

Abstract
Sirtuin 6 (Sirt6), an NAD+-dependent deacylase, has emerged as a promising target for aging-related diseases and cancer. Advancing the medicinal chemistry of Sirt6 modulators is crucial for the development of chemical probes aimed at unraveling the intricate biological functions of Sirt6 and unlocking its therapeutic potential. A proprietary DNA-encoded library yielded Sirt6 inhibitor 2-Pr, displaying remarkable inhibitory activity and isoform-selectivity, and featuring a chemical structure distinct from reported Sirt6 modulators. In this study, we explore the inhibitory mechanism of 2-Pr, evaluating the impact of chemical modifications and presenting a crystal structure of the Sirt6/ADP-ribose/2-Pr complex. Notably, co-crystal structure analysis reveals an unexpected and unprecedented binding mode of Sirt6, with 2-Pr spanning the acyl channel of the enzyme, extending into the acetyl-lysine binding pocket, and reaching toward the C-site. This unique binding mode guides potential avenues for developing potent and selective Sirt6 inhibitors.
Keywords: Sirt6 inhibition, Crystal structure, Deacylase, DNA-encoded chemical libraries, Acyl binding pocket
Graphical Abstract
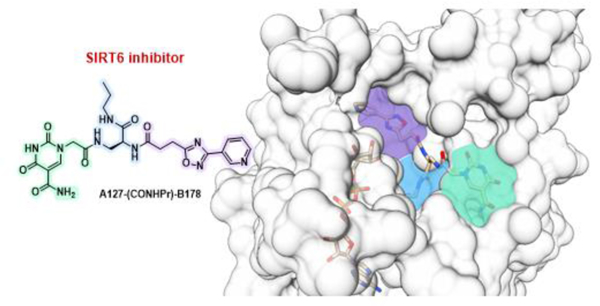
This study explores the mechanism and medicinal chemistry of 2-Pr, a highly potent and selective inhibitor of the protein deacylase Sirtuin 6. We evaluate the impact of chemical modifications and present a crystal structure of the Sirt6/ADP-ribose/2-Pr complex, which reveals an unexpected and unprecedented binding mode. This unique binding mode guides potential avenues for developing potent and selective Sirt6 inhibitors.
Introduction
Sirtuins form Class III of the protein lysine deacylase family (also known as histone deacetylases or HDACs) and play pivotal roles in various cellular processes, including energy metabolism and cell proliferation.[1] Their dependence on NAD+ as a co-substrate makes Sirtuins metabolic sensors in cell signaling pathways.[2] Sirtuins are implicated in the control of cellular senescence and organismal lifespan, and they are considered therapeutic targets for various aging-related diseases.[3]
Among the seven mammalian Sirtuin isoforms (Sirt1-7), Sirt1, Sirt6, and Sirt7 predominantly reside within the cell nucleus.[3] Many small molecule modulators have been identified for Sirt1, the most extensively studied isoform and potential therapeutic target for inflammatory diseases.[4] In contrast, few potent and specific compounds are available for Sirt6, an isoform regulating chromatin homeostasis, transcription factors, and stress response proteins.[5] Sirt6 overexpression can extend the lifespan of male mice, and Sirt6 suppresses several aging-related phenotypes and can promote apoptosis in various cancer types.[6] Because of this therapeutic relevance, there is considerable interest in small molecule modulators that effectively and selectively target Sirt6.[5]
Sirt6 exhibits a conserved Sirtuin catalytic core comprising ~270 amino acids, flanked by N- and C-terminal extensions that are specific to this isoform and contribute to its association with chromatin.[6,7] Within the core, the active site is formed by an interface between a large α/β Rossmann-fold subdomain and a structurally more diverse, small Zn2+-binding module. During Sirtuin catalysis, the nicotinamide (NAM) moiety from NAD+ is placed in the conserved C-pocket and its glycosidic bond cleaved, while the carbonyl oxygen of the acyl group attacks the ribose C1 position, resulting in an alkylamidate intermediate. Rearrangement into a bicyclic intermediate and eventual hydrolysis yield the end products: a deacylated peptide and 2′-O-acyl-ADP-ribose.[8] Amino acid variations between Sirtuin isoforms lead to differing preferences in substrate sequence and acyl modifications. Notably, the extended acyl binding pocket of Sirt6 allows it to hydrolyze long-chain fatty acyl groups in vitro more efficiently than acetyl groups.[8a] Sirt6 indeed promotes TNF-α secretion through its demyristoylation, but a broader role of this activity remains to be established.[8a] Although the in vitro deacetylase activity of Sirt6 is low for unknown reasons, it effectively deacetylates histones and other target proteins in vivo to elicit physiological effects.
