

ABSTRACT
The estrogen signalling pathway is highly dynamic and primarily mediated by estrogen receptors (ERs) that transcriptionally regulate the expression of target genes. While transcriptional functions of ERs have been widely studied, their roles in RNA biology have not been extensively explored. Here, we reveal a novel biological role of ER alpha (ERα) in mRNA 3’ end processing in breast cancer cells, providing an alternative mechanism in regulating gene expression at the post-transcriptional level. We show that ERα activates poly(A) specific ribonuclease (PARN) deadenylase using in vitro assays, and that this activation is further increased by tumour suppressor p53, a factor involved in mRNA processing. Consistent with this, we confirm ERα-mediated activation of nuclear deadenylation by PARN in samples from MCF7 and T47D breast cancer cells that vary in expression of ERα and p53. We further show that ERα can form complex(es) with PARN and p53. Lastly, we identify and validate expression of common mRNA targets of ERα and PARN known to be involved in cell invasion, metastasis and angiogenesis, supporting the functional overlap of these factors in regulating gene expression in a transactivation-independent manner. Together, these results show a new regulatory mechanism by which ERα regulates mRNA processing and gene expression post-transcriptionally, highlighting its contribution to unique transcriptomic profiles and breast cancer progression.
KEYWORDS: Breast cancer, deadenylation, estrogen receptor alpha (ERα), mRNA 3’ end processing, PARN deadenylase, post-transcriptional regulation
Introduction
Breast cancer is the second most commonly diagnosed cancer among women, Bray et al. [1]. Long-term hormonal exposure and deregulation of factors involved in hormone and growth factor signalling pathways are main drivers for breast cancer development and progression. Estrogen is an essential hormone involved in mammary gland development and reproductive organ function in females, and signalling is predominantly mediated by estrogen receptors (ERs), we recommend the following reviews on estrogen receptor signalling mechanisms in breast cancer [2–5]. ERs are members of the nuclear receptor superfamily of transcription factors, whose activity is largely regulated by their binding to estrogen. While there are two well-characterized ERs: ERα and ERβ, approximately 70% of breast tumours express ERα and are classified as ER-positive (ER+) [2].
Functional studies of ERα have largely focused on its DNA binding activity and transcriptional regulation. Upon binding to estrogen, ERα dimerizes and translocates to the nucleus whereby it can bind directly to estrogen response elements (EREs) within promoter regions of target genes or indirectly to cofactors, inducing a cascade of events that regulate transcriptional activation or repression of target genes [2–6]. Additionally, estrogen non-genomic and estrogen-independent activation of ERα have been described [7,8] and more recently, its role as a non-canonical RNA binding protein (RBP) has emerged [9–11]. Studies have shown that ERα can interact with target mRNAs via the RNA binding motif in the hinge region [12–14], and it has been suggested that RNAs might bridge ERα and its interactors to form a complex [9]. Moreover, ERα’s RBP function was shown to be critical for breast cancer progression and is independent of its known DNA binding activity [11]. More specifically, Xu et al. [11] demonstrated that ERα binds target transcripts involved in the adaptive response to stress by their 3’ untranslated regions (3’UTRs) during tumour development, resulting in their alternative splicing and translation. To date, ERα’s potential role in mRNA 3’ end processing has not been explored and is the focus of this study.
Eukaryotic mRNAs undergo extensive processing prior to their nuclear export as mature mRNAs. With the exception of histone mRNAs, all mRNAs undergo cleavage and polyadenylation steps at their 3’ end, determining their fate, as poly(A) tails are crucial in regulating mRNA stability, translation initiation and subcellular localization [15,16]. Under different cellular conditions, the highly regulated process of deadenylation, or removal of poly(A) tails, occurs to control mRNA steady-state levels and gene expression [17,18]. Therefore, 3’ end processing regulates gene expression through balancing the biosynthesis and turnover of mRNAs [16]. In mammalian cells, deadenylation is the initial and rate-limiting step in mRNA decay, ultimately affecting the transcriptome [18]. Deadenylases catalyse the degradation of mRNAs in the 3’ −5’ direction. While there are three predominant deadenylases in eukaryotes, poly(A) specific ribonuclease (PARN) is the major 3’ exoribonuclease identified in the nucleus [19].
We have previously shown that PARN deadenylase can functionally interact with factors involved in mRNA 3’ processing such as cleavage stimulation factor 50 [20], tumour suppressors BRCA1/BARD1 and p53 [21,22], Argonaute-2 [23], nucleolin [24], microtubule associate protein tau, and prolyl isomerase Pin 1 [25]. We identified dynamic interactions between these factors that resulted in either cleavage/polyadenylation inhibition and/or PARN-mediated deadenylation activation, affecting gene expression under varying cellular conditions. Interestingly, we described a feedback loop between PARN and p53 whereby under nonstress conditions, PARN controls TP53 mRNA stability and UV-induced increase in p53 activates PARN deadenylase to regulate gene expression during the DNA damage response (DDR) in a transactivation-independent manner [21,23]. p53 has also been functionally associated to the ERα regulatory pathway [26–28].
Here, we show that ERα can form complex(es) with factors involved in nuclear mRNA 3’ end processing, specifically PARN and p53, and activates PARN-mediated nuclear deadenylation in MCF7 and T47D ERα(+) breast cancer cell lines. We confirm the ERα-mediated activation of PARN using in vitro deadenylation assays with recombinant proteins and by ERα depletion and selective estrogen receptor degrader (SERD) fulvestrant treatment. Lastly, we validate common mRNA targets of ERα and PARN involved in tumorigenesis, cell invasion and metastasis in a panel of breast cancer cells, supporting the functional overlap of these factors in regulating transcript levels in a transactivation-independent manner. Together, we report a novel role for ERα in regulating mRNA 3’ end processing contributing to changes in gene expression.
