

Abstract
The effect of past environmental changes on the demography and genetic diversity of natural populations remains a contentious issue and has rarely been investigated across multiple, phylogenetically distant species. Here, we perform comparative population genomic analyses and demographic inferences for seven widely distributed and ecologically contrasting European forest tree species based on concerted sampling of 164 populations across their natural ranges. For all seven species, the effective population size, Ne, increased or remained stable over many glacial cycles and up to 15 million years in the most extreme cases. Surprisingly, the drastic environmental changes associated with the Pleistocene glacial cycles have had little impact on the level of genetic diversity of dominant forest tree species, despite major shifts in their geographic ranges. Based on their trajectories of Ne over time, the seven tree species can be divided into three major groups, highlighting the importance of life history and range size in determining synchronous variation in genetic diversity over time. Altogether, our results indicate that forest trees have been able to retain their evolutionary potential over very long periods of time despite strong environmental changes.
Subject terms: Genetic variation, Natural variation in plants, Population genetics, Evolutionary genetics
European forest tree species experienced strong climatic fluctuations over Quaternary. In spite of these pronounced environmental changes, population genomic analyses reveal that major forest tree species managed to retain their evolutionary potential over the period.
Introduction
Extant northern temperate and boreal tree species have existed for millions of years and survived multiple glacial cycles. Micro- and macrofossil data indicate that these tree species have undergone huge range changes and large fluctuations in their census population sizes (Nc) over time1. This was largely corroborated by numerous phylogeographic studies2. Yet, today most tree species harbor large amounts of genetic diversity3 and they have been shown to respond rapidly, both genetically and demographically, to recent environmental challenges such as the Little Ice Age4 or the Last Glacial Maximum (LGM)5,6. While these rapid responses to new selection pressures are consistent with their current large population sizes, high realized outcrossing rates, and efficient gene flow, they may seem paradoxical in view of the large census size changes suggested by the fossil records. Unfortunately, we still lack a comprehensive view of the impact of past demographic changes on the effective population size (Ne), the key evolutionary parameter defining the genetic diversity and efficacy of selection7. For example, did Ne fluctuate strongly through time or, on the contrary, was it retained and stable over repeated glacial cycles despite changes in Nc? Were changes in Ne primarily driven by climatic events or do they also reflect intrinsic biological characteristics such as life history or physiological properties? In the former case one would expect a high synchronicity in changes across multiple species independently of their biological properties while, in the latter case, one would expect species to form categories according to their patterns of intraspecific diversity changes and shared biological properties8,9.
In Europe, the LGM which occurred ~27,000 to 19,000 years ago, and ensuing Holocene recolonizations have often been assumed to be the main drivers of the current distribution of intraspecific genetic diversity, with southern populations being typically more diverged than those from the more northern core range10,11. Earlier analyses of the demographic histories of European forest tree species generally relied on organellar markers whose polymorphisms are informative on a shorter time span than nuclear markers since in monoecious species their effective population size is half of that for nuclear markers and in dioecious a quarter12,13. Quite naturally, outcomes were interpreted from the perspective of only the most recent glacial period (i.e., LGM)11. Further, inferences based on organellar markers that behave as a single locus and, in most cases, are maternally inherited and only disperse via seeds, have limited relevance for nuclear genetic diversity wherein most of the genetic variation lies. Genome re-sequencing combined with coalescence-based demographic methods allowed inferring the demographic history of forest trees and its timescale well beyond the LGM, up to millions of years. However, most studies so far address single species or focus on inferring the timing of divergence and the extent of gene flow between populations or closely related species14–18. Congruence between population history and glacial oscillations has been observed in some species14,19 but remained far from being conclusive in others20,21. A general conclusion on the drivers of temporal changes in genetic diversity across species cannot, however, be drawn from the compilation of these studies, or even from the reanalysis of the data they present, due to the heterogeneity of sampling strategies, genomic sources of polymorphism, and numbers of loci.
Here, we carried out a comprehensive demographic inference of seven major European tree species, distributed from the boreal to the Mediterranean regions (Table 1), based on a common strategy both for sampling populations across Europe and for sequencing genomic regions. All seven species are wind-pollinated, three are conifers (Picea abies, Pinus pinaster, and Pinus sylvestris) and four are angiosperms (Betula pendula, Fagus sylvatica, Populus nigra, and Quercus petraea). We conducted targeted nuclear DNA sequencing (~10,000 species-specific probes that covered ~3 Mbp of largely orthologous sequences) on a total of 3407 adult trees collected from 19 to 26 locations per species (~25 individuals each) across large parts of their natural ranges (Figs. 1, S1, Supplementary Text, Table 1, S1, Supplementary Data 1)22. We first conducted a comprehensive survey of the distribution of current genetic diversity in all seven species and used state-of-the-art coalescent approaches to reconstruct changes in Ne over multiple glacial cycles and test for synchronous changes across species.
Table 1.
Biological characteristics and genetic summary statistics for seven European tree species
| Species | Biological characteristics | Hybridization with | Min. flowering age (years) | Max. age known (years) | FST | π4 (per bp) | IBD (slope, 10−3) |
|---|---|---|---|---|---|---|---|
|
Betula pendula (Silver birch) |
• deciduous • temperate to boreal • wind pollination • wind seed dispersal • large and continuous range |
B. platyphylla B. pubescens |
10–25 | 90–150 | 0.03a | 0.0036 | 4 |
|
Fagus sylvatica (European beech) |
• deciduous • temperate • wind pollination • animal seed dispersal • large and continuous range |
F. orientalis | 40–50 | 150–300 | 0.05a | 0.0050 | 15 |
|
Populus nigra (Black poplar) |
• deciduous • Mediterranean to temperate • wind pollination • water and wind seed dispersal • vegetative and sexual reproduction • intermediate and discontinuous range (riparian) |
P. nigra ‘Italica’ P. deltoides P. trichocarpa P. maximowiczii Populus sp. hybrid cultivars |
4–10 | 100–400 | 0.16a | 0.0032 | 125 |
|
Quercus petraea (Sessile oak) |
• deciduous • temperate • wind pollination • animal seed dispersal • large and continuous range |
Q. robur Q. pubescens Q. pyrenaica |
40–100 | >1000 | 0.04a | 0.0072 | 9 |
|
Picea abies (Norway spruce) |
• conifer • temperate to boreal • wind pollination • wind seed dispersal • large and discontinuous range |
P. obovata | 20–40 | 200–300 | 0.06a | 0.0048 | 33 |
|
Pinus pinaster (Maritime pine) |
• conifer • Mediterranean • wind pollination • wind seed dispersal • limited and discontinuous range |
6–20 | 120–250 | 0.13a | 0.0027 | 59 | |
|
Pinus sylvestris (Scots pine) |
• conifer • temperate to boreal • wind pollination • wind seed dispersal • large and continuous range |
P. mugo P. uliginosa |
15–30 | 400–750 | 0.01a | 0.0039 | 3 |
Age information was retrieved from the European atlas of tree species https://forest.jrc.ec.europa.eu/en/european-atlas/ and the European forest genetic resources program (EUFORGEN).
Notes: mean pairwise genetic differentiation (FST).
aone-sided p-value for among population variation based on AMOVA was 0.01 for all species; pairwise nucleotide diversity per site at four-fold sites (π4); Isolation by distance (IBD) is represented by the slope of the regression of FST /(1- FST) over the logarithm of the distance (km) between pairs of populations.
Fig. 1. Admixture, isolation by distance, genetic diversity and divergence patterns of the seven studied European tree species.

A–G Pie charts represent population average admixture coefficients. Four genetic clusters are shown to visualize genetic structure, except for Pinus sylvestris (K = 1; B) and Populus nigra (K = 7; F) (see Figs. S3–S16 for other cluster numbers). An admixture bar plot of all individuals is shown at the bottom of each panel (country codes are explained in Table S1). Background maps82 represent species’ ranges. Inset graphs show patterns of isolation by distance. H Nucleotide diversity at four-fold sites, π4, per base pair as a function of latitude. I Population-specific scaled differentiation estimated as the average of the ratios of pairwise Fst over pairwise distance for all population pairs as a function of π4. Different symbols and colors represent populations of the different species with their respective trend lines. Species codes are explained in Table S1. The few populations from Russia are not represented here and can be found in Figs. S1, S12. (AT Austria, BA Bosnia and Herzegovina, BY Belarus, CH Switzerland, DE Germany, ES Spain, FI Finland, FR France, GB Great Britain, GR Greece, IT Italy, LT Lithuania, MA Morocco, NO Norway, PL Poland, PT Portugal, RO Romania, RU Russia, SE Sweden, SI Slovenia, TN Tunisia, UA Ukraine, BP Betula pendula, FS Fagus sylvatica, PA Picea abies, PO Populus nigra, PP Pinus pinaster, PS Pinus sylvestris, QP Quercus petraea). Source data are provided as a Source Data file-1.