Limited options are currently available for pharmacological modulation of Sirt6 activity. Fatty acids[9] and the flavonoid quercetin[10] activate Sirt6-dependent deacetylation but require high concentrations. More efficient activation has been achieved with synthetic pyrrolo[1,2-a]quinoxalines,[11] the quercetin derivative cyanidin,[12] and MDL-801[13] and MDL-811.[14] These molecules bind to the Sirt6 catalytic core, partially occupying the NAM site and the unique Sirt6 acyl channel. Their mechanism of action remains unclear, however, hindering efficient improvement. Inhibition of Sirt6 activity can be achieved with the pan-Sirtuin inhibitor NAM, but with limited potency and selectivity, and with increased potency using the NAM analog, pyrazinamide.[15] Slightly better potency was also obtained with salicylate derivatives,[16] and cyclic peptides enabled potent Sirt6 inhibition but without isoform specificity.[17] JYQ-42 also shows potent effects (IC50 = 2.3 μM), presumably via an allosteric site,[18] while trichostatin A (TSA) and some quercetin derivatives like catechin gallate (GC) inhibit Sirt6 with low micromolar IC50 values (2.0–5.4 μM) by targeting the acyl binding channel.[12] However, TSA and quercetin derivatives also affect various other targets, including inhibition of class I and II HDACs with nanomolar potency for TSA.
In the present study, we explore the inhibitory mechanism of 2-Pr, a potent and selective Sirt6 inhibitor recently identified by some of us. We evaluate chemical modifications of 2-Pr and present a crystal structure of a Sirt6/ADP-ribose/2-Pr complex, revealing an unexpected and unprecedented binding mode. Our results provide potential avenues for developing potent and selective Sirt6 inhibitors.
Results and Discussion
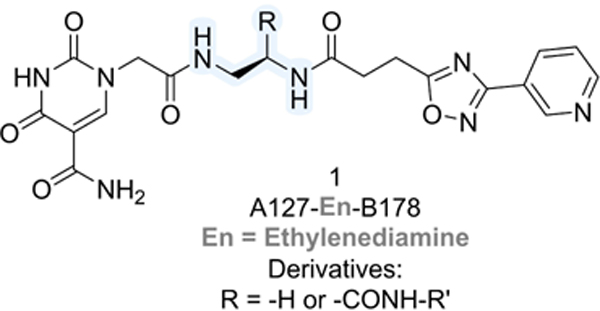
Attempting to identify new chemotypes of Sirt6 inhibitors, we previously conducted a DNA-encoded chemical library (DECL)[19] screen for this enzyme. We used a NAD+-mimicking DECL (NADEL), a focused 2-cycle library with 58,302 compounds, that has a homogeneous composition, low truncation rates, and allows for in-depth sequencing following selections.[20] The results from the screen resulted in the discovery of a unique compound with a 5-aminocarbonyl-uracil and a 3-pyrinidyl-1,2,4-oxadiazole moiety (2-Pr; Table 1).[20] The molecule inhibited Sirt6 with an IC50 = 8.9 μM in a peptide demyristoylation assay, and consistent with activity in cells, 2-Pr induced DNA-damage markers and elicited a dose-dependent reduction in TNF-α export levels.[20] Since NADEL was designed to mimic NAD+, we anticipated compound 1 to interact with the pocket of Sirt6 binding the co-factor, and docking studies corroborated this assumption (Figure S14d in Ref.[20]). Based on such a binding mode, we postulated that the full linker might extend into the fatty-acid binding pocket of Sirt6.[8a] The tested molecules lacked the C12-linker used to attach the molecule to DNA (R in structure in Table 1), and it was therefore reasonable to assume that the inclusion of hydrophobic groups at the linker attachment site could enhance the potency of these molecules. Indeed, derivatives of the DECL hit featuring a propylcarboxamide at the linker attachment site (2-Pr) exhibited superior Sirt6 inhibition compared to a derivative with a methylcarboxamide (2-Me), and the removal of the alkyl group (R=−H) led to loss of activity (Table 1).[20]
Table 1.