Results
Early response to estrogen treatment involves activation of nuclear deadenylation in MCF7 cells
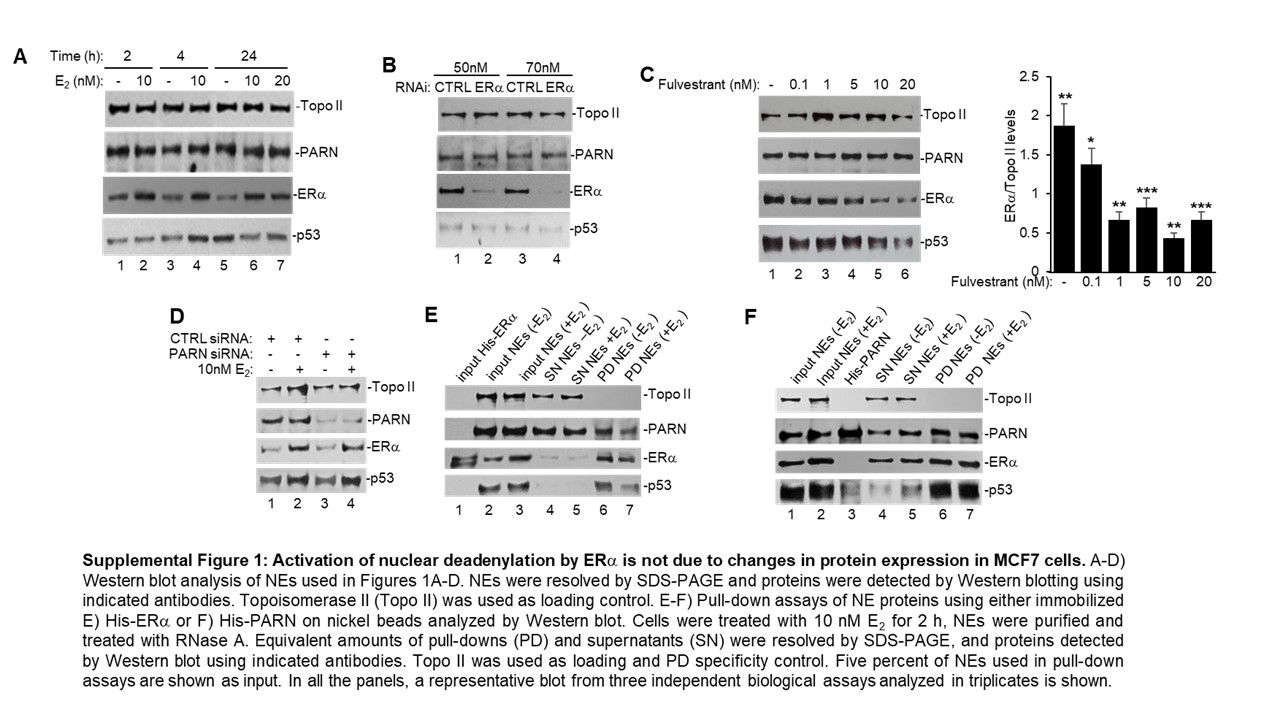
Recent studies have shown that ERα can bind to the 3’UTR of target transcripts involved in adaptive stress responses and regulate mRNA biology, such as alternative splicing and translation [11]. Here, we investigated whether the estrogen signalling pathway plays a regulatory role in mRNA 3’ end processing, specifically deadenylation. We first performed in vitro deadenylation assays using a radiolabeled/capped L3(A30) RNA substrate as previously described [21] and nuclear extracts (NEs) from MCF7 cells treated with 17β-estradiol (E2) (Figure 1A). To ensure that changes in nuclear deadenylation were due to E2 treatment and not from tissue culture conditions, we serum starved cells for 24 h in a phenol red-free medium and used 10% charcoal stripped FBS for treatment. Interestingly, after 2 h of E2 treatment, nuclear deadenylation was induced (Figure 1A, compare lanes 2 to 1) at a timepoint where there is an increase in ERα but not p53 protein levels (Supplemental Figure S1A). A further increase in nuclear deadenylation was observed after 4 h of treatment (compare lanes 4 to 3), when an increase in both ERα and p53 was detected (Supplemental Figure S1A). PARN deadenylase expression did not change at either timepoint with treatment. While at 24 h we observed high levels of deadenylation (Figure 1A, lane 5) and induction of p53 protein expression in non-treated cells (Supplementary Figure S1A), 24-h E2 treatment reduced deadenylation (Figure 1A, lane 6) and p53 expression (Supplementary Figure S1A). The high basal deadenylation observed in untreated cells is consistent with the previously described stress-induced increase in deadenylation [20,21], which may be due to serum starvation and not a response of the estrogen signalling pathway. Therefore, we focused all subsequent studies on the 2 h early treatment timepoint. This data is the first to show that E2 treatment induces nuclear deadenylation in MCF7 breast cancer cells at early time points, suggesting that the estrogen signalling pathway plays a role in mRNA 3’ end processing.
Figure 1.
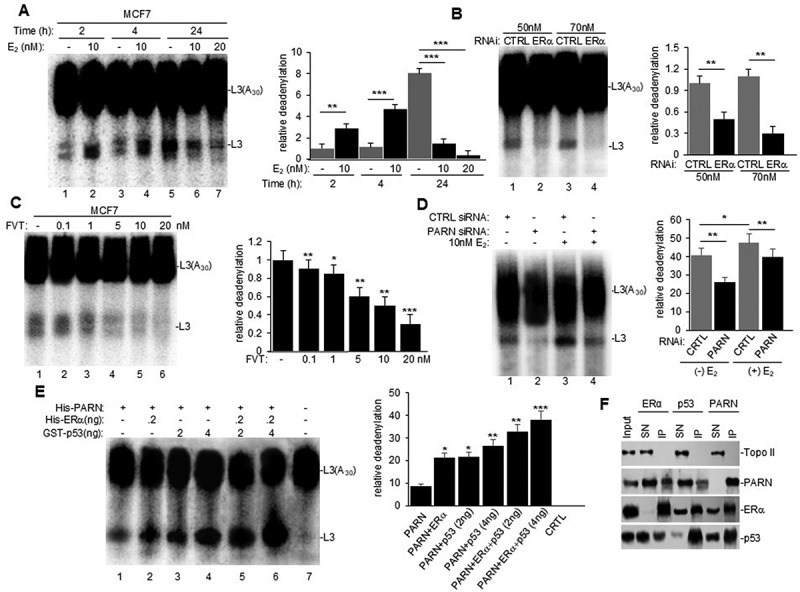
Estrogen receptor alpha (ERα) is an activator of PARN-mediated nuclear deadenylation in MCF7 (ERα+) cells. (A) nuclear extracts (NEs) for cells treated with different concentrations of 17β-estradiol (E2) for the indicated times were used in in vitro deadenylation assays with radiolabeled capped L3(A30) RNA substrate. Purified RNA was analysed by denaturing PAGE. Left panel: representative deadenylation reactions from three independent biological assays are shown. Positions of the polyadenylated RNA L3(A30) and the L3 deadenylated product are indicated. Right panel: bar graph of relative deadenylation (RD) is shown. (B–C) in vitro deadenylation assays using NEs from cells treated with (B) control (CTRL) or ERα siRNA for 24 h or (C) with increasing concentrations of fulvestrant for 2 h (FVT) were performed and analysed as in (A). (D) MCF7 cells were treated with either CTRL or PARN siRNA and subsequently treated with vehicle or E2. NEs were used for in vitro deadenylation as performed and analysed in (A). E) Cell-free deadenylation assays were performed in the presence of radiolabeled capped L3(A30) RNA substrates, limiting amount of his-PARN deadenylase and his-ERα and increasing amounts of GST-p53. Conditions for deadenylation assays were performed as in (A). F) NEs from untreated cells were used in endogenous reciprocal co-immunoprecipitation (e-ip) assays with polyclonal ERα, PARN, or p53 antibodies. NEs were treated with RNase A. Equivalent amounts of pellets (IP) and supernatants (SN) were resolved by SDS-PAGE, and proteins were detected by Western blot. Topo II was used as loading and IP specificity control. Ten percent of the NEs used in the e-ip assays are shown as input. All figures show representative deadenylation reactions and Western blot analyses from at least three independent biological assays analysed by triplicate (n = 3). Experiments with two groups were analysed using two-tailed unpaired Student’s t-test. The p-values are indicated as *(<0.01), **(<0.001) and ***(<0.0001).
ERα is an activator of PARN-mediated nuclear deadenylation in MCF7 cells
Next, we determined whether E2-mediated activation of nuclear deadenylation depended on ERα expression. We analysed NEs from MCF7 cells that were depleted of ERα by siRNA treatment or treated with the SERD fulvestrant, which prevents ERα nuclear localization and function [2,10]. In vitro deadenylation reactions using NEs from each condition showed that both siRNA-mediated depletion of ERα (Figures 1B) and fulvestrant-mediated inhibition of ERα function (Figures 1C) resulted in dose-dependent reductions of nuclear deadenylation. Western blot analysis confirmed decreases in ERα levels by siRNA (Supplementary Figure S1B) and fulvestrant (Supplementary Figure S1C) treatments, but neither treatment affected PARN protein levels. A significant decrease in p53 levels was detected only at the highest concentration of fulvestrant. Taken together, these results suggest that ERα is an activator of nuclear mRNA deadenylation in MCF7 cells independently of estrogen treatment.
As PARN is the major nuclear deadenylase identified in mammalian cells, we investigated whether this observed estrogen pathway-mediated induction of deadenylation in nuclear fractions is dependent on PARN deadenylase expression. Consistent with Devany et al. [21], siRNA-mediated depletion of PARN resulted in a decrease in nuclear deadenylation in MCF7 cells when compared to CTRL siRNA treatment (Figure 1D, compare lanes 1 to 2). Interestingly, the addition of E2 treatment with PARN depletion decreased the estrogen-induced activation of deadenylation (compare lanes 3 and 4). Western blot analysis confirmed siRNA-mediated knockdown of PARN expression in both non-treated and E2 treated conditions (Suplementary Figure S1D). While we cannot discard the possibility that other nuclear deadenylase complexes might also be involved, our results indicate that the estrogen-mediated activation of nuclear deadenylation is dependent on PARN deadenylase expression. Moreover, we further confirmed that PARN deadenylase activity was activated by ERα using in vitro deadenylation assays. As in Cevher et al. [20], cell-free assays were performed using limiting amounts of recombinant His-PARN and recombinant His-tagged full-length ERα and/or increasing amounts of GST-tagged p53 (Figure 1E). Consistent with previous studies, increasing amounts of GST-p53 activated His-PARN deadenylase activity [21]. Addition of full-length ERα-induced PARN deadenylase activity, and this activity was further increased by p53 (Figure 1E), demonstrating an additive effect of ERα and p53 in activating nuclear deadenylation. Together, these results indicate that ERα activates PARN-mediated deadenylation and suggest a novel transactivation-independent role/function of ERα.