In this work, we show that past glacial and interglacial cycles did not have a major impact on genetic diversity of common European tree species. All seven species show signs of recent population growth and species cluster in both diversity-divergence spectrum and based on their demographic trajectories. Importantly, this last clustering does not reflect their phylogenetic closeness, but instead is likely the consequence of shared ecological and biological characteristics.
Results
Patterns of genetic diversity do not reflect phylogeny or environmental preferences
Overall, current genetic diversity and structure in the seven species (Fig. 1) reflected neither phylogeny nor current environmental preferences. The patterns likely followed from a combination of biological and ecological characteristics as well as range-limit constraints (biotic or abiotic). With respect to genetic diversity and genetic differentiation among populations, the seven species can be divided into four sets: highly genetically structured and low diversity P. pinaster and P. nigra; moderately structured, and intermediate diversity F. sylvatica and P. abies; moderately structured, high diversity Q. petraea; and finally, panmictic and moderate diversity P. sylvestris and B. pendula (Fig. 1I).
Nucleotide diversity at four-fold degenerate, synonymous sites (π4) ranged from 0.0027 to 0.0072 per bp across the seven species (Table 1, Supplementary data 2), as is typical of outcrossing trees23, and was remarkably similar among populations of a given species (Fig. 1H). Quercus petraea had the highest genetic diversity and it increased towards the north. Also, boreal species (P. abies, P. sylvestris and B. pendula) exhibited slightly higher diversity at high latitudes, whereas genetic diversity tended to decrease northwards for the temperate species P. nigra, F. sylvatica and P. pinaster (Fig. 1H, Supplementary Data 2). Thus, the geographic distribution of genetic diversity did not consistently follow the south–north latitudinal gradient that is often considered as a proxy for postglacial recolonization history24.
Genetic differentiation between populations (FST) was low for most species, except for P. pinaster and P. nigra (Table 1). Isolation-by-distance at the range scale was significant for most species, likely reflecting the distance-dependency of wind-mediated pollen dispersal over mere population-level genetic drift (Fig. 1A–G). However, the level of divergence was not uniform across the species’ distributions. Generally, the most genetically divergent populations were found at southern latitudes (Fig. S2), similarly to what Petit et al.11 found across several angiosperm trees. This result is also supported by the spatial distribution of ancestry proportions and principal component analysis (PCA) (Figs. 1A–G, S3–S23, Supplementary Data 2). Additionally, populations at higher elevations were genetically more differentiated from the rest of the range (Fig. S2, Supplementary Data 3). Globally, genetic structure coincided with the main discontinuities in the species’ distributions, but with considerable variation across species (Figs. 1A–G, S3–S16).
Main divergence events largely predate the last glacial maximum in all seven species
To compare divergence and demographic events across species, we analyzed different subsets of the data with, in each case, simple and consistent modeling choices and methodology. The origin and timing of the divergence between populations (Table 1) was studied using demographic models implemented in fastsimcoal225,26. To estimate the timeframe of population separation we analyzed in each species two non-admixed populations representing the main southern and northern clusters (Supplementary Data 4, Fig. S24). In all species, divergence models with migration had better support than models without (Supplementary Data 4), and the estimated divergence times between major clusters largely predated the LGM, extending from 0.6 Mya (middle Quaternary) up to 17 Mya within the Miocene (Fig. S25, Supplementary Data 4–6). Hence, for all species the formation of the main genetic groups was an outcome of demographic events having occurred over multiple glacial cycles, and, importantly, these groups were preserved through glacial cycles despite extensive gene flow. Consequently, the overall pattern of genetic differentiation better reflects topography and other persistent barriers to movement of populations, instead of recent divergence during the LGM. For example, the mountains of southern Europe (e.g., Pyrenees, Alps) could have driven the recurrent formation of sky islands, i.e., isolated high-elevation regions to which cold-adapted species repeatedly shifted during interglacial periods, resulting in higher divergence between southern high-elevation populations than between populations at lower altitudes or populations at more northern latitudes27.
General increase in Ne over multiple glacial cycles
To infer the timescale of changes in Ne in the seven species and over many glacial cycles, we used Stairway Plot 228, a composite likelihood method that infers changes in Ne over time from site frequency spectra (SFS). Because Stairway Plot 2 is model-flexible and inferences can be biased towards complex models, with more demographic events occurring (overfitting), we tested the robustness of the results with the more constrained fastsimcoal2 2-epoch model. SFS-based inference of Ne trajectory measures changes in coalescence rates of gene genealogies, which depend on historical changes in Nc affecting Ne but also on barriers to gene flow in structured populations and the way gene genealogies are sampled (29 and references therein). To account for the effect of sampling and population structure on demographic inferences30, we conducted analyses at the species, population, and one-sample-per-population levels. With the last level, the analysis was focused on the dominating collecting phase of the genealogy31,32.
All estimates of past and present Ne were in the range of tens or hundreds of thousands; these values are much smaller than any reasonable estimate of the study species’ current census sizes (Nc), which is in the scale of billions of individuals for most species33,34. While the observed ratios were lower than usual estimates of Ne/Nc, they fit with the trend of species with large Nc displaying below-average estimates of Ne/Nc35.
All species, except F. sylvatica, showed an excess of rare variants in the SFS (Fig. 2) revealing a global signal of ancient population growth (from 0.6 Mya in P. sylvestris to 15 Mya in Q. petraea). This signal was consistent across sampling schemes and the two inference methods (Figs. 3A–B, S26) and is in line with earlier studies on, e.g., P. abies and P. sylvestris17,36. Strikingly, very few populations exhibited a signal of decreasing Ne through time, and those populations were often disconnected from the rest of the range and, thus, likely to have experienced stronger genetic drift (Fig. S27, Supplementary Data 7). The magnitude of increase in Ne varied across species and was largest for P. sylvestris (from ~5000 to 500,000) and weakest for F. sylvatica, for which the proportional increase was only two-fold (from 100,000 to 200,000).
Fig. 2. Folded site frequency spectra (fSFS) of all seven studied species (inset) and of the remaining six species after excluding Pinus sylvestris (with adjusted scale in the y axis).
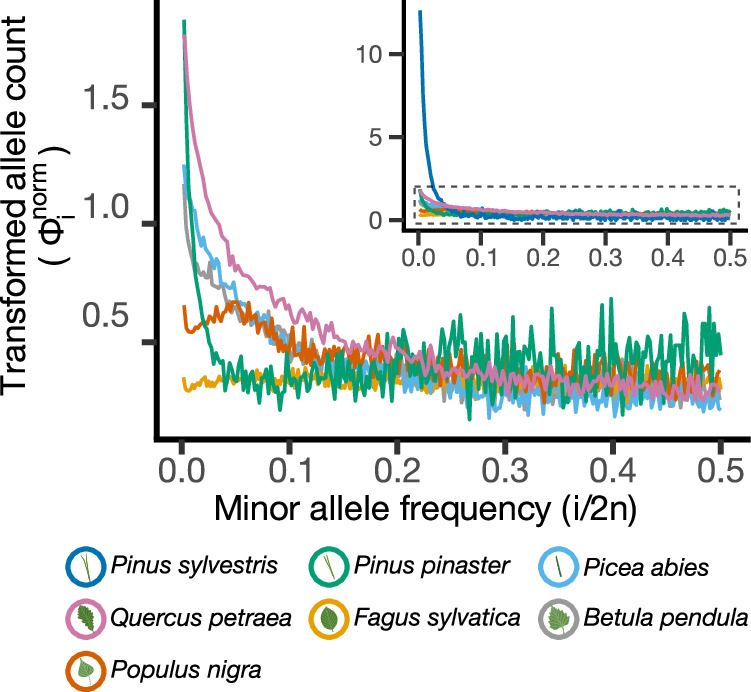
The SFS were transformed and normalized following83 (see Materials & Methods) so that they are flat under the standard neutral model, facilitating the visualization of the SFS that depart from expectations of the standard neutral model84,85. Source data are provided as a Source Data file-1.
Fig. 3. Demographic change across time for the seven tree species.