Impact of size and hydrophobicity of carboxamide derivatives derived from the DECL hit (A127/B178) on Sirt6 deacylase activity.
| ||
|---|---|---|
| Cmpd | Substituent R = | Inhibition c = 10 μM |
| 1 | H | 0% |
| 2-Me |

|
40%[a] |
| 2-Pr |
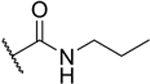
|
60% (IC50 = 8.9 μM)[b] pIC50 (5.1 ± 0.03) |
| 2-MeOEt |
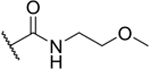
|
9% |
| 2-PhEt |
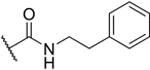
|
29% |
| 2-Bn |

|
30%[a] |
| 2-Pip |

|
28% |
| 2-Ph |
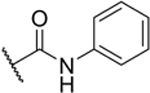
|
53% (IC50 = 9.3 μM) pIC50 (5.0 ± 0.05) |
| 2-CyBu |
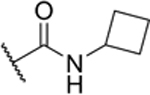
|
64% (IC50 = 8.2 μM) pIC50 (5.1 ± 0.05) |
| 3 |

|
3% |
| Nicotinamide (1000 μM) | 92% | |
Sirt6 activity measured at c = 40 μM.
Previously measured IC50 = 6.7 μM Ref.[20] The biochemical assay used to assess the in vitro activity of the compounds was done in duplicate and repeated twice on different days.
Interested in exploring the structure-activity relationship at this position in more detail, we synthesized a series of carboxamide derivatives of compound 1 (Table 1). These molecules were prepared through a late-stage amidation approach, starting with a common carboxylic acid precursor and various amines (reaction scheme shown in the SI, Scheme S1 and Figures S1 and S2). The synthesis of the 2-COOH precursor involved three sequential steps, encompassing a first amide coupling between H-Dap(Boc)-OMe and 3-(3-(pyridin-3-yl)-1,2,4-oxadiazol-5-yl)propanoic acid to give the Boc-protected intermediate, that followed by Boc deprotection and a second amide coupling with 2-(5-carbamoyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)acetic acid yield the ester compound, which was submitted to hydrolysis to afford 2-COOH. The inhibitory activity of these molecules against Sirt6 was assessed using a fluorogenic Sirt6 activity assay based on peptide demyristoylation (BPS Bioscience) at a concentration of 10 μM (Table 1).
The introduction of a methoxyethyl amide group (2-MeOEt) led to a decrease in activity (9% inhibition at 10 μM), consistent with the presence of a hydrophobic site at the linker attachment point. Surprisingly, larger substituents like phenylethyl (2-PhEt; 29% inhibition at 10 μM) and benzyl carboxamide (2-Bn; 30% inhibition at 40 μM), which were expected to enhance binding, decreased Sirt6 inhibition compared to 2-Pr (60% inhibition at 10 μM). This result suggests that the binding site is sensitive to larger modifications.
The experiments further indicated that the –CONH– group may play a role in Sirt6 binding, as the introduction of a piperidine group (2-Pip) was not well-tolerated (28% inhibition at 10 μM). Similarly, a phenyl derivative lacking the amide bond (3), synthesized in three steps from (R)-tert-Butyl (2-amino-2-phenylethyl)carbamate (Scheme S2 in the SI), exhibited poor inhibition of Sirt6 (3% inhibition at 10 μM). Most potent Sirt6 inhibition, comparable to that of 2-Pr, was achieved with molecules with phenyl (2-Ph) and cyclobutyl (2-CyBu) substituents (53% and 64% inhibition at 10 μM, respectively) on the carboxamide. Measurement of the IC50 values confirmed that the potency of 2-Ph (9.3 μM) and 2-CyBu (8.2 μM) was comparable to that of 2-Pr (8.9 μM). The IC50 value measured here for 2-Pr is slightly higher than measured previously (IC50 = 6.7 μM[20]) with the difference being within the experimental error associated with IC50 measurements (Figures S3–S5 in the SI). In conclusion, our investigation into the structure-activity relationship revealed that Sirt6 favors medium-sized carboxamide substituents at the linker position of compound 1.
