

Summary
The characterization of somatic genomic variation associated with the biology of tumors is fundamental for cancer research and personalized medicine, as it guides the reliability and impact of cancer studies and genomic-based decisions in clinical oncology. However, the quality and scope of tumor genome analysis across cancer research centers and hospitals are currently highly heterogeneous, limiting the consistency of tumor diagnoses across hospitals and the possibilities of data sharing and data integration across studies. With the aim of providing users with actionable and personalized recommendations for the overall enhancement and harmonization of somatic variant identification across research and clinical environments, we have developed ONCOLINER. Using specifically designed mosaic and tumorized genomes for the analysis of recall and precision across somatic SNVs, insertions or deletions (indels), and structural variants (SVs), we demonstrate that ONCOLINER is capable of improving and harmonizing genome analysis across three state-of-the-art variant discovery pipelines in genomic oncology.
Keywords: somatic variant calling, benchmarking, cancer genomics, bioinformatics, benchmarking data, oncology
Graphical abstract
Highlights
-
•
Heterogeneity in cancer genome analysis affects diagnosis and sharing of data
-
•
ONCOLINER is a solution to align somatic variant analysis across centers
-
•
ONCOLINER provides recommendations to improve somatic variant calling
-
•
Here, we also provide adapted benchmark datasets to assess somatic variant calling
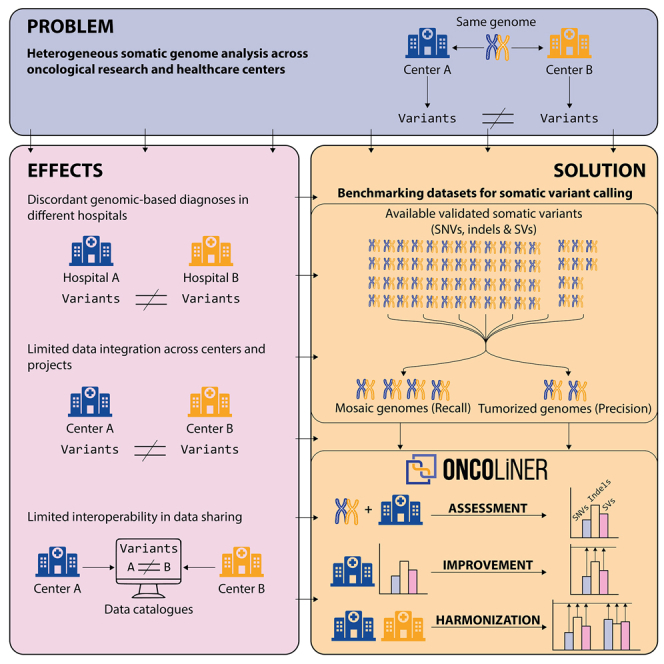
Current methods to identify somatic variants in oncological research and healthcare are highly heterogeneous across centers. Martín et al. present a new paradigm for assessing, improving, and harmonizing somatic variant calling across genomic oncology centers. This will allow the sharing of oncology data and provide consistency in diagnosis across hospitals.
Introduction
Understanding how somatic genomic variation drives the biology of tumors is the foundation of modern personalized oncology. The characterization of somatic changes in cancer genomes has already uncovered hundreds of tumor-associated genes that can potentially be used as diagnosis, prognosis, and treatment markers.1,2,3,4 For this reason, the analysis of tumor genomes has become a critical step within cancer genomic research and for its downstream clinical translation into personalized medicine protocols.
This has motivated the development of multiple somatic variant calling solutions over the past years, providing a wide catalog of different available tools and methods, each of them typically focused on specific types and sizes of variants.5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22 Furthermore, the combination of different variant callers into complex pipelines has proven to be the best solution for different types of analyses in both research and clinical settings.23,24,25,26 Selecting the best-performing tools and deciding how to best combine them to maximize recall and precision of all types of variant discovery, as well as their precise characteristics (e.g., tumor allele frequency, ploidy, exact break-junction sequence, etc.), are critical and challenging steps when developing genome analysis pipelines, as they require know-how as well as high-quality benchmarking information.27,28,29,30 In addition, pipelines can be designed to prioritize recall or precision depending on the scenario of application, such as research or healthcare.
Despite the publication of many benchmarking efforts, their results are usually descriptive and difficult to translate into practical decisions for the design and development of variant calling pipelines. This is due, for example, to the tendency to generate and use theoretical scenarios of application, combined with full in silico benchmarking datasets, which do not properly capture the nature of the biological behavior, noise, and variation of the data. That, together with the limited availability of real benchmarking data for somatic variant detection, defines the current scenario where variant calling across most of the different research and clinical centers is often limited and highly heterogeneous. This is particularly severe when considering the analysis of whole-genome sequencing (WGS) and the identification of structural variants (SVs), which are critical for cancer genomic studies. In this context, an increasing number of initiatives worldwide are aiming at introducing the sequencing and analysis of whole genomes as a routine for healthcare in genomic oncology, facing the challenge of genome analysis pipeline design and implementation.31,32,33,34,35,36,37 It is therefore necessary to have actionable solutions in place, adjusted to the specific needs and scenarios that require variant calling pipeline design, development, or improvement.
Current limitations in somatic variant calling have a direct impact on the quality and scope of downstream variant interpretation in research studies and clinical applications that impede the possibility of sharing, combining, and integrating data and results across different research groups and centers. In fact, growing national and worldwide efforts toward designing and building multicentric research ecosystems,38,39 operating under federated or centralized schemes, require output harmonization of their different analysis platforms at both the level of quality and the scope of the variant calling, as well as at the level of standards that allow interoperability. Despite the high number of global initiatives to standardize and harmonize the management of biomedical data such as GA4GH40 and ELIXIR (https://elixir-europe.org/), limited efforts have been devoted to the harmonization and standardization of analysis pipelines. The existing heterogeneity across different cancer research and clinical centers currently frustrates any attempt to integrate data and results, restricting the possibilities of new scientific discoveries, as well as generating potential discordant tumor reports from different healthcare centers. This heterogeneity also limits the chances for interoperability at other levels, for example through variant-based data discovery engines (e.g., Beacon from GA4GH), which ideally also require homogeneous and consistent variant data across centers. Because the adoption and maintenance of identical reference pipelines across many research centers is not a realistic solution for harmonization within growing federated (decentralized) data-sharing scenarios, we need applicable and practical solutions for the improvement and harmonization of somatic genome analysis across these data environments. This will allow us to answer more ambitious biomedical research questions and will enhance and globalize genomic oncology.
With this objective, we have designed a new actionable benchmarking paradigm, implemented as the ONCOLINER platform, a modular, configurable, and easy-to-use software solution that provides users with direct and personalized recommendations for building, improving, and harmonizing somatic variant calling pipelines from whole-genome short-read sequencing data within and across research and clinical oncology centers. The recommendations for improvement and harmonization are based on quality standards measured through common accuracy metrics using a comprehensive set of validated somatic variants. We have also processed and integrated these experimentally validated somatic variant data into more accessible benchmarking datasets that, in contrast to traditional in silico simulations, capture real data noise and variability impacting the calling of SNVs, insertions or deletions (indels), and SVs. We here demonstrate that the combination of these developments and resources significantly improved and harmonized real somatic variant calling pipelines that represent current research and clinical scenarios in genomic oncology.
Results
Large data integration efforts in cancer research1,4,21,41,42 have already highlighted significant heterogeneity in performance, quality, and scope of short-read-based genome analysis across different centers. With the ultimate aim of providing actionable and personalized solutions for the improvement and harmonization of genome analysis across cancer research centers and hospitals, we have first assessed the extent to which existing somatic variant calling pipelines need improvement and harmonization. For this, we have measured and compared the analysis performance (recall and precision) of three selected state-of-the-art variant calling pipelines from three active genome analysis research centers (A, B, and C). As recall and precision values reflect different properties of the variant calling, their measurement also requires specific and dedicated benchmarking datasets and scenarios.
