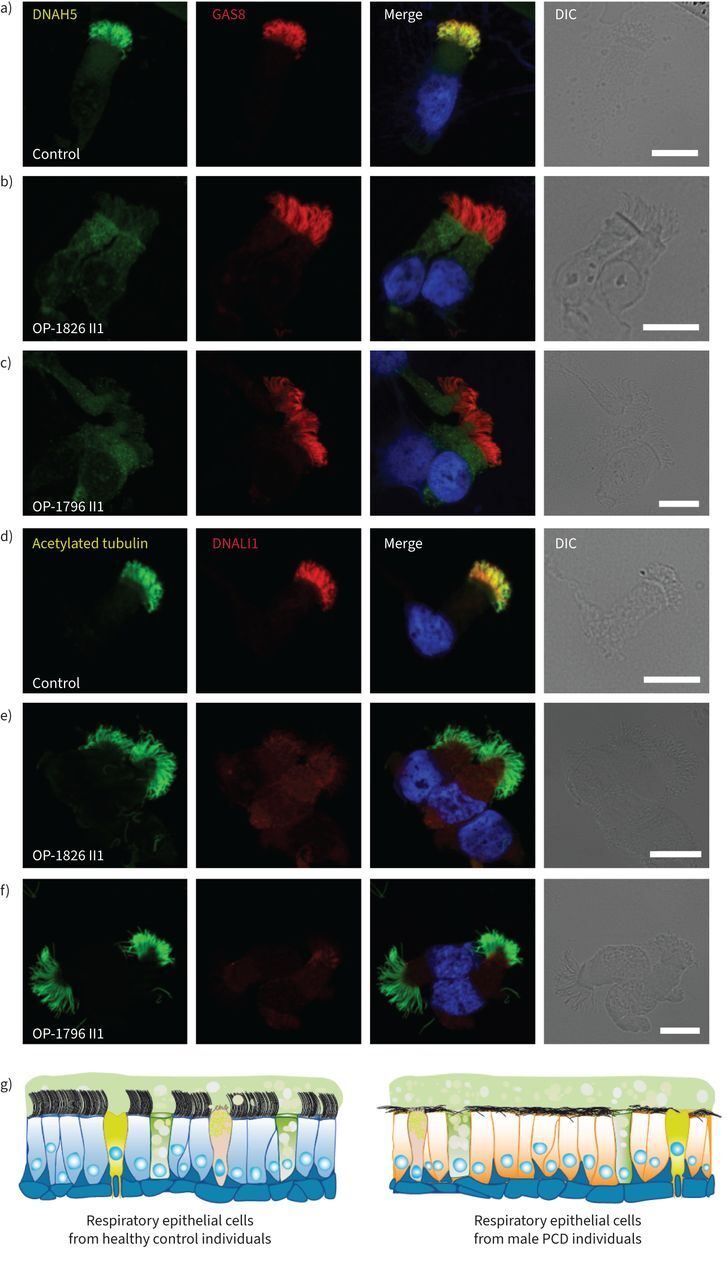
Hemizygous pathogenic variants in DNAAF6 result in outer and inner dynein arm defects in primary ciliary dyskinesia (PCD)-affected males. a–c) Respiratory epithelial cells from control individuals and male PCD individuals OP-1826 II1 and OP-1796 II1 with a hemizygous pathogenic DNAAF6 variant were double-labelled with antibodies directed against the outer dynein arm protein DNAH5 (green) and GAS8 (red). Both proteins colocalise (yellow) along the ciliary axonemes in cells from the unaffected control (a). In cells from PCD individuals OP-1826 II1 (b) and OP-1796 II1 (c), DNAH5 was completely absent or severely reduced from the ciliary axonemes of all cells analysed. d–f) Respiratory epithelial cells from a control and male PCD individuals OP-1826 II1 and OP-1796 II1 with hemizygous pathogenic DNAAF6 variants were double-labelled with antibodies directed against acetylated tubulin (green) and the inner dynein arm protein DNALI1 (red). Both proteins colocalise (yellow) along the ciliary axonemes in all cells from the unaffected control (d). In cells from the male PCD individuals OP-1826 II1 (e) and OP-1796 II1 (f), DNALI1 was completely absent or severely reduced in the ciliary axonemes of all cells analysed. Nuclei were stained with Hoechst 33342 (blue). g) Schematic summarising the immunofluorescence microscopy results. In all control cells (blue), cytoplasmic preassembly of dynein arms was not altered and all cells showed normal axonemal composition of inner (DNALI1) and outer (DNAH5) dynein arms. In contrast, in male PCD individuals with hemizygous DNAAF6 pathogenic variants, all cells show abnormal composition of dynein arms (yellow). Scale bars: 10 μm. DIC: differential interference contrast.
FIGURE 5.

Pooled statistical analysis of immunofluorescence (IF) microscopy findings using antibodies directed against outer dynein arm (ODA) component DNAH5 (a) and the inner dynein arm (IDA) protein DNALI1 (b) in control respiratory cells, cells from female carriers with a heterozygous pathogenic DNAAF6 variant (OP-1796 I2, OP-1796 I3, OP-1826 I2, OP-3577 I2 and OP-2835 I2) and male primary ciliary dyskinesia (PCD) individuals with a hemizygous pathogenic DNAAF6 variant (OP-1796 II1, OP-1826 II1, OP-3577 II1, OP-2835 II1 and OP-2835 II2). a) Analyses of ciliated respiratory cells of females with a heterozygous pathogenic DNAAF6 variant revealed that 54.3±10% (range 38–70%, table 1) show abnormal axonemal localisation of DNAH5 when compared to control respiratory cells (1.8±0.67%). In male PCD individuals with a hemizygous pathogenic DNAAF6 variant, almost all respiratory cells showed absence of DNAH5 from the ciliary axonemes (99.8±0.37%). b) Analyses of ciliated respiratory cells of female carriers with a heterozygous DNAAF6 pathogenic variant revealed that 45.2±13% (range 32–66%, table 1) show abnormal localisation of DNALI1 when compared to control respiratory cells (2.4±1.6%). In male PCD individuals with hemizygous pathogenic DNAAF6 variants, almost all respiratory cells showed axonemal absence of DNALI1 (99.6±0.4%). For male (n=1) and female controls (n=2) and for OP-1796 I2, OP-1796 I3, OP-1796 II1, OP-1826 I2, OP-1826 II1, OP-3577 I2 and OP-3577 II1, a total of 500 cells per IF staining were counted. For OP-2835 II1, 200 cells were counted. For OP-2835 I2 and OP-2835 II2, 75 cells were counted. Error bars denote standard deviation. p-values were generated by the ANOVA two-tailed tests. ***: p<0.001.
TEM
To further analyse the ciliary defect resulting from heterozygous pathogenic variants in DNAAF6, we performed TEM with samples from OP-1796 I3. In total, 581 ciliary axonemes were analysed. Our findings confirm that heterozygous pathogenic DNAAF6 variants result in a mosaic of cells with and without axonemal ODA defects (figure 6 and supplementary figure S14). Furthermore, ultrastructural analyses found ciliary cross-sections without ODAs (n=391, 67%) but also ciliary cross-sections with normal ODA localisation (n=190, 33%), consistent with our IF microscopy findings (table 1) and skewed XCI in OP-1796 I3.
FIGURE 6.

Heterozygous pathogenic DNAAF6 variants in female carriers result in a mosaic of cells with and without axonemal outer dynein arm (ODA) defects. Transmission electron microscopy analysis of ciliary cross-sections from a) a control and b) female individual OP-1796 I3 carrying a heterozygous DNAAF6 variant show the absence of ODAs in more than half of the ciliated cells (n=391 of 581 sections, 67%) from the female carrier. Red arrows mark ODAs. A total of 581 ciliary cross-sections from OP-1796 I3 were analysed. White scale bars: 500 nm; black scale bars: 250 nm.
HVMA
HVMA of nasal respiratory epithelial cells to assess ciliary beating in male PCD individuals carrying hemizygous DNAAF6 variants showed that all respiratory cells were lined with immotile cilia (supplementary videos 1 and 2), consistent with the observed axonemal ODA defects by IF microscopy (figure 4). In the female carriers, patches with motile and immotile cilia were observed (supplementary videos 3–7 and supplementary table S2), consistent with variable XCI and our IF microscopy findings. In OP-1796 I3, 340 of 597 (57%) multiciliated cells carried immotile cilia, consistent with skewed XCI and also supported by IF and TEM findings. In the other female carriers who were analysed by HVMA (OP-2835 II2 and OP-1826 I2), patches of multiciliated cells with motile and immotile cilia were observed. However, due to the low number of cells counted, no quantification was performed.
Measurement of ciliary clearance in vitro and in vivo
Next, we tested whether respiratory cilia of DNAAF6-mutant cells can generate a directed fluid flow. To mimic the process of ciliary clearance in vitro, we added fluorescent particles to the apical compartment of respiratory cells in ALI culture from healthy control individuals; female carriers OP-1796 I2, OP-1796 I3, OP-1826 I2 and OP-3577 I2 with the heterozygous pathogenic DNAAF6 variant; and PCD male individuals OP-1796 II1, OP-1826 II1, OP-2835 II1 and OP-3141 II1 carrying hemizygous pathogenic DNAAF6 variants. Consistent with PCD and aberrant ciliary clearance, mutant cilia from PCD male individuals with hemizygous pathogenic DNAAF6 variants were not able to transport fluorescent particles along the surface of the differentiated epithelium (figure 7 and supplementary figure S15) and ciliary clearance significantly differed from healthy controls (p=0.0003). In contrast, respiratory cilia from healthy controls and carriers of heterozygous pathogenic DNAAF6 variants were able to generate a directed fluid flow (figure 7). However, the velocity of this flow was significantly slower than that of the healthy controls (p=0.009). Two female carriers (OP-1796 I2 and OP-3577 I2) had in vitro ciliary transport velocities in the lower normal range also consistent with IF findings. OP-1796 I3 showed the lowest in vitro ciliary transport velocity (figure 7), consistent with skewed XCI that was also supported by IF, TEM and HVMA findings. OP-1826 I2 also had low in vitro ciliary transport velocity, also consistent with IF findings.
FIGURE 7.

Air–liquid interface cultures of respiratory epithelial cells from female carriers with heterozygous pathogenic DNAAF6 variants are able to generate a directed fluid flow while respiratory epithelial cells from male individuals with hemizygous pathogenic DNAAF6 variants are unable to produce a fluid flow. a) After complete cell differentiation and ciliation (30 days after airlift), fluorescent particles were added to the apical compartments of the respiratory epithelial cells to assess ciliary clearance capacity. Tracking videos are represented as z-stack projections, while the transport direction of each particle is summarised in polargraphs. b) Respiratory cells from a control group (n=19) and female carriers with heterozygous pathogenic DNAAF6 variants (OP-1796 I2, OP-1796 I3, OP-1826 I2 and OP-3577 I2) transported fluorescent particles in a linear direction along the cell layer. The velocity of this flow was significantly slower than that of the healthy controls (p=0.009, t-test). In contrast, the particle transport was non-oriented and significantly slower in male primary ciliary dyskinesia (PCD) individuals carrying hemizygous pathogenic DNAAF6 variants (OP-1796 II1, OP-1826 II1, OP-2835 II1 and OP-3141 II1; p=0.0003, t-test). Circles indicate values measured with 2 µm beads while triangles indicate values measured with 0.5 µm beads. Because two different bead sizes were used for the analyses, values were normalised against the mean value of the healthy control group. Exact values are shown in supplementary figure S15. The significantly reduced particle transport measured in the male PCD individuals (dashed red line) is a thermal-driven background flow (Brownian movement). c) Measurement of the ciliary beat frequency of healthy controls (8.5 Hz), female carriers with heterozygous variants in DNAAF6 (OP-1796 I2=7.6 Hz, OP-1796 I3=6.3 Hz, OP-1826 I2=7.7 Hz, OP-3577 I2=7.5 Hz) and male PCD individuals with hemizygous pathogenic DNAAF6 variants (0 Hz). There was a subtle reduction in the ciliary beat frequency in female carriers with heterozygous DNAAF6 variants compared to healthy controls (p=0.04, t-test). In total, 30 videos per individuals were analysed for statistical evaluation and 253 particles were tracked per video on average from three different cell culture inserts from each individual. d) Schematic summarising the particle tracking results. Ciliated cells from female control individuals (blue) were able to produce a directed flow. In contrast, in male PCD individuals with pathogenic DNAAF6 variants (orange), all cells display cilia immotility due to lack of functional outer and inner dynein arms and the absence of directed particle transport.
To measure pulmonary ciliary clearance in vivo, we performed bronchoscintigraphy in the female carriers OP-1796 I2, OP-1796 I3 and OP-1826 I2 with a heterozygous pathogenic DNAAF6 variant and in the PCD male individual OP-1796 II1 carrying a hemizygous pathogenic DNAAF6 variant. In the heterozygous female carriers, PRMC showed lung retention in the lower range of normal limits, with retention in the airways during the first 2 h of examination that was normal (z-score <1.65). There was no sign of increased airway retention in OP-1796 I2, but abnormal retention in OP-1796 I3 and slightly abnormal retention in the right lung in OP-1826 I2 after 24 h (supplementary table S3). TBV (normal range: 2–6 mm·min−1) was normal in OP-1796 I2 (2.5 mm·min−1) and OP-1826I2 (3 mm·min−1), but slightly abnormal in OP-1796 I3 (1.7 mm·min−1) (supplementary table S3, supplementary figure S16 and supplementary video 9). The subjects did not cough during the test. Thus, female carriers OP-1796 I3 and OP-1826 I2 showed slightly reduced in vivo pulmonary mucociliary clearance consistent with the reduced in vitro ciliary clearance velocity, probably caused by skewed XCI. In contrast, PCD individual OP-1796 II1 had abnormal lung retention after 1 h, 2 h and 24 h (z-score >1.65) and a TBV of 0 mm·min−1 (supplementary video 10). Also, nasal mucociliary clearance (NMC) velocity was 0 mm·min−1 (supplementary table S3 and supplementary figure S14). In fact, there was limited mucus transport in periods without coughing, while coughing could partly compensate for the reduced ciliary clearance.
Please see the supplementary file for additional results.
Discussion
We and others previously described the first non-syndromic X-linked PCD variant caused by DNA variants in PIH1D3/DNAAF6 [6, 7]. Through careful analyses of family pedigrees, we identified four additional families consistent with an X-linked inheritance pattern (figure 2). Using WES, targeted gene panel sequencing and Sanger sequencing, we identified additional deletions and point mutations in DNAAF6 (figure 2). The 28 kb deletion identified in the family OP-1796/OP-1826 comprises the first noncoding exon of DNAAF6 and the first two noncoding exons of the adjacent gene NUP62CL (figure 2a and supplementary figure S1). The deletions of the two noncoding exons of PIH1D3/DNAAF6 were not detectable by WES. However, WES is frequently used for PCD diagnostics [23]. This finding has implications for genetic diagnostics because WES usually captures only coding exons of genes in a genomic region; deletion of noncoding exons that might affect further splicing will be missed. Geneticists should be aware of this problem.
All affected male PCD individuals carrying hemizygous DNAAF6 variants showed typical PCD symptoms (table 1). HVMA and particle tracking analyses revealed immotile respiratory cilia that were not able to generate in vitro and in vivo ciliary clearance (figure 7, supplementary videos 1 and 2, supplementary table S3 and supplementary figure S16). IF microscopy findings are consistent with previous reports [6, 7]. Interestingly, three male infants died during early infancy due to unexplained cardiorespiratory failure (figure 2). Possibly, those infants had PCD and congenital heart disease or early respiratory failure. Early infant death in PCD has been reported but has not been studied in detail [24].
DNAAF6 is located on the X-chromosome and, in female individuals, one of their two X-chromosomes is transcriptionally silenced to compensate for a double dose of X-linked genes [25]. Consistent with variable XCI we found different degrees of defects in the multiciliated cells of the female carriers. IF microscopy showed that 38–70% of respiratory ciliated cells presented with an absence of ODAs and 32–66% presented with an absence of IDAs (table 1, figures 3 and 5 and supplementary figures S5, S7, S9, S11 and S13). In vitro measurement of ciliary clearance by particle tracking showed that the remaining functional cilia of the female carriers were able to generate a directed flow, contrasting with the findings in affected male individuals (no flow). However, flow was significantly slower than that of the healthy controls (p=0.009; figure 7). Two female carriers (OP-1796 I2 and OP-3577 I2) had in vitro ciliary transport velocities in the lower normal range consistent with IF findings. Individual OP-1796 I2 also had normal PRMC. However, OP-1796 I3 showed the lowest in vitro ciliary transport velocity (figure 7), consistent with skewed XCI and supported by IF, TEM and HVMA findings. OP-1826 I2 also had low in vitro ciliary transport velocity, also consistent with IF findings. OP-1796 I3 and OP-1826 I2 showed subtle abnormalities in in vivo PRMC (supplementary table S3). Interestingly, besides mild respiratory symptoms (table 1), female carriers did not have symptoms consistent with PCD. Only OP-1796 I3, who had the lowest proportion of functioning multiciliated cells (30% normal ODA composition), reported mild respiratory symptoms with chronic bronchitis, recurrent pneumonia and chronic wet cough (normal results on chest computed tomography).
The current BEAT PCD expert consensus guideline for TEM diagnosis considers the absence of ODAs in >50% of cross-sections as a class 1 hallmark defect pathognomonic for PCD [26]. Thus, our findings have implications for PCD diagnostics because by chance about half of female carriers will have more than 50% ODA-affected cilia, as present in OP-1796 I3 (figure 6 and supplementary figure S14). This is also consistent with our IF data showing that four of six female carriers had >50% abnormal cilia (table 1). The observed deviation of abnormal TEM (67%) and abnormal IF (70%) findings in the female carrier OP-1796 I3 probably resulted from a skewed XCI. This might also explain the differences in ciliary particle transport and in PRMC measurement observed between the female carriers.
Interestingly, female carriers of DNAAF6 variants reported ectopic pregnancies and spontaneous abortions (figure 2). One woman died owing to complications of ectopic pregnancy. It has been speculated that, owing to dysfunction of motile cilia within the fallopian tubes, female PCD individuals might have an increased risk for infertility, ectopic pregnancies and spontaneous abortion. However, the few studies addressing this clinical finding in female PCD individuals could not support an increased risk [27, 28]. Our findings indicate that female DNAAF6 carriers might carry a higher risk for ectopic pregnancies and spontaneous abortions.
A limitation to our study is the small number of individuals per group, especially for in vivo PRMC measurement (three heterozygous female carriers, one hemizygous PCD individual) because pathogenic variants in DNAAF6 are rare.
Our results also suggest that clinical analyses of female carriers of heterozygous pathogenic DNAAF6 variants should be considered in cases of respiratory symptoms consistent with PCD. It has been shown that skewness of XCI is relatively common and increases with age and can be tissue-specific [13, 14, 29, 30]. Therefore, skewed XCI can lead to an imbalanced inactivation of the wild-type X chromosome, resulting in more defective respiratory cilia which might cause PCD. Such an imbalanced inactivation of the wild-type X chromosome has previously been reported in female Duchenne muscular dystrophy patients [15–17]. Very recently, a study by Thomas et al. [31] showed that in female carriers of pathogenic DNAAF6 variants, skewed inactivation (80%) results in PCD symptoms, in one case even causing bronchiectasis. In our study, in one female carrier (OP-1796 I3) we found 70% of cells were defective. This female carrier presented slightly abnormal bolus transport measured by PRMC and mild respiratory symptoms (table 1 and supplementary table S3) but no bronchiectasis, indicating that having 30% of ciliated respiratory cells functioning might be sufficient to provide ciliary clearance of the airway to avoid severe PCD symptoms. Based on the published findings and our findings, clinical monitoring of female carriers of variants in DNAAF6 should be considered because skewness of XCI increases with age [26, 27].
Hemizygous male PCD individuals did not have any in vitro or in vivo ciliary clearance. However, despite 38–70% of the respiratory ciliated cells of female heterozygous pathogenic DNAAF6 variant carriers having immotile cilia, these individuals showed no or only mild respiratory symptoms. We conclude that it is not necessary to have 100% of respiratory multiciliated cells functioning to ensure effective ciliary clearance. A range of 30–62% of multiciliated cells functioning is sufficient to provide either normal or only a subtle reduction in in vitro and in vivo ciliary clearance. Thus, precision medicine approaches probably do not have to completely correct all multiciliated airway cells to improve ciliary clearance in PCD caused by pathogenic DNAAF6 variants.
Shareable PDF
Acknowledgement
We thank the PCD-affected individuals and their families for their participation. We thank A. Borgscheiper, S. Helms, M. Herting, L. Schwiddessen, F.J. Seesing, S. Sivalingam and M. Tekaat (Department of General Pediatrics, University Children’s Hospital Muenster, Muenster, Germany) for excellent technical work. Several authors of this manuscript are members of the European Reference Network for Rare Respiratory Diseases (ERN-LUNG).
Footnotes
Ethics approval: Signed and informed consent was obtained from individuals fulfilling the diagnostic criteria of PCD and from family members according to protocols approved by the institutional ethics review boards of the University Hospitals of Muenster and Copenhagen.
Conflict of interest: The authors have no potential conflicts of interest to declare.
This article has an editorial commentary: https://doi.org/10.1183/13993003.01573-2024
Support statement: This work was supported by the Deutsche Forschungsgemeinschaft (DFG OM6/7, OM6/8, OM6/10, OM6/14 and CRU 326 (subproject OM6/11) (H. Omran), OL450/1 (H. Olbrich)), and by the IZKF Muenster (to H. Omran: Om2/009/12, Om/015/16 and Om2/010/20, Om2/014/24). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. Funding information for this article has been deposited with the Crossref Funder Registry.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-01441-2023.Supplement (2.5MB, pdf)
Supplementary video 1. Analysis of ciliary beating pattern of respiratory cilia from OP-1796 II1 by high-speed videomicroscopy. ERJ-01441-2023.Video_1 (29.4MB, avi)
Supplementary video 2. Analysis of ciliary beating pattern of respiratory cilia from OP-3141 II1 by high-speed videomicroscopy. ERJ-01441-2023.Video_2 (29.4MB, avi)
Supplementary video 3. Analysis of ciliary beating pattern of respiratory cilia from OP-1796 I2 by high-speed videomicroscopy. ERJ-01441-2023.Video_3 (29.4MB, avi)
Supplementary video 4. Analysis of ciliary beating pattern of respiratory cilia from OP-1796 I3 by high-speed videomicroscopy. ERJ-01441-2023.Video_4 (29.4MB, avi)
Supplementary video 5. Analysis of ciliary beating pattern of respiratory cilia from OP-1826 I2 by high-speed videomicroscopy. ERJ-01441-2023.Video_5 (58.8MB, avi)
Supplementary video 6. Analysis of ciliary beating pattern of respiratory cilia from OP-2835 II2 by high-speed videomicroscopy. ERJ-01441-2023.Video_6 (29.4MB, avi)
Supplementary video 7. Analysis of ciliary beating pattern of respiratory cilia from OP-3577 I2 by high-speed videomicroscopy. ERJ-01441-2023.Video_7 (29.4MB, avi)
Supplementary video 8. Analysis of ciliary beating pattern of control respiratory cilia by high-speed videomicroscopy. ERJ-01441-2023.Video_8 (29.4MB, avi)
Supplementary video 9. Pulmonary radioaerosol mucociliary clearance measurements for in vivo radiolabelled tracer in OP-1796 I2. ERJ-01441-2023.Video_9 (2.1MB, avi)
Supplementary video 10. Pulmonary radioaerosol mucociliary clearance measurements for in vivo radiolabelled tracer in OP-1796 II1. ERJ-01441-2023.Video_10 (2.3MB, avi)
References
- 1.Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol 2007; 8: 880–893. doi: 10.1038/nrm2278 [DOI] [PubMed] [Google Scholar]
- 2.Wallmeier J, Nielsen KG, Kuehni CE,et al. Motile ciliopathies. Nat Rev Dis Primers 2020; 6: 77. doi: 10.1038/s41572-020-0209-6 [DOI] [PubMed] [Google Scholar]
- 3.Nielsen KG, Holgersen MG, Crowley S, et al. Chronic airway disease in primary ciliary dyskinesia-spiced with geno-phenotype associations. Am J Med Genet C Semin Med Genet 2022; 190: 20–35. doi: 10.1002/ajmg.c.31967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Desai PB, Dean AB, Mitchell DR. Cytoplasmic preassembly and trafficking of axonemal dyneins. In: King SM, ed. Dyneins: Structure, Biology and Disease (Second Edition). London, Academic Press, 2018; pp. 140–161. [Google Scholar]
- 5.Braschi B, Omran H, Witman GB, et al. Consensus nomenclature for dyneins and associated assembly factors. J Cell Biol 2022; 221: e202109014. doi: 10.1083/jcb.202109014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paff T, Loges NT, Aprea I,et al. Mutations in PIH1D3 cause X-linked primary ciliary dyskinesia with outer and inner dynein arm defects. Am J Hum Genet 2017; 100: 160–168. doi: 10.1016/j.ajhg.2016.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olcese C, Patel MP, Shoemark A, et al. X-linked primary ciliary dyskinesia due to mutations in the cytoplasmic axonemal dynein assembly factor PIH1D3. Nat Commun 2017; 8: 14279. doi: 10.1038/ncomms14279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aprea I, Raidt J, Höben IM, et al. Defects in the cytoplasmic assembly of axonemal dynein arms cause morphological abnormalities and dysmotility in sperm cells leading to male infertility. PLoS Genet 2021; 17: e1009306. doi: 10.1371/journal.pgen.1009306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kobbernagel HE, Buchvald FF, Haarman EG, et al. Efficacy and safety of azithromycin maintenance therapy in primary ciliary dyskinesia (BESTCILIA): a multicentre, double-blind, randomised, placebo-controlled phase 3 trial. Lancet Respir Med 2020; 8: 493–505. doi: 10.1016/S2213-2600(20)30058-8 [DOI] [PubMed] [Google Scholar]
- 10.Paff T, Omran H, Nielsen KG,et al. Current and future treatments in primary ciliary dyskinesia. Int J Mol Sci 2021; 22: 9834. doi: 10.3390/ijms22189834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graves JA. Evolution of vertebrate sex chromosomes and dosage compensation. Nat Rev Genet 2016; 17: 33–46. doi: 10.1038/nrg.2015.2 [DOI] [PubMed] [Google Scholar]
- 12.Galupa R, Heard E. X-chromosome inactivation: a crossroads between chromosome architecture and gene regulation. Annu Rev Genet 2018; 52: 535–566. doi: 10.1146/annurev-genet-120116-024611 [DOI] [PubMed] [Google Scholar]
- 13.Shvetsova E, Sofronova A, Monajemi R, et al. Skewed X-inactivation is common in the general female population. Eur J Hum Genet 2019; 27: 455–465. doi: 10.1038/s41431-018-0291-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharp A, Robinson D, Jacobs P. Age- and tissue-specific variation of X chromosome inactivation ratios in normal women. Hum Genet 2000; 107: 343–349. doi: 10.1007/s004390000382 [DOI] [PubMed] [Google Scholar]
- 15.Giliberto F, Radic CP, Luce L, et al. Symptomatic female carriers of Duchenne muscular dystrophy (DMD): genetic and clinical characterization. J Neurol Sci 2014; 336: 36–41. doi: 10.1016/j.jns.2013.09.036 [DOI] [PubMed] [Google Scholar]
- 16.Schänzer A, Rau I, Kress W, et al. Duchenne muscular dystrophy in a 4-year-old girl due to heterozygous frame shift deletion of the dystrophin gene and skewed X-inactivation. Klin Padiatr 2012; 224: 256–258. doi: 10.1055/s-0032-1304626 [DOI] [PubMed] [Google Scholar]
- 17.Carsana A, Frisso G, Intrieri M, et al. A 15-year molecular analysis of DMD/BMD: genetic features in a large cohort. Front Biosci (Elite Ed) 2010; 2: 547–558. [DOI] [PubMed] [Google Scholar]
- 18.Lucas JS, Barbato A, Collins SA,et al. European Respiratory Society guidelines for the diagnosis of primary ciliary dyskinesia. Eur Respir J 2017; 49: 1601090. doi: 10.1183/13993003.01090-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sisson JH, Stoner JA, Ammons B,et al. All-digital image capture and whole-field analysis of ciliary beat frequency. J Microsc 2003; 211: 103–111. doi: 10.1046/j.1365-2818.2003.01209.x [DOI] [PubMed] [Google Scholar]
- 21.Mortensen J, Lange P, Nyboe J,et al. Lung mucociliary clearance. Eur J Nucl Med 1994; 21: 953–961. doi: 10.1007/BF00238119 [DOI] [PubMed] [Google Scholar]
- 22.Marthin JK, Holgersen MG, Nielsen KG,et al. Pulmonary radioaerosol mucociliary clearance assessment: searching for genotype-specific differences and potential as an outcome measure in primary ciliary dyskinesia. ERJ Open Res 2023; 9: 00685-2023. doi: 10.1183/23120541.00685-2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gileles-Hillel A, Mor-Shaked H, Shoseyov D,et al. Whole-exome sequencing accuracy in the diagnosis of primary ciliary dyskinesia. ERJ Open Res 2020; 6: 00213-2020. doi: 10.1183/23120541.00213-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harrison MJ, Shapiro AJ, Kennedy MP. Congenital heart disease and primary ciliary dyskinesia. Paediatr Respir Rev 2016; 18: 25–32. [DOI] [PubMed] [Google Scholar]