To further improve this family of Sirt6 inhibitors, we explored the molecular details of Sirt6 inhibition by 2-Pr by solving the crystal structure of a human Sirt6/ADP-ribose/2-Pr complex. The structure was refined at 1.75 Å resolution to R/Rfree values of 16.7/20.2% (Figure 1a, Table 2) and the ligand was well defined by electron density (Figure S6a in the SI). The complex structure reveals that 2-Pr, designed as a NAD+-mimetic compound, occupies the Sirt6-specific acyl substrate binding channel rather than the NAD+ cofactor binding site, which is mostly occupied by ADP-ribose (Figure 1a and b; SI, Figure S6b and c). The 3-pyrinidyl-1,2,4-oxadiazole moiety of 2-Pr is positioned in the acetyl-Lys accommodating site. The diamide linker extends into the channel connecting acetyl-Lys site and the C-site, and the propylcarboxamide mimics the end of the myristoyl substrate tail, which curls toward the C-site. The 5-aminocarbonyl-uracil moiety is placed in the Sirt6-specific, wide surface opening of the acyl channel, which is largely covered by a larger helix in other Sirtuin isoforms. This insight in the 2-Pr binding mode rationalizes its isoform specificity, which should mostly stem from the modified uracil, and suggests a non-competitive inhibition mode with respect to NAD+ in contrast to what was predicted from prior docking analyses (see above).
Figure 1.
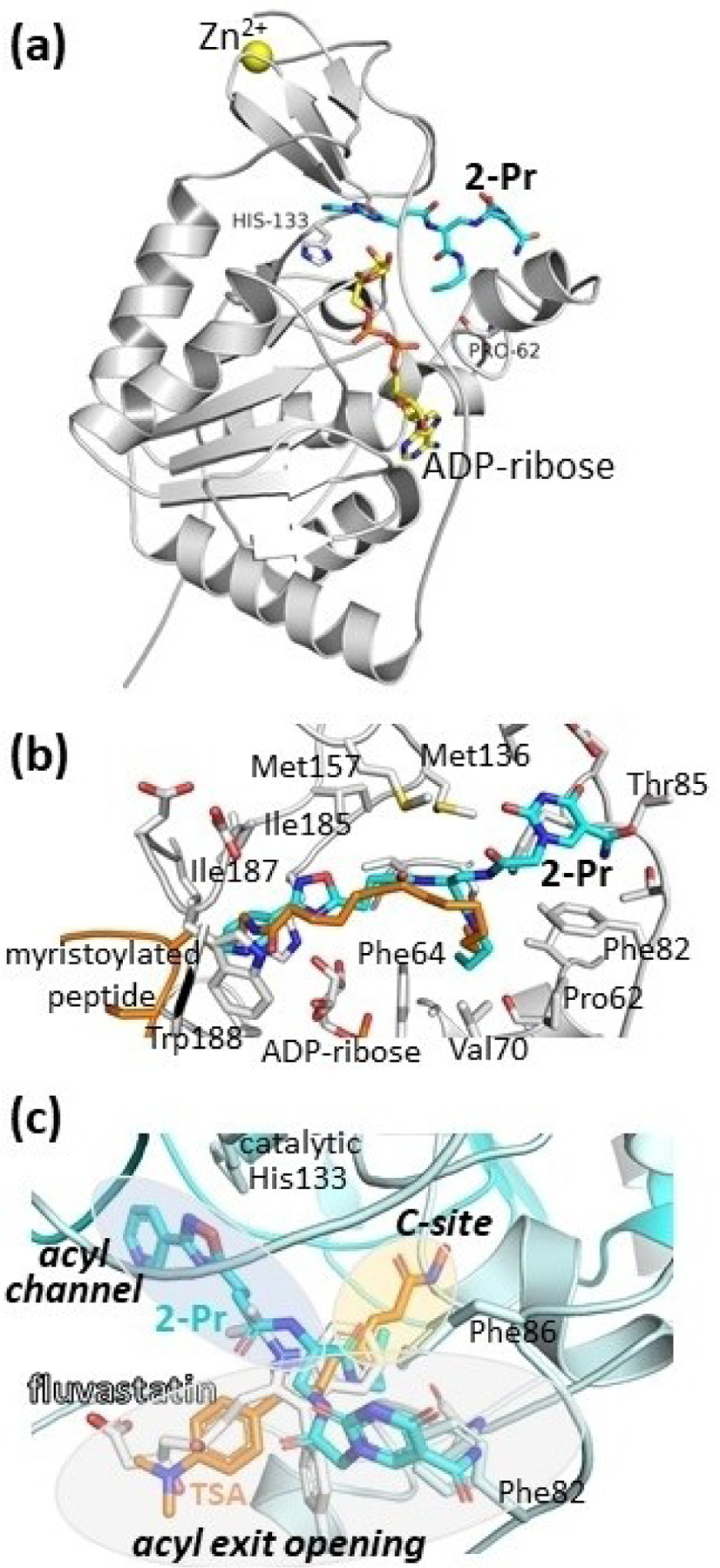
Crystal structure of a Sirt6/ADP-ribose/2-Pr complex. (a) Overall view, with His133 indicating the active site. (b) Closeup showing the Sirt6 residues interacting with 2-Pr (see Figure S6 in the SI for a scheme showing the complete interaction details). A Sirt6/myristoyl-peptide complex (PDB ID 3ZG6[8a]) indicates the acyl-Lys binding region. (c) Overlay of Sirt6/2-Pr with Sirt6 complexes (protein of these complexes not shown for clarity) with TSA (PDB: 6HOY[23]) and fluvastatin (PDB 6ZU4.[25] Binding pockets are highlighted and labeled in italics. An overlay also including CG (PDB 6QCJ[24]) and MDL- 801 (PDB 6XVG[26] is shown in SI Figure S6d.