To measure the recall (sensitivity) performance of these pipelines, we used a comprehensive collection of reported and validated somatic variants encapsulated within mosaic tumor-normal samples. These samples have been designed to retain the recall assessment properties of the original samples while reducing the analysis burden considerably (Figure 1A). Tumor and normal mosaic samples preserve the intrinsic noise and properties from the sample preparation (e.g., purity) and sequencing (e.g., insert sizes). To produce these mosaics, all the original sample reads mapped around a 2 kb window surrounding the coordinates of each validated variant are inserted into a WGS genome simulated from the GRCh37 reference while removing the artificial reads that overlap the span of the window. The same process is equally applied to the tumor and normal samples. For this study, we have built four mosaic genomes selecting a total of 32,267 validated variants (with non-overlapping genomic positions), distributed across 47 selected tumor-normal samples from the Pilot-504 and 12 from the HMF-129 datasets. These variants include 15,000 SNVs, 13,896 indels (composed of insertions, duplications, and deletions with a length less than 100 bp), and 3,371 SVs (i.e., variants with length greater than 100 bp). SVs are further subclassified in sizes and classes defined through their breakpoints, resulting in 736 translocations, 667 inversions, 1,318 deletions, and 650 duplications. While all the variants of the 12 HMF-12 samples could be integrated into a single mosaic tumor-normal pair with 1,754 SVs, the heterogeneity of insert sizes across the 47 PCAWG Pilot-50 tumor-normal samples forced us to group them in three mosaic samples with consistent insert sizes and an average of 10,171 variants each, including SNVs, indels, and SVs (see Figures S1–S4). The variant allele frequency (VAF) of the mosaic genome variants ranges from 5% to 98%, following the original distribution of the PCAWG samples (see Figures S5 and S6). Within the scope of these variants and using a wide and representative collection of available somatic variant callers, we demonstrate that the mosaic genomes retain practically identical recall assessment properties to the original benchmarking datasets (Figure 1B).
Figure 1.

Generation of the mosaic tumor/normal genome pairs
(A) To assess the levels of recall for somatic analysis pipelines, we have designed mosaic tumor-normal genome pairs, which condense the benchmarking information of a total of 59 (47 + 12) validated tumor-normal pairs into only four resulting mosaic pairs. This is achieved by literally transferring (copying) a 2 kb region of mapped reads around each validated variant of the original genomes into a canvas genome generated by simulated reads from the human reference genome (GRCh37). Whereas HMF-12 samples have homogeneous insert sizes and all the variants could be condensed into a single tumor-normal mosaic genome pair, the heterogeneity across samples of the PCAWG-Pilot dataset forced us to generate three sample pairs with similar ranges of insert sizes (see Figure S1). This avoids conflicts from some variant callers that depend on and need to generate internal decision criteria based on read pair distances. Mosaic genomes reproduce the exact same genomic context for variant detection as in the original samples. In fact, in (B), we show that these mosaic genomes retain the recall benchmarking properties of the original source samples. The results that we obtained using 12 different variant callers on the recall for SNVs, indels, and SVs are practically identical between the 59 original samples (yellow line) and the derived mosaic genomes (blue line). During the quality check of the data, we discarded two outlier samples from the 49 available tumor-normal pairs from the PCAWG-Pilot dataset. Defined by the original data, this benchmarking set considers effective sequencing coverages ranging from 27× to 62× for the normal samples and from 27× to 149× for the tumor samples (see Figure S2), as well as variants with a defined variant allele frequency (VAF) larger than 1% (see Figure S6).
On the other hand, to measure the reliability of the calling, we have calculated the precision performance of pipelines A, B, and C using tumorized genomes (Figure 2). In contrast to real or fully simulated benchmarking genomes, tumorized samples are hybrids that can accurately capture precision values and false positive rates. To balance the control of the variants and, at the same time, provide a real testing scenario to evaluate false positive calls, we combined samples from the GIAB project (NA12878 and HG002)43 with a collection of synthetically reproduced true somatic variants (11,987 SNVs, 1,397 indels, and 239 SVs) from the PCAWG consensus callsets.4 Tumorized samples contain only 0.2% of reads that are modified to accurately represent the sequence of the variant, while the other 99.8% of reads remain unaltered. Importantly, in contrast to mosaics, which may retain germline variants within somatic variant windows and require controlled access agreements, tumorized samples can be shared openly within the community as described by the GIAB project.43
Figure 2.

Design and construction of the tumorized tumor-normal genome pairs
For an accurate assessment of the precision and to calculate the rate of false positives during somatic variant discovery, we have generated tumorized genomes. These consist of real WGS samples from the GIAB project43 with synthetic cancer somatic variants extracted from the PCAWG consensus callsets.4 For each introduced variant, a subset of reads in the tumor sample are modified to represent the variant. The number of modified reads depends on the depth of the region where the variant is located and the selected VAF (see Figure S6). This method is implemented in GenomeVariator, a wrapper tool that enhances the functionalities of BAMSurgeon.25,26,44 The high coverage of these samples (300×) allows the generation of tumor and normal genomes with a different composition of reads, recreating real tumor-normal analysis scenarios. Furthermore, the fact that only 0.2% of the reads have been modified to reconstruct the variants in the tumor samples of the tumorized genome pairs makes these samples ideal for an accurate evaluation of precision, as they retain 99.8% of the original sequencing and mapping properties. In order to avoid potential sample bias, we have generated two tumor-normal samples with the same validated variants: one derived from the NA12878 GIAB sample and the other from the HG002 GIAB sample.
Analysis of heterogeneity
To precisely assess the extent to which pipelines A, B, and C need improvement and harmonization, we tested them against the four mosaics and the two tumorized benchmarking samples described above and extracted recall and precision values for each variant type and size. An initial inspection of the results already shows some degree of heterogeneity and possibilities for improvement for some pipelines across different variant types and sizes (see Figure S7). As a signal of how different two or more pipelines perform against the same benchmark samples, we have defined a performance heterogeneity score (PHS), reflecting the differences between their performances, as of recall and precision (Figure 3A). Moreover, we also studied the functional and clinical impact that these pipelines are able to capture and how heterogeneous this is across centers by evaluating the fraction of coding genes, including cancer drivers,2 that are affected by non-synonymous variants (i.e., gene discordance ratio [GDR]).
Figure 3.

Comparative study of somatic variant analysis across different research and clinical scenarios
(A) A graphic and mathematical representation of the underlying rationale for the definition of PHS. This score measures the degree of heterogeneity in somatic variant calling performance across two or more centers, independently of the overall quality of each pipeline. It is calculated as the normalized average distance from each pipeline to the centroid in a Euclidean space delimited by recall and precision values. PHS values of 0% indicate no heterogeneity, whereas those of 100% indicate maximum heterogeneity.
(B) The results for the top four variant types and sizes by PHS for centers A, B, and C, representative of real and active genome analysis pipelines. SVs are grouped by type, such as deletions (DEL), duplications (DUP), translocations (TRA) and inversions (INV) and by size (from 101 to 500 bp and more than 500 bp). Performance parameters, including recall and precision, were obtained using a validated set of variants with a total of 38,947 SNVs, 16,688 indels, and 3,851 SVs of different subtypes and sizes. The samples only contained inversions above 500 bp, so smaller ones could not be assessed (see Figures 1 and 2).
(C) The PHSs for all variant types.
(D) The effect of this calling heterogeneity on the functional reach and impact of the analysis in the form of the fraction of discordant genes identified as mutated by at least one center and missed by another (GDR).