ERα, p53 and PARN form a complex in MCF7 cells independently of estrogen treatment
To further analyse the role of ERα in PARN-mediated nuclear deadenylation, we examined the physical association of ERα with PARN deadenylase and p53 through reciprocal endogenous co-immunoprecipitation (co-IP) assays using NEs from MCF7 cells and specific antibodies against ERα, PARN, p53 (Figure 1F). Extracts were treated with RNase A to verify that the interactions were not due to an RNA tethering effect. The reciprocal co-IPs revealed that ERα, PARN and p53 can form (a) complex (es) in samples from MCF7 cells independently of estrogen treatment (Figure 1F). Consistent with this, complementary pull-down assays using recombinant protein derivatives and NEs from MCF7 showed that His-ERα pulled down PARN and p53 (Supplementary Figure S1E) and His-PARN pulled-down ERα and p53 (Supplementary Figure S1F). Together, these results indicate that ERα interacts with PARN and p53 to form (a) complex(es) in nuclear fractions independently of estrogen treatment, and this may be a mechanism that activates PARN-mediated deadenylation.
ERα expression in breast cancer cells correlates with higher PARN-mediated nuclear deadenylation
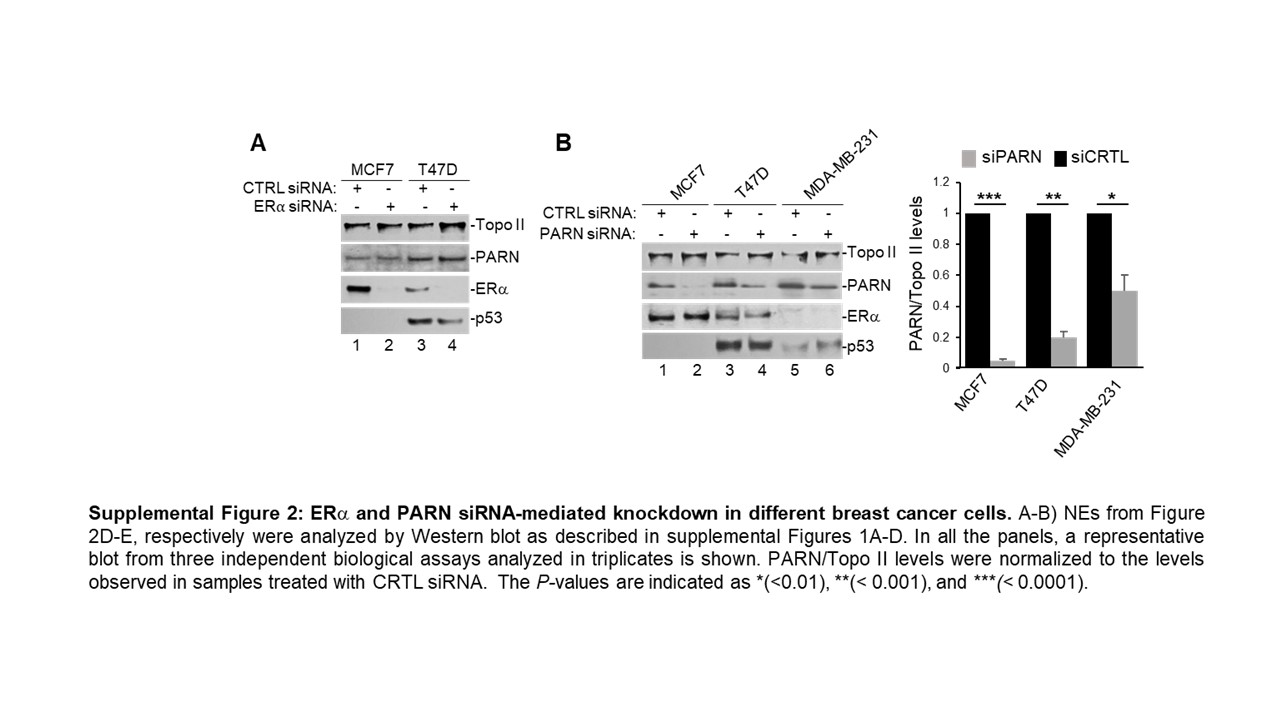
To further understand the ERα-mediated nuclear activation of deadenylation, we analysed NEs from a panel of breast cancer cell lines: MCF7 (ERα+, WT p53), T47D (ERα+, 580C > T mut p53) and MDA-MB-231 (ERα-, 839 G>A mut p53), and performed in vitro deadenylation assays as described above (Figure 2A). As p53 and ERα activities are mutually regulated (reviewed in [29]), these cell lines allow us to distinguish p53 and ERα effect on nuclear deadenylation. Western blot analysis confirmed ERα expression levels in MCF7 and T47D and its absence in MDA-MB-231 cells (Figure 2B). Additionally, both WT and mutant p53 were detected by immunoblot. Interestingly, we observed differential expression of the main nuclear deadenylase PARN when compared to topoisomerase II (Topo II) loading control, with MDA-MB-231 expressing the highest levels compared to MCF7 and T47D (Figure 2B and inputs in 2F-H). As higher PARN levels result in higher nuclear deadenylation, to analyse whether ERα activates nuclear deadenylation in this cell-line panel, we normalized the levels deadenylation to PARN levels. Interestingly, when nuclear deadenylation levels (Figure 2A) were normalized to PARN expression (Figure 2B), samples from ERα(+) cells (MCF7 and T47D) showed higher nuclear deadenylation than ERα(-) cells (MDA-MB-231) (Figure 2C). These results indicate the ERα expression plays a role in activating nuclear deadenylation and that p53 mutant (839 G>A) expressed in MDA-MB-231 cells cannot rescue PARN-mediated deadenylation. Confirming ERα-mediated activation of nuclear deadenylation, the siRNA-mediated depletion of ERα (Supplementary Figure S2A) decreased nuclear deadenylation in MCF7 and T47D cells (Figure 2D).
Figure 2.
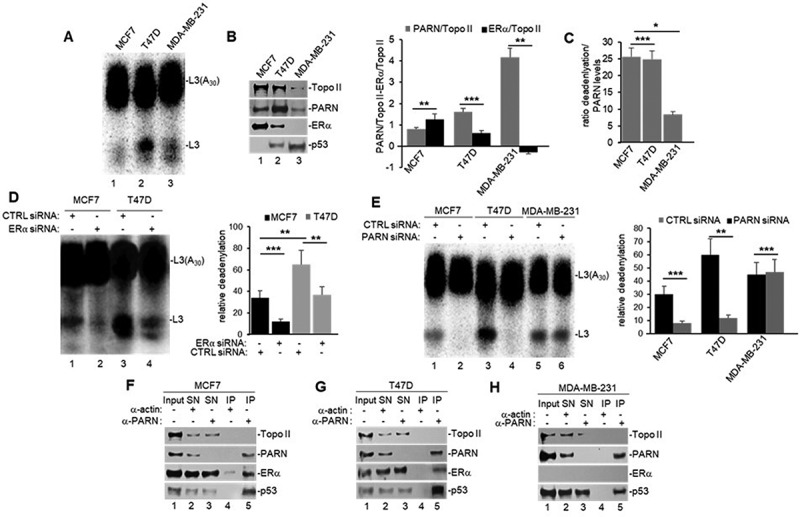
ERα activates PARN-mediated nuclear deadenylation in (ERα+) cells (MCF7 and T47D), but not (ERα-) cells (MDA-MB-231). A) in vitro deadenylation assay and B) Western blot analysis using NEs from untreated breast cancer cells, prepared and analysed as in Figure 1A. Quantifications of PARN or ERα expression in B) is shown in a bar graph and normalized to Topo II loading control. C) normalization of PARN-mediated deadenylations shown in A) to PARN protein levels shown in B) for each cell line. D) in vitro deadenylation assay using NEs from cells treated with either CTRL or ERα siRNA for 24 h were performed as in 2A. E) in vitro deadenylation assay using NEs from breast cancer cells treated with either CTRL or PARN siRNA were analysed as in 2A. F–H) NEs from breast cancer cells were used in endogenous co-IP assays with anti-PARN and IgG antibodies, and performed as in Figure 1E. All figures show representative deadenylation reactions and Western blot analyses from at least three independent biological assays analysed by triplicate (n = 3). Experiments with two groups were analysed using two-tailed unpaired Student’s t-test. The p-values are indicated as *(<0.01) and ***(<0.0001).