Table 2.
Data collection and refinement statistics for crystal structure of a Sirt6/ADPr/2-Pr complex.
| Sirt6/ADPr/2-Pr | |
|---|---|
| Data collection[a] | |
| Space group | P63 |
| Cell dim. a = b, c (Å) | 91.47, 144.61 |
| Resolution (Å)[b] | 45.73–1.75 (1.86–1.75) |
| Unique Reflections | 68965 |
| I/σI[b] | 12.39 (0.89) |
| Redundancy[b] | 9.5 (9.2) |
| Completeness (%)[b] | 100 (99.9) |
| R meas [b] | 0.13 (2.63) |
| CC1/2[b] | 0.999 (0.349) |
| Refinement | |
| Resolution (Å)[b] | 45.73–1.75 (1.79–1.75) |
| No. reflections | 66815 |
| Rwork/Rfree (%) | 16.7/20.2 |
| Twin fractions -[c] | 0.57/0.43 |
| No. atoms | 4656 |
| B-factors | |
| Protein | 39.2 |
| Ligands and solvent | 38.4 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.010 |
| Bond angles (°) | 1.911 |
Data collected at BESSY beamline MX 14.1.[21]
Highest-resolution shell is shown in parentheses.
Twinning was detected through L-tests with POINTLESS (twin law −k, −h, −l) and twin fractions were determined during amplitude-based twin refinement with Refmac.
Importantly, the Sirt6/2-Pr complex structure reveals a binding mode that differs from all other Sirt6 modulators with known binding mode. The linker and propyl group of 2-Pr overlay well with the head groups of the Sirt6 activators MDL-801 and Fluvastatin and the Sirt6 inhibitors catechin gallate (CG) and Trichostatin A (TSA), which all fill the entrance to the C-site (Figure 1c; SI, Figure S6d). Only TSA enters even deeper into the C-site. All compounds have in common that they then point roughly out of the proximal acyl exit opening, interacting with different parts of its edge. 2-Pr shares this general mode but exploits a different region of the exit funnel edge, interacting with Phe82 and Phe86 (Figures 1b and c; S6d). This interaction should be dominant for the Sirt6 specificity (see above) and likely also contributes to its potency. Unique to 2-Pr, the compound also extends in the other direction, deep into the acetyl-Lys site (Figure 1c). This site is not filled by other Sirt6 ligands but is exploited by ligands to other Sirtuin isoforms, such as the inhibitor SirReal in Sirt2.[22] 2-Pr now shows how to add this pocket to the binding site of Sirt6 ligands exploiting the very prominent Sirt6 binding pocket at the C-site entrance.
An intriguing aspect of the Sirt6/2-Pr complex structure, in addition to the unexpected pyrimidine position and simultaneous use of acyl and Lys channel, is the positioning of the linker attachment site from the DNA-based screen toward the rather closed C-site. In DECL technology it is typically assumed that the linker points towards the solvent,[19a] and one would rather expect it to protrude out of the molecule via the wider acyl channel. The crystal structure corrects this assumption and now allows rational design of compounds. It is possible that the compound, with its long linker to the DNA, was able to enter its binding site via the NAD+ binding site rather than the wider acyl channel opening or acetyl-Lys exit, respectively. Indeed, the two Sirtuin core subunits are known to undergo a closure movement upon binding of either substrate or NAD+ cosubstrate,[4b,27] indicating a more open state for the NAD+ site in the apo state of Sirt6, which should allow such a binding route. An alternative explanation could be that the interaction of the molecule when attached to the DNA is different than a small molecule without the linker. The surprising binding mode of 2-Pr enabled us to rationalize the effects of compound modification on the potency of Sirt6 inhibition. The positioning of the inhibitor’s amide linker (–CONH–Pr) in the acyl binding site, which also accommodates part of the aliphatic linker of TSA, illustrates why medium-sized, hydrophobic modifications at this compound position, such as a CyBu group, are well tolerated, whereas larger groups such as phenyl groups with additional linkers are less well tolerated. In summary, the co-crystal structure of 2-Pr and Sirt6 provides detailed insights in pocket geometry, ligand pose, and binding interactions and reveals potential interaction partners for the design of improved Sirt6 modulators.