Overall, the three pipelines together could correctly identify 28,644 variants (13,834 SNVs, 11,704 indels, and 3,106 SVs) of the 32,267 original truth variants from all four mosaic samples. From the correctly identified variants, only 19,805 (10,052 SNVs, 7,273 indels, and 2,480 SVs) were concordantly identified by the three centers, whereas 8,839 (3,782 SNVs, 4,431 indels, and 626 SVs) were missed by at least one of the pipelines. Pairwise comparisons between pipelines A-B, A-C, and B-C showed 7,612, 3,616, and 6,450 discordant variants, respectively. Translating these differences into potential functional and clinical impact, from a total of 3,217 genes with somatic coding mutations, 2,624 were found by all three pipelines. Conversely, 593 (18%) of them, including 24 coding cancer drivers, were missed or excluded by at least one of the pipelines. Among these, we found genes that are key in decision-making processes within tumor boards for the diagnosis, treatment, and prognosis of different cancer types (ONCOKB45,46). For example, the gene KIT, which codes for a receptor tyrosine kinase, is a proto-oncogene and a US Food and Drug Administration-approved therapy target of kinase inhibitor drug groups.47,48,49,50 Similarly, among the discordant variants, we found a translocation disrupting the RARA gene, a retinoic acid receptor that is found translocated as a gene fusion in certain types of leukemia. In particular, some RARA mutations are standard diagnostic biomarkers for acute promyelocytic leukemia.50,51 Another important discordance was a translocation affecting CCND3. Alterations in CCND3 are used as prognostic biomarkers of various hematologic malignancies.50,52 Lastly, other genes, used as support for the diagnosis of different cancer types, such as FOXP1 and NOTCH2, also presented mutations that were discordantly detected between the three pipelines.50
Overall, PHSs among the three pipelines range from 1.92% to 36.53%, showing heterogeneity across all variant types and sizes (Figure 3B). We also observe differences in PHSs affected by precision and recall heterogeneity values asymmetrically. For SNVs, in agreement with previous studies,53 we find a significant performance heterogeneity (PHS = 11.71%), mostly affecting recall values (Figure 3B), which translates into 3,782 discordant variants from all 13,834 SNVs identified by at least one center, as well as 58 discordant coding and 4 cancer driver genes. We also detected a degree of heterogeneity (PHS = 4.14%) for indels, resulting from the discordance of 4,431 variants that have been missed by at least one center, affecting 39 coding and 4 cancer driver genes. Finally, the calling of SVs also shows low concordance across centers, with 16 discordant cancer driver genes disrupted by SVs in total (PHS = 6.63%). Particularly, the 100–500 bp range shows the two highest PHS values, with 36.53% for duplications and 29.22% for deletions, translating into 95 duplications and 68 deletions of the total 626 SVs differently identified by these pipelines, respectively (Figure 3C). Conversely, deletions and duplications with lengths above 500 bp present the two lowest PHSs (1.92% and 3.7%, respectively). This demonstrates that the detection of SVs within the 100–500 bp range remains challenging with short-read technologies. Altogether, duplications and deletions in the 100–500 bp range cause 47 coding and 1 cancer driver and 34 coding and 4 cancer discordant genes, respectively (Figure 3D). Interestingly, discordant false positive deletions produce significant differences in precision across pipelines, whereas duplications of the same size differ mostly in recall (see Figure 3B).
ONCOLINER solution
To overcome these quantitative and qualitative differences across centers, and driven by the specific needs of each of the pipelines, we designed and implemented a solution called ONCOLINER to assist in the development and improvement of somatic genome analysis pipelines for cancer genomics research and clinical oncology. ONCOLINER provides users with multiple functionalities encapsulated in different interoperable modules, covering the assessment, improvement, and harmonization of already operational variant calling pipelines, to the de novo generation of optimized pipelines (Figure 4). In brief, this tool first analyzes targeted pipelines, and then makes a diagnosis based on calling performances and harmonization levels, to finally provide improved and harmonized solutions in the form of specific and actionable recommendations, such as adding or intersecting with additional variant callers.
Figure 4.

Conceptual and functional map of ONCOLINER
Left of the image, a box represents the different functional modules and basic elements of ONCOLINER. In our use case, the defined benchmarking genomes are composed of three mosaic and two tumorized tumor-normal genome pairs that have been designed to capture recall and precision performance, respectively (see Figures 1 and 2). The three modules in orange correspond to the three main functionalities, assessment, improvement, and harmonization, which are described on the right part of the image. In addition, shown in the blue box on the bottom, ONCOLINER also provides different standalone solutions for the improvement and harmonization of genome analysis in additional contexts. In the middle of the image, a user interface (GUI) allows interaction with the platform and the selection of the best solution for each specific clinical or research need.
The first step of ONCOLINER requires users to execute their analysis pipelines against the benchmarking data, corresponding in this case to the four mosaics and the two tumorized samples. After entering the pipeline’s results (in VCF format), the assessment module first calculates basic performance metrics, such as recall and precision, across a wide range of variant types and sizes (see Figure S7), highlighting parts that can be improved. Next, targeting these limitations, the improvement module provides specific recommendations that increase detection power or precision for each of the variant types and sizes. These recommendations are based on the current knowledge in state-of-the-art somatic variant detection, obtained through thorough testing and benchmarking of 12 selected variant callers and their combinations over the mosaic and tumorized benchmarking genomes. Recommendations targeting the improvement of recall values for certain pipelines and variant types involve adding one or more of these variant callers, whereas recommendations to lower the rate of false positives and improve precision involve recommending intersections. As different research and clinical scenarios prioritize either recall or precision differently, ONCOLINER generates different possibilities for improvement, from which users can select the most suitable option for their needs. Finally, if more than one pipeline has been provided, then the harmonization module enables selecting the recommendations for improvement that also maximize the harmonization across centers, quantitatively and qualitatively.
Along with ONCOLINER, we also provide additional standalone software solutions to meet multiple needs associated with improving, standardizing, and harmonizing genome analysis across centers. For example, to ensure interoperable interpretation and representation of variants from VCF files, we developed VariantExtractor as a library used by ONCOLINER and also as a standalone package. VariantExtractor reads and interprets SNV, indel, and SV records by applying a set of consistent rules across all VCFs. This also facilitates downstream analysis with no information losses, especially for SVs, as there are different ways to encode the same variant that are biologically identical but very different in the VCF format (see Figure S8). In addition, we also provide GenomeVariator and GenomeMosaicMaker for the generation of custom tumorized and mosaic genomes, respectively. These tools can be used to generate genomes adapted to specific needs for benchmarking data.
Beyond the functionalities aiming at improving and harmonizing existing pipelines, we also include PipelineDesigner, a standalone tool that helps users to find the best strategy to combine and merge specific variant callers to maximize recall and precision over all variant types. Using PipelineDesigner, we designed and implemented a de novo variant calling pipeline with the combination of the best-performing variant callers of this study (see Table S1) that can be readily adopted. To facilitate the application of the recommendations from both ONCOLINER and PipelineDesigner, we provide each variant caller we used in a container and the necessary tools for merging and combining their results to provide a consistent framework for applying improvement and harmonization recommendations. In addition, to increase flexibility and ensure future applicability over a wider range of variant calling benchmarking scenarios, we allowed ONCOLINER to function with other provided benchmarking datasets and other reference variant callers, adapting to the specific needs of the user.
Application to a real research scenario
In order to prove ONCOLINER’s functional applicability to a common research scenario, we have used pipelines A, B, and C. First, the assessment module calculated and provided all variant calling metrics for each pipeline and each variant type, including affected coding and cancer driver genes, from the analysis of the tumorized and mosaic genomes (see Figures S9–S11). Next, the improvement module provided recommendations that enhance recall (at least 5%), precision (at least 5%), or F1-score. For example, ONCOLINER’s recommendation of adding GRIDSS2 to pipelines A and C improved their recall for detection of SVs from 86% to 91% and from 78% to 89%, respectively. This recommendation also increased the number of discovered mutated protein-coding genes for both pipelines by 277 and 274 and cancer driver genes by 12 and 8 for A and C, respectively. The recommended intersection of pipeline A with mutect2 (from GATK) and SAGE removed 1,741 false positives for SNVs and increased the precision from 93% to 99.99% (see Figures S12 and S13).