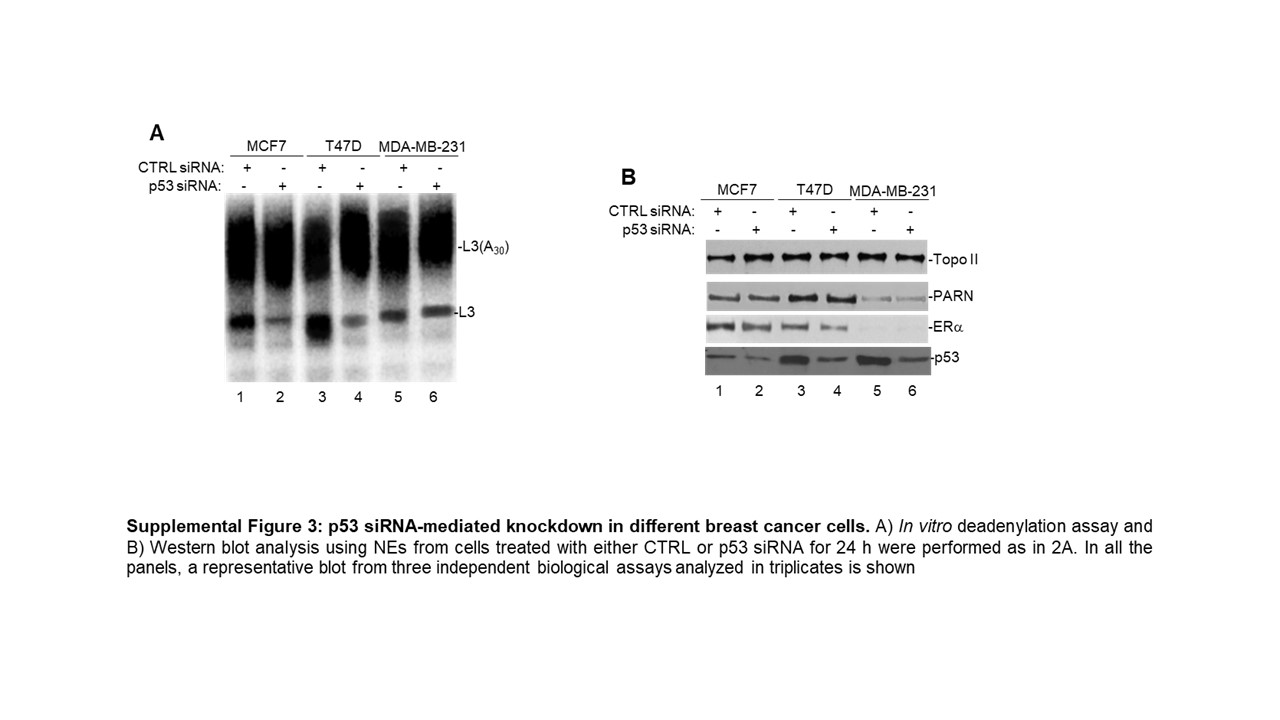
To confirm that PARN is the deadenylase involved in this ERα-mediated nuclear activation of deadenylation, we performed siRNA-mediated knockdown of PARN in this panel of breast cancer cells. PARN knockdown decreased nuclear deadenylation in NEs from MCF7 and T47D cells, however no changes were observed in samples from MDA-MB-231 (Figure 2E), indicating that the basal levels of nuclear deadenylation observed in MDA-MB-231 might be PARN independent. Consistent with this, siRNA-mediated knockdown of p53 decreased the levels of nuclear deadenylation in samples from MCF7 and T47D cells but not from MDA-MB-231 cells (Suplementary Figure S3SA), suggesting that mutant p53 expressed in T47D can activate PARN-mediated deadenylation. Western blot analysis confirmed PARN (Supplementary Figure S2B) and p53 (Supplementary Figure S3B) knockdowns in all three cell lines. Additionally, endogenous co-IPs confirmed the nuclear complex(es) formation of ERα, PARN and p53 in MCF7 and T47D (Figures 2F–G) and of PARN and p53 in MDA-MB-231 (Figure 2H), consistent with the previously described direct interaction of PARN and p53 in colorectal cancer cells [21]. Together, these results indicate that ERα can activate PARN-mediated nuclear deadenylation in ERα(+) breast cancer cells. While further studies are necessary to determine the details of this mechanism, our data indicate that nuclear deadenylation in ERα(-) cells may rely on nuclear deadenylases other than PARN.
ERα and PARN regulate expression of common mRNA targets involved in cell invasion, metastasis and angiogenesis
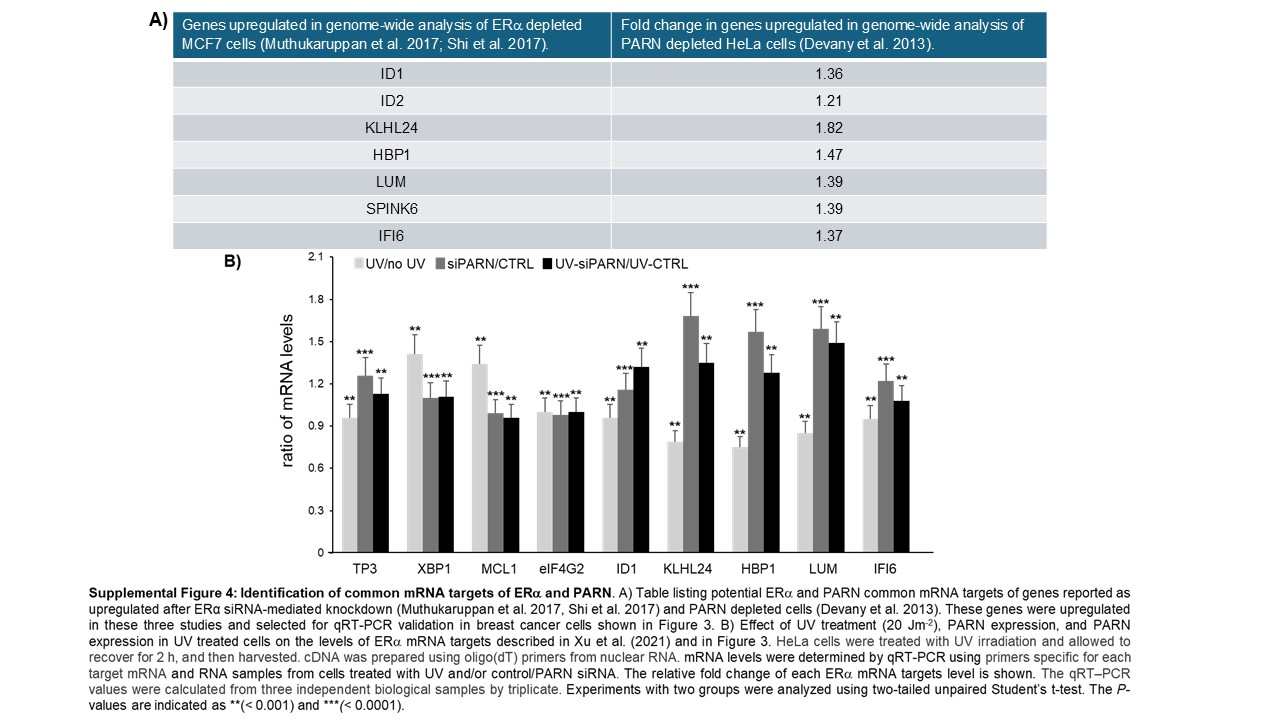
Our functional assays indicate that ERα can form complex(es) with mRNA 3’ processing factors and regulate nuclear PARN deadenylase activity in ERα(+) breast cancer cells (Figures 1–2), suggesting an overlapping role of ERα and PARN in regulating gene expression in a transactivation-independent manner. To further explore this, we identified common mRNA targets of ERα and PARN and validated their expression on the panel of breast cancer cells used in Figure 2. As PARN is a deadenylase and decreases the steady-state levels of its targets [21], we focused our study to identify genes that were upregulated upon PARN and ERα depletion. We analysed published datasets by Muthukaruppan et al. [30] and Shi et al. [31] to identify ERα mRNA targets and a microarray analysis by Devany et al. [21] to identify PARN mRNA targets [21,30,31]. We identified seven potential common targets comparing these datasets under ERα/PARN regulation: KLHL24, HBP1, ID1, ID2, SPINK6, LUM and IFI6 (Supplementary Figure S3A). Interestingly, these upregulated targets are AU-rich element (ARE)-containing mRNAs, which is a characteristic of PARN targets [17,18].