The complex structure indicates that modifications of the amide linker to simultaneously occupy the C site and acyl channel extension may have the potential to enhance potency and selectivity of our Sirt6 inhibitor class, but also other compound families. We were particularly intrigued by the prospect of introducing moieties that could extend into the C-site of Sirt6, emulating the interactions of NAM with protein residues within this pocket. Previous research has indicated that the inhibitory activity of TSA involves the binding of its hydroxamate group to this site, establishing interactions with amino acids responsible for recognizing the carboxamide group of NAM.[23] Docking calculations with a small set of derivatives encouraged us to attach hydroxamate groups to the phenyl and cyclobutyl rings of 2-Ph and 2-CyBu, respectively. These modified molecules (5 and 7; Table 3) were synthesized from their respective ester derivatives (4 and 6; Table 3) through treatment with hydroxylamine in the final step (Scheme S3 in the SI). Regrettably, the hydroxylamine-containing molecules failed to inhibit Sirt6-mediated demyristoylation at a concentration of 10 μM (Table 3). In contrast, the ester compounds 4 and 6 exhibited only slightly reduced affinity relative to the unmodified derivatives (2-Ph, 2-CyBu) indicating that substituents are tolerated, and that modification of these groups is a valid strategy for future lead optimization.
Table 3.
Effect of carboxamide derivatives of compound 1 on Sirt6 myristoylated activity.

| |||
|---|---|---|---|
| Cmpd | R = Carboxamides derivatives |
R1 = | Inhibition c = 10 μM |
| 4 |

|

|
29% |
| 5 |
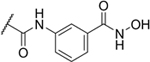
|

|
1% |
| 6 |
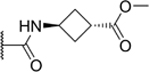
|
|
34% |
| 7 |

|

|
1% |
| 8 |

|

|
38% (IC50 = 26 μM)[a] pIC50 (4.6 ± 0.1) |
| Nicotinamide (1000 μM) | 92% | ||
Based on an inhibition curve ending with 60% inhibition at 50 μM compound; higher concentrations not tested.
We further explored the effect of modification of the carboxamide on the uracil ring. In the crystal structure, the amide group forms a hydrogen bond with Thr84, prompting our curiosity regarding whether alkylation of the carboxamide would disrupt this binding interaction. A derivative of compound 1 featuring a propylcarboxamide modification (8) was synthesized (Scheme S4 in the SI) and the potential of 8 to inhibit Sirt6 was assessed. A modest decrease in activity was observed, with a 38% inhibition of Sirt6 at 10 μM and an IC50 value of 26 μM based on an inhibition curve ending with 60% inhibition at 50 μM compound (higher concentrations not tested; Figure S7 in the SI). These findings suggest that while modifications at this position may not be privileged to enhance the potency of Sirt6 inhibitors, small alkyl groups are indeed tolerated.
Conclusions
In conclusion, our study revealed that the presented family of Sirt6 inhibitors adopt a binding mode that is distinct from those of other Sirt6 inhibitors. The molecules span the Sirt6-specific acyl substrate binding channel extending into the acetyl-lysine binding pocket and a shallow binding site at the acyl channel exit. The alkylcarboxamide groups occupy a hydrophobic channel towards the Sirt6 binding site for NAD+, which explains the observed preference for medium-sized hydrophobic substituents at this position. The insights obtained through structure elucidation and medicinal chemistry studies will be useful to guide the development of highly potent and selective Sirt6 inhibitors, and possibly also for the development of activators with improved potency.
Supplementary Material
Acknowledgements
We thank Paul Sebahar from the University of Utah Synthesis Core Facility for providing intermediate 2-COOH and the staff at BESSY for excellent technical support. NMR spectroscopy was performed using the Health Sciences Center Cores facility at the University of Utah. Accurate mass measurements were performed by the University of Utah, Department of Chemistry, Mass Spectrometry Facility. Open Access funding enabled and organized by Projekt DEAL.