Finally, the harmonization module evaluated and selected those recommendations for improvement that also minimized heterogeneity scores across centers. For example, among multiple choices with similar outcomes (see Figures S14 and S15), the addition of GRIDSS2 to pipelines A and C not only improved their recall as described above but also reduced PHSs from 6.63% to 2.57% for SVs, with notable effects in duplications between 101 and 500 bp decreasing PHSs from 36.53% to 4.03%. Moreover, this harmonization option decreased the GDR for SVs from 17.50% to 2.09% (notably from 87.04% to 3.64% for 100–500 bp duplications), which translated into 436 less discordant affected genes across pipelines. Overall, prioritizing PHSs, the recommended strategies for harmonization and improvement made consistent a total of 5,987 true variants (2,234 SNVs, 3,219 indels, and 534 SVs) out of the initial 8,839 discordant variants across the three centers and also recovered variants that affected 499 protein-coding and 21 cancer driver genes, including the five actionable genes previously missed by at least one of the three pipelines (Table 1). Despite considerably improving performance and homogeneity across centers, a total of 2,852 true variants remained discordant after a first iteration with ONCOLINER, including 94 affected genes and three drivers. The improvement and harmonization reached here apply to these specific pipelines. We expect even higher improvement and harmonization levels across research centers and hospitals that, for example, require the de novo inclusion of WGS and SVs into their protocols.
Table 1.
Pipeline heterogeneity and performance after ONCOLINER
| SNV | Indel | SV | |
|---|---|---|---|
| Discordant variants | 1,548 (2,234 ↓) | 1,212 (3,219 ↓) | 92 (534 ↓) |
| Discordant genes | 22 (36 ↓) | 12 (27 ↓) | 60 (436 ↓) |
| Discordant driver genes | 2 (2 ↓) | 1 (3 ↓) | 0 (16 ↓) |
| GDR, % | 9 (15 ↓) | 8 (19 ↓) | 2 (15 ↓) |
| PHS, % | 4 (7 ↓) | 4 (0 = ) | 3 (4 ↓) |
| Average F1-score, % | 92 (3 ↑) | 87 (7 ↑) | 90 (2 ↑) |
Discussion
Naturally, the ultimate value of benchmarking efforts during the development and improvement of genome analysis pipelines critically depends on the quality and scope of the reference (truth) set of validated variants. They will determine the reach of the assessment and its final level of trust. Unfortunately, there are only a handful of available and suitable patient-derived datasets with enough numbers and varieties of validated somatic variants in WGS for building and calibrating somatic variant identification and classification pipelines. Of these, we have used the PCAWG-Pilot4 (SNVs, indels, and SVs) and the HMF-129 (SVs) datasets, both generated in benchmarking contexts of somatic variant calling. While the HMF dataset is more homogeneous and internally consistent, the PCAWG set derives from samples collected, processed, and sequenced in multiple centers with different quality standards, such as tumor purity, insert size, and sequencing coverage. This affects not only the mosaic strategy, which requires homogeneous insert sizes, but also the scope of this study. For this reason, we cannot assure that pipelines calibrated with these specific datasets will actually translate in calling improvement when applied outside their sample purity, sequencing error rate, coverage, and insert size ranges (see Figure 1). Considering these limitations, we plan to improve further benchmarking datasets by generating publicly accessible tumor-normal benchmarking genomes for the evaluation of recall and precision of somatic variant calling in a single run.
At the same time, the identification and validation methods used to generate truth sets of variants also determine the value and reach of benchmarking studies. For instance, at the level of variant type, our benchmarking datasets cover SNVs, indels, and breakpoint-definable SVs (deletions, inversions, translocations, and duplications) but do not include, for example, large insertions and coverage-derived copy-number variants. In addition, other data quality issues can also affect the reliability of pipeline assessment. Among these, even validated somatic variation datasets still contain a certain amount of germline contamination and sequencing errors recalled as low-VAF SNVs. This could slightly generate underestimations of recall values, even in this study. Upcoming efforts aiming to generate more accurate, customized, and targeted benchmarking datasets for other potential applications will surely improve the usability and quality of the functionalities of ONCOLINER.
The reach of ONCOLINER is determined by the collection of preselected variant callers used to generate recommendations for improvement and harmonization. Although we have selected twelve variant callers based on their acceptance and use within the community, we cannot discard that other variant callers could, in fact, outperform this set and generate better recommendations for improvement and harmonization. For this reason, together with customized benchmarking datasets, we have also allowed the inclusion by the user of additional variant callers into the platform to be able to improve the calling in general or to target specific types of variants not included here. Among other benchmarking datasets that can be used with ONCOLINER, we can find high-quality tumor-normal pairs in previous studies.23,30 Nevertheless, even considering the limitations of the datasets used as the ground truth, ONCOLINER managed to substantially improve and harmonize the performance of the pipelines of the three centers, leading to the recovery of mutations on five discordant clinically actionable cancer genes. Thus, further benchmarking efforts applying this paradigm shift with curated gold-standard variants will be able to generate actionable recommendations for health centers.
Taking these results together, we present and validate a new concept for the benchmarking of somatic variant discovery with actionable recommendations to users for improving and harmonizing across centers the identification of somatic variants associated with cancer. The application of ONCOLINER to align genome analysis across research centers and hospitals can provide consistency in the diagnosis and selection of treatment within primary care, as well as for the possibility of improving scientific discovery by allowing an interoperable integration and sharing of cancer genomics datasets within emerging federated data spaces around the world.
Limitations of the study
There are two major aspects of ONCOLINER that can have generic and specific limitations, with potential consequences for users. One relies on the quality of the benchmarking dataset, which determines the scope and the reliability of all functionalities of ONCOLINER. Benchmarking datasets with low quality or low diversity of variants will result in inaccurate and poor performance assessments, which will also affect all improvement and harmonizing recommendations. For our study, we have taken two specific datasets4,9 that cover SNVs, indels, and SVs that have passed different rounds of quality check, but we cannot discard, for example, that a fraction of variants labeled as somatic within original datasets are, in fact, germline. Other sources of limitations rely on the sequencing and preprocessing methods that have been used on those original benchmark datasets, which also determine the scope of application of ONCOLINER. To solve these limitations, users can provide their own benchmarking dataset with specific parts of their methodology considered. Finally, other limitations rely on the algorithm and implementation of ONCOLINER, which has been designed to offer full functionality within low computational requirements. This compromise forced us to allow the prioritization of the harmonization to rely on the improvement of either the recall or the precision but not both at the same time. Although we do not expect much impact during implementations, this can sometimes result in suboptimal harmonization recommendations for precision and require follow-up executions of ONCOLINER.
Resource availability
Lead contact
Further information and requests should be directed to and will be fulfilled by the lead contact, David Torrents (david.torrents@bsc.es).
Material availability
No materials were generated in this study.
Data and code availability
-
•
The interactive HTML report generated by ONCOLINER for centers A, B and C has been deposited at a custom HTTP server and is publicly available as of the date of publication. URL is listed in the key resources table.
-
•
Tumorized genomes CRAM and VCF files have been deposited at ENA and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
Mosaic tumor-normal genome pairs CRAM files and Gold Standard VCF files from PCAWG-Pilot have been deposited at ICGC Data Portal, and accession numbers are listed in the key resources table. They are available upon request if access is granted.
-
•
Mosaic tumor-normal genome pairs CRAM files and Gold Standard VCF files from HMF-12 have been deposited at EGA, and accession numbers are listed in the key resources table. They are available upon request if access is granted.
-
•
All original code for ONCOLINER, PipelineDesigner, GenomeVariator and GenomeMosaicMaker has been deposited at GitHub and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
All original code for VariantExtractor has been deposited at GitHub and is publicly available as of the date of publication. DOI is listed in the key resources table. VariantExtractor is also available through PyPi.
-
•
All the variant callers included in this study are listed and can be downloaded as Singularity containers from GitHub and are publicly available as of the date of publication. URL is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
BSC-CNS discloses support for the research of this work from the European Union’s Horizon 2020 research and innovation programme under EUCANCan (grant agreement no. 825835), the Instituto de Salud Carlos III (ISCIII) and “Unión Europea NextGenerationEU/Mecanismo para la Recuperación y la Resilencia (MRR)/PRTR” under project PMP21/00015, the Departament de Recerca i Universitats de la Generalitat de Catalunya (code: 2021 SGR 01626), and the Science and Innovation Spanish Ministry under project BenchSV (PID2020-119797RB-I00/AEI/10.13039/501100011033). Institut Curie discloses support for the research of this work from the European Union’s Horizon 2020 research and innovation programme under EUCANCan (grant agreement no. 825835) and Cancéropôle Île-de-France (grant GENOPROFILE - RIC2021). The German Cancer Research Center (DKFZ) received funding from the European Union’s Horizon 2020 research and innovation programme and the Canadian Institutes of Health Research under the grant agreement no. 825325. CNAG institutional support was from the Spanish Instituto de Salud Carlos III, Fondo de Investigaciones Sanitarias, and cofunded with ERDF funds (PI19/01772); the Spanish Ministry of Science and Innovation through the Instituto de Salud Carlos III; the 2014–2020 Smart Growth Operating Program and institutional co-financing with the European Regional Development Fund (MINECO/FEDER, BIO2015-71792-P); and the Generalitat de Catalunya through the Departament de Salut, Departament d’Empresa i Coneixement. We acknowledge Hartwig Medical Foundation for their help and contributions to different parts of the study. We specifically thank and highly appreciate the valuable contribution of Matias Mendeville. In addition, we would like to express our sincere gratitude to the Genome in a Bottle Consortium (GIAB), the International Cancer Genome Consortium (ICGC), and The Cancer Genome Atlas (TCGA) for making high-quality data accessible through secure protocols, which have been instrumental to the success of this study.
Author contributions
D.T. and P.H. contributed to study conception and design and jointly directed the work. D.T., P.H., B.B., L.D.S., S.C.-G., and I.G. were principal investigators and contributed to study initiation. R.M., N.G., F.J., L.F., H.d.S., M.A., T.G., M.P., A.F., A.G., L.E., and R.R. contributed to the study methodology. All authors had unrestricted access to final study data and were responsible for data interpretation, preparation of the manuscript, and the decision to submit for publication. The manuscript was written and compiled by D.T., P.H., R.M., N.G., F.J., and L.F. R.M. and N.G. have accessed and verified the data. All authors attest to study completeness and the accuracy of the data and data analysis and approved the final version of the manuscript.
Declaration of interests
The authors declare no competing interests.
STAR★Methods
Key resources table
Method details
Benchmarking datasets: Mosaic and tumorized genomes
Experimentally validated variants are the best solution to properly benchmark variant calling algorithms and pipelines. Validated variant sets are highly valuable for providing realistic data in various fields, especially for cancer research. However, due to the difficulty of the validation process samples with validated variants are scarce. Additionally, existing validated datasets tend to be large and require computationally intensive processing due to the inclusion of numerous samples, even when they contain few validated variants. This computational demand poses challenges for researchers working with limited resources. To address this issue, we produced two alternative approaches in Mosaic and Tumorized genomes. We also provide the necessary open-source tools to generate them, GenomeVariator and GenomeMosaicMaker, intended for researchers interested in using these approaches for their studies.
First, Mosaic genomes provide a condensed representation of the complete benchmarking datasets, conveying the same information but significantly decreasing computational burden. Nevertheless, Mosaic genomes pose limitations. For once, the availability of restricted access benchmarking datasets due to privacy concerns or other confidentiality clauses impedes open access to Mosaic genomes. Additionally, source benchmarking datasets often lack control over false positives, affecting reliability for precision assessment on Mosaic genomes. To complement Mosaics and overcome these limitations, the Tumorized genomes were developed. Tumorized genomes address the need for patient data protection by ensuring that the genomes are de-identified, eliminating the need for bureaucratic processes that can impede progress in cancer research. By utilizing Tumorized genomes, researchers gain absolute control over the features and variants included in these datasets, allowing precise and controlled analysis. The method to bring these approaches into real datasets for our study is detailed below.
The short-read sequencing data needed to discover somatic variants in a sample consists of a pair of tumor-normal samples mapped to a reference genome. The ground for constructing the tumor-normal datasets comprising a Mosaic genome are the original reads sequenced from validated datasets such as the PCAWG-Pilot and HMF-12, and the validated variants within them. The first step is to estimate the average depth, read length, and insert sizes of each of the validated samples. Then, a canvas WGS dataset is simulated with these values from the same reference genome to which the samples are mapped. For our read simulations, we used the ART Illumina software.56 Finally, read-alignments from the original tumor-normal samples overlapping with a 2kb window centered around each of the validated variants are inserted into the canvas genome. The simulated reads in these regions from the canvas genome are discarded in order to remove discrepancies in read depths or lengths. Thus, Mosaic genomes provide a realistic representation of the original somatic variants. We implemented this method as the GenomeMosaicMaker tool.
The HMF-12 samples contain a collection of experimentally validated short indels and SVs. These experiments were carried out after variant discovery using GRIDSS2, Manta, and Strelka.9 Only SVs were used to construct the HMF-12 Mosaic genome because short indel calls presented inconsistent coordinates between the variant callers mentioned earlier. Then, we analyzed the features of the reads from each sample finding that all of them are homogeneous regarding sequencing depth, and lengths of reads and inserts. Thus, we generated one single Mosaic compiling all HMF-12 samples into a canvas, simulated from the GRCh37 reference with median (Mdn) read length values of 150 bp, and Mdn insert length of 500 bp with a standard deviation (SD) of 125 bp. The read depth of the normal Mosaic sample was 32x, and the tumor sample was 100x.
The PCAWG-Pilot data originates from different laboratories using different sequencing protocols and machines. Although the variant discovery and validation pipelines were homogenized, the original experimental conditions made it impossible to compile all samples into a single Mosaic. To find a consistent number of Mosaic genomes for this data, the following procedure was performed. First, we estimated the insert size median (Mdn) and standard deviation (SD) values for each sample. Using this information, we performed K-means clustering iteratively trying different cluster numbers to find the most consistent grouping for these samples. Then, for each configuration, we executed the variant callers and computed their performance metrics. These results were compared to their performances on the original samples, proving that the best results were obtained with the three Mosaic representations. Finally, these three Mosaics were generated with read depths of 40x for the normal samples and 60x for the tumor samples (see Figures S3 and S4). To select the better suited mapping method for the benchmarking and further evaluate the potential effect of different read aligners on ONCOLINER recommendations, the assessment process was performed over Mosaic genomes aligned with three state-of-the-art algorithms in BWA-MEM (v.0.7.17),58 Bowtie2 (v.2.5.3),59 and Hisat2 (v.2.2.1).60 Based on these results BWA-MEM was chosen for the generation of the final Mosaics (see Figure S16). Overall, the construction process of the Mosaics from both datasets highlights the importance of careful control of sequencing features such as insert sizes, depths, and even mapping strategies to ensure the reliability of the Mosaic genome to represent real variation from the original samples.
In contrast to the Mosaic genome approach, a Tumorized genome provides a benchmark to test the precision of variant discovery. To overcome data sharing limitations, publicly available read mappings are used as the base. First, they are carefully split into the tumor-normal sets, ensuring that there are no duplicated reads between them, and that they come from different libraries, mimicking real scenarios where normal and tumoral samples are processed and sequenced independently. To reach the desired coverage for the benchmarking genome, balanced random downsampling is applied to maintain the original proportions of the read libraries. Then, for each variant in the validated VCF, a subset of reads in the tumoral sample is modified to represent the variant. The number of modified reads depends on the depth of the region where the variant is located and the provided VAF (see Figure S6). These modified reads are remapped to the original reference genome. Therefore, the Tumorized genome will only contain somatic variants that were inserted into the reads (see Figure S17), eliminating external false positives while avoiding artificial variations in read depth. This method is implemented in GenomeVariator, a wrapper tool that enhances the functionalities of BAMSurgeon25,26,44 to facilitate the construction of Tumorized genomes.
The Tumorized genomes (60x Tumoral - 40x Normal) used in this study were constructed from 300x depth Illumina reads from two real WGS samples from the GIAB project: NA12878 and AshkenazimTrio son HG002,43 mapped to the GRCh37 reference using BWA-MEM.55 The SNV, indel, and SV-validated variants from the PCAWG Consensus Callsets were simulated into the tumoral sample. To ensure the Tumorized genome contains a realistic amount of variants, the number of simulated variants of each type was set to the median number of total variants of the same type in the original samples, discovered by all callers. The number of SVs was set to double the median and overlapping SVs were excluded to avoid undesired results, prioritizing by SV type (SV type priority order: translocation, inversion, duplication, and deletion).