We next validated the expression of the identified mRNA targets by using nuclear RNA samples of cells depleted of ERα and PARN expression and analysed their steady-state levels by qRT-PCR. siRNA-mediated depletion of ERα resulted in a reduction of the endogenous ESR1 levels in MCF7 and T47D (Figure 3A–B, respectively, left panels) and undetectable levels as expected in MDA-MB-231, whereas siRNA-mediated depletion of PARN resulted in a reduction of endogenous PARN levels in all three cell lines (Figure 3A–C, right panels). ERα depletion in MCF7 and T47D resulted in upregulation of ID1, KLHL24, HBP1, LUM and IFI6 mRNA levels (Figure 3A–B, left panels). Lack of ERα expression in MDA-MB-231 resulted in detectable basal expression of ID1, KLHL24, HBP1, LUM and IFI6 mRNA levels (Figure 3C, left panel). PARN depletion also showed upregulation of ID1, KLHL24, HBP1 and LUM mRNAs (Figure 3A–C, right panels). While these results validated the common mRNA targets of PARN and ERα previously identified in genome-wide studies [21,30,31], some variations were observed in different cell lines after PARN and ERα depletion. For example, upregulation of IF6 was observed only in T47D and MDA-MB-231, upregulation of ID2 was observed only in MCF7 and MDA-MB-231, and SPINK6 was downregulated in all three cell lines. Interestingly, ID1, KLHL24, HBP1 and LUM play roles in tumorigenesis, cell invasion and metastasis in breast cancer [32–44]. Future studies are necessary as we cannot discard the possibility that the variations among these cell lines are also due to the gain or loss of functions of mutant p53 in this PARN-mediated regulation of gene expression. Together, these results indicate a functional overlap of ERα and PARN in the regulation of the steady-state levels of common mRNA targets under different cellular conditions and in different breast cancer cell types, providing further evidence for a transactivation-independent mechanism in regulating gene expression by ERα.
Figure 3.
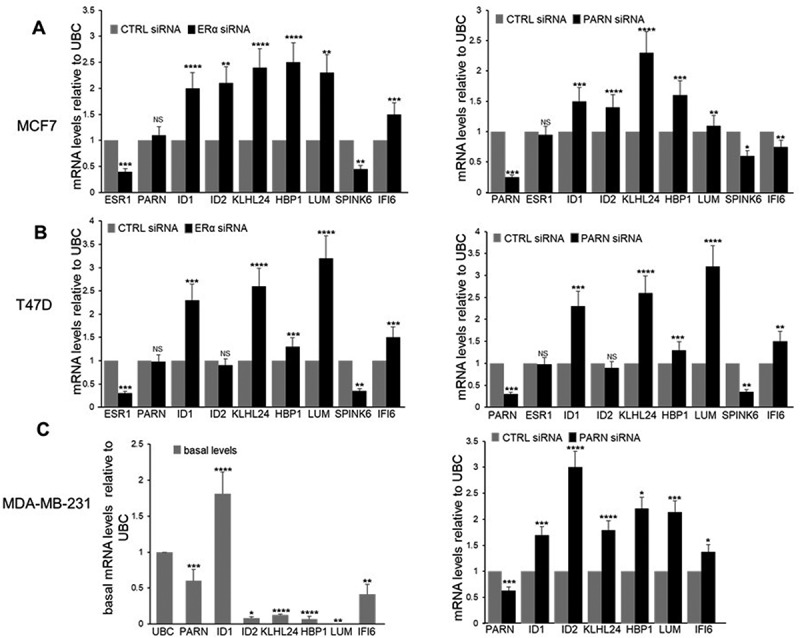
Common mRNA targets of ERα and PARN suggests functional overlap in regulating nuclear deadenylation and gene expression in a cell specific manner. A-C) endogenous common mRNA transcripts of ERα and PARN analysed by qRT-PCR using RNA samples from breast cancer cells depleted of ERα (A–B, left panels) or PARN (A–C, right panels) by siRNA treatment. The levels of transcripts observed for each condition were normalized to the levels of ubiquitin C (UBC) from cells treated with control (CTRL) siRNA. Fold changes were calculated using the ΔΔCT method. Left panel, C) basal endogenous transcripts analysed by qRT-pcr using RNA samples from untreated ERα(-) breast cancer cells were normalized to UBC. Representative qRT-PCR analysis from at least three independent biological samples analysed as triplicates is shown. Experiments with two groups were analysed using two-tailed unpaired Student’s t-test. The p-values are indicated as *(<0.01), **(<0.001), ***(<0.0001) and ****(<0.00001).
Discussion
This is the first report of a novel ERα function in regulating post-transcriptional activity of PARN-deadenylase in breast cancer cells, thereby providing a transactivation-independent mechanism to control gene expression. Based on these data, we propose a mechanistic working model whereby ERα affects gene expression under non-stress conditions by regulating PARN deadenylase in ERα(+) breast cancer cells either by recruiting ERα to mRNA 3’ processing complex(es), including PARN and p53, or by direct binding of ERα to the 3’ UTR of mRNA targets and recruitment of these mRNA 3’ processing factors (Figure 4). Consistent with this idea, studies have shown that ERα binds to the 3’UTR region of target RNAs via its RNA binding motif located in the hinge region, and ERα has been described as a non-canonical RBP helping mediate RNA–protein complexes in the nucleus of breast cancer cells [9,11–14]. Although mechanistic details of these ERα-RNA binding functions in mRNA metabolism have not been described, Xu et al. [11] demonstrated that this binding mediates alternative splicing of XBP1 and translation of eIF4G2 and MCL1 mRNAs, which are two cellular functions controlled by mRNA deadenylation [16].
Figure 4.