Footnotes
Supporting information for this article is available on the WWW under https://doi.org/10.1002/cmdc.202400273
Supporting Information Summary
The authors have cited additional references within the Supporting Information.[28–36]
Conflict of Interests
CS is a member of the scientific advisory board of Ovibio.
Data Availability Statement
Coordinates and diffraction data for the Sirt6/2-Pr complex have been deposited with the wwPDB (https://www.rcsb.org/) under accession code 9G7H.
References
- [1].Wu QJ, Zhang TN, Chen HH, Yu XF, Lv JL, Liu YY, Liu YS, Zheng G, Zhao JQ, Wei YF, Guo JY, Liu FH, Chang Q, Zhang YX, Liu CG, Zhao YH, Signal. Transduct. Target Ther 2022, 7, 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Houtkooper RH, Pirinen E, Auwerx J, Nat. Rev. Mol. Cell Biol 2012, 13, 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Morris BJ, Free Radic. Biol. Med 2013, 56, 133–171. [DOI] [PubMed] [Google Scholar]
- [4].a) Hubbard BP, Sinclair DA, Trends Pharmacol. Sci 2014, 35, 146–154; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dai H, Sinclair DA, Ellis JL, Steegborn C, Pharmacol. Ther 2018, 188, 140–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Fiorentino F, Mai A, Rotili D, J. Med. Chem 2021, 64, 9732–9758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tasselli L, Zheng W, Chua KF, Trends Endocrinol. Metab 2017, 28, 168–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sanders BD, Jackson B, Marmorstein R, Biochim. Biophys. Acta 2010, 1804, 1604–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Jiang H, Khan S, Wang Y, Charron G, He B, Sebastian C, Du J, Kim R, Ge E, Mostoslavsky R, Hang HC, Hao Q, Lin H, Nature 2013, 496, 110–113; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pan PW, Feldman JL, Devries MK, Dong A, Edwards AM, Denu JM, J. Biol. Chem 2011, 286, 14575–14587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Feldman JL, Baeza J, Denu JM, J. Biol. Chem 2013, 288, 31350–31356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rahnasto-Rilla M, Kokkola T, Jarho E, Lahtela-Kakkonen M, Moaddel R, ChemBioChem 2016, 17, 77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Xu J, Shi S, Liu G, Xie X, Li J, Bolinger AA, Chen H, Zhang W, Shi PY, Liu H, Zhou J, Eur. J. Med. Chem 2023, 246, 114998; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) You W, Rotili D, Li TM, Kambach C, Meleshin M, Schutkowski M, Chua KF, Mai A, Steegborn C, Angew. Chem. Int. Ed. Engl 2017, 56, 1007–1011. [DOI] [PubMed] [Google Scholar]
- [12].Rahnasto-Rilla M, Tyni J, Huovinen M, Jarho E, Kulikowicz T, Ravichandran S, Ferrucci ABVL, Lahtela-Kakkonen M, Moaddel R, Sci. Rep 2018, 8, 4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Huang Z, Zhao J, Deng W, Chen Y, Shang J, Song K, Zhang L, Wang C, Lu S, Yang X, He B, Min J, Hu H, Tan M, Xu J, Zhang Q, Zhong J, Sun X, Mao Z, Lin H, Xiao M, Chin YE, Jiang H, Xu Y, Chen G, Zhang J, Nat. Chem. Biol 2018, 14, 1118–1126. [DOI] [PubMed] [Google Scholar]
- [14].Shang J, Zhu Z, Chen Y, Song J, Huang Y, Song K, Zhong J, Xu X, Wei J, Wang C, Cui L, Liu CY, Zhang J, Theranostics 2020, 10, 5845–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cen Y, Youn DY, Sauve AA, Curr. Med. Chem 2011, 18, 1919–1935. [DOI] [PubMed] [Google Scholar]
- [16].a) Damonte P, Sociali G, Parenti MD, Soncini D, Bauer I, Boero S, Grozio A, Holtey MV, Piacente F, Becherini P, Sanguineti R, Salis A, Damonte G, Cea M, Murone M, Poggi A, Nencioni A, Del Rio A, Bruzzone S, Bioorg. Med. Chem 2017, 25, 5849–5858; [DOI] [PubMed] [Google Scholar]; b) Parenti MD, Grozio A, Bauer I, Galeno L, Damonte P, Millo E, Sociali G, Franceschi C, Ballestrero A, Bruzzone S, Del Rio A, Nencioni A, J. Med. Chem 2014, 57, 4796–4804. [DOI] [PubMed] [Google Scholar]