Variant caller selection
We originally selected 18 candidate programs for somatic variant identification, covering different calling algorithms, strategies, and scopes (SNV, indels, and SVs). From these 12 were selected for our study based on usability and efficiency: SvABA5 (version 1.1.0), Delly6 (version 1.1.6), mutect2 (from GATK7 4.2.6.1), Strelka28 (version 2.9.10), GRIDSS29 (version 2.13.2), MuSE11 (version 2.0), Manta10 (version 1.6.0), SAGE21 (version 3.0), cgpPindel12 (version 3.9.0), cgpCaVEManWrapper13 (version 1.16.0), Shimmer14 and BRASS22 (version 6.3.4). The other 6 variant callers were excluded due to: excessive execution time, non-standard input requirements, not maintained, or with merged germline-somatic variants outputs. The excluded tools were Lancet61 (version 1.1.0), Platypus15 (version 0.8.1.1), Lumpy16 (version 0.3.1), breakdancer17 (version 1.4.5), SomaticSniper18 (version 1.0.5.0) and Seurat19 (version 2.5).
Assessment module
The ONCOLINER assessment module compares the discovered variants from the results of the input pipelines against the validated variants. Comparisons depend on the variant types and sizes. In particular, SNVs and small indels are considered true positives if their chromosome, coordinate, and alternate allele exactly match with a ground truth variant. To consider SVs as true positives their breakends must be located within a 100 bp window of the breakends of a gold standard SV, and their orientation must be equal. Conversely, even though large insertions are not present in the Mosaic or Tumorized datasets, their length would be compared instead of their second breakend. The assessment module counts true positive, false positive, and false negative calls from the evaluated pipeline to estimate recall, and precision using the Mosaics and Tumorized genomes, respectively. Then, the F-score is computed to provide a combined accuracy measure (see Figures S9–S11).
To assess the functional performance of the pipelines, every true positive variant inherits precomputed gene annotations of their matched gold standard variant. This avoids the need to annotate every incoming test VCF. The same method is used to identify affected cancer-driver genes. VEP57 was used to annotate SNVs and indels, avoiding annotations without functional phenotypes. For SV annotation, VEP could not process multiple variants due to their VCF representation. To properly annotate SVs we developed scripts based on VariantExtractor, where each gene that intersected the span of an SV was added to the corresponding VCF record. In particular, for inversions and translocations, only the genes that intersected their breakpoints were considered as affected thereby disrupting the open reading frame of the gene. Finally, the source for the annotation of protein-coding genes to SVs was the Ensembl GHRCh37 transcriptome (v87),54 and the cancer driver genes were collected from the IntOGen Catalog (release 2023.05.31).2
Improvement module
The ONCOLINER improvement module follows the assessment step and provides recommendations based on the performance evaluation of the input pipelines and the 12 selected variant callers. Specifically, the recommendations are the best combinations of variant callers to integrate into the pipeline to maximize performance metrics. The first step is to perform both the union and the intersection of the pipeline calls with the callers. Then, performance metrics are calculated for these merged results, and combinations are sorted based on them. In addition to a list of all possible combinations provided as a CSV file, the interactive GUI shows the most relevant recommendations. This allows the user to sort them by any of the metrics between recall, precision, F1-score, or even by the number of affected protein-coding or cancer-driver genes. To improve visualization of the most relevant recommendations, they are filtered by selecting the one in the top 5% for each performance metric, prioritizing those with the least number of callers. This follows the rationale that a better recommendation minimizes the cost of adding too many tools to a pipeline and the effort of going through redundant combinations (see Figure S18).
Harmonizer module
The ONCOLINER harmonizer module follows the improvement step generating recommendations to bring the performance of all input pipelines closer while maintaining the best possible performance. To achieve this, pipeline heterogeneity is quantified into two metrics, the Performance Heterogeneity Score (PHS) and Gene Discordance Ratio (GDR). The PHS is estimated from plotting precision-recall ranges in a Euclidean space, where pipelines are represented by their respective performance coordinates. Considering (recall, precision) as a pipeline point from the set of n pipelines, a centroid (i.e., the gravity center) c is the point that minimizes the sum of its square distance to all . Then, the PHS is computed as the normalized mean of all . In other words, the PHS is the normalized average Euclidean distance from the position of each pipeline to the centroid or the theoretical maximum homogeneity point. Equation 1 shows the calculation of the PHS.
| (Equation 1) |
To measure functional impact heterogeneity in variant discovery we created the Gene Discordance Ratio (GDR). The GDR is calculated as the complement of the proportion between the variant-affected genes found by every pipeline (intersection) over the total number of variant-affected genes even if only detected by one of the pipelines (union). Hence, a GDR value closer to 1 would imply a high level of heterogeneity in functional impact between the pipelines. This metric follows Equation 2 where represents the number of genes affected by the discovered variants from the i-th pipeline.
| (Equation 2) |
To achieve comprehensive harmonization, this module first prioritizes the best recommendations to maximize performance for each of the pipelines. Then, it minimizes PHS and GDR to decrease heterogeneity in accuracy and functional impact. This priority order avoids optimized homogenization where the distance from a pipeline to the centroid could be 0, but would sometimes worsen accuracy. As the harmonization module works with the performance enhancements generated by the improvement module, it will highlight recommendations where heterogeneity decreases, but most importantly where performance improves in all possible homogenization scenarios. Finally, following the filtering logic of the improvement step, the user visualizes non-redundant recommendations for harmonizing the pipelines that minimize the effort of adding too many variant callers (see Figure S19; Table S2).
Published: August 30, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2024.100639.
