

Abstract
Ferroptosis is a form of regulated cell death, characterized by excessive iron-dependent lipid peroxidation. Biochemically, ferroptosis can be selectively induced by erastin through glutathione depletion or through inhibition of glutathione peroxidase 4 by RSL3, which leads to accumulation of cytotoxic lipid reactive oxygen species (ROS). Protein disulfide isomerase (PDI) was recently shown to mediate erastin/RSL3-induced ferroptosis and thus also become a new target for protection against chemically-induced ferroptosis. The present study aims to identify endogenous compounds that can protect against erastin/RSL3-induced ferroptotic cell death. We find that 2-hydroxyestrone, 2-hydroxyestradiol, 4-hydroxyestrone and 4-hydroxyestradiol, four major endogenous catechol estrogens, are effective inhibitors of PDI, and can strongly protect against chemically-induced ferroptotic cell death in cultured HT22 mouse hippocampal neuronal cells. The CETSA assay showed that these catechol estrogens can bind to PDI in live cells. PDI knockdown attenuates the protective effect of these catechol estrogens against chemically-induced ferroptosis. Mechanistically, inhibition of PDI’s catalytic activity by catechol estrogens abrogates erastin/RSL3-induced dimerization of nitric oxide synthase, thereby preventing the subsequent accumulation of cellular nitric oxide, ROS and lipid-ROS, and ultimately ferroptotic cell death. In addition, joint treatment of cells with catechol estrogens also abrogates erastin/RSL3-induced upregulation of nitric oxide synthase protein levels, which also contributes to the cytoprotective effect of the catechol estrogens. In conclusion, the present study demonstrates that the catechol estrogens are protectors of HT22 neuronal cells against chemically-induced ferroptosis, and inhibition of PDI’s catalytic activity by these estrogens contributes to a novel, estrogen receptor-independent mechanism of cytoprotection.
Keywords: Protein disulfide isomerase, Catechol estrogens, Nitric oxide synthase, Nitric oxide, Reactive oxygen species, Lipid reactive oxygen species, Cytoprotective action
Subject terms: Cell death, Mechanism of action, Cell biology
Introduction
Ferroptosis is a form of iron-dependent regulated cell death1–4 and is morphologically distinct from apoptosis-associated changes1,2,5,6. Many studies have shown that ferroptosis is closely related to many human diseases, such as ischemic brain injury, ischemic heart disease, renal failure and drug-induced liver injury7–9. In addition, there is also evidence linking ferroptotic neuronal death to neurodegeneration10,11. Biochemically, ferroptosis is usually associated with glutathione (GSH) depletion and/or inhibition of glutathione peroxidase 4 (GPX4), which is an antioxidant enzyme that quenches phospholipid hydroperoxides12. Erastin, a selective inhibitor of the cystine-glutamate antiporter (system Xc‒), induces ferroptosis by blocking the influx of extracellular cystine5. Depletion of cellular cystine leads to blockage of the cellular synthesis of cysteine and GSH, and ultimately, GSH depletion3,13–15. GSH depletion is associated with GPX4 inhibition and accumulation of iron-dependent lipid reactive oxygen species (lipid-ROS)11,16–19. In comparison, RSL3 induces ferroptosis by directly inhibiting GPX4, resulting in accumulation of lipid-ROS4,6,13,19. In addition, RSL3 can also directly inhibit the catalytic activity of thioredoxin reductase 1 (TrxR1)20. Recently, we have shown that inhibition of TrxR1 by RSL3 leads to activation (i.e., formation of the disulfide bond in its active site) of protein disulfide isomerase A1 (PDI), which then contributes to RSL3-induced ferroptosis through activation of the PDI → NOS → NO → ROS/lipid-ROS pathway21.
Protein disulfide isomerase (PDI or PDIA1) is a ubiquitous dithiol/disulfide oxidoreductase of the thioredoxin superfamily22–24. Primarily localized in the endoplasmic reticulum of mammalian cells, a small fraction of PDI is also found in the nucleus, cytosol, mitochondria, plasma membrane and extracellular space25. PDI is involved in protein processing by catalyzing the formation of intra- and inter-molecular disulfide bridges in proteins26. PDI in its reduced state can be oxidized (i.e., formation of a disulfide bond in its catalytic site), which is catalyzed by the endoplasmic reticulum oxidoreductin 1 (ERO1)27,28, and the oxidized PDI can be reduced by TrxR129. Our earlier studies have shown that while inhibition of ERO1 (which inhibits PDI activation) partially protects against erastin-induced ferroptosis30,31, inhibition of TrxR1 (which favors PDI in its oxidized state) enhances RSL3-induced ferroptosis21.
Previous studies have shown that the accumulation of cellular ROS and lipid-ROS is a hallmark of chemically-induced ferroptosis32–34. In a recent study, we demonstrated that PDI plays an important role in driving chemically-induced ferroptosis in immortalized HT22 mouse hippocampal neuronal cells by catalyzing the dimerization of nitric oxide synthase (NOS) and subsequently nitric oxide (NO) accumulation, which then leads to accumulation of cellular ROS and lipid-ROS21,31. In addition, earlier we have also shown that PDI is required for nNOS dimerization during glutamate-induced GSH depletion, which ultimately leads to O2·‒ accumulation and oxidative cytotoxicity in HT22 cells35. Inhibition of PDI effectively abrogated nNOS dimerization, which is associated with a strong protection against chemically-induced, GSH depletion-associated ferroptosis21,31,35,36. These lines of evidence highlight PDI as an important mediator of GSH depletion-associated ferroptosis and also an important target for protection against ferroptotic cell death.
Estrone (E1) and 17β-estradiol (E2) are major endogenous estrogens, and they are metabolized via hydroxylation to their respective catechol estrogen derivatives (i.e., 2-OH-E1, 2-OH-E2, 4-OH-E1 and 4-OH-E2)37,38. The metabolic reactions are catalyzed mostly by cytochrome P450 enzymes present in the liver (e.g., CYP3A4 and CYP1A2) and in extrahepatic tissues (e.g., CYP1A1 and CYP1B1)37. These catechol estrogens can be further O-methylated by catechol-O-methyltransferase (COMT) to form respective mono-methoxy metabolites, such as 2-methoxyestrone, 2-methoxyestradiol, 4-methoxyestrone and 4-methoxyestradiol37–39.
Previous studies have shown that some of the endogenously-formed estrogen metabolites, including those mentioned above, exhibit certain biological actions that are not mediated by the classical estrogen receptors α and β40,41. For instance, previously we have shown that 4-OH-E1, one of the catechol estrogen metabolites38, has a strong cytoprotective effect against glutamate-induced neuronal death42 and erastin-induced ferroptosis36, and the cytoprotective effect of 4-OH-E1 is independent of the estrogen receptors36. The strong cytoprotective effect of 4-OH-E1 has also been demonstrated in vivo42. These earlier observations are the basis as well as impetus for our present study, which sought to study the protective effect of 2-OH-E1, 2-OH-E2, 4-OH-E1 and 4-OH-E2, four endogenous catechol estrogens, against erastin/RSL3-induced ferroptosis in cultured neuronal cells, with a focus on determining the mechanism of their neuroprotective actions. We find that 2-OH-E1, 2-OH-E2, 4-OH-E1 and 4-OH-E2 each have a strong protective effect against erastin/RSL3-induced ferroptosis in HT22 cells. Mechanistically, these catechol estrogens selectively target and inhibit PDI’s enzymatic activity, and abrogate PDI-mediated nNOS and iNOS dimerization and NO accumulation, which then leads to reduction in cellular ROS and lipid-ROS accumulation. In addition, these catechol estrogens also abrogate erastin/RSL3-induced increase in cellular nNOS and iNOS protein levels. It is believed that these effects of catechol estrogens jointly contribute to their strong protective effect against chemically-induced ferroptosis in cultured HT22 neuronal cells.
Methods and materials
Chemicals and reagents
2-OH-E1 (#E1130-000), 2-OH-E2 (#E2470-000), 4-OH-E1 (#E1170-000), 4-OH-E2 (#E2500-000), 2-methoxyestrone (#E1148-000), 4-methoxyestrone (#E1175-000), 2-methoxyestradiol (#E2490-000) and 4-methoxyestradiol (#E2510-000) were obtained from Steraloids Inc. (Newport, RI, USA). E1 (#986888) was obtained from J&K Scientific (San Jose, CA, USA). E2 (#3301) and cystamine (#108318) were from Sigma-Aldrich (St Louis, MO, USA). E1, E2 and estrogen metabolites were dissolved in pure ethanol to prepare their stock solutions (usually at 20 mM). Erastin (#S7262), RSL3 (#SML2234), Ferrostatin-1 (#S7243), CPZ (#S5749) and SMT (#S3631) were purchased from Selleck Chemicals (Houston, TX, USA) and dissolved in dimethyl sulfoxide (DMSO). Diaminofluorescein-FM diacetate (DAF-FM-DA, #S0019) and diaminofluorescein-2 diacetate (DCF-DA, #S0033) were purchased from Beyotime Biotechnology (Shanghai, China), and C11-BODIPY-581/591 (#D3861) from Thermo Fishers (Waltham, MA, USA). The anti-PDI (#3501) and anti-β-actin (#4970) antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA); anti-nNOS (#ab1376) and anti-iNOS (#ab3523) antibodies were from Abcam (Waltham, MA, USA). PDI-siRNAs and nNOS-siRNAs (their sequences described earlier31) were purchased from Ruibo Biotechnology (GuangZhou, China). iNOS-siRNAs (#SC36092) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA).