A Change in effective population size (Ne) through time (million years ago, Mya), inferred with Stairway Plot 2 (lines, one-sample-per-population dataset; see Supplementary Data 7) or with fastsimcoal2 (diamonds, 2-epoch model and one-sample-per-population dataset; see Supplementary Data 5 and 6). The median changes in Ne are reported from both methods. B A zoom-in of (A) focusing on the 0–1.8 Mya period. Blue shaded rectangles delineate glacial periods. Timing of the glacial periods was inferred from composite CO2 records publicly available at (https://www.ncei.noaa.gov/). C Heatmap based on Kendall’s correlation coefficients computed from changes in Ne through time between each pair of species. The order of species along the x-axis is the same as that along the y-axis. Blue and red colors represent negative and positive correlation values, respectively. Source data are provided as a Source Data file-1.
Crucially, these patterns suggest that the overall genetic diversity of each species, or equivalently for neutral loci their Ne, has been maintained even during the massive range contractions caused by glacial advances. In other words, dominant forest trees with large ranges, large Nc, and efficient gene flow have retained genetic diversity over long periods of time, despite regional extinctions during ecologically unfavorable periods. This contrasts with previous studies that generally tended to consider the LGM as the major cause of genetic change in forest tree species in Europe. Simulations of unstructured populations undergoing cyclic 10-fold demographic changes confirm that Nc fluctuations do not translate in fluctuating SFS-based Ne trajectories when Ne remains large (>100,000) and generation time is long (>=15 years) but rather suggest an ancient decay of Ne (Figs. S28–S31).
Synchronicity and idiosyncrasy in changes in Ne
Across the seven species, the changes in Ne over time were not entirely species-specific (Figs. 3C, S32 and Table S2). Instead, three groups of species could be distinguished based on their demographic trajectories (Fig. 3C). A first group included the three boreal species (P. abies, P. sylvestris and B. pendula) and the riparian P. nigra, a second group comprised the two major temperate broadleaves (F. sylvatica and Q. petraea), and the only Mediterranean conifer, P. pinaster, constituted a group of its own. Interestingly, this grouping differs from the one established above based on current nucleotide diversity and population genetic structure. Neither patterns of Ne of individual species nor synchronous changes align well with the known glacial and interglacial periods (Fig. 3). This contrasts with some earlier results of individual forest tree species (e.g., refs. 14, 19, 37), but is in line with other comparative studies8,9. The overall lack of correlation between changes in Ne and climatic oscillations suggest that forest tree populations remained highly interconnected and acted as a single, large metapopulation, whose Ne was less affected by climatic fluctuations than Nc. Still, repeated synchronous phases of slight decrease in Ne can be detected despite the global increasing trends, likely being the signature of shared, recurrent and possibly delayed influence of glacial cycles on these metapopulations (Fig. S32 and Table S2).
Discussion
Current population genetic diversity is the result of long-term processes
Our study demonstrates that these tree species have been able to retain their evolutionary potential through multiple glacial cycles. This is in agreement with recent studies showing the ability of tree species to rapidly respond to environmental challenges4,6 and to swiftly colonize new areas as they become suitable38. This potential likely reflects their unique biological features. Very large and genetically connected populations along with long generation time—hence, a limited number of generations with elevated drift—allowed forest tree species to retain genetic diversity through time, despite intermittent, substantial geographical range contractions. This diversity has taken shape and accumulated over very long periods of time, involving multiple glacial–interglacial cycles. Similarly, an investigation of the Distribution of Fitness Effects (DFE) of new mutations for a subset of the same data showed differences in DFE between species but no differences among populations within species. This finding also suggests that differences in DFE parameters accrue over long time periods39 and essentially reflect the collecting phase of the coalescent process of a many-demes model31,32. It is worth pointing out that the seven species considered in this study are all widely distributed, relatively abundant, and ecologically important species of European forests. As such, they are interesting and important models to study the effects of environmental factors on the evolutions of European forests across space and time. However, they do not form a representative sample of the modern European tree flora. Furthermore, current species are those that survived past mass extinctions. At the Pliocene-Pleistocene transition the climate in the northern hemisphere changed dramatically with the onset of the glacial-interglacial periods, resulting in large scale extinction of trees, especially in Europe40. The modern flora represents less than 30% of the tree genera present during the Tertiary41,42. Extinctions eliminated deterministically cold sensitive species43, and following episodes of selection further favored species sharing invasiveness attributes44 (prolificity, competitive ability, dispersal) that facilitated locally the replacement of the extinct species.
While the use of pollen and macrofossil records as well as low-resolution uni-parentally inherited organellar DNA markers in previous studies revealed post-LGM migration patterns11, these approaches did not give a complete understanding of the dynamics of genetic diversity over time, simply because both data types cover a too short time period. Pollen records are informative on past plant population movements but chronologies of tree pollen records are limited in time45, often uncertain especially at the species level46, and heterogeneous across space47. Further, they comprise no information on within-species genetic relationships. Long-lived organisms, such as forest trees, have fewer generations per glacial cycle than annual and herbaceous species, and therefore will, for the same number of generations, experience a larger number of glacial cycles. Simulations of demographic changes following glacial cycles show that inferred Ne trajectory of annuals can capture the demographic fluctuations of the last glacial-interglacial cycle. However, it is not the case for organisms with much longer generation time like trees (Figs. S28–S31). On the evolutionary time scale, glacial cycles are shorter and recur faster for species with longer generation time and larger Ne. Hence, as was indeed observed, the current structure of genetic diversity reasonably reflects the impact of many glacial cycles (e.g., refs. 48, 49). Importantly, our results explain how tree species that have survived repeated glacial cycles were able to retain genetic diversity and, hence, a capacity to respond to new environmental challenges. However, genetic stability across millions of years does not exclude drastic changes in the short term, e.g., in species distributions and local abundances, which can have major impacts on ecosystem and forest functions.
Since we focused on inferring these demographic events based on current genetic diversity, we disregarded individuals with a high degree of admixture. However, in most plant species, including trees, hybridization contributes significantly to genetic diversity14,50,51. Among species included here, this primarily holds for Q. petraea52, which shows high nucleotide diversity (Fig. 1H) among the seven species we studied, but it may also be relevant for others that show hybridization at least in parts of their ranges or have hybridized in the past (Table 1). For demographic inference, hybridization introduces signals of even older evolutionary events and leads to elevated estimates of Ne. For predicting population responses to climate change, more information on groups of closely related species will be essential, especially as introgression can be important in environmental adaptation53.
Changes in Ne are not only driven by glacial cycles
The genetic diversity of all seven species was maintained across long periods of time: most of the time, Ne either increased or remained stable. However, we did not observe a single shared dynamic of changes in Ne across the seven species, which would be expected if glacial cycles were the main drivers. Instead, there were three clear species groups based on Ne change over time (Fig. 3C). In this respect, our results are congruent with previous studies that also showed a tendency to low levels of overall synchronicity among species and even populations within species (8,9 and references therein). Bai et al. 8 carried out a comparative demographic study in walnut species and showed that the timing and amplitude of changes in Ne differed among species. They concluded that the population histories of these walnut species were not driven by extrinsic environmental changes alone and that different species responded idiosyncratically to similar environmental challenges8. Similarly, ref. 9 reconstructed the trajectories of Ne of three palm species and four Annonaceae tree species from African rainforest. Here too, evolutionary responses were largely asynchronous and individualistic. Interestingly, the three palm species had large Ne (around 500,000) that increased regularly through time, as observed for our seven temperate forest tree species. This was in contrast to the four Annonaceae species, whose Ne were significantly smaller (around 50,000) and fluctuated strongly over the same time period. In the present study, the peculiarity of P. pinaster likely mirrors its more southern distribution as well as its highly fragmented range compared to the other six species while the synchronicity between Q. petraea and F. sylvatica likely finds its origin in similarities in both biology (e.g., generation time, ecological niche, dispersal) and geographical distribution. The recovered demographic histories of Q. petraea and F. sylvatica go back much further in time (i.e., ~32 and ~17 Mya, respectively) than for the other studied species. The depth of the Q. petraea genealogy and its relatively large Ne are likely a consequence of continuous hybridization with other abundant and closely related white oak species54, combined with long generation times. Fagus sylvatica may have had small, secondary refugia outside the core refugial areas, maintaining local reservoirs of genetic diversity across glacial cycles47,55. The synchronicity of the three boreal species also reflects some similarities in both biology and geographical distribution, although they also show marked differences in population genetic estimates. One explanation for the grouping of P. nigra with the boreal species could be that as a riparian species it was less affected by global climatic patterns and could have survived glacial times in microrefugia close to rivers, also in colder regions. Finally, it is worth pointing out that the two pine species, P. pinaster and P. sylvestris, were very distinct from each other, despite similar mutation rates, rates of adaptive evolution and generation times, reflecting their different ecological characteristics and geographic ranges5,56.
