

Abstract
Among the aromatic heterocycle rings, pyrazole –a five-membered ring with two adjacent nitrogen atoms in its structure has been postulated as a potent candidate in the pharmacological context. This moiety is an interesting therapeutic target covering a broad spectrum of biological activities due to its presence in many natural substances.
Hence, the potential of the pyrazole derivatives as antitumor agents has been explored in many investigations, showing promising results in some cases. In this sense, breast cancer, which is already the leading cause of cancer mortality in women in some countries, has been the topic selected for this review, which covers a range of different research from the earliest studies published in 2003 to the most recent ones in 2021.
Keywords: Breast, cancer, heterocycle, pyrazole, tumor, HDI
1. INTRODUCTION
It is well known that cancer is the second cause of mortality in the world after cardiovascular diseases [1]. Every year, millions of people get this disease, and half lose their lives. Although chemotherapy is one of the most effective and extensive treatments to combat this malady, the lack of selectivity of the drugs joined to the secondary effects caused by them or the development of drug-resistance experimented by the patients reduces the efficacy of this approach [2]. Consequently, this supposes a big problem of health in many developed countries, and a great number of research groups have focused their efforts on the study of this mortal illness [3-12]. Therefore, there is a current need for discovering new techniques and drug candidates overall due to the high mortality rates [13-15].
1.1. Breast Cancer
Among the different types of cancer, which overall represent approximately 30% of premature deaths among the adult population aged 30-69 [16], breast cancer is postulated to be the predominant form of cancer in the female population, involving more than 50% of the reported cases of this disease [17].
Although hereditary and genetic factors such as personal or family history of breast or ovarian cancer and inherited mutations (in BRCA1, BRCA2 and other breast cancer susceptibility genes) account for 5-10% of breast cancer cases, studies in relation to migration processes have shown that non-hereditary factors are the main drivers of international and inter-ethnic differences. This has been demonstrated in comparative studies of low-risk populations that have migrated to high-risk populations, the movement of which has led to increased breast cancer incidence rates in subsequent generations [18].
These increased incidence rates in countries with higher HDI (Human Development Index, a summary measure of average achievement in dimensions of human development: a long and healthy life, being knowledgeable and having a decent standard of living) are associated with a higher prevalence of known risk factors. On the other hand, high incidence rates in transition countries are related to a higher prevalence of known risk factors concerning menstruation, reproduction, nutrition or anthropometry, among others [19].
However, knowledge of this disease in terms of geographical or temporal variations in rates and specific etiological factors is still limited. Looking at different geographical areas, it can be observed that countries, where incidence rates have historically been low have experienced a marked increase during the last decades (e.g., in South American, African or Asian countries) [20]. These trends are probably due to a combination of demographic factors related to social and economic development. Similarly, among other aspects, the decline in incidence in the early 2000s in countries such as the United States, Canada, the United Kingdom and France was attributed to a decrease in the use of postmenopausal hormone treatment following the report of the Women's Health Initiative study linking the use of postmenopausal hormones with an increased risk of breast cancer [21].
The major risk factors for breast cancer are not easily modifiable because they derive from endogenous and prolonged hormonal exposures, although prevention by reducing alcohol consumption, engaging in physical activity, enhancing breastfeeding during the postpartum period or limiting hormone therapy may be beneficial.
1.2. Heterocycle-Based Drugs
Within the plethora of anticancer drugs, those based on heterocycles have received special attention [22-27]. Among them, the pyrazole core is present in many of these potential active compounds (Fig. 1), and new synthetic methods have been developed in the last two decades to achieve a great scope of this interesting family of targets [28].
Fig. (1).

Examples of pyrazole-based biologically active compounds.
In view of its large pharmacological scope, the pyrazole skeleton is considered a privileged scaffold in medicinal chemistry because of its properties as anti-influenza [29], antiviral [30-32], anti-inflammatory [33], antitubercular, antimicrobial, antioxidant, antidepressant, and insecticidal agent [34-36]. Related to cancer treatment, Crizotinib and Ruxolitinib [37] or Celecoxib [38-40] are important pyrazole-based anticancer drugs [41, 42]. Interestingly, there are also pyrazole-based drugs commercially available such as those described in Fig. (2) [43], among others.
Fig. (2).

Examples of pyrazole-based drugs are approved for commercialization [43].
Like many other drugs, pyrazoles exhibit activity due to their inhibitory capacity of diverse pivotal targets directly involved in the mechanism of cancer. In this work, we will also consider different aspects related to the role of pyrazole in the mechanism of cellular death. It is also remarkable that the rational design of new pyrazole scaffolds could be of interest in new drug discovery.
We would like to give the readers a broad overview of the plethora of pyrazole structures synthesized in the last two decades (2003-2021), emphasizing their utility as privileged scaffolds in the context of breast cancer, as previously mentioned, one of the most widespread cancers in women [44]. In this way, various representative examples of highly substituted pyrazole scaffolds and their pharmacological activity as potential anticancer drugs are illustrated and discussed in this work.
2. PYRAZOLES AS PROMISING ANTITUMOR AGENTS AGAINST BREAST CANCER
Hereafter, a great number of studies related to the different pyrazole derivatives with interesting results as antitumor agents are reported. Some of the sections in which this review has been structured are their capacity as kinase inhibitors, angiogenic agents, and those that act on estrogen-dependent factors or studies based on the synergistic effect with other structures of interest. Although the tumor lines studied in most research are diverse (and will be mentioned), only the results focused on breast cancer cells will be highlighted in this revision.
2.1. Role as Kinase Inhibitors
Biological functions in eukaryotes are regulated by phosphorylation and dephosphorylation of proteins by kinases and phosphatases, respectively [45]. Therefore, the blocking of kinases is considered for possible therapies against cancer as well as inflammatory diseases. Hence, the human genome encodes many of these proteins, some of which are recognized as disease-relevant, and small molecules targeting the ATP-binding site in these enzymes may inhibit the phosphorylation selectively, despite its ubiquitous nature [46, 47].
Moreover, selective inhibitors can be modelled by optimizing the peripheral groups, with the pyrazole moiety being a favored structure targeting kinases [48-51]. Over the last decade, much research has highlighted the use of pyrazole derivatives as kinase inhibitors with a broad spectrum of biological properties with a high pharmacological interest in different areas, like those referred to antimicrobial activity and neurodegenerative disorders or even cancer, among others [52].
In this context, Nielsen and co-workers carried out the synthesis of highly functionalized pyrazole carboxamides and carboxylic acids by saponification and transamidation, respectively, of ester-functionalized pyrazoles [53]. Following this synthetic protocol, pyrazole scaffolds were tuned to optimize protein kinase inhibition, being tested on AKT1, CK2, PKA, PKCα and SAPK2a (p38) (each compound was studied using a final concentration of 10 μM). The most promising inhibitor compounds are depicted in Fig. (3). Thus, ester 1a showed significant inhibition of AKT1 and SAPK2a (p38) kinases, reducing activity by 40% and 35%, respectively (Fig. 3A), while carboxylic acid 1b inhibited AKT1 by 46% and PKA by 82% (Fig. 3A). Additionally, the most promising demonstration of inhibitory activity was observed for amide 1c with a lipophilic attachment, which inhibits several kinases (AKT1 by 61%, CK2 by 42% and PKA by 84%), indicating its multi-targeting approach (Fig. 3, A).
Fig. (3).

Pyrazoles 1 with protein kinase inhibition (A) and antitumoral activity (B) [53].
It is known that a drug targeting, for instance, AKT, is expected to affect cell growth and viability. Therefore, to further evaluate the physiological effect of these compounds, the authors first analyzed human MCF-7 breast cancer cells by phase-contrast microscopy either untreated or incubated for 48 h with the tested product at concentrations of 50 and 100 μM. The morphological effects revealed that treatment with 1a and 1c had a clear inhibitory effect on cell growth (especially at the higher concentration), while treatment of the cells with 1b caused no change in cell growth (Fig. 3B, (a)). However, since no large numbers of floating dead cells were observed under the microscope for none of the compounds, it seems they did not cause severe cell death.
Later, the crystal violet binding method was used to quantify the effect of the selected inhibitors on cell growth (Fig. 3B, (b)). Then, incubation of cells for 24 h with ester (1a) at 50 and 100 μM concentrations resulted in a ∼50% decrease in viability, reaching up to 24% of cells remaining with 100 μM after 72 h; although the best result corresponded to amide 1c, which induced the loss of 90% of cells under the same conditions (100 μM after 72 h). However, carboxylic acid 1b did not lead to a significant alteration. Since crystal violet staining does not distinguish between actively dividing and quiescent cells, the reduction in cell numbers could result from cytotoxic and cytostatic effects.
Moreover, the distribution of cells was analyzed in different phases of the MCF-7 cell cycle before and after treatment with 100 μM of these pyrazole derivatives for 24 h and 48 h (Fig. 3B, (c)). In this case, only incubation with 1c for 48 h produced cell cycle arrest (S phase: 7.7% (control) → 20.3% (treated cells) and G2/M phase: 11.1% (control cells) → 22.4% (treated cells)), while the other two compounds 1a and 1b did not cause any variation in it. According to these data, the effect of amide 1c over cell viability can be attributed to arrested cells, while ester 1a influence can be associated to deceleration of cell metabolism.
In a study reported in 2010 by Lee’s group, 1H- and 2H-pyrazole derivatives were designed and synthesized for an extensive structure-activity relationship study [54] on the bases of the potent B-Raf inhibitor GDC-0879 (Fig. 1) [55]. In Fig. (4), six compounds out of thirteen (2a-f) showed very good activity in preliminary assays at 10 μM and were derived in subsequent assays to determine the IC50 in 60 cell lines. In particular, excellent values were obtained for six breast cancer cell lines (Fig. 4), which in some cases were in the nanomolar range (i.e., 26 nM for 2c in MDA-MB-468).
Fig. (4).

Selected compounds 2a-f to carry out the 60-cell-line screening at the National Cancer Institute (NCI), Bethesda, Maryland, USA and IC50 values in μM for six different cell lines of breast cancer [54].
The results obtained from the in vitro assays, together with the structure-activity relationship (SAR) correlation studies, revealed that to obtain the best activity, the compounds should be substituted at the 4-position of the pyrazole with a 4-(dimethylamino) phenyl group (2a, 2c and 2d) at the 2-position of the terminal pyridyl group, followed by 4-phenoxyphenyl (2e) and 4-acetyl (2b) substitution. The acetonitrile group at the 2-position of the pyrazole ring (2H isomers) was also enhanced over the 1-position. In addition, the effect of demethylation of the methoxy group (2a) to produce the hydroxyl analogues (2d-e) was not constant in the derivatives, suggesting the possibility of variable binding modes of these compounds at their target site or the presence of different targets, whose binding mode at each molecule would be different.
Moreover, following the interest in kinase inhibition and the understanding of the mechanism of action, compound 2c, which had the highest potency in almost all cancer cell lines, was tested at 10 μM concentration on a panel of 54 kinases at Reaction Biology Corporation, showing very good selectivity for FLT3 kinase with an inhibition percentage of 82.3%. This would mean that this compound could be used as a good new lead for developing selective FLT3 kinase inhibitors [56-58]. However, the moderate potency of the compound on FLT3 kinase (IC50 = 1.74 μM) could not justify its strong and broad-spectrum anticancer activity, and this suggested the presence of other underlying mechanisms that (in addition to moderate FLT3 kinase) might control the anticancer activity.
As well-known, the epidermal growth factor receptor (EGFR) kinase plays a crucial role in signal transduction pathways that regulate cell division, differentiation and migration. As usual, its abnormal function implies the development of illness. Thus, the overexpression of EGFR has been observed in many human cancers, such as bladder, breast, colon and lung cancers [59, 60]. In this context, small molecules may inhibit the kinase activity of EGFR by binding the ATP site of the intracellular tyrosine kinase domain to block tyrosine kinase (TK) autophosphorylation. This is a traditional area of research for developing anticancer drugs [61, 62] and many EGFR inhibitors have been used to treat human cancers [63-65].
Among other interesting structures, compounds containing pyrazole and cinnamyl moieties have shown high inhibitory activity against EGFR [66-70]. In particular, cinnamic acid ester derivatives have been postulated as structures with potential antitumor activity [71, 72]. For instance, Qian and co-workers found some metronidazole derivatives 3 to be potent inhibitors of EGFR and HER-2 (Fig. 5), as well as they presented good antiproliferative activity against MCF-7 cell line, such as compounds 3a-b which reached values of IC50 of 0.36 and 0.98 μM, respectively [73].
Fig. (5).

Cinnamic acid metronidazole ester derivatives 3 were evaluated as anticancer agents, and IC50 values for the two most active molecules (3a-b) against the MCF-7 cell line [73].
A few years later, the same group performed the synthesis and biological evaluation of other series of pyrazole-cinnamoyl derivatives 4 (Fig. 6) [74]. Antiproliferative activity against MCF-7 (breast cancer) of the synthesized compounds was also tested in this case, showing remarkable effects. In addition, it was found that most of these derivatives presented good inhibitory activity against EGFR and HER-2. Among them, compound 4ae exhibited the most potent activity (IC50 = 0.30 μM (MCF-7), IC50 = 0.21 μM (EGFR) and IC50 = 1.08 μM (HER-2)) (Fig. 6).
Fig. (6).

Structure-activity relationship study of pyrazole-cinnamyl derivatives 4 [74].
Structure-activity relationship studies were performed by modification of the parent compound to determine how these substituents might affect the activity. Inspection of the chemical structures of compounds suggested that they could be divided into two subunits: The A-ring and the B-ring (Fig. 6). The introduction of an electron-donating group in both could enhance its antiproliferative activity, and the order of potency was OMe > Me. At the same time, compounds with electron-withdrawing groups in ring B caused a decrease in the activity.
The authors further performed a molecular modeling study to explore the interaction with the active site of EGFR/HER-2, taking as a reference compound 4ae, which had provided the best inhibition values. It was found to bind very efficiently to the ATP binding site of EGFR through hydrophobic interactions and stabilized by a hydrogen bond and a π-σ interaction. Thus, the oxygen atom (OMe group) at the A-ring was able to form a hydrogen bond with the amino hydrogen of Lys A: 721 (N-HꞏꞏꞏO = 2.477 Ȧ; 130.6˚). A π-σ interaction between Leu694 and the benzene ring (bond length: 2.48 Ȧ) was also confirmed. Similarly, it was seen that compound 4ae bound to the active site of HER-2 via hydrogen bonding through the formation of a hydrogen bond between that oxygen atom and the amino hydrogen of Met A: 801 (N-HꞏꞏꞏO = 2.278 Ȧ; 145.8˚).
Among the most recent research, Shahsavari’s group has focused on triple-negative breast cancer [75], which is approximately 15-20% of all breast carcinomas and is associated with an earlier age of onset, an aggressive clinical course and a poor prognosis [76].
During this study, the activity of a series of 1,3-diaryl-5-(3,4,5-trimethoxyphenyl)-1H-pyrazoles (5) and 1,3-diaryl-5-(3,4,5-trimethoxyphenyl)-4,5-dihydro-1H-pyrazoles (6) was evaluated against MDA-MB-468, the triple-negative human breast cancer cell line, and the in vitro cytotoxicity assays revealed its dose- and time-dependent anti-proliferative properties (Fig. 7). 3-(4-Methoxyphenyl)-1-(p-tolyl)-5-(3,4,5-trimethoxyphe-nyl)-4,5-dihydro-1H-pyrazole (6f) resulted in being the most active compound against MDA-MB-468, with IC50 values of 14.97 μM and 6.45 μM, after 24 and 48 hours, respectively, compared to the positive control (Paclitaxel, IC50 values of 49.90 μM and 25.19 μM). This compound also induces the minimum toxicity on normal fibroblast cell lines (AGO1522), with IC50 values of 28.74 μM and 20.33 μM, after 24 and 48 hours, respectively.
Fig. (7).