- [17].Liu J, Zheng W, Org. Biomol. Chem 2016, 14, 5928–5935. [DOI] [PubMed] [Google Scholar]
- [18].Zhang Q, Chen Y, Ni D, Huang Z, Wei J, Feng L, Su JC, Wei Y, Ning S, Yang X, Zhao M, Qiu Y, Song K, Yu Z, Xu J, Li X, Lin H, Lu S, Zhang J, Acta Pharm. Sin. B 2022, 12, 876–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Franzini RM, Randolph C, J. Med. Chem 2016, 59, 6629–6644; [DOI] [PubMed] [Google Scholar]; b) Peterson AA, Liu DR, Nat. Rev. Drug Discov 2023, 22, 699–722; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gironda-Martinez A, Donckele EJ, Samain F, Neri D, ACS Pharmacol. Transl. Sci 2021, 4, 1265–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yuen LH, Dana S, Liu Y, Bloom SI, Thorsell AG, Neri D, Donato AJ, Kireev D, Schuler H, Franzini RM, J. Am. Chem. Soc 2019, 141, 5169–5181. [DOI] [PubMed] [Google Scholar]
- [21].Mueller U, Darowski N, Fuchs MR, Forster R, Hellmig M, Paithankar KS, Puhringer S, Steffien M, Zocher G, Weiss MS, J. Synchrotron Radiat 2012, 19, 442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rumpf T, Schiedel M, Karaman B, Roessler C, North BJ, Lehotzky A, Olah J, Ladwein KI, Schmidtkunz K, Gajer M, Pannek M, Steegborn C, Sinclair DA, Gerhardt S, Ovadi J, Schutkowski M, Sippl W, Einsle O, Jung M, Nat. Commun 2015, 6, 6263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].You W, Steegborn C, J. Med. Chem 2018, 61, 10922–10928. [DOI] [PubMed] [Google Scholar]
- [24].You W, Zheng W, Weiss S, Chua KF, Steegborn C, Sci. Rep 2019, 9, 19176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].You W, Steegborn C, ACS Med. Chem. Lett 2020, 11, 2285–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].You W, Steegborn C, Nat. Chem. Biol 2021, 17, 519–521. [DOI] [PubMed] [Google Scholar]
- [27].Moniot S, Shutkowski M, Steegborn C, J. Struct. Biol 2013, 182, 136–143. [DOI] [PubMed] [Google Scholar]
- [28].a) Kabsch W, Acta Crystallogr. D Biol. Crystallogr 2010, 66, 125–132; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sparta KM, Krug M, Heinemann U, Mueller U, Weiss MS, J. Appl. Crystallogr 2016, 49, 1085–1092. [Google Scholar]
- [29].Mccoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ, J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS, Acta Crystallogr. D 2011, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Emsley P, Cowtan K, Acta Crystallogr. D 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- [32].Murshudov GN, Vagin AA, Dodson EJ, Acta Crystallogr. D 1997, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- [33].Padilla JE, Yeates TO, Acta Crystallogr. D Biol. Crystallogr 2003, 59, 1124–1130. [DOI] [PubMed] [Google Scholar]
- [34].Beillard A, Bhurruth-Alcor Y, Bouix-Peter C, Bouquet K, Chambon S, Clary L, Harris CS, Millois C, Mouis G, Ouvry G, Pierre R, Reitz A, Tomas L, Tetrahedron Lett 2016, 57, 2165–2170. [Google Scholar]
- [35].a) Accetta A, Corradini R, Sforza S, Tedeschi T, Brognara E, Borgatti M, Gambari R, Marchelli R, J. Med. Chem 2009, 52, 87–94; [DOI] [PubMed] [Google Scholar]; b) Maslova AA, Matyugina ES, Snoeck R, Andrei G, Kochetkov SN, Khandazhinskaya AL, Novikov MS, Molecules 2020, 25, 3350–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Laskowski RA, Swindells MB, J. Chem. Inf. Model 2011, 51, 2778–2786. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates and diffraction data for the Sirt6/2-Pr complex have been deposited with the wwPDB (https://www.rcsb.org/) under accession code 9G7H.