Ne estimates and their timing scale according to the assumed mutation rates. We used the best current estimates obtained from forest trees based (see detail below). However, it is possible that the scaling of events may extend or compress across the timeline as more precise estimates of mutation rates and generation time become available. It is noteworthy that the actual Ne trajectories are not affected by the mutation rate, just their scaling.
The present study highlights the existence of some commonalities in the Ne trajectories of seven major European forest tree species. Firstly, the Ne of all seven species we analyzed showed a mostly monotonic growth over a large part of the Quaternary. Hence, the genetic diversity of large tree metapopulations has been strikingly resilient to the drastic environmental changes during which the species experienced regional extinctions and extensive shifts in species distributions. Secondly, the trajectories of Ne through time were correlated within groups of species sharing ecological and biogeographical properties, but not across all species. This supports the idea that changes in Ne are not solely driven by climatic events but also reflect species’ shared biological characteristics such as life history or physiological properties. For instance, two species, such as B. pendula and P. abies, can have rather different extant population genetic structure across their similar distribution ranges, and yet have highly similar Ne trajectories.
Finally, it has been suggested that understanding individual species’ responses to past climatic oscillations is critical to predict their ability to cope with climate change9. Our results, in contrast, suggest that species’ idiosyncratic responses, while highly desirable to understand for other purposes, may not be required for predicting their evolutionary response to ongoing rapid climate change. However, recovering the evolutionary response of a wider spectrum of species would be needed to establish a reliable typology of demographic histories and their effects on genetic diversity.
Methods
Sampling
We sampled seven tree species of considerable economic and/or ecological importance in Europe: B. pendula, F. sylvatica, P. nigra, P. abies, P. pinaster, P. sylvestris and Q. petraea. For each species, we sampled a minimum of 20 populations (Table S1, Supplementary Data 1) from across the species’ natural distribution ranges (Fig. S1). Sampling was carried out within the framework of the EU Horizon 2020 project GenTree. The majority of the sampled populations are the same as reported in ref. 22, with additional samples reported in Table S1. Sampling principles and details are described in ref. 22. We dried the samples with silica gel and collated them in a single lab per species, where we extracted DNA (see Table S3 for details). We eluted the DNA in water, quality-checked it by UV spectrophotometry, and treated it with RNAse. We then sent all DNA extracts to IGA Technology Services (Udine, Italy) for targeted sequencing.
Sequencing and SNP calling
We focused on a limited part of the genome (3 Mbp) using targeted sequencing. The targeted regions consisted of orthologous genes involved in putative functions of interest, species-specific candidate genes, and randomly selected genes (Table S4). We first established a list of 2639 genes involved in functions of interest (e.g., response to stress, immune response, circadian clock, detection of abiotic stimulus) in Arabidopsis thaliana using a term search in Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (Supplementary Data 8). For each species independently, we then identified putative orthologs to those 2639 genes using a reciprocal best hit approach based on protein sequences (Blastp, BLAST v.2.5.0 +). Finally, we used Orthofinder v.1.1.457 on the complete set of putative orthologs to define orthogroups across the seven species. The Quercus robur reference genome was used to identify orthologs for Q. petraea, a closely related species with which it often hybridizes. The Populus trichocarpa reference genome was used for P. nigra. We selected 811 best orthogroups that included at least one gene for at least six of the seven tree species (Supplementary Data 9). For some species, we also included a variable number of other orthogroups including genes that were found only in a reduced set of species (e.g., 59 additional genes across the three conifer species; Supplementary Data 10). For each species, we then completed the ortholog list with genes of interest and randomly selected genes to reach up to 6 Mbp of sequence to serve as a template for probe definition. Starting from the 6 Mbp of sequence mentioned above, Roche designed a set of uniquely mapping probes based on either a reference genome or coding sequence (CDS) data coordinates (Table S4), relying on its custom probe design pipeline (454 Life Sciences, a Roche company, Branford, CT, USA) including the SSAHA algorithm58. We then restricted candidate probes to cover 3 Mbp of sequence, prioritizing probes covering best-ortholog genes (Table S5).
To estimate the quality of genomic DNA, we quantified random samples from each 96-well plate using a Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA, USA) and a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). We quantified all 4754 samples using the GloMax Explorer System (Promega Corporation, Madison, WI, USA) and prepared libraries for target enrichment using the SeqCap EZ – HyperPlus kit (Roche Sequencing Solutions, Pleasanton, CA, USA) with 100 ng/µl of input DNA, following the manufacturer’s instructions. For P. abies we conducted a second round of library preparation using 200 ng/µL of input DNA. We evaluated library size using the Bioanalyzer High Sensitivity DNA assay (Agilent Technologies, Santa Clara, CA, USA) and quantified libraries using a Qubit 2.0 Fluorometer. We sequenced the libraries on a HiSeq 2500 (125 cycles per read) for P. abies and B. pendula and on a NovaSeq 6000 (Illumina, San Diego, CA, USA; 150 cycles per read) for the remaining species, in both cases working in paired-end mode. We used Illumina bcl2fastq v.2.20 for base calling and demultiplexing, and we used ERNE v.1.4.659 and Cutadapt60 for quality and adapter trimming, both with default parameters.
For mapping we used BWA mem v.0.7.1761 and samtools v.1.762 against the available reference genome of the same or closely related species, adding the mitochondrial and chloroplast genomes when those were missing from the reference (Tables S6–S7, see https://github.com/GenTree-h2020-eu/GenTree/blob/master/Alignment_commands.txt for exact commands). In brief, we removed reads mapping on the organellar genomes, multiple mapping reads using samtools, marked duplicates with Picard and removed them. We maintained only a relevant portion of the genome with sufficient depth (>5n, where n = sample size) for the next step to speed up the SNP calling stage, which can be computationally demanding for large, fragmented genomes.
We performed SNP calling using the software package GATK v.4.0.10.059. Briefly, we ran HaplotypeCaller in GVCF mode to call potential variant sites at the single-sample level, then used GenomicsDBImport and GenotypeGVCFs to perform joint genotyping on the entire cohort of samples. Command lines are available at https://github.com/GenTree-h2020-eu/GenTree/blob/master/SNP_calling_commands.txt.
QC and SNP filtering
We conducted initial quality control of the SNP data using PCA and ADMIXTURE. We removed non-desired samples, e.g., misidentified species or obvious hybrids. We also removed samples with excessive amounts of missing data ( 60% for all species except B. pendula where the threshold was 20%) or extreme values of heterozygosity ( 6% of the calls heterozygous) for P. nigra and B. pendula. Jupyter notebooks of initial quality control are available at https://github.com/GenTree-h2020-eu/GenTree/tree/master/cervantesarango/JupyterNotebooks.
We retained bi-allelic variants and followed GATK recommendations to exclude poorly supported SNPs with scores QD < 0.25, QUAL < 20, SOR > 3.0, MQ < 30, MQRankSum < −12.5 and/or ReadPosRankSum < −8.0. Filtering code is available at https://github.com/GenTree-h2020-eu/GenTree/tree/master/cervantesarango/GATK_rawSNPs_to_v2. We identified putative false SNPs derived from paralogs mapping to a single location in the reference genome. We based the identification on heterozygote excess (H > 0.6) and deviation from the expected read ratio (D < −20 or D > 20) using the HDplot method https://github.com/GenTree-h2020-eu/GenTree/tree/master/rellstab63.
We further utilized the location information of each SNP to delineate regions with an especially large proportion of paralog-derived SNPs
| 1 |
and excluded all additional polymorphic positions included in those regions (https://github.com/GenTree-h2020-eu/GenTree/tree/master/kastally/paralog_window_filtering).
In addition to the variant-level filtering described above, we applied genotype-level filtering. We reported genotype calls with depth (DP) < 8 or genotype quality (GQ) < 20 as missing data to ensure high-quality genotypes64, and we filtered out SNPs with >50% missing calls to produce the v.5.3.2 VCF files.
To estimate the size of the portion of the genome sequenced with sufficient quality and depth across individual of each species independently (available genome, Table S7), we applied the same filtering procedure to the monomorphic positions, removing positions with DP < 8 and GQ < 20. Additionally, we excluded the same areas enriched with paralogs. The exact limit of those areas was defined at mid-distance between the last paralog and the next retained SNP.
Site-based annotation (4-fold degenerate, 2–3-fold degenerate and 0-fold degenerate sites) of detected SNPs was completed using the python script NewAnnotateRef.py available at https://github.com/fabbyrob/science/blob/master/pileup_analyzers/NewAnnotateRef.py.
SNPs were classified as intergenic, intron, stop, up and down using ANNOVAR65 (Tables S8 and S9).
SFS scaling
Inference of demographic history can be done using SFS28,66. The SFS must be estimated on a sample of a given size for all sites, however, in real datasets sample size varies among sites due to missing data. We therefore used the SNP set v.5.3.2 (see Supplementary Methods), removed SNPs with >50% of missing data in any population and down-sampled the SFS for each population or subset used for demographic inference (see demographic analyses section for details) to half the initial sample size. The source code for resampling the SFS is available at https://github.com/GenTree-h2020-eu/GenTree/tree/master/kastally/sfs_resampling. The SFSs produced were then used in downstream analyses (see below).
Population genetic structure and isolation-by-distance
To characterize the main genetic clusters among populations, we used SNP from dataset v.6.3.2 with only putatively neutral SNPs (i.e., 4-fold degenerate sites or located in introns or intergenic regions), pruned for SNPs in high linkage disequilibrium and excluding singletons (plink v.1.9.), to compute ancestry components (Q score) for each individual using ADMIXTURE v.1.367. We performed an unsupervised analysis of individual ancestry proportions based on maximum likelihood and implemented 20 replicates for each K value (1–12) to assess cross-validation errors. Then, we averaged the Q scores of individuals for each population and visualized the geographic distribution of the genetic groups identified in ADMIXTURE using the raster R package v.3.1.567.
We calculated genetic distances with dataset v.6.3.1 based on pairwise FST values68, implemented with the stamppFst function of the StAMPP R package v.1.6.369. We then performed a hierarchical analysis of molecular variance (AMOVA), using the poppr.amova function of the poppr R package v.2.9.370, to assess the partitioning of genetic variation: (i) between populations, (ii) between individuals within populations, and (iii) within individuals. We tested levels of significance using the randtest function of poppr.
We quantified the pattern of isolation-by-distance (IBD) with dataset v.6.3.1 by regressing the genetic distance between pairs of populations (FST68) over the natural logarithm of the geographic distance between populations, following Rousset’s71 approach for a two-dimensional stepping-stone model.
| 2 |
For each pair of populations i, we estimated FST using vcftools (v.0.1.13)72. xi is the geodesic distance separating the pair of populations in km (geosphere R package v.1.5-1073), β is the slope of the regression, α is the intercept, and ε is the error term.
Principal component analysis
In order to obtain a general picture of the main population structure of each species, we conducted a PCA with EIGENSOFT and default parameters (v.7.2.0, https://github.com/DreichLab/EIG). For each species we used all populations included in the dataset v5.3 and kept only 4-fold, intronic and intergenic SNPs pruned for high linkage disequilibrium. The whole procedure was repeated using dataset v.6.3.1 but removing the most divergent populations to visualize more subtle population structure.
Scaling FST and computing population-specific FST
To investigate the change in population differentiation along latitude, longitude and elevation, we computed the average population-specific FST scaled by the natural logarithm of the geographic distance. For each focal population, we computed the average pairwise FST divided by the average distance (km) between each pair of populations in which the focal population was involved.
Measuring genetic diversity
We estimated the pairwise nucleotide diversity, π, for 4-fold (π4) and 0-fold sites (π0) as well as for silent sites (πs, comprising intergenic, intronic and 4-fold sites). Based on the allele frequencies obtained from the SFS after down-sampling per species and per population based on SNP set v.5.3.2 (Table S10), we estimated the expected heterozygosity for each polymorphic site and summed the resulting values over all segregating sites. To obtain a π value per site, we divided this sum by the total number of sites, including monomorphic sites:
| 3 |
where n is the sample size, i.e., the number of allele copies, S is the number of segregating sites, pji is the allele frequency at a polymorphic site i for the jth allele, and L is the total number of sites (polymorphic and monomorphic) in a class. Nucleotide diversity was estimated from and averaged over 1000 resampled replicates using https://github.com/GenTree-h2020-eu/GenTree/blob/master/kastally/sfs_resampling/vcf2sfs_resample.R and https://github.com/GenTree-h2020-eu/GenTree/blob/master/kbudde/pi_from_fsfs.R.
Demographic modeling
To infer past changes in the effective population size (Ne), we applied two approaches based on the SFS: model-based estimates using fastsimcoal266 and model-free estimates with Stairway Plot 2 v.2.1.128. For both approaches, we used folded and rescaled SFS (see above) based on 4-fold, intergenic and intronic sites (SNP set v5.3.2. without 0-fold sites). For the estimates using Stairway Plot 2, we applied different hierarchical levels and subsamples. First, we made stairway plots by pooling all the samples together by species, to maximize the power to detect relatively recent demographic events by using large sample sizes74. Considering that the SFS resulted from pooling all the samples together, different populations and especially population genetic clusters were not always equally represented, due to unequal missing data across populations and unequal representation (in terms of populations and individuals) of different clusters. To investigate the impact of hierarchical population clustering on the stairway plots, we reran Stairway Plot 2 at different hierarchical levels: (i) one sample per population (to account for unequal representation of populations) and (ii) separately for each population.
For the four broad-leaved species, we used the mutation rate of 7.77 × 10-9 per site per generation75 estimated for Prunus, which is close to estimates of the mutation rate per generation of 7 × 10-9 for A. thaliana76 and Silene latifolia77. To scale estimates of timing from generations to years, we assumed a generation time of 60 years for Q. petraea and F. sylvatica, and 15 years for P. nigra and B. pendula. For the three conifers, we used a mutation rate of 2.7 × 10-8 per site per generation78 which is consistent with earlier, divergence-based estimates, assuming a 25-year generation time17,79. We ran all stairway plot estimates using 67% of the sites for training and 200 resamplings from the SFS. We calculated breakpoints following the suggestions in the stairway plot manual, i.e., at n/4, n/2, n*¾ and n-2 with n indicating the sample size.
To confirm the results obtained with Stairway Plot 2, we used fastsimcoal225,66 to infer the past demographic history of each species. We used the same folded SFSs (fSFS) as for Stairway Plot 2. We explored three single population models (Fig. S24): an equilibrium model (standard neutral model, SNM), a model with one demographic change (epoch-2) and a model with two demographic changes (epoch-3). For all parameters (Ne at each step and each time of events, specifically: NCUR = most recent Ne; NANC = ancestral Ne; NBOT = Ne after the first demographic event in the 3-epoch model; TBOT = time of the first demographic event; TENDBOT: time of the second demographic event in the 3-epoch model), we set a log prior with a range of 10 to 107. We first inferred the best model by running 100 independent runs for each of the three models using fastsimcoal2 with 106 simulations (-n 1,000,000), a minimum of 10 conditional maximization algorithm (ECM) cycles for likelihood computations (-l 10), 40 ECM cycles for parameter estimations (-L 40), and a minimum of one allele count for parameter estimation (–minSFScount 1; default value). For each model, we then identified the run with the best likelihood score. We computed the AIC score for each run, and we compared the AIC values of different models. Finally, following Excoffier et al.66, we computed confidence intervals for each parameter, selecting the parameter values of the best run for both 2-epoch and 3-epoch models to simulate 100 SFSs using fastsimcoal2. We then ran, for each of those 100 SFSs, 100 independent runs of fastsimcoal2 using the same settings as before, obtaining 100 sets of parameters for each model, which we used to compute confidence intervals for each model parameter. We used this same approach for each species, using the SFS computed over all samples and the SFS computed using only one haplotype per population.
To further test if more realistic models would better fit the observed data and impact our inference of past demographic changes, we tested four models of demographic changes including events of divergence. For these models, we subset, for each species, samples from two locations, a southern and a northern one (we used admixture results to make sure that the samples came from two distinct gene pools; Figs. S3–S16). From these sets, we explored the same three models, with no divergence events and pooling all samples together regardless of origin, and four models of divergence differing regarding whether migration was possible after the divergence and including or not a demographic change before the divergence event. We used fastsimcoal2 to assess models, using the fSFS of all samples pooled together for reference models, and the joint fSFS of the population pairs for models of divergence. We conducted the simulations and inference with fastsimcoal2 by running 100 independent runs with 100,000 simulations each (-n 100,000), the same number of ECM cycles as used previously (-l 10 -L 40) and in folded mode (-m). We explored all parameters (including all or some of the following for the divergence models: NCUR = Ne before the split but after the demographic event; NANC = ancestral Ne; NPOP1 = Ne after the split for population 1; NPOP2 = Ne after the split for population 2; TDIV = time of the split; TSEP = time of the demographic event; N1M21 = effective migration rate from population 1 to population 2; N2M12 = effective migration rate from population 2 to population 1; and the same parameters as above for the panmictic models) with a logunif prior between 10 and 109, except for the parameters related to the number of effective migrants (the product between the migration rate and the effective population size, Ne × m), which used logunif priors set between 10-4 and 104. Finally, we compared the seven models using the likelihood and AIC score of the single best run out of 100, to identify the best model for each species and its associated parameters.