Cell culture and cell viability assay
The HT22 murine hippocampal neuronal cells were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China), cultured in DMEM supplied with 10% (v/v) fetal bovine serum and 1% penicillin and streptomycin, and incubated at 37 °C under 5% CO2. Cells were sub-cultured when they reached approximately 80% confluence.
The MTT reduction assay was used to determine the change in gross cell viability. HT22 cells were usually seeded in 96-well plates at a density of 2000 cells/well 24 h prior to receiving various experimental treatments. To test the potential protective effect of a compound, the cells were jointly treated with erastin or RSL3 ± the compound for 24 h. Afterwards, MTT at a final concentration of 0.5 mg/mL was added to each well, and incubated for 2.5 h at 37 °C under 5% CO2. After incubation, the MTT-containing medium was removed and 100 μL DMSO was added to each well to dissolve the MTT formazan. Absorbance of the MTT formazan was measured with a UVmax microplate reader (Molecular Device, Palo Alto, CA, USA) at the 560-nm wavelength.
Fluorescence microscopy
Fluorescence microscopy was used to visualize cellular accumulation of NO and ROS under different drug treatments. HT22 cells were seeded in 24-well plates at a density of 5 × 104 cells/well, and then they were treated with erastin or RSL3 ± selected test compounds for indicated length of time. After the cells were washed with HBSS twice, they were stained with cell-permeant fluorescent probe DAF-FM-DA (10 μM, for detection of cellular NO) or DCFH-DA (10 μM, for detection of total cellular ROS) dissolved in 200 μL DMEM without of phenol red and serum, and incubated at 37 °C for an additional 20 min. Then the cells were washed twice with HBSS, and the fluorescence images were taken using a fluorescence microscope (AXIO, Carl Zeiss Corporation, Germany).
Analytical flow cytometry
For quantitation of the cellular levels of NO, ROS and lipid-ROS by a flow cytometer, cells were plated in 6-well plates at a density of 15 × 104 cells/well. Following drug treatment for indicated length of time, cells were trypsinized, collected and suspended in phosphate-buffered saline (PBS). Cells were then centrifuged, and the resulting cell pellets were resuspended in DMEM (without phenol red and serum) containing DAF-FM-DA (10 μM, for NO detection), DCFH-DA (10 μM, for ROS detection) or BODIPY-581/591-C11 (10 μM, for lipid-ROS detection). After 20-min incubation at 37 °C, the cells were washed three times with HBSS to remove any remaining fluorescent dyes. Levels of cellular NO, ROS and lipid-ROS were measured using a flow cytometer (Beckman Coulter, Brea, CA, USA) and the results were analyzed using the FlowJo software (FlowJo, LLC, Ashland, USA).
Confocal microscopy
For visualizing subcellular localization of lipid-ROS, confocal microscopy was used. Cells were seeded at a density of 5 ⨯ 104 per well on coverslips placed inside the 24-well plates. Twenty-four h later, cells were treated with different compounds as indicated. Coverslips were then washed in HBSS and incubated in HBSS containing BODIPY-581/591-C11 (10 μM) for 20 min at 37 ℃ under 5% CO2. Coverslips were then mounted on microscope slides for visualization. Slides were imaged using a LSM 900 confocal laser scanning microscope (LSM 900; Carl Zeiss, Oberkochen, Germany), and images were analyzed with the Zen software (Carl Zeiss).
Immunoblot analysis
After treatment of cells with erastin or RSL3 for different durations as indicated, the cells were collected by trypsinization and centrifugation, and then lysed on ice for 30 min in the RIPA buffer containing 1% protease inhibitor cocktail. Protein concentrations were determined using the BCA assay kit (ThermoFisher, MA, USA). For analysis of total nNOS and iNOS (including both monomeric and dimeric forms of nNOS), samples were heated at 95 °C for 5 min with a reducing buffer before loaded onto the gel. To analyze the monomeric and dimeric forms of nNOS and iNOS, samples were prepared with a non-reducing buffer and were not heated, and the temperature of the gel was maintained at below 15 °C during electrophoresis. Proteins were then separated using 10% agarose gel (for analysis of total NOS protein) or 6% agarose gel (for analysis of monomeric and dimeric NOS proteins), and transferred to PVDF membranes. The membranes were then incubated for 1 h in 5% skim milk at room temperature and incubated with primary antibody overnight. Afterwards, the membranes were washed three times with TBST at room temperature (10 min each time). The membranes were then incubated with the secondary antibody (Tanon 5200, Shanghai, China).
Here it should be noted that for the detection of monomer and dimer NOS proteins, the full-length membranes were used for hybridization with the NOS antibody. Accordingly, the whole blot images were shown in the Supplementary File. In comparison, for the detection for total NOS, PDI and β-actin, the membranes were cut before hybridization with different antibodies; as such, only the corresponding areas of the membrane were shown for each of these three proteins.
Cellular thermal shift assay (CETSA)
To determine the binding interaction of the cellular PDI protein with each of the catechol estrogens, CETSA was performed according to the published method43. Briefly, after the cells were treated with the test compound or the dissolving vehicle for 45 min, they were washed with ice-cold PBS, harvested by trypsinization, centrifuged, and re-suspended in PBS supplemented with the complete protease inhibitor cocktail and the same concentrations of the test compound. Equal amounts of cell suspensions were aliquoted into 0.2 mL PCR microtubes. Subsequently, the cell suspension aliquots were heated individually at the indicated temperatures for 3 min (Ristretto Thermal Cycler; VWR, Darmstadt, Germany), followed by cooling at room temperature for 3 min. Finally, the cells were lysed using three cycles of freeze-and-thawing, and the soluble fractions were isolated by centrifugation and analyzed for PDI protein by SDS-PAGE followed by Western blotting as described above. For isothermal dose–response CETSA (ITDRCETSA), the change in the levels of the PDI protein (normalized to the β-actin control) was plotted as a function of temperature to generate the PDI melting curves for the different treatments.
siRNA transfection
For siRNA transfection, the procedures were described in our earlier study31. Briefly, the siRNAs (at a final concentration of 60 nM) for targeted genes (PDI, iNOS, and nNOS) were transfected into HT22 cells 24 h after seeding with Lipofectamine RNA iMAX (Invitrogen). Twenty-four h after siRNA transfection, cells were treated with selected drugs, and subsequently processed for cell viability measurement, fluorescence imaging and immunoblot analysis.
Statistical analysis
All quantitative experiments and data described in this study were repeated at least three times, and similar observations were made. The data were usually presented as mean ± S.D. of multiple replicate measurements of one selected experiment. Statistical analysis and comparisons were performed with one-way ANOVA using the GraphPad Prism 7 (GraphPad Software, La Jolla, CA, USA). Statistical significance was denoted by P < 0.05 (* or #) and P < 0.01 (** or ##) for significant and very significant differences, respectively. In most cases, * and ** denote the comparison for statistical significance between the control group (cells treated with vehicle only) and the cells treated with a chemical, whereas # and ## denote the comparison between the cells treated with the ferroptosis inducer alone (such as erastin or RSL3) and the cells jointly treated with a ferroptosis inducer + a modulating compound.
Results
Catechol estrogens protect against erastin-induced oxidative ferroptosis
Cytoprotective activity Earlier studies reported that erastin can induce ferroptotic cell death in HT22 mouse hippocampal neuronal cells in culture31,44. These observations were confirmed in this study by showing that erastin elicited a dose- and time-dependent reduction in cell viability in HT22 cells (Fig. 2A,B). Erastin-induced cell death in HT22 cells could be strongly rescued by Fer-1 (Fig. 2C), confirming that erastin induces ferroptotic cell death. We chose to use 0.8 μM erastin for subsequent cytoprotective experiments as erastin at this concentration effectively reduced cell viability by 80–90% (some variations were noted from experiments to experiments which were conducted at different times).
Fig. 2.