Supplemental information
References
- 1.International Cancer Genome Consortium. Hudson T.J., Anderson W., Artez A., Barker A.D., Bell C., Bernabé R.R., Bhan M.K., Calvo F., Eerola I., et al. International network of cancer genome projects. Nature. 2010;464:993–998. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martínez-Jiménez F., Muiños F., Sentís I., Deu-Pons J., Reyes-Salazar I., Arnedo-Pac C., Mularoni L., Pich O., Bonet J., Kranas H., et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer. 2020;20:555–572. doi: 10.1038/s41568-020-0290-x. [DOI] [PubMed] [Google Scholar]
- 3.Tate J.G., Bamford S., Jubb H.C., Sondka Z., Beare D.M., Bindal N., Boutselakis H., Cole C.G., Creatore C., Dawson E., et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47:D941–D947. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium Pan-cancer analysis of whole genomes. Nature. 2020;578:82–93. doi: 10.1038/s41586-020-1969-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wala J.A., Bandopadhayay P., Greenwald N.F., O'Rourke R., Sharpe T., Stewart C., Schumacher S., Li Y., Weischenfeldt J., Yao X., et al. SvABA: genome-wide detection of structural variants and indels by local assembly. Genome Res. 2018;28:581–591. doi: 10.1101/gr.221028.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rausch T., Zichner T., Schlattl A., Stütz A.M., Benes V., Korbel J.O. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28:i333–i339. doi: 10.1093/bioinformatics/bts378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim S., Scheffler K., Halpern A.L., Bekritsky M.A., Noh E., Källberg M., Chen X., Kim Y., Beyter D., Krusche P., Saunders C.T. Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods. 2018;15:591–594. doi: 10.1038/s41592-018-0051-x. [DOI] [PubMed] [Google Scholar]
- 9.Cameron D.L., Baber J., Shale C., Valle-Inclan J.E., Besselink N., van Hoeck A., Janssen R., Cuppen E., Priestley P., Papenfuss A.T. GRIDSS2: comprehensive characterisation of somatic structural variation using single breakend variants and structural variant phasing. Genome Biol. 2021;22 doi: 10.1186/s13059-021-02423-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen X., Schulz-Trieglaff O., Shaw R., Barnes B., Schlesinger F., Källberg M., Cox A.J., Kruglyak S., Saunders C.T. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32:1220–1222. doi: 10.1093/bioinformatics/btv710. [DOI] [PubMed] [Google Scholar]
- 11.Fan Y., Xi L., Hughes D.S.T., Zhang J., Zhang J., Futreal P.A., Wheeler D.A., Wang W. MuSE: accounting for tumor heterogeneity using a sample-specific error model improves sensitivity and specificity in mutation calling from sequencing data. Genome Biol. 2016;17 doi: 10.1186/s13059-016-1029-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raine K.M., Hinton J., Butler A.P., Teague J.W., Davies H., Tarpey P., Nik-Zainal S., Campbell P.J. cgpPindel: Identifying somatically acquired insertion and deletion events from paired end sequencing. Curr. Protoc. Bioinformatics. 2015;52:15.7.1–15.7.12. doi: 10.1002/0471250953.bi1507s52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones D., Raine K.M., Davies H., Tarpey P.S., Butler A.P., Teague J.W., Nik-Zainal S., Campbell P.J. cgpCaVEManWrapper: Simple execution of CaVEMan in order to detect somatic single nucleotide variants in NGS data. Curr. Protoc. Bioinformatics. 2016;56:15.10.1–15.10.18. doi: 10.1002/cpbi.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hansen N.F., Gartner J.J., Mei L., Samuels Y., Mullikin J.C. Shimmer: detection of genetic alterations in tumors using next-generation sequence data. Bioinformatics. 2013;29:1498–1503. doi: 10.1093/bioinformatics/btt183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rimmer A., Phan H., Mathieson I., Iqbal Z., Twigg S.R.F., WGS500 Consortium. Wilkie A.O.M., McVean G., Lunter G. Integrating mapping-assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet. 2014;46:912–918. doi: 10.1038/ng.3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Layer R.M., Chiang C., Quinlan A.R., Hall I.M. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 2014;15:R84. doi: 10.1186/gb-2014-15-6-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fan X., Abbott T.E., Larson D., Chen K. BreakDancer: Identification of genomic structural variation from paired-end read mapping. Curr. Protoc. Bioinformatics. 2014;45:15.6.1–15.6.11. doi: 10.1002/0471250953.bi1506s45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larson D.E., Harris C.C., Chen K., Koboldt D.C., Abbott T.E., Dooling D.J., Ley T.J., Mardis E.R., Wilson R.K., Ding L. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics. 2012;28:311–317. doi: 10.1093/bioinformatics/btr665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Christoforides A., Carpten J.D., Weiss G.J., Demeure M.J., Von Hoff D.D., Craig D.W. Identification of somatic mutations in cancer through bayesian-based analysis of sequenced genome pairs. BMC Genom. 2013;14:302. doi: 10.1186/1471-2164-14-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moncunill V., Gonzalez S., Beà S., Andrieux L.O., Salaverria I., Royo C., Martinez L., Puiggròs M., Segura-Wang M., Stütz A.M., et al. Comprehensive characterization of complex structural variations in cancer by directly comparing genome sequence reads. Nat. Biotechnol. 2014;32:1106–1112. doi: 10.1038/nbt.3027. [DOI] [PubMed] [Google Scholar]
- 21.Martínez-Jiménez F., Movasati A., Brunner S.R., Nguyen L., Priestley P., Cuppen E., Van Hoeck A. Pan-cancer whole-genome comparison of primary and metastatic solid tumours. Nature. 2023;618:333–341. doi: 10.1038/s41586-023-06054-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nik-Zainal S., Davies H., Staaf J., Ramakrishna M., Glodzik D., Zou X., Martincorena I., Alexandrov L.B., Martin S., Wedge D.C., et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534:47–54. doi: 10.1038/nature17676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alioto T.S., Buchhalter I., Derdak S., Hutter B., Eldridge M.D., Hovig E., Heisler L.E., Beck T.A., Simpson J.T., Tonon L., et al. A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nat. Commun. 2015;6 doi: 10.1038/ncomms10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nadeu F., Royo R., Massoni-Badosa R., Playa-Albinyana H., Garcia-Torre B., Duran-Ferrer M., Dawson K.J., Kulis M., Diaz-Navarro A., Villamor N., et al. Detection of early seeding of richter transformation in chronic lymphocytic leukemia. Nat. Med. 2022;28:1662–1671. doi: 10.1038/s41591-022-01927-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ewing A.D., Houlahan K.E., Hu Y., Ellrott K., Caloian C., Yamaguchi T.N., Bare J.C., P'ng C., Waggott D., Sabelnykova V.Y., et al. Combining tumor genome simulation with crowdsourcing to benchmark somatic single-nucleotide-variant detection. Nat. Methods. 2015;12:623–630. doi: 10.1038/nmeth.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee A.Y., Ewing A.D., Ellrott K., Hu Y., Houlahan K.E., Bare J.C., Espiritu S.M.G., Huang V., Dang K., Chong Z., et al. Combining accurate tumor genome simulation with crowdsourcing to benchmark somatic structural variant detection. Genome Biol. 2018;19 doi: 10.1186/s13059-018-1539-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olson N.D., Wagner J., Dwarshuis N., Miga K.H., Sedlazeck F.J., Salit M., Zook J.M. Variant calling and benchmarking in an era of complete human genome sequences. Nat. Rev. Genet. 2023;24:464–483. doi: 10.1038/s41576-023-00590-0. [DOI] [PubMed] [Google Scholar]
- 28.Cameron D.L., Di Stefano L., Papenfuss A.T. Comprehensive evaluation and characterisation of short read general-purpose structural variant calling software. Nat. Commun. 2019;10 doi: 10.1038/s41467-019-11146-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cortés-Ciriano I., Gulhan D.C., Lee J.J.-K., Melloni G.E.M., Park P.J. Computational analysis of cancer genome sequencing data. Nat. Rev. Genet. 2021;23:298–314. doi: 10.1038/s41576-021-00431-y. [DOI] [PubMed] [Google Scholar]
- 30.Fang L.T., Zhu B., Zhao Y., Chen W., Yang Z., Kerrigan L., Langenbach K., de Mars M., Lu C., Idler K., et al. Establishing community reference samples, data and call sets for benchmarking cancer mutation detection using whole-genome sequencing. Nat. Biotechnol. 2021;39:1151–1160. doi: 10.1038/s41587-021-00993-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schipper L.J., Samsom K.G., Snaebjornsson P., Battaglia T., Bosch L.J.W., Lalezari F., Priestley P., Shale C., van den Broek A.J., Jacobs N., et al. Complete genomic characterization in patients with cancer of unknown primary origin in routine diagnostics. ESMO Open. 2022;7 doi: 10.1016/j.esmoop.2022.100611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Samsom K.G., Schipper L.J., Roepman P., Bosch L.J., Lalezari F., Klompenhouwer E.G., de Langen A.J., Buffart T.E., Riethorst I., Schoenmaker L., et al. Feasibility of whole-genome sequencing-based tumor diagnostics in routine pathology practice. J. Pathol. 2022;258:179–188. doi: 10.1002/path.5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Samsom K.G., Bosch L.J.W., Schipper L.J., Roepman P., de Bruijn E., Hoes L.R., Riethorst I., Schoenmaker L., van der Kolk L.E., Retèl V.P., et al. Study protocol: Whole genome sequencing implementation in standard diagnostics for every cancer patient (wide) BMC Med. Genom. 2020;13 doi: 10.1186/s12920-020-00814-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Horak P., Klink B., Heining C., Gröschel S., Hutter B., Fröhlich M., Uhrig S., Hübschmann D., Schlesner M., Eils R., et al. Precision oncology based on omics data: The nct heidelberg experience. Int. J. Cancer. 2017;141:877–886. doi: 10.1002/ijc.30828. [DOI] [PubMed] [Google Scholar]