To test whether our results may be explained by a lack of statistical power, we tested the ability of Stairway Plot 2 to recover demographic cycles for a set of parameter values relevant to our data and model species: generation time (1, 15, 25 and 60 years per generation), size of the sampled genome (1 Mbp or 6 Mbp), mutation rates (2.7 × 10-8 or 7.7 × 10-9 per site per generation), and current Ne (103, 105, 106; the latest demographic event being an expansion, it is the highest Ne). The intensity of the expansion/contraction of population size was set to 10-fold, and we implemented 10 events (i.e., 5 cycles of an expansion event followed by a contraction event) following the expected times of the glacial / interglacial periods (i.e., a demographic expansion 15 kya, a decline 120 kya, then following a period of 120 kya, expansion or decline events successively, until a final decline about 1 Mya). Each event was modeled as an instantaneous change of Ne with Ne remaining stable in-between. These models were simulated with fastsimcoal2 to produce fSFS using a sample size of 20 haploid genomes. We then ran demographic analyses with Stairway Plot 2 on the simulated fSFS using the settings used with the empirical fSFS (4 breakpoints fixed at sample sizes following manual instructions and 200 simulations) (Figs. S28–S31). We also report nucleotide diversity (π) of each fSFS generated.
To assess the influence that unaccounted for population genetic structure may have on our analyses, we compare a set of three demographic inferences with Stairway plot 2 for each of the seven species. For the first one we artificially mixed two populations, the second and third ones correspond to the analysis of each population separately (Fig. S33). For each species, we selected the same pairs of populations as for the demographic modeling analyses (see above). In most cases, the inference is consistent across the mixed and separate analyses, suggesting that the level of population genetic structure we have in our data has limited effects.
Synchronicity analysis
We then assessed whether the dynamics of changes in Ne over time were species-specific or synchronous across species. To do so, we first computed Kendall’s correlation coefficients between the output of Stairway Plot 2 for each pair of species and then investigated their covariance across the seven species using a heatmap (heatmap3 R package, v.1.1.9 https://cran.r-project.org/package=heatmap3 with default parameters, Fig. 3C). This global approach allowed us to quantify the synchronous pattern in change in Ne over time between some species and test whether it was primarily driven by the global increase in Ne over the period studied. This approach captures the main trend but will not allow the identification of specific periods of high synchronicity between several species. In particular, it will miss decreases in Ne that could be expected to be associated with glacial periods. To specifically test whether periods over which the species experienced a decrease in Ne showed higher synchronicity than expected given the actual change in Ne over time for the various species, we used a randomization approach. This analysis was conducted independently for the two groups of species showing the highest synchronicity F. sylvatica and Q. petraea on the one hand, and P. abies, B. pendula, P. sylvestris and P. nigra, on the other hand.
More specifically, the Stairway Plot 2 output consists of a succession of intervals, defined by a given Ne estimate and two different time points. To simplify this, for each species independently, each interval was represented by a pair of values: the unique Ne value characteristic of the interval and the midpoint of the two time points. We excluded the most recent time point of the output of Stairway Plot 2 as it does not correspond to a proper step. As the time points at which Stairway Plot 2 provides estimates of Ne are species-specific, we inferred Ne at every time point in the joint dataset by considering for each species independently the value of the closest Ne estimated by Stairway Plot 2. To smooth random fluctuations between time points and to mitigate the effects associated with small deviations to our estimates of generation time and mutation rate, we averaged the changes in Ne between two time points (ΔNe = Ne(t) - Ne(t+1)) over 250 consecutive time points, using sliding windows (μΔNe). Finally, we used a randomization approach to detect periods of time during which the pattern of synchronicity in Ne change is stronger than what would be expected given species-specific change in Ne over time; i.e., periods over which a larger number of species experienced a decrease in Ne than expected if changes in Ne were independently distributed across time in each species. For each species independently, we first randomized the vector of ΔNe values and averaged the randomized values over 250 consecutive time points using sliding windows (μΔNe), as we did for the observed data. From the randomized time series, we recorded the maximum number of species experiencing a decrease simultaneously, as well as the longest span over which synchronicity was conserved (i.e., the maximum number of consecutive positive ΔNe). We repeated the whole procedure 10,000 times and compared the observed values with the 95% percentile of the distribution of the maximum values obtained through the 10,000 simulations (Table S2 and Fig. S32).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Source data
Acknowledgements
Computations were made possible using resources from projects SNIC 2017/7-328, SNIC 2020/15-107 and SNIC 2021/5-540 of the Swedish National Infrastructure for Computing (SNIC) at UPPMAX, partially funded by the Swedish Research Council through grant agreement no. 2018-05973. The CSC – IT Center for Science (Finland), the Genotoul Bioinformatics platform at Toulouse (France) and the Genetic Diversity Centre at ETH Zurich (Switzerland) are acknowledged for generous computational and storage resources. This work also benefited from the Scientific Compute Cluster at GWDG, the joint data center of the Max Planck Society for the Advancement of Science (MPG) and the University of Göttingen (Germany). We also thank all GenTree partners, the Slovenian Forestry Institute (Slovenian Research and Innovation Agency grant P4-0107 and LIFEGENMON) and the Natural Resources Institute Finland (LUKE) for their contribution to sampling. This work was funded by European Union’s Horizon 2020 Research and Innovation Programme grant agreement no. 676876; Academy of Finland grant nr. 287431 (T.P.); Swiss Secretariat for Education, Research and Innovation (SERI) contract no. 6.0032 (F.G.); Spanish Ministry of Agriculture, Fisheries and Food (MAPAMA) grant no. AEG 17-048 (D.G.); European Agricultural Fund for Rural Development (EAFRD) (D.G.); Swedish Research Council for Sustainable Development (FORMAS) grant nos. 2016-00780, 2020-01456 (M.L.); Spanish Ministry of Economy and Competitiveness (MINECO) contract n. PTA2015-10836-I (S.O.); LIFEGENMON (LIFE13 ENV/SI/000148) (M.W.); The Slovenian Research Agency, research core funding no. P4-0107 (M.W.); Biocenter Oulu (S.C.).
Author contributions
Conceptualization: St.C., S.C.G.M., D.G., M.L., P.M., T.P., and G.G.V. Methodology: K.B.B., P.F.R., S.C.G.M., C.K., M.L., P.M., S.P., T.P., C.R., and S.S. Software: K.B.B., B.D., C.K., S.P., T.P., S.S., and I.S. Validation: Sa.C., V.J., P.M., T.P., and S.S. Formal Analysis: F.B., K.B.B., Sa.C., B.D., P.F.R., S.C.G.M., D.I.O., V.J., C.K., I.L.K., P.M., L.O., S.P., C.P., T.P., C.R., O.R., and I.S. Investigation: F.B., S.C.G.M., D.G., F.G., V.J., M.L., I.L.K., P.M., S.O., L.O., C.P., T.P., C.R., I.S., and M.W. Resources: B.D., P.F.R., S.C.G.M., D.G., F.G., M.L., S.O., L.O., C.P., C.R., and I.S., Data curation: B.D., Sa.C., V.J., C.K., P.M., L.O., and T.P. Visualization: B.D., C.K., P.M., I.S., and M.W. Funding acquisition: St.C., B.F., S.C.G.M., D.G., F.G., M.L., S.O., L.O., T.P., G.G.V., and M.W. Project administration: St.C., B.F., S.C.G.M., F.G., M.L., L.O., T.P., and G.G.V. Supervision: F.B., S.C.G.M., F.G., V.J., M.L., L.O., C.P., T.P., C.R., G.G.V., and M.W. Writing – original draft: K.B.B., Sa.C., B.D., S.C.G.M., V.J., C.K., M.L., P.M., L.O., S.P., T.P., C.R., O.R., and I.S. Writing – review & editing: F.B., K.B.B., B.D., B.F., P.F.R., S.C.G.M., F.G., V.J., C.K., M.L., I.L.K., P.M., L.O., C.R., S.S., I.S., G.G.V., and M.W.
Peer review
Peer review information
Nature Communications thanks Olivier Hardy and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Funding
Open access funding provided by Uppsala University.
Data availability
The short read data generated in this study have been deposited to NCBI BioProjects under accession codes PRJNA602465, PRJNA602466, PRJNA602467, PRJNA602468, PRJNA602470, PRJNA602471, PRJNA602473. The vcf, ped and map -files generated in this study are available in Data INRAE at 10.57745/DV2X0M80. Source data are provided as a Source Data file. Source data are provided with this paper.
Code availability
Code is available at: 10.5281/zenodo.794387681.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Pascal Milesi, Chedly Kastally, Benjamin Dauphin, Sandra Cervantes, Martin Lascoux, Tanja Pyhäjärvi.
A list of authors and their affiliations appears at the end of the paper.
Contributor Information
Pascal Milesi, Email: pascal.milesi@scilifelab.uu.se.
Martin Lascoux, Email: martin.lascoux@ebc.uu.se.
Tanja Pyhäjärvi, Email: tanja.pyhajarvi@helsinki.fi.
On behalf of the GenTree Consortium:
Pascal Milesi, Chedly Kastally, Benjamin Dauphin, Sandra Cervantes, Francesca Bagnoli, Katharina B. Budde, Stephen Cavers, Bruno Fady, Patricia Faivre-Rampant, Santiago C. González-Martínez, Delphine Grivet, Felix Gugerli, Véronique Jorge, Isabelle Lesur Kupin, Dario I. Ojeda, Sanna Olsson, Lars Opgenoorth, Sara Pinosio, Christophe Plomion, Christian Rellstab, Odile Rogier, Simone Scalabrin, Ivan Scotti, Giovanni G. Vendramin, Marjana Westergren, Martin Lascoux, and Tanja Pyhäjärvi
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-52612-y.
References
- 1.Birks, H. J. B. & W, T. in European Atlas ofForest TreeSpecies (eds San-Miguel-Ayanz, J., de Rigo, D., Caudullo, G., Houston Durrant, T. & Mauri, A.) (Publication Office of the European Union, Luxembourg, 2016).
- 2.Lascoux, M., Palmé, A. E., Cheddadi, R. & Latta, R. G. Impact of ice ages on the genetic structure of trees and shrubs. Philos. Trans. R. Soc. Lond. Ser. B: Biol. Sci.359, 197–207 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kremer, A. How well can existing forests withstand climate change in: Climate Change and Forest Genetic Diversity: Implications for Sustainable Forest Management in Europe.pp. 3–17. (eds. Koskela, J., Buck, A. & Teissier du Cros, E.), (Bioversity International, Rome, 2007).
- 4.Saleh, D. et al. Genome-wide evolutionary response of European oaks during the Anthropocene. Evolu. Lett.6, 4–20 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alberto, F. J. et al. Potential for evolutionary responses to climate change - evidence from tree populations. Glob. Change Biol.19, 1645–1661 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li, L. et al. Teasing apart the joint effect of demography and natural selection in the birth of a contact zone. N. Phytol.236, 1976–1987 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waples, R. S. What is Ne, anyway. J. Heredity113, 371–379 (2022). [DOI] [PubMed] [Google Scholar]
- 8.Bai, W. et al. Demographically idiosyncratic responses to climate change and rapid Pleistocene diversification of the walnut genus Juglans (Juglandaceae) revealed by whole‐genome sequences. N. Phytol.217, 1726–1736 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Helmstetter, A. J., Béthune, K., Kamdem, N. G., Sonké, B. & Couvreur, T. L. P. Individualistic evolutionary responses of Central African rain forest plants to Pleistocene climatic fluctuations. Proc. Natl Acad. Sci. USA117, 32509–32518 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taberlet, P., Fumagalli, L., Wust-Saucy, A. & Cosson, J. Comparative phylogeography and postglacial colonization routes in Europe. Mol. Ecol.7, 453–464 (1998). [DOI] [PubMed] [Google Scholar]
- 11.Petit, R. J. et al. Glacial refugia: Hotspots but not melting pots of genetic diversity. Science300, 1563–1565 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Ballard, J. W. O. & Whitlock, M. C. The incomplete natural history of mitochondria. Mol. Ecol.13, 729–744 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Doyle, J. J. Defining coalescent genes: theory meets practice in organelle phylogenomics. Syst. Biol.71, 476–489 (2022). [DOI] [PubMed] [Google Scholar]
- 14.Liu, S. et al. Demographic history and natural selection shape patterns of deleterious mutation load and barriers to introgression across Populus genome. Mol. Biol. Evol.39, msac008 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olsson, S. et al. Evolutionary history of the mediterranean Pinus halepensis-brutia species complex using gene-resequencing and transcriptomic approaches. Plant Mol. Biol.106, 367–380 (2021). [DOI] [PubMed] [Google Scholar]
- 16.Mayol, M. et al. A multiscale approach to detect selection in nonmodel tree species: Widespread adaptation despite population decline in Taxus baccata L. Evolu. Appl.13, 143–160 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen, J. et al. Genomic data provide new insights on the demographic history and the extent of recent material transfers in Norway spruce. Evolu. Appl.12, 1539–1551 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hou, Z. & Li, A. Population genomics reveals demographic history and genomic differentiation of Populus davidiana and Populus tremula. Front. Plant Sci.11, 553736 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salojärvi, J. et al. Genome sequencing and population genomic analyses provide insights into the adaptive landscape of silver birch. Nat. Genet.49, 904–912 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Cai, M., Wen, Y., Uchiyama, K., Onuma, Y. & Tsumura, Y. Population genetic diversity and structure of ancient tree populations of Cryptomeria japonica var. sinensis based on RAD-seq data. Forests11, 1192 (2020). [Google Scholar]
- 21.Capblancq, T. et al. Whole‐exome sequencing reveals a long‐term decline in effective population size of red spruce (Picea rubens). Evolu. Appl.13, 2190–2205 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Opgenoorth, L. et al. The GenTree Platform: growth traits and tree-level environmental data in 12 European forest tree species. GigaScience10, giab010 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen, J., Glémin, S. & Lascoux, M. Genetic diversity and the efficacy of purifying selection across plant and animal species. Mol. Biol. Evol.34, 1417–1428 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Eckert, C. G., Samis, K. E. & Lougheed, S. C. Genetic variation across species’ geographical ranges: the central–marginal hypothesis and beyond. Mol. Ecol.17, 1170–1188 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Excoffier, L. & Foll, M. Fastsimcoal: a continuous-time coalescent simulator of genomic diversity under arbitrarily complex evolutionary scenarios. Bioinformatics27, 1332–1334 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Excoffier, L. et al. fastsimcoal2: demographic inference under complex evolutionary scenarios. Bioinformatics37, 4882–4885 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirao, A. S. et al. Genetic diversity within populations of an arctic-alpine species declines with decreasing latitude across the Northern Hemisphere. J. Biogeogr.44, 2740–2751 (2017). [Google Scholar]
- 28.Liu, X. & Fu, Y.-X. Stairway Plot 2: demographic history inference with folded SNP frequency spectra. Genome Biology21, 280 (2020). [DOI] [PMC free article] [PubMed]
- 29.Mazet, O. & Noûs, C. Population genetics: coalescence rate and demographic parameters inference. Peer Community J.3, e53 (2023). [Google Scholar]
- 30.Chikhi, L., Sousa, V. C., Luisi, P., Goossens, B. & Beaumont, M. A. The confounding effects of population structure, genetic diversity and the sampling scheme on the detection and quantification of population size changes. Genetics186, 983–995 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wakeley, J. & Aliacar, N. Gene genealogies in a metapopulation. Genetics159, 893–905 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wakeley, J. Nonequilibrium migration in human history. Genetics153, 1863–1871 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petit, R. J. & Hampe, A. Some evolutionary consequences of being a tree. Annu. Rev. Ecol. Evol. Syst.37, 187–214 (2006). [Google Scholar]
- 34.Pyhäjärvi, T., Kujala, S. T. & Savolainen, O. 275 years of forestry meets genomics in Pinus sylvestris. Evolu. Appl.13, 11–30 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palstra, F. P. & Fraser, D. J. Effective/census population size ratio estimation: a compendium and appraisal. Ecol. Evol.2, 2357–2365 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tyrmi, J. S. et al. Genomics of clinal local adaptation in Pinus sylvestris under continuous environmental and spatial genetic setting.G3: Genes, Genomes Genet.10, 2683–2696 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jaramillo-Correa, J. P. et al. Evolutionary rate and genetic load in an emblematic Mediterranean tree following an ancient and prolonged population collapse. Mol. Ecol.29, 4797–4811 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Dial, R. J., Maher, C. T., Hewitt, R. E. & Sullivan, P. F. Sufficient conditions for rapid range expansion of a boreal conifer. Nature608, 546–551 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.James, J. et al. Between but not within-species variation in the distribution of fitness effects. Mol. Biol. Evol.40, msad228 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rull, V. Quaternary ecology, evolution, and biogeography (Academic Press, 2020).