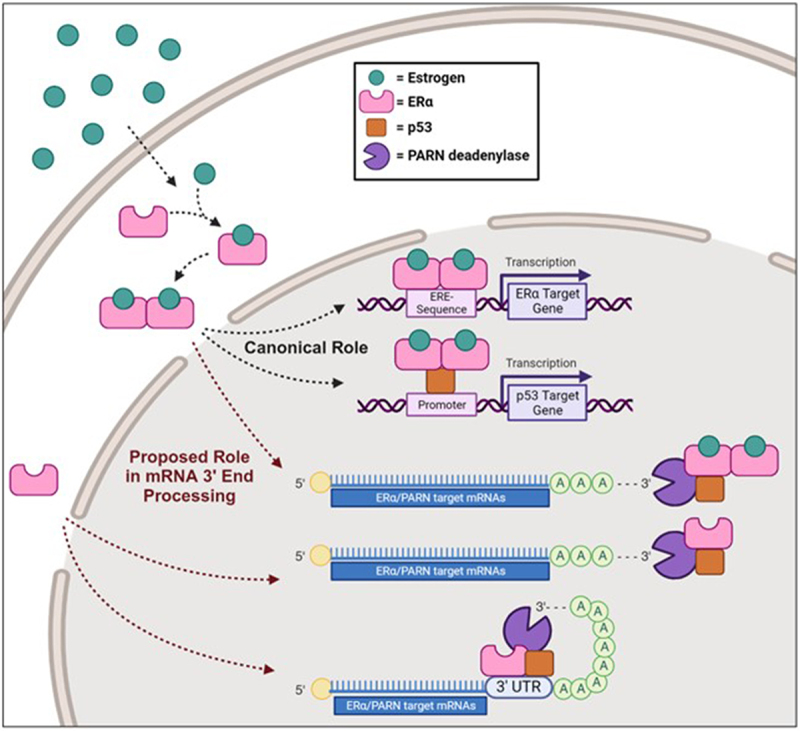
Proposed mechanism of ERα in PARN-mediated deadenylation. In the canonical signalling pathway, estrogen binds to ERα in the cytosol, leading to its dimerization, nuclear translocation and activation of transcription to regulate gene expression. More specifically, ERα can bind either directly to estrogen response elements (EREs) within the promoter region of its target genes to directly regulate transcription, or to co-factors, to indirectly regulate transcription of its targets. Here we show p53 as an example of a cofactor. We propose a novel transactivation-independent mechanism of ERα to regulate gene expression by activating PARN deadenylase. In ERα(+) breast cancer cells, ERα can form (a) complex(es) with 3’ processing factors, such as PARN and p53, and activate nuclear deadenylation of its mRNA targets. Alternatively, ERα might bind directly to the 3’UTR of these mRNA targets resulting in the recruitment of mRNA 3’ processing factors and activation of PARN deadenylase activity.
Analysing genome-wide studies [21,30,31], we identified and then validated ID1, KLHL24, HBP1 and LUM as common mRNA targets of ERα and PARN on a panel of breast cancer cells under non-stress conditions (Figure 3), supporting the functional overlap of ERα and PARN in regulating common transcript levels. ID1 encodes for an inhibitor of DNA binding 1 protein, which might be a potential prognostic marker and therapeutic target for breast cancer as its expression is associated with chemoresistance, tumorigenesis, invasiveness and metastasis [38,40,42,43]. KLHL24 encodes a cullin 3–RBX1 ubiquitin ligase substrate receptor, which is differentially expressed in breast cancer under varying conditions suggesting involvement in an autophagy signature and promoting tumorigenesis [32,37,39]. HBP1 encodes the HMG-box protein 1 transcription factor that might play a role as a tumour suppressor in breast cancer and in miRNA-mediated triple negative breast cancer cell proliferation [33,41,44]. LUM encodes lumican, a proteoglycan that exhibits anticancer activity in breast cancer affecting cell adhesion, inhibiting metastatic features of epithelial mesenchymal transition cells and attenuating proliferation, migration and invasion [34–36]. As these mRNA targets have been described to play roles in tumorigenesis, cell invasion and metastasis in breast cancer, together our data further provides a functional connection between mRNA 3’ end processing and breast cancer progression. Although subsequent studies are warranted to identify additional mRNA targets, the data presented here provide new insights into post-transcriptional regulation of steady-state levels and gene expression of specific transcripts by ERα through its role in nuclear deadenylation. The identity of the mRNA targets regulated by the ERα/PARN duo might be cell-specific and depend on specific genetic backgrounds as well as cellular conditions.
Xu and colleagues [11] demonstrated that ERα binds target transcripts involved in the adaptive response to stress during tumour development modifying their mRNA by processes related to deadenylation [15,16]. Consistent with that study, our microarray data [21] reveals an increase in XBP1 and MCL1 mRNA levels after UV treatment that is lost after PARN depletion (Supplementary Figure S4), suggesting that PARN deadenylase plays a role in this stress-induced response. Interestingly, the levels of the ERα/PARN target transcripts described in this study (IDl1, KLHL24, HBP1, LUM and IF6) decrease after UV treatment, and this is reversed by PARN depletion. However, under stress conditions, p53 might play a more relevant role in nuclear deadenylation as ERα induces its expression [26] and stress-induced p53 further activates PARN deadenylation [21,23]. While future studies are necessary to address whether these results reflect a direct or indirect role of PARN in controlling the mRNA levels of these genes after UV treatment, together these results further support the connection between the ERα pathway and mRNA processing in stress conditions.
Our study demonstrated that ERα can bind to mRNA 3’ processing factors PARN and p53 resulting in the activation of PARN-mediated deadenylation in ERα(+) breast cancer cells under non-stress conditions, and that common mRNA targets involved in breast cancer cell invasion and metastasis can be regulated by both ERα and PARN. Together, these data further support the functional overlap of ERα/PARN/p53 regulating gene expression in in a transactivation-independent manner. This is also consistent with previously described functional interactions between p53 and ERα [26–28]. While future studies are needed to further understand ERα function in RNA biology, this current study indicates that ERα not only regulates gene expression transcriptionally but also post-transcriptionally. As 70–80% of breast cancers are identified as ER(+) and deadenylation is a widespread mechanism controlling gene expression, this novel ERα regulatory mechanism of gene expression will help us comprehend unique breast cancer profiles to improve individualized treatments and therapies.
Materials and methods
Tissue culture methods
MCF7 and MDA-MB-231 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with sodium pyruvate (Corning) supplemented with 10% fetal bovine serum (FBS-ThermoFisher) and 1% penicillin/streptomycin (PS, ThermoFisher). T47D cells were cultured in RPMI-1640 medium (Corning) supplemented with 0.2 U/mL of human insulin (Sigma), 10% FBS and 1% PS. Serum starvation culture consisted of phenol red-free DMEM and 1% PS without FBS. 17-β estradiol (Sigma Aldrich), Fulvestrant (SelleckChem, cat# S1191) or DMSO (FisherSci) vehicle were prepared in phenol red-free DMEM supplemented with 10% charcoal-stripped FBS and 1% PS.
Compounds, treatments and DNA damaging agent
MCF7, T47D and MDA-MB-231 cells were plated in 10 cm dishes, allowed to adhere overnight, serum starved for 24 h, and treated with 17-β estradiol (Sigma Aldrich), Fulvestrant (SelleckChem, cat# S1191) or DMSO (FisherSci) vehicle for times outlined in each figure. Nuclear extracts were prepared after indicated time points and used for deadenylation and immunoblotting assays. Cells were treated with UV irradiation (20 Jm−2) and allowed to recover for 2 h before microarray analysis as in [21].
ERα, PARN and p53 siRNA-mediated depletion
Lipofectamine RNAiMAX (ThermoFisher) was used according to the manufacturer’s instructions to deliver 50 nM ERα siRNA (Horizon Discovery, Dharmacon, cat# D-003401-01-0020), 100 nM p53 siRNA (Horizon Discovery, Dharmacon, cat# L-003329-00-0020), or 100 nM PARN siRNA (Horizon Discovery, Dharmacon, cat# D-011348-04-0050) to cells plated at 50–60% confluency. ERα-siRNA treated cells were harvested 24-h post-transfection. p53 and PARN siRNA treatments were repeated for an additional 24 h and cells were harvested 48 h after initial transfection. Nuclear extracts were prepared from harvested cells and used for deadenylation assays and western blot. siGENOME non-targeting siRNA #2 (Horizon Discovery, Dharmacon siRNA, cat# D-001210-02-50) was used as control. Each experiment was performed at least three times with three biological replicates.
Nuclear extracts (NE) preparation
Nuclear extracts were immediately prepared from harvested MCF7, T47D and MDA-MB-231 cells as described [21].
Immunoblotting
Samples were analysed as in [25]. Blots were incubated with antibodies against ERα (Santa Cruz Biotechnology, cat# sc-8002), PARN (Abnova, cat# H-00005073-BP) and p53 (Santa Cruz Biotechnology, cat# sc-126). Topo II (Santa Cruz) was used as loading control.