Inhibitory growth on breast cancer cells (MDA-MB-468) and toxicity on normal fibroblast cell line (AGO1522) of synthesized compounds 5a-d and 6a-g [75].
Additionally, a cell cycle study was performed after treatment with 14.97 μM of the most active compound (6f) after 24 h, concluding that the arrest occurred during the S phase. The cell death assays determined a significant increase in the early apoptotic phase in treated cells, concluding that apoptosis, and not necrosis, was the mechanism triggered by these compounds. Once this was known, the role of reactive oxygen species (ROS) in the induction of apoptosis was studied. The results of these assays led to the conclusion that ROS plays an important role in the apoptosis process. In addition, the fluorometric assay kit was used to measure the activity of caspase 3 as the main protease in the signaling of the apoptosis process, and a significant increase in its activity was observed in the cells treated with the candidate compound 6f compared to the control.
2.2. Estrogen Dependant Anti-Cancer Derivatives
Estrogens control the normal function of female sex organs, secondary sex characteristics, and mammary glands. Moreover, some breast cancers are estrogen-dependent, where the binding of estrogens to their receptors activates the transcription of their target genes, the last ones responsible for the proliferation of cancer cells in the tumor [77]. In this context, therapy based on anti-estrogens is used in several diseases, such as breast malignancies [78], including estrogen receptor antagonists, such as tamoxifen [79, 80] and toremifene [81, 82]. The discovery of the effect of cyclooxygenase-2 (COX-2) inhibitors, such as celecoxib (Fig. 2) or meloxicam [83] in many types of malignancies, especially in breast cancer [84-86], made 1,5-diphenylpyrazoles became important structures to work with.
Motivated by this background, Farag and co-workers were interested in synthesizing several 4-cyano-1,5-diphenylpyrazoles linked to different heterocyclic ring systems (7a-k) and acyl hydrazide derivatives (8a-i) at the 3-position (Fig. 8) to test their anti-estrogenic effects in vivo and to evaluate their cytotoxic properties against estrogen-dependent tumors in vitro [87].
Fig. (8).

General structures of 4-cyano-1,5-diphenylpyrazole derivatives 7 and 8 evaluated [87].
The in vitro assays were performed on seven breast tumor cell lines, namely MDA/MB-231 ATTC, MCF-7, T-47D, NCI/ADR-RES, HS-578T, MDA/MB-435, MiDA-N and BT549, as well as on additional ovarian cancer cells (6 lines), which were also addressed in this study.
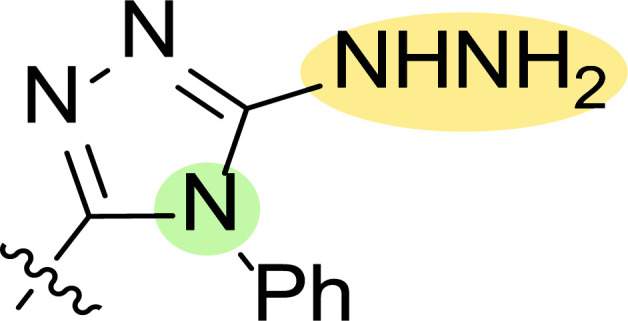
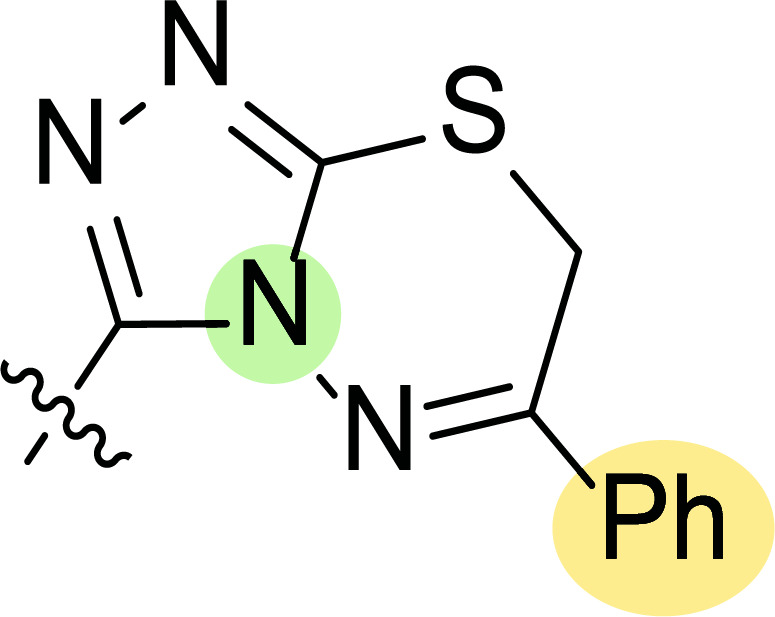
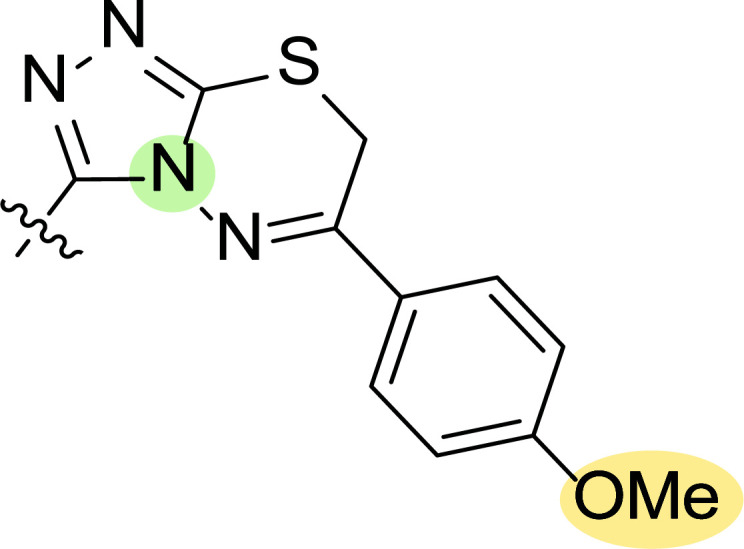
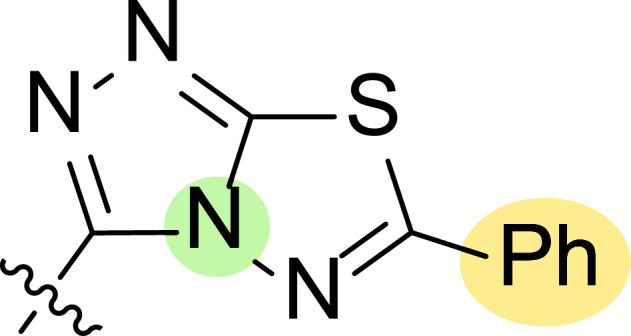
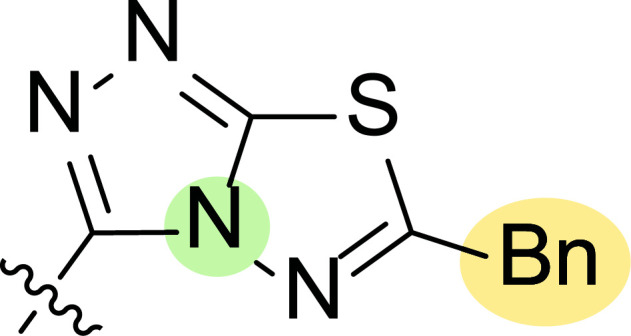
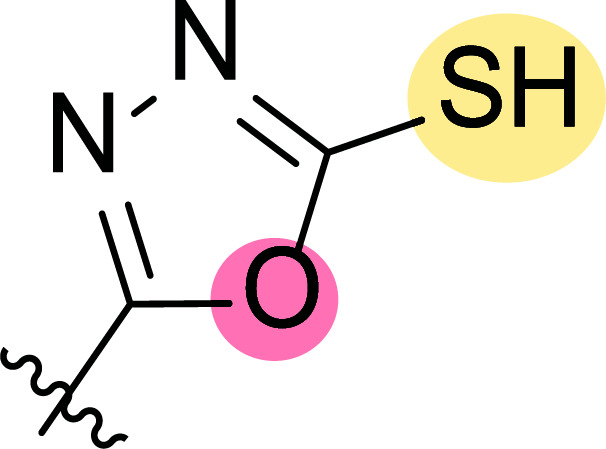
Tables 1 and 2 collect the results as growth inhibitory concentration (GI50) values for all the breast cancer lines tested, showing good activity (below 10 μM) in red. Regarding the effect of the structure, some correlations were found and are summarized as follows. Analyzing first the results obtained for a big family of triazoles (7a-i, Table 1), it is worth mentioning how variations on mercaptotriazole derivative 7a affect, which revealed good activity (2.48-6.68 μM) against five out of eight cell lines. Concerning the substitution on the N, increasing lipophilicity with a phenyl group (7b) resulted in total inactivity, while a more hydrophilic substituent, such as an amino group (7c), improved the activity against only one of the cell lines (HS-578T, 8.94 μM) for which compound 7a was inactive.
Table 1. Molecular structures 7a-k and in vitro results for different breast cancer cell lines (Growth inhibitory concentration (GI50, µM)).
| Compounds 7a-k |
MDA
MB-231 ATTC |
MCF-7 | T-47D |
NCI
ADR-RES |
HS-578T |
MDA
MB-435 |
MiDA-N | BT 549 | |
|---|---|---|---|---|---|---|---|---|---|
| 7a |
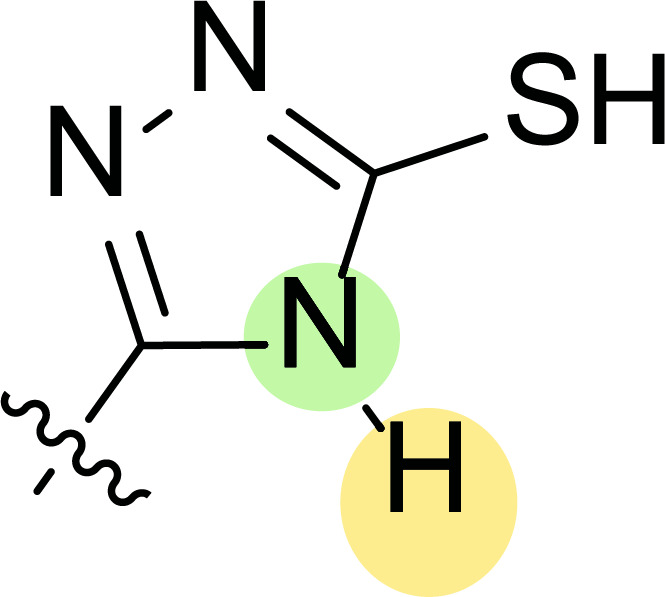
|
6.27 | IA* | 6.68 | 2.48 | IA* | 2.64 | 2.58 | IA* |
| 7b |
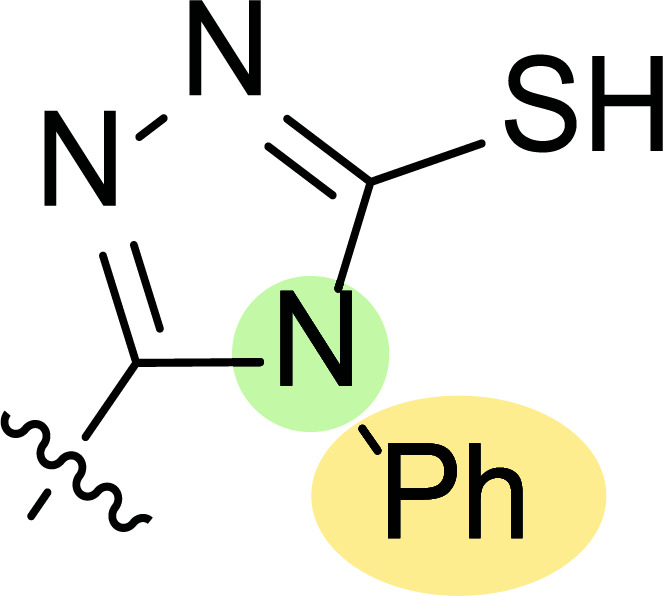
|
IA* | IA* | IA* | IA* | IA* | IA* | IA* | IA* |
| 7c |
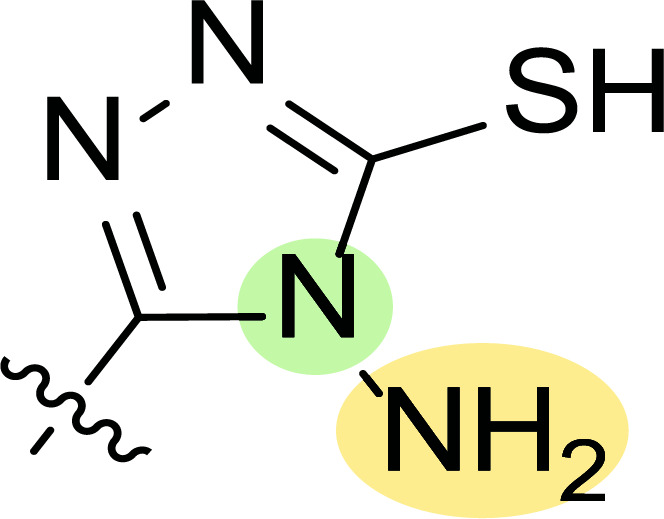
|
IA* | 23.4 | 39.9 | IA* | 8.94 | 2.93 | 5.00 | IA* |
| 7d |
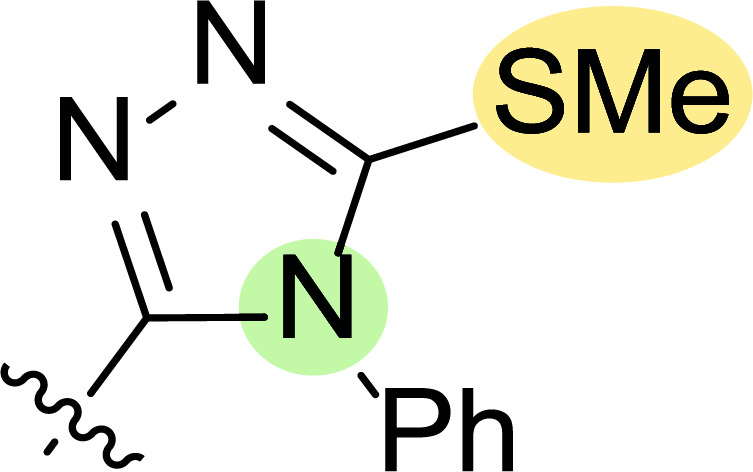
|
IA* | 68.0 | 36.3 | IA* | 69.8 | 41.8 | 73.8 | 44.8 |
| 7e |
|
IA* | IA* | IA* | IA* | IA* | IA* | IA* | IA* |
| 7f |
|
13.2 | 19.8 | 9.08 | 28.0 | IA* | 48.0 | 73.0 | 51.6 |
| 7g |
|
5.49 | IA* | IA* | 8.4 | 9.23 | 1.23 | 1.19 | IA* |
| 7h |
|
IA* | IA* | IA* | IA* | IA* | IA* | IA* | IA* |
| 7i |
|
52.1 | 41.6 | 42.6 | 74.7 | 8.94 | 9.23 | 5.49 | IA* |
| 7j |
|
5.84 | 8.53 | 18.0 | 99.8 | IA* | 55.8 | 93.8 | IA* |
| 7k |
|
38.4 | 38.0 | 72.0 | 99.0 | 98.0 | IA* | 55.4 | 26.2 |
Note: *IA: Inactive; GI50 >100 µM.
Table 2. Molecular structures (8a-i) and in vitro results for different breast cancer cell lines (Growth inhibitory concentration (GI50, µM)).
| Compounds 8a-i |
MDA
MB-231 ATTC |
MCF-7 | T-47D |
NCI
ADR-RES |
HS-578T |
MDA
MB-435 |
MiDA-N |
BT
549 |
|
|---|---|---|---|---|---|---|---|---|---|
| 8a |
|
61.2 | 85.8 | NT** | NT** | NT** | NT** | 1.34 | 9.13 |
| 8b |
|
IA* | 99.8 | 45.6 | IA* | 61.8 | 42.8 | 34.8 | 37.7 |
| 8c |
|
4.90 | IA* | 63.4 | IA* | 8.04 | 9.30 | 2.86 | IA* |
| 8d |
|
6.21 | IA* | 2.45 | 2.76 | IA* | 2.75 | IA* | IA* |
| 8e |
|
23.8 | 19.8 | 54.8 | 98.0 | IA* | 39.9 | 23.8 | 11.8 |
| 8f |
|
7.36 | 4.72 | 62.5 | 7.4 | 6.72 | 8.62 | 7.00 | 4.12 |
| 8g |
|
28.0 | 40.8 | 22.2 | IA* | 61.8 | 61.0 | 56.8 | 11.8 |
| 8h |
|
IA* | IA* | IA* | IA* | IA* | IA* | IA* | IA* |
| 8i |
|
IA* | 79.8 | 26.5 | 28.0 | IA* | IA* | 26.5 | 11.8 |
*IA: Inactive; GI50 >100 µM, **NT: not tested.
Additional changes on the inactive molecule 7b, such as methylation of the thiol group (7d), caused little improvement in the activity, getting GI50 up to 36.3 μM, whereas produced no effect in the case of replacement of the thiol group by a hydrazine moiety (7e), which was also totally inactive. The idea of generating fused ring systems (7f-i) out of the amino and thiol groups of molecule 7c had different effects depending on the cell line. Hence, in the case of the two derivatives with a second 5-membered ring (7h-i), aromatic substitution (Ph) rendered a totally inactive compound (7h), while aliphatic substitution (Bn) involved improvement only for two of the cell lines (MDA MB-231 ATTC and NCI ADR-RES), keeping similar activity for the other ones. The other two derivatives with a second 6-membered ring (7f-g) both contain aromatic substitution; a curious complementarity is observed in the case of a phenyl group (7f), compared to parent molecule 7c, meaning that the most sensitive cells (HS-578T, MDA MB-435 and MiDA-N) for the later (7c), are the most resistant ones for the former (7f). Exactly the same occurs when the comparison is between 7f and 7i. In general, introducing a methoxy group in the para-position of the phenyl substituent (7g) translates into the best activity, except against MCF-7, T-47D and BT549, compared to both its parent molecule 7c and its homologous 7f. In fact, the best activity within all newly synthesized compounds is achieved in derivative 7g with GI50 values of 1.19 and 1.23 μM against MiDA-N and MDA MB-435, respectively.
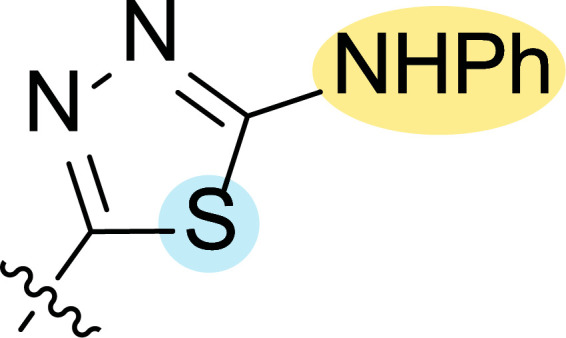
Finally, the change of the N of the triazole by another heteroatom was examined. In this context, the activity of oxadiazole 7k is worst compared to its homologous triazole 7a, except against MCF-7, HS-578T and BT549. Thiadiazole 7j, which is not exactly comparable with 7a because of its different substitution at the C-position (7j: NHPh and 7a: SH), gave good inhibition in the case of MDA MB-231 ATTC and MCF-7 (5.84 and 8.53 μM, respectively).
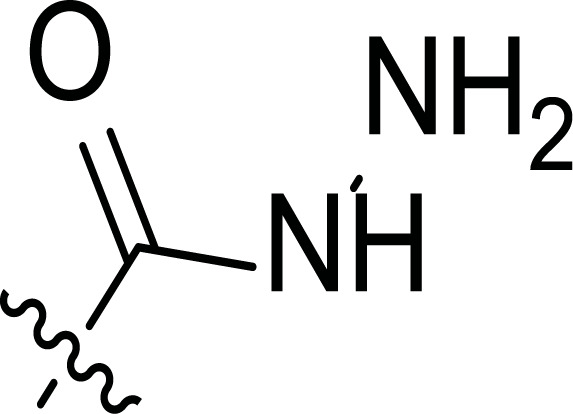
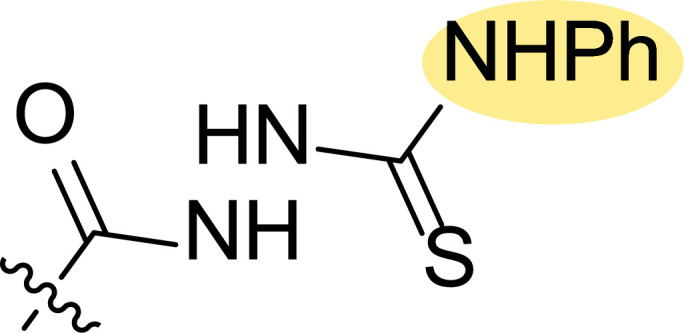
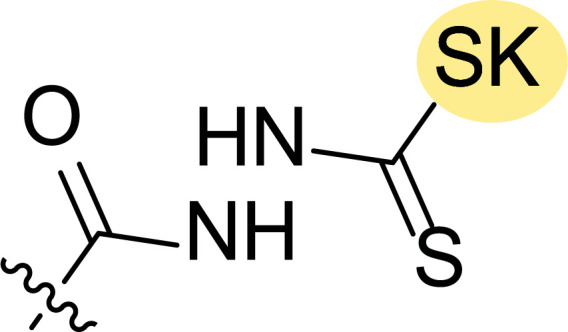
The second line of structural optimization was carried out based on the remarkable cytotoxicity of the carboxylic acid hydrazide derivative 8a (GI50 1.34 μM against MiDA-N, Table 2). Thus, for instance, the thiosemicarbazide derivative 8b was found to have moderate to weak activity, whereas the potassium salt 8c provided good inhibition between 2.86 and 9.30 μM against MDA MB-231 ATTC, HS-578T, MDA MB-435 and MiDA-N cell lines.
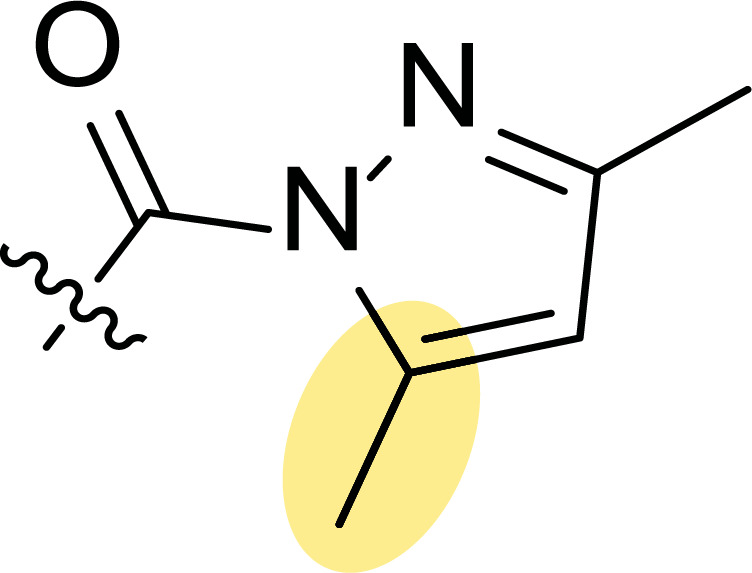
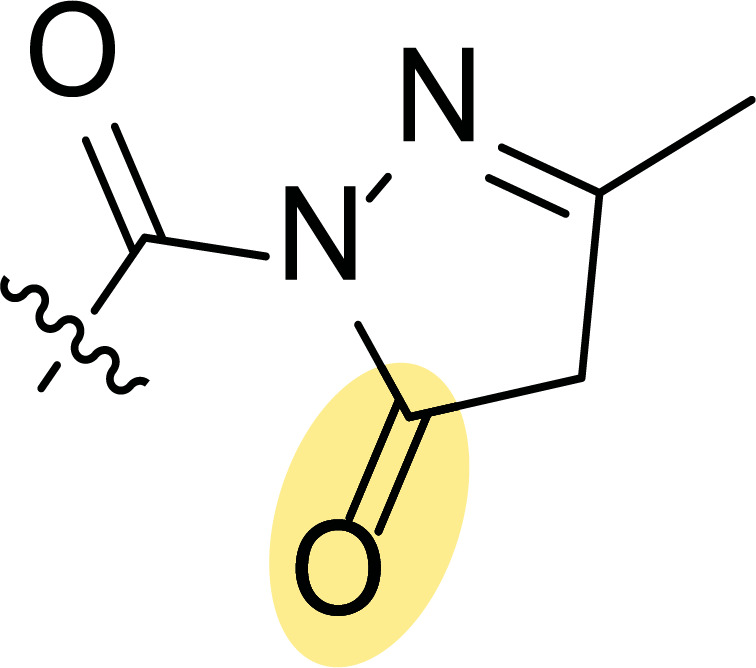
Additionally, the pyrazole derivative 8d seemed cytotoxic against MDA MB-231 ATTC, T-47D, NCI ADR-RES and MDA MB-435 lines (6.21, 2.45, 2.76 and 2.75 µM, respectively), while in general, its homologous pyrazolone 8e had a moderate to weak cytotoxicity.
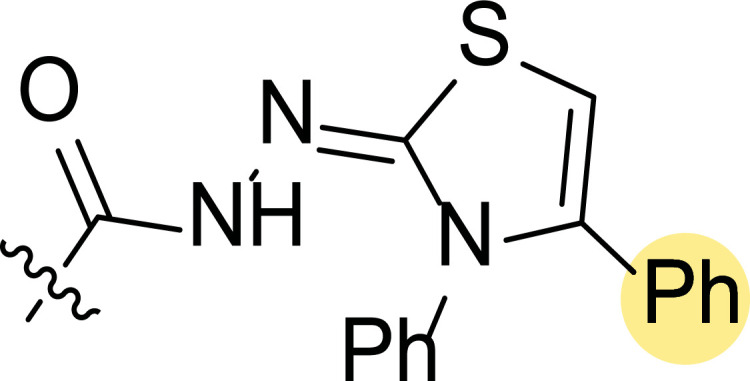
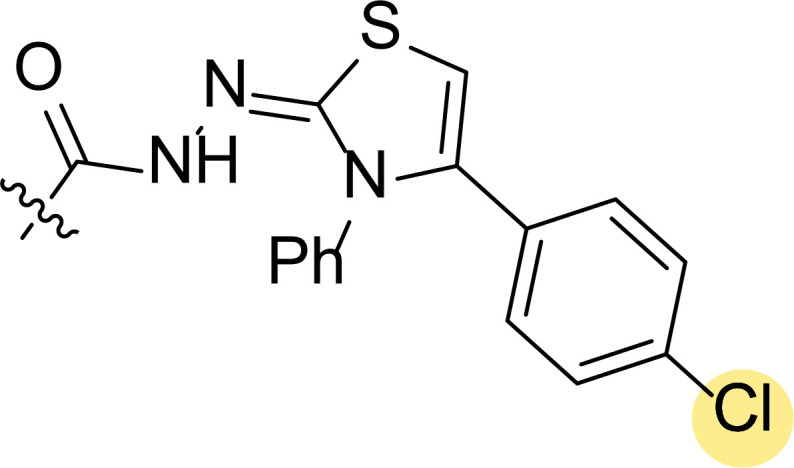
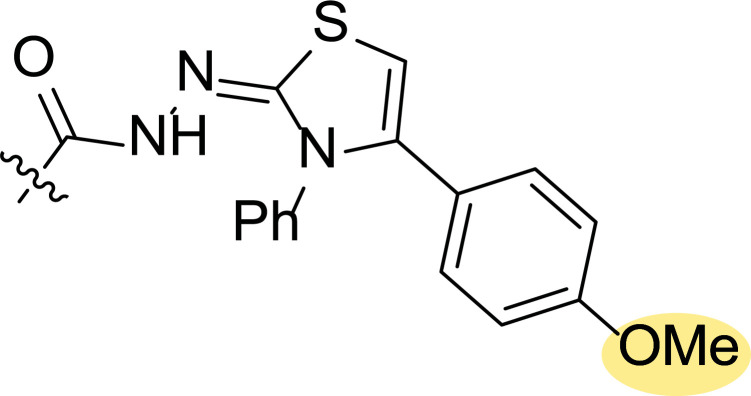
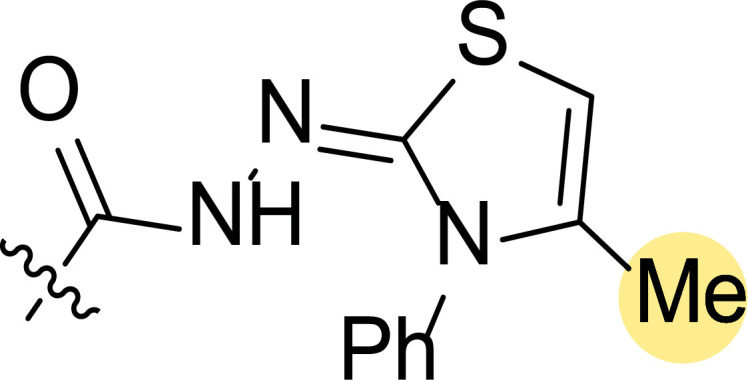
Finally, the authors also explored incorporating a 5-membered ring containing S and N in the 2-N position of the acyl hydrazide (8f-i). Thus, the integration of phenylthiazolidene led to compound 8f with the broadest scope, showing GI50 values ranging between 4.12 and 8.62 µM for all the cell lines except T-47D (62.5 µM) and either replacing the phenyl group with a methyl group (8i) or incorporating a chlorine atom at the para-position of it (8g) allowed the improvement of the activity against this cell line (26.5 and 22.2 µM, respectively), being the inhibition worse for the rest of tested cell lines. Curiously, substitution in the para-position of the phenyl ring with a methoxy group turned the compound (8h) inactive.
Acute in vivo toxicity studies were performed on a representative sample of the novel compounds synthesized. Rats were orally treated with different doses of an aqueous suspension of a very fine powder of the tested compounds. Under these conditions, as shown in Table 3, the tested compounds were found to be highly safe (LD50 values) for albino rats compared to the reference drug letrozole [88].
Table 3. Acute toxicity (LD50) of the synthesized compounds and letrozole.
| Compounds | LD50 (mg/kg) |
|---|---|
| Letrozole | 252.61 ± 0.11 |
| 7a | 434.71 ± 0.13 |
| 7b | 122.48 ± 0.13 |
| 7k | 323.06 ± 0.11 |
| 8a | 332.11 ± 0.13 |
| 8b | 311.52 ± 0.12 |
| 8c | 431.85 ± 0.13 |
| 8d | 873.06 ± 0.19 |
| 8e | 234.71 ± 0.13 |
Additionally, many pathological complications (breast cancer, endometrial cancer, osteoporosis, etc.) originate in aberrant expression of estrogen receptor alpha (ER-α). It plays a key role in breast cancer initiation and progression and confers chemoresistance to cancer cells by regulating the expression of anti-apoptotic proteins (Bcl2) [89]. The overexpression of estrogen receptors is responsible for 70% of breast cancer cases, named estrogen receptor-positive tumors [90, 91].
On the base that pyrazoles are good ligands for estrogen receptors, Rangappa and coworkers have been working in the last years on the synthesis and study of the antitumoral activity of novel derivatives. From a series of novel 1-(4-methoxybenzyl)-3-cyclopropyl-1H-pyrazol-5-amine derivatives 9a-h, some compounds exhibited interesting growth inhibitory effects against MCF-7 cell line (Fig. 9) [92]. Structure-activity relationship studies revealed that electron donating groups on the phenyl ring, such as 3-methoxy (9b), 3-methyl (9g), and 4-tert-butyl (9d), provided significant activity, whereas electrons withdrawing groups, such as 4-nitro (9a), 4-chloro (9c), 2-fluoro (9e) and 3-bromo (9f) delivered poor activity.
Fig. (9).

1-(4-Methoxybenzyl)-3-cyclopropyl-1H-pyrazol-5-amine derivatives 9a-h and IC50 (μM) values against MCF-7 [92].
Later in 2017, these authors prepared 1-aryl-3,5-bis(het)aryl pyrazole derivatives with complementary regioselectivity: 10a-e versus 11a-e, and also evaluated in vitro their activity against several breast cancer cell lines, such as MCF-7 and T47D (Fig. 10) [93].
Fig. (10).

1-Aryl-3,5-bis(het)aryl pyrazole derivatives 10a-e and 11a-e and IC50 (μM) values against MCF-7 and T47D [93, 94].
Compound 11d showed the highest antiproliferative effects from this series, blocking cells in the sub-G1 phase of the cell cycle and promoting apoptosis. In addition, it was concluded that the compound decreased mitochondrial membrane potential, induced DNA fragmentation and chromatin condensation, and necessitated caspases cleavage in breast cancer cell lines [85].
Authors continued their work searching for new anti-estrogens or selective estrogen receptor modulators (SERMs) and published in 2018 further studies using compound 11d (Fig. 10) [94]. Molecular docking studies showed that fixes efficiently to the ligand binding domain of ER-α via hydrophobic interactions with a receptor pocket [95]. Additionally, in vivo assays were performed in Sprague-Dawley (SD) rats, carriers of a DMBA-induced mammary gland tumor. Twenty-five days after inoculation, animals were sacrificed, and the mammary glands were removed for evaluation. Treatment with the drug candidate reduced tumor size, indicating its ER-α antagonistic effect.
2.3. Pyrazoles with Anti-Angiogenic Activity
The growth of blood vessels (a process known as angiogenesis) is essential for many physiological procedures (embryogenesis, organ development, wound healing, etc.) [96, 97]. However, unregulated angiogenesis has been associated with different diseases, such as cancer, retinopathy, rheumatoid arthritis, psoriasis, atherosclerosis and hemangioma [98-101]. Antiangiogenic therapy targets activated endothelial cells and have gained increasing interest for its various advantages over targeted therapy. In addition, targets for anti-angiogenic therapy are easily accessible by systemic administration, as 10 out of 100 new tumor cells require at least one new endothelial cell (one gram of tumor contains approximately 20 million endothelial cells and 100 million to 1 billion tumor cells). Therefore, inhibiting angiogenesis and stopping the growth of an endothelial cell could have an amplified effect on tumor cells [102].
In this sense, and taking into account the good results achieved in terms of apoptosis (the programmed cell death that maintains the balance between cell survival and cell death) for some tumor cell lines previously reported using structures such as celecoxib and SC-236 (Figs. 1 and 2) [103-105], Abadi and co-workers carried out in 2003 the synthesis of a battery of 1,3,4-trisubstituted pyrazole derivatives. With a similar substitution pattern, those new structures proved to be potential antitumors and antiangiogenic agents [106].
The prepared compounds were evaluated in preliminary assays in 3 tumor cell lines (MCF-7 (breast), NCI-H460 (lung) and SF268 (central nervous system)) with a 10–4 M drug concentration, and the results were provided as a percentage of cell growth after 48 h of incubation. Of thirty-four compounds tested, 12a-b (Fig. 11) were the most active in MCF-7, being compound 12a the best one which provided a cell growth inhibition of 99%.
Fig. (11).

Selected compounds 12a-b as the most active against MCF-7 cancer cell line [106].
A second test was performed for both compounds in in vitro screening, which provides the effect on cell growth in a panel of approximately 60 human tumor lines. These lines were divided into subpanels according to different representative tumor cell lines such as leukemia, ovary, prostate, breast or central nervous system. Although no comparatives were performed with normal cells, the ratio obtained by dividing the mean graph mid-point (MG-MID) (μM) of the full panel of the compound by the MG-MID (μM) of its subpanel is considered an indicator of selectivity. Ratios between 3 and 6 indicate moderate selectivity, while those above 6 indicate high selectivity towards the corresponding subpanel [107-109]. Compound 12a exhibited significant anticancer activity with a broad spectrum of action, although without being selective towards a specific tumor line.
Furthermore, subsequent studies determined that these 1,3,4-trisubstituted pyrazole derivatives showed interesting antiangiogenetic activity, even in compounds that did not present cytotoxicity in the preliminary assays.
In 2010 the group of Liekens and Haroutounian reported the synthesis of novel fused pyrazolo [4,3-c]quinolone derivatives as potential anti-angiogenic agents, combining in a single backbone the known pharmacophore quinoline ring with a substituted pyrazole. In the course of this investigation, the authors also evaluated the cytotoxicity against various tumor cell lines, including MFC-7 (breast) (Fig. 12) [110]. Therefore, in vitro tumor cell proliferation inhibition studies were performed for hydroxyl pyrazolocarboxaldehydes 13a-e and hydroxyl pyrazoloquinolin-4-ones 14a-c. Notably, most of the tested compounds showed moderate IC50 values for the MCF-7 line between 20 and 50 μM, being compound 14a the most promising, with an IC50 value of 5.8 μM.
Fig. (12).

Cytostatic activity of compounds 13 and 14 [110].
The advantages associated with steroid-based chemotherapeutics [111-113], including non-toxicity, reduced vulnerability to multidrug resistance (MDR) and high bioavailability, led to a growing interest in the modification of steroid molecules for their biological properties. This was demonstrated by different ring modification studies of steroid molecules involving the A- and D-rings, in which the incorporation of heteroatoms (N or O) was reported to enhance the biological activities of these molecules, including antimicrobial, anti-inflammatory, hypotensive, hypocholesterolemic and diuretic activities [114-117]. Among them, the natural steroid pregnenolone is a precursor of other hormones such as cortisone, estrogen, testosterone and progesterone [118, 119]. Under these conditions, in 2012, Mohareb’s group used it as a template to develop new anticancer compounds [120, 121]. Modification of the D-ring by reaction with cyanoacetylhydrazine gave rise to the hydrazone derivative (non-shown), which finally underwent heterocyclization reactions to give the pyrazoles 15a-f (Fig. 13), pyridine (non-shown), thiazole (non-shown) and thiophene (non-shown) derivatives of pregnenolone [120]. In a parallel article, the same authors modified this precursor by reaction with 1-phenyl-3-thiosemicarbazide to give the thiosemicarbazone derivatives (non-shown), which, under the same hypothesis as above, were subjected to heterocyclization reactions to give the derivatives thiazolyl hydrazonoandrostane (non-shown) and pyrazolyl semicarbazidoandrostane derivatives 16a-d (Fig. 13) [121].
Fig. (13).

Pyrazole derivatives 15a-f and 16a-d synthesized from pregnenolone and their in vitro cytotoxic results [120, 121].
The cytotoxicity of the newly synthesized heterocyclic steroids was studied against three human tumor cell lines, namely non-small cell lung cancer (NCI-H460), central nervous system cancer (SF-268) and breast adenocarcinoma (MCF-7). The in vitro cytotoxicity assays showed little selectivity towards the different tumor lines studied, although very good GI50 values were obtained. The data for the pyrazole derivatives of both studies against the MCF-7 line range between 0.01 and 50.1 μM, as shown in Fig. (13). In fact, some of the tested compounds reached the nanomolar range and, in some cases, showed much higher inhibitory effects towards the three tumor cell lines than the reference drug, doxorubicin (MCF-7: 0.04 μM). Regarding the structure-activity relationship, it is possible to make some comments. For instance, in the case of pyrazoles 15a-f, it seems to be important the substitution of the phenyl group at the 5-position of pyrazole moiety, being the highly electronegative chlorine atom, the group that provided better GI50 values (15c-d: 0.02 and 0.01 μM), in comparison with the methoxy group (15e-f: 4.8 and 2.2 μM) and the unsubstituted phenyl ring (15a-b: 30.0 and 22.3 μM). On the other hand, in the case of pyrazoles 16c-d, their higher inhibitory effect may be due to the presence of an electron-withdrawing OH group –again at the 5-position of pyrazole moiety– (0.01 and 0.02 μM), instead of a methyl group (16a-b: 50.1 and 38.0 μM).
2.4. Candidates with Anti-Invasive Effects
Up to now, cancer treatment has mainly focused on growth reduction. Surgery, radiotherapy, and chemotherapy have all proven to be very effective in tackling tumor volume. However, cancer patients usually do not die from growth effects but the sequelae of invasion and metastasis.
The molecular mechanisms of invasion are not completely understood, but important contributions have been ascribed to proteases secreted by cancer cells, which can degrade the surrounding extracellular matrix (ECM) to cancer cell motility factors and factors that disrupt cell-cell adhesion [122, 123]. In view of the molecular cross-talks between cancer cells and host cells, receptor signaling through cytoplasmic tyrosine and serine/threonine kinases has been recognized as an important study target in cancer research. It was this growing interest in tissue invasion, a hallmark of malignant tumors responsible for poor prognosis in untreated cancer patients, that encouraged the group of Parmar to search for a new anti-invasive treatment, screening a total of ninety-five compounds from different classes of heterocyclic scaffolds, including lactones, coumarins, chromans and pyrazoles, among others [124]. These derivatives were tested against organotypic confrontation cultures of invasive human mammary carcinoma MCF-7/6 cells with embryonic chicken heart fragments in vitro. The assay consisted of three-dimensional confrontations between tumor cells and normal tissue.
In control cultures treated with solvent, MCF-7/6 cells invaded the PHF (precultured heart fragments) after 8 days of incubation. Many compounds showed no inhibition of invasion, and histologically their cultures were similar to controls. Other compounds were anti-invasive at the 10 µM concentration. However, only three of the 95 compounds, including pyrazol (17) (Fig. 14), inhibited invasion of MCF-7/6 cells at a concentration as low as 1 µM. Unfortunately, none of the compounds was active at 0.1 µM. Furthermore, sulforhodamine B (SRB) assay showed no inhibition of MCF-7/6 cell growth by these three compounds, concluding that the compounds exhibited anti-invasive activity but were not cytotoxic to cancer cells.
Fig. (14).

Pyrazole derivative 17 with anti-invasive activity [124].
Also, before performing the invasion assay on the synthesized compounds, several anti-invasive agents that inhibited both invasion and growth of tumor cells were screened. One of these compounds interfered with a common molecular target in mitosis and directional migration, namely vinca alkaloids, which bind to tubulin and prevent the assembly of the mitotic spindle and the cytoplasmic microtubule complex [125]. Another of the selected compounds was cytotoxic, with the anti-invasive effect resulting from a reduction in the number of intrinsically invasive cells in the challenged cultures.
However, the anti-invasive compounds in the study only limited invasion without affecting growth, which meant that their activity was not a result of cytotoxicity. These selective invasion inhibitors are structures of interest to understand which targets are involved in invasion mechanisms and which are involved in growth. The E-cadherin/catenin complex, for example, which is located in the cell membrane of normal epithelial cells, prevents epithelia from growing beyond their normal tissue boundaries [126]. In invasive tumors derived from epithelial tissues, the expression of this complex is down-regulated [127]. The complex in MCF-7/6 cells is inactive but is sensitive to functional regulation by several compounds shown to possess anti-invasive activity. Tangeretin, a citrus methoxyflavone, can increase E-cadherin-dependent cell-cell adhesion between MCF-7/6 cells and inhibit their invasion in the chicken heart invasion assay. This mechanism of action could be excluded for the anti-invasive compounds in the study, as no stimulation of cell aggregation was observed in vitro.
In summary, it was proved that the tested compounds did not inhibit tumor cell growth and did not have an effect through activation of the MCF-7/6 E-cadherin/catenin complex.
In addition, the broad battery of pyrazole-derived compounds synthesized allowed a further structure-activity study. Thirteen of the sixteen pyrazolyl acrylonitriles tested showed anti-invasive activity at 10 µM on MCF-7/6 cells in the chicken heart invasion assay. Moreover, among the five pyrazolyl acrylonitriles synthesized with the 3,4-difluorophenyl substituent at the 3-position, three of them showed anti-invasive activity at a concentration of 10 µM, but only compound 17 showed the activity at a concentration of 1 µM.
These results revealed that the difluorophenyl moiety at the 3-position and the 4-chlorophenyl moiety at the 5-position of the pyrazole in compound 17 enhanced the anti-invasive activity of acrylonitriles.
2.5. Synergistic Effects Related to Structure-Activity Relationship (SAR) Studies
The growing interest in the biological activity of pyrazole derivatives and the study of pharmacophore groups in the cancer field led Rostom and co-workers to prepare compounds with a 4-hydroxypyrazole core and different heterocyclic systems such as triazoles and oxadiazoles, linked to it [128]. In this way, they aimed to achieve a chemotherapeutic synergy, increasing the selectivity and decreasing the toxicity of the substances. It seems that the substituent at the 4-position of the 1,2,4-triazolin-3-thiones influences the antitumor activity, as well as in the case of the 1,3,4-oxadiazoles this is closely related to the nature of the substituent at 2-position. Thus, from the battery of synthesized compounds, 1,2,4-triazolin-3-thione 18 (4-cyclohexyl substituted) and 1,3,4-oxadiazoles 19 (2-one substituted) and 20 (2-phenyl substituted) (Fig. 15), presented very good GI50 values for all the breast cancer lines tested: MCF-7, NCI/ADR-RES, HS 578T and MDA-MB-435, showing results below 0.01 μM in some cases. Among them, compound 20 showed an amazing profile of activity towards the rest of the cancer cell lines studied (26 lines) with GI50 values lying in the nanomolar concentration range (< 0.01 μM).
Fig. (15).

Compounds 18, 19 and 20 proved to be the most active ones in Rostom’s study [128].
Interestingly, for further in vitro studies, compounds 18 and 19 were selected by the National Cancer Institute (NCI) Development Therapeutic Program and compound 20 was taken to the NCI Biological Evaluation Committee.
In 2005, Lee’s group became interested in heterocyclic cores restricted to a limited number of conformations and hindered in rotation by the substituents [129]. This led them to synthesize pyrazole oxime ether derivatives and to evaluate their inhibitory potential against different tumor cell lines through sulforhodamine B (SBR) assay [130]. Cytotoxicity results were shown as a percentage of growth inhibition after adding the different compounds at 1 µM. Among the synthesized compounds differently substituted with alkyl or aromatic groups in the R1 and R2 positions, phenoxy in R3 and benzyl or 2-aminoethyl group in R4, only three of them 21a-c (with R1, R2 = Me) had a moderate activity close to 40% inhibition at 1 μM against MCF-7 (Fig. 16).
Fig. (16).

Pyrazole oxime ether derivatives 21 as potential antitumor agents [129].
Analyzing the structure-activity relationship, a decrease in activity was observed when a larger group replaced the methyl group. Compounds with a benzyl group at the R4 position showed potent cytotoxic activity against a wide range of tumor cell lines, which led to further investigation of the modification at the R3 position of the pyrazole framework, although it was not tested in breast cancer lines.
It is known that compounds based on a tricyclic planar structure consisting entirely or partially of anthraquinone, anthrapyrazole or acridine show interesting cytostatic and antitumor properties [131]. Furthermore, the presence of a five- or six-membered heterocyclic ring fused to the anthraquinone or acridine moiety can increase the activity and overcome the multidrug resistance of tumor cells [132, 133], there has been a growing interest in the development of these structures. Pouli’s group has worked on the synthesis and biological studies of pyrano(thio)xanthenone derivatives [134-136], possessing structural similarity to the pyranoacridone alkaloid acronycine (Fig. 17), which showed promising antitumor properties in several murine solid tumor models [137] and was used as a lead for the synthesis of pharmacologically significantly improved analogues [138, 139]. A study of the structure-activity relationship in the pyranoxanthenone derivatives showed that substitution of the 6-methoxy group with a flexible (N,N-dialkylamino)ethylamino side chain substitution caused a strong enhancement of anti-proliferative activity towards the murine leukemia cell line L1210 (22, Fig. 17) [140]. Furthermore, pyrazole-fused homologues of these molecules (pyran moiety: 23 [141] and imidazole ring: 24 [142], (Fig. 17) were also synthesized and studied.
Fig. (17).

Structures of acronycine and prepared xanthenone derivatives 22-24 [140-142].
The results showed that incorporating a pyrazole ring in a xanthenone ring significantly increased antiproliferative activity. Fig. (17) presents the lowest IC50 (µM) values obtained against the MDA-MB-231 breast cancer cell line for these pyrazole-fused xanthenone amino derivatives.
At the same time, Bandgar and coworkers synthesized a library of 3,5-diaryl pyrazole derivatives 25 to test them against five tumor cell lines, including breast cancer (Fig. 18) [143]. The screening method selected to carry out these assays was the fluorescence test with propidium iodide [144]. Dyes such as propidium iodide (PI), which bind to DNA, provide a quick and accurate tool to quantify total nuclear DNA. The intensity of the PI fluorescence signal is directly proportional to the amount of DNA in each cell. Propidium iodide cannot penetrate an intact membrane, so the cells must first be permeabilized; therefore, there must be a cellular damaged.
Fig. (18).

3,5-Diaryl pyrazole derivatives 25 synthesized for the biological assays [143].
The best results regarding anticancer activity for the MCF-7 cell line were found with those compounds with the R substitution at positions 2 or 4 in the aromatic ring, preferably with Cl or Br substituents. Subsequent structure-activity studies revealed that only the halosubstituents (Cl and Br) were directly implicated in enhancing the anticancer activity. In contrast, fluorine, methyl and methoxy groups as substituents produced a marked decrease in activity (63% inhibition in the case of 2-Cl (25a) versus 15% for 2-F (25e), and 11% for 2-CH3 (25f) substitution, Fig. 18).
However, it is not just the binding of the pyrazole structure to organic compounds that have been of interest in studying its biological properties. It is well known that ferrocene core has provided a versatile scaffold for the preparation of interesting structures useful in many areas such as catalysis, materials science, or bioinorganic chemistry [145-148]. In addition, numerous studies support that combining a ferrocenyl moiety with a heterocycle could increase their biological activity or give rise to new properties [149, 150]. Moreover, it has been demonstrated that ferrocenes could act in the activation of reactive oxygen species (ROS) formed in tumor cells, partially contributing to the antiproliferative effects of cancer cells [151-153]. On the other hand, ferrocenium salts were the first type of organometallic compounds reported to have antiproliferative properties, and nowadays, many ferrocene-based compounds are used as therapeutics [154, 155].
In this context, Ruan and co-workers, encouraged by their previous works [156, 157] and the properties of ferrocene derivatives, developed a variety of ferrocenes containing the pyrazole moiety 26 and 27 to study their activity as antitumor agents (Fig. 19) [158].
Fig. (19).

Ferrocene derivatives 26 and 27 used for the in vitro assays [158].
In vitro assays were performed to study three tumor cell lines, including breast cancer MDA-MB-45, using 5-fluorouracil and cisplatin as positive controls (Fig. 19). Although none of the synthesized compounds displayed IC50 values lower than these references, the results were below 9 μg/mL in most cases. In particular, compounds 26e,h,l showed the strongest inhibitory activities against the MDA-MB-45 cell line, with IC50 values lower than 6 μg/mL. Therefore, these promising results, together with the low toxicity and the possibility of oral administration, positioned these compounds as good candidates for developing new anti-tumor drugs.
In search for new bioactive compounds, the pharmacophore hybridization strategy has been proven effective in modern medicinal chemistry since combining two molecules with different mechanisms of action often results in improved effects [159, 160]. In this context, and inspired by hybrid molecules containing benzimidazole (an interesting heterocycle for its biological properties and applications in medicine [161-163]) with enhanced anticancer activities [164, 165], Kamal, Shukla and coworkers undertook the synthesis of pyrazole derivatives 28 which incorporate that structural motif to produce promising new anticancer agents (Fig. 20) [166].
Fig. (20).

In vitro antiproliferative activity (IC50) of pyrazolo-benzimidazole derivatives 28 [166].
The authors assessed the cytotoxicity of the prepared hybrids against HaCaT (normal keratinocyte cells) and three different cancer cell lines: A549 (lung), HeLa (cervix) and MCF-7 (breast, Fig. 20). In general, growth inhibition was highly satisfactory, finding in many cases better results than those of controls used such as 5-fluorouracil (IC50 = 2.36 μM) and nocodazole (IC50 = 1.6 μM). In the case of the breast cancer line, values of IC50 below 1 μM were reached for several compounds (28d,l,p). In addition, the line corresponding to human epidermal keratinocyte provided numbers above 50 μM in most of the assays, which indicated selectivity of the pyrazoles 28 towards cancer cells in contrast to normal HaCaT cells.
Structure-activity relationships generally showed that mono-substitution with an F, Cl, Br and OMe group on the benzimidazole B ring exhibited more potent activities than CF3 groups and Me. In addition, di-substitution with Cl and Me groups demonstrated a lower inhibitory effect. On the other hand, it seems that substitution on the A ring did not influence significantly.
Moreover, the authors conducted further mechanistic studies with MCF-7 cells using the most potent compounds. For instance, it was found that the number of viable MCF-7 cells was significantly reduced after treatment with these compounds with concentrations corresponding to their IC50 for 48 h, compared to control cells.
Additional evaluation of the long-term cytotoxicity by measuring the ability of a single cell to grow into a colony revealed that these compounds inhibited MCF-7 cell colony formation, and all of them reduced clonogenic survival by about 50%.
As a potential metastatic measurement, the migration capacity of cancer cells is also a property of interest to be estimated. In this case, after treatment with pyrazolo-benzimidazoles, the amount of invasive breast cancer cells penetrating the respective wound areas was inhibited.
In addition, flow cytometry assays indicated that cell cycle arrest in the G0/G1 phase contributed to cytotoxicity. Studies conducted to elucidate the responsible mechanisms concluded that these compounds could decrease the levels of cyclins and cyclin-dependent kinases (CDKs), cell cycle regulators, leading to cell cycle arrest and, consequently, inhibiting the cell growth.
The apoptotic effect was studied through morphological changes found in MCF-7 cells treated with the IC50 concentration of the corresponding compounds for 24 h and after staining with Hoechst 33242. While control cells treated with DMSO showed uniformly dispersed chromatin, 25-34% of the cells treated with the different compounds showed apoptotic features, including chromatin condensation and nuclear degradation. Moreover, MCF-7 cells were treated with the IC50 concentration of the different compounds for 24 hours and chromosomal DNA was extracted to perform DNA fragmentation assays which may give extra information about the apoptosis-inducing effect of these products. The DNA-smeared ladder pattern observed was compatible with DNA breaks at multiple positions across the chromosomal DNA. Further contributions to the apoptotic effect of these compounds were identified, such as the 44-62% loss of mitochondrial membrane potential (DΨm) and the 2-4 times increased levels of reactive oxygen species (ROS) in the mitochondria, exhibited by compound-treated cells compared to control cells.
A failure to regulate apoptosis can lead to disease, including cancer. During this process, cysteine protease enzymes, called caspases, are activated, making caspase 3 a key regulator. Using those proteins as a target to induce apoptosis in cancer cells has emerged as an attractive strategy in cancer therapy [167]. Moreover, the introduction of a trifluoromethyl group, which increases the lipophilicity of the final products, has been of collective interest during the last decades for its effect on pharmacological properties [168]. Increased absorption across cell membranes improves the selectivity, efficacy and bioavailability of the compound in the body [169]. In addition, incorporating an azo group has also been of interest following the discovery of its ability to raise the biological activity of heterocyclic compounds [170].
Given these comments, Fayed group has recently designed a new class of pyrazoles 29 and pyrazolopyrimidines 30 bearing trifluoromethyl and azo groups into their molecular structures to examine the effect of this combination on the anticancer activity of the system [171]. In particular, the authors studied three different cancer cell lines: HepG-2 (liver), HCT-116 (colon) and MCF-7 (breast). Fig. (21) shows the predicted and experimental activity expressed as IC50 values obtained for the latter cancer cell line. The efficacy of the most active compounds (29, 30e and 30g) in activating caspase 3 in the MCF-7 cell line was tested to elucidate the pathway leading to cell death through these new derivatives. Thus, treatment of the cells with 29 and 30e resulted in an almost 5-fold increase in the level of caspase 3 compared to the control, whereas in the case of 30g, this increase was only 3.6-fold, suggesting a caspase-dependent pathway to explain the activity of these compounds (Fig. 21).
Fig. (21).

Molecular structures of 29 and 30a-h, and results of the activity against MCF-7 cells: predicted activity, experimental activity and caspase 3 enzyme assay [171].
The further analyses of a structure-activity relationship based on modeling studies led to conclude that a CN group at the 6-position, a (substituted)phenyl group at the 5-position and a free NH2 group at the 2-position of pyrazolopyrimidine-containing compounds 30, as well as the latter group at 3-position of the non-fused pyrazole scaffold 29, could be a requirement for anticancer activity. Moreover, these studies confirmed the importance of fluorine building blocks in substituting the pyrazolopyrimidine scaffold.
An important aspect to consider in medicine is the concept of “drug repurposing” as an alternative to developing drugs from the ground up [172]. This approach provides the opportunity to find new drugs from pre-existing ones or molecules with proven good activities, which is a substantial time and cost saver. With this premise, Zheng, Zhu and coworkers decided to synthesize novel derivatives (31 and 32) of Sorafenib, bis-aryl urea with remarkable biological properties [173, 174], replacing this group with a pyrazole moiety to evaluate the influence of the change on the cytotoxicity against several cancer cell lines (A549, HepG2, MCF-7, and PC-3) and kinases (VEGFR-2/KDR, BRAF, CRAF, c-Met, EGFR and Flt-3) (Fig. 22) [175].
Fig. (22).

Sorafenib molecule-based substitution pattern developed [175].
The results of the biological tests showed that the substitution of the urea fraction by the pyrazole moiety did benefit the antitumor activity. Moreover, the different substitutions in the ring also involved a change in the cytotoxicity of the final compound. Regarding the results obtained for the breast cancer line MCF-7, among other observations, it was concluded that the presence of Br in the 3-position of the aromatic ring (31a) or the lack of substituent (31d) was the most effective pattern through which the best IC50 values were obtained (1.96 and 1.24 μM, respectively), even lower than Sorafenib (Fig. 22). However, strongly electro-attracting groups, such as NO2 in the 3-position (31b), decreased activity (Fig. 22).
To further explore the mode of binding of these compounds to vascular endothelial growth factor (VEGFR-2), molecular simulation studies were performed with compound 31a. These analyses showed that the mode of binding of the synthesized compound 31a produced a new H-bond with the ASN923 residues of the carbonyl group of the N-methylpicolinamide that was not present in the reference compound, playing an important role in the enhancement of the inhibitory potency.
CONCLUSION
It has been proven that the pyrazole core and the infinite number of derivatives to which it can be linked represent a privileged structure of great pharmacological interest.
Their action as kinase inhibitors or potent antitumor agents against breast cancer lines (MCF-7 or MDA-MB-231, among others) is highly effective, reaching in some cases a level of efficacy compared to current treatments. The activity of these derivatives has not only been reflected in breast tumors but also in other cancer cell lines such as HeLa (cervix) or A549 (lung) and even as antimicrobial agents, reflecting the wide spectrum of biological activities that can be achieved.
Among the many publications collected, we highlight the preclinical in vivo assays with some of them, and the promising results obtained, encouraging us to continue working on these structures, providing a new pathway towards effective and less invasive therapies.
ACKNOWLEDGEMENTS
S.A. thanks the DGA (Aragon Government) for a predoctoral fellowship.
LIST OF ABBREVIATIONS
- HDI
Human Development Index
- SD
Sprague-Dawley
- MDR
Multidrug Resistance
- ECM
Extracellular Matrix
- SRB
Sulforhodamine B
- ROS
Reactive Oxygen Species
- CDKs
Cyclin-Dependent Kinases
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
This research was funded by Agencia Estatal de Investigación (AEI), project PID2020-117455GB-I00/AEI/10.13039/501100011033; RED2018-102471-T/AEI/10.13039/501100011033 and Gobierno de Aragón-Fondo Social Europeo (Research Group E07_20R).
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Miller K.D., Nogueira L., Mariotto A.B., Rowland J.H., Yabroff K.R., Alfano C.M., Jemal A., Kramer J.L., Siegel R.L. Cancer treatment and survivorship statistics, 2019. CA Cancer J. Clin. 2019;69(5):363–385. doi: 10.3322/caac.21565. [DOI] [PubMed] [Google Scholar]
- 2.Wu Q., Yang Z., Nie Y., Shi Y., Fan D. Multi-drug resistance in cancer chemotherapeutics: Mechanisms and lab approaches. Cancer Lett. 2014;347(2):159–166. doi: 10.1016/j.canlet.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 3.Brown S.B., Brown E.A., Walker I. The present and future role of photodynamic therapy in cancer treatment. Lancet Oncol. 2004;5(8):497–508. doi: 10.1016/S1470-2045(04)01529-3. [DOI] [PubMed] [Google Scholar]
- 4.Peer D., Karp J.M., Hong S., Farokhzad O.C., Margalit R., Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007;2(12):751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 5.Dasari S., Tchounwou P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014;740:364–378. doi: 10.1016/j.ejphar.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.West A.C., Johnstone R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Invest. 2014;124(1):30–39. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao J. Cancer stem cells and chemoresistance: The smartest survives the raid. Pharmacol. Ther. 2016;160:145–158. doi: 10.1016/j.pharmthera.2016.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keibler M.A., Wasylenko T.M., Kelleher J.K., Iliopoulos O., Vander Heiden M.G., Stephanopoulos G. Metabolic requirements for cancer cell proliferation. Cancer Metab. 2016;4:16. doi: 10.1186/s40170-016-0156-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nilsson A., Nielsen J. Genome scale metabolic modeling of cancer. Metab. Eng. 2017;43(Pt B):103–112. doi: 10.1016/j.ymben.2016.10.022. [DOI] [PubMed] [Google Scholar]
- 10.Hason M., Bartůněk P. Zebrafish models of cancer-new insights on modeling human cancer in a non-mammalian vertebrate. Genes (Basel) 2019;10(11):935. doi: 10.3390/genes10110935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andrei L., Kasas S., Ochoa Garrido I., Stanković T., Suárez Korsnes M., Vaclavikova R., Assaraf Y.G., Pešić M. Advanced technological tools to study multidrug resistance in cancer. Drug Resist. Updat. 2020;48:100658. doi: 10.1016/j.drup.2019.100658. [DOI] [PubMed] [Google Scholar]
- 12.Afshar N., English D.R., Thursfield V., Mitchell P.L., Te Marvelde L., Farrugia H., Giles G.G., Milne R.L. Differences in cancer survival by sex: A population-based study using cancer registry data. Cancer Causes Control. 2018;29(11):1059–1069. doi: 10.1007/s10552-018-1079-z. [DOI] [PubMed] [Google Scholar]
- 13.Vanneman M., Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer. 2012;12(4):237–251. doi: 10.1038/nrc3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jeon J.S., Bersini S., Gilardi M., Dubini G., Charest J.L., Moretti M., Kamm R.D. Human 3D vascularized organotypic microfluidic assays to study breast cancer cell extravasation. Proc. Natl. Acad. Sci. USA. 2015;112(1):214–219. doi: 10.1073/pnas.1417115112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaushik A.K., De Berardinis R.J. Applications of metabolomics to study cancer metabolism. Rev. Can. 2018;1870(1):2–14. doi: 10.1016/j.bbcan.2018.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.WHO. Who report on cancer: Setting priorities, investing wisely and providing care for all. 2020. Available from: https://www.who.int/publications/i/item/9789240001299.
- 17.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018;68(6):394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 18.Ziegler R.G., Hoover R.N., Pike M.C., Hildesheim A., Nomura A.M., West D.W., Wu-Williams A.H., Kolonel L.N., Horn-Ross P.L., Rosenthal J.F., Hyer M.B. Migration patterns and breast cancer risk in Asian-American women. J. Natl. Cancer Inst. 1993;85(22):1819–1827. doi: 10.1093/jnci/85.22.1819. [DOI] [PubMed] [Google Scholar]
- 19.Brinton L.A., Gaudet M.M., Gierach G.L. Breast cancer. In: Thun M.J., Linet M.S., Cerhan J.R., Haiman C.A., Schottenfeld D., editors. Cancer Epidemiology and Prevention. 4th ed. Oxford University Press:; New York: 2018. pp. 861–888. [DOI] [Google Scholar]
- 20.Bray F., McCarron P., Parkin D.M. The changing global patterns of female breast cancer incidence and mortality. Breast Cancer Res. 2004;6(6):229–239. doi: 10.1186/bcr932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rossouw J.E., Anderson G.L., Prentice R.L., LaCroix A.Z., Kooperberg C., Stefanick M.L., Jackson R.D., Beresford S.A.A., Howard B.V., Johnson K.C., Kotchen J.M., Ockene J. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results From the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288(3):321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 22.Ali I., Lone M.N., Al-Othman Z.A., Al-Warthan A., Sanagi M.M. Heterocyclic scaffolds: Centrality in anticancer drug development. Curr. Drug Targets. 2015;16(7):711–734. doi: 10.2174/1389450116666150309115922. [DOI] [PubMed] [Google Scholar]
- 23.Martins P., Jesus J., Santos S., Raposo L.R., Roma-Rodrigues C., Baptista P.V., Fernandes A.R. Heterocyclic anticancer compounds: Recent advances and the paradigm shift towards the use of nanomedicine’s tool box. Molecules. 2015;20(9):16852–16891. doi: 10.3390/molecules200916852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lang D.K., Kaur R., Arora R., Saini B., Arora S. Nitrogen-containing heterocycles as anticancer agents: An overview. Anticancer. Agents Med. Chem. 2020;20(18):2150–2168. doi: 10.2174/1871520620666200705214917. [DOI] [PubMed] [Google Scholar]
- 25.Auria-Luna F., Marqués-López E., Romanos E., Fernández-Moreira V., Gimeno M.C., Marzo I., Herrera R.P. Novel ureido-dihydropyridine scaffolds as theranostic agents. Bioorg. Chem. 2020;105:104364. doi: 10.1016/j.bioorg.2020.104364. [DOI] [PubMed] [Google Scholar]
- 26.Rodrigues J.M., Calhelha R.C., Nogueira A., Ferreira I.C.F.R., Barros L., Queiroz M.R.P. Synthesis of novel methyl 7-[(hetero)arylamino]thieno[2,3-b]pyrazine-6-carboxylates and antitumor activity evaluation: Effects in human tumor cells growth, cell cycle analysis, apoptosis and toxicity in non-tumor cells. Molecules. 2021;26(16):4823. doi: 10.3390/molecules26164823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lenis-Rojas O.A., Cordeiro S., Horta-Meireles M., Fernández J.A.A., Fernández Vila S., Rubiolo J.A., Cabezas-Sainz P., Sanchez L., Fernandes A.R., Royo B. N- Heterocyclic carbene iron complexes as anticancer agents: In vitro and in vivo biological studies. Molecules. 2021;26(18):5535. doi: 10.3390/molecules26185535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fustero S., Sánchez-Roselló M., Barrio P., Simón-Fuentes A. From 2000 to mid-2010: A fruitful decade for the synthesis of pyrazoles. Chem. Rev. 2011;111(11):6984–7034. doi: 10.1021/cr2000459. [DOI] [PubMed] [Google Scholar]
- 29.Shih S-R., Chu T-Y., Reddy G.R., Tseng S-N., Chen H-L., Tang W-F., Wu M-S., Yeh J-Y., Chao Y-S., Hsu J.T., Hsieh H-P., Horng J-T. Pyrazole compound BPR1P0034 with potent and selective anti-influenza virus activity. J. Biomed. Sci. 2010;17(13):13. doi: 10.1186/1423-0127-17-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hashem A.I., Youssef A.S.A., Kandeel K.A., Abou-Elmagd W.S.I. Conversion of some 2(3H)-furanones bearing a pyrazolyl group into other heterocyclic systems with a study of their antiviral activity. Eur. J. Med. Chem. 2007;42(7):934–939. doi: 10.1016/j.ejmech.2006.12.032. [DOI] [PubMed] [Google Scholar]
- 31.Rashad A.E., Hegab M.I., Abdel-Megeid R.E., Micky J.A., Abdel-Megeid F.M.E. Synthesis and antiviral evaluation of some new pyrazole and fused pyrazolopyrimidine derivatives. Bioorg. Med. Chem. 2008;16(15):7102–7106. doi: 10.1016/j.bmc.2008.06.054. [DOI] [PubMed] [Google Scholar]
- 32.Morsy A.R.I., Ramadan S.K., Elsafty M.M. Synthesis and antiviral activity of some pyrrolonyl substituted heterocycles as additives to enhance inactivated Newcastle disease vaccine. Med. Chem. Res. 2020;29:979–988. doi: 10.1007/s00044-020-02538-z. [DOI] [Google Scholar]
- 33.Chandna N., Kumar S., Kaushik P., Kaushik D., Roy S.K., Gupta G.K., Jachak S.M., Kapoor J.K., Sharma P.K. Synthesis of novel celecoxib analogues by bioisosteric replacement of sulfonamide as potent anti-inflammatory agents and cyclooxygenase inhibitors. Bioorg. Med. Chem. 2013;21(15):4581–4590. doi: 10.1016/j.bmc.2013.05.029. [DOI] [PubMed] [Google Scholar]
- 34.Steinbach G., Lynch P.M., Phillips R.K., Wallace M.H., Hawk E., Gordon G.B., Wakabayashi N., Saunders B., Shen Y., Fujimura T., Su L.K., Levin B., Godio L., Patterson S., Rodriguez-Bigas M.A., Jester S.L., King K.L., Schumacher M., Abbruzzese J., DuBois R.N., Hittelman W.N., Zimmerman S., Sherman J.W., Kelloff G. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N. Engl. J. Med. 2000;342(26):1946–1952. doi: 10.1056/NEJM200006293422603. [DOI] [PubMed] [Google Scholar]
- 35.Padmaja A., Payani T., Reddy G.D., Padmavathi V. Synthesis, antimicrobial and antioxidant activities of substituted pyrazoles, isoxazoles, pyrimidine and thioxopyrimidine derivatives. Eur. J. Med. Chem. 2009;44(11):4557–4566. doi: 10.1016/j.ejmech.2009.06.024. [DOI] [PubMed] [Google Scholar]
- 36.Ramadan S.K., El-Helw E.A.E. Synthesis and antimicrobial activity evaluation of some novel heterocycles derived from chromonyl-2(3H)-furanone. J. Chem. Res. 2018;42(6):332–336. doi: 10.3184/174751918X15295796734379. [DOI] [Google Scholar]
- 37.Bronson J., Dhar M., Ewing W., Lonberg N. Chapter thirty-one -to market, to market-2011. Annu. Rep. Med. Chem. 2012;47:499–569. doi: 10.1016/B978-0-12-396492-2.00031-X. [DOI] [Google Scholar]
- 38.Hsu A-L., Ching T-T., Wang D-S., Song X., Rangnekar V.M., Chen C-S. The cyclooxygenase-2 inhibitor celecoxib induces apoptosis by blocking Akt activation in human prostate cancer cells independently of Bcl-2. J. Biol. Chem. 2000;275(15):11397–11403. doi: 10.1074/jbc.275.15.11397. [DOI] [PubMed] [Google Scholar]
- 39.Williams C.S., Watson A.J., Sheng H., Helou R., Shao J., DuBois R.N. Celecoxib prevents tumor growth in vivo without toxicity to normal gut: Lack of correlation between in vitro and in vivo models. Cancer Res. 2000;60(21):6045–6051. [PubMed] [Google Scholar]
- 40.Kulp S.K., Yang Y-T., Hung C-C., Chen K-F., Lai J-P., Tseng P-H., Fowble J.W., Ward P.J., Chen C-S. 3-phosphoinositide-dependent protein kinase-1/Akt signaling represents a major cyclooxygenase-2-independent target for celecoxib in prostate cancer cells. Cancer Res. 2004;64(4):1444–1451. doi: 10.1158/0008-5472.CAN-03-2396. [DOI] [PubMed] [Google Scholar]
- 41.Nitulescu G.M., Draghici C., Missir A.V. Synthesis of new pyrazole derivatives and their anticancer evaluation. Eur. J. Med. Chem. 2010;45(11):4914–4919. doi: 10.1016/j.ejmech.2010.07.064. [DOI] [PubMed] [Google Scholar]
- 42.Ramadan S.K., El-Helw E.A.E., Sallam H.A. Cytotoxic and antimicrobial activities of some novel heterocycles employing 6-(1,3-diphenyl-1H-pyrazol-4-yl)-4-oxo-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile. Heterocycl. Commun. 2019;25(1):107–115. doi: 10.1515/hc-2019-0008. [DOI] [Google Scholar]
- 43.FDA. Drug approval date and data were obtained from the following sources. Available from: https://www.ema. europa.eu/
- 44.Torre L.A., Islami F., Siegel R.L., Ward E.M., Jemal A. Global cancer in women: Burden and trends. Cancer Epidemiol. Biomarkers Prev. 2017;26(4):444–457. doi: 10.1158/1055-9965.EPI-16-0858. [DOI] [PubMed] [Google Scholar]
- 45.Manning G., Whyte D.B., Martinez R., Hunter T., Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 46.Noble M.E.M., Endicott J.A., Johnson L.N. Protein kinase inhibitors: Insights into drug design from structure. Science. 2004;303(5665):1800–1805. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- 47.Cherry M., Williams D.H. Recent kinase and kinase inhibitor X-ray structures: Mechanisms of inhibition and selectivity insights. Curr. Med. Chem. 2004;11(6):663–673. doi: 10.2174/0929867043455792. [DOI] [PubMed] [Google Scholar]
- 48.Furet P., Meyer T., Strauss A., Raccuglia S., Rondeau J-M. Structure-based design and protein X-ray analysis of a protein kinase inhibitor. Bioorg. Med. Chem. Lett. 2002;12(2):221–224. doi: 10.1016/S0960-894X(01)00715-6. [DOI] [PubMed] [Google Scholar]
- 49.Ikuta M., Kamata K., Fukasawa K., Honma T., Machida T., Hirai H., Suzuki-Takahashi I., Hayama T., Nishimura S. Crystallographic approach to identification of cyclin-dependent kinase 4 (CDK4)-specific inhibitors by using CDK4 mimic CDK2 protein. J. Biol. Chem. 2001;276(29):27548–27554. doi: 10.1074/jbc.M102060200. [DOI] [PubMed] [Google Scholar]
- 50.Sawyer J.S., Beight D.W., Britt K.S., Anderson B.D., Campbell R.M., Goodson T., Jr, Herron D.K., Li H-Y., McMillen W.T., Mort N., Parsons S., Smith E.C., Wagner J.R., Yan L., Zhang F., Yingling J.M. Synthesis and activity of new aryl- and heteroaryl-substituted 5,6-dihydro-4H-pyrrolo[1,2-b]pyrazole inhibitors of the transforming growth factor-β type I receptor kinase domain. Bioorg. Med. Chem. Lett. 2004;14(13):3581–3584. doi: 10.1016/j.bmcl.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 51.Sawyer J.S., Anderson B.D., Beight D.W., Campbell R.M., Jones M.L., Herron D.K., Lampe J.W., McCowan J.R., McMillen W.T., Mort N., Parsons S., Smith E.C.R., Vieth M., Weir L.C., Yan L., Zhang F., Yingling J.M. Synthesis and activity of new aryl- and heteroaryl-substituted pyrazole inhibitors of the transforming growth factor-β type I receptor kinase domain. J. Med. Chem. 2003;46(19):3953–3956. doi: 10.1021/jm0205705. [DOI] [PubMed] [Google Scholar]
- 52.El-Gamal M.I., Zaraei S-O., Madkour M.M., Anbar H.S. Evaluation of substituted pyrazole-based kinase inhibitors in one decade (2011-2020): Current status and future prospects. Molecules. 2022;27(1):330. doi: 10.3390/molecules27010330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Persson T., Yde C.W., Rasmussen J.E., Rasmussen T.L., Guerra B., Issinger O-G., Nielsen J. Pyrazole carboxamides and carboxylic acids as protein kinase inhibitors in aberrant eukaryotic signal transduction: Induction of growth arrest in MCF-7 cancer cells. Org. Biomol. Chem. 2007;5(24):3963–3970. doi: 10.1039/b711279c. [DOI] [PubMed] [Google Scholar]
- 54.El-Deeb I.M., Lee S.H. Design and synthesis of new potent anticancer pyrazoles with high FLT3 kinase inhibitory selectivity. Bioorg. Med. Chem. 2010;18(11):3961–3973. doi: 10.1016/j.bmc.2010.04.029. [DOI] [PubMed] [Google Scholar]
- 55.Shankar D.B., Li J., Tapang P., Owen McCall J., Pease L.J., Dai Y., Wei R-Q., Albert D.H., Bouska J.J., Osterling D.J., Guo J., Marcotte P.A., Johnson E.F., Soni N., Hartandi K., Michaelides M.R., Davidsen S.K., Priceman S.J., Chang J.C., Rhodes K., Shah N., Moore T.B., Sakamoto K.M., Glaser K.B. ABT-869, a multitargeted receptor tyrosine kinase inhibitor: Inhibition of FLT3 phosphorylation and signaling in acute myeloid leukemia. Blood. 2007;109(8):3400–3408. doi: 10.1182/blood-2006-06-029579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schmidt-Arras D., Schwäble J., Böhmer F-D., Serve H. Flt3 receptor tyrosine kinase as a drug target in leukemia. Curr. Pharm. Des. 2004;10(16):1867–1883. doi: 10.2174/1381612043384394. [DOI] [PubMed] [Google Scholar]
- 57.Gazit A., Yee K., Uecker A., Böhmer F-D., Sjöblom T., Östman A., Waltenberger J., Golomb G., Banai S., Heinrich M.C., Levitzki A. Tricyclic quinoxalines as potent kinase inhibitors of PDGFR kinase, Flt3 and Kit. Bioorg. Med. Chem. 2003;11(9):2007–2018. doi: 10.1016/S0968-0896(03)00048-8. [DOI] [PubMed] [Google Scholar]
- 58.Mahboobi S., Uecker A., Cénac C., Sellmer A., Eichhorn E., Elz S., Böhmer F-D., Dove S. Inhibition of FLT3 and PDGFR tyrosine kinase activity by bis(benzo[b]furan-2-yl)methanones. Bioorg. Med. Chem. 2007;15(5):2187–2197. doi: 10.1016/j.bmc.2006.12.011. [DOI] [PubMed] [Google Scholar]
- 59.Woodburn J.R. The epidermal growth factor receptor and its inhibition in cancer therapy. Pharmacol. Ther. 1999;82(2-3):241–250. doi: 10.1016/S0163-7258(98)00045-X. [DOI] [PubMed] [Google Scholar]
- 60.Wells A. Tumor invasion: Role of growth factor-induced cell motility. Adv. Cancer Res. 2000;78:31–101. doi: 10.1016/S0065-230X(08)61023-4. [DOI] [PubMed] [Google Scholar]
- 61.Bridges A.J. The rationale and strategy used to develop a series of highly potent, irreversible, inhibitors of the epidermal growth factor receptor family of tyrosine kinases. Curr. Med. Chem. 1999;6(9):825–843. doi: 10.2174/092986730609220401151141. [DOI] [PubMed] [Google Scholar]
- 62.Boschelli D.H. Small molecule inhibitors of receptor tyrosine kinases. Drugs Future. 1999;24(5):515–537. doi: 10.1358/dof.1999.024.05.858622. [DOI] [Google Scholar]
- 63.Liu Y., Gray N.S. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2006;2(7):358–364. doi: 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- 64.Prossnitz E.R., Arterburn J.B., Smith H.O., Oprea T.I., Sklar L.A., Hathaway H.J. Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Annu. Rev. Physiol. 2008;70:165–190. doi: 10.1146/annurev.physiol.70.113006.100518. [DOI] [PubMed] [Google Scholar]
- 65.Sawai A., Chandarlapaty S., Greulich H., Gonen M., Ye Q., Arteaga C.L., Sellers W., Rosen N., Solit D.B. Inhibition of Hsp90 down-regulates mutant epidermal growth factor receptor (EGFR) expression and sensitizes EGFR mutant tumors to paclitaxel. Cancer Res. 2008;68(2):589–596. doi: 10.1158/0008-5472.CAN-07-1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wei F., Zhao B-X., Huang B., Zhang L., Sun C-H., Dong W-L., Shin D-S., Miao J-Y. Design, synthesis, and preliminary biological evaluation of novel ethyl 1-(2′-hydroxy-3′-aroxypropyl)-3-aryl-1H-pyrazole-5-carboxylate. Bioorg. Med. Chem. Lett. 2006;16(24):6342–6347. doi: 10.1016/j.bmcl.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 67.Das J., Pany S., Panchal S., Majhi A., Rahman G.M. Binding of isoxazole and pyrazole derivatives of curcumin with the activator binding domain of novel protein kinase C. Bioorg. Med. Chem. 2011;19(21):6196–6202. doi: 10.1016/j.bmc.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 68.Xie Y-S., Pan X-H., Zhao B-X., Liu J-T., Shin D-S., Zhang J-H., Zheng L-W., Zhao J., Miao J-Y. Synthesis, structure characterization and preliminary biological evaluation of novel 5-alkyl-2-ferrocenyl-6,7-dihydropyrazolo [1,5-a]pyrazin-4(5H)-one derivatives. J. Organomet. Chem. 2008;693(7):1367–1374. doi: 10.1016/j.jorganchem.2008.01.043. [DOI] [Google Scholar]
- 69.Insuasty B., Tigreros A., Orozco F., Quiroga J., Abonía R., Nogueras M., Sanchez A., Cobo J. Synthesis of novel pyrazolic analogues of chalcones and their 3-aryl-4-(3-aryl-4,5-dihydro-1H-pyrazol-5-yl)-1-phenyl-1H-pyrazole derivatives as potential antitumor agents. Bioorg. Med. Chem. 2010;18(14):4965–4974. doi: 10.1016/j.bmc.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 70.Li D-D., Lv P-C., Zhang H., Zhang H-J., Hou Y-P., Liu K., Ye Y-H., Zhu H-L. The combination of 4-anilinoquinazoline and cinnamic acid: A novel mode of binding to the epidermal growth factor receptor tyrosine kinase. Bioorg. Med. Chem. 2011;19(16):5012–5022. doi: 10.1016/j.bmc.2011.06.044. [DOI] [PubMed] [Google Scholar]
- 71.Cárdenas M., Marder M., Blank V.C., Roguin L.P. Antitumor activity of some natural flavonoids and synthetic derivatives on various human and murine cancer cell lines. Bioorg. Med. Chem. 2006;14(9):2966–2971. doi: 10.1016/j.bmc.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 72.Dallavalle S., Cincinelli R., Nannei R., Merlini L., Morini G., Penco S., Pisano C., Vesci L., Barbarino M., Zuco V., De Cesare M., Zunino F. Design, synthesis, and evaluation of biphenyl-4-yl-acrylohydroxamic acid derivatives as histone deacetylase (HDAC) inhibitors. Eur. J. Med. Chem. 2009;44(5):1900–1912. doi: 10.1016/j.ejmech.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 73.Qian Y., Zhang H-J., Zhang H., Xu C., Zhao J., Zhu H-L. Synthesis, molecular modeling, and biological evaluation of cinnamic acid metronidazole ester derivatives as novel anticancer agents. Bioorg. Med. Chem. 2010;18(14):4991–4996. doi: 10.1016/j.bmc.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 74.Zhang W-M., Xing M., Zhao T-T., Ren Y-J., Yang X-H., Yang Y-S., Lv P-C., Zhu H-L. Synthesis, molecular modeling and biological evaluation of cinnamic acid derivatives with pyrazole moieties as novel anticancer agents. RSC Advances. 2014;4(70):37197–37207. doi: 10.1039/C4RA05257A. [DOI] [Google Scholar]
- 75.Ashourpour M., Mostafavi Hosseini F., Amini M., Saeedian Moghadam E., Kazerouni F., Arman S.Y., Shahsavari Z. Pyrazole derivatives induce apoptosis via ROS generation in the triple negative breast cancer cells, MDA-MB-468. Asian Pac. J. Cancer Prev. 2021;22(7):2079–2087. doi: 10.31557/APJCP.2021.22.7.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tao Z., Shi A., Lu C., Song T., Zhang Z., Zhao J. Breast cancer: Epidemiology and etiology. Cell Biochem. Biophys. 2015;72(2):333–338. doi: 10.1007/s12013-014-0459-6. [DOI] [PubMed] [Google Scholar]
- 77.Hong Y., Cho M., Yuan Y-C., Chen S. Molecular basis for the interaction of four different classes of substrates and inhibitors with human aromatase. Biochem. Pharmacol. 2008;75(5):1161–1169. doi: 10.1016/j.bcp.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 78.Leonard G.D., Swain S.M. Ductal carcinoma in situ, complexities and challenges. J. Natl. Cancer Inst. 2004;96(12):906–920. doi: 10.1093/jnci/djh164. [DOI] [PubMed] [Google Scholar]
- 79.Gusberg S.B. Tamoxifen for breast cancer: Associated endometrial cancer. Cancer. 1990;65(7):1463–1464. doi: 10.1002/1097-0142(19900401)65:7<1463:AID-CNCR2820650702>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 80.Neven P., Vergote I. Should tamoxifen users be screened for endometrial lesions? Lancet. 1998;351(9097):155–157. doi: 10.1016/S0140-6736(05)78216-7. [DOI] [PubMed] [Google Scholar]
- 81.Wiseman L.R., Goa K.L. Toremifene. A review of its pharmacological properties and clinical efficacy in the management of advanced breast cancer. Drugs. 1997;54(1):141–160. doi: 10.2165/00003495-199754010-00014. [DOI] [PubMed] [Google Scholar]
- 82.Diasio R.B. The role of dihydropyrimidine dehydrogenase (DPD) modulation in 5-FU pharmacology. Oncology (Williston Park) 1998;12(10) Suppl. 7:23–27. [PubMed] [Google Scholar]
- 83.Naruse T., Nishida Y., Ishiguro N. Synergistic effects of meloxicam and conventional cytotoxic drugs in human MG-63 osteosarcoma cells. Biomed. Pharmacother. 2007;61(6):338–346. doi: 10.1016/j.biopha.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 84.Dang C.T., Dannenberg A.J., Subbaramaiah K., Dickler M.N., Moasser M.M., Seidman A.D., D’Andrea G.M., Theodoulou M., Panageas K.S., Norton L., Hudis C.A., Phase I.I. Phase II study of celecoxib and trastuzumab in metastatic breast cancer patients who have progressed after prior trastuzumab-based treatments. Clin. Cancer Res. 2004;10(12 Pt 1):4062–4067. doi: 10.1158/1078-0432.CCR-03-0463. [DOI] [PubMed] [Google Scholar]
- 85.Reardon D.A., Quinn J.A., Vredenburgh J., Rich J.N., Gururangan S., Badruddoja M., Herndon J.E., II, Dowell J.M., Friedman A.H., Friedman H.S. Phase II trial of irinotecan plus celecoxib in adults with recurrent malignant glioma. Cancer. 2005;103(2):329–338. doi: 10.1002/cncr.20776. [DOI] [PubMed] [Google Scholar]
- 86.Csiki I., Morrow J.D., Sandler A., Shyr Y., Oates J., Williams M.K., Dang T., Carbone D.P., Johnson D.H. Targeting cyclooxygenase-2 in recurrent non-small cell lung cancer: A phase II trial of celecoxib and docetaxel. Clin. Cancer Res. 2005;11(18):6634–6640. doi: 10.1158/1078-0432.CCR-05-0436. [DOI] [PubMed] [Google Scholar]
- 87.Farag A.M., Mayhoub A.S., Eldebss T.M.A., Amr A-G.E., Ali K.A.K., Abdel-Hafez N.A., Abdulla M.M. Synthesis and structure-activity relationship studies of pyrazole-based heterocycles as antitumor agents. Arch. Pharm. (Weinheim) 2010;343(7):384–396. doi: 10.1002/ardp.200900176. [DOI] [PubMed] [Google Scholar]
- 88.Weil C.S. Tables for convenient calculation of median-effective dose (LD50 or ED50) and instructions in their use. Biometrics. 1952;8(3):249–263. doi: 10.2307/3001557. [DOI] [Google Scholar]
- 89.Giuliano M., Hu H., Wang Y-C., Fu X., Nardone A., Herrera S., Mao S., Contreras A., Gutiérrez C., Wang T., Hilsenbeck S.G., De Angelis C., Wang N.J., Heiser L.M., Gray J.W., López-Tarruella S., Pavlick A.C., Trivedi M.V., Chamness G.C., Chang J.C., Osborne C.K., Rimawi M.F., Schiff R. Upregulation of ER signaling as an adaptive mechanism of cell survival in HER2-positive breast tumors treated with anti-HER2 therapy. Clin. Cancer Res. 2015;21(17):3995–4003. doi: 10.1158/1078-0432.CCR-14-2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dunnwald L.K., Rossing M.A., Li C.I. Hormone receptor status, tumor characteristics, and prognosis: A prospective cohort of breast cancer patients. Breast Cancer Res. 2007;9(1):R6. doi: 10.1186/bcr1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ali S., Coombes R.C. Estrogen receptor alpha in human breast cancer: Occurrence and significance. J. Mammary Gland Biol. Neoplasia. 2000;5(3):271–281. doi: 10.1023/A:1009594727358. [DOI] [PubMed] [Google Scholar]
- 92.Raju H., Chandrappa S., Prasanna D.S., Ananda H., Nagamani T.S., Byregowda S.M., Rangappa K.S. Synthesis, characterization and in-vitro antiproliferative effects of novel 5-amino pyrazole derivatives against breast cancer cell lines. Anti-Cancer Drug Discov. 2011;6(2):186–195. doi: 10.2174/157489211795328459. [DOI] [PubMed] [Google Scholar]
- 93.Ananda H., Sharath Kumar K.S., Nishana M., Hegde M., Srivastava M., Byregowda R., Choudhary B., Raghavan S.C., Rangappa K.S. Regioselective synthesis and biological studies of novel 1-aryl-3, 5-bis (het) aryl pyrazole derivatives as potential antiproliferative agents. Mol. Cell. Biochem. 2017;426(1-2):149–160. doi: 10.1007/s11010-016-2887-7. [DOI] [PubMed] [Google Scholar]
- 94.Ananda H., Sharath Kumar K.S., Sudhanva M.S., Rangappa S., Rangappa K.S. A trisubstituted pyrazole derivative reduces DMBA-induced mammary tumor growth in rats by inhibiting estrogen receptor-α expression. Mol. Cell. Biochem. 2018;449(1-2):137–144. doi: 10.1007/s11010-018-3350-8. [DOI] [PubMed] [Google Scholar]
- 95.Vajdos F.F., Hoth L.R., Geoghegan K.F., Simons S.P., LeMotte P.K., Danley D.E., Ammirati M.J., Pandit J. The 2.0 A crystal structure of the ERalpha ligand-binding domain complexed with lasofoxifene. Protein Sci. 2007;16(5):897–905. doi: 10.1110/ps.062729207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Folkman J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971;285(21):1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 97.Liekens S., De Clercq E., Neyts J. Angiogenesis: Regulators and clinical applications. Biochem. Pharmacol. 2001;61(3):253–270. doi: 10.1016/S0006-2952(00)00529-3. [DOI] [PubMed] [Google Scholar]
- 98.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438(7070):932–936. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 99.Folkman J. Angiogenesis: An organizing principle for drug discovery? Nat. Rev. Drug Discov. 2007;6(4):273–286. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 100.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995;1(1):27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 101.Wu Y., Sun W.L., Feng J.F. Antiangiogenic therapy in the management of breast cancer. Asia Pac. J. Clin. Oncol. 2013;9(2):110–116. doi: 10.1111/j.1743-7563.2012.01569.x. [DOI] [PubMed] [Google Scholar]
- 102.Kerbel R.S. Clinical trials of antiangiogenic drugs: Opportunities, problems, and assessment of initial results. J. Clin. Oncol. 2001;19(18) Suppl.:45S–51S. [PubMed] [Google Scholar]
- 103.Leahy K.M., Koki A.T., Masferrer J.L. Role of cyclooxygenases in angiogenesis. Curr. Med. Chem. 2000;7(11):1163–1170. doi: 10.2174/0929867003374336. [DOI] [PubMed] [Google Scholar]
- 104.Connolly E.M., Harmey J.H., O’Grady T., Foley D., Roche-Nagle G., Kay E., Bouchier-Hayes D.J. Cyclo-oxygenase inhibition reduces tumour growth and metastasis in an orthotopic model of breast cancer. Br. J. Cancer. 2002;87(2):231–237. doi: 10.1038/sj.bjc.6600462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Blanke C.D. Celecoxib with chemotherapy in colorectal cancer. Oncology (Williston Park) 2002;16(4) Suppl. 3:17–21. [PubMed] [Google Scholar]
- 106.Abadi A.H., Eissa A.A.H., Hassan G.S. Synthesis of novel 1,3,4-trisubstituted pyrazole derivatives and their evaluation as antitumor and antiangiogenic agents. Chem. Pharm. Bull. (Tokyo) 2003;51(7):838–844. doi: 10.1248/cpb.51.838. [DOI] [PubMed] [Google Scholar]
- 107.Boyd M.R., Paull K.D. Some practical considerations and applications of the national cancer institute in vitro anticancer drug discovery screen. Drug Dev. Res. 1995;34(2):91–109. doi: 10.1002/ddr.430340203. [DOI] [Google Scholar]
- 108.Monks A., Scudiero D., Skehan P., Shoemaker R., Paull K., Vistica D., Hose C., Langley J., Cronise P., Vaigro-Wolff A., Gray-Goodrich M., Campbell H., Mayo J., Boyd M. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991;83(11):757–766. doi: 10.1093/jnci/83.11.757. [DOI] [PubMed] [Google Scholar]
- 109.Grever M.R., Schepartz S.A., Chabner B.A. The National Cancer Institute: Cancer drug discovery and development program. Semin. Oncol. 1992;19(6):622–638. [PubMed] [Google Scholar]
- 110.Christodoulou M.S., Liekens S., Kasiotis K.M., Haroutounian S.A. Novel pyrazole derivatives: Synthesis and evaluation of anti-angiogenic activity. Bioorg. Med. Chem. 2010;18(12):4338–4350. doi: 10.1016/j.bmc.2010.04.076. [DOI] [PubMed] [Google Scholar]
- 111.Elmegeed G.A., Khalil W.K.B., Mohareb R.M., Ahmed H.H., Abd-Elhalim M.M., Elsayed G.H. Cytotoxicity and gene expression profiles of novel synthesized steroid derivatives as chemotherapeutic anti-breast cancer agents. Bioorg. Med. Chem. 2011;19(22):6860–6872. doi: 10.1016/j.bmc.2011.09.033. [DOI] [PubMed] [Google Scholar]
- 112.El-Far M., Elmegeed G.A., Eskander E.F., Rady H.M., Tantawy M.A. Novel modified steroid derivatives of androstanolone as chemotherapeutic anti-cancer agents. Eur. J. Med. Chem. 2009;44(10):3936–3946. doi: 10.1016/j.ejmech.2009.04.020. [DOI] [PubMed] [Google Scholar]
- 113.Chiang K-C., Yeh C-N., Chen H-Y., Lee J.M., Juang H-H., Chen M-F., Takano M., Kittaka A., Chen T.C. 19-Nor-2α-(3-hydroxypropyl)-1α,25-dihydroxyvitamin D3 (MART-10) is a potent cell growth regulator with enhanced chemotherapeutic potency in liver cancer cells. Steroids. 2011;76(13):1513–1519. doi: 10.1016/j.steroids.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 114.Troisi L., Florio S., Granito C. Chemoselective construction of novel steroid derivatives. Steroids. 2002;67(8):687–693. doi: 10.1016/S0039-128X(02)00032-6. [DOI] [PubMed] [Google Scholar]
- 115.Mohareb R.M., Elmegeed G.A., Abdel-Salam O.M.E., Doss S.H., William M.G. Synthesis of modified steroids as a novel class of non-ulcerogenic, anti-inflammatory and anti-nociceptive agents. Steroids. 2011;76(10-11):1190–1203. doi: 10.1016/j.steroids.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 116.Banday A.H., Mir B.P., Lone I.H., Suri K.A., Kumar H.M. Studies on novel D-ring substituted steroidal pyrazolines as potential anticancer agents. Steroids. 2010;75(12):805–809. doi: 10.1016/j.steroids.2010.02.014. [DOI] [PubMed] [Google Scholar]
- 117.Shinkawa T., Nakajima H., Nishijima K., Yamasaki F., Kato K., Ohzawa N., Mizota M. A novel quinolinone diuretic, M12285, and its activation mechanism through sulfate conjugation. Eur. J. Pharmacol. 1992;219(2):217–224. doi: 10.1016/0014-2999(92)90299-J. [DOI] [PubMed] [Google Scholar]
- 118.Maurice T., Urani A., Phan V-L., Romieu P. The interaction between neuroactive steroids and the σ1 receptor function: Behavioral consequences and therapeutic opportunities. Brain Res. Brain Res. Rev. 2001;37(1-3):116–132. doi: 10.1016/S0165-0173(01)00112-6. [DOI] [PubMed] [Google Scholar]
- 119.Vajda F.J.E. Neuroprotection and neurodegenerative disease. J. Clin. Neurosci. 2002;9(1):4–8. doi: 10.1054/jocn.2001.1027. [DOI] [PubMed] [Google Scholar]
- 120.Mohareb R.M., Al-Omran F. Reaction of pregnenolone with cyanoacetylhydrazine: Novel synthesis of hydrazide-hydrazone, pyrazole, pyridine, thiazole, thiophene derivatives and their cytotoxicity evaluations. Steroids. 2012;77(14):1551–1559. doi: 10.1016/j.steroids.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 121.Mohareb R.M., Wardakhan W.W., Elmegeed G.A., Ashour R.M.S. Heterocyclizations of pregnenolone: Novel synthesis of thiosemicarbazone, thiophene, thiazole, thieno[2,3-b]pyridine derivatives and their cytotoxicity evaluations. Steroids. 2012;77(14):1560–1569. doi: 10.1016/j.steroids.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 122.Stetler-Stevenson W.G., Aznavoorian S., Liotta L.A. Tumor cell interactions with the extracellular matrix during invasion and metastasis. Annu. Rev. Cell Biol. 1993;9:541–573. doi: 10.1146/annurev.cb.09.110193.002545. [DOI] [PubMed] [Google Scholar]
- 123.Van Aken E., De Wever O., Correia da Rocha A.S., Mareel M. Defective E-cadherin/catenin complexes in human cancer. Virchows Arch. 2001;439(6):725–751. doi: 10.1007/s004280100516. [DOI] [PubMed] [Google Scholar]
- 124.Parmar V.S., Sharma N.K., Husain M., Watterson A.C., Kumar J., Samuelson L.A., Cholli A.L., Prasad A.K., Kumar A., Malhotra S., Kumar N., Jha A., Singh A., Singh I. Himanshu; Vats, A.; Shakil, N.A.; Trikha, S.; Mukherjee, S.; Sharma, S.K.; Singh, S.K.; Kumar, A.; Jha, H.N.; Olsen, C.E.; Stove, C.P.; Bracke, M.E.; Mareel, M.M. Synthesis, characterization and in vitro anti-invasive activity screening of polyphenolic and heterocyclic compounds. Bioorg. Med. Chem. 2003;11(6):913–929. doi: 10.1016/S0968-0896(02)00539-4. [DOI] [PubMed] [Google Scholar]
- 125.Mareel M.M., De Mets M. Effect of microtubule inhibitors on invasion and on related activities of tumor cells. Int. Rev. Cytol. 1984;90:125–168. doi: 10.1016/S0074-7696(08)61489-8. [DOI] [PubMed] [Google Scholar]
- 126.Takeichi M. Morphogenetic roles of classic cadherins. Curr. Opin. Cell Biol. 1995;7(5):619–627. doi: 10.1016/0955-0674(95)80102-2. [DOI] [PubMed] [Google Scholar]
- 127.Birchmeier W., Behrens J. Cadherin expression in carcinomas: Role in the formation of cell junctions and the prevention of invasiveness. Biochim. Biophys. Acta. 1994;1198(1):11–26. doi: 10.1016/0304-419X(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 128.Rostom S.A.F., Shalaby M.A., El-Demellawy M.A. Polysubstituted pyrazoles, part 5. Synthesis of new 1-(4-chlorophenyl)-4-hydroxy-1H-pyrazole-3-carboxylic acid hydrazide analogs and some derived ring systems. A novel class of potential antitumor and anti-HCV agents. Eur. J. Med. Chem. 2003;38(11-12):959–974. doi: 10.1016/j.ejmech.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 129.Park H-J., Lee K., Park S-J., Ahn B., Lee J-C., Cho H., Lee K-I. Identification of antitumor activity of pyrazole oxime ethers. Bioorg. Med. Chem. Lett. 2005;15(13):3307–3312. doi: 10.1016/j.bmcl.2005.03.082. [DOI] [PubMed] [Google Scholar]
- 130.Rubinstein L.V., Shoemaker R.H., Paull K.D., Simon R.M., Tosini S., Skehan P., Scudiero D.A., Monks A., Boyd M.R. Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J. Natl. Cancer Inst. 1990;82(13):1113–1118. doi: 10.1093/jnci/82.13.1113. [DOI] [PubMed] [Google Scholar]
- 131.Krapcho A.P., Menta E., Oliva A., Di Domenico R., Fiocchi L., Maresch M.E., Gallagher C.E., Hacker M.P., Beggiolin G., Giuliani F.C., Pezzoni G., Spinelli S. Synthesis and antitumor evaluation of 2,5-disubstituted-indazolo[4, 3-gh]isoquinolin-6(2H)-ones (9-aza-anthrapyra-zoles). J. Med. Chem. 1998;41(27):5429–5444. doi: 10.1021/jm9804432. [DOI] [PubMed] [Google Scholar]
- 132.Bontemps-Gracz M.M., Kupiec A., Antonini I., Borowski E. The ability to overcome multidrug resistance of tumor cell lines by novel acridine cytostatics with condensed heterocyclic rings. Acta Biochim. Pol. 2002;49(1):87–92. doi: 10.18388/abp.2002_3824. [DOI] [PubMed] [Google Scholar]
- 133.Stefańska B., Bontemps-Gracz M.M., Antonini I., Martelli S., Arciemiuk M., Piwkowska A., Rogacka D., Borowski E. 2,7-Dihydro-3H-pyridazino[5,4,3-kl]acridin-3-one derivatives, novel type of cytotoxic agents active on multidrug-resistant cell lines. Synthesis and biological evaluation. Bioorg. Med. Chem. 2005;13(6):1969–1975. doi: 10.1016/j.bmc.2005.01.023. [DOI] [PubMed] [Google Scholar]
- 134.Ghirtis K., Pouli N., Marakos P., Skaltsounis A-L., Leonce S., Gaignard D.H., Atassi G. Synthesis and conformational analysis of some new Pyrano[2,3-c]xanthen-7-one and Pyrano[3,2-b]xanthen-6-one derivatives with cytotoxic activity. Heterocycles. 2000;53(1):93–106. doi: 10.3987/COM-99-8727. [DOI] [Google Scholar]
- 135.Ghirtis K., Pouli N., Marakos P., Skaltsounis A-L., Leonce S., Atassi G., Caignard D.H. Design and synthesis of some new pyranoxanthenones with cytotoxic activity. J. Heterocycl. Chem. 2001;38(1):147–152. doi: 10.1002/jhet.5570380121. [DOI] [Google Scholar]
- 136.Kostakis I.K., Pouli N., Marakos P., Mikros E., Skaltsounis A-L., Leonce S., Atassi G., Renard P. Synthesis, cytotoxic activity, NMR study and stereochemical effects of some new pyrano[3,2-b]thioxanthen-6-ones and pyrano[2,3-c]thioxanthen-7-ones. Bioorg. Med. Chem. 2001;9(11):2793–2802. doi: 10.1016/S0968-0896(01)00130-4. [DOI] [PubMed] [Google Scholar]
- 137.Svoboda G.H., Poore G.A., Simpson P.J., Boder G.B. Alkaloids of Acronychia baueri schott I. isolation of the alkaloids and a study of the antitumor and other biological properties of acronycine. J. Pharm. Sci. 1966;55(8):758–768. doi: 10.1002/jps.2600550803. [DOI] [PubMed] [Google Scholar]
- 138.Michel S., Gaslonde T., Tillequin F. Benzo[b]acronycine derivatives: A novel class of antitumor agents. Eur. J. Med. Chem. 2004;39(8):649–655. doi: 10.1016/j.ejmech.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 139.Elomri A., Mitaku S., Michel S., Skaltsounis A-L., Tillequin F., Koch M., Pierré A., Guilbaud N., Léonce S., Kraus-Berthier L., Rolland Y., Atassi G. Synthesis and cytotoxic and antitumor activity of esters in the 1,2-dihydroxy-1,2-dihydroacronycine series. J. Med. Chem. 1996;39(24):4762–4766. doi: 10.1021/jm9602975. [DOI] [PubMed] [Google Scholar]
- 140.Kostakis I., Ghirtis K., Pouli N., Marakos P., Skaltsounis A-L., Leonce S., Caignard D.H., Atassi G. Synthesis and cytotoxic activity of 2-dialkylaminoethylamino substituted xanthenone and thioxanthenone derivatives. Farmaco. 2000;55(6-7):455–460. doi: 10.1016/S0014-827X(00)00068-9. [DOI] [PubMed] [Google Scholar]
- 141.Kostakis I.K., Magiatis P., Pouli N., Marakos P., Skaltsounis A.L., Pratsinis H., Léonce S., Pierré A. Design, synthesis, and antiproliferative activity of some new pyrazole-fused amino derivatives of the pyranoxanthenone, pyranothioxanthenone, and pyranoacridone ring systems: A new class of cytotoxic agents. J. Med. Chem. 2002;45(12):2599–2609. doi: 10.1021/jm011117g. [DOI] [PubMed] [Google Scholar]
- 142.Giannouli V., Kostakis I.K., Pouli N., Marakos P., Kousidou O.Ch., Tzanakakis G.N., Karamanos N.K. Design, synthesis, and evaluation of the antiproliferative activity of a series of novel fused xanthenone aminoderivatives in human breast cancer cells. J. Med. Chem. 2007;50(7):1716–1719. doi: 10.1021/jm061410m. [DOI] [PubMed] [Google Scholar]
- 143.Bandgar B.P., Totre J.V., Gawande S.S., Khobragade C.N., Warangkar S.C., Kadam P.D. Synthesis of novel 3,5-diaryl pyrazole derivatives using combinatorial chemistry as inhibitors of tyrosinase as well as potent anticancer, anti-inflammatory agents. Bioorg. Med. Chem. 2010;18(16):6149–6155. doi: 10.1016/j.bmc.2010.06.046. [DOI] [PubMed] [Google Scholar]
- 144.Dengler W.A., Schulte J., Berger D.P., Mertelsmann R., Fiebig H.H. Development of a Propidium Iodide fluorescence assay for proliferation and cytotoxicity assays. Anticancer Drugs. 1995;6(4):522–532. doi: 10.1097/00001813-199508000-00005. [DOI] [PubMed] [Google Scholar]
- 145.Hu P., Zhao K-Q., Xu H-B. (4-Hydroxybenzylidene)-4-ferrocenylaniline. Molecules. 2001;6(12):M251. doi: 10.3390/M251. [DOI] [Google Scholar]
- 146.Togni A., Halterman R.L. Metallocenes. Weinheim, Germany: Wiley-VCH Verlag GmbH; 1998. Eds.; . [DOI] [Google Scholar]
- 147.Drent E. Ferrocene: Homogenous catalysis, organic synthesis, material. Weinheim: VCH; 1995. [Google Scholar]
- 148.Stepnicka P. Ferrocenes: Ligands, Material and Biomolecules. NJ, USA: John Wiley and Sons; 2008. Ed.; . [Google Scholar]
- 149.Bruijnincx P.C.A., Sadler P.J. New trends for metal complexes with anticancer activity. Curr. Opin. Chem. Biol. 2008;12(2):197–206. doi: 10.1016/j.cbpa.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yu H., Shao L., Fang J. Synthesis and biological activity research of novel ferrocenyl-containing thiazole imine derivatives. J. Organomet. Chem. 2007;692(5):991–996. doi: 10.1016/j.jorganchem.2006.10.059. [DOI] [Google Scholar]
- 151.Tabbì G., Cassino C., Cavigiolio G., Colangelo D., Ghiglia A., Viano I., Osella D. Water stability and cytotoxic activity relationship of a series of ferrocenium derivatives. ESR insights on the radical production during the degradation process. J. Med. Chem. 2002;45(26):5786–5796. doi: 10.1021/jm021003k. [DOI] [PubMed] [Google Scholar]
- 152.Hillard E., Vessières A., Thouin L., Jaouen G., Amatore C. Ferrocene-mediated proton-coupled electron transfer in a series of ferrocifen-type breast-cancer drug candidates. Angew. Chem. Int. Ed. 2005;45(2):285–290. doi: 10.1002/anie.200502925. [DOI] [PubMed] [Google Scholar]
- 153.Hamels D., Dansette P.M., Hillard E.A., Top S., Vessières A., Herson P., Jaouen G., Mansuy D. Ferrocenyl quinone methides as strong antiproliferative agents: Formation by metabolic and chemical oxidation of ferrocenyl phenols. Angew. Chem. Int. Ed. Engl. 2009;48(48):9124–9126. doi: 10.1002/anie.200903768. [DOI] [PubMed] [Google Scholar]
- 154.van Staveren D.R., Metzler-Nolte N. Bioorganometallic chemistry of ferrocene. Chem. Rev. 2004;104(12):5931–5985. doi: 10.1021/cr0101510. [DOI] [PubMed] [Google Scholar]
- 155.Köpf-Maier P., Köpf H., Neuse E.W. Ferrocenium salts—the first antineoplastic iron compounds. Angew. Chem. Int. Ed. Engl. 1984;23(6):456–457. doi: 10.1002/anie.198404561. [DOI] [Google Scholar]
- 156.Sun M-L., Ruan B-F., Zhang Q., Liu Z-D., Li S-L., Wu J-Y., Jin B-K., Yang J-X., Zhang S-Y., Tian Y-P. Synthesis, crystal structures, electrochemical studies and anti-tumor activities of three polynuclear organotin(IV) carboxylates containing ferrocenyl moiety. J. Organomet. Chem. 2011;696(20):3180–3185. doi: 10.1016/j.jorganchem.2011.06.045. [DOI] [Google Scholar]
- 157.Huang X-F., Tang J-F., Ji J-L., Wang X-L., Ruan B-F. Synthesis, characterization and antitumor activity of novel amide derivatives containing ferrocenyl pyrazol-moiety. J. Organomet. Chem. 2012;706-707:113–123. doi: 10.1016/j.jorganchem.2012.02.001. [DOI] [Google Scholar]
- 158.Huang X-F., Wang L-Z., Tang L., Lu Y-X., Wang F., Song G-Q., Ruan B-F. Synthesis, characterization and antitumor activity of novel ferrocene derivatives containing pyrazolyl-moiety. J. Organomet. Chem. 2014;749:157–162. doi: 10.1016/j.jorganchem.2013.08.043. [DOI] [Google Scholar]
- 159.Viegas-Junior C., Danuello A., da Silva Bolzani V., Barreiro E.J., Fraga C.A.M. Molecular hybridization: A useful tool in the design of new drug prototypes. Curr. Med. Chem. 2007;14(17):1829–1852. doi: 10.2174/092986707781058805. [DOI] [PubMed] [Google Scholar]
- 160.Gediya L.K., Njar V.C. Promise and challenges in drug discovery and development of hybrid anticancer drugs. Expert Opin. Drug Discov. 2009;4(11):1099–1111. doi: 10.1517/17460440903341705. [DOI] [PubMed] [Google Scholar]
- 161.Shah K., Chhabra S., Shirvastava S.K., Mishra P. Benzimidazole: A promising pharmacophore. Med. Chem. Res. 2013;22:5077–5104. doi: 10.1007/s00044-013-0476-9. [DOI] [Google Scholar]
- 162.Sarhan A.A.O., Al-Dhfyan A., Al-Mozaini M.A., Adra C.N., Aboul-Fadl T. Cell cycle disruption and apoptotic activity of 3-aminothiazolo[3,2-a]benzimidazole-2-carboni-trile and its homologues. Eur. J. Med. Chem. 2010;45(6):2689–2694. doi: 10.1016/j.ejmech.2010.02.025. [DOI] [PubMed] [Google Scholar]
- 163.El Rashedy A.A., Aboul-Enein H.Y. Benzimidazole derivatives as potential anticancer agents. Mini Rev. Med. Chem. 2013;13(3):399–407. doi: 10.2174/138955713804999847. [DOI] [PubMed] [Google Scholar]
- 164.Husain A., Rashid M., Shaharyar M., Siddiqui A.A., Mishra R. Benzimidazole clubbed with triazolo-thiadiazoles and triazolo-thiadiazines: New anticancer agents. Eur. J. Med. Chem. 2013;62:785–798. doi: 10.1016/j.ejmech.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 165.Paul K., Bindal S., Luxami V. Synthesis of new conjugated coumarin-benzimidazole hybrids and their anticancer activity. Bioorg. Med. Chem. Lett. 2013;23(12):3667–3672. doi: 10.1016/j.bmcl.2012.12.071. [DOI] [PubMed] [Google Scholar]
- 166.Reddy T.S., Kulhari H., Reddy V.G., Bansal V., Kamal A., Shukla R. Design, synthesis and biological evaluation of 1,3-diphenyl-1H-pyrazole derivatives containing benzimidazole skeleton as potential anticancer and apoptosis inducing agents. Eur. J. Med. Chem. 2015;101:790–805. doi: 10.1016/j.ejmech.2015.07.031. [DOI] [PubMed] [Google Scholar]
- 167.Cotter T.G. Apoptosis and cancer: The genesis of a research field. Nat. Rev. Cancer. 2009;9(7):501–507. doi: 10.1038/nrc2663. [DOI] [PubMed] [Google Scholar]
- 168.Aggarwal R., Kumar R., Kumar S., Garg G., Mahajan R., Sharma J. Synthesis and antibacterial activity of some 5-hydroxy-5-trifluoromethyl-4,5-dihydropyrazol-1-thiocar-boxamides, 3-trifluoromethylpyrazol-1-thiocarboxamides and 4-aryl-2-(5(3)-trifluoromethyl-1-pyrazolyl)thiazoles. J. Fluor. Chem. 2011;132(11):965–972. doi: 10.1016/j.jfluchem.2011.07.029. [DOI] [Google Scholar]
- 169.Usachev B.I., Obydennov D.L., Röschenthaler G-V., Sosnovskikh V.Y. 2-Cyano-6-(trifluoromethyl)-4H-pyran-4-one: A novel versatile CF3-containing building block. J. Fluor. Chem. 2012;137:22–26. doi: 10.1016/j.jfluchem.2012.01.006. [DOI] [Google Scholar]
- 170.Aggarwal R., Bansal A., Mittal A. Synthesis and antimicrobial activity of 3-(2-thienyl)-4-arylazo-5-hydroxy-5-trifluoromethyl-Δ2-isoxazolines and 3-(2-thienyl)-4-aryla-zo-5-trifluoromethylisoxazoles. J. Fluor. Chem. 2013;145:95–101. doi: 10.1016/j.jfluchem.2012.10.005. [DOI] [Google Scholar]
- 171.Fayed E.A., Eissa S.I., Bayoumi A.H., Gohar N.A., Mehany A.B.M., Ammar Y.A. Design, synthesis, cytotoxicity and molecular modeling studies of some novel fluorinated pyrazole-based heterocycles as anticancer and apoptosis-inducing agents. Mol. Divers. 2019;23(1):165–181. doi: 10.1007/s11030-018-9865-9. [DOI] [PubMed] [Google Scholar]
- 172.Ashburn T.T., Thor K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004;3(8):673–683. doi: 10.1038/nrd1468. [DOI] [PubMed] [Google Scholar]
- 173.Lu W., Li P., Shan Y., Su P., Wang J., Shi Y., Zhang J. Discovery of biphenyl-based VEGFR-2 inhibitors. Part 3: Design, synthesis and 3D-QSAR studies. Bioorg. Med. Chem. 2015;23(5):1044–1054. doi: 10.1016/j.bmc.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 174.Kamal A., Faazil S., Ramaiah M.J., Ashraf M., Balakrishna M., Pushpavalli S.N.C.V.L., Patel N., Pal-Bhadra M. Synthesis and study of benzothiazole conjugates in the control of cell proliferation by modulating Ras/MEK/ERK-dependent pathway in MCF-7 cells. Bioorg. Med. Chem. Lett. 2013;23(20):5733–5739. doi: 10.1016/j.bmcl.2013.07.068. [DOI] [PubMed] [Google Scholar]
- 175.Wang M., Xu S., Lei H., Wang C., Xiao Z., Jia S., Zhi J., Zheng P., Zhu W. Design, synthesis and antitumor activity of Novel Sorafenib derivatives bearing pyrazole scaffold. Bioorg. Med. Chem. 2017;25(20):5754–5763. doi: 10.1016/j.bmc.2017.09.003. [DOI] [PubMed] [Google Scholar]