- 41.Eiserhardt, W. L., Borchsenius, F., Eiserhardt WL, B. F., Plum, C. M., Ordonez, A. & Svenning, J. C. Climate‐driven extinctions shape the phylogenetic structure of temperate tree floras. Ecol. Lett.18, 263–272 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Latham, R. E. & Ricklefs, R. E. Global patterns of tree species richness in moist forests: energy-diversity theory does not account for variation in species richness. Oikos67, 325–333 (1993). [Google Scholar]
- 43.Svenning, J. C. Deterministic Plio‐Pleistocene extinctions in the European cool‐temperate tree flora. Ecol. Lett.6, 646–653 (2003). [Google Scholar]
- 44.Lamarque, L. J., Delzon, S. & Lortie, C. J. Tree invasions: a comparative test of the dominant hypotheses and functional traits. Biol. Invasions13, 1969–1989 (2011). [Google Scholar]
- 45.Birks, H. J. B. Contributions of Quaternary botany to modern ecology and biogeography. Plant Ecol. Divers.12, 189–385 (2019). [Google Scholar]
- 46.Petit, R. J. et al. Identification of refugia and post-glacial colonisation routes of European white oaks based on chloroplast DNA and fossil pollen evidence. For. Ecol. Manag.156, 49–74 (2002). [Google Scholar]
- 47.Magri, D. et al. A new scenario for the Quaternary history of European beech populations: palaeobotanical evidence and genetic consequences. N. Phytol.171, 199–221 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Shalev, T. J. et al. The western redcedar genome reveals low genetic diversity in a self-compatible conifer. Genome Res.32, 1952–1964 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gugerli, F. et al. A range‐wide postglacial history of Swiss stone pine based on molecular markers and palaeoecological evidence. J. Biogeogr.50, 1049–1062 (2023). [Google Scholar]
- 50.Whitham, T. G., Morrow, P. A. & Potts, B. M. Conservation of hybrid plants. Science254, 5033 (1991). [DOI] [PubMed] [Google Scholar]
- 51.Fu, R. et al. Genome-wide analyses of introgression between two sympatric Asian oak species. Nat. Ecol. Evol.6, 924–935 (2022). [DOI] [PubMed] [Google Scholar]
- 52.Petit, R. J., Bodénès, C., Ducousso, A., Roussel, G. & Kremer, A. Hybridization as a mechanism of invasion in oaks. N. Phytol.161, 151–164 (2004). [Google Scholar]
- 53.Suarez-Gonzalez, A., Lexer, C. & Cronk, Q. C. B. Adaptive introgression: a plant perspective. Biol. Lett.14, 20170688 (2018). [DOI] [PMC free article] [PubMed]
- 54.Leroy, T. et al. Extensive recent secondary contacts between four European white oak species. N. Phytol.214, 865–878 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Magri, D. Patterns of post‐glacial spread and the extent of glacial refugia of European beech (Fagus sylvatica). J. Biogeogr.35, 450–463 (2008). [Google Scholar]
- 56.Grivet, D. et al. High rate of adaptive evolution in two widespread European pines. Mol. Ecol.26, 6857–6870 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Emms, D. M. & Kelly, S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol.16, 1–14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ning, Z., Cox, A. J. & Mullikin, J. C. SSAHA: a fast search method for large DNA databases. Genome Res.11, 1725–1729 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McKenna, A. et al. The genome analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res.20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J.17, 10–12 (2011). [Google Scholar]
- 61.Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997 (2013).
- 62.Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience10, giab008 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McKinney, G. J., Waples, R. K., Seeb, L. W. & Seeb, J. E. Paralogs are revealed by proportion of heterozygotes and deviations in read ratios in genotyping-by-sequencing data from natural populations. Mol. Ecol. Resour.17, 656–669 (2017). [DOI] [PubMed] [Google Scholar]
- 64.Carson, A. R. et al. Effective filtering strategies to improve data quality from population-based whole exome sequencing studies. BMC Bioinform.15, 1–15 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res.38, e164–e164 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Excoffier, L., Dupanloup, I., Huerta-Sánchez, E., Sousa, V. C. & Foll, M. Robust demographic inference from genomic and SNP data. PLoS Genet.9, e1003905 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Alexander, D. H. & Lange, K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinform.12, 1–6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weir, B. S. & Cockerham, C. C. Estimating F-statistics for the analysis of population structure. Evolution38, 1358–1370 (1984). [DOI] [PubMed] [Google Scholar]
- 69.Pembleton, L. W., Cogan, N. O. I. & Forster, J. W. St AMPP: an R package for calculation of genetic differentiation and structure of mixed‐ploidy level populations. Mol. Ecol. Resour.13, 946–952 (2013). [DOI] [PubMed] [Google Scholar]
- 70.Kamvar, Z. N., Tabima, J. F. & Grünwald, N. J. Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ2, e281 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rousset, F. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics145, 1219–1228 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Danecek, P. et al. The variant call format and VCFtools. Bioinformatics27, 2156–2158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hijmans R. J. Spherical Trigonometry, R package geosphere version 1.5–10. (2019).
- 74.Keinan, A. & Clark, A. G. Recent explosive human population growth has resulted in an excess of rare genetic variants. Science336, 740–743 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xie, Z. et al. Mutation rate analysis via parent–progeny sequencing of the perennial peach. I. A low rate in woody perennials and a higher mutagenicity in hybrids. Proc. R. Soc. B: Biol. Sci.283, 20161016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ossowski, S. et al. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science327, 92–94 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Krasovec, M., Chester, M., Ridout, K. & Filatov, D. A. The mutation rate and the age of the sex chromosomes in Silene latifolia. Curr. Biol.28, 1832–1838.e4 (2018). [DOI] [PubMed] [Google Scholar]
- 78.Hanlon, V. C. T., Otto, S. P. & Aitken, S. N. Somatic mutations substantially increase the per-generation mutation rate in the conifer Picea sitchensis. Evol. Lett.3, 348–358 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Willyard, A., Syring, J., Gernandt, D. S., Liston, A. & Cronn, R. Fossil calibration of molecular divergence infers a moderate mutation rate and recent radiations for Pinus. Mol. Biol. Evol.24, 90–101 (2007). [DOI] [PubMed] [Google Scholar]
- 80.Pyhäjärvi, T. et al. Gene sequence variation data for 3407 adult individuals from 164 range-wide populations of 7 widely distributed European forest tree species, 10.57745/DV2X0M (2023).
- 81.PyhaTanja et al. GenTree-h2020-eu/GenTree: GenTree - Milesi et al. 2023 v1.0 (v1.0), https://zenodo.org/records/7943876 (2023).
- 82.Caudullo, G., Welk, E. & San-Miguel-Ayanz, J. Chorological data for the main European woody species. Mendeley DataV18, https://data.mendeley.com/datasets/hr5h2hcgg4/18 (2024). [DOI] [PMC free article] [PubMed]
- 83.Lapierre, M., Lambert, A. & Achaz, G. Accuracy of demographic inferences from the site frequency spectrum: the case of the Yoruba population. Genetics206, 439–449 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nawa, N. & Tajima, F. Simple method for analyzing the pattern of DNA polymorphism and its application to SNP data of human. Genes Genet. Syst.83, 353–360 (2008). [DOI] [PubMed] [Google Scholar]
- 85.Achaz, G. Frequency spectrum neutrality tests: one for all and all for one. Genetics183, 249–258 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The short read data generated in this study have been deposited to NCBI BioProjects under accession codes PRJNA602465, PRJNA602466, PRJNA602467, PRJNA602468, PRJNA602470, PRJNA602471, PRJNA602473. The vcf, ped and map -files generated in this study are available in Data INRAE at 10.57745/DV2X0M80. Source data are provided as a Source Data file. Source data are provided with this paper.
Code is available at: 10.5281/zenodo.794387681.