

Abstract
Muscle wasting is a serious complication in heart failure patients. Oxidative stress and inflammation are implicated in the pathogenesis of muscle wasting. Oxidative stress leads to the formation of toxic lipid peroxidation products, such as 4-hydroxy-2-nonenal (HNE), which covalently bind with proteins and DNA and activate atrophic pathways. Whether the formation of lipid peroxidation products and metabolic pathways that remove these toxic products are affected during heart failure-associated skeletal muscle wasting has never been studied. Male C57BL/6J mice were subjected to sham and transverse aortic constriction (TAC) surgeries for 4, 8 or 14 weeks. Different skeletal muscle beds were weighed, and the total cross-sectional area of the gastrocnemius muscle was measured via immunohistochemistry. Muscle function and muscle stiffness were measured by a grip strength meter and atomic force microscope, respectively. Atrophic and inflammatory marker levels were measured via qRT‒PCR. The levels of acrolein and HNE-protein adducts, aldehyde-removing enzymes, the histidyl dipeptide-synthesizing enzyme carnosine synthase (CARNS), and amino acid transporters in the gastrocnemius muscle were measured via Western blotting and qRT‒PCR. Histidyl dipeptides and histidyl dipeptide aldehyde conjugates in the Gastrocnemius and soleus muscles were analyzed by LC/MS–MS. Body weight, gastrocnemius muscle and soleus muscle weights and the total cross-sectional area of the gastrocnemius muscle were decreased after 14 weeks of TAC. Heart weight, cardiac function, grip strength and muscle stiffness were decreased in the TAC-operated mice. Expression of the atrophic and inflammatory markers Atrogin1 and TNF-α, respectively, was increased ~ 1.5–2fold in the gastrocnemius muscle after 14 weeks of TAC (p < 0.05 and p = 0.004 vs sham). The formation of HNE and acrolein protein adducts was increased, and the expression of the aldehyde-removing enzyme aldehyde dehydrogenase (ALDH2) was decreased in the gastrocnemius muscle of TAC mice. Carnosine (sham: 5.76 ± 1.3 vs TAC: 4.72 ± 0.7 nmol/mg tissue, p = 0.04) and total histidyl dipeptide levels (carnosine and anserine; sham: 11.97 ± 1.5 vs TAC: 10.13 ± 1.4 nmol/mg tissue, p < 0.05) were decreased in the gastrocnemius muscle of TAC mice. Depletion of histidyl dipeptides diminished the aldehyde removal capacity of the atrophic gastrocnemius muscle. Furthermore, CARNS and TAUT protein expression were decreased in the atrophic gastrocnemius muscle. Our data reveals that reduced expression of ALDH2 and depletion of histidyl dipeptides in the gastrocnemius muscle during heart failure leads to the accumulation of toxic aldehydes and might contribute to muscle wasting.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13395-024-00354-2.
Keywords: Anserine, Atrophy, Autophagy , Heart failure, Muscle wasting, Ubiquitin‒proteasome pathway
Introduction
Heart failure is a multifaceted and life-threatening syndrome that affects up to 1–2% of the global population. Comorbidities such as skeletal muscle wasting, anemia, chronic kidney disease and chronic obstructive pulmonary disease are extremely common in this patient population [1, 2]. Among these comorbidities, skeletal muscle wasting has emerged as a serious complication for patients, with both reduced and preserved ejection fractions [2]. The prevalence of muscle wasting in heart failure patients is approximately 20–50%, which leads to reduced functional capacity, frequent hospital visits, and increased mortality [1, 3, 4]. Despite the armamentarium of drugs available for the treatment of heart failure, such as angiotensin convertase inhibitors and beta-blockers, they provide only minor benefits in reversing muscle wasting [5, 6]. Although testosterone therapy improves exercise capacity in heart failure patients, [7–9] however it has not been tested in patients with muscle wasting. Nonetheless, given the impact of muscle wasting on increasing mortality and reducing quality of life, effective interventions to reverse this debilitating condition in heart failure patients are urgently needed. Therefore, a deeper understanding of the mechanisms contributing to heart failure-induced muscle wasting is essential.
The causes of muscle wasting in heart failure patients are multifactorial and include immune activation, neurohormonal derangements, reduced blood flow, oxidative stress, reduced anabolism, and increased catabolism [1, 10–13]. Oxidative stress within the muscle is one of the most common and principal factors involved in muscle wasting [13]. In atrophic muscle, oxidative stress activates the ubiquitin‒proteasome system (UPS) proteolytic pathway, the main mechanism involved in protein degradation [14, 15]. Oxidative stress induces autophagy, a lysosomal pathway that maintains cell homeostasis by removing damaged cellular components. However, under pathological conditions such as fasting, hypoxia, and exercise, autophagy is increased in association with muscle wasting [16–18]. In addition to influencing catabolic pathways, oxidative stress also reduces anabolic pathways by oxidizing the specific cysteine residues of phosphorylases, such as protein kinase A, which activates AKT [19]. While oxidative stress is closely associated with muscle wasting, antioxidant interventions in heart failure patients are unable to improve cardiac function [20, 21]; consequently, no attempts have been made to examine their effects on muscle wasting.
In biological tissues, excessive generation of reactive oxygen species (ROS) oxidizes membrane lipids, forming several toxic lipid peroxidation products, such as acrolein and 4-hydroxy-2-nonenal (4HNE) [22–24]. These toxic aldehydes contain reactive carbonyl groups that react with amino acid residues, such as lysine in proteins and nucleophilic sites of DNA, thus giving rise to a multitude of aldehyde-modified proteins and DNA adducts [25–27]. The formation of lipid peroxidation products has been widely reported in numerous oxidative stress-associated pathologies, such as atherosclerosis and ischemia‒reperfusion [28–30]. Both lipid peroxidation products and aldehyde-modified proteins trigger muscle wasting pathways, such as inflammation, autophagy, and the UPS [25, 31]. In tissues, lipid peroxidation products are rapidly oxidized or reduced by enzymes such as aldehyde dehydrogenase (ALDH2) and aldose reductase (AKR1B1), respectively. Lipid peroxidation products are also removed by binding with the nucleophile glutathione and further reduced by the reductive transformation catalyzed by AKR1B1 [32, 33]. Additionally, these toxic products are removed by forming conjugates with endogenous histidyl dipeptides, such as carnosine (β-alanine-histidine) [23, 34, 35].
Nonetheless, whether lipid peroxidation products are generated and the metabolic pathways that eliminate them in skeletal muscle during heart failure-induced muscle wasting have never been studied. Therefore, in this study, we subjected C57BL/6J mice to different durations of pressure overload model of heart failure and examined which muscle bed undergoes atrophy with the progression of heart failure. Furthermore, we measured, whether the aldehyde-modified protein adducts and the aldehyde removal ability of the muscle, including the expression of aldehyde-removing enzymes and histidyl dipeptide levels, is affected in atrophic muscle.
Methods
Animal housing and maintenance
C57BL/6J male mice were obtained from Jackson Laboratory (Bar Harbor, Maine) and maintained on normal chow in a pathogen-free facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. All procedures were approved by the University of Louisville Institutional Animal Care and Use Committee. Only C57BL/6J male mice at 12 weeks of age were used for all the experiments.
Animal surgeries
Male C57BL/6J mice were subjected to sham and transverse aortic constriction (TAC) surgeries as described previously [25, 36]. Briefly, following anesthesia (i.p. 50 mg/kg sodium pentobarbital and 50 mg/kg ketamine hydrochloride), the mice were orally intubated and ventilated (oxygen supplement to the room-air inlet) with a mouse ventilator (Hugo Sachs). The aorta was visualized following an intercostal incision. A 7–0 nylon suture was used to loop around the aorta between the brachiocephalic and left common carotid arteries. The suture was tied around a 27-gauge needle placed adjacent to the aorta to constrict the aorta to a reproducible diameter. The needle was removed, and the chest was closed in layers. The mice were extubated upon recovery from spontaneous breathing. Analgesia (ketoprofen, 5 mg/kg) was provided prior to recovery and at 24 and 48 h postsurgery. Sham mice were subjected to the same procedure as the TAC cohort, except that the suture was not tied. The mice in this study were exposed to HEPA- and charcoal-filtered room air (6 h/day, 5 days/week) as previously described [37].
Echocardiography
Cardiac function was measured via echocardiography via a VisualSonics Vevo 3100 as described previously [25, 36]. Briefly, the mice were anesthetized with 2% isoflurane, and the LV end-diastolic area, end-diastolic average wall thickness and end-diastolic volume (EDV), end-systolic area (LVESA), end-systolic volume (ESV), and ejection fraction (EF) were recorded and calculated [25, 36].
Muscle strength measurement
Grip strength measurements were performed as described previously [38, 39]. Before grip strength was assessed, the mice were weighed and allowed to acclimatize for 5 min. Each mouse was allowed to grasp the total paw bull-bar assembly, and in a separate experiment, the forepaw pull bar assembly was performed. Each mouse was gently pulled back by its tail until it could no longer grasp the bar. The force at the time of release was recorded as the peak tension, and each mouse was tested 5 times with a delay of 2–40 s to obtain forelimb–hindlimb grip strength measurements. The mean peak tension was calculated from the recordings and normalized to the body weight.
Histidyl dipeptide and histidyl dipeptide aldehyde conjugate measurements
Following 14 weeks of sham or TAC surgery, all the muscle beds, including the gastrocnemius, soleus, tibialis anterior, and extensor digitorum longus, were isolated from the sham- and TAC-operated mice. Soleus and gastrocnemius muscles were analyzed for the presence of histidyl dipeptides and histidyl dipeptide aldehyde conjugates via UPLC‒ESI‒MS/MS as described previously [35, 40].
Protein extraction and immunoblotting
Gastrocnemius muscle from the sham and TAC mice was homogenized in lysis buffer and centrifuged, and the supernatants were analyzed via Western blotting as described previously [25]. Immunoblots were developed using anti-acrolein (1:1000, LSBio), anti-HNE (1:1000; Abcam), anti-AKR1B1 (1:1000, ABclonal), anti-ALDH2 (1:1000; NOVUSBIO), anti-CARNS1 (1:1000; COSMOBIO), anti-TAUT (1:1000; ABclonal), and anti-PEPT2 (1:1000; NOVUSBIO) antibodies. The immunoblot images were captured with the Bio-Rad ChemiDoc Imaging system. Band intensity was quantified by using Image Quant TL software and normalized to Amido-black staining.
RNA isolation and quantitative real-time PCR
Total RNA from the gastrocnemius and soleus muscles was isolated via a Qiagen Fibrous Tissue RNA Mini Kit, and the purity of the RNA was analyzed via Nanodrop One (Thermo Fisher Scientific) as described previously [25]. Briefly, cDNA was generated from 2 µg of RNA via Syber green SuperScript™ IV VILO™ Master Mix (Thermo Fisher), and PCR was performed via a standard procedure with QuantStudio5 from Applied Biosystems. The expression of the genes encoding Atrogin1, Murf1, TNF-α, IL-6, CARNS, TAUT, and PEPT2 was determined via quantitative RT‒PCR. The results were normalized to those of the 18S ribosome and expressed according to the comparative Ct method, where the Ct values of the genes of interest were compared to those of the controls.
Atomic force microscopy (AFM) measurements
To generate elasticity maps of the isolated skeletal muscle independent of any neurohormonal activation, AFM measurements were performed as described previously [41]. Briefly, the experiments were performed via AFM (Nanowizard 4, JPK Instruments, Berlin, Germany) combined with an inverted optical microscope (AxioObserver ZI, Zeiss), driven by JPK Nanowizard software 6.0. Data were acquired in peak force spectrometry mode via MLCT-Bio cantilevers with a spring constant of 0.8 N/m (Bruker, Santa Barbara, CA, USA). The cantilever spring constant and sensitivity were calibrated before each experiment via the thermal fluctuation method of the JPK software, yielding spring constant values ranging from 0.018 to 0.025 N/m. Cryosections of the gastrocnemius muscle from C57BL/6J mice subjected to sham (n = 5) or TAC (n = 5) for 14 weeks were used for the experiment. Frozen tissue slides were fixed on the AFM sample stage. Bright-field imaging was used to identify the area of interest in the frozen tissue sample. Visual control of the immobility of a slide was performed via an optical microscope coupled with an AFM instrument. This ensured that only peak force spectrometry files were recorded under static conditions. The cantilever tip was subsequently engaged in an area of interest to scan an area of 16 × 16 µm. The AFM tip was scanned on the area of interest of the frozen section by applying a force trigger of 2nN with a 3 µm Z ramp at 80 µm/s.
Cross-sectional area of muscle fibers
The gastrocnemius muscle tissue from C57BL/6J sham and TAC mice (n = 5) was isolated, flash frozen in liquid nitrogen, mounted in embedding medium and sectioned with a microtome cryostat. To assess tissue morphology, 10 µm thick transverse sections were stained with anti(α)-Laminin antibody (1:250, Sigma, L9393) in blocking buffer and incubated overnight at 4 °C. The next day, the sections were washed with freshly prepared goat anti-rabbit IgG (H + L) Alexa Flour™ 488 (1:1000, Invitrogen, A-11034) in blocking buffer at room temperature for 1 h. Finally, the sections were counterstained with ProLong™ Gold Antifade mountant with DAPI. Images were captured with a Keyence BZ-X800 microscope, and the total cross-sectional area was measured via the Keyence Hybrid and Macro Cell Count application.
Statistical analysis
The data are presented as the means ± SEMs. The data from the sham and TAC groups were analyzed via one-way analysis of variance followed by Bonferroni correction or Student’s t test. Statistical significance was accepted at p < 0.05.
Results
Chronic heart failure induces muscle wasting
To determine the time for the onset of muscle wasting during the progression of heart failure, we performed sham and transverse aortic constriction (TAC) surgeries for durations of 4, 8 and 14 weeks. No change in body weight was observed after 4 and 8 weeks of TAC (Fig. 1), whereas after 14 weeks of TAC, body weight was significantly lower than that of the sham mice (Fig. 1). Heart weights at 4, 8 and 14 weeks were significantly greater in TAC- than in sham- operated mice (Fig. 1). Echocardiographic analysis at 14 weeks revealed that, compared with sham, TAC mice developed significant left ventricular dilation (end-diastolic volume, EDV; sham: 50 ± 13.0 vs. TAC: 99 ± 43, p = 0.001; and end-systolic volume, ESV; sham: 16 ± 7.6 vs. TAC: 75 ± 43 µL, p = 0.0002), a decrease in the ejection fraction (% EF; sham: 68 ± 5.7 vs. TAC: 28 ± 13.0, p = 0.00001), and fractional shortening (FS; sham: 42 ± 6.3 vs. TAC: 13 ± 8.5%, p = 0.0001) (Table 1). Furthermore, the left ventricular internal diameter in diastole (LVIDd; sham: 3.5 ± 0.3 vs. TAC: 4.8 ± 0.8 mm, p = 0.0001), left ventricular internal diameter in systole (LVIDs; sham: 2.0 ± 0.4 vs. TAC: 4.3 ± 1.0 mm, p = 0.0003), were greater, stroke volume (SV; sham: 33.6 ± 6.5 vs. TAC: 23.9 ± 4.2 µL, p = 0.0005) and cardiac output (CO; sham: 18.3 ± 3.7 vs. TAC: 13.4 ± 2.1 mL/min, p = 0.001; Table 1) were decreased, in the TAC than in the sham mice.
Fig. 1.

Transverse aortic constriction (TAC)-induced heart failure decreases body weight and increases heart weight. C57BL/6J mice were subjected to sham or TAC surgeries for 4, 8 or 14 weeks. (A) Body weight and (B) heart weight were normalized to tibia length. Data are presented as mean ± SEM; n = 6–10 mice in each group; *p < 0.05 vs sham
Table 1.
Echocardiographic parameters of mice subjected to sham or transverse aortic constriction surgeries for 14 weeks
| Parameters | Sham | TAC | p value |
|---|---|---|---|
| Body Mass (g) | 32.7 ± 1.62 | 30.9 ± 1.64 | 0.0198 |
| IVRT (ms) | 11.6 ± 1.5 | 16.1 ± 1.7 | 2E-06 |
| Heart Rate (BPM) | 544.2 ± 25 | 565 ± 53 | |
| LVESV (uL) | 16.6 ± 7.6 | 75.8 ± 43.6 | 0.0002 |
| LVEDV (uL) | 50.2 ± 13.7 | 99.8 ± 43.9 | 0.001 |
| SV (uL) | 33.6 ± 6.5 | 23.9 ± 13.0 | 0.0005 |
| Ejection Fraction (%) | 68.0 ± 5.7 | 28 ± 13.0 | 0.0001 |
| Cardiac Output (mL/min) | 18.3 ± 3.7 | 13.4 ± 2.1 | 0.0013 |
| LVAW;d (mm) | 1.1 ± 0.1 | 1.2 ± 0.1 | 0.5151 |
| LVAW;s (mm) | 1.7 ± 0.1 | 1.5 ± 0.1 | 0.0023 |
| LVID;d (mm) | 3.5 ± 0.3 | 4.8 ± 0.8 | 0.0001 |
| LVID;s (mm) | 2.0 ± 0.4 | 4.3 ± 1.0 | 0.0003 |
| FS (%) | 42.0 ± 6.3 | 13.0 ± 8.5 | 0.0001 |
Data are presented as the mean ± SEM, n = 10 samples in each group
Four weeks after TAC, the weights of different muscle beds, including the gastrocnemius (sham: 17.05 ± 2.25 vs TAC: 17.63 ± 5.93 mg/mm), soleus (sham: 4.65 ± 0.63 vs TAC: 5.68 ± 1.76 mg/mm), tibialis anterior (sham: 9.37 ± 1.82 vs TAC: 11.22 ± 2.25 mg/mm), and extensor digitorum longus (EDL; sham: 1.30 ± 0.18 vs TAC: 1.36 ± 0.27 mg/mm) muscles, remained unchanged compared with sham mice (Suppl. Figure 1 A‒D). Similarly, after 8 weeks of TAC, the gastrocnemius (sham: 17.46 ± 2.44 vs TAC: 16.90 ± 1.99 mg/mm), soleus (sham: 5.03 ± 0.51 vs TAC: 4.56 ± 0.57 mg/mm), tibialis anterior (sham: 9.83 ± 1.16 vs TAC: 9.30 ± 1.10 mg/mm), and EDL muscle weights (sham: 1.23 ± 0.20 vs TAC: 1.27 ± 0.17 mg/mm, Suppl. Figure 1 E–H) were unchanged compared with sham mice. However, after 14 weeks of TAC, the weights of the gastrocnemius muscle (sham: 15.34 ± 2.0 vs TAC: 12.79 ± 2.0 mg/mm, p = 0.02) and soleus muscle (sham: 4.24 ± 0.80 vs TAC 3.50 ± 0.62 mg/mm, p = 0.04) were lower than sham mice (Fig. 2A and B), whereas the tibialis anterior (sham: 5.75 ± 1.04 vs TAC: 5.82 ± 1.20 mg/mm) and EDL muscle weights (sham: 1.39 ± 0.22 vs TAC: 1.25 ± 0.15 mg/mm) remained unchanged between the sham and TAC mice (Fig. 2C and D). Taken together, these findings suggest that advanced heart failure leads to muscle wasting by targeting the gastrocnemius and soleus muscles.
Fig. 2.

Transverse aortic constriction (TAC)-induced heart failure decreases muscle weight. C57BL/6J mice were subjected to sham and TAC surgeries for 14 weeks. (A) Weights of the gastrocnemius (gastroc) muscle, (B) soleus muscle, (C) tibialis anterior muscle, and (D) extensor digitorium longus (EDL) muscle were normalized to tibia length. Data are presented as the mean ± SEM, n = 10 samples in each group, *p < 0.05 vs sham
Progression of atrophy-related genes in skeletal muscle during heart failure
Given that gastrocnemius and soleus muscle weights are affected primarily in heart failure model mice, we next examined the progression of atrophy-related genes over time. Consistent with the observations that gastrocnemius muscle weight was unchanged after 4 and 8 weeks of TAC, expression of the Atrogin1 and Trim63/MURF1 genes remained unchanged at these time points (Suppl. Figure 2 A-B), whereas after 14 weeks of TAC, Atrogin1 expression was increased ~ 1.5-fold (Fig. 3, p < 0.05) compared with sham mice. The expression of Trim63/MURF1 tended to increase in the atrophic gastrocnemius muscle but was not statistically significant (Fig. 3, p = 0.08). Although soleus muscle weight was decreased after 14 weeks of TAC, atrophic markers remained unchanged between the sham and TAC mice (Suppl. Figure 2C).
Fig. 3.
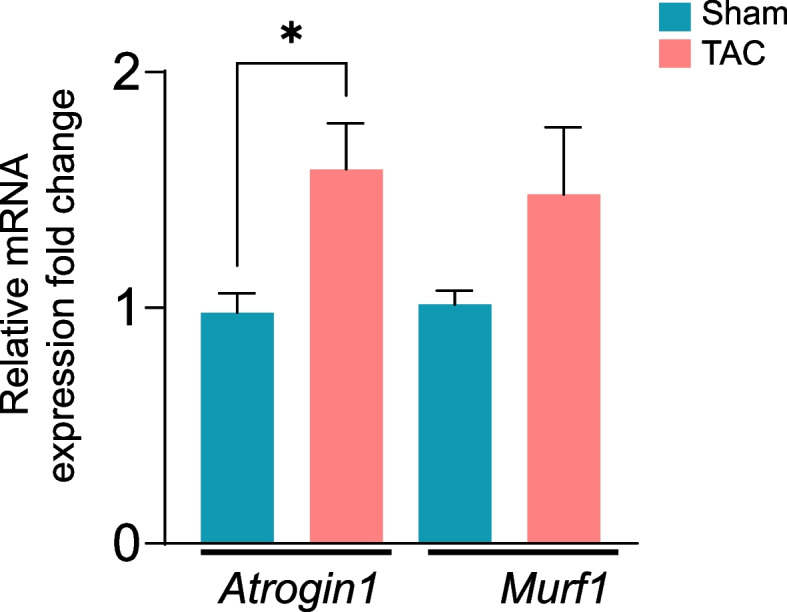
Transverse aortic constriction (TAC)-induced heart failure increases atrophy-related gene expression in the gastrocnemius muscle. mRNA levels of the atrophic genes Atrogin1 and Murf1 in the gastrocnemius muscle after 14 weeks of sham or TAC surgeries. Data are presented as the mean ± SEM, n = 10 samples in each group. *p < 0.05 vs sham
The cross-sectional area is decreased in atrophic gastrocnemius muscle
Muscle wasting during heart failure is associated with a decrease in the cross-sectional area [42, 43]. Given that the gastrocnemius muscle was affected primarily after 14 weeks of TAC (Fig. 2A), we next compared the total cross-sectional area of the gastrocnemius muscle between the sham and TAC mice. Our results revealed that the average cross-sectional area of the gastrocnemius muscle was significantly smaller in the TAC than in the sham mice (sham: 24.3 ± 7.7 vs TAC: 13.1 ± 7.2 µm2, p = 0.04; Fig. 4).
Fig. 4.

Fiber size of the gastrocnemius muscle is reduced during chronic heart failure. Total cross-sectional area of the gastrocnemius muscle was analyzed by immunohistochemistry in C57BL/6J mice subjected to sham or transverse aortic constriction surgeries for 14 weeks. (A) Representative photomicrographs of transverse gastrocnemius muscle sections after anti-laminin staining; scale bar, 50 µm. (B) Total cross-sectional area normalized to total cell counts. Data are presented as the mean ± SEM; n = 5 samples in each group; *p = 0.04 vs sham
Grip strength is decreased in heart failure mice
In heart failure patients, muscle strength is decreased [44], and these patients exhibit lower handgrip and quadricep strengths [4, 45]. To examine whether muscle strength is also affected in mice during heart failure, we measured the muscle function of the forelimbs and total grip strength (forelimb and hindlimbs) with a grip strength meter. Compared with those of sham mice, forelimb strength (sham: 2.81 ± 0.36 vs TAC: 1.52 ± 0.27 N, p = 0.0001; Fig. 5A) and total grip strength (sham: 3.56 ± 0.29 vs TAC: 2.64 ± 0.32 N, p = 0.0001; Fig. 5B) were lower after 14 weeks of TAC, suggesting a decline in muscle function.
Fig. 5.
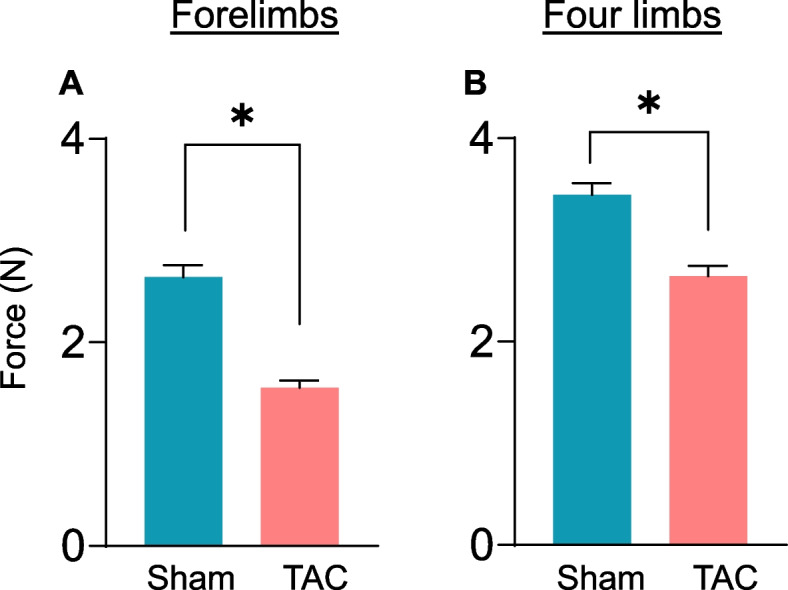
Heart failure reduces muscle function. Muscle function was measured with a grip strength meter for the forelimbs and four limbs (forelimb and hindlimbs) of mice subjected to sham and transverse aortic constriction surgeries for 14 weeks. (A) Forelimb muscle strength and (B) total muscle strength (forelimb and hind limb) muscle strength. Data are presented as the mean ± SEM; n = 9–10 samples in each group; *p = 0.0001 vs sham
Atomic force microscopy (AFM) analysis of the atrophic gastrocnemius muscle
To examine whether heart failure could affect the elastic properties of muscle, we measured muscle stiffness and force production with AFM in tissue sections, which is closer to natural tissue environment, and independent of any neurohormonal interactions. Slices of the gastrocnemius muscles were cut longitudinally into 10 µm thick sections, immobilized on microscope glass slides and incubated in PBS. Approximately 200–300 individual measurements were performed on these muscle sections to determine the elasticity changes on the basis of force versus displacement measurements via AFM. Plotting the Young’s modulus, a measure of muscle stiffness in response to the applied load, revealed that the gastrocnemius muscle from the sham mice yielded Young’s modulus values of 15.19 ± 6.31 kPa, whereas those of the TAC mice were ~ 2–3fold lower (6.17 ± 3.32 kPa, p = 0.03; Fig. 6), indicating lower resistance to deformation in the atrophic muscle of the heart failure mice.
Fig. 6.
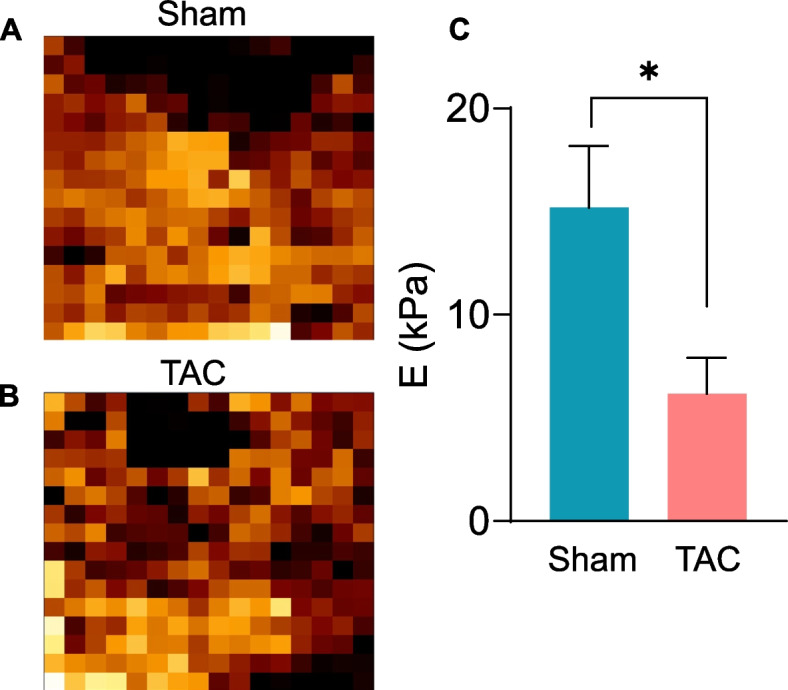
Heart failure reduces the elasticity of the gastrocnemius muscle. Muscle elasticity in the gastrocnemius muscle of mice subjected to sham or transverse aortic constriction surgeries for 14 weeks was measured via atomic force microscopy (AFM). Representative images of gastrocnemius muscle sections from (A) sham and (B) TAC mice were used for calculating force maps via Young’s modulus. (C) Data are presented as the mean ± SEM, n = 5 samples in each group, *p = 0.03 vs sham.
Heart failure increases tumor necrosis factor-alpha (TNF-α) expression in the gastrocnemius muscle
The levels of inflammatory cytokines, such as TNFα and interleukin-6 (IL-6), are increased in the skeletal muscle of heart failure patients [46]. Next, to examine the progression of heart failure-related inflammatory cytokines, we measured the gene expression of TNF-α, interleukin-6 (IL-6) and interleukin-1β (IL-1β) in the gastrocnemius muscle at 4, 8 and 14 weeks after sham or TAC surgeries. The expression of these inflammatory cytokines was unchanged after 4 and 8 weeks of TAC (Suppl. Figure 3 A-B). Consistent with the onset of atrophy in the gastrocnemius muscle at 14 weeks after TAC, TNF-α expression was increased ~ 1–2fold (p = 0.004), whereas the levels of IL-6 and IL-1β remained unchanged compared with those in sham mice (Fig. 7).
Fig. 7.
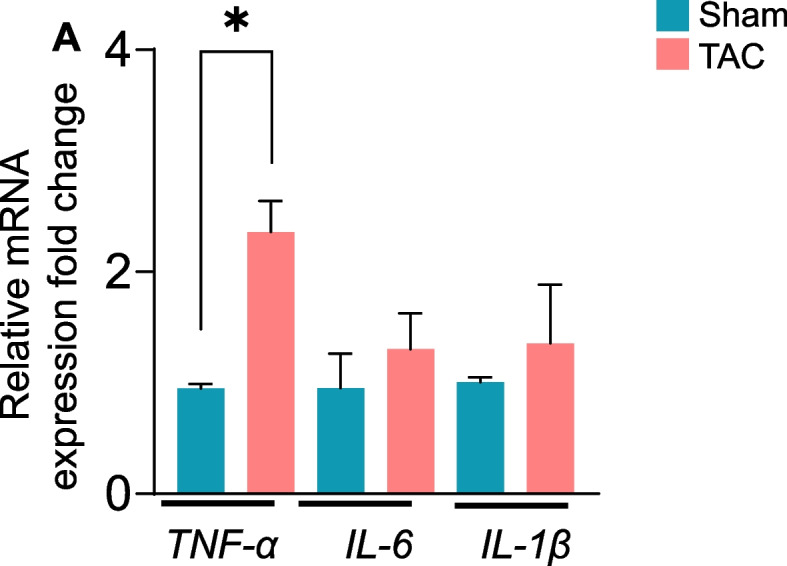
Heart failure increases tumor necrosis factor-α (TNF-α) expression in the gastrocnemius muscle. mRNA levels of the inflammatory-related genes TNF-α, IL-6, and IL-1β in the gastrocnemius muscle of C57BL/6J mice that were subjected to sham or transverse aortic constriction (TAC) surgeries for 14 weeks. Data are presented as the mean ± SEM; n = 10 samples in each group; *p = 0.04 vs sham
Heart failure causes carbonyl stress in the gastrocnemius muscle
To examine whether lipid peroxidation products, the downstream effectors of reactive oxygen species (ROS), are accumulate in muscle during heart failure, gastrocnemius muscle collected after 14 weeks of sham and TAC was immunoblotted with anti-HNE and anti-acrolein antibodies. The formation of acrolein (Fig. 8A) and HNE (Fig. 8B) protein adducts was increased ~ 2–3fold in the gastrocnemius muscle of TAC compared with sham mice (p < 0.05; Fig. 8C). In conclusion, these results suggest that uncontrolled generation of ROS in skeletal muscle during heart failure increases the formation of aldehyde protein adducts in the atrophic gastrocnemius muscle.
Fig. 8.

Heart failure increases the accumulation of aldehyde protein adducts in the gastrocnemius muscle. Representative Western blots of (A) acrolein and (B) HNE protein adducts are accumulated in the gastrocnemius muscle of C57BL/6J mice subjected to sham or transverse aortic constriction (TAC) surgeries for 14 weeks. (C) Bar graph showing the intensity of the bands normalized to amido-black (AB). Data are shown as the mean ± SEM; n = 5 samples in each group; *p < 0.05 vs sham
Aldehyde removal pathways are diminished in the atrophic gastrocnemius muscle
To investigate whether the metabolic processes that eliminate lipid peroxidation products are affected during heart failure, we compared the expression of the enzymes aldose reductase (AKR1B1) and aldehyde dehydrogenase (ALDH2) in the gastrocnemius muscle following 14 weeks of sham and TAC. The expression of AKR1B1 remained unchanged (Fig. 9A), whereas ALDH2 expression was decreased (~ 1.5-fold, p = 0.02; Fig. B) in the TAC compared with the sham mice (Fig. 9C).
Fig. 9.

Heart failure decreases the expression of aldehyde dehydrogenase (ALDH2) in the gastrocnemius muscle. Gastrocnemius muscles from C57BL/6J mice subjected to sham or transverse aortic constriction surgeries for 14 weeks were analyzed by Western blotting. Representative blots for (A) aldose reductase (AKR1B1) and (B) aldehyde dehydrogenase (ALDH2) are shown. (C) Bar graph shows the intensity of bands normalized to amido black (AB). Data are presented as the mean ± SEM; n = 5 samples in each group; *p = 0.04 vs sham
Skeletal muscle is a reservoir for histidyl dipeptides, which bind with different reactive aldehydes [40]. Using LC‒MS/MS, we measured the levels of carnosine and anserine in the gastrocnemius muscle. Compared with those in sham mice, carnosine levels in the gastrocnemius muscle were significantly lower after 14 weeks of TAC (sham: 5.76 ± 1.3 vs TAC: 4.72 ± 0.75 nmol/mg tissue, p = 0.04; Fig. 10A). Although anserine levels also decreased, the difference did not reach statistical significance (sham: 6.20 ± 1.08 vs TAC: 5.42 ± 0.82 nmol/mg tissue, p = 0.07; Fig. 10B). Total histidyl dipeptide levels (carnosine and anserine) were significantly lower in the gastrocnemius muscle of TAC compared with sham mice (sham: 11.97 ± 1.5 vs TAC: 10.13 ± 1.4 nmol/mg tissue, p < 0.05; Fig. 10C). In the soleus muscle, carnosine, anserine and total histidyl dipeptide levels remained unchanged between the sham and TAC mice (Fig. 10D-F).
Fig. 10.

Histidyl dipeptide levels are depleted in the atrophic gastrocnemius muscle. Levels of different histidyl dipeptides in the gastrocnemius and soleus muscles after 14 weeks of sham and transverse aortic constriction surgeries. Levels of (A) carnosine, (B) anserine, and (C) total histidyl dipeptides in the gastrocnemius muscle measured by LC–MS/MS. Levels of (D) carnosine, (E) anserine, and (F) total histidyl dipeptides in the soleus muscle. Data are presented as the mean ± SEM; n = 9–10 samples in each group; *p < 0.05 vs sham
Next, to determine whether the depletion of histidyl dipeptides in skeletal muscle affects the removal of lipid peroxidation products, we measured carnosine aldehyde conjugates; carnosine-propanal, carnosine-propanol, carnosine-HNE and carnosine-DHN, via LC‒MS/MS. The levels of carnosine-propanal in the gastrocnemius muscle of TAC mice tended to decrease but were unable to reach statistical significance (carnosine propanal: sham: 24.12 ± 2.3 vs TAC: 21.30 ± 3.0 pmol/mg tissue, p = 0.07; Suppl. Figure 4 A and B). Carnosine-propanol, carnosine-HNE, and carnosine-DHN remained unchanged between the sham and TAC (Suppl. Figure 4 C and D). Taken together, these results show that both the enzymatic and nonenzymatic pathways that remove reactive aldehydes are diminished in the atrophic gastrocnemius muscle.
Histidyl dipeptide synthesis and transport are decreased in the gastrocnemius muscle during heart failure
Finally, to determine how histidyl dipeptide synthesis in the gastrocnemius muscle decreases during heart failure, we compared the expression of carnosine synthase (CARNS) and amino acid transporters (TAUT and PEPT2) between sham and TAC mice. The expression of CARNS and TAUT was decreased ~ 1.5-fold (p = 0.02, Fig. 11A) and ~ 2.0-fold (p < 0.01, Fig. 11B), respectively, whereas PEPT2 remained unchanged in the gastrocnemius muscle of TAC compared with the sham mice (Fig. 11C and D). To determine whether the decrease in CARNS and TAUT occurred at the mRNA level, we performed RT‒PCR and found that CARNS, PEPT2, and TAUT expression was unchanged between the sham and TAC mice (Suppl. Figure 5). Taken together, these results suggest that a decrease in histidyl dipeptide synthesis and the transport of amino acids could contribute to diminishing histidyl dipeptide levels in the atrophic gastrocnemius muscle during heart failure.
Fig. 11.

The expression of carnosine synthase (CARNS) and TAUT is decreased in the gastrocnemius muscle of heart failure model mice. Gastrocnemius muscle collected after 14 weeks of sham and transverse aortic constriction surgeries was analyzed by Western blotting for (A) CARNS, (B) taurine transporter (TAUT) and (C) peptide transporter (PEPT2). (D) Bar graph shows the band intensity normalized to amido-black. Data are presented as mean ± SEM; n = 5 samples in each group; *p < 0.05 vs sham
Discussion
Muscle wasting is a frequent comorbidity in patients with heart failure that negatively affects their prognosis [47]. The predominant feature of muscle wasting is an imbalance between catabolic and anabolic processes, with catabolism outpacing the latter. Within the skeletal muscle of heart failure patients, the activity of antioxidative enzymes is decreased, and inflammatory cytokines are accumulated [46, 48]. A key consequence of oxidative stress and inflammation in muscle is the onset of muscle wasting. Several studies in heart failure patients have targeted oxidative stress and inflammatory cytokines to improve functional outcomes, resulting in very limited success [20, 21, 49]. Prolonged oxidative stress inflicts direct damage to lipids, especially polyunsaturated fatty acids, forming highly toxic lipid peroxidation products, such as acrolein and 4-hydroxynonenal (4HNE) [22–24]. These toxic reactive aldehydes react with cellular or tissue proteins forming aldehyde adducts [25–27]. Previous work has shown that deficiency of aldehyde dehydrogenase (ALDH2), the enzyme that detoxifies lipid peroxidation products, promotes age-related muscle atrophy, suggesting that downstream effectors of oxidative stress could trigger muscle wasting [50]. However, whether lipid peroxidation products are generated during heart failure and pathways that metabolize these toxic products are affected in muscle have not been studied. An in-depth examination of these metabolic pathways in skeletal muscle under heart failure conditions could pave the way for development of novel therapeutics to address muscle wasting in heart failure patients. In the present study, we determined the timeline for the onset of muscle wasting in mice subjected to pressure overload model of heart failure and reported that chronic heart failure leads to gastrocnemius muscle atrophy. Muscle wasting in these mice was associated with a decrease in muscle strength and stiffness, indicating both quantitative and qualitative abnormalities in skeletal muscle during heart failure. The formation of aldehyde protein adducts increased, and the expression of aldehyde-removing enzyme ALDH2 was decreased in the atrophic gastrocnemius muscle. The levels of endogenous histidyl dipeptides, especially carnosine, which binds with different reactive aldehydes, were decreased in the gastrocnemius muscle of heart failure model mice. The protein expression of enzyme CARNS, which synthesizes carnosine, and the amino acid transporter TAUT was decreased in the gastrocnemius muscle of these mice, suggesting that both the synthesis and transport of essential amino acids for histidyl dipeptide synthesis could contribute to histidyl dipeptide depletion in atrophic muscle. The distinct decrease in histidyl dipeptide synthesis and ALDH2 in the gastrocnemius muscle suggests that derangements in these aldehyde removal pathways might be specifically involved in the increased formation of aldehyde protein adducts and thus trigger muscle wasting during heart failure.
Muscle wasting is a serious complication that affects a large proportion of heart failure patients. In this patient population, the prevalence of muscle wasting is ~ 20% greater than that in age-matched normal individuals [4]. Recent report has shown that the prevalence of muscle wasting is also greater in younger heart failure patients [51]. Muscle wasting and impaired skeletal muscle function following heart failure play key roles in the development of exercise intolerance, fatigue and poor quality of life [3, 4, 52, 53]. In this study, long-term TAC-induced heart failure decreased body weight, gastrocnemius and soleus muscle weights, suggesting the impact of heart failure progression on muscle wasting. Although we did not weigh the adipose tissue, a previous report showed that after 12 weeks of TAC-induced heart failure, the inguinal and abdominal fat contents are reduced [54]. Therefore, the decreases in both adipose and muscle weights may have contributed to the decrease in body weight observed in the TAC-treated mice. Our results contrast with those of previous work by Szaroszyk et al. [54], who reported that TAC-induced heart failure for 12 weeks reduced the weights of all muscles, including the quadriceps, gastrocnemius, triceps, and soleus. This discrepancy could be due to the handling of the mice. In our study, both the sham- and TAC-operated mice were exposed to HEPA- and charcoal-filtered room air [37]. In addition to the decrease in muscle weight, additional signs of muscle atrophy in heart failure model mice showed that after 14 weeks of TAC, skeletal muscle strength was reduced. AFM measurements, which provide sensitive detection of the elasticity and properties of individual fibers, revealed that muscle elasticity in heart failure mice was decreased. Similarly, Gene expression of the muscle-specific ubiquitin ligase Atrogin1, a marker of atrophy, was increased, and the total cross-sectional area of the gastrocnemius muscle was decreased. A previous report in chronic heart failure patients revealed that TNF-α expression in muscle is elevated compared with that in healthy controls [46]. In the present study, TNF-α expression in the muscle coincided with the onset of atrophy. This obvious link between diminished muscle function, increased expression of TNF-α and muscle wasting supports a role of local inflammation in heart failure-induced muscle atrophy. Nonetheless, TAC-induced heart failure decreased body weight, induced muscle wasting, diminished muscle strength and elasticity, and increased the expression of atrophy- and inflammatory-related genes, suggesting that the progressive heart failure model of TAC in mice replicates muscle wasting syndrome in heart failure patients.
The mechanisms by which heart failure induces muscle wasting are not clear, and currently, no therapies are available that can stop this condition in heart failure patients. One of the common features associated with muscle wasting is the release of atrophic factors, such as angiotensin II, from diseased tissue. Patients with heart failure have increased levels of circulating angiotensin, which induces oxidative stress by binding through its AT1 receptor [55–57]. Increased levels of oxidative stress markers have been documented in the skeletal muscle of chronic heart failure patients, which is correlated with reduced exercise capacity and lower antioxidant levels [48]. While the formation of reactive oxygen species (ROS) is tightly controlled in biological systems, the deregulation of redox homeostasis is a common pathogenic mechanism associated with age, heart failure and cancer-related muscle loss [58]. When ROS formation increases, antioxidant defenses become overwhelmed, resulting in the production of a wide variety of lipid peroxidation products, such as acrolein and 4-hydroxy-2-nonenal (4-HNE), which can covalently bind with proteins and DNA [25]. In this study, we found that the levels of acrolein and HNE-protein adducts were increased in the gastrocnemius muscle of TAC-operated mice, indicating that heart failure overwhelms both redox and aldehyde removal homeostasis in skeletal muscle, thus resulting in the accumulation of 4HNE- and acrolein-modified proteins. Previous work has shown that acrolein induces myotube atrophy and inhibits myogenic differentiation in myoblasts [59]. Acrolein exposure also decreases muscle weight and retards muscle regeneration in mice [60]. Similarly, increased formation of HNE protein adducts was reported in the gastrocnemius muscle of mice with increasing age [61], and preventing the accumulation of HNE in the gastrocnemius muscle alleviated muscle atrophy [62]. In this context, the accumulation of acrolein and HNE protein adducts in skeletal muscle could aggravate muscle wasting during heart failure. Previous reports have shown that aldehyde-modified proteins behave as damage-associated molecular patterns (DAMPs) that alarm the immune system by inducing adaptive immune responses [63]. In particular, different human pathologies associated with oxidative stress, such as atherosclerosis, involving increased formation of aldehyde-modified proteins, activates adaptive immune responses [64–66]. Our results showing that TNF-α expression was increased in atrophic skeletal muscle suggest that the formation of aldehyde-modified DAMPs might activate inflammation in atrophic skeletal muscle under heart failure conditions. Therefore, future studies are warranted to determine the contribution of these aldehyde-modified DAMPs to immune-modulating activities and muscle atrophy under heart failure conditions.
Recent reports have shown that a missense single nucleotide polymorphism in the aldehyde dehydrogenase 2 (ALDH2) gene, rs671 (ALDH2*2), increases 4-HNE formation in skeletal muscle and promotes muscle atrophy [67]. ALDH2 deficiency also promotes age-related muscle atrophy, increases the formation of HNE adducts [50], and treatment with antioxidants such as Vit. E and Chlorella ameliorate the genetic and age-induced risk of atrophy [62, 67]. On the other hand, overexpression of ALDH2 in skeletal muscle reverses oxidative stress and muscle atrophy caused by exhaustive exercise [68]. We investigated whether the accumulation of aldehyde-modified proteins in the gastrocnemius during heart failure is also associated with the derangement of mechanisms that remove reactive aldehydes. Our results showing that ALDH2 expression was decreased and that the formation of aldehyde protein adducts was increased in the gastrocnemius muscle of heart failure model mice support the view that the decrease in the ALDH2 expression could contribute to the accumulation of aldehyde-modified proteins and consequently lead to muscle wasting. Extensive evidence shows that the activation of ALDH2 by a small-molecular-weight activator of ALDH2 prevents the accumulation of aldehydes in ischemic tissues and protects against acute ischemic injury in the heart and brain [69–71]. Therefore, activation of ALDH2 by a selective ALDH2 activator may remove toxic aldehydes from skeletal muscle and protect against heart failure-induced muscle wasting.
In skeletal muscle, especially the gastrocnemius muscle, high levels of histidyl dipeptides, such as carnosine and anserine, are present [40]. Among these histidyl dipeptides, carnosine is present in humans, whereas anserine is found in rodents [35, 40]. These dipeptides are synthesized by the enzymes CARNS and carnosine methyltransferase [35, 72–74]. Histidyl dipeptides exhibit unique chemistry, where the amino group of the β-alanine can bind with reactive aldehydes via Michael adducts or Schiff’s base. They also exhibit the ability to quench reactive oxygen species, buffer the intracellular pH and chelate first transition metals [75, 76]. Among all the nucleophiles present in skeletal muscle, only histidyl dipeptide levels can be increased either by exercise or by supplementation with the precursor amino acid β-alanine [40]. Because of their multifunctionality and the ease with which these dipeptides can be increased in different tissues, β-alanine supplementation is widely used to improve exercise capacity [40, 77]. Previously, we showed that increasing carnosine levels in the skeletal muscle of humans via β-alanine supplementation enhances the removal of reactive aldehydes from skeletal muscle [40]. Furthermore, carnosine levels are decreased in the skeletal muscle of cancer cachexia patients [78]. Recent reports have shown that carnosine supplementation improves the exercise capacity of heart failure patients and glucose homeostasis in type 2 diabetics [79, 80]. Therefore, given the multitude of benefits of carnosine associated with maintaining skeletal muscle function, our observations revealed that carnosine and total histidyl dipeptides were depleted in the gastrocnemius muscle of chronic heart failure mice, suggests these dipeptides are essential for skeletal muscle health during heart failure. Interestingly, our results revealed that the weight of soleus muscle was decreased in heart failure model mice; however, no changes in histidyl dipeptides or atrophic markers were observed, suggesting that specific depletion of carnosine in the gastrocnemius muscle could be the underlying cause of muscle atrophy induced by heart failure.
Histidyl dipeptide homeostasis within skeletal muscle is maintained by a complex array of transporters, such as TAUT and PEPT, synthesis by CARNS and binding with lipid peroxidation products [78, 81]. We found that the expression of the enzyme CARNS and transporter TAUT was decreased in the gastrocnemius muscle of heart failure model mice, suggesting that both the synthesis of carnosine and the transport of amino acids needed for carnosine synthesis could contribute to histidyl dipeptide depletion during heart failure. Interestingly, decreases in CARNS and TAUT protein expression were not mimicked at the mRNA level, suggesting that CARNS and TAUT might be targets of protein degradation machinery activated during muscle wasting. Nonetheless, how CARNS and TAUT protein expression are decreased needs to be studied. Paralleling the decrease in carnosine synthesis was the decrease in the removal of reactive aldehydes in the gastrocnemius muscle of heart failure model mice. Overall, both the enzymatic and nonenzymatic aldehyde removal pathways become defective in skeletal muscle during heart failure and thus could contribute to triggering muscle wasting.
Conclusion
In conclusion, our results suggest that, TAC-induced chronic heart failure in mice causes muscle wasting. Lipid peroxidation products, the downstream toxic products of oxidative stress, are accumulated and result in the formation of acrolein and HNE-modified protein adducts in the gastrocnemius muscle of TAC mice. In addition, processes of aldehyde removal become defective and endogenous histidyl dipeptides are depleted in the atrophic gastrocnemius muscle. Since dipeptides form conjugates with reactive aldehydes, and these dipeptides can be replenished in the muscle by supplementation [40, 82, 83], therefore our findings opens an opportunity to test how these dipeptides contribute to muscle wasting and whether maintaining or increasing their synthesis in the skeletal muscle could prevent or reverse muscle wasting during heart failure.
Supplementary Information
Supplementary Material 1: Fig. 1. Muscle weights after 4 and 8 weeks of sham and transverse aortic constriction (TAC) surgery. Weights of different muscle beds that were normalized to tibia length, including the gastrocnemius muscle after, (A) 4 and (E) 8 weeks, the soleus muscle after, (B) 4 and (F) 8 weeks, the tibialis anterior muscle after, (C) 4 and (G) 8 weeks, and the extensor digitorium longus (EDL) after, (D) 4 and (H) 8 weeks of sham and TAC surgeries. The data are presented as the mean ± SEM, n = 6 samples in each group. Fig. 2. Atrophy-related genes in the gastrocnemius muscle after sham and transverse aortic constriction (TAC) surgeryies. mRNA levels of Atrogin1 in the gastrocnemius muscle of C57BL/6J mice after (A) 4 and (B) 8 weeks of sham and TAC surgery. (C) mRNA levels of Atrogin 1 and Murf1 in the soleus muscle after 14 weeks of sham and TAC surgery. Data are presented as the mean ± SEM; n = 6–7 samples in each group. Fig. 3. Expression of inflammatory genes in the gastrocnemius muscle after sham and transverse aortic constriction (TAC) surgeries. TNF-α, IL-6 and IL-1β mRNA levels in the gastrocnemius muscle of C57BL/6J mice after (A) 4 and (B) 8 weeks of sham or TAC surgeries. Data are presented as the mean ± SEM, n = 6–7 samples in each group, p > 0.05 vs sham. Fig. 4. Carnosine aldehyde conjugates in the gastrocnemius muscle after sham and transaortic constriction (TAC) surgeries. Gastrocnemius muscles collected after 14 weeks of sham or TAC surgeries was analyzed by LC‒MS/MS for different carnosine aldehyde conjugates. Levels of (A) carnosine propanal, (B) carnosine HNE, (C) carnosine propanol and (D) carnosine DHN. Data are presented as the mean ± SEM, n = 9–10 samples in each group. Fig. 5. Gene expression of carnosine synthase (CARNS), human taurine transporter (TAUT) and human peptide (PEPT2) in the gastrocnemius muscle after sham and transverse aortic constriction surgeries. Relative mRNA levels of CARNS, TAUT and PEPT2 in the gastrocnemius muscle after 14 weeks of sham or TAC surgeries. Data are presented as the mean ± SEM, n = 9–10 samples in each group.
Acknowledgements
We thank the Bioanalytical Core for biochemical analysis and the Imaging Core for their technical support.
Conflict of interest disclosures
None.
Abbreviations
- TAC
Transverse aortic constriction
- ROS
Reactive oxygen species
- AKR1B1
Aldose reductase
- ALDH2
Aldehyde dehydrogenase
- CARNS
Carnosine synthase
- HNE
4-Hydroxy-2-nonenal
- TAUT
Taurine transporter
- TNF-α
Tumor necrosis factor-alpha
- IL-6
Interleukin 6
- IL-1β
Interleukin-1β
Authors' contributions
SPB, IJ, SS, conceptualized and designed the study. MC, BD, YN, PL and DH collected samples, sample processing, data acquisition and analysis. AB and SS were responsible for interpretation. All authors reviewed and contributed to the manuscript.
Funding
This work was supported by the NIH grant R21ES033334 and 5P42ES023716.
Availability of data and materials
All data generated for the manuscript will be available from the corresponding author on request.
Data availability
No datasets were generated or analysed during the current study.
Declarations
Ethics approval and consent to participate
Approvals for all procedures were provided by the University of Louisville Institutional Animal Care and Use Committee.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.von Haehling S, Ebner N, Dos Santos MR, Springer J, Anker SD. Muscle wasting and cachexia in heart failure: mechanisms and therapies. Nat Rev Cardiol. 2017;14:323–41. 10.1038/nrcardio.2017.51. [DOI] [PubMed] [Google Scholar]
- 2.Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, Gonzalez-Juanatey JR, Harjola VP, Jankowska EA, et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail. 2016;18:891–975. 10.1002/ejhf.592. [DOI] [PubMed] [Google Scholar]
- 3.Bekfani T, Pellicori P, Morris DA, Ebner N, Valentova M, Steinbeck L, Wachter R, Elsner S, Sliziuk V, Schefold JC, et al. Sarcopenia in patients with heart failure with preserved ejection fraction: Impact on muscle strength, exercise capacity and quality of life. Int J Cardiol. 2016;222:41–6. 10.1016/j.ijcard.2016.07.135. [DOI] [PubMed] [Google Scholar]
- 4.Fulster S, Tacke M, Sandek A, Ebner N, Tschope C, Doehner W, Anker SD, von Haehling S. Muscle wasting in patients with chronic heart failure: results from the studies investigating co-morbidities aggravating heart failure (SICA-HF). Eur Heart J. 2013;34:512–9. 10.1093/eurheartj/ehs381. [DOI] [PubMed] [Google Scholar]
- 5.Hryniewicz K, Androne AS, Hudaihed A, Katz SD. Partial reversal of cachexia by beta-adrenergic receptor blocker therapy in patients with chronic heart failure. J Card Fail. 2003;9:464–8. 10.1016/s1071-9164(03)00582-7. [DOI] [PubMed] [Google Scholar]
- 6.Lainscak M, Keber I, Anker SD. Body composition changes in patients with systolic heart failure treated with beta blockers: a pilot study. Int J Cardiol. 2006;106:319–22. 10.1016/j.ijcard.2005.01.061. [DOI] [PubMed] [Google Scholar]
- 7.Pugh PJ, Jones TH, Channer KS. Acute haemodynamic effects of testosterone in men with chronic heart failure. Eur Heart J. 2003;24:909–15. 10.1016/s0195-668x(03)00083-6. [DOI] [PubMed] [Google Scholar]
- 8.Malkin CJ, Pugh PJ, West JN, van Beek EJ, Jones TH, Channer KS. Testosterone therapy in men with moderate severity heart failure: a double-blind randomized placebo controlled trial. Eur Heart J. 2006;27:57–64. 10.1093/eurheartj/ehi443. [DOI] [PubMed] [Google Scholar]
- 9.Caminiti G, Volterrani M, Iellamo F, Marazzi G, Massaro R, Miceli M, Mammi C, Piepoli M, Fini M, Rosano GM. Effect of long-acting testosterone treatment on functional exercise capacity, skeletal muscle performance, insulin resistance, and baroreflex sensitivity in elderly patients with chronic heart failure a double-blind, placebo-controlled, randomized study. J Am Coll Cardiol. 2009;54:919–27. 10.1016/j.jacc.2009.04.078. [DOI] [PubMed] [Google Scholar]
- 10.Okutsu M, Call JA, Lira VA, Zhang M, Donet JA, French BA, Martin KS, Peirce-Cottler SM, Rembold CM, Annex BH, et al. Extracellular superoxide dismutase ameliorates skeletal muscle abnormalities, cachexia, and exercise intolerance in mice with congestive heart failure. Circ Heart Fail. 2014;7:519–30. 10.1161/CIRCHEARTFAILURE.113.000841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Egerman MA, Glass DJ. Signaling pathways controlling skeletal muscle mass. Crit Rev Biochem Mol Biol. 2014;49:59–68. 10.3109/10409238.2013.857291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glass DJ. Signaling pathways perturbing muscle mass. Curr Opin Clin Nutr Metab Care. 2010;13:225–9. 10.1097/mco.0b013e32833862df. [DOI] [PubMed] [Google Scholar]
- 13.Abrigo J, Elorza AA, Riedel CA, Vilos C, Simon F, Cabrera D, Estrada L, Cabello-Verrugio C. Role of oxidative stress as key regulator of muscle wasting during cachexia. Oxid Med Cell Longev. 2018;2018: 2063179. 10.1155/2018/2063179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sandri M. Protein breakdown in muscle wasting: role of autophagy-lysosome and ubiquitin-proteasome. Int J Biochem Cell Biol. 2013;45:2121–9. 10.1016/j.biocel.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bilodeau PA, Coyne ES, Wing SS. The ubiquitin proteasome system in atrophying skeletal muscle: roles and regulation. Am J Physiol Cell Physiol. 2016;311:C392-403. 10.1152/ajpcell.00125.2016. [DOI] [PubMed] [Google Scholar]
- 16.Dobrowolny G, Aucello M, Rizzuto E, Beccafico S, Mammucari C, Boncompagni S, Belia S, Wannenes F, Nicoletti C, Del Prete Z, et al. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 2008;8:425–36. 10.1016/j.cmet.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 17.Rahman M, Mofarrahi M, Kristof AS, Nkengfac B, Harel S, Hussain SN. Reactive oxygen species regulation of autophagy in skeletal muscles. Antioxid Redox Signal. 2014;20:443–59. 10.1089/ars.2013.5410. [DOI] [PubMed] [Google Scholar]
- 18.Rodney GG, Pal R, Abo-Zahrah R. Redox regulation of autophagy in skeletal muscle. Free Radic Biol Med. 2016;98:103–12. 10.1016/j.freeradbiomed.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cross JV, Templeton DJ. Regulation of signal transduction through protein cysteine oxidation. Antioxid Redox Signal. 2006;8:1819–27. 10.1089/ars.2006.8.1819. [DOI] [PubMed] [Google Scholar]
- 20.Cleland JG, Coletta AP, Freemantle N, Velavan P, Tin L, Clark AL. Clinical trials update from the American College of Cardiology meeting: CARE-HF and the remission of heart failure, Women’s Health Study, TNT, COMPASS-HF, VERITAS, CANPAP, PEECH and PREMIER. Eur J Heart Fail. 2005;7:931–6. 10.1016/j.ejheart.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 21.Lonn E, Bosch J, Yusuf S, Sheridan P, Pogue J, Arnold JM, Ross C, Arnold A, Sleight P, Probstfield J, et al. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial. JAMA. 2005;293:1338–47. 10.1001/jama.293.11.1338. [DOI] [PubMed] [Google Scholar]
- 22.Baba SP, Hellmann J, Srivastava S, Bhatnagar A. Aldose reductase (AKR1B3) regulates the accumulation of advanced glycosylation end products (AGEs) and the expression of AGE receptor (RAGE). Chem Biol Interact. 2011;191:357–63. 10.1016/j.cbi.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baba SP, Hoetker JD, Merchant M, Klein JB, Cai J, Barski OA, Conklin DJ, Bhatnagar A. Role of aldose reductase in the metabolism and detoxification of carnosine-acrolein conjugates. J Biol Chem. 2013;288:28163–79. 10.1074/jbc.M113.504753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Conklin DJ, Guo Y, Jagatheesan G, Kilfoil PJ, Haberzettl P, Hill BG, Baba SP, Guo L, Wetzelberger K, Obal D, et al. Genetic deficiency of glutathione S-transferase P increases myocardial sensitivity to ischemia-reperfusion injury. Circ Res. 2015;117:437–49. 10.1161/CIRCRESAHA.114.305518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baba SP, Zhang D, Singh M, Dassanayaka S, Xie Z, Jagatheesan G, Zhao J, Schmidtke VK, Brittian KR, Merchant ML, et al. Deficiency of aldose reductase exacerbates early pressure overload-induced cardiac dysfunction and autophagy in mice. J Mol Cell Cardiol. 2018;118:183–92. 10.1016/j.yjmcc.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barrera G, Pizzimenti S, Ciamporcero ES, Daga M, Ullio C, Arcaro A, Cetrangolo GP, Ferretti C, Dianzani C, Lepore A, et al. Role of 4-hydroxynonenal-protein adducts in human diseases. Antioxid Redox Signal. 2015;22:1681–702. 10.1089/ars.2014.6166. [DOI] [PubMed] [Google Scholar]
- 27.Liu X, Lovell MA, Lynn BC. Detection and quantification of endogenous cyclic DNA adducts derived from trans-4-hydroxy-2-nonenal in human brain tissue by isotope dilution capillary liquid chromatography nanoelectrospray tandem mass spectrometry. Chem Res Toxicol. 2006;19:710–8. 10.1021/tx0502903. [DOI] [PubMed] [Google Scholar]
- 28.Barski OA, Xie Z, Baba SP, Sithu SD, Agarwal A, Cai J, Bhatnagar A, Srivastava S. Dietary carnosine prevents early atherosclerotic lesion formation in apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2013;33:1162–70. 10.1161/ATVBAHA.112.300572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Srivastava S, Vladykovskaya E, Barski OA, Spite M, Kaiserova K, Petrash JM, Chung SS, Hunt G, Dawn B, Bhatnagar A. Aldose reductase protects against early atherosclerotic lesion formation in apolipoprotein E-null mice. Circ Res. 2009;105:793–802. 10.1161/CIRCRESAHA.109.200568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaiserova K, Srivastava S, Hoetker JD, Awe SO, Tang XL, Cai J, Bhatnagar A. Redox activation of aldose reductase in the ischemic heart. J Biol Chem. 2006;281:15110–20. 10.1074/jbc.M600837200. [DOI] [PubMed] [Google Scholar]
- 31.Hill BG, Haberzettl P, Ahmed Y, Srivastava S, Bhatnagar A. Unsaturated lipid peroxidation-derived aldehydes activate autophagy in vascular smooth-muscle cells. Biochem J. 2008;410:525–34. 10.1042/BJ20071063. [DOI] [PubMed] [Google Scholar]
- 32.Srivastava S, Dixit BL, Cai J, Sharma S, Hurst HE, Bhatnagar A, Srivastava SK. Metabolism of lipid peroxidation product, 4-hydroxynonenal (HNE) in rat erythrocytes: role of aldose reductase. Free Radic Biol Med. 2000;29:642–51. [DOI] [PubMed] [Google Scholar]
- 33.Srivastava S, Watowich SJ, Petrash JM, Srivastava SK, Bhatnagar A. Structural and kinetic determinants of aldehyde reduction by aldose reductase. Biochemistry. 1999;38:42–54. 10.1021/bi981794l. [DOI] [PubMed] [Google Scholar]
- 34.Blancquaert L, Baba SP, Kwiatkowski S, Stautemas J, Stegen S, Barbaresi S, Chung W, Boakye AA, Hoetker JD, Bhatnagar A, et al. Carnosine and anserine homeostasis in skeletal muscle and heart is controlled by beta-alanine transamination. J Physiol. 2016;594:4849–63. 10.1113/JP272050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao J, Conklin DJ, Guo Y, Zhang X, Obal D, Guo L, Jagatheesan G, Katragadda K, He L, Yin X, et al. Cardiospecific Overexpression of ATPGD1 (Carnosine Synthase) increases histidine dipeptide levels and prevents myocardial ischemia reperfusion injury. J Am Heart Assoc. 2020;9: e015222. 10.1161/JAHA.119.015222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watson LJ, Facundo HT, Ngoh GA, Ameen M, Brainard RE, Lemma KM, Long BW, Prabhu SD, Xuan YT, Jones SP. O-linked beta-N-acetylglucosamine transferase is indispensable in the failing heart. Proc Natl Acad Sci U S A. 2010;107:17797–802. 10.1073/pnas.1001907107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zelko IN, Dassanayaka S, Malovichko MV, Howard CM, Garrett LF, Uchida S, Brittian KR, Conklin DJ, Jones SP, Srivastava S. Chronic benzene exposure aggravates pressure overload-induced cardiac dysfunction. Toxicol Sci. 2021;185:64–76. 10.1093/toxsci/kfab125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gallot YS, Bohnert KR, Straughn AR, Xiong G, Hindi SM, Kumar A. PERK regulates skeletal muscle mass and contractile function in adult mice. FASEB J. 2019;33:1946–62. 10.1096/fj.201800683RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Straughn AR, Kakar SS. Withaferin A ameliorates ovarian cancer-induced cachexia and proinflammatory signaling. J Ovarian Res. 2019;12:115. 10.1186/s13048-019-0586-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoetker D, Chung W, Zhang D, Zhao J, Schmidtke VK, Riggs DW, Derave W, Bhatnagar A, Bishop DJ, Baba SP. Exercise alters and beta-alanine combined with exercise augments histidyl dipeptide levels and scavenges lipid peroxidation products in human skeletal muscle. J Appl Physiol (1985). 2018. 10.1152/japplphysiol.00007.2018. [DOI] [PMC free article] [PubMed]
- 41.Kammoun M, Ternifi R, Dupres V, Pouletaut P, Meme S, Meme W, Szeremeta F, Landoulsi J, Constans JM, Lafont F, et al. Development of a novel multiphysical approach for the characterization of mechanical properties of musculotendinous tissues. Sci Rep. 2019;9:7733. 10.1038/s41598-019-44053-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carvalho RF, Castan EP, Coelho CA, Lopes FS, Almeida FL, Michelin A, de Souza RW, Araujo JP Jr, Cicogna AC, Dal Pai-Silva M. Heart failure increases atrogin-1 and MuRF1 gene expression in skeletal muscle with fiber type-specific atrophy. J Mol Histol. 2010;41:81–7. 10.1007/s10735-010-9262-x. [DOI] [PubMed] [Google Scholar]
- 43.Cunha TF, Bechara LR, Bacurau AV, Jannig PR, Voltarelli VA, Dourado PM, Vasconcelos AR, Scavone C, Ferreira JC, Brum PC. Exercise training decreases NADPH oxidase activity and restores skeletal muscle mass in heart failure rats. J Appl Physiol. 1985;2017(122):817–27. 10.1152/japplphysiol.00182.2016. [DOI] [PubMed] [Google Scholar]
- 44.Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, Jatoi A, Loprinzi C, MacDonald N, Mantovani G, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. 2011;12:489–95. 10.1016/S1470-2045(10)70218-7. [DOI] [PubMed] [Google Scholar]
- 45.Emami A, Saitoh M, Valentova M, Sandek A, Evertz R, Ebner N, Loncar G, Springer J, Doehner W, Lainscak M, et al. Comparison of sarcopenia and cachexia in men with chronic heart failure: results from the studies investigating co-morbidities aggravating heart failure (SICA-HF). Eur J Heart Fail. 2018;20:1580–7. 10.1002/ejhf.1304. [DOI] [PubMed] [Google Scholar]
- 46.Gielen S, Adams V, Mobius-Winkler S, Linke A, Erbs S, Yu J, Kempf W, Schubert A, Schuler G, Hambrecht R. Anti-inflammatory effects of exercise training in the skeletal muscle of patients with chronic heart failure. J Am Coll Cardiol. 2003;42:861–8. 10.1016/s0735-1097(03)00848-9. [DOI] [PubMed] [Google Scholar]
- 47.Narumi T, Watanabe T, Kadowaki S, Takahashi T, Yokoyama M, Kinoshita D, Honda Y, Funayama A, Nishiyama S, Takahashi H, et al. Sarcopenia evaluated by fat-free mass index is an important prognostic factor in patients with chronic heart failure. Eur J Intern Med. 2015;26:118–22. 10.1016/j.ejim.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 48.Linke A, Adams V, Schulze PC, Erbs S, Gielen S, Fiehn E, Mobius-Winkler S, Schubert A, Schuler G, Hambrecht R. Antioxidative effects of exercise training in patients with chronic heart failure: increase in radical scavenger enzyme activity in skeletal muscle. Circulation. 2005;111:1763–70. 10.1161/01.CIR.0000165503.08661.E5. [DOI] [PubMed] [Google Scholar]
- 49.Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT, Anti TNFTACHFI. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart failure (ATTACH) trial. Circulation. 2003;107:3133–40. 10.1161/01.CIR.0000077913.60364.D2. [DOI] [PubMed] [Google Scholar]
- 50.Kasai A, Jee E, Tamura Y, Kouzaki K, Kotani T, Nakazato K. Aldehyde dehydrogenase 2 deficiency promotes skeletal muscle atrophy in aged mice. Am J Physiol Regul Integr Comp Physiol. 2022;322:R511–25. 10.1152/ajpregu.00304.2021. [DOI] [PubMed] [Google Scholar]
- 51.Hajahmadi M, Shemshadi S, Khalilipur E, Amin A, Taghavi S, Maleki M, Malek H, Naderi N. Muscle wasting in young patients with dilated cardiomyopathy. J Cachexia Sarcopenia Muscle. 2017;8:542–8. 10.1002/jcsm.12193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.von Haehling S, Steinbeck L, Doehner W, Springer J, Anker SD. Muscle wasting in heart failure: an overview. Int J Biochem Cell Biol. 2013;45:2257–65. 10.1016/j.biocel.2013.04.025. [DOI] [PubMed] [Google Scholar]
- 53.Zamboni M, Rossi AP, Corzato F, Bambace C, Mazzali G, Fantin F. Sarcopenia, cachexia and congestive heart failure in the elderly. Endocr Metab Immune Disord Drug Targets. 2013;13:58–67. 10.2174/1871530311313010008. [DOI] [PubMed] [Google Scholar]
- 54.Szaroszyk M, Kattih B, Martin-Garrido A, Trogisch FA, Dittrich GM, Grund A, Abouissa A, Derlin K, Meier M, Holler T, et al. Skeletal muscle derived Musclin protects the heart during pathological overload. Nat Commun. 2022;13:149. 10.1038/s41467-021-27634-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Anker SD, Steinborn W, Strassburg S. Cardiac cachexia. Ann Med. 2004;36:518–29. 10.1080/07853890410017467. [DOI] [PubMed] [Google Scholar]
- 56.Adigun AQ, Ajayi AA. The effects of enalapril-digoxin-diuretic combination therapy on nutritional and anthropometric indices in chronic congestive heart failure: preliminary findings in cardiac cachexia. Eur J Heart Fail. 2001;3:359–63. 10.1016/s1388-9842(00)00146-x. [DOI] [PubMed] [Google Scholar]
- 57.Chamberlain JS. ACE inhibitor bulks up muscle. Nat Med. 2007;13:125–6. 10.1038/nm0207-125. [DOI] [PubMed] [Google Scholar]
- 58.Moulin M, Ferreiro A. Muscle redox disturbances and oxidative stress as pathomechanisms and therapeutic targets in early-onset myopathies. Semin Cell Dev Biol. 2017;64:213–23. 10.1016/j.semcdb.2016.08.003. [DOI] [PubMed] [Google Scholar]
- 59.Rom O, Kaisari S, Aizenbud D, Reznick AZ. The effects of acetaldehyde and acrolein on muscle catabolism in C2 myotubes. Free Radic Biol Med. 2013;65:190–200. 10.1016/j.freeradbiomed.2013.06.024. [DOI] [PubMed] [Google Scholar]
- 60.Chen HJ, Wang CC, Chan DC, Chiu CY, Yang RS, Liu SH. Adverse effects of acrolein, a ubiquitous environmental toxicant, on muscle regeneration and mass. J Cachexia Sarcopenia Muscle. 2019;10:165–76. 10.1002/jcsm.12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Braga M, Sinha Hikim AP, Datta S, Ferrini MG, Brown D, Kovacheva EL, Gonzalez-Cadavid NF, Sinha-Hikim I. Involvement of oxidative stress and caspase 2-mediated intrinsic pathway signaling in age-related increase in muscle cell apoptosis in mice. Apoptosis. 2008;13:822–32. 10.1007/s10495-008-0216-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nakashima Y, Ohsawa I, Nishimaki K, Kumamoto S, Maruyama I, Suzuki Y, Ohta S. Preventive effects of Chlorella on skeletal muscle atrophy in muscle-specific mitochondrial aldehyde dehydrogenase 2 activity-deficient mice. BMC Complement Altern Med. 2014;14: 390. 10.1186/1472-6882-14-390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moghaddam AE, Gartlan KH, Kong L, Sattentau QJ. Reactive carbonyls are a major Th2-inducing damage-associated molecular pattern generated by oxidative stress. J Immunol. 2011;187:1626–33. 10.4049/jimmunol.1003906. [DOI] [PubMed] [Google Scholar]
- 64.Di Gioia M, Spreafico R, Springstead JR, Mendelson MM, Joehanes R, Levy D, Zanoni I. Endogenous oxidized phospholipids reprogram cellular metabolism and boost hyperinflammation. Nat Immunol. 2020;21:42–53. 10.1038/s41590-019-0539-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ngwenyama N, Kirabo A, Aronovitz M, Velazquez F, Carrillo-Salinas F, Salvador AM, Nevers T, Amarnath V, Tai A, Blanton RM, et al. Isolevuglandin-Modified Cardiac Proteins Drive CD4+ T-Cell Activation in the Heart and Promote Cardiac Dysfunction. Circulation. 2021;143:1242–55. 10.1161/CIRCULATIONAHA.120.051889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miller YI, Choi SH, Wiesner P, Fang L, Harkewicz R, Hartvigsen K, Boullier A, Gonen A, Diehl CJ, Que X, et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res. 2011;108:235–48. 10.1161/CIRCRESAHA.110.223875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kobayashi H, Nakamura S, Sato Y, Kobayashi T, Miyamoto K, Oya A, Matsumoto M, Nakamura M, Kanaji A, Miyamoto T. ALDH2 mutation promotes skeletal muscle atrophy in mice via accumulation of oxidative stress. Bone. 2021;142: 115739. 10.1016/j.bone.2020.115739. [DOI] [PubMed] [Google Scholar]
- 68.Zhang Q, Zheng J, Qiu J, Wu X, Xu Y, Shen W, Sun M. ALDH2 restores exhaustive exercise-induced mitochondrial dysfunction in skeletal muscle. Biochem Biophys Res Commun. 2017;485:753–60. 10.1016/j.bbrc.2017.02.124. [DOI] [PubMed] [Google Scholar]
- 69.Fu SH, Zhang HF, Yang ZB, Li TB, Liu B, Lou Z, Ma QL, Luo XJ, Peng J. Alda-1 reduces cerebral ischemia/reperfusion injury in rat through clearance of reactive aldehydes. Naunyn Schmiedebergs Arch Pharmacol. 2014;387:87–94. 10.1007/s00210-013-0922-8. [DOI] [PubMed] [Google Scholar]
- 70.Woods C, Shang C, Taghavi F, Downey P, Zalewski A, Rubio GR, Liu J, Homburger JR, Grunwald Z, Qi W, et al. In Vivo Post-Cardiac Arrest Myocardial Dysfunction Is Supported by Ca2+/Calmodulin-Dependent Protein Kinase II-Mediated Calcium Long-Term Potentiation and Mitigated by Alda-1, an Agonist of Aldehyde Dehydrogenase Type 2. Circulation. 2016;134:961–77. 10.1161/CIRCULATIONAHA.116.021618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gomes KM, Campos JC, Bechara LR, Queliconi B, Lima VM, Disatnik MH, Magno P, Chen CH, Brum PC, Kowaltowski AJ, et al. Aldehyde dehydrogenase 2 activation in heart failure restores mitochondrial function and improves ventricular function and remodelling. Cardiovasc Res. 2014;103:498–508. 10.1093/cvr/cvu125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Drozak J, Chrobok L, Poleszak O, Jagielski AK, Derlacz R. Molecular identification of carnosine N-methyltransferase as chicken histamine N-methyltransferase-like protein (hnmt-like). PLoS ONE. 2013;8: e64805. 10.1371/journal.pone.0064805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Drozak J, Piecuch M, Poleszak O, Kozlowski P, Chrobok L, Baelde HJ, de Heer E. UPF0586 Protein C9orf41 Homolog Is Anserine-producing Methyltransferase. J Biol Chem. 2015;290:17190–205. 10.1074/jbc.M115.640037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Drozak J, Veiga-da-Cunha M, Vertommen D, Stroobant V, Van Schaftingen E. Molecular identification of carnosine synthase as ATP-grasp domain-containing protein 1 (ATPGD1). J Biol Chem. 2010;285:9346–56. 10.1074/jbc.M109.095505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boldyrev AA, Aldini G, Derave W. Physiology and pathophysiology of carnosine. Physiol Rev. 2013;93:1803–45. 10.1152/physrev.00039.2012. [DOI] [PubMed] [Google Scholar]
- 76.Aldini G, Granata P, Carini M. Detoxification of cytotoxic alpha, beta-unsaturated aldehydes by carnosine: characterization of conjugated adducts by electrospray ionization tandem mass spectrometry and detection by liquid chromatography/mass spectrometry in rat skeletal muscle. J Mass Spectrom. 2002;37:1219–28. 10.1002/jms.381. [DOI] [PubMed] [Google Scholar]
- 77.Baguet A, Koppo K, Pottier A, Derave W. Beta-alanine supplementation reduces acidosis but not oxygen uptake response during high-intensity cycling exercise. Eur J Appl Physiol. 2010;108:495–503. 10.1007/s00421-009-1225-0. [DOI] [PubMed] [Google Scholar]
- 78.Posa DK, Miller J, Hoetker D, Ramage MI, Gao H, Zhao J, Doelling B, Bhatnagar A, Wigmore SJ, Skipworth RJE, et al. Skeletal muscle analysis of cancer patients reveals a potential role for carnosine in muscle wasting. J Cachexia Sarcopenia Muscle. 2023. 10.1002/jcsm.13258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.de Courten B, Jakubova M, de Courten MP, Kukurova IJ, Vallova S, Krumpolec P, Valkovic L, Kurdiova T, Garzon D, Barbaresi S, et al. Effects of carnosine supplementation on glucose metabolism: Pilot clinical trial. Obesity (Silver Spring). 2016;24:1027–34. 10.1002/oby.21434. [DOI] [PubMed] [Google Scholar]
- 80.Lombardi C, Carubelli V, Lazzarini V, Vizzardi E, Bordonali T, Ciccarese C, Castrini AI, Dei Cas A, Nodari S, Metra M. Effects of oral administration of orodispersible levo-carnosine on quality of life and exercise performance in patients with chronic heart failure. Nutrition. 2015;31:72–8. 10.1016/j.nut.2014.04.021. [DOI] [PubMed] [Google Scholar]
- 81.Everaert I, De Naeyer H, Taes Y, Derave W. Gene expression of carnosine-related enzymes and transporters in skeletal muscle. Eur J Appl Physiol. 2013;113:1169–79. 10.1007/s00421-012-2540-4. [DOI] [PubMed] [Google Scholar]
- 82.Derave W, Ozdemir MS, Harris RC, Pottier A, Reyngoudt H, Koppo K, Wise JA, Achten E. beta-Alanine supplementation augments muscle carnosine content and attenuates fatigue during repeated isokinetic contraction bouts in trained sprinters. J Appl Physiol. 1985;2007(103):1736–43. 10.1152/japplphysiol.00397.2007. [DOI] [PubMed] [Google Scholar]
- 83.Everaert I, Stegen S, Vanheel B, Taes Y, Derave W. Effect of beta-alanine and carnosine supplementation on muscle contractility in mice. Med Sci Sports Exerc. 2013;45:43–51. 10.1249/MSS.0b013e31826cdb68. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material 1: Fig. 1. Muscle weights after 4 and 8 weeks of sham and transverse aortic constriction (TAC) surgery. Weights of different muscle beds that were normalized to tibia length, including the gastrocnemius muscle after, (A) 4 and (E) 8 weeks, the soleus muscle after, (B) 4 and (F) 8 weeks, the tibialis anterior muscle after, (C) 4 and (G) 8 weeks, and the extensor digitorium longus (EDL) after, (D) 4 and (H) 8 weeks of sham and TAC surgeries. The data are presented as the mean ± SEM, n = 6 samples in each group. Fig. 2. Atrophy-related genes in the gastrocnemius muscle after sham and transverse aortic constriction (TAC) surgeryies. mRNA levels of Atrogin1 in the gastrocnemius muscle of C57BL/6J mice after (A) 4 and (B) 8 weeks of sham and TAC surgery. (C) mRNA levels of Atrogin 1 and Murf1 in the soleus muscle after 14 weeks of sham and TAC surgery. Data are presented as the mean ± SEM; n = 6–7 samples in each group. Fig. 3. Expression of inflammatory genes in the gastrocnemius muscle after sham and transverse aortic constriction (TAC) surgeries. TNF-α, IL-6 and IL-1β mRNA levels in the gastrocnemius muscle of C57BL/6J mice after (A) 4 and (B) 8 weeks of sham or TAC surgeries. Data are presented as the mean ± SEM, n = 6–7 samples in each group, p > 0.05 vs sham. Fig. 4. Carnosine aldehyde conjugates in the gastrocnemius muscle after sham and transaortic constriction (TAC) surgeries. Gastrocnemius muscles collected after 14 weeks of sham or TAC surgeries was analyzed by LC‒MS/MS for different carnosine aldehyde conjugates. Levels of (A) carnosine propanal, (B) carnosine HNE, (C) carnosine propanol and (D) carnosine DHN. Data are presented as the mean ± SEM, n = 9–10 samples in each group. Fig. 5. Gene expression of carnosine synthase (CARNS), human taurine transporter (TAUT) and human peptide (PEPT2) in the gastrocnemius muscle after sham and transverse aortic constriction surgeries. Relative mRNA levels of CARNS, TAUT and PEPT2 in the gastrocnemius muscle after 14 weeks of sham or TAC surgeries. Data are presented as the mean ± SEM, n = 9–10 samples in each group.
Data Availability Statement
All data generated for the manuscript will be available from the corresponding author on request.
No datasets were generated or analysed during the current study.