Differential protective effect of catechol estrogens, their parent hormones and the monomethoxy estrogen metabolites against erastin-induced cell death in HT22 cells. (A–B) Effect of erastin on cell viability (MTT assay, n = 5) (A) and gross cell morphology (B). The cells were treated with different concentrations of erastin for 24 h (A), or treated with 0.8 μM erastin for different durations (B). (C) Protective effect of Fer-1 against erastin-induced cell death (MTT assay, n = 4). Cells were treated for 24 h with erastin (0.8 μM) ± Fer-1 (1 μM). Note that in this experiment (which conducted separately at a different time), the cells experienced a reduced sensitivity to erastin-induced cytotoxicity. (D–G) Cells were treated with erastin (0.8 µM) ± a catechol estrogen (2-OH-E1, 2-OH-E2, 4-OH-E1 or 4-OH-E2) at indicated concentrations for 24 h, and cell viability was determined by MTT assay (n = 5). (H, I) Comparison of the protective effect of E1 and E2 against erastin-induced cytotoxicity. Cells were treated with different concentrations of E1 (H) or E2 (I) for 24 h, and cell viability was determined by MTT assay (n = 5). (J–M) Comparison of the protective effect of the mono-methoxy estrogen metabolites (2-MeO-E1, 2-MeO-E2, 4-MeO-E1 and 4-MeO-E2) against erastin-induced cytotoxicity. Cells were treated with different concentrations of the estrogen metabolites for 24 h, and cell viability was determined by MTT assay (n = 5). (N) Protective effect of cystamine against erastin-induced ferroptotic cell death (MTT assay, n = 5). (O) Protective effect of CPZ and SMT against erastin-induced cell death (MTT assay, n = 5). Cells were treated for 24 h with erastin (0.8 μM) ± CPZ (20 μM) or SMT (100 μM). Note that in this experiment (which conducted separately at a different time), the cells experienced a reduced sensitivity to erastin-induced cytotoxicity. The quantitative data are presented as mean ± S.D. * or #, P < 0.05; ** or ##, P < 0.01. n.s., not significant. (* or #denote statistical significance in comparison with the control group or the erastin alone group, respectively).
Next, we determined the protective effect of four catechol estrogens (2-OH-E1, 2-OH-E2, 4-OH-E1 and 4-OH-E2; structures shown in Fig. 1) against erastin-induced ferroptosis. We found that each catechol estrogen exerted a strong protection against erastin-induced ferroptosis in HT22 cells. For instance, joint treatment of cells with erastin + 2-OH-E1 or 2-OH-E2 (at 1.25–10 μM) effectively rescued the cells from erastin-induced cytotoxicity (Fig. 2D,E), and the maximal protection (i.e., cell viability reaching ~ 80% of the control group cell viability) was seen at 2.5–5 μM concentrations. Similarly, joint treatment of cells with erastin + 4-OH-E1 or 4-OH-E2 (at 1.25–10 μM) also exerted a strong protection against erastin-induced cytotoxicity (Fig. 2F,G), with maximal protection (cell viability reaching ~ 80% of the control) seen at 5 μM. It is of note that these four catechol estrogens when present alone at concentrations ≥ 5 μM also elicited significant cytotoxicity in the cells (Fig. 2D–G).
Fig. 1.
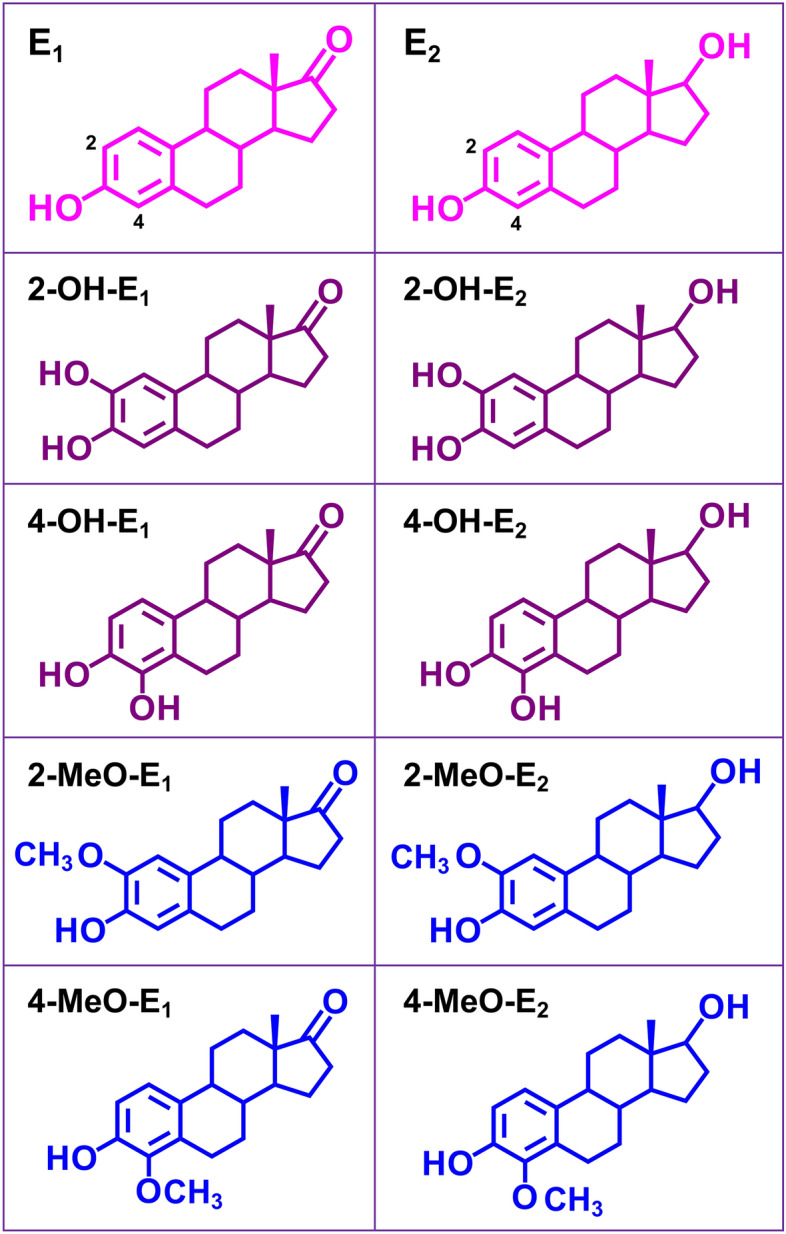
Chemical structures of the four catechol estrogens (2-OH-E1, 2-OH-E2, 4-OH-E1 and 4-OH-E2), their parent hormones (E1 and E2) and the monomethoxy estrogen metabolites (2-MeO-E1, 2-MeO-E2, 4-MeO-E1 and 4-MeO-E2) tested in this study. E1 and E2 can be hydroxylated to their catechol estrogens catalyzed by cytochrome P450 enzymes present in the liver (e.g., CYP3A4 and CYP1A2) and extrahepatic tissues (e.g., CYP1A1 and CYP1B1). These catechol estrogens can be further O-methylated by catechol-O-methyltransferase (COMT) to form respective monomethoxy metabolites.
Different from the observations made with these four catechol estrogens, a similarly-strong protective effect was not observed when HT22 cells were jointly treated with the parent hormones E1 and E2 at the same concentrations (Fig. 2H,I). Moreover, three monomethoxy metabolites of E1 and E2 (i.e., 2-methoxyestrone, 4-methoxyestrone and 4-methoxyestradiol; structures shown in Fig. 1) also did not display a similarly-strong protective effect against erastin-induced ferroptosis at the same concentrations (Fig. 2J–M), except 2-methoxyestradiol which exhibited approximately 50% protection against erastin-induced ferroptosis (Fig. 2K).
In this study, we also tested, for comparison, the protective effect of cystamine (a known PDI inhibitor45) against erastin-induced ferroptosis in HT22 cells under the same experimental conditions30. Cystamine had approximately 85% protection against erastin-induced ferroptosis when it was present at 6.25 μM, and a complete protection was seen at 50 μM (Fig. 2N). In addition, we found that CPZ (an nNOS inhibitor46) and SMT (an iNOS inhibitor47) each showed a strong protective effect against erastin-induced cell death (Fig. 2O).
Effect on NO, ROS and lipid-ROS accumulation Our recent studies have shown that erastin-induced ferroptosis is associated with accumulation of cellular NO, which is followed by accumulation of cellular ROS and lipid-ROS30,31. Using analytical flow cytometry, we confirmed the time-dependent change in cellular NO and ROS levels following treatment with erastin (Fig. 3A,G). It was confirmed that the erastin-induced increase in cellular ROS accumulation was slightly behind the rise in cellular NO accumulation.
Fig. 3.