Purification of recombinant proteins
Plasmid encoding His-PARN was provided by Dr. A. Virtanen (Uppsala University) and purified as previously described [20,21,25]. Commercially available purified His-ERα recombinant protein (Abcam catalogue #: ab82606) and purified GST-p53 recombinant protein (Millipore-Sigma Aldrich, catalogue #: 14–865) were used.
Deadenylation assays using nuclear extracts
Preparation of 32P-labelled L3(A30) substrates and conditions for deadenylation assays were as described [20]. Each experiment was performed at least three times with three biological replicates. Relative deadenylation was determined by the relative intensity of each band and expressed as L3 fragment/(L3(A30)+ L3 fragment)] × 100 and expressed as means ± standard deviation as previously described [20,21,25]. Quantifications were calculated by Image J software (http://rsb.info.nih.gov/ij/).
In vitro deadenylation assays
Conditions for in vitro deadenylation assays were performed as previously described [20]. Deadenylation assays with recombinant protein derivatives His-PARN, His-ERα, or GST-p53 were carried at 30°C for 90 min. Each experiment was performed at least three times with three biological replicates and results from independent samples were quantified by Image J.
Statistical methods
Statistical significance between experimental groups were calculated using GraphPad Prism 9.0. Results from in vitro deadenylation assays and qRT-PCR were performed in three or more independent biological samples analysed by triplicate, presented as mean ± standard deviation. Experiments with two groups were analysed using a two-tailed unpaired Student’s t-test. In the presented data, one (*), two (**), three (***) and four (****) corresponded to p < 0.01, p < 0.001, p < 0.0001 and p < 0.00001, respectively.
Endogenous immunoprecipitation (IP) assays
IPs conditions were as described [25]. NEs were IPed with antibodies against ERα polyclonal (Cell Signaling, cat# 8644S), PARN polyclonal (Bethyl Laboratories, cat# A303-562A), or p53 polyclonal (Santa Cruz Biotechnology, cat# FL-393) using protein A-magnetic beads (Millipore, PureProteome cat# LSKMAGA10) per manufacturer’s instructions.
Pull-down assays
Pull-down assays using His-PARN or His-ERα were as described [25].
Endogenous mRNA real-time reverse transcription polymerase chain reaction (qRT-PCR)
Total nuclear RNA was purified from MCF7, T47D and MDA-MB-231 cells using the RNeasy kit (Qiagen) according to the manufacturer’s directions. qRT-PCR reactions were adapted from Baquero et al. [25]. RNA concentrations were determined using NanoDrop (ThermoFisher). Equivalent amounts (2 μg) of purified RNA were used as a template to synthesize cDNA using GoScript Reverse Transcriptase and oligo-d(T)s primer as in [45]. Primers for PARN, ID1, ID2, KLHL24, HBP1, SPINK6, LUM and IFI6 used in the qRT–PCR reactions were generated using Integrated DNA Technologies. PARN: forward primer 5’- TGTCCTGTCACGATTCCTGAG −3’, reverse primer: CCGGTACATGGCTCTAAATCCAA −3’. ID1: forward primer 5’- CCAGAACCGCAAGGTGAG −3’, reverse 5’- GGTCCCTGATGTAGTCGATGA-3’. ID2: forward primer 5’- ATGAAAGCCTTCAGTCCCGT-3’, reverse 5’- TTCCATCTTGCTCACCTTCTT-3’. KLHL24: forward primer 5’-TGAGAAGACCACTGTTACACGAGC-3’, reverse 5’-CCTTGGGGACATCATTTCATTC-3’. HBP1: forward primer 5′-ATCATCTCCTGTACACATCATAGC-3′, reverse 5′-CATAGAAAGGGTGGTCCAGCTTAC-3′. SPINK: forward primer 5′-ATGAAACTGTCAGGCATG-3′, reverse 5′-TCAGCATTTTCCAGGATG-3′. LUM: forward primer 5’- CCACCACACCTGACAGAGT −3’, reverse 5’- CAAGTTGATTGACCTCCAGG −3’. IFI6: forward primer 5’- CTGGTCTGCGATCCTGAATG −3’, reverse 5’- AGAGGTTCTGGGAGCTGCTG − 3’. Relative mRNA levels were calculated using the ΔΔCT method as described [21,25]. Each experiment was performed at least three times with three biological replicates.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was supported by National Cancer Institute, National Institutes of Health (NIH), 1U54CA221704-01A (to SV).
Funding Statement
The work was supported by the National Institutes of Health [1U54CA221704-01A].
Disclosure statement
SV is currently employed by AstraZeneca and declares no conflict with this work.
Authors contributions
Conceived and designed the experiments: S.V. and F.E.K. Performed the experiments: S.V., A.Y., Y.Q.X., D.M.N. and A.R. Wrote and edited the paper: S.V. and F.E.K.
Data availability statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15476286.2024.2413821
References
- [1].Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J Clin. 2024;74(3):229–263. doi: 10.3322/caac.21834 [DOI] [PubMed] [Google Scholar]
- [2].Clusan L, Ferriere F, Flouriot G, et al. A basic review on estrogen receptor signaling pathways in breast cancer. Int J Mol Sci, 24(7). 2023;24(7):6834. doi: 10.3390/ijms24076834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Fuentes N, Silveyra P.. Estrogen receptor signaling mechanisms. Adv Protein Chem Struct Biol. 2019;116:135–170. doi: 10.1016/bs.apcsb.2019.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Miziak P, Baran M, Blaszczak E, et al. Estrogen receptor signaling in breast cancer. Cancers (Basel). 2023;15(19):4689. doi: 10.3390/cancers15194689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yasar P, Ayaz G, User SD, et al. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod Med Biol. 2017;16(1):4–20. doi: 10.1002/rmb2.12006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Simões J, Amado FM, Vitorino R, et al. A meta-analysis to evaluate the cellular processes regulated by the interactome of endogenous and over-expressed estrogen receptor alpha. Oncoscience. 2015;2(5):487–496. doi: 10.18632/oncoscience.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cignarella A, Boscaro C, Albiero M, et al. Post-transcriptional and epigenetic regulation of estrogen signaling. J Pharmacol Exp Ther. 2023;386(3):288–297. doi: 10.1124/jpet.123.001613 [DOI] [PubMed] [Google Scholar]
- [8].Thiebaut C, Vlaeminck-Guillem V, Tredan O, et al. Non-genomic signaling of steroid receptors in cancer. Mol Cell Endocrinol. 2021;538:111453. doi: 10.1016/j.mce.2021.111453 [DOI] [PubMed] [Google Scholar]
- [9].Nassa G, Giurato G, Salvati A, et al. The RNA-mediated estrogen receptor alpha interactome of hormone-dependent human breast cancer cell nuclei. Sci Data. 2019;6(1):173. doi: 10.1038/s41597-019-0179-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Singh A, Mishra R, Mazumder A. Breast cancer and its therapeutic targets: a comprehensive review. Chem Biol Drug Des. 2024;103(1):e14384. doi: 10.1111/cbdd.14384 [DOI] [PubMed] [Google Scholar]
- [11].Xu Y, Huangyang P, Wang Y, et al. Eralpha is an RNA-binding protein sustaining tumor cell survival and drug resistance. Cell. 2021;184(20):5215–5229 e5217. doi: 10.1016/j.cell.2021.08.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lanz RB, McKenna NJ, Onate SA, et al. A steroid receptor coactivator, SRA, functions as an RNA and Is present in an SRC-1 complex. Cell. 1999;97(1):17–27. doi: 10.1016/S0092-8674(00)80711-4 [DOI] [PubMed] [Google Scholar]
- [13].Soota D, Saravanan B, Mann R, et al. 2023. doi: 10.1101/2023.08.10.552751 [DOI] [Google Scholar]