- 35.Worst B.C., van Tilburg C.M., Balasubramanian G.P., Fiesel P., Witt R., Freitag A., Boudalil M., Previti C., Wolf S., Schmidt S., et al. Next-generation personalised medicine for high-risk paediatric cancer patients – the inform pilot study. Eur. J. Cancer. 2016;65:91–101. doi: 10.1016/j.ejca.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 36.Lejeune C., Amado I.F., DEFIDIAG study group FHU Translad and Aviesan. Binquet C., Deleuze J.-F., Delmas C., Dollfus H., Esperou H., Faivre L., Frebourg T., et al. Valuing genetic and genomic testing in france: current challenges and latest evidence. J. Community Genet. 2022;13:477–485. doi: 10.1007/s12687-020-00503-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lévy Y. Genomic medicine 2025: France in the race for precision medicine. Lancet. 2016;388:2872. doi: 10.1016/s0140-6736(16)32467-9. [DOI] [PubMed] [Google Scholar]
- 38.Solary E., Blanc P., Boutros M., Girvalaki C., Locatelli F., Medema R.H., Nagy P., Tabernero J. UNCAN.eu, a european initiative to UNderstand CANcer. Cancer Discov. 2022;12:2504–2508. doi: 10.1158/2159-8290.cd-22-0970. [DOI] [PubMed] [Google Scholar]
- 39.Bates M. The cancer moonshot enters a new phase. IEEE Pulse. 2022;13:2–5. doi: 10.1109/mpuls.2022.3227807. [DOI] [PubMed] [Google Scholar]
- 40.Rehm H.L., Page A.J.H., Smith L., Adams J.B., Alterovitz G., Babb L.J., Barkley M.P., Baudis M., Beauvais M.J.S., Beck T., et al. Ga4gh: International policies and standards for data sharing across genomic research and healthcare. Cell Genom. 2021;1 doi: 10.1016/j.xgen.2021.100029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Priestley P., Baber J., Lolkema M.P., Steeghs N., de Bruijn E., Shale C., Duyvesteyn K., Haidari S., van Hoeck A., Onstenk W., et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature. 2019;575:210–216. doi: 10.1038/s41586-019-1689-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tikellis G., Dwyer T., Paltiel O., Phillips G.S., Lemeshow S., Golding J., Northstone K., Boyd A., Olsen S., Ghantous A., et al. The international childhood cancer cohort consortium (i4c): A research platform of prospective cohorts for studying the aetiology of childhood cancers. Paediatr. Perinat. Epidemiol. 2018;32:568–583. doi: 10.1111/ppe.12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zook J.M., Catoe D., McDaniel J., Vang L., Spies N., Sidow A., Weng Z., Liu Y., Mason C.E., Alexander N., et al. Extensive sequencing of seven human genomes to characterize benchmark reference materials. Sci. Data. 2016;3 doi: 10.1038/sdata.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.adamewing L.,M., Rapsssito, Mjko1210, SebastianHollizeck, Xia C., Cook D.E., GILLET-Markowska A., Richter D., Hammerbacher J., St. John J., et al. 2021. Dami Rebergen, and Zhmz90. BAMSurgeon. URL: [DOI] [Google Scholar]
- 45.Chakravarty D., Gao J., Phillips S.M., Kundra R., Zhang H., Wang J., Rudolph J.E., Yaeger R., Soumerai T., Nissan M.H., et al. Oncokb: A precision oncology knowledge base. JCO Precis. Oncol. 2017;2017:1–16. doi: 10.1200/po.17.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suehnholz S.P., Nissan M.H., Zhang H., Kundra R., Nandakumar S., Lu C., Carrero S., Dhaneshwar A., Fernandez N., Xu B.W., et al. Quantifying the expanding landscape of clinical actionability for patients with cancer. Cancer Discov. 2024;14:49–65. doi: 10.1158/2159-8290.cd-23-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gajiwala K.S., Wu J.C., Christensen J., Deshmukh G.D., Diehl W., DiNitto J.P., English J.M., Greig M.J., He Y.-A., Jacques S.L., et al. Kit kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients. Proc. Natl. Acad. Sci. USA. 2009;106:1542–1547. doi: 10.1073/pnas.0812413106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heinrich M.C., Blanke C.D., Druker B.J., Corless C.L. Inhibition of kit tyrosine kinase activity: A novel molecular approach to the treatment of kit-positive malignancies. J. Clin. Oncol. 2002;20:1692–1703. doi: 10.1200/jco.2002.20.6.1692. [DOI] [PubMed] [Google Scholar]
- 49.Bauer S., Duensing A., Demetri G.D., Fletcher J.A. Kit oncogenic signaling mechanisms in imatinib-resistant gastrointestinal stromal tumor: Pi3-kinase/akt is a crucial survival pathway. Oncogene. 2007;26:7560–7568. doi: 10.1038/sj.onc.1210558. [DOI] [PubMed] [Google Scholar]
- 50.Zehir A., Benayed R., Shah R.H., Syed A., Middha S., Kim H.R., Srinivasan P., Gao J., Chakravarty D., Devlin S.M., et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017;23:703–713. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lo-Coco F., Avvisati G., Vignetti M., Thiede C., Orlando S.M., Iacobelli S., Ferrara F., Fazi P., Cicconi L., Di Bona E., et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N. Engl. J. Med. 2013;369:111–121. doi: 10.1056/nejmoa1300874. [DOI] [PubMed] [Google Scholar]
- 52.Wang H., Nicolay B.N., Chick J.M., Gao X., Geng Y., Ren H., Gao H., Yang G., Williams J.A., Suski J.M., et al. The metabolic function of cyclin d3–cdk6 kinase in cancer cell survival. Nature. 2017;546:426–430. doi: 10.1038/nature22797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garcia-Prieto C.A., Martínez-Jiménez F., Valencia A., Porta-Pardo E. Detection of oncogenic and clinically actionable mutations in cancer genomes critically depends on variant calling tools. Bioinformatics. 2022;38:3181–3191. doi: 10.1093/bioinformatics/btac306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cunningham F., Amode M.R., Barrell D., Beal K., Billis K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fitzgerald S., et al. Ensembl 2015. Nucleic Acids Res. 2015;43:D662–D669. doi: 10.1093/nar/gku1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H., Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang W., Li L., Myers J.R., Marth G.T. ART: a next-generation sequencing read simulator. Bioinformatics. 2012;28:593–594. doi: 10.1093/bioinformatics/btr708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R.S., Thormann A., Flicek P., Cunningham F. The ensembl variant effect predictor. Genome Biol. 2016;17 doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Langmead B., Salzberg S.L. Fast gapped-read alignment with bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim D., Paggi J.M., Park C., Bennett C., Salzberg S.L. Graph-based genome alignment and genotyping with hisat2 and hisat-genotype. Nat. Biotechnol. 2019;37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Narzisi G., Corvelo A., Arora K., Bergmann E.A., Shah M., Musunuri R., Emde A.-K., Robine N., Vacic V., Zody M.C. Genome-wide somatic variant calling using localized colored de bruijn graphs. Commun. Biol. 2018;1 doi: 10.1038/s42003-018-0023-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The interactive HTML report generated by ONCOLINER for centers A, B and C has been deposited at a custom HTTP server and is publicly available as of the date of publication. URL is listed in the key resources table.
-
•
Tumorized genomes CRAM and VCF files have been deposited at ENA and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
Mosaic tumor-normal genome pairs CRAM files and Gold Standard VCF files from PCAWG-Pilot have been deposited at ICGC Data Portal, and accession numbers are listed in the key resources table. They are available upon request if access is granted.
-
•
Mosaic tumor-normal genome pairs CRAM files and Gold Standard VCF files from HMF-12 have been deposited at EGA, and accession numbers are listed in the key resources table. They are available upon request if access is granted.
-
•
All original code for ONCOLINER, PipelineDesigner, GenomeVariator and GenomeMosaicMaker has been deposited at GitHub and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
All original code for VariantExtractor has been deposited at GitHub and is publicly available as of the date of publication. DOI is listed in the key resources table. VariantExtractor is also available through PyPi.
-
•
All the variant callers included in this study are listed and can be downloaded as Singularity containers from GitHub and are publicly available as of the date of publication. URL is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.