- 25.Lyon MF. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 1961; 190: 372–373. doi: 10.1038/190372a0 [DOI] [PubMed] [Google Scholar]
- 26.Shoemark A, Boon M, Brochhausen C,et al. International consensus guideline for reporting transmission electron microscopy results in the diagnosis of primary ciliary dyskinesia (BEAT PCD TEM Criteria). Eur Respir J 2020; 55: 1900725. doi: 10.1183/13993003.00725-2019 [DOI] [PubMed] [Google Scholar]
- 27.Raidt J, Werner C, Menchen T,et al. Ciliary function and motor protein composition of human fallopian tubes. Hum Reprod 2015; 30: 2871–2880. doi: 10.1093/humrep/dev227 [DOI] [PubMed] [Google Scholar]
- 28.Vanaken GJ, Bassinet L, Boon M, et al. Infertility in an adult cohort with primary ciliary dyskinesia: phenotype–gene association. Eur Respir J 2017; 50: 1700314. doi: 10.1183/13993003.00314-2017 [DOI] [PubMed] [Google Scholar]
- 29.Christensen K, Kristiansen M, Hagen-Larsen H,et al. X-linked genetic factors regulate hematopoietic stem-cell kinetics in females. Blood 2000; 95: 2449–2451. doi: 10.1182/blood.V95.7.2449 [DOI] [PubMed] [Google Scholar]
- 30.Mengel-From J, Lindahl-Jacobsen R, Nygaard M,et al. Skewness of X-chromosome inactivation increases with age and varies across birth cohorts in elderly Danish women. Sci Rep 2021; 11: 4326. doi: 10.1038/s41598-021-83702-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thomas L, Cuisset L, Papon JF, et al. Skewed X-chromosome inactivation drives the proportion of DNAAF6-defective airway motile cilia and variable expressivity in primary ciliary dyskinesia. J Med Genet 2024; 61: 595–604. doi: 10.1136/jmg-2023-109700 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This one-page PDF can be shared freely online.
Shareable PDF ERJ-01441-2023.Shareable (1,006.7KB, pdf)
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-01441-2023.Supplement (2.5MB, pdf)
Supplementary video 1. Analysis of ciliary beating pattern of respiratory cilia from OP-1796 II1 by high-speed videomicroscopy. ERJ-01441-2023.Video_1 (29.4MB, avi)
Supplementary video 2. Analysis of ciliary beating pattern of respiratory cilia from OP-3141 II1 by high-speed videomicroscopy. ERJ-01441-2023.Video_2 (29.4MB, avi)
Supplementary video 3. Analysis of ciliary beating pattern of respiratory cilia from OP-1796 I2 by high-speed videomicroscopy. ERJ-01441-2023.Video_3 (29.4MB, avi)
Supplementary video 4. Analysis of ciliary beating pattern of respiratory cilia from OP-1796 I3 by high-speed videomicroscopy. ERJ-01441-2023.Video_4 (29.4MB, avi)
Supplementary video 5. Analysis of ciliary beating pattern of respiratory cilia from OP-1826 I2 by high-speed videomicroscopy. ERJ-01441-2023.Video_5 (58.8MB, avi)
Supplementary video 6. Analysis of ciliary beating pattern of respiratory cilia from OP-2835 II2 by high-speed videomicroscopy. ERJ-01441-2023.Video_6 (29.4MB, avi)
Supplementary video 7. Analysis of ciliary beating pattern of respiratory cilia from OP-3577 I2 by high-speed videomicroscopy. ERJ-01441-2023.Video_7 (29.4MB, avi)
Supplementary video 8. Analysis of ciliary beating pattern of control respiratory cilia by high-speed videomicroscopy. ERJ-01441-2023.Video_8 (29.4MB, avi)
Supplementary video 9. Pulmonary radioaerosol mucociliary clearance measurements for in vivo radiolabelled tracer in OP-1796 I2. ERJ-01441-2023.Video_9 (2.1MB, avi)
Supplementary video 10. Pulmonary radioaerosol mucociliary clearance measurements for in vivo radiolabelled tracer in OP-1796 II1. ERJ-01441-2023.Video_10 (2.3MB, avi)