- [14].Steiner HR, Lammer NC, Batey RT, et al. An extended DNA binding domain of the estrogen receptor alpha directly interacts with RNAs in vitro. Biochemistry. 2022;61(22):2490–2494. doi: 10.1021/acs.biochem.2c00536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mandel CR, Bai Y, Tong L. Protein factors in pre-mRNA 3’-end processing. Cell Mol Life Sci. 2008;65(7–8):1099–1122. doi: 10.1007/s00018-007-7474-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Murphy MR, Kleiman FE. Connections between 3’ end processing and DNA damage response: Ten years later. Wiley Interdiscip Rev RNA. 2020;11(2):e1571. doi: 10.1002/wrna.1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cevher MA, Kleiman FE. Connections between 3’-end processing and DNA damage response. Wiley Interdiscip Rev RNA. 2010;1(1):193–199. doi: 10.1002/wrna.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhang X, Virtanen A, Kleiman FE. To polyadenylate or to deadenylate: that is the question. Cell Cycle. 2010;9(22):4437–4449. doi: 10.4161/cc.9.22.13887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Goldstrohm AC, Wickens M. Multifunctional deadenylase complexes diversify mRNA control. Nat Rev Mol Cell Biol. 2008;9(4):337–344. doi: 10.1038/nrm2370 [DOI] [PubMed] [Google Scholar]
- [20].Cevher MA, Zhang X, Fernandez S, et al. Nuclear deadenylation/polyadenylation factors regulate 3’ processing in response to DNA damage. Embo J. 2010;29(10):1674–1687. doi: 10.1038/emboj.2010.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Devany E, Zhang X, Park JY, et al. Positive and negative feedback loops in the p53 and mRNA 3’ processing pathways. Proc Natl Acad Sci USA. 2013;110(9):3351–3356. doi: 10.1073/pnas.1212533110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Nazeer FI, Devany E, Mohammed S, et al. Inhibits mRNA 3’ processing through its interaction with the CstF/BARD1 complex. Oncogene. 2011;30(27), 3073-3083. 53. doi: 10.1038/onc.2011.29 [DOI] [PubMed] [Google Scholar]
- [23].Zhang X, Devany E, Murphy MR, et al. PARN deadenylase is involved in miRNA-dependent degradation of TP53 mRNA in mammalian cells. Nucleic Acids Res. 2015;43(22):10925–10938. doi: 10.1093/nar/gkv959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang X, Xiao S, Rameau RD, et al. Nucleolin phosphorylation regulates PARN deadenylase activity during cellular stress response. RNA Biol. 2018;15(2):251–260. doi: 10.1080/15476286.2017.1408764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Baquero J, Varriano S, Ordonez M, et al. Nuclear Tau, p53 and Pin1 regulate PARN-Mediated deadenylation and gene expression. Front Mol Neurosci. 2019;12:242. doi: 10.3389/fnmol.2019.00242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Berger CE, Qian Y, Liu G, et al. Target of estrogen receptor (ER) α, modulates DNA damage-induced growth suppression in er-positive breast cancer cells. J Biol Chem. 2012. 53;287(36):30117–30127. doi: 10.1074/jbc.M112.367326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liu W, Konduri SD, Bansal S, et al. Estrogen receptor-α binds p53 tumor suppressor protein directly and represses its function *. J Biol Chem. 2006;281(15):9837–9840. doi: 10.1074/jbc.C600001200 [DOI] [PubMed] [Google Scholar]
- [28].Sayeed A, Konduri SD, Liu W, et al. Estrogen receptor α inhibits p53-mediated transcriptional repression: implications for the regulation of Apoptosis. Cancer Res. 2007;67(16):7746–7755. doi: 10.1158/0008-5472.Can-06-3724 [DOI] [PubMed] [Google Scholar]
- [29].Berger CE, Qian Y, Chen X. The p53-estrogen receptor loop in cancer. Curr Mol Med. 2013;13(8):1229–1240. doi: 10.2174/15665240113139990065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Muthukaruppan A, Lasham A, Woad KJ, et al. Multimodal assessment of estrogen receptor mRNA profiles to quantify estrogen pathway activity in breast tumors. Clin Breast Cancer. 2017;17(2):139–153. doi: 10.1016/j.clbc.2016.09.001 [DOI] [PubMed] [Google Scholar]
- [31].Shi Y, Ye P, Long X. Differential expression profiles of the transcriptome in breast cancer cell lines revealed by next generation sequencing. Cell Physiol Biochem. 2017;44(2):804–816. doi: 10.1159/000485344 [DOI] [PubMed] [Google Scholar]
- [32].Böckers M, Paul NW, Efferth T. Organophosphate ester tri-o-cresyl phosphate interacts with estrogen receptor α in MCF-7 breast cancer cells promoting cancer growth. Toxicol Appl Pharmacol. 2020;395:114977. doi: 10.1016/j.taap.2020.114977 [DOI] [PubMed] [Google Scholar]
- [33].Escamilla-Powers JR, Daniel CJ, Farrell A, et al. The tumor suppressor protein HBP1 is a novel c-myc-binding protein that negatively regulates c-myc transcriptional activity. J Biol Chem. 2010;285(7):4847–4858. doi: 10.1074/jbc.M109.074856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Karamanou K, Franchi M, Onisto M, et al. Evaluation of lumican effects on morphology of invading breast cancer cells, expression of integrins and downstream signaling. FEBS J. 2020;287(22):4862–4880. doi: 10.1111/febs.15289 [DOI] [PubMed] [Google Scholar]
- [35].Karamanou K, Franchi M, Piperigkou Z, et al. Lumican effectively regulates the estrogen receptors-associated functional properties of breast cancer cells, expression of matrix effectors and epithelial-to-mesenchymal transition. Sci Rep. 2017;7(1):45138. doi: 10.1038/srep45138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Karamanou K, Franchi M, Vynios D, et al. Epithelial-to-mesenchymal transition and invadopodia markers in breast cancer: lumican a key regulator. Semin Cancer Biol. 2020;62:125–133. doi: 10.1016/j.semcancer.2019.08.003 [DOI] [PubMed] [Google Scholar]
- [37].Mascia F, Mazo I, Alterovitz WL, et al. In search of autophagy biomarkers in breast cancer: receptor status and drug agnostic transcriptional changes during autophagy flux in cell lines. PLOS ONE. 2022;17(1):e0262134. doi: 10.1371/journal.pone.0262134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Roschger C, Cabrele C. The id-protein family in developmental and cancer-associated pathways. Cell Commun And Signaling. 2017;15(1):7. doi: 10.1186/s12964-016-0161-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Savci-Heijink CD, Halfwerk H, Koster J, et al. Association between gene expression profile of the primary tumor and chemotherapy response of metastatic breast cancer. BMC Cancer. 2017;17(1):755. doi: 10.1186/s12885-017-3691-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wazir U, Jiang WG, Sharma AK, et al. The mRNA expression of inhibitors of DNA binding-1 and -2 is associated with advanced tumour stage and adverse clinical outcome in human breast cancer. Anticancer Res. 2013;33(5):2179–2183. [PubMed] [Google Scholar]
- [41].Yi B, Wang S, Wang X, et al. CRISPR interference and activation of the microRNA-3662-HBP1 axis control progression of triple-negative breast cancer. Oncogene. 2022;41(2):268–279. doi: 10.1038/s41388-021-02089-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhao Y, Luo A, Li S, et al. Inhibitor of Differentiation/DNA binding 1 (ID1) inhibits Etoposide-induced Apoptosis in a c-Jun/c-fos-dependent manner*. J Biol Chem. 2016;291(13):6831–6842. doi: 10.1074/jbc.M115.704361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhao Z, Bo Z, Gong W, et al. Inhibitor of differentiation 1 (Id1) in cancer and cancer therapy. Int J Med Sci. 2020;17(8):995–1005. doi: 10.7150/ijms.42805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zhou Y, Zhang T, Wang S, et al. Targeting of HBP1/TIMP3 axis as a novel strategy against breast cancer. Pharmacol Res. 2023;194:106846. doi: 10.1016/j.phrs.2023.106846 [DOI] [PubMed] [Google Scholar]
- [45].Murphy MR, Ramadei A, Doymaz A, et al. Long non-coding RNA generated from CDKN1A gene by alternative polyadenylation regulates p21 expression during DNA damage response. Nucleic Acids Res. 2023;51(21):11911–11926. doi: 10.1093/nar/gkad899 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper.