

Summary
Atopic dermatitis (AD) is a chronic itchy inflammatory disease of the skin. Genetic studies have identified multiple risk factors linked to the disease; however, most of the studies have been derived from European and East Asian populations. The admixed African American (AA) genome may provide an opportunity to discovery ancestry-specific loci involved in AD susceptibility. Herein, we present joint analysis of ancestry and genotype effects followed by validation using differential gene expression analysis on AD using 726 AD-affected individuals and 999 non-AD control individuals from the AA population, genotyped using Multi-Ethnic Global Array (MEGA) followed by imputation using the Consortium on Asthma among African Ancestry Populations in the Americas (CAAPA) reference panel. The joint analysis identified two novel AD-susceptibility loci, rs2195989 in gene ANGPT1 (8q23.1) and rs62538818 in the intergenic region between genes LURAP1L and MPDZ (9p23). Admixture mapping (AM) results showed potential genomic inflation, and we implemented genomic control and identified five ancestry-of-origin loci with European ancestry effects. The multi-omics functional prioritization of variants in AM signals prioritized the loci SLAIN2, RNF39, and FOXA2. Genome-wide association study (GWAS) identified variants significantly associated with AD in the AA population, including SGK1 (rs113357522, odds ratio [OR] = 2.81), EFR3A (rs16904552, OR = 1.725), and MMP14 (rs911912, OR = 1.791). GWAS variants were common in the AA but rare in the European population, which suggests an African-ancestry-specific risk of AD. Four genes (ANGPT1, LURAP1L, EFR3A, and SGK1) were further validated using qPCR from AD and healthy skin. This study highlighted the importance of genetic studies on admixed populations, as well as local ancestry and genotype-ancestry joint effects to identify risk loci for AD.
Keywords: atopic dermatitis, African American, admixed population, local and global ancestry, admixture mapping, genome-wide association, joint analysis, fine mapping
Graphical abstract
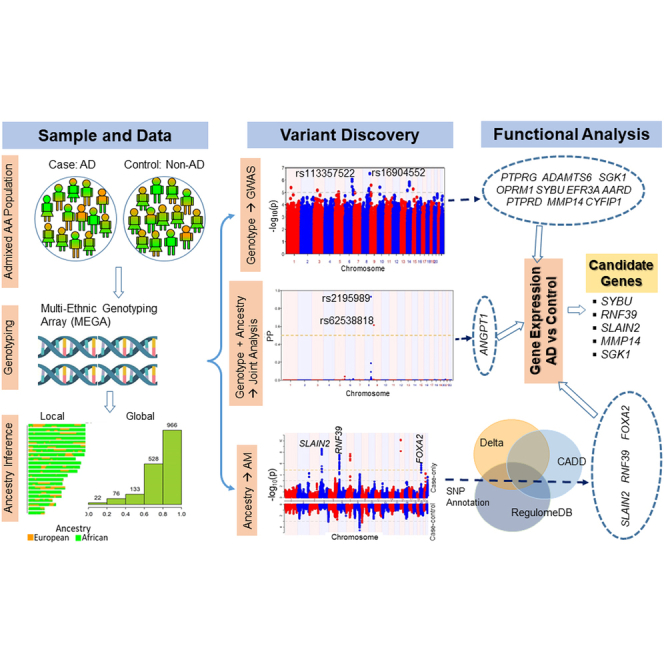
The study leverages the genetics of admixed African American samples with atopic dermatitis to identify disease risk loci. The results highlight the importance of admixed populations for testing ancestry via admixture mapping, genotype via association mapping, and joint admixture and association testing to identify ancestry-specific and shared risk loci.
Introduction
Atopic dermatitis (AD) is a chronic itchy inflammatory skin disease characterized by clinical symptoms including rash, severe itchiness, oozing and crusting, and skin pain. Physiologically, it is a multifactorial disease involving defective skin barrier function, immune dysregulation, environmental exposures, and viral infections.1 It is one of the most common skin diseases affecting children and adversely impacts the quality of life of affected individuals and their families.2 AD often starts during infancy and progresses into other atopic diseases, including food allergy, asthma, and allergic rhinitis, a phenomenon known as the atopic march.3 The prevalence and clinical presentation of AD varies worldwide, ranging from 0.2% to 24.6% depending on geographic location and ethnicity.4 The single most predictive factor of AD development is a positive family history of AD or allergic diseases. Parental history of AD is associated with 3- to 5-fold increase in the risk of AD among offspring.5 Twin studies showed up to 86% concordance rate in monozygotic twins and approximately 22% concordance in dizygotic twins.6,7 The heritability of AD was estimated to be ∼75%, underscoring the genetic basis of AD.8,9
Several well-powered genome-wide association studies (GWASs) have identified and replicated genetic-susceptibility variants associated with AD. The genetic variations in the epidermal differentiation complex on chromosome 1q21.3 that encodes the filaggrin (FLG [MIM: 134950]) gene were reported by GWAS and candidate gene approaches to be associated with AD. Several loss-of-function mutations in the FLG gene associated with the defects in the skin barrier functions are the most studied known genetic risk factors for AD.10 The defective skin barriers lead to increased transepidermal water loss and skin dehydration, and increased penetration of allergens and infectious microbes into the skin, which trigger the AD symptoms.11 Recently, a genetic variant in SPRR2B (MIM: 182268) was found to be associated with AD risk and the risk showed an increase with the presence of the loss-of-function FLG mutation.12 Other well-established GWAS associations are CD207 (MIM: 604862) on chromosome 2p13.3, the cytokine cluster on chr5q31.1 including IL13 (MIM: 147683) and IL4 (MIM: 147780), and the MHC region on 6p21.3. GWAS meta-analysis on African American (AA) participants identified a single locus near RORC (MIM: 602943) in chromosome 1.13 Candidate gene analysis showed variants in TSLP (MIM: 607003) and IL7R (MIM: 146661) were associated with AD in European and AA participants.14 Immunochip analyses have also identified intergenic regions on chromosome 4q27 and chromosome 16p13.13, and ZNF652 (MIM: 613907) on 17q21.32 as risk loci associated with AD.15 Grosche et al. identified multiple exonic variants in DUSP1 (MIM: 600714), NOTCH4 (MIM: 164951), and SLC9A4 (MIM: 600531) associated with AD.16 Several large-scale GWASs and meta-analyses have identified shared genetic risk among AD, asthma, and hay fever.17,18,19 Mucha et al. identified several rare protein-coding variations in genes IL4R (MIM: 147781), IL13 and JAK1 (MIM: 147795), JAK2 (MIM: 147796), and TYK2 (MIM: 176941) that were associated with AD.20 Genomic findings to date account for ∼30% of AD heritability21 and much more remains to be discovered.
Many epidemiological studies have found racial and ethnic variations in AD prevalence. In the US, data from the 2003 National Survey of Children’s Health involving more than 102,000 families demonstrated an overall prevalence of AD of 10.7% of children and that AD is more prevalent (15.9% vs. 9.7%) in AA children than European American (EA) children.22 After adjusting for potential confounders/covariates, African ancestry was still significantly associated with a higher prevalence of AD compared to European ancestry (odds ratio [OR], 1.70, p = 0.005).22 A similarly disparity in prevalence of AD was found between Black Caribbean children (16.3%) vs. White children (8.7%) was observed in a UK study based on dermatologist examination.23 A clinic-based study in south-east London found AD to be the most frequently diagnosed dermatologic condition in Black children, affecting 36.5% of those seen.24 A multi-country cross-sectional study also found high prevalence, up to 23%, of AD among several African countries.25 Studies also demonstrated that children of African descendants are three times more likely than White children to attend a dermatology visit during which AD is diagnosed, even though they are significantly less likely to seek dermatological care.26 A recent study on two large AA cohorts showed that self-identified race of AA was associated with 2-fold increase in AD prevalence; however, global African ancestry as determined by the overall proportion of genome from the African population was not associated with AD.27 In addition to epidemiologic data, results from immuno-histochemistry and transcriptomic studies revealed that AD in the AA population manifests as a distinct clinical subtype demarcated by significantly higher allergen-specific serum immunoglobulin (Ig) E and attenuated T helper (Th) 1 and Th17 molecular signatures, which discriminates it from EA and Asian molecular AD subtypes.28,29 Lesional skin samples from AA patients can be characterized by decreased expression of innate immune, Th1-related, and Th17-related markers but increased dendritic cell infiltration compared to EA and Asian patient samples.28 Taken together, multiple lines of evidence from epidemiologic, transcriptomic, and immunologic studies indicate that African ancestry is significantly associated with increase in prevalence of AD in the AA population who carry genetic contributions from parental genomes coming from African and European lineages. Genomic analysis could therefore unravel the molecular basis underlying the discordant prevalence and molecular subtype of AD in AA and other racial groups leading to improved personalized care for AD.
The major drawback in the genetics studies of AD so far is that the AD-associated genetic variations are based on studies with participants of European and East Asian populations and there is a lack of representation of other ancestral populations, such as African, Native Americans, and recently admixed populations such as AA and Latinos. A 2015 GWAS used samples from multi-ancestry populations, but only ∼2% of cases and <1% of control individuals were from AA ancestry.30 The genomes of admixed populations consist of mosaics of chromosomal segments from the founding populations. Even though GWAS can be used to unbiasedly detect disease-associated variants in admixed populations adjusted for genetic ancestry, it does not leverage the allele frequency differences across the founding populations. In admixed populations, the causal loci may be inherited in higher proportion from the founding population with higher disease prevalence. Admixture mapping (AM) leverages the admixture pattern in the admixed samples to detect abundance of the ancestry-of-origin loci associated with the disease. AM is more powerful for detecting the association when there are differences in disease prevalence and allele frequency across the founding populations.31 Because of methodological issues, most researchers avoid mixed race or minorities in their analysis. The lack of racial or ethnic diversity in AD genetic research limits the discovery of ancestry-specific variants and potentially exaggerates the disparity in precision diagnosis and medical care for the under-represented groups. In this study, we analyzed 1,725 samples (726 AD-affected individuals and 999 non-AD control individuals) from the AA population genotyped using Illumina’s Multi-Ethnic Global Array (MEGA). We use AM, which is a complementary statistical approach to GWAS, to leverage local ancestry estimates in the admixed population. We also use GWAS to identify ancestry-shared variants associated with AD in the AA population. We use a joint GWAS-AM analysis to identify risk variants that otherwise would be missed when studied separately.32 To identify potential casual variants associated with AD, we perform fine mapping and functional prioritization.
Materials and methods
Study design and population
The study cohort consisted of 2,687 participants selected from two local cohorts, the Greater Cincinnati Pediatric Clinical Repository (GCPCR)33 and the Cincinnati Genomic Control Cohort (GCC),34 at the Cincinnati Children’s Hospital Medical Center (CCHMC). The majority of the samples were from the greater Cincinnati metro area and recruited through the main campus as well as through the satellite locations. Clinical history including AD, skin conditions, asthma, allergy, and demographic information was ascertained through hospital visits, emergency visits, and inpatient visits. Cases were selected based on the reported medical history of AD. Control individuals consisted of subjects with a history of asthma, food allergy, and allergic rhinitis but with no history of AD or eczema. Parental informed consent was obtained for all participants under 18 years of age in the study for the purpose of DNA collection and genotyping, and from patients aged 18 and older. The study was approved by the institutional review boards at CCHMC.
Genotyping
Genotyping was performed using MEGA, which contains SNP sets tailored toward admixed ancestry.35 MEGA maximizes coverage and captures the genomic architecture of the AA population. Genotypes were called using the Genetrain2 algorithm in Illumina Genome Studio software following the Illumina’s top-bottom strand orientation. Genotype data were available for over 1.43 million variants aligned with the genome assembly GRCh37.
Quality control
Quality control (QC) analysis was performed in two stages: prior to imputation and post imputation. During the first stage, participants with sub-optimal genotype missing rate of ≥10% were removed. Also, duplicates or monozygotic twins were determined using identity by descent (IBD) score >0.90 and only the sample with lowest missing rate was retained. Sex checks were performed and samples with discrepancy between predicted and reported sex were also removed. Out of the 2,687 samples with genotype data available, 2,382 samples passed the filtering criteria. In the next stage, SNPs were filtered for the sub-optimal missing of ≥5%, and significant deviation from Hardy-Weinberg Equilibrium (HWE) in controls at p < 1E−5. Genetically related samples were determined using IBD statistics from PLINK 2 (https://www.cog-genomics.org/plink/2.0/). IBD analysis was conducted using a set of LD-pruned SNPs and IBD score ≥ 0.25 was used as a cutoff for filtering potential duplicate and cryptic related samples. IBD score ≥ 0.25 potentially removes first- and second-degree relations.36 Among the related samples, the sample with the lowest missing rate was selected, and others were removed from the analysis. With the IBD filtering criteria, 105 related samples were identified and further removed from the analysis. Principal-component analysis (PCA) was performed, and the top five PCs were extracted. For both IBD and PCA analyses, variants were pruned for linkage disequilibrium (LD) and only variants with LD < 0.2 were used. All QC analyses were conducted using PLINK 2.37
Genotype imputation
Imputation was carried out across the autosomal chromosomes using the Michigan Imputation Server, which implemented the minimac4 algorithm.38 Strand-aligned genotype data were loaded into the server. Imputation was conducted using the Consortium on Asthma among African Ancestry Populations in the Americas (CAAPA) reference panel.39 The output files with imputation dosage were converted to plink binary files using the “make-bed” option, resulting in the hard-call binary plink files that were subsequently used for the association analysis. All bi-allelic variants with imputation quality threshold of Rsq score ≥0.3 were reported.
Local ancestry estimates
The chromosomes of admixed participants consist of a mosaic of chromosomal blocks from the ancestral populations, which are called local ancestry blocks. Since the true ancestries are unknown, ancestry at each locus or block could be inferred computationally based on appropriate reference ancestral populations. Local ancestry for the participants was inferred by modeling AA samples as a two-way admixture between African and European populations that occurred approximately eight generations prior.40 Inference was carried out using RFMix v2, which uses a supervised conditional random forest method to optimally infer the ancestries of the alleles at a marker locus.41 The CEU (n = 99) and YRI (n = 108) panels from the 1000 Genomes Project III were used as the reference populations for the European and African ancestry. The sample genotype data were checked for strand alignment using coform-gt (https://faculty.washington.edu/browning/conform-gt.html). After removing the SNPs that did not conform to the alignment of the reference panel, samples were phased using Beagle with the African and European populations as the reference panels.42 The genetic map files were downloaded from the Beagle site (http://bochet.gcc.biostat.washington.edu/beagle/genetic_maps/). The number of generations since admixture was set to be eight and inference was carried out for each autosomal chromosome under the Expectation Maximization (EM) option with five iterations. Inference was carried out for each autosomal chromosome.
Global ancestry estimates
Global ancestry, the proportions of genomic contribution from the ancestral populations in the entire genome of an admixed sample, was conducted using RFMix v2 with CEU and YRI as the reference populations for European and African ancestry; the genomic proportions of European and African ancestries were estimated for each autosomal chromosome.41 Global ancestry proportion for each sample was estimated using the weighted sum of the chromosomal ancestry estimates where weights were proportional to the size of the chromosomes. Samples with global African-ancestry proportion <0.02 were removed from further analysis.
AM
To identify the association between AD and African ancestry, AM was performed using case-only and case-control analyses.43 Estimation of local and global ancestry were carried out as described before. In the case-only analysis, each sample’s local ancestry at a marker locus was compared to the respective global ancestry. In case-control analysis, deviations in local ancestries between the AD and control individuals were tested. The case-only and case-control AM approaches are described below. Superscript "c” and “d” are used to represent cases and controls, respectively, unless otherwise defined.
Let and be the proportion of African ancestry at a marker locus l and and be the global ancestry of i-th case and j-th control, respectively. Let and be the number of cases and controls.
Case only: define as average local ancestry at marker locus l and as average global ancestry among all cases. The test statistics for case only is defined as , where is the standard error.
Case-control: define as average local ancestry at marker locus l and as average global ancestry among all control individuals. The test statistics for case-control analysis is defined as , where is the standard error.
For large , both and are approximated with the standard normal distribution.
Post hoc power analysis
The statistical power of the study was calculated using the Power Analysis in Multi-ancestry Admixture Mapping (PAMAM) web tool.44 First, we estimated the testing burden using the R package STEAM.45 We followed the analytical approximation with # of generations since admixture, g = 8, and the map file thinned to have a variant selected for each 0.2 cM chunk. We selected these parameters to match the default setting used for the local ancestry estimation in the RFMix V2. The analysis resulted in a genome-wide significant p value threshold of 1.47E−5. Next, we used the PAMAM for the power estimation, with sample size of 726 cases and 999 controls, a significance threshold of p < 1.47E−5, and the known African ancestry proportion of 0.78. Accordingly, this study achieved >80% power to detect an ancestral OR >1.7.
Genomic inflation factor adjustment
Both AM and GWAS results were evaluated for the potential genomic inflation factors. Genomic control (GC) test statistics was used to correct the genomic inflation.46 Briefly, the test statistics were adjusted using the GC factor λ estimated as the ratio of median of the test statistic to the median of with 1 degree of freedom. Finally, the adjusted test statistics were used to compute the inflation-adjusted p value.
Fine-mapping analysis and SNP prioritization
A functional-mapping study of the significant AM region was conducted. All the SNP variants mapped to the significant AM regions with minor-allele frequency (MAF) >0.01 and imputation quality score (Rsq) >0.3 were accessed from the genome-wide imputed data. SNPs were tested for the association with AD using logistic regression test adjusted for age, sex, PCs, and the global ancestry. For each AM locus, the SNP with strongest association was reported. For each AM locus, the significance threshold used was p < 0.05/n, where n was determined after LD-pruning with LD cutoff of r2 = 0.3.
To prioritize potential functional variants in the AM loci, SNPs were annotated using multi-omics functional annotation tools. First, the allele frequency difference , with allele frequency of the African () and European () were accessed from the 1000 Genomes project. Functional annotation of the variants with ≥ 0.25 was performed with CADD47 and RegulomeDB.48 The Variant Effect Predictor (VEP) from Ensembl (http://grch37.ensembl.org/info/docs/tools/vep/index.html) was used for the variant annotation including the CADD score and allele frequencies, whereas RegulomeDB 2.0.3 (https://regulomedb.org/regulome-search) was used for the RegulomeDB score. Overlap of variants with CADD score ≥14 and RegulomeDB score ≤3 are considered as the top prioritized variants associated to AD.
Genome-wide association analysis
Genome-wide association analysis was performed under the logistic regression framework, between the binary AD phenotype (Y) and the genotype (X) adjusted for the covariates (W), which includes, sex, age, and top five PCs as , where 's are the regression coefficients, and is the normally distributed error terms. GWAS was performed using PLINK 2.37 By default, PLINK 2 performs logistic regression analysis with “firth-fallout” option, which allows firth regression if the logistic regression fails to converge. Variants were filtered for MAF < 0.01, missing rate >0.05, and HWE < 1E−5 in controls. GWAS significance threshold of 5E−8 was used to assess the significant association of genotype with AD.
Replication of known AD GWAS signals
Variants associated with AD in prior GWASs were accessed from the NHGRI-EBI GWAS Catalog.49 With p < 1E−5, 258 variants associated with AD were identified. Using LDlinkR,50 LD variants within ±100 kb distance with r2 ≥ 0.3 in the AFR population from the 1000 Genomes Project III were extracted and overlapped with the GWAS results. For each catalog SNP, the GWAS SNP with strongest p value was identified. Catalog SNPs were pruned when multiple variants were tagged with the same GWAS SNP. The pruning resulted in 173 AD-associated loci with at least one GWAS SNP within ±100 kb and r2 ≥ 0.3. Replication significance was assessed at p < 0.05/173 = 2.9E−4.
Joint association and admixture analysis
Association-admixture joint analysis has potential to capture the complementary signals from the genotype-based association mapping and ancestry-based admixture mapping and hence detect signals that are only marginally significant in individual analysis alone. We performed the association-ancestry joint analysis on the childhood AA samples using the Bayesian mixed SNP and admixture (BMIX) testing approach.32 The BMIX approach combines genotype and ancestry information. BMIX uses the posterior probabilities from the AM as a prior for the association and computes the posterior probability (PP) for the genotype test. The test relies on the user-specified information on the multiple testing burden of admixture and association mapping. Following the analytical approaches of BMIX, we estimated n = 1240.5 for the AM. For the association mapping, we used the n = 345450.3 as the number of testing burden as suggested in the BMIX. The BMIX analysis was conducted for the genotype markers after the QC filtering- MAF < 0.05, HWE p ≤ 1E−5 in control and ≤ 1E−8 in case, and genotype missing rate of ≥0.05. SNPs with posterior probability of joint BMIX test greater than 0.5 are considered significant.
Gene expression analysis
Validation of the risk loci were performed using the publicly available gene expression data from Gene expression Omnibus (GEO: GSE32924).51 Data consisted of gene expression profiles from skin biopsy samples of lesional and non-lesional skins from 12 AD-affected individuals and eight control individuals. The expression was accessed using the Affymetrix human U133Aplus2 arrays (Affymetrix, Santa Clara, CA). The AD samples consisted of moderate-to-severe patients aged 16–81 years. Details on sample preparation and QCs are described elsewhere.51 Differential gene expression analyses comparing lesional vs. control and non-lesional vs. controls were performed with NetworkAnalyst 3.0 using the limma algorithm.52 Differential gene expression results for the targeted prioritized genes were assessed for significance at false discovery rate (FDR)-adjusted p (q value) <0.05 and an absolute fold change ≥1.5.
qPCR analysis
Validation of the GWAS genes was conducted using real-time qPCR technique. Target genes for the validation were identified using GWAS and the joint ancestry-genotype analysis. Total blood collected from the Mechanisms of Progression of Atopic Dermatitis to Asthma in Children (MPAACH) participants that were diagnosed with AD (n = 6) and from control individuals (n = 3) was used to extract RNA. The MPAACH study was approved by the CCHMC Institutional Review Board (IRB) under protocol number 2016-5842, informed consent was provided by all subjects, and 800 μL of total blood was used for RNA extraction using the RiboPure RNA Blood Extraction Protocol (catalog no. AM1928, Thermo Fisher Scientific, Waltham, MA).
Extracted RNA was checked for quality and quantity using Thermo Scientific Nanodrop OneC Microvolume UV-Vis Spectrophotometer (Thermo Fisher Scientific) and Agilent TapeStation 4150 System (Agilent Technologies, Santa Clara, CA), respectively. RNA integrity number (RIN) values for the RNA samples were 8.00–9.00. A higher RIN value indicates a higher degree of integrity. Then 250 ng of total RNA was reverse transcribed using SuperScript IV VILO Master Mix (catalog no. 11756500, Thermo Fisher Scientific). For qPCR analysis, the resulting cDNA was run on the QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific) using TaqMan gene expression assays and TaqMan Fast Advanced Master Mix (Thermo Fisher Scientific) per manufacturer’s instructions. Comparative critical cycle (Ct) values for analysis were obtained from Applied Biosystems Analysis Software and processed in the Thermo Fisher Connect Software (Thermo Fisher Scientific). The fold change expression levels of the target genes were normalized to the 18s and the relative abundance was calculated by the 2-ΔΔCT method.53
Experimental values are expressed as means ± SEM of three independent RNA samples derived from healthy control individual and six from AD patients. GraphPad Prism version 9.5 (Boston, MA) was used to determine the statistical differences between healthy control individuals and AD patients. Data analysis was performed using non-parametric Mann-Whitney tests. A p value < 0.05 was considered statistically significant when comparing the gene expression between healthy control individuals with AD patients.
Functional pathway and network analysis
To understand the biological role of the AR-associated genes, over-representation enrichment analysis for functional pathways was conducted using the ConsensusPathDB (http://cpdb.molgen.mpg.de/).54 AD-associated genes identified from the AM, GWAS, and joint analysis were used for the analysis. Significant pathways and gene ontology terms were assessed at FDR-adjusted p < 0.05.
Results
Demographic characteristics
In total 1,725 samples (726 AD-affected individuals and 999 non-AD control individuals) passed all QC criteria and global African-ancestry proportion ≥0.02 and were selected for the study. AD-affected individuals were significantly younger than the control individuals (average ages: AD-affected individuals, 8.04 years vs. control individuals, 9.81, p < 0.001). Females represented 46.46% of cases compared to 41.97% among control individuals, but the difference was not significant. There was no difference in the proportion of asthma status among AD-affected individuals and control individuals; however, allergic rhinitis (AR) and food allergy (FA) were observed in higher proportion among AD-affected individuals than control individuals (Table 1).
Table 1.
Demographic information
| Characteristics | Total (n = 1725) | Cases (n = 726) | Control (n = 999) | p value |
|---|---|---|---|---|
| Female, count (%) | 757 (43.88%) | 338 (46.56%) | 419 (41.94%) | 0.063 |
| Mean age in years (±SD) | 9.08 (5.77) | 8.01 (4.84) | 9.85 (6.25) | <0.001 |
| Mean African ancestry (±SD) | 0.757 (0.16) | 0.761 (0.16) | 0.753 (0.17) | 0.328 |
| Comorbidities: | ||||
| Asthma | 1216(70.5%) | 516 (71.07%) | 700 (70.07%) | 0.69 |
| AR | 863 (50%) | 398 (54.78%) | 465 (45.81%) | <0.01 |
| FA | 181 (10.5%) | 109 (15.35%) | 72 (7.09%) | <0.001 |
AR, allergic rhinitis; FA, food allergy.
The genome of AA individuals consisted of European and African ancestry with varied composition. Figure 1A shows the variation of the global African-ancestry proportion among AD-affected and control individuals in comparison to the reference individuals of CEU and YRI panels from the 1000 Genomes project. The overall distribution of the global African ancestry was skewed left with over 86% of the samples (1,494/1,725 = 86.6%) having greater than 60% of their genomes derived from the African ancestry (Figure 1B). The average global African ancestry of the whole cohort was 0.757. The global African ancestry among cases was marginally higher than that for the control individuals, but the difference was not statistically significant (p = 0.328; Table 1). The first PC (PC1) explained most of the global African-ancestry variation with correlation coefficient (r) = 0.997 (Figure 1C).
Figure 1.

Ancestry estimation
(A) Distribution of global ancestry for two reference populations (CEU and YRI) and samples in case and control cohorts (orange, European ancestry; green, African ancestry).
(B) Histogram showing the distribution of African ancestry in sample.
(C) Correlation between African ancestry and PC1.
AM
To identify the ancestry-specific loci associated with AD in AA, we performed AM using case-only and case-control approaches. The Miami plot showed the results from both analysis (Figure S1). The quantile-quantile (QQ) plot showed both AM results were inflated with genomic inflation factors of 1.22 and 1.56 for case-control and case-only analysis, respectively (Figure S2). Both AM results were adjusted for the genomic inflation factor as described in the “materials and methods” section. Prior to the adjustment of genomic inflations, case-only analysis identified 10 significant loci, whereas case-control analysis identified one significant locus with p <1.47E−5 (Figure S1). After the adjustment, five of the 10 AM loci remained significant (Figure 2; Table 2A). The top AM peak was observed at chromosome 15q11.2 (case-only adjusted p = 6.24E−10). The locus is a gene-rich region including multiple genes from the Immunoglobulin Heavy Diversity (IGHD) family and POTE Ankyrin Domain family. All five significant loci were enriched for European ancestry as suggested by the negative Z score. AM locus at chromosome 7q11.23 was also marginally significant at p < 0.05 in case-control analysis. No locus reached the significant p value threshold in case-control analysis, with the strongest association observed at locus 8q23.1–2 (case-control adjusted p = 2.75E−5; Table 2A). The locus 8q23.1-2 spanned about a 1.3-Mb region and was enriched for the European ancestry. Two additional loci, 4q34.3 and 9q23, reached suggestive significance level of adjusted p < 0.001 in case-control analysis (Table 2B). The locus 4q34.3 was enriched for African ancestry. All three loci reached marginal association of adjusted p < 0.05 in case-only analysis. AM analyses identified potential AD loci with European ancestry effect. However, we found high inflation factor and further research would be required to validate the findings.
Figure 2.

Miami plot of AM result
The upper section shows results from case-only analysis. The lower section shows results from case-control analysis. The x axis represents chromosome position. The y axis represents p value. Orange horizontal line represent genome-wide significance at p < 1.47E−5 with adjustment for genomic inflation. Gray dashed horizontal lines represent suggestive association at p-value < 1E-3.
Table 2.
AM loci associated with AD in AA samples
| Locus | START | END | Z_cc | P_cc | Z_c | P_c | P_c_adj | P_cc_adj |
|---|---|---|---|---|---|---|---|---|
| A. Case-only AM results | ||||||||
| 4p12-q12 | 48226267 | 49003382 | −0.88 | 0.38 | −7.44 | 9.77E−14 | 2.40E−09 | 0.43 |
| 6p22.1-21.33 | 29696849 | 31319586 | −0.06 | 0.95 | −6.93 | 4.33E−12 | 2.81E−08 | 0.96 |
| 7q11.23 | 76910730 | 77044434 | −2.73 | 6.32E−3 | −6.99 | 2.66E−12 | 2.05E−08 | 0.014 |
| 15q11.2 | 20083584 | 22560396 | 1.54 | 0.123 | −8.154 | 3.51E−16 | 6.24E−11 | 0.163 |
| 20p11.21 | 22487052 | 22672462 | −0.138 | 0.89 | −6.119 | 9.42E−10 | 9.31E−07 | 0.91 |
| B. Case-control AM results | ||||||||
| 4q34.3 | 181489069 | 181576044 | 3.698 | 2.17E−04 | 2.64 | 8.36E−03 | 3.45E−02 | 8.24E−04 |
| 8q23.1–2 | 109871639 | 111194968 | −4.636 | 3.55E−06 | −4.958 | 7.12E−07 | 7.04E−05 | 2.75E−05 |
| 9p23 | 12925921 | 12942772 | −3.784 | 1.54E−04 | −3.24 | 1.19E−03 | 9.37E−04 | 6.21E−03 |
A. Significant AM loci in case-only analysis with the inflation-adjusted case-only p value (P_c_adj) <1.47E−5, the genome-wide significance threshold for AM. B. Results from case-control analysis with inflation-adjusted p value <0.001. START, starting base pair position;, END, ending base pair position; Z_cc, case-control test statistics; Z_c, case-only test statistics; P_cc, case-control p value; P_cc_adj, inflation-adjusted case-control p value; P_c, case-only p value; P_c_adj, inflation-adjusted case-only p value.
Fine mapping of AM loci
SNP variants mapped to the AM loci were tested for the allelic association with AD. All SNPs that passed the imputation Rsq ≥ 0.3 and QC criteria (MAF ≥ 0.01, HWE ≥ 1E-5 in control, missing rate <0.05) were tested for allelic association using the logistic regression test, adjusted for age, sex, and top five PCs. Table 3A shows the strongest association within each AM locus. Multiple testing adjustment was performed for each locus independently using the number of LD-pruned SNPs tested in each locus. No significant association was observed at the five significant AM loci, but SNPs with marginal association at p < 0.01 were identified at each AM locus.
Table 3.
Fine mapping and variant prioritization of AM loci associated with AD in AA samples
| A. Fine mapping of AM loci | ||||||||
|---|---|---|---|---|---|---|---|---|
| SNP | CHR | POS | OR | P_assoc | P_thrs | P_cc_adj | P_c_adj | |
| rs74864956 | 4 | 48432376 | 0.557 | 3.92E−3 | 2.35E−5 | 0.43 | 2.40E−09 | |
| rs3130569 | 6 | 31102955 | 1.268 | 3.24E−3 | 3.24E−5 | 0.96 | 2.81E−08 | |
| rs148783322 | 7 | 76919259 | 1.474 | 2.33E−3 | 4.81E−4 | 0.014 | 2.05E−08 | |
| rs563250505 | 15 | 22357139 | 1.723 | 3.86E−3 | 8.47E−4 | 0.163 | 6.24E−11 | |
| rs79548864 | 20 | 22654011 | 1.463 | 7.56E−3 | 4.46E−4 | 0.91 | 9.31E−07 | |
| B. Prioritization of AM loci | ||||||||
| SNP | CHR | POS | AFR_AF | EUR_AF | DELTA | CADD | RDB | GENE |
| rs375259 | 4 | 48343550 | 0.056 | 0.3439 | 0.2879 | 16.65 | 2b | SLAIN2 |
| rs3807033 | 6 | 30043955 | 0.4425 | 0.0825 | 0.36 | 18.8 | 1f | RNF39 |
| rs77580993 | 6 | 30059592 | 0.5068 | 0.1889 | 0.3179 | 17.42 | 2b | – |
| rs7765810 | 6 | 30063496 | 0.5091 | 0.1779 | 0.3312 | 17.96 | 1a | – |
| rs2277764 | 20 | 22566124 | 0.351 | 0.0606 | 0.2904 | 17.77 | 2b | FOXA2 |
A. Fine mapping association test. CHR, chromosome; POS, base-pair position; OR, odds ratio; P_assoc, association p value; P_thrs, p value threshold for significant association after multiple testing adjustment; P_cc_adj, inflation-adjusted case-control p value; P_c_adj, inflation-adjusted case-only p value. B. Prioritized variants in AM loci. AFR_AF and EUR_AF, Minor allele frequency in African and European populations, respectively; RDB, RegulomeDB score; CADD, CADD score; DELTA, absolute difference in AFR_AF and EUR_AF.
Functional prioritization of variants in AM loci
To identify the putative genetic variants underlying the AM regions, we examined all variants mapped to each significant AM region using the 1000 Genomes Project. Variants were annotated and prioritized using CADD score, allele frequency difference (), and RegulomeDB score (as detailed in the “materials and methods” section). The approach identified five variants with the prioritization criteria CADD ≥ 14, ≥ 0.25, and RegulomeDB ≤ 3 (Table 3B). On locus 6p22.1-21.33, three variants were identified in the intergenic region upstream of gene RNF39 (MIM: 607524). Similarly, a 5′ UTR variant in gene SLAIN2 (MIM: 610492) and a regulatory variant in a promotor region near gene FOXA2 (MIM: 600288) were prioritized at chromosome 4 and chromosome 20, respectively. The multi-omics functional prioritization approach identified 3 AM loci associated with AD and highlights loci encoding the genes SLAIN2, RNF39, and FOXA2 as candidate genes associated with AD in AA.
Genome-wide association analysis
GWAS was performed on 6 million variants with MAF > 0.01 using logistic regression, adjusted for age, sex, and PC1–PC5 (PC1 is highly correlated with global ancestry). No locus reached genome-wide significance at p ≤ 5E−8 (Figure 3). At the suggestive significance level of 1E−6, the analysis identified two variants associated with AD in AA (Table 4). The strongest signals were observed at intronic variant rs16904552 (p = 2.94E−7, OR = 1.744, 95% confidence interval [CI] = (1.41, 2.157)) in the gene EFR3A (MIM: 611798) on chromosome 8. The second signal was observed at intronic variant rs113357522 (p = 8.61E−7, OR = 2.717, 95% CI = (1.825, 4.047)) in the gene SGK1 (MIM: 602958) on chromosome 6. GWAS results showed 11 additional loci with suggestive evidence of association at the p < 1E−5 (Table 4).
Figure 3.

AD GWAS results
(A) Manhattan plot of AD GWAS. Gray horizontal lines represent significance signals at p < 1E−5.
(B) QQ plot. λ = 1.013 is the genomic inflation factor.
Table 4.
GWAS results of AD in AA samples
| SNP | CHR | POS | A1 | OR | OR CI (95%) | p | GENE |
|---|---|---|---|---|---|---|---|
| rs111541801 | 1 | 63559620 | C | 1.858 | (1.428, 2.418) | 4.04E−06 | – |
| rs141216295 | 3 | 61773755 | T | 2.826 | (1.798, 4.441) | 6.67E−06 | PTPRG |
| rs11950228 | 5 | 64680454 | C | 0.62 | (0.502, 0.766) | 9.27E−06 | ADAMTS6 |
| rs113357522 | 6 | 134505244 | C | 2.717 | (1.825, 4.047) | 8.61E−07 | SGK1 |
| rs681243 | 6 | 154427741 | C | 1.376 | (1.198, 1.58) | 6.41E−06 | OPRM1 |
| rs77076745 | 8 | 110683128 | T | 0.631 | (0.516, 0.772) | 7.36E−06 | SYBU |
| rs6998067 | 8 | 117951521 | T | 1.422 | (1.223, 1.654) | 4.92E−06 | AARD |
| rs16904552 | 8 | 132942296 | G | 1.744 | (1.41, 2.157) | 2.94E−07 | EFR3A |
| rs16925550 | 9 | 10227095 | A | 0.408 | (0.281, 0.593) | 2.61E−06 | PTPRD |
| rs60796647 | 10 | 31099687 | G | 2.626 | (1.744, 3.953) | 3.78E−06 | – |
| rs911912 | 14 | 23314675 | C | 1.761 | (1.376, 2.253) | 6.76E−06 | MMP14 |
| rs177731 | 14 | 59379946 | T | 1.906 | (1.466, 2.479) | 1.45E−06 | – |
| rs59722874 | 15 | 22941055 | A | 0.431 | (0.3, 0.619) | 5.35E−6 | CYFIP1 |
A1, risk allele; CI, confidence interval .
Joint analysis of ancestry and genotype
A joint ancestry-genotype analysis was performed using the BMIX approach. The joint analysis accounts for association signals condition upon the local ancestry at each marker. The posterior probability at each genotyped marker is shown in Figure S3. The analysis identified two novel risk variants for AD susceptibility in AA (Figure 4; Table 5). The first significant association was observed at the intron variant rs2195989 in gene ANGPT1 (MIM: 601667) on chromosome 8 with the posterior probability (PP) = 0.932. The GWAS analysis showed the locus has moderate evidence of association with AD (p = 5.71E−4, OR = 1.314) and AM analysis showed the region is also moderately associated with AD with European ancestry as a risk factor (P_cc = 5.76E−5) (Figure 4). Gene ANGPT1 encodes angiopoietin-1, which plays roles in angiogenesis, inflammation, and tumor growth.55 Using the GTEx portal, the variant is found to be expression quntitative trait loci (eQTL) with the target gene ANGPT1 in muscle-skeletal tissue. The second joint association was found at rs62538818 (PP = 0.614) in the intergenic region between genes LURAP1L (MIM: 616130) and MPDZ (MIM: 603785). The locus overlaps with a promoter flanking region that is active in the dermal fibroblast, lung, and multiple other cell/tissue types. Dermal fibroblasts are part of skin connective tissue and play an essential role during cutaneous wound healing.56 Both GWAS and AM analyses showed the variant was moderately associated with AD (GWAS, p = 1.22E−5, OR = 1.774; AM case-control, p = 2.58E−4). Local ancestry distribution at both loci showed the higher African-ancestry proportion among control individuals than among affected individuals (Figure 4).
Figure 4.

Regional plot of region with significant BMIX SNPs
(A) SNP rs2195989 at chromosome 8q23.1.
(B) SNP rs62538818 at chromosome 9p23. The upper part shows the results from AM analysis (red dots, case-only AM result; green dot, case-control AM result). The middle section shows local ancestry distribution in cases (red), control (green), and global ancestry (orange). The lower section shows GWAS results. Vertical dashed line marks the BMIX SNP.
Table 5.
Joint analysis of ancestry and genotype effects on AD in AA samples
| CHR | POS | SNP | PP | Z_cc | P_cc | Z_c | P_c | P_gwas | OR | GENE |
|---|---|---|---|---|---|---|---|---|---|---|
| 8 | 108485912 | rs2195989 | 0.932 | −4.026 | 5.76E−5 | −4.02 | 5.81E−5 | 5.71E−04 | 1.314 | ANGPT1 |
| 9 | 12924211 | rs62538818 | 0.614 | −3.654 | 2.58E−4 | −2.922 | 3.48E−3 | 1.22E−05 | 1.774 | – |
_cc, case-control; _c, case only; P_gwas, p.value from GWAS; PP, posterior probability
Replication of known AD GWAS signals
Variants associated with AD in prior GWASs were accessed from the NHGRI-EBI GWAS Catalog. With p < 1E−5, 258 variants were identified to be associated with AD. Variants were pruned when multiple variants were identified within ±100 kb distance and the SNP with strongest p value was assigned as the tag SNP for the locus. The pruning resulted in 173 AD-associated GWAS loci. Replication significance was assessed at p < 0.05/173 = 2.90E−4. Four variants were replicated in AM analysis (Figure 5). Three variants (rs200291258, rs28752924, and rs9368677) are in the locus 6p21.33 (P_c = 5.69E−11). The variants are in pairwise moderate correlation in YRI population based on the 1000 Genomes Project III. The fourth variant rs2050190 (gene TSBP1 (MIM: 618151)) is in the locus 6p21.32 (P_c = 0.00028). Both AM loci, 6p21.33 and 6p21.32, showed enriched European ancestry proportions. SNP rs28752924 was found to be associated with AD in European populations,57 while rs200291258, rs9368677, and rs2050190 were found be associated with AD in East Asian populations.58,59,60 At p <0.05, 28 loci showed ancestry effects based on the case-only AM analysis (Figure 5). The strongest GWAS replication was identified at the intronic rs10519584 in gene RNF150 (p = 5.11E−05, OR = 1.638). The variant rs10519584 was also found to be associated with AD in a study of 240 AA samples.61 At p = 0.05, nominal replication was detected at 41 additional loci (Figure 5).
Figure 5.

Replication of existing AD-risk variants reported in GWAS Catalog
The outer track shows the AD-risk SNPs from the GWAS Catalog and the purple dots with varying shade represent the p value. Second layer showed the GWAS p value of the SNPs with dark red segments showing the significant replication at p < 2.9E−4 and lighter red segments showing marginal replication at p < 0.05. The innermost layer shows the AM case-only p value with dark orange segments representing significant replication.
Validation of AD target genes using gene expression analysis
Annotations of the variants from the GWAS, AM, and joint analysis using the VEP (http://grch37.ensembl.org/Tools/VEP) identified 14 target genes associated with AD (Table S1). The expression of the genes on 12 samples of lesional AD skins were compared with skin samples from eight control individuals. At FDR < 0.05 and fold change ≥1.5, four genes (SYBU, SGK1, SLAIN2, RNF39, and MMP14 [MIM: 600754]) were identified to be significantly differentially expressed in lesional AD skin (Table 6) Four of these (SGK1, SLAIN2, RNF39, and MMP14) were also significantly differentially expressed in non-lesional skin against controls.
Table 6.
Differential gene expression analysis of top genes associated with AD in AA samples
| Gene | FC_L | P_L | Q_L | FC_NL | P_NL | Q_NL |
|---|---|---|---|---|---|---|
| SYBU | 2.187 | 1.90E−04 | 1.65E−03 | 1.34 | 5.41E-03 | 2.24E-02 |
| SGK1 | −1.996 | 6.55E−05 | 7.73E−04 | −1.52 | 1.56E−02 | 4.80E−02 |
| RNF39 | 1.829 | 9.32E−03 | 2.90E−02 | 2.202 | 2.15E−03 | 1.18E−02 |
| SLAIN2 | 1.677 | 1.72E−04 | 1.55E−03 | 1.706 | 1.31E−04 | 1.94E−03 |
| MMP14 | 1.56 | 1.75E−02 | 4.70E−02 | 1.718 | 4.18E−03 | 1.88E−02 |
| EFR3A | 1.359 | 2.32E−03 | 1.01E−02 | 1.395 | 1.28E−03 | 8.34E−03 |
| ANGPT1 | 1.303 | 2.78E−01 | 3.96E−01 | −1.26 | 1.89E−01 | 3.05E−01 |
| PTPRG | 1.128 | 5.72E−01 | 6.78E−01 | 1.204 | 3.38E−01 | 4.71E−01 |
| CYFIP1 | 1.054 | 5.16E−01 | 6.28E−01 | 1.094 | 1.62E−01 | 2.72E−01 |
FC, Fold change; P, p value; Q, false discovery rate adjusted p value; L, lesional skin; NL, non-lesional skin.
qPCR validation analysis
To further validate the genes associated with AD, qPCR analysis was performed using mRNA expression on control individuals (n = 3) and moderately AD-affected individuals (n = 6). Table S2 provides the AD severity, sex, and race of the MPAACH participants used for this study. For the analysis, two GWAS genes, SGK1 and EFR3A, and two genes from the joint analysis, ANGPT1 and LURAP1L, were selected. Table S3 provides the TaqMan probes and the assay ID for the selected genes and housekeeping gene used for this study. Analysis showed that all four genes were significantly upregulated in AD patients compared to control individuals at p = 0.05 (Figure 6).
Figure 6.

Expression levels of select AD-associated genes
(A) SGK1, (B) EFR3A, (C) ANGPT1, and (D) LURAP1L. All genes were upregulated in AD patients when compared to healthy control individuals. Experiments were done on RNA isolated from blood using three healthy control individuals and six participants diagnosed with AD. ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Functional enrichment analysis
Fourteen genes identified from AM, GWAS, and joint analysis were used in the functional enrichment analysis (Table S1). Over-representation analysis identified two enriched pathways including immune-related pathways PI3K-Akt signaling pathway (Table 7). Enriched Geno Ontology (GO) terms are shown in Table S4. AD-associated genes were enriched for several biological processes related to the nervous system (such as neurogenesis, nervous system development, and generation of neurons), system development, and cell differentiation.
Table 7.
Functional enrichment analysis
| Pathway | Overlapped genes | p value | q value |
|---|---|---|---|
| Transcriptional regulation by MECP2 | SGK1; OPRM1 | 6.14E−4 | 4.91E−3 |
| PI3K-Akt signaling pathway | SGK1; ANGPT1 | 1.74E−2 | 4.63E−2 |
| Signaling by receptor tyrosine kinases | CYFIP1; SGK1 | 2.90E−2 | 5.81E−2 |
| Signal transduction | OPRM1; ANGPT1; CYFIP1; SGK1 | 4.04E−2 | 6.46E−2 |
Table shows the pathways enriched for the AD-associated genes.
Discussion
In this study, we performed AM, GWAS, and joint analysis on the AA population using 1,725 samples (726 AD cases and 999 controls). AM, GWAS, and the joint analysis revealed multiple novel risk loci associated with AD in AA population that were not identified in the prior studies with samples of European ancestry. The main finding of the study are the two novel loci associated with AD in AA that are discovered using the ancestry-genotype joint analysis. The two loci are an intronic variant rs2195989 in gene ANGPT1 on chromosome 8q23.1 and an intergenic regulatory region variant rs62538818 in chromosome 9p23. Additionally, GWAS results identified 13 loci associated with AD in AA at p < 1E−5. AM identified five loci where local genetic ancestry was associated with AD, all of which showed European ancestry as a disease risk factor. Further validation of the AD loci using a gene expression analysis showed four loci encoding genes SYBU, SGK1, RNF39, SLAIN2, and MMP14 were differentially expressed in lesional skin of individuals with AD. GWAS replicated the locus RNF150, which was identified in the study with AA samples.61 AD loci were enriched for two pathways, including PI3K-Akt signaling pathway and multiple gene ontology terms.
The joint analysis combines the effects from both genotype and ancestry at SNP level in admixed populations and allows us to detect loci that may not be detected with either approach when performed separately. Both variants exhibited moderate evidence of association with AD when the genotype and the ancestry effects were considered separately using GWAS and AM, respectively. The strongest joint signal was observed at the intronic variant rs2195989 in gene ANGPT1 on chromosome 8q23.1 with posterior probability 0.764. Both genetic and ancestry effects are moderate at the locus with admixture signal showing European ancestry effect with p < 5.76E−05 and genetic effect with GWAS p < 5.71E−04, respectively. The gene ANGPT1 encodes a secreted glycoprotein involved in angiogenesis and tumor growth55 and hereditary angioedema.62 The SNP rs2195989 is found to be eQTL with the gene in muscle-skeletal tissue in the GTEx portal (https://gtexportal.org/home/). Signals on chromosome 8 have a strong signal buildup in case-only analysis. Similar signal buildup trends were observed for case-control analysis (Figure 2). Such signal buildup in both approaches is less likely to be false positive and could indicate functionality. Further validation using more sample size will be relevant. The next variant, SNP rs62538818 in 9p23, showed moderate admixture effect with p = 2.58E−04 and genetic effect (p = 1.22E−06). The SNP overlaps with promoter flanking region ENSR00001447046, which is active in multiple cell/tissue types, including dermal fibroblast, lung, spleen, and stomach. SNP rs2195989 is more common in the African population than the European population (risk allele C, allele frequency: 0.28 [AFR] vs. 0.18 [EUR]) while the SNP rs62538818 is more common in the European population than the African population (risk allele G, allele frequency: 0.02 [AFR] vs. 0.31 [EUR]). Ancestry analysis showed the regions were enriched for European ancestry, so the observed genetic effects at the loci were likely driven by the European-ancestry-specific risk factors.
The AM analysis showed loci with a higher proportion of European ancestry contributed to the AD-susceptibility risk in AA individuals. African-ancestry-specific effect was observed at 6p21.32-21.31, but the locus did not reached significance after the adjustment for genomic inflation. The study also showed that the average proportion of African ancestry across the genome (global ancestry) was not significantly associated with AD (p = 0.328), which is consistent with the literature.27 However, the genome of admixed individuals such as AA consisted of the mosaic of chromosomal segments of ancestry (local ancestry) from parental populations and the test of global ancestry alone would be insufficient to account for the variation in the local ancestry that may harbor the ancestry-specific disease susceptibility risk. This study highlights the importance of conducting local ancestry analysis using AM approach to discover the ancestry-specific association in the admixed populations.
GWAS results showed that the genomic inflation is minimal (λ = 1.013, Figure 3B), which suggested that the population structure that may have existed in the genotype data is corrected or controlled during the QC steps and GWAS analysis by using the top five PCs. However, inflations were observed in the AM results with genomic inflation factors of 1.22 and 1.56 for case-control and case-only analysis, respectively (Figure S2). To investigate the effect of covariates and PCs on the inflation of AM result, we performed case-control AM analysis under the logistic regression framework with copies of African-origin allele as a predictor and sex, age, and PCs as covariates. AM results from the analysis did not materialistically change from our initial analysis and the genomic inflation factor remained same (data not shown). This suggests that inclusion of PCs has no effect on the model. We further investigated the extent of the admixture LD among the local ancestry loci using PLINK. Using admixture LD cutoff = 0.8, only 1,308 variants remained and, with cutoff = 0.3, only 241 variants remained. This showed that the local ancestry loci were highly correlated. The observed inflation in the AM analysis could potentially be driven by the high level of local admixture LD. LD analysis on SNP data is very common and reference LD panels for ancestry-matched sample data can be obtained, and methods such as LDSC63 have been developed to quantify the LD-induced inflation. However, similar adjustment of AM results would require a suitable reference panel for the admixture LD for the AA population.
We performed fine mapping analysis of the AM region using association test and functional annotation of variants mapped to the regions. The association test allowed us to identify the variants with genetic effects potentially contributing to the ancestry effects. Association test of SNPs showed some evidence of allelic effects of variants in the regions at p < 0.01. The moderate association may suggest the presence of ancestry-specific allelic effects that the data may have limited power to detect in the association analysis. SNP annotation using multi-omics functional tools such as CADD and RegulomeDB identified variants with strong evidence for potential regulatory functions and substantial difference in the allele frequency differences among the ancestral (African and European) populations. Three prioritized variants in the region 6p22.1-21.33 near gene RNF39 were found to be eQTL with genes in the region including RNF39 in whole blood, HLA-V in skin, whole blood, and lung. Prioritized variant rs375259 in chromosome 4 near gene SLAIN2 was found to be eQTL to the gene in multiple tissues including colon, esophagus, and lung.
GWAS identified two loci associated to AD in AA at p < 1E−6. The strongest association was observed at the variant rs113357522, intronic to gene SGK1 in chromosome 6q23. The lead variant rs113357522 on gene SGK1 is a low-frequency African-specific variant, potentially exhibiting African-ancestry effects. The gene is ubiquitously expressed and encodes a membrane protein that affects cell wall composition and plays an important role in cellular stress response.64 SGK1 expression is modulated by hormones such as gluco- and mineralocorticoids and has been linked to psychiatric disorders, including major depression.65 Variants in the gene are found to be associated with psychiatric disorder, protein level measurement, and electrocardiography. The second variant discovered from the GWAS is an intronic variant in the gene EFR3A at chromosome 8. The gene encodes a peripheral membrane protein and regulates localization of phosphatidylinositol 4-kinase (PI4K) to the plasma membrane.66,67 The lead variant at this locus rs16904552 is common in African and East Asian populations (MAF: AFR = 0.17, EAS = 0.17) but rare in the European population (MAF < 0.01). This suggests that the study identified GWAS loci with potentially African-ancestry-specific effects. Additional loci associated with AD in AA were identified at p < 1E−5, including MMP14. An animal model showed lack of MMP14 in dermal fibroblast was linked to skin homeostasis and wound repair.68 The lead variant rs911912 is common in the African population (AFR MAF = 0.11) but rare in the European populations. GWAS based on the European/East Asian samples may have failed to detect the loci due to the low frequency or due to African-ancestry-specific effect.
Gene expression profiling identified SYBU, RNF39, SLAIN2, MMP14, and SGK1 genes with significantly differentially expressed in lesional skin of AD compared with non-AD control individuals with adjusted p < 0.05 and fold change ≥1.5. Expression of SYBU, SLAIN2, RNF39, and MMP14 were downregulated in lesional skin of AD-affected individuals. Exploring the GTEx portal for the potential eQTL and sQTL effects, rs375259 showed sQTL effects on the expression of SLAIN2 in several tissues, including in the skin, and eQTL effects in lung, esophagus, and adipose. Similarly, rs3807033 showed eQTL effects on the expression of RNF39 in lung and whole blood. MMP14 gene was also found to be significantly downregulated in lesional skin of AD-affected individuals. MMP14 is associated to skin homeostasis and repair68 and also relevant to lipoedema, an estrogen-dependent disorder of adipose tissue.69 The GWAS gene SGK1 is significantly upregulated in lesional skin.
The present study has notable strengths and limitations. This is the first AA genetic mapping AD study using the joint analysis of AM and GWAS. The joint analysis combines the ancestry and genetic effects at loci to detect disease-associated risk variants that are not detected when studied separately. To capture the African-ancestry-specific variants, samples were genotyped using MEGA and genotype imputation was carried out using the CAAPA reference panel. The ancestry-specific genotyping and imputation improve the power for disease-gene mapping under both AM and GWAS approaches to identify ancestry-specific AD-risk variants in AA. Multi-omic features including in silico regulatory annotations and transcriptomic analysis prioritized the potential functional variants associated with AD and would provide a list of risk variants for further study of pathogenesis of AD. First, this study did not include socioenvironmental or geographic attributes that might influence or confound the ancestry in disease prevalence of AD. Second, sample size is not large enough to detect the variants with smaller effects as our strongest association at rs16904552 did not reach the GWAS cutoff p value of 5E−8, even with an odds ratio of 1.744. Third, even though the control group consisted of samples with no history of AD, it may have other comorbid atopic disorders such as asthma, FA, and AR that share the risk genes.19,70 Fourth, the source of the genomic inflation in the AM results was not fully explored in the study, and further research would be required to account for residual inflation that could have arisen due to stratification within ancestral populations and to validate the findings. It should be also noted that the accuracy of local ancestry inference for AM depends on reference panels (e.g., sample size, representativeness) and software (e.g., RFMix v2). Nevertheless, this study presents comprehensive disease-gene mapping in an AA cohort using GWAS, AM, and joint analysis and highlights the novel discovery of ancestry-specific AD-risk variants.
In summary, we have identified multiple target variants and genes associated with AD in the AA population. The joint analysis of AM and GWAS identified novel genetic risk loci of AD, an intronic variant rs2195989 in gene ANGPT1 and a regulatory variants rs62538818 in chromosome 9, in the AA population. The ANGPT1 gene is known to be involved in angiogenesis, tumor growth, and hereditary angioedema. AM analysis showed that both African and European ancestry contributed to the genetic risks of AD in AA. AM loci 6p21.32-21.31 was enriched for African ancestry while remaining loci were enriched for the European ancestry. Two GWAS loci, rs113357522 (SGK1) and rs16904552 (EFR3A), are found to be common in the African population but rare in the European population. AD-susceptibility loci RNF39, SLAIN2, MMP14, and SGK1 were differentially expressed in lesional skin from AD-affected individuals. Further biological validation on the role of the discovered variants in AD using independent AA samples is needed. The current euro-centric genomics studies only provide incomplete genetic architecture of human diseases including AD and, hence, hinder the translation of genetic research into clinical practices and therapeutic discovery.71,72 Hence, there is an urgent need to increase representation of diverse ancestry in genomic research and to conduct ancestry-specific assessment of pathogenic variants. Our study highlights the need for population-specific genomic resources and creation of multi-ancestry cohorts for future studies.
Data and code availability
Access to the data is available for collaboration with the lead contact with appropriate institutional approval. Data from the study cohort are not publicly available as they contain information that could compromise participant consent and confidentiality.
Acknowledgments
This work was supported by the National Institutes of Health (NIH) grants R01 HG011411 (to T.B.M.) and U19AI070235 (to G.K.K.H.).
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xhgg.2024.100350.
Web resources
PLINK 2, https://www.cog-genomics.org/plink/2.0/
RFMix v2, https://github.com/slowkoni/rfmix.
Beagle, https://faculty.washington.edu/browning/beagle/beagle.html.
Conform-gt, https://faculty.washington.edu/browning/conform-gt.html.
Ensembl, http://grch37.ensembl.org/index.html.
VEP, http://grch37.ensembl.org/info/docs/tools/vep/index.html.
GWAS Catalog, https://www.ebi.ac.uk/gwas/home.
LDlinkR, https://cran.r-project.org/web/packages/LDlinkR/index.html.
RegulomeDB, https://www.regulomedb.org/regulome-search/
GTEx, https://gtexportal.org/home/
ConsensusPathDB, http://cpdb.molgen.mpg.de/
Supplemental information
BP, biological process; CC, cellular component; MF, molecular function.