Catechol estrogens abrogate erastin-induced accumulation of NO and ROS in HT22 cells. (A) Time-dependent accumulation of cellular NO after the cells were treated with 0.8 μM erastin (flow cytometry). Each value is the mean ± S.D. (n = 3). (B) Abrogation of erastin-induced NO accumulation by catechol estrogens (fluorescence microscopy). The cells were treated with erastin (0.8 μM) ± each catechol estrogen (2.5 µM 2-OH-E1, 2.5 µM 2-OH-E2, 5 µM 4-OH-E1 or 5 µM 4-OH-E2). (C–F) Abrogation of erastin-induced NO accumulation by each catechol estrogen at 8 h after erastin exposure (flow cytometry). The concentrations of erastin and each catechol estrogen metabolite used here were the same as in (B). Each value is the mean ± S.D. (n = 3). (G) Time-dependent accumulation of cellular ROS after the cells were treated with 0.8 μM erastin (flow cytometry). Each value is the mean ± S.D. (n = 3). (H) Abrogation of erastin-induced ROS accumulation by catechol estrogens (fluorescence microscopy). The cells were treated with erastin (0.8 μM) ± each catechol estrogen (2.5 µM 2-OH-E1, 2.5 µM 2-OH-E2, 5 µM 4-OH-E1 or 5 µM 4-OH-E2). (I–L) Abrogation of erastin-induced ROS accumulation by each catechol estrogen at 8 h after erastin exposure (flow cytometry). The concentrations of erastin and each catechol estrogen metabolite used here were the same as in (H). Each value is the mean ± S.D. (n = 3). The quantitative data are presented as mean ± S.D. * or #, P < 0.05; ** or ##, P < 0.01. n.s., not significant. (* or #denote statistical significance in comparison with the control group or the erastin alone group, respectively).
Next, we jointly used the analytical flow cytometry and fluorescence microscopy to determine the change in cellular NO levels in HT22 cells treated with erastin ± a catechol estrogen (2-OH-E1, 2-OH-E2, 4-OH-E1 or 4-OH-E2) (Fig. 3B–F). While treatment of cells with 0.8 μM erastin alone increased cellular NO levels, joint treatment with each catechol estrogen strongly abrogated erastin-induced NO accumulation (Fig. 3B–F). Similarly, while the ROS levels in erastin-treated HT22 cells were significantly increased, joint treatment of cells with a catechol estrogen (2-OH-E1, 2-OH-E2, 4-OH-E1 or 4-OH-E2) almost completely abrogated erastin-induced accumulation of cellular ROS (Fig. 3H–L).
Accumulation of cellular lipid-ROS is a hallmark of ferroptotic cell death6,16,48. Our earlier study showed that treatment of HT22 cells with erastin increased cellular lipid-ROS accumulation in a time-dependent manner31, and this observation was confirmed in this study (Supplementary Fig. S1A). In addition, we observed that erastin-induced accumulation of cellular lipid-ROS was completely abrogated by joint treatment of the cells with each of the four catechol estrogens (Fig. 4A–E).
Fig. 4.

Catechol estrogens abrogate erastin-induced accumulation of lipid-ROS in HT22 cells. (A–D) Abrogation of erastin-induced lipid-ROS accumulation by catechol estrogens at 8 h after erastin exposure (flow cytometry). The cells were treated with erastin (0.8 μM) ± a catechol estrogen (2.5 µM 2-OH-E1, 2.5 µM 2-OH-E2, 5 µM 4-OH-E1 or 5 µM 4-OH-E2) for 8 h. Each value is the mean ± S.D. (n = 3). (E) Abrogation of erastin-induced lipid-ROS accumulation by catechol estrogens (confocal microscopy). The concentrations of erastin and each catechol estrogen metabolite used here were the same as in (A–D) The quantitative data are presented as mean ± S.D. * or #, P < 0.05; ** or ##, P < 0.01. n.s., not significant. (* or #denote statistical significance in comparison with the control group or the erastin alone group, respectively).
In summary, we demonstrate that each of the four endogenous catechol estrogens can strongly protect HT22 cells against erastin-induced ferroptotic cell death, and this protection is associated with a strong abrogation of erastin-induced accumulation of cellular NO, ROS and lipid-ROS by catechol estrogens.
Catechol estrogens protect against RSL3-induced ferroptosis
Cytoprotective activity RSL3 is another commonly-used ferroptosis inducer18,48. RSL3 induces ferroptosis by inhibiting GPX4 (which result in lipid-ROS accumulation4,6,19) and by inhibiting TrxR1 (which results in PDI activation and NOS dimerization)20,21. In this study, we also compared the protective effect of catechol estrogens against RSL3-induced ferroptosis in cultured HT22 cells.
Based on MTT assay and gross morphological analysis, we confirmed that RSL3 could induce cell death in HT22 cells in a dose- and time-dependent manner and with very high potency (Fig. 5A,B). RSL3-induced cell death was effectively rescued by Fer-1 (Fig. 5C), which is in agreement with ferroptotic cell death. The 80-nM RSL3 concentration was selected to evaluate the cytoprotective effect of catechol estrogens.
Fig. 5.

Differential protective effect of catechol estrogens, their parent hormones and the monomethoxy estrogen metabolites against RSL3-induced cell death in HT22 cells. (A–B) Effect of RSL3 on cell viability (MTT assay; (A)) and gross cell morphology (B). Cells were treated for 24 h with different concentrations of RSL3 (A), or treated with 80 nM RSL3 for different durations (B). Each value is the mean ± S.D. (n = 5). (C) Protective effect of Fer-1 against RSL3-induced cell death (MTT assay, n = 4). Cells were treated for 24 h with RSL3 (80 nM) ± Fer-1 (1 μM). Note that in this experiment (conducted separately at a different time), the cells experienced a reduced sensitivity to RSL3 cytotoxicity. (D–G) Cells were treated with RSL3 (80 nM) ± a catechol estrogen (2-OH-E1, 2-OH-E2, 4-OH-E1 or 4-OH-E2) at indicated concentrations for 24 h, and cell viability was determined by MTT assay. Each value is the mean ± S.D. (n = 5). (H, I) Comparison of the protective effect of E1 and E2 against RSL3-induced cytotoxicity (MTT assay). Cells were treated with different concentrations of E1 (H) or E2 (I) for 24 h. Each value is the mean ± S.D. (n = 5). (J–M) Comparison of the protective effect of the monomethoxy estrogen metabolites against RSL3-induced cytotoxicity (MTT assay). Cells were treated with different concentrations of the estrogen metabolites for 24 h. Each value is the mean ± S.D. (n = 5). (N) Protective effect of cystamine against RSL3-induced ferroptotic cell death. The cell viability was determined by MTT assay. Each value is the mean ± S.D. (n = 5). (O) Protective effect of CPZ and SMT against RSL3-induced cell death (MTT assay, n = 5). Cells were treated for 24 h with RSL3 (80 nM) ± CPZ (20 μM) or with RSL3 (80 nM) ± SMT (100 μM). Note that in this experiment (which conducted separately at a different time), the cells experienced a reduced sensitivity to erastin-induced cytotoxicity. The quantitative data are presented as mean ± S.D. * or #, P < 0.05; ** or ##, P < 0.01. n.s., not significant. (* or #denote statistical significance in comparison with the control group or the RSL3 alone group, respectively).
We found that each of the four catechol estrogens dose-dependently protected HT22 cells against RSL3-induced ferroptosis, with maximal protection (cell viability reaching ~ 80% of the control viability) seen at 5 μM (Fig. 5D–G). Similar to the observations made with erastin, the parent hormones E1 and E2 failed to exert a similarly-strong protective effect against RSL3-induced cell death (Fig. 5H,I). Three of the monomethoxy estrogen metabolites (i.e., 2-methoxyestrone, 4-methoxyestrone and 4-methoxyestradiol) had a markedly weaker protective effect against RSL3-induced cell death (Fig. 5J–M), except 2-methoxyestradiol which showed a relatively stronger protective effect against RSL3-induced cytotoxicity (Fig. 5K).
When cystamine was analyzed for comparison, it also displayed a strong protection against RSL3-induced ferroptosis (Fig. 5N). The apparent potency of cystamine in protection against RSL3-induced ferroptosis is lower than its protection against erastin-induced ferroptosis (compare Fig. 5N with Fig. 2N), indicating that PDI likely plays a more dominant upstream role in mediating erastin-induced cell death. In addition, we found that while CPZ exerted a modest protective effect against RSL3-induced cell death, SMT exerted a markedly stronger protective effect (Fig. 5O).
Effect on NO, ROS and lipid-ROS accumulation In this study, the change in cellular NO, ROS and lipid-ROS levels was also determined in HT22 cells treated with RSL3 alone or in combination with each catechol estrogen. It is of considerable interest to determine whether NO accumulation is also involved in RSL3-induced ferroptosis given the fact that NO accumulation is upstream of ROS and lipid-ROS accumulation based on observations with erastin-induced ferroptosis30,31. Flow cytometric analyses of RSL3-treated cells showed that RSL3-treated cells displayed a rapid time-dependent rise in cellular NO levels (Fig. 6A). Joint treatment of cells with RSL3 + a catechol estrogen (2-OH-E1, 2-OH-E2, 4-OH-E1 or 4-OH-E2) strongly abrogated RSL3-induced accumulation of cellular NO (Fig. 6B–F). Similarly, flow cytometric analyses showed that treatment of HT22 cells with RSL3 also elicited a time-dependent increase in cellular ROS levels (Fig. 6G). Joint treatment of cells with RSL3 + each catechol estrogen abrogated RSL3-induced accumulation of cellular ROS (Fig. 6H–L).
Fig. 6.

Catechol estrogen metabolites suppress RSL3-induced accumulation of NO and ROS in HT22 cells. (A) Time-dependent induction of NO accumulation in cells treated with 80 nM RSL3 for different durations as indicated (flow cytometry). (B) Abrogation of RSL3-induced NO accumulation by catechol estrogens (fluorescence microscopy). The cells were treated with RSL3 (80 nM) ± each catechol estrogen (5 µM) for 6 h. (C–F) Abrogation of 80 nM RSL3-induced NO accumulation by each catechol estrogen at 6 h after RSL3 exposure (flow cytometry). The concentration of each catechol estrogen used here was the same as in (B). Each value is the mean ± S.D. (n = 3). (G) Time-dependent induction of ROS accumulation in cells treated with 80 nM RSL3 for different durations as indicated (flow cytometry). (H) Abrogation of RSL3-induced ROS accumulation by catechol estrogens (fluorescence microscopy). The cells were treated with RSL3 (80 nM) ± each catechol estrogen (5 µM). (I–L) Abrogation of RSL3-induced ROS accumulation by each catechol estrogen at 6 h after RSL3 exposure (flow cytometry). The concentrations of RSL3 and each catechol estrogen used here were the same as in (H). Each value is the mean ± S.D. (n = 3). The quantitative data are presented as mean ± S.D. * or #, P < 0.05; ** or ##, P < 0.01. n.s., not significant. (* or #denote statistical significance in comparison with the control group or the RSL3 alone group, respectively).
Analytical flow cytometry showed that treatment of HT22 cells with RSL3 increased cellular lipid-ROS accumulation in a time-dependent manner (Supplementary Fig. S1B). Confocal microscopic analysis confirmed that RSL3 markedly increased cellular lipid-ROS accumulation (Fig. 7E); moreover, RSL3-induced lipid-ROS accumulation was abrogated by joint treatment of cells with RSL3 + each catechol estrogen (Fig. 7A–D).
Fig. 7.

Catechol estrogens abrogate RSL3-induced accumulation of lipid-ROS in HT22 cells. (A–D) Abrogation of RSL3-induced lipid-ROS accumulation by catechol estrogens at 6 h after RSL3 exposure (flow cytometry). The cells were treated with RSL3 (80 nM) ± each catechol estrogen (5 µM) for 6 h. Each value is the mean ± S.D. (n = 3). (E) Abrogation of RSL3-induced lipid-ROS accumulation by catechol estrogens (confocal microscopy). The concentrations of RSL3 and each catechol estrogen metabolite used here were the same as in (A–D). The quantitative data are presented as mean ± S.D. * or #, P < 0.05; ** or ##, P < 0.01. n.s., not significant. (* or #denote statistical significance in comparison with the control group or the RSL3 alone group, respectively).
In summary, RSL3 induces ferroptotic death through induction of cellular NO, ROS and lipid-ROS accumulation in HT22 cells. Joint treatment of cells with each catechol estrogen abrogates RSL3-induced accumulation of cellular NO, ROS and lipid-ROS, and this effect contributes to the protective action of catechol estrogens against RSL3-induced ferroptosis.
Catechol estrogens can bind directly to PDI in live HT22 cells
The results described above prompted us to identify the potential cellular target that might mediate the protective effect of catechol estrogens against chemically-induced ferroptotic cell death in cultured HT22 cells. Our earlier studies have shown that PDI is an important cellular protein that can mediate chemically-induced ferroptotic cell death30,31. In addition, our recent study showed that 4-OH-E1 protects erastin-induced cell death through binding to PDI and inhibition of its enzymatic activity36. In the present study, therefore, we sought to determine whether these catechol estrogens can bind to PDI in HT22 cells to mediate the cytoprotective action against chemically-induced ferroptosis.
We performed the cellular thermal shift assay (CETSA) to evaluate the binding interaction between each catechol estrogen and PDI in live HT22 cells. Based on Western blot analysis of PDI stability, a thermal shift associated with PDI was observed in HT22 cells jointly treated with each catechol estrogen (2-OH-E1, 2-OH-E2, 4-OH-E1 or 4-OH-E2) compared to vehicle-treated control cells. The change in the Tm50 values (the temperature at which 50% of the proteins are precipitated by thermal denaturation) was determined to reflect the relative binding affinity of catechol estrogens to PDI. In the absence of a catechol estrogen, PDI has a Tm50 value of 55 °C, but the presence of each catechol estrogen increased its Tm50 to varying degrees (Fig. 8A–E). Specifically, when 2-OH-E1, 2-OH-E2 or 4-OH-E2 was present, the Tm50 values for PDI were increased to approximately 58 °C, but when 4-OH-E1 was present, the Tm50 value for PDI was increased to approximately 59 °C (Fig. 8A–E).
Fig. 8.

Catechol estrogens increase the thermal stability of PDI. (A) The thermal stability of PDI (CETSA) in vehicle-treated control intact cells or in cells treated with each of the catechol estrogens (10 μM, n = 3). The corresponding unprocessed Western blot images are shown in Supplementary Fig. S4. (B–E) The relative intensities of the protein bands in (A) were quantified using the ImageJ software. For the CETSA curves of each catechol estrogen, the band intensities were normalized to the respective controls at 45 °C. Each measurement had three replicates with similar observations.
To provide further support for the above experimental observations, the isothermal dose–response CETSA (ITDRCETSA) was also performed. The isothermal stability of PDI treated with different concentrations of catechol estrogens at 60 °C further demonstrated that the PDI protein in live HT22 cells was stabilized by the presence of catechol estrogens in a concentration-dependent manner (Fig. 9A–E). The CETSA curves reached the plateaus and showed comparable stabilization patterns when higher concentrations of the catechol estrogens were present.
Fig. 9.

The ITDRCETSA analysis of PDI protein in intact cells treated with each of the catechol estrogens. (A) The ITDRCETSA analysis of PDI protein in intact cells at 60 °C in response to increasing concentrations of each of the four catechol estrogens (2-OH-E1, 2-OH-E2 4-OH-E1 and 4-OH-E2). The corresponding unprocessed Western blot images are shown in Supplementary Fig. S5. (B–E) The relative intensities of the protein bands in (A) were normalized to β-actin. Each measurement had three replicates, and the data is the mean ± S.D. (n = 3).
Together, these results demonstrate that the catechol estrogens can directly bind to PDI protein and increase its stability in live HT22 cells in culture.
PDI mediates the protective effect of catechol estrogens against chemically-induced ferroptosis
To provide support for the hypothesis that inhibition of PDI’s catalytic activity by catechol estrogens plays an important role in protecting HT22 cells against chemically-induced ferroptosis, we transfected HT22 cells with the PDI-siRNAs to selectively knock down its expression (Fig. 10A–G). The effectiveness of PDI knockdown was assessed by Western blot analysis of cellular PDI protein levels (Fig. 10A). We found that PDI knockdown reduced erastin-induced ferroptosis in HT22 cells (Fig. 10C,F), and the cytoprotective action of each catechol estrogen was also partially abrogated by PDI knockdown (Fig. 10B,C,E,F). Selective PDI knockdown similarly reduced RSL3-induced ferroptosis (Fig. 10D,G), and the protective effect of catechol estrogens against RSL3-induced ferroptotic cell death was also diminished by PDI knockdown (Fig. 10B,D,E,G). Together, these results suggest that PDI is a cellular protein involved in mediating the protective action of catechol estrogens against erastin/RSL3-induced ferroptosis.
Fig. 10.

PDI knockdown attenuates the protective effect of catechol estrogens against erastin-or RSL3-induced ferroptosis. (A) The efficiency of PDI knockdown was confirmed by Western blotting analysis of cellular PDI protein levels. The corresponding unprocessed Western blot images are shown in Supplementary Fig. S6. (B) Effect of each catechol estrogen alone on cell viability in cells transfected with the control siRNAs (siCon). Cell viability was determined by MTT assay (n = 5). (C, D) Protective effect of four catechol estrogens (2-OH-E1, 2-OH-E2 4-OH-E1 and 4-OH-E2) against chemically-induced cell death in cells transfected with the control siRNAs (siCon). The cells were jointly treated with 0.8 μM erastin (C) or 80 nM RSL3 (D) ± each of the catechol estrogen for 24 h. Cell viability was determined by MTT assay (n ≥ 4). (E) Effect of each catechol estrogen alone on cell viability in cells transfected with PDI-specific siRNAs (siPDI). Cell viability was determined by MTT assay (n = 5). (F, G) Protective effect of four catechol estrogens (2-OH-E1, 2-OH-E2 4-OH-E1 and 4-OH-E2) against chemically-induced cell death in cells transfected with PDI-specific siRNAs (siPDI). The cells were jointly treated with 0.8 μM erastin (F) or 80 nM RSL3 (G) ± each of the catechol estrogen for 24 h. Cell viability was determined by MTT assay (n ≥ 4). The quantitative data are presented as mean ± S.D. * or #, P < 0.05; ** or ##, P < 0.01. n.s., not significant. (* or #denote statistical significance in comparison with the control group or the erastin or RSL3 alone group, respectively).
Our recent studies have shown that treatment of cultured cells with erastin strongly induces total cellular nNOS and iNOS protein levels and the formation of respective dimers30,31. In this study, the change in total cellular nNOS and iNOS protein levels and their respective dimer formation was also observed in erastin-treated HT22 cells (Fig. 11A–H). In addition, we determined whether catechol estrogens can effectively abrogate chemically-induced increase in total cellular nNOS and iNOS protein levels as well as their respective dimer protein levels (Fig. 11A–H). The non-reducing SDS-PAGE which preserves the disulfide bonds in nNOS and iNOS homodimers was used to determine the change in nNOS and iNOS dimer levels. We found that erastin-induced increase in nNOS and iNOS homodimers was abrogated by joint treatment of the cells with catechol estrogens (Fig. 11A–H). Similarly, it was recently reported that treatment of HT22 cells with RSL3 can also lead to increases in total cellular nNOS and iNOS protein levels and their respective dimer formation21. These changes were confirmed in this study. We found that the total nNOS and iNOS protein levels were increased following exposure to RSL3 (Fig. 11I–P). In addition, joint treatment of cells with catechol estrogens effectively abrogated RSL3-induced nNOS and iNOS dimer formation (Fig. 11I–P).
Fig. 11.

Effect of catechol estrogens on cellular total nNOS and iNOS levels and on their dimerization. (A–H) Abrogation of erastin-induced increase in total nNOS (A–D) and iNOS (E–H) protein levels (including both monomer and dimer NOS) and their respective dimers by each of the catechol estrogens. Cells were treated with erastin (0.8 μM) ± each catechol estrogen (2.5 μM for 2-OH-E1, 2-OH-E2 and 5 μM for 4-OH-E1 or 4-OH-E2) for 8 h. The corresponding unprocessed Western blot images are shown in Supplementary Fig. S7–S10. (I–P) Abrogation of RSL3-induced increase in total nNOS (I–L) and iNOS (M–P) protein levels (including both monomer and dimer NOS) and their respective dimers by each of the catechol estrogens. Cells were treated with RSL3 (80 nM) ± each catechol estrogen (5 μM) for 6 h. The corresponding unprocessed Western blot images are shown in Supplementary Fig. S11–S14.
Together, these results indicated that catechol estrogens can directly inhibit PDI-mediated NOS dimerization, and this effect is reflected by the observations that catechol estrogens can abrogate chemically-induced accumulation of cellular NO, ROS and lipid-ROS and ferroptotic cell death.
Discussion
In a recent study31, we have shown that PDI plays an important role in mediating erastin-induced ferroptosis in HT22 cells through catalyzing the conversion of NOS monomer to its homodimer, which subsequently leads to accumulation of NO, ROS and lipid-ROS, and ultimately ferroptotic cell death. In addition, we have further shown that PDI also plays an important role in mediating RSL3-induced ferroptotic cell death in HT22 cells because RSL3 is a strong inhibitor of TrxR120, and inhibition of TrxR1 activity would keep more PDI molecules in the catalytically-active oxidized form and thus facilitate NOS dimerization and ferroptotic cell death21,29. The discovery of an important role of PDI in mediating both erastin- and RSL3-induced ferroptotic cell death indicates that PDI may also be an important therapeutic target for protection against ferroptosis-related cell death21,30,31,36. Using the HT22 cells as an in vitro model, the present study seeks to determine whether some of the endogenously-formed estrogen metabolites are inhibitors of PDI and can protect against chemically-induced ferroptosis. We find that the four catechol estrogens tested in this study, i.e., 2-OH-E1, 2-OH-E2, 4-OH-E1 and 4-OH-E2, each exhibit a strong protection against erastin/RSL3-induced ferroptosis in HT22 cells. In comparison, E1, E2 and their monomethylated metabolites did not exhibit a similarly-strong protective effect against chemically-induced ferroptosis when present at the same concentrations. Mechanistically, we find that catechol estrogens elicit their neuroprotection in HT22 cells by directly targeting PDI and subsequently inhibiting PDI-mediated conversion of nNOS and iNOS monomers to the catalytically-active homodimers, and ultimately, prevents chemically-induced ferroptosis. As discussed below, this mechanistic explanation is supported by some experimental observations:
First, by using the cellular thermal shift assay (CETSA), we show that catechol estrogens can directly bind to PDI in live HT22 cells by causing an increase in the Tm50 values of PDI. In addition, the isothermal dose–response CETSA (ITDRCETSA) further shows that the PDI protein in HT22 cells is stabilized by the presence of each catechol estrogen in a concentration-dependent manner. These observations in live HT22 cells are in agreement with our recent surface plasmon resonance (SPR) assay showing that these catechol estrogens can bind to purified wild-type PDI-His256 with apparent Kd in the low nM range (unpublished data). In addition, when the enzymatic activity of purified wild-type PDI-His256 and mutant PDI-Ala256 proteins is assayed in vitro, we find that the presence of each catechol estrogen can effectively inhibit the catalytic activity of the wild-type PDI-His256 but not the mutant PDI-Ala256 (unpublished data). Collectively, these results indicate that catechol estrogens can bind to PDI (through its His256) and then inhibit its catalytic activity.
Second, our recent studies showed that pharmacological inhibition of PDI (such as the use of cystamine) can effectively suppress the formation of NOS dimers in erastin/RSL3-treated cells, and abrogates the subsequent NO, ROS and lipid-ROS accumulation30,31, which is accompanied by protection against ferroptotic death as observed in this study. In this study, it is confirmed that joint treatment of cells with cystamine can strongly abrogate both erastin—and RSL3-induced cell death (with close to 100% protection) in HT22 cells. Cystamine is a very simple chemical (i.e., HN2-CH3-CH2-S‒S-CH2-CH2-NH2) which is widely-used as a PDI inhibitor. The strong protective effect of cystamine against erastin/RSL3-induced cell death cannot be due to any of its free radical-scavenging activity since this small chemical is not a reducing agent at all; in fact, it is a weak oxidizing agent (like the oxidized glutathione GSSG). The inhibition of PDI activity by cystamine is due to its ability to covalently modify the free thiol group(s) in PDI’s catalytic site, resulting in the formation of PDI-S-S-CH2CH2-NH2 (i.e., one of PDI’s free thiol groups in the catalytic site is covalently modified). The dramatic protective effect elicited by cystamine not only highlights the important upstream role of PDI in mediating erastin/RSL3-induced ferroptotic cell death, but it also highlights its importance as a mechanistic target for ferroptosis protection.
Interestingly, the siRNA-mediated PDI depletion is only found to be associated with a partial protection against erastin/RSL3-induced ferroptotic death. Furthermore, PDI knockdown only partially abrogates the cytoprotective action of catechol estrogens. This rather modest protection by PDI knockdown against erastin/RSL3-induced cell death may be due to two factors: Firstly, it is observed that PDI knockdown per se (which involves the global reduction in cellular PDI levels) is associated with significant cytotoxicity. This cytotoxic effect may partially contribute to the blunted protective effect of PDI knockdown against erastin/RSL3-induced cell death. Second, the relatively low efficiency of the transient PDI knockdown may not last the whole duration of the cell viability assay (72 h in total). Consistent with incompletion PDI knockdown, it is observed that while the protective effect of 2-OH-E1 and 2-OH-E2 is markedly reduced by PDI knockdown, a small but significant protection is still seen.
Third, it is known that during the development of chemically-induced ferroptosis, the upregulated nNOS and iNOS proteins and PDI-mediated conversion of the NOS monomers to their dimers in HT22 cells would jointly contribute to elevated NO accumulation. We find that joint treatment of HT22 cells with catechol estrogens abrogates erastin/RSL3-induced upregulation of nNOS and iNOS and PDI-mediated dimerization of nNOS and iNOS.
It was reported earlier35 that iNOS was barely detected in untreated control HT22 cells. However, in the present study it was found that treatment of HT22 cells with erastin or RSL3 each elicited a strong induction of both nNOS and iNOS expression, and these increases are abrogated by joint treatment of cells with catechol estrogens. It is believed that iNOS may contribute more importantly to NO accumulation in HT22 cells treated with erastin or RSL3. Offering support for this hypothesis, we find that joint treatment of HT22 cells with SMT (an iNOS inhibitor47) exerts a stronger protection against erastin/RSL3-induced ferroptotic cell death in HT22 cells than does CPZ (an nNOS inhibitor46).
Fourth, our recent study has shown that erastin-induced ferroptosis in HT22 cells is driven by NO accumulation, which is followed by ROS and lipid-ROS accumulation31. A time-dependent sequential increase in cellular NO, ROS and lipid-ROS is confirmed in this study. In addition, we find that the catechol estrogens almost completely abrogate erastin-induced accumulation of cellular NO, ROS and lipid-ROS.
In the case of RSL3, it induces ferroptosis jointly through GPX4 inhibition18 and TrxR1 inhibition20,21. Our recent study has shown that TrxR1 inhibition facilitates PDI activation and PDI-mediated NOS dimerization, which then leads to accumulation of NO, ROS and lipid-ROS, and ultimately ferroptosis21. As expected, the cellular NO, ROS and lipid-ROS levels are significantly increased in RSL3-treated cells, and these changes are almost completely abolished by joint treatment with catechol estrogens. In addition, the high-efficacy protection by cystamine (a covalent inhibitor of PDI) against RSL3-induced cell death as observed in this study as well as in our earlier study21 also offers support for the notion that PDI is an important cellular target for the neuroprotective actions of catechol estrogens.
Fifth, it is of note that earlier studies have shown that catechol estrogens have antioxidant activity. For instance, catechol estrogens were reported to have antioxidant capacity due to their ability to bind iron or reduce peroxyl radicals by donating hydrogen from their catecholic hydroxyl groups49–51. Lacort et al. reported that E1 and 2-OH-E2 and 4-OH-E2 can reduce microsomal lipid peroxidation in rat liver52. In addition, E1 and E2, which are the parent hormones of 2-OH-E1, 2-OH-E2, 4-OH-E1 and 4-OH-E2, can modulate the activity and expression of various antioxidant enzymes, such as superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPX), enhancing the cell’s defense against ROS53. On the other hand, catechol estrogens are well known for their prooxidant potentials54. The cytotoxic effect of catechol estrogens is associated either with their ability to directly produce semiquinone/quinone radicals or indirectly through their redox cycling to produce ROS54. Therefore, while the antioxidant activity of catechol estrogens may partially contribute to their protective effect against chemically-induced ferroptosis, its overall contribution likely is relatively small. On the other hand, the prooxidant effect of catechol estrogens may contribute to the cytotoxicity which is readily seen when higher concentrations of these catechol estrogens are present alone.
It is of interest to note that we have observed in this study (Supplementary Figs. S2, S3) as well as in our recent study55 that the use of a strong ROS scavenger (such as Trolox or Fer-1) can also lead to reduced NOS dimer formation. However, this effect does not negate the role of PDI in mediating GSH depletion-associated NOS dimerization and ferroptosis. Mechanistically, it is speculated that the presence of a strong antioxidant (e.g., Trolox or Fer-1) will be associated with reduced levels of cellular ROS and lipid-ROS, and this reduction would help restore cellular GSH levels. Higher cellular GSH levels would then keep more PDI proteins in the reduced state (i.e., PDI’s disulfide bond in its active site will be in the reduced form), thereby reducing NOS dimerization. Since catechol estrogens have antioxidant activity, their ROS-scavenging ability is believed to also partially contribute to the reduced PDI activation and reduced NOS dimerization.
Lastly, it is of note that estrogens have recently been implicated in several earlier studies in displaying distinct physiological and pathophysiological functions via the ER-independent mechanisms36,42,56,57. For instance, some endogenous estrogen metabolites were reported to exhibit a distinct lipid-lowering effect that is not associated with their parent hormone E1 and E2 or with their binding affinity to ERα or ERβ56. Similarly, the results of our present study demonstrate that catechol estrogens can strongly protect cells against chemically-induced ferroptosis through inhibition of PDI and subsequent abrogation of cellular NO, ROS and lipid-ROS accumulation36. In addition, the protective effect of catechol estrogens is markedly stronger than their parent hormones E1 and E2, despite that fact that these catechol estrogens have much weaker estrogenic activity at the ERs than E1 and E256,58. Similarly, the catechol estrogens also have stronger neuroprotective effect than their respective O-methylated metabolites (i.e., 2-methoxyestrone, 4-methoxyestrone, 2-methoxyestradiol and 4-methoxyestradiol). It is of note that our earlier study showed that 4-OH-E1 is more effective than E2 in an in vivo animal model for protection against kainic acid-induced neuronal damage42. It will be of considerable interest to determine in the future whether alterations in the rate of metabolic conversion of endogenous E2 and E1 to their respective catechol derivatives are associated with an altered risk of developing neurodegenerative conditions in humans.
Conclusion
As depicted in Fig. 12, the results of our present study demonstrate that PDI is a cellular target that mediates the protective effect of catechol estrogens against chemically-induced ferroptosis. Mechanistically, PDI is involved in catalyzing the activation of NOS, which then leads to accumulation of cellular NO, ROS and lipid-ROS, and ultimately ferroptosis. The mechanistic understanding gained in this study with the mouse hippocampal neuronal cells may also aid in understanding the ER-independent cytoprotective actions of endogenous estrogens and their metabolic derivatives in other cell types.
Fig. 12.
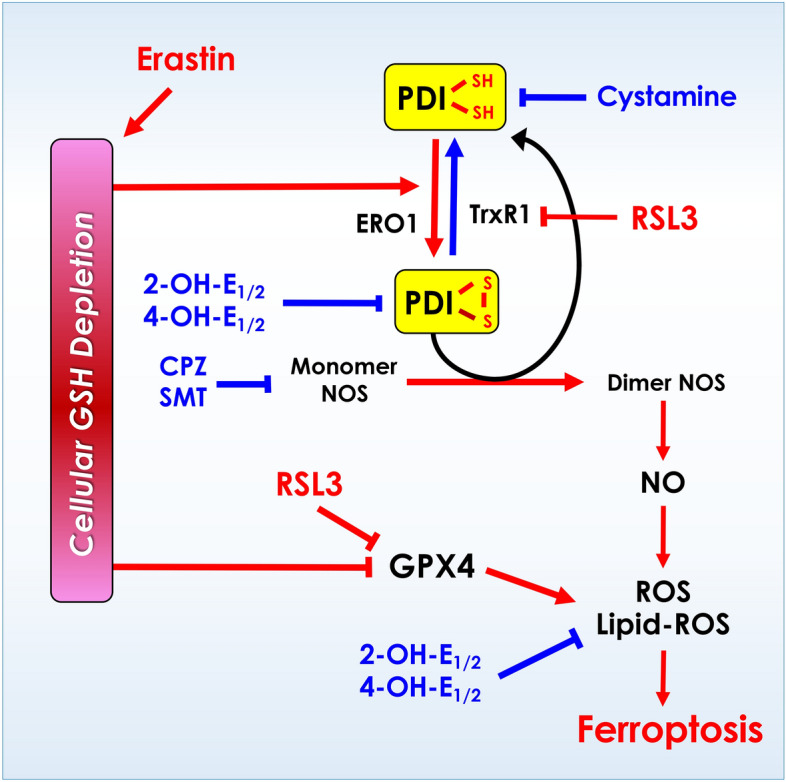
Schematic depiction of the mechanism underlying estrogen receptor-independent cytoprotective action of catechol estrogens. It is proposed that PDI plays an important role in mediating erastin- and RSL3-induced ferroptotic cell death through PDI-mediated NOS dimerization (activation), which then leads to cellular accumulation of NO, ROS and lipid-ROS, and ultimately ferroptotic cell death. Inhibition of PDI by catechol estrogens (2-OH-E1, 2-OH-E2, 4-OH-E1 and 4-OH-E2) mediates their protective action against chemically-induced ferroptosis in cultured HT22 neuronal cells.
Supplementary Information
Author contributions
H.X.: Methodology, Validation, Formal analysis, Investigation, Writing—Original Draft, Visualization; H.M.J: Formal analysis, Investigation, Visualization; Z.B.T: Conceptualization, Methodology, Writing—Review & Editing, Supervision, Funding acquisition.
Funding
This study was supported, in part, by research grants from Shenzhen Peacock Plan (No. KQTD2016053117035204), Shenzhen Key Laboratory of Steroid Drug Discovery and Development (No. ZDSYS20190902093417963), the National Natural Science Foundation of China (No. 81630096), Shenzhen Bay Laboratory (No. SZBL2019062801007), the Stable Funding Support Program for Shenzhen Institutions of High Learning, and the Longgang District Science and Technology Bureau’s Key Laboratory Program.
Data availability
All data generated or analyzed during this study are included in this article.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-74742-5.
References
- 1.Stockwell, B. R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell185(14), 2401–2421 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang, W. S. & Stockwell, B. R. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol.26(3), 165–176 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hirschhorn, T. & Stockwell, B. R. The development of the concept of ferroptosis. Free Radic. Biol. Med.133, 130–143 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen, X. et al. Ferroptosis: Machinery and regulation. Autophagy17(9), 2054–2081 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dixon, S. J. et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell149(5), 1060–1072 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conrad, M. et al. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev.32(9–10), 602–619 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Skouta, R. et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc.136(12), 4551–4556 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, J. et al. The multifaceted role of ferroptosis in liver disease. Cell Death Differ.29(3), 467–480 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu, J. et al. Ferroptosis in liver disease: New insights into disease mechanisms. Cell Death Discov.7(1), 276 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Do, B. et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis.94, 169–178 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Chen, L. et al. Ablation of the ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J. Biol. Chem.290(47), 28097–28106 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li, F. J. et al. System X(c) (-)/GSH/GPX4 axis: An important antioxidant system for the ferroptosis in drug-resistant solid tumor therapy. Front. Pharmacol.13, 910292 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dixon, S. J. et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife3, e02523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stockwell, B. R. et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell171(2), 273–285 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dolma, S. et al. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell3(3), 285–296 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Ursini, F. & Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med.152, 175–185 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Yang, W. S. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell156(1–2), 317–331 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sui, X. et al. RSL3 drives ferroptosis through GPX4 inactivation and ROS production in colorectal cancer. Front. Pharmacol.9, 1371 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Friedmann Angeli, J. P. et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol16(12), 1180–1191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheff, D. M. et al. The ferroptosis inducing compounds RSL3 and ML162 are not direct inhibitors of GPX4 but of TXNRD1. Redox Biol.62, 102703 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hou, M. J., Huang, X. & Zhu, B. T. Mechanism of RSL3-Induced Ferroptotic Cell Death in HT22 Cells: Contributing Role of Protein Disulfide Isomerase. Acta Biochim Biophys Sin (Shanghai), In Press (2024).
- 22.Tasanen, K. et al. Characterization of the human gene for a polypeptide that acts both as the beta subunit of prolyl 4-hydroxylase and as protein disulfide isomerase. J. Biol. Chem.263(31), 16218–16224 (1988). [PubMed] [Google Scholar]
- 23.Pihlajaniemi, T. et al. Molecular cloning of the beta-subunit of human prolyl 4-hydroxylase. This subunit and protein disulphide isomerase are products of the same gene. EMBO J.6(3), 643–649 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Freedman, R. B., Hirst, T. R. & Tuite, M. F. Protein disulphide isomerase: Building bridges in protein folding. Trends Biochem. Sci.19(8), 331–336 (1994). [DOI] [PubMed] [Google Scholar]
- 25.Ohba, H., Harano, T. & Omura, T. Intracellular and intramembranous localization of a protein disulfide isomerase in rat liver. J. Biochem.89(3), 889–900 (1981). [DOI] [PubMed] [Google Scholar]
- 26.Cai, H., Wang, C. C. & Tsou, C. L. Chaperone-like activity of protein disulfide isomerase in the refolding of a protein with no disulfide bonds. J. Biol. Chem.269(40), 24550–24552 (1994). [PubMed] [Google Scholar]
- 27.Benham, A. M. et al. Ero1-PDI interactions, the response to redox flux and the implications for disulfide bond formation in the mammalian endoplasmic reticulum. Philos. Trans R Soc. Lond. B Biol. Sci.368(1617), 20110403 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sevier, C. S. & Kaiser, C. A. Ero1 and redox homeostasis in the endoplasmic reticulum. Biochim. Biophys. Acta1783(4), 549–556 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Mustacich, D. & Powis, G. Thioredoxin reductase. Biochem. J.346(1), 1–8 (2000). [PMC free article] [PubMed] [Google Scholar]
- 30.Wang, H., Wang, P. & Zhu, B. T. Mechanism of erastin-induced ferroptosis in MDA-MB-231 human breast cancer cells: Evidence for a critical role of protein disulfide isomerase. Mol. Cell Biol.42(6), e0052221 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hou, M. J., Wang, P. & Zhu, B. T. Biochemical mechanism of erastin-induced ferroptotic cell death in neuronal cells. Acta Biochim. Biophys. Sin. (Shanghai)55(5), 853–865 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuang, F. et al. Oxidative damage and antioxidant defense in ferroptosis. Front. Cell Dev. Biol.8, 586578 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu, Y. et al. Ferroptosis: A cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell Death Discov.7(1), 193 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang, L. M. et al. Analysis and identification of oxidative stress-ferroptosis related biomarkers in ischemic stroke. Sci. Rep.14(1), 3803 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okada, K., Fukui, M. & Zhu, B. T. Protein disulfide isomerase mediates glutathione depletion-induced cytotoxicity. Biochem. Biophys. Res. Commun.477(3), 495–502 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Wang, H. et al. Strong protection by 4-hydroxyestrone against erastin-induced ferroptotic cell death in estrogen receptor-negative human breast cancer cells: Evidence for protein disulfide isomerase as a mechanistic target for protection. Biochemistry63, 984–999 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martucci, C. P. & Fishman, J. P450 enzymes of estrogen metabolism. Pharmacol. Ther.57(2–3), 237–257 (1993). [DOI] [PubMed] [Google Scholar]
- 38.Zhu, B. T. et al. Dietary administration of an extract from rosemary leaves enhances the liver microsomal metabolism of endogenous estrogens and decreases their uterotropic action in CD-1 mice. Carcinogenesis19(10), 1821–1827 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Zhu, B. T. Catechol-O-methyltransferase (COMT)-mediated methylation metabolism of endogenous bioactive catechols and modulation by endobiotics and xenobiotics: Importance in pathophysiology and pathogenesis. Curr. Drug Metab.3(3), 321–349 (2002). [DOI] [PubMed] [Google Scholar]
- 40.Voltan, G. et al. Role of estrogen and estrogen receptor in GH-secreting adenomas. Int. J. Mol. Sci.24(12), 9920 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rej, R. K. et al. Targeting the estrogen receptor for the treatment of breast cancer: Recent advances and challenges. J. Med. Chem.66, 8339–8381 (2023). [DOI] [PubMed] [Google Scholar]
- 42.Choi, H. J. et al. 4-hydroxyestrone, an endogenous estrogen metabolite, can strongly protect neuronal cells against oxidative damage. Sci. Rep.10(1), 7283 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jafari, R. et al. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc.9(9), 2100–2122 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Xuan, W. et al. Propofol protects against erastin-induced ferroptosis in HT-22 cells. J. Mol. Neurosci.72(9), 1797–1808 (2022). [DOI] [PubMed] [Google Scholar]
- 45.Fujita, I. et al. Cystamine-mediated inhibition of protein disulfide isomerase triggers aggregation of misfolded orexin-A in the Golgi apparatus and prevents extracellular secretion of orexin-A. Biochem. Biophys. Res. Commun.489(2), 164–170 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Palacios, M. et al. Chlorpromazine inhibits both the constitutive nitric oxide synthase and the induction of nitric oxide synthase after LPS challenge. Biochem. Biophys. Res. Commun.196(1), 280–286 (1993). [DOI] [PubMed] [Google Scholar]
- 47.Balaganur, V. et al. Effect of S-methylisothiourea, an inducible nitric oxide synthase inhibitor, in joint pain and pathology in surgically induced model of osteoarthritis. Connect. Tissue Res.55(5–6), 367–377 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Imai, H. et al. Lipid peroxidation-dependent cell death regulated by GPx4 and ferroptosis. Curr. Top. Microbiol. Immunol.403, 143–170 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Markides, C. S., Roy, D. & Liehr, J. G. Concentration dependence of prooxidant and antioxidant properties of catecholestrogens. Arch. Biochem. Biophys.360(1), 105–112 (1998). [DOI] [PubMed] [Google Scholar]
- 50.Nakano, M. et al. Novel and potent biological antioxidants on membrane phospholipid peroxidation: 2-hydroxy estrone and 2-hydroxy estradiol. Biochem. Biophys. Res. Commun.142(3), 919–924 (1987). [DOI] [PubMed] [Google Scholar]
- 51.De La Cruz, J. P. et al. Role of the catechol group in the antioxidant and neuroprotective effects of virgin olive oil components in rat brain. J. Nutr. Biochem.26(5), 549–555 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Lacort, M. et al. Protective effect of estrogens and catecholestrogens against peroxidative membrane damage in vitro. Lipids30(2), 141–146 (1995). [DOI] [PubMed] [Google Scholar]
- 53.Xiang, D. et al. Protective effects of estrogen on cardiovascular disease mediated by oxidative stress. Oxid. Med. Cell Longev.2021, 5523516 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bukato, K. et al. Endogenous estrogen metabolites as oxidative stress mediators and endometrial cancer biomarkers. Cell Commun. Signal22(1), 205 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhu, Y. et al. Protein disulfide isomerase plays a crucial role in mediating chemically-induced, glutathione depletion-associated hepatocyte injury in vitro and in vivo. Cell Commun. Signal.22, 431 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang, P. & Zhu, B. T. Unique effect of 4-hydroxyestradiol and its methylation metabolites on lipid and cholesterol profiles in ovariectomized female rats. Eur. J. Pharmacol.800, 107–117 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Corti, C. et al. Novel endocrine therapies: What is next in estrogen receptor positive, HER2 negative breast cancer?. Cancer Treat. Rev.117, 102569 (2023). [DOI] [PubMed] [Google Scholar]
- 58.Zhu, B. T. et al. Quantitative structure-activity relationship of various endogenous estrogen metabolites for human estrogen receptor alpha and beta subtypes: Insights into the structural determinants favoring a differential subtype binding. Endocrinology147(9), 4132–4150 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this article.