References
- 1.Boothe W.D., Tarbox J.A., Tarbox M.B. Atopic Dermatitis: Pathophysiology. Adv. Exp. Med. Biol. 2024;1447:21–35. doi: 10.1007/978-3-031-54513-9_3. [DOI] [PubMed] [Google Scholar]
- 2.Drucker A.M., Wang A.R., Li W.Q., Sevetson E., Block J.K., Qureshi A.A. The Burden of Atopic Dermatitis: Summary of a Report for the National Eczema Association. J. Invest. Dermatol. 2017;137:26–30. doi: 10.1016/j.jid.2016.07.012. [DOI] [PubMed] [Google Scholar]
- 3.Gupta J., Johansson E., Bernstein J.A., Chakraborty R., Khurana Hershey G.K., Rothenberg M.E., Mersha T.B. Resolving the etiology of atopic disorders by using genetic analysis of racial ancestry. J. Allergy Clin. Immunol. 2016;138:676–699. doi: 10.1016/j.jaci.2016.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Odhiambo J.A., Williams H.C., Clayton T.O., Robertson C.F., Asher M.I., ISAAC Phase Three Study Group Global variations in prevalence of eczema symptoms in children from ISAAC Phase Three. J. Allergy Clin. Immunol. 2009;124:1251–1258.e23. doi: 10.1016/j.jaci.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 5.Wadonda-Kabondo N., Sterne J.A.C., Golding J., Kennedy C.T.C., Archer C.B., Dunnill M.G.S., ALSPAC Study Team Association of parental eczema, hayfever, and asthma with atopic dermatitis in infancy: birth cohort study. Arch. Dis. Child. 2004;89:917–921. doi: 10.1136/adc.2003.034033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Larsen F.S., Holm N.V., Henningsen K. Atopic dermatitis. A genetic-epidemiologic study in a population-based twin sample. J. Am. Acad. Dermatol. 1986;15:487–494. [PubMed] [Google Scholar]
- 7.Schultz Larsen F. Atopic dermatitis: a genetic-epidemiologic study in a population-based twin sample. J. Am. Acad. Dermatol. 1993;28:719–723. doi: 10.1016/0190-9622(93)70099-f. [DOI] [PubMed] [Google Scholar]
- 8.Brown S.J., Elias M.S., Bradley M. Genetics in Atopic Dermatitis: Historical Perspective and Future Prospects. Acta Derm. Venereol. 2020;100:adv00163. doi: 10.2340/00015555-3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elmose C., Thomsen S.F. Twin Studies of Atopic Dermatitis: Interpretations and Applications in the Filaggrin Era. J. Allergy. 2015;2015:902359. doi: 10.1155/2015/902359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palmer C.N.A., Irvine A.D., Terron-Kwiatkowski A., Zhao Y., Liao H., Lee S.P., Goudie D.R., Sandilands A., Campbell L.E., Smith F.J.D., et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat. Genet. 2006;38:441–446. doi: 10.1038/ng1767. [DOI] [PubMed] [Google Scholar]
- 11.Kim B.E., Leung D.Y.M. Significance of Skin Barrier Dysfunction in Atopic Dermatitis. Allergy Asthma Immunol. Res. 2018;10:207–215. doi: 10.4168/aair.2018.10.3.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Debinska A., Danielewicz H., Boznański A., Matusiak Ł., Szepietowski J.C. A small proline-rich protein (SPRR) gene variant contributes to atopic eczema and eczema-associated asthma susceptibility. Postepy Dermatol Alergol. 2022;39:965–971. doi: 10.5114/ada.2022.113145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Almoguera B., Vazquez L., Mentch F., March M.E., Connolly J.J., Peissig P.L., Linneman J.G., Plaza-Serón M.D.C., Pino-Yanes M., Burchard E.G., et al. Novel locus for atopic dermatitis in African Americans and replication in European Americans. J. Allergy Clin. Immunol. 2019;143:1229–1231. doi: 10.1016/j.jaci.2018.10.038. [DOI] [PubMed] [Google Scholar]
- 14.Gao P.S., Rafaels N.M., Mu D., Hand T., Murray T., Boguniewicz M., Hata T., Schneider L., Hanifin J.M., Gallo R.L., et al. Genetic variants in thymic stromal lymphopoietin are associated with atopic dermatitis and eczema herpeticum. J. Allergy Clin. Immunol. 2010;125:1403–1407.e4. doi: 10.1016/j.jaci.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ellinghaus D., Baurecht H., Esparza-Gordillo J., Rodríguez E., Matanovic A., Marenholz I., Hübner N., Schaarschmidt H., Novak N., Michel S., et al. High-density genotyping study identifies four new susceptibility loci for atopic dermatitis. Nat. Genet. 2013;45:808–812. doi: 10.1038/ng.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grosche S., Marenholz I., Esparza-Gordillo J., Arnau-Soler A., Pairo-Castineira E., Rüschendorf F., Ahluwalia T.S., Almqvist C., Arnold A., Australian Asthma Genetics Consortium AAGC, et al. Rare variant analysis in eczema identifies exonic variants in DUSP1, NOTCH4 and SLC9A4. Nat. Commun. 2021;12:6618. doi: 10.1038/s41467-021-26783-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Margaritte-Jeannin P., Budu-Aggrey A., Ege M., Madore A.M., Linhard C., Mohamdi H., von Mutius E., Granell R., Demenais F., Laprise C., et al. Identification of OCA2 as a novel locus for the co-morbidity of asthma-plus-eczema. Clin. Exp. Allergy. 2022;52:70–81. doi: 10.1111/cea.13972. [DOI] [PubMed] [Google Scholar]
- 18.Johansson Å., Rask-Andersen M., Karlsson T., Ek W.E. Genome-wide association analysis of 350 000 Caucasians from the UK Biobank identifies novel loci for asthma, hay fever and eczema. Hum. Mol. Genet. 2019;28:4022–4041. doi: 10.1093/hmg/ddz175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferreira M.A., Vonk J.M., Baurecht H., Marenholz I., Tian C., Hoffman J.D., Helmer Q., Tillander A., Ullemar V., van Dongen J., et al. Shared genetic origin of asthma, hay fever and eczema elucidates allergic disease biology. Nat. Genet. 2017;49:1752–1757. doi: 10.1038/ng.3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mucha S., Baurecht H., Novak N., Rodríguez E., Bej S., Mayr G., Emmert H., Stölzl D., Gerdes S., Jung E.S., et al. Protein-coding variants contribute to the risk of atopic dermatitis and skin-specific gene expression. J. Allergy Clin. Immunol. 2020;145:1208–1218. doi: 10.1016/j.jaci.2019.10.030. [DOI] [PubMed] [Google Scholar]
- 21.Brown S.J. What Have We Learned from GWAS for Atopic Dermatitis? J. Invest. Dermatol. 2021;141:19–22. doi: 10.1016/j.jid.2020.05.100. [DOI] [PubMed] [Google Scholar]
- 22.Shaw T.E., Currie G.P., Koudelka C.W., Simpson E.L. Eczema prevalence in the United States: data from the 2003 National Survey of Children's Health. J. Invest. Dermatol. 2011;131:67–73. doi: 10.1038/jid.2010.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Williams H.C., Pembroke A.C., Forsdyke H., Boodoo G., Hay R.J., Burney P.G. London-born black Caribbean children are at increased risk of atopic dermatitis. J. Am. Acad. Dermatol. 1995;32:212–217. doi: 10.1016/0190-9622(95)90128-0. [DOI] [PubMed] [Google Scholar]
- 24.Child F.J., Fuller L.C., Higgins E.M., Du Vivier A.W. A study of the spectrum of skin disease occurring in a black population in south-east London. Br. J. Dermatol. 1999;141:512–517. doi: 10.1046/j.1365-2133.1999.03047.x. [DOI] [PubMed] [Google Scholar]
- 25.Ait-Khaled N., Odhiambo J., Pearce N., Adjoh K.S., Maesano I.A., Benhabyles B., Bouhayad Z., Bahati E., Camara L., Catteau C., et al. Prevalence of symptoms of asthma, rhinitis and eczema in 13- to 14-year-old children in Africa: the International Study of Asthma and Allergies in Childhood Phase III. Allergy. 2007;62:247–258. doi: 10.1111/j.1398-9995.2007.01325.x. [DOI] [PubMed] [Google Scholar]
- 26.Janumpally S.R., Feldman S.R., Gupta A.K., Fleischer A.B., Jr. In the United States, blacks and Asian/Pacific Islanders are more likely than whites to seek medical care for atopic dermatitis. Arch. Dermatol. 2002;138:634–637. doi: 10.1001/archderm.138.5.634. [DOI] [PubMed] [Google Scholar]
- 27.Abuabara K., You Y., Margolis D.J., Hoffmann T.J., Risch N., Jorgenson E. Genetic ancestry does not explain increased atopic dermatitis susceptibility or worse disease control among African American subjects in 2 large US cohorts. J. Allergy Clin. Immunol. 2020;145:192–198.e11. doi: 10.1016/j.jaci.2019.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanyal R.D., Pavel A.B., Glickman J., Chan T.C., Zheng X., Zhang N., Cueto I., Peng X., Estrada Y., Fuentes-Duculan J., et al. Atopic dermatitis in African American patients is T(H)2/T(H)22-skewed with T(H)1/T(H)17 attenuation. Ann. Allergy Asthma Immunol. 2019;122:99–110.e6. doi: 10.1016/j.anai.2018.08.024. [DOI] [PubMed] [Google Scholar]
- 29.Brunner P.M., Guttman-Yassky E. Racial differences in atopic dermatitis. Ann. Allergy Asthma Immunol. 2019;122:449–455. doi: 10.1016/j.anai.2018.11.015. [DOI] [PubMed] [Google Scholar]
- 30.Paternoster L., Standl M., Waage J., Baurecht H., Hotze M., Strachan D.P., Curtin J.A., Bønnelykke K., Tian C., Takahashi A., et al. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat. Genet. 2015;47:1449–1456. doi: 10.1038/ng.3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patterson N., Hattangadi N., Lane B., Lohmueller K.E., Hafler D.A., Oksenberg J.R., Hauser S.L., Smith M.W., O'Brien S.J., Altshuler D., et al. Methods for high-density admixture mapping of disease genes. Am. J. Hum. Genet. 2004;74:979–1000. doi: 10.1086/420871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shriner D., Adeyemo A., Rotimi C.N. Joint ancestry and association testing in admixed individuals. PLoS Comput. Biol. 2011;7:e1002325. doi: 10.1371/journal.pcbi.1002325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Butsch Kovacic M., Biagini Myers J.M., Lindsey M., Patterson T., Sauter S., Ericksen M.B., Ryan P., Assa'ad A., Lierl M., Fischer T., et al. The Greater Cincinnati Pediatric Clinic Repository: A Novel Framework for Childhood Asthma and Allergy Research. Pediatr. Allergy Immunol. Pulmonol. 2012;25:104–113. doi: 10.1089/ped.2011.0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin L.J., Murrison L.B., Butsch Kovacic M. Building a Population Representative Pediatric Biobank: Lessons Learned From the Greater Cincinnati Childhood Cohort. Front. Public Health. 2020;8:535116. doi: 10.3389/fpubh.2020.535116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DeWan A.T., Egan K.B., Hellenbrand K., Sorrentino K., Pizzoferrato N., Walsh K.M., Bracken M.B. Whole-exome sequencing of a pedigree segregating asthma. BMC Med. Genet. 2012;13:95. doi: 10.1186/1471-2350-13-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anderson C.A., Pettersson F.H., Clarke G.M., Cardon L.R., Morris A.P., Zondervan K.T. Data quality control in genetic case-control association studies. Nat. Protoc. 2010;5:1564–1573. doi: 10.1038/nprot.2010.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M., Lee J.J. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Das S., Forer L., Schönherr S., Sidore C., Locke A.E., Kwong A., Vrieze S.I., Chew E.Y., Levy S., McGue M., et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016;48:1284–1287. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mathias R.A., Taub M.A., Gignoux C.R., Fu W., Musharoff S., O'Connor T.D., Vergara C., Torgerson D.G., Pino-Yanes M., Shringarpure S.S., et al. A continuum of admixture in the Western Hemisphere revealed by the African Diaspora genome. Nat. Commun. 2016;7:12522. doi: 10.1038/ncomms12522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen G., Shriner D., Zhou J., Doumatey A., Huang H., Gerry N.P., Herbert A., Christman M.F., Chen Y., Dunston G.M., et al. Development of admixture mapping panels for African Americans from commercial high-density SNP arrays. BMC Genom. 2010;11:417. doi: 10.1186/1471-2164-11-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maples B.K., Gravel S., Kenny E.E., Bustamante C.D. RFMix: a discriminative modeling approach for rapid and robust local-ancestry inference. Am. J. Hum. Genet. 2013;93:278–288. doi: 10.1016/j.ajhg.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Browning B.L., Tian X., Zhou Y., Browning S.R. Fast two-stage phasing of large-scale sequence data. Am. J. Hum. Genet. 2021;108:1880–1890. doi: 10.1016/j.ajhg.2021.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gautam Y., Altaye M., Xie C., Mersha T.B. AdmixPower: Statistical Power and Sample Size Estimation for Mapping Genetic Loci in Admixed Populations. Genetics. 2017;207:873–882. doi: 10.1534/genetics.117.300312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gautam Y., Ghandikota S., Chen S., Mersha T.B. PAMAM: Power analysis in multiancestry admixture mapping. Genet. Epidemiol. 2019;43:831–843. doi: 10.1002/gepi.22216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grinde K.E., Brown L.A., Reiner A.P., Thornton T.A., Browning S.R. Genome-wide Significance Thresholds for Admixture Mapping Studies. Am. J. Hum. Genet. 2019;104:454–465. doi: 10.1016/j.ajhg.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Devlin B., Roeder K., Wasserman L. Genomic control, a new approach to genetic-based association studies. Theor. Popul. Biol. 2001;60:155–166. doi: 10.1006/tpbi.2001.1542. [DOI] [PubMed] [Google Scholar]
- 47.Rentzsch P., Witten D., Cooper G.M., Shendure J., Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886–D894. doi: 10.1093/nar/gky1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boyle A.P., Hong E.L., Hariharan M., Cheng Y., Schaub M.A., Kasowski M., Karczewski K.J., Park J., Hitz B.C., Weng S., et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–1797. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sollis E., Mosaku A., Abid A., Buniello A., Cerezo M., Gil L., Groza T., Güneş O., Hall P., Hayhurst J., et al. The NHGRI-EBI GWAS Catalog: knowledgebase and deposition resource. Nucleic Acids Res. 2023;51:D977–D985. doi: 10.1093/nar/gkac1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Myers T.A., Chanock S.J., Machiela M.J. LDlinkR: An R Package for Rapidly Calculating Linkage Disequilibrium Statistics in Diverse Populations. Front. Genet. 2020;11:157. doi: 10.3389/fgene.2020.00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suarez-Farinas M., Tintle S.J., Shemer A., Chiricozzi A., Nograles K., Cardinale I., Duan S., Bowcock A.M., Krueger J.G., Guttman-Yassky E. Nonlesional atopic dermatitis skin is characterized by broad terminal differentiation defects and variable immune abnormalities. J. Allergy Clin. Immunol. 2011;127:954–964.e1-4. doi: 10.1016/j.jaci.2010.12.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou G., Soufan O., Ewald J., Hancock R.E.W., Basu N., Xia J. NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019;47:W234–W241. doi: 10.1093/nar/gkz240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 54.Herwig R., Hardt C., Lienhard M., Kamburov A. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat. Protoc. 2016;11:1889–1907. doi: 10.1038/nprot.2016.117. [DOI] [PubMed] [Google Scholar]
- 55.Poto R., Cristinziano L., Modestino L., de Paulis A., Marone G., Loffredo S., Galdiero M.R., Varricchi G. Neutrophil Extracellular Traps, Angiogenesis and Cancer. Biomedicines. 2022;10:431. doi: 10.3390/biomedicines10020431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kisiel M.A., Klar A.S. Isolation and Culture of Human Dermal Fibroblasts. Methods Mol. Biol. 2019;1993:71–78. doi: 10.1007/978-1-4939-9473-1_6. [DOI] [PubMed] [Google Scholar]
- 57.Sliz E., Huilaja L., Pasanen A., Laisk T., Reimann E., Mägi R., FinnGen. Estonian Biobank Research Team. Hannula-Jouppi K., Peltonen S., et al. Uniting biobank resources reveals novel genetic pathways modulating susceptibility for atopic dermatitis. J. Allergy Clin. Immunol. 2022;149:1105–1112.e9. doi: 10.1016/j.jaci.2021.07.043. [DOI] [PubMed] [Google Scholar]
- 58.Sakaue S., Kanai M., Tanigawa Y., Karjalainen J., Kurki M., Koshiba S., Narita A., Konuma T., Yamamoto K., Akiyama M., et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 2021;53:1415–1424. doi: 10.1038/s41588-021-00931-x. [DOI] [PubMed] [Google Scholar]
- 59.Hirota T., Takahashi A., Kubo M., Tsunoda T., Tomita K., Sakashita M., Yamada T., Fujieda S., Tanaka S., Doi S., et al. Genome-wide association study identifies eight new susceptibility loci for atopic dermatitis in the Japanese population. Nat. Genet. 2012;44:1222–1226. doi: 10.1038/ng.2438. [DOI] [PubMed] [Google Scholar]
- 60.Tanaka N., Koido M., Suzuki A., Otomo N., Suetsugu H., Kochi Y., Tomizuka K., Momozawa Y., Kamatani Y., Biobank Japan Project, et al. Eight novel susceptibility loci and putative causal variants in atopic dermatitis. J. Allergy Clin. Immunol. 2021;148:1293–1306. doi: 10.1016/j.jaci.2021.04.019. [DOI] [PubMed] [Google Scholar]
- 61.DeVore S.B., Stevens M.L., He H., Biagini J.M., Kroner J.W., Martin L.J., Khurana Hershey G.K. Novel role for caspase recruitment domain family member 14 and its genetic variant rs11652075 in skin filaggrin homeostasis. J. Allergy Clin. Immunol. 2022;149:708–717. doi: 10.1016/j.jaci.2021.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lopez Lera A. Pathophysiology and underlying mechanisms in hereditary angioedema. Balkan Med. J. 2021;38:82–88. doi: 10.4274/balkanmedj.galenos.2020.2020.10.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bulik-Sullivan B.K., Loh P.R., Finucane H.K., Ripke S., Yang J., Schizophrenia Working Group of the Psychiatric Genomics Consortium. Patterson N., Daly M.J., Price A.L., Neale B.M. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015;47:291–295. doi: 10.1038/ng.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tomishige N., Noda Y., Adachi H., Shimoi H., Yoda K. SKG1, a suppressor gene of synthetic lethality of kex2Deltagas1Delta mutations, encodes a novel membrane protein that affects cell wall composition. Yeast. 2005;22:141–155. doi: 10.1002/yea.1206. [DOI] [PubMed] [Google Scholar]
- 65.Dattilo V., Amato R., Perrotti N., Gennarelli M. The Emerging Role of SGK1 (Serum- and Glucocorticoid-Regulated Kinase 1) in Major Depressive Disorder: Hypothesis and Mechanisms. Front. Genet. 2020;11:826. doi: 10.3389/fgene.2020.00826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chung J., Nakatsu F., Baskin J.M., De Camilli P. Plasticity of PI4KIIIalpha interactions at the plasma membrane. EMBO Rep. 2015;16:312–320. doi: 10.15252/embr.201439151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bojjireddy N., Guzman-Hernandez M.L., Reinhard N.R., Jovic M., Balla T. EFR3s are palmitoylated plasma membrane proteins that control responsiveness to G-protein-coupled receptors. J. Cell Sci. 2015;128:118–128. doi: 10.1242/jcs.157495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kumper M., Zamek J., Steinkamp J., Pach E., Mauch C., Zigrino P. Role of MMP3 and fibroblast-MMP14 in skin homeostasis and repair. Eur. J. Cell Biol. 2022;101:151276. doi: 10.1016/j.ejcb.2022.151276. [DOI] [PubMed] [Google Scholar]
- 69.Kruglikov I.L., Joffin N., Scherer P.E. The MMP14-caveolin axis and its potential relevance for lipoedema. Nat. Rev. Endocrinol. 2020;16:669–674. doi: 10.1038/s41574-020-0395-z. [DOI] [PubMed] [Google Scholar]
- 70.Guo H., An J., Yu Z. Identifying Shared Risk Genes for Asthma, Hay Fever, and Eczema by Multi-Trait and Multiomic Association Analyses. Front. Genet. 2020;11:270. doi: 10.3389/fgene.2020.00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Graham S.E., Clarke S.L., Wu K.H.H., Kanoni S., Zajac G.J.M., Ramdas S., Surakka I., Ntalla I., Vedantam S., Winkler T.W., et al. The power of genetic diversity in genome-wide association studies of lipids. Nature. 2021;600:675–679. doi: 10.1038/s41586-021-04064-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Duncan L., Shen H., Gelaye B., Meijsen J., Ressler K., Feldman M., Peterson R., Domingue B. Analysis of polygenic risk score usage and performance in diverse human populations. Nat. Commun. 2019;10:3328. doi: 10.1038/s41467-019-11112-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
BP, biological process; CC, cellular component; MF, molecular function.
Data Availability Statement
Access to the data is available for collaboration with the lead contact with appropriate institutional approval. Data from the study cohort are not publicly available as they contain information that could compromise participant consent and